
A simple, efficient and selective catalyst for closed-loop recycling of PEF in situ towards a circular materials economy approach†
Shaowei
Wu
abc,
Lu
Li
 *ac,
Lei
Song
ac,
Guannan
Zhou
ac,
Lixin
Liu
ac,
Hailan
Kang
*b,
Guangyuan
Zhou
ac and
Rui
Wang
ac
*ac,
Lei
Song
ac,
Guannan
Zhou
ac,
Lixin
Liu
ac,
Hailan
Kang
*b,
Guangyuan
Zhou
ac and
Rui
Wang
ac
aDivision of Energy Materials (DNL 22), Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, 116023, China. E-mail: lilu528@dicp.ac.cn
bKey Laboratory for Rubber Elastomer of Liaoning Province, Shenyang University of Chemical Technology, Shenyang, China. E-mail: kanghailan@syuct.edu.cn
cLiaoning Key Laboratory of Speciality Polymers, Dalian, China
First published on 14th November 2024
Abstract
Developing plastics from biomass and performing chemical recycling are two essential strategies in circular materials economy. Herein, we present an innovative technique for the closed-loop, in situ chemical recycling of bio-derived poly(ethylene 2,5-furandicarboxylate) (PEF), utilizing the exceptional capabilities of monodisperse nano γ-Ga2O3 with tunable oxygen vacancy density. This framework enables seamless cycling of bio-based plastics from polymerization to de-polymerization and re-polymerization, promoting a sustainable polymer economy. The introduction of oxygen vacancy defects in the structure of gallium oxide, a low toxicity and transparent metal oxide, is considered to be an effective strategy for improving catalytic activity. The polymerization process was controlled by using novel oxygen vacancy-defective Ga2O3, which catalyzed the reaction between bio-based 2,5-furandicarboxylic acid and ethylene glycol to produce high molecular weight PEF. (Mn = 41 kg mol−1). This PEF can then undergo efficient in situ glycolysis, achieving complete de-polymerization under moderate conditions without the need for external catalysts. The glycolysis derivatives of PEF can be directly re-polymerized to polyester rPEF, achieving a significant molecular weight (Mn = 43 kg mol−1) and a remarkable yield (93%). Notably, γ-Ga2O3 with nano oxygen vacancy defects exhibits the ability to selectively de-polymerize PEF within composite material systems containing commercial PET. This research highlights the significant utility of a green catalyst in in situ closed-loop recycling processes.
Introduction
Increasing waste volumes and greenhouse gas emissions have demonstrated that the current linear plastics economy, characterized by extraction, usage, and disposal, compromises global sustainability and circularity goals.1 Enhancing the circularity of plastics is thus essential for transitioning to a future centered on sustainable materials. This necessitates developing effective recycling and reuse strategies. Among these, chemical recycling, which transforms waste polymers into valuable chemicals through processes like solvent decomposition or chemical breakdown, has garnered significant attention.2 To date, chemical recycling strategies have been widely applied to polyester plastics.3–9 These strategies not only provide an effective means to mitigate environmental pollution from plastic waste but also offer a new way to recover valuable chemicals,10–14 thus creating a recycling process aligned with the vision of circular plastics economy.15–18Polyesters, characterized by their polar main-chain ester bonds, already play a crucial role in today's polymer plastics.19,20 They are poised to become even more significant in a future sustainability-focused circular plastics economy21 due to their ability to undergo chemical or biological cleavage of the main-chain ester bond on demand, offering enhanced recycling options for discarded plastics.22,23 In addition to chemical recycling, developing polymer materials derived from biomass resources represents another effective strategy for achieving sustainable resource utilization as part of a sustainable economy. Among the promising bio-based polymers, poly(ethylene 2,5-furandicarboxylate) (PEF) stands out due to its low permeability,24 high-performance properties25 and compatibility with existing processing and recycling infrastructures.26 Thus, PEF is expected to be a more sustainable alternative to traditional fossil-based aromatic polyesters such as PET.27,28
The preparation and recycling of PEF are crucial for its future applications. A range of catalysts have been reported to support PEF production, including tetrabutyl titanate,29,30 titanium(IV) isopropoxide,31 tin(II) and alkyl tin(IV) salts,26,32 antimony oxide,33 zinc acetate and aluminum acetylacetonate.34 Although effective, these catalysts have notable drawbacks. Titanium-based catalysts exhibit poor stability and produce darker-colored products, while organotin compounds are highly toxic, limiting a material's further application in biomedicine and applications involving food contact.35,36 Among various catalysts, metal oxide catalysts are extensively utilized in transesterification reactions due to their high catalytic activity, stability, and strong resistance to hydrolysis. Research has demonstrated that the acidic sites on metal oxide catalysts, such as metal cations, can readily interact with hydroxyl oxygen and ester bond oxygen to form multi-ring structures. These multi-ring structures exhibit strong coordination capabilities, which can redistribute the charges on their groups, rendering the ester carbonyl carbon positively charged. This positive charge accelerates nucleophilic reactions between groups, thereby enhancing the reaction rate.37 Oxygen vacancy defects, a common type of point defect, are widely present in metal oxides. They influence the local geometric and electronic structure of the material, generating acidic sites that serve as numerous catalytic active sites. By modulating the density of oxygen vacancy defects on the surface of metal oxides, it is possible to quantitatively control the number of surface acidic sites, thereby regulating the activity of polymerization reactions. Thus, the abundant acidic sites and oxygen vacancy defects on the surface of metal oxides offer a promising means for achieving efficient synthesis and de-polymerization of PEF.
Research on chemical recycling strategies for PEF is increasing annually, with processes such as methanolysis,6,38 glycolysis39,40 and hydrolysis41,42 being reported. Among these, glycolysis is favored due to its mild reaction conditions and the ease of separating the target product bis(hydroxyethyl)-2,5-furandicarboxylate (BHEFDC), which can be used for re-polymerization. Gabirondo et al.39 reported the glycolysis of commercial PEF (Mn = 12.4 kg mol−1) using an organocatalyst system (DBU![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :benzoic acid = 1:1) at 180 °C with 5 wt% catalyst, achieving 92% conversion to BHEFDC and some BHEFDC oligomers in 5 h. The purified BHEFDC was re-polymerized via melt polycondensation and solid-state polymerization, producing rPEF with Mn = 11.2 kg mol−1. Agostinho et al.40 used a eutectic solvent (DES, urea and Zn(OAc)2 = 4:1) to recycle low molecular weight PEF (catalyzed by titanium butoxide, viscosity 0.268 dl g−1). At 180 °C with 2 wt% DES, 85% glycolysis conversion to BHEFDC was achieved in 1 hour. Without purification, rPEF (viscosity 0.409 dl g−1) was obtained via direct re-polymerization using vacuum and heat, with DES still present. However, current de-polymerization studies of polyesters typically require substantial additional amounts of specific catalysts, which hinder catalyst recycling within the closed-loop system, complicate the overall process, and lead to increased costs. This issue is likely due to the disparity in activity and selectivity between the catalysts used for polyester synthesis and those used for de-polymerization.
:benzoic acid = 1:1) at 180 °C with 5 wt% catalyst, achieving 92% conversion to BHEFDC and some BHEFDC oligomers in 5 h. The purified BHEFDC was re-polymerized via melt polycondensation and solid-state polymerization, producing rPEF with Mn = 11.2 kg mol−1. Agostinho et al.40 used a eutectic solvent (DES, urea and Zn(OAc)2 = 4:1) to recycle low molecular weight PEF (catalyzed by titanium butoxide, viscosity 0.268 dl g−1). At 180 °C with 2 wt% DES, 85% glycolysis conversion to BHEFDC was achieved in 1 hour. Without purification, rPEF (viscosity 0.409 dl g−1) was obtained via direct re-polymerization using vacuum and heat, with DES still present. However, current de-polymerization studies of polyesters typically require substantial additional amounts of specific catalysts, which hinder catalyst recycling within the closed-loop system, complicate the overall process, and lead to increased costs. This issue is likely due to the disparity in activity and selectivity between the catalysts used for polyester synthesis and those used for de-polymerization.
In this work, we present a novel in situ closed-loop recycling strategy for bio-based PEF, leveraging the potential of monodisperse γ-Ga2O3 with tunable oxygen vacancy defect density as an innovative and highly efficient green catalyst. The chemistry described here is inspired by the catalytic versatility of Ga2O3, which includes its low toxicity,43–48 transparent properties,49 and low Ga–O bond dissociation energy, facilitating the formation of controllable oxygen-vacancy defects and potential catalytic activity.50 This catalytic system enables controlled polymerization and efficient de-polymerization of PEF in a continuous cycle, whilst promoting its re-polymerization into rPEF. Experimental results and DFT calculations confirm the feasible reaction pathways for both PEF synthesis and glycolysis on the catalyst surface. Additionally, under this strategy, PEF can also undergo selective glycolysis within mixed polyester systems, leading to a truly practical sustainable circular economy approach.
Results and discussion
Synthesis and characterization of nano γ-Ga2O3 with oxygen vacancy defects
We synthesized sub-stable Ga2O3via thermal injection and calcination using anhydrous gallium chloride, ethylene glycol, and deionized water (Fig. 1A). The experimental details are described in the ESI.† This method is simpler, more cost-effective, and more suitable for large-scale industrial production compared to hydrothermal methods. X-ray powder diffraction (XRD) was performed on Ga2O3 catalyst samples synthesized via 3 h, 6 h, and 12 h reflux reactions to study their crystal structures. As depicted in Fig. 1B, all Ga2O3 catalysts exhibited similar XRD patterns, with characteristic diffraction peaks at 36.2° and 64.1°, corresponding to the (311) and (440) crystals of cubic spinel-type γ-Ga2O3 with defects (JCPDS#20-0426).51 This similarity suggests that the duration of the reflux reaction does not significantly affect the crystal structure. The broad, well-defined peaks indicate that the synthesized γ-Ga2O3 exhibits low crystallinity and consists of small-sized particles or very tiny microcrystals. The solution exhibited a Tyndall effect under UV light at the reaction endpoint (Fig. S1 in the ESI†), indicating the presence of small γ-Ga2O3 particles. To study nanoparticle morphology, transmission electron microscopy (TEM) was employed for direct observation. TEM images (Fig. 1C–E) reveal that γ-Ga2O3-n, where n represents the reflux reaction time in hours, is monodisperse and granular with irregular edges, with particle sizes ranging from a few nanometers to several dozen nanometers.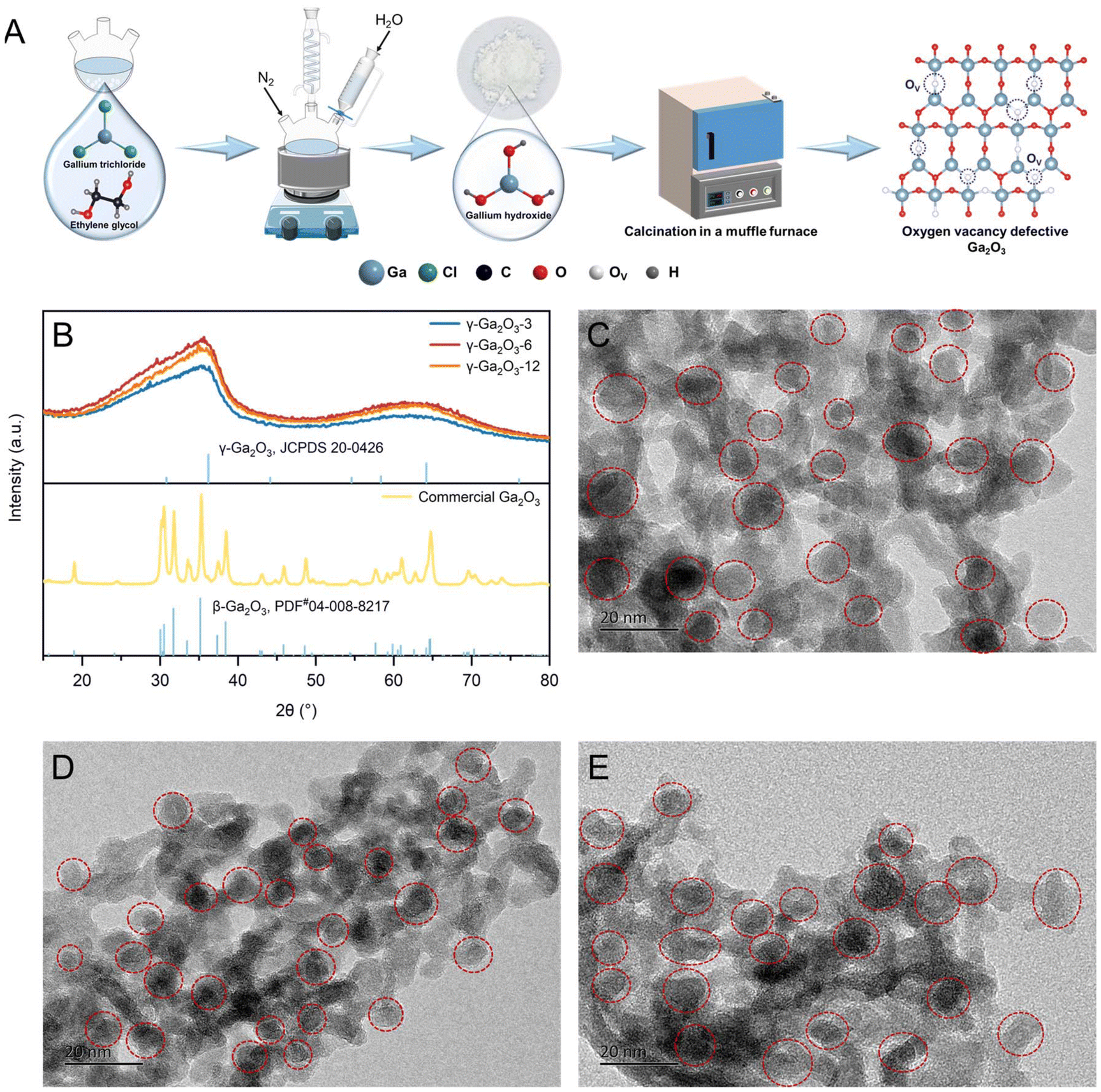 | ||
| Fig. 1 (A) Preparation process of Ga2O3 with oxygen vacancy defects; (B) XRD patterns of γ-Ga2O3-n and commercial Ga2O3 catalysts (n represents the reflux reaction time in hours); and TEM images of (C) γ-Ga2O3-3, (D) γ-Ga2O3-6, and (E) γ-Ga2O3-12. | ||
Oxygen vacancies in the γ-Ga2O3 samples were detected during synthesis via electron paramagnetic resonance (EPR) analysis at low temperatures. The strong EPR signals in γ-Ga2O3-n samples at g = 2.003 are characteristic of oxygen vacancies with trapped electrons (Fig. 2C).52 A stronger characteristic signal of single-electron captured oxygen vacancies (OV) was noted in γ-Ga2O3-6 compared to γ-Ga2O3-3 and γ-Ga2O3-12. However, γ-Ga2O3-3 and γ-Ga2O3-12 exhibited similar EPR intensity levels.
 | ||
| Fig. 2 (A) O 1s spectra, (B) Ga 2p spectra, (C) EPR spectra of γ-Ga2O3-n and β-Ga2O3 catalysts and (D) NH3-TPD profiles of γ-Ga2O3-n catalysts (n represents the reflux reaction time in hours). | ||
We analyzed catalyst samples for oxygen, defects, and metal states using X-ray photoelectron spectroscopy (XPS). Distinct Ga 2p and O 1s signals were identified in the catalyst samples, with no impurity peaks detected. The XPS spectra of O 1s (Fig. 2A) in the γ-Ga2O3-n samples can be split into three peaks: lattice oxygen (OL, 530.6 eV), vacant oxygen (OV, 532.2 eV), and hydroxyl oxygen (OOH−, 531.5 eV).53 The peak area ratio of OV to OL in γ-Ga2O3-6 is 0.59, which is higher than those in γ-Ga2O3-3 (0.36) and γ-Ga2O3-12 (0.41) (Table S1 in the ESI†). This indicates a higher concentration of oxygen vacancy defects in γ-Ga2O3-6, which aligns well with the results from the EPR spectra. In addition, the ratios of OOH− to OV in γ-Ga2O3 samples—specifically, γ-Ga2O3-3 (0.75), γ-Ga2O3-6 (0.38), and γ-Ga2O3-12 (0.60) —exhibit a distinct trend. This variation indicates a decrease in the OOH− content and a substantial increase in OV, suggesting that thermal treatment induces a dehydroxylation reaction, leading to the formation of oxygen-vacancy defects within the gallium oxide structure. However, no information on the OV was observed in either the XPS or EPR spectrum of the commercial Ga2O3 sample, suggesting that the commercial Ga2O3 has no capacity for bearing surface OV (Fig. 2A and C). This leads to its poor catalytic performances. Furthermore, the OV peak of the γ-Ga2O3-n material with oxygen vacancy defects exhibits a considerable blue-shift in its binding energy compared to β-Ga2O3 with oxygen vacancy-free defects (Fig. 2A). This shift can be attributed to the significant influence of oxygen vacancies on the electron density and chemical potential of the OL sites.54 The interaction between the OV and the surrounding gallium and oxygen atoms alters the electronic properties of the material, resulting in the observed blue-shift in the OL peak's binding energy. This alteration in binding energy can significantly impact the catalytic activity, as it is directly related to the ease of electron transfer in chemical reactions. Additionally, the observed blue-shift indicates that the γ-Ga2O3-n material with oxygen vacancy defects may exhibit enhanced electron-attracting properties and acid strength compared to its counterparts, making it a promising candidate as a catalyst for polyester synthesis.
The Ga 2p3/2 and Ga 2p1/2 XPS peaks of β-Ga2O3 are centered at binding energies of 1117.6 eV and 1144.4 eV, respectively (Fig. 2B), which is characteristic of Ga3+–O bonds in Ga2O3.55 In contrast, the Ga 2p XPS spectra of γ-Ga2O3−x with non-stoichiometric defects exhibit different features, where the Ga 2p3/2 and Ga 2p1/2 peaks shift to higher binding energies at 1118.0 eV and 1144.8 eV, respectively, representing a shift of 0.4 eV (Table S2 in the ESI†). Upon conducting a thorough analysis, it is reasonable to deduce that the positive shift in the Ga 2p energy level is attributable to the significant interplay between Ga3+ and oxygen vacancies. This interaction pattern is analogous to the lattice contraction induced by oxygen vacancies in titanium dioxide (TiO2). Specifically, in TiO2 crystals, the presence of oxygen vacancies induces a contraction in the lattice structure, facilitated by the strong interaction between oxygen vacancies and Ti4+ ions.56–59 This lattice adjustment subsequently influences the catalytic properties of the material, including photocatalytic efficiency and electron mobility. Analogously, in γ-Ga2O3, the removal of an O atom causes the nearest Ga atoms to relax away from the vacancy, strengthening their bonding with the surrounding lattice. This outward relaxation decreases the overlap of the Ga dangling bonds and shortens the Ga–O bond length. The Ga3+–O bond binding energy increases to maintain the crystal structure and stability, potentially impacting the material's catalytic performance. Therefore, a thorough investigation into the mechanism behind the Ga 2p level shift is crucial for enhancing the performance and application potential of Ga2O3 catalysts.
The surface acidity of the γ-Ga2O3 catalyst was measured using temperature-programmed desorption of NH3 (NH3-TPD). All three γ-Ga2O3-n catalysts exhibited two distinct NH3 desorption peaks. The first peak, observed in the low temperature range (298 K to 473 K), corresponded to weak acid centers. The second peak, detected in the medium temperature range (473 K to 673 K), indicated medium–strong acidic centers (Fig. 2D). This suggests that γ-Ga2O3-n catalysts possess both weak and medium–strong acidic sites on their surfaces. The surface acidity of the γ-Ga2O3 catalysts can be effectively controlled and adjusted by varying the duration of the reflux reaction (Table S3 in the ESI†), and the total acidic site abundance followed a specific order: Ga2O3-6 > Ga2O3-12 > Ga2O3-3. The highest concentration of surface acidic sites in Ga2O3-6 is attributed to its robust capacity for single-electron captured oxygen vacancies, as predicted in our previous analyses.
A thermal gravimetric analyzer (TGA) was used to assess the thermal stability of the catalysts by measuring the percentage of mass loss at high temperatures (Fig. S2 in the ESI†). The TGA curves of the three catalysts show minimal mass loss between 30 °C and 600 °C, indicating that the nano γ-Ga2O3 catalysts with defects exhibit high thermal stability below 600 °C with negligible heat loss. Notably, the TGA curves display a sharp decline after 600 °C, corresponding to the phase transition from γ-Ga2O3 to β-Ga2O3.60 These results demonstrate that the synthesized γ-Ga2O3 catalysts maintain stability under the thermal conditions required for polyester synthesis.
In situ catalytic PEF closed-loop chemical recovery performance
The γ-Ga2O3-n catalysts were employed for the polymerization of renewable 2,5-furandicarboxylic acid (FDCA) and ethylene glycol (EG) through a direct esterification method that involves a two-step process: esterification followed by polycondensation. The esterification was performed under a nitrogen atmosphere at temperatures of 190 °C for 1 h, 200 °C for 1 h, and 210 °C for 1 h, respectively. Subsequently, the polycondensation was conducted at 240 °C for 3 hours under high vacuum to yield PEF. The results of γ-Ga2O3-6 with oxygen-vacancy defects and the conventional polyester catalyst tetrabutyl titanate (TBT) in synthesizing PEF are shown in Fig. 3A and B and Table 1. The 1H NMR spectra of PEFs revealed chemical shifts at 4.69 ppm (c) and 4.17 ppm (d), attributed to the methylene proton in the diethylene glycol furandicarboxylate (DEGF) unit of PEF (Fig. 3A). This unit, a by-product of etherification side reactions involving EG or hydroxyethyl ester end-groups, has its molar fraction (φDEGF) calculated accordingly (eqn (1)). Consequently, the catalysts γ-Ga2O3-6 and TBT can be found to exhibit similar selectivity (φDEGF, 4.90% vs. 4.40%) in catalyzing the PEF synthesis reaction. The FTIR spectrum shown in Fig. 3C confirmed the expected PEF structure, with characteristic peaks evident for the furan's![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH stretching at 3116 cm−1, the ester carbonyl CO stretching vibration at 1720 cm−1, and the C–O stretching of the ester carbonyl group at 1265 cm−1. Differential scanning calorimetry (DSC) and TGA results (Table 1 and Fig. S4 in the ESI†) indicated that the thermal properties of PEF synthesized using γ-Ga2O3-6 as the catalyst, such as the glass transition temperature (Tg), 5% degradation temperature (Td-5%), and maximum degradation temperature (Td-max), are comparable to those of PEF synthesized using TBT as the catalyst. It is noteworthy that PEF catalyzed by γ-Ga2O3-6 exhibited lower yellowness and higher transmittance (Fig. 3D), suggesting that γ-Ga2O3-6 not only achieves slightly better catalytic activity compared to TBT but also significantly ameliorates the yellow-brown coloration issue of PEF products, resulting in a product with enhanced optical properties.
CH stretching at 3116 cm−1, the ester carbonyl CO stretching vibration at 1720 cm−1, and the C–O stretching of the ester carbonyl group at 1265 cm−1. Differential scanning calorimetry (DSC) and TGA results (Table 1 and Fig. S4 in the ESI†) indicated that the thermal properties of PEF synthesized using γ-Ga2O3-6 as the catalyst, such as the glass transition temperature (Tg), 5% degradation temperature (Td-5%), and maximum degradation temperature (Td-max), are comparable to those of PEF synthesized using TBT as the catalyst. It is noteworthy that PEF catalyzed by γ-Ga2O3-6 exhibited lower yellowness and higher transmittance (Fig. 3D), suggesting that γ-Ga2O3-6 not only achieves slightly better catalytic activity compared to TBT but also significantly ameliorates the yellow-brown coloration issue of PEF products, resulting in a product with enhanced optical properties.| φDEGF = Ad/(Ad + Ab) × 100% | (1) |
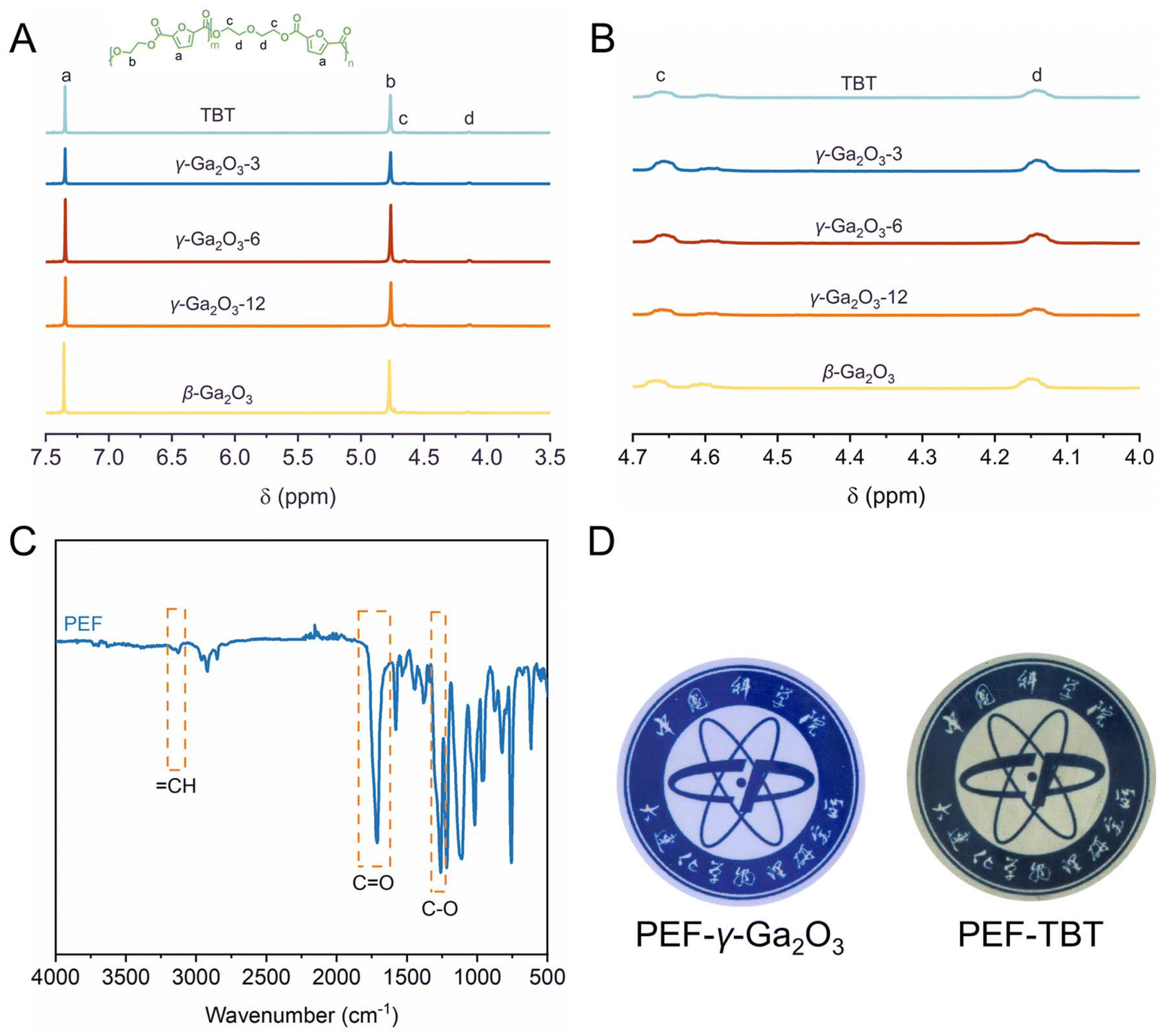 | ||
| Fig. 3 (A) 1H NMR of PEFs catalyzed by TBT, nano γ-Ga2O3-n and β-Ga2O3 with oxygen-vacancy defects (the inset is a structural formula of PEF); (B) 1H NMR local magnification (4.7–4.0 ppm) of PEFs; (C) FTIR spectrum of PEF catalyzed by nano γ-Ga2O3 with oxygen-vacancy defects; and (D) appearance of the synthesized PEFs catalyzed by γ-Ga2O3 and TBT, respectively. | ||
| Samples | Catalysts | Concentration of catalyst (ppm) | M n (kg mol−1) | Đ | φ DEGF (%) | T g ( °C) | T d-5% ( °C) | T d-max ( °C) | Yield (%) |
|---|---|---|---|---|---|---|---|---|---|
|
a Reaction conditions: transesterification stage, under a nitrogen atmosphere at temperatures of 190 °C for 1 h, 200 °C for 1 h, and 210 °C for 1 h, respectively; polycondensation stage, 240 °C for 3 h under high vacuum; molar ratio of EG to FDCA was 1.7:1.
b
Đ: polydispersity index of PEFs determined by gel permeation chromatography.
c Diethylene glycol furandicarboxylate (DEGF) unit content in PEF determined by 1H NMR and calculated by using eqn (1).
d TBT: traditional polyester catalyst tetrabutyl titanate.
e β-Ga2O3: commercial gallium oxide with oxygen vacancy-free defects.
|
|||||||||
| PEF-1 | TBTd | 1600 | 38 | 1.77 | 4.40 | 86.3 | 375.5 | 417.3 | 87.5 |
| PEF-2 | γ-Ga2O3-3 | 880 | 35 | 1.60 | 5.88 | 84.6 | 370.6 | 425.6 | 85.3 |
| PEF-3 | γ-Ga2O3-6 | 880 | 41 | 1.79 | 4.90 | 86.0 | 374.1 | 424.4 | 88.1 |
| PEF-4 | γ-Ga2O3-12 | 880 | 39 | 1.81 | 5.37 | 85.8 | 370.2 | 424.1 | 85.1 |
| PEF-5 | β-Ga2O3e | 880 | 24 | 1.46 | 6.00 | 80.0 | 373.5 | 418.0 | 83.0 |
To investigate the relationship between the density of oxygen vacancy defects in γ-Ga2O3 and its catalytic activity, we compared the performance of γ-Ga2O3-n catalysts with commercial β-Ga2O3, which lacks oxygen-vacancy defects (Fig. 1B, 2A and C). All γ-Ga2O3-n catalysts demonstrated activity (Fig. 3A, B and Table 1). The number average molecular weights (Mn) of PEF catalyzed by γ-Ga2O3-3, γ-Ga2O3-6, and γ-Ga2O3-12 were 35 kg mol−1, 41 kg mol−1, and 39 kg mol−1, respectively. This trend aligns with the initial increase and subsequent decrease in the density of surface oxygen vacancy defects and the concentration of surface acidic sites on γ-Ga2O3, demonstrating a positive correlation between these two characteristics of the catalyst and its catalytic activity, as previously predicted. Notably, the Mn of PEF catalyzed by β-Ga2O3 was significantly lower than that achieved by γ-Ga2O3-n catalysts, attributable to the lower surface acidity and absence of oxygen vacancies in β-Ga2O3 (Fig. 2A and Table S3 in the ESI†). This result suggests that oxygen-vacancy defects can indeed increase the number of acidic sites on nano-oxides, thereby enhancing catalytic activity. Among the γ-Ga2O3-n catalysts, γ-Ga2O3-6, which is monodisperse and has the highest surface acidity, exhibits the best catalytic activity.
In the catalytic glycolysis study, we conducted an in situ glycolysis of PEF, synthesized using the γ-Ga2O3-6 catalyst. This approach obviates the requirement for additional catalysts during the glycolysis of the polyester material in ethylene glycol solution, enhancing process efficiency. We discovered that in situ glycolysis of PEF can be efficiently facilitated by using trace amounts of the nano γ-Ga2O3 catalyst with residual oxygen-vacancy defects in PEF at 180 °C for 2 h under atmospheric pressure. A comparison of the reaction solution before and after glycolysis reveals that the PEF powder/glycol mixture became clear and transparent after 2 h of reaction, indicating the near-complete de-polymerization of PEF powder (Fig. S6 in the ESI†). To determine the degradation efficiency, we analyzed the original solution after the glycolysis reaction using high-performance liquid chromatography (HPLC). Fig. S10† confirms that trace amounts of γ-Ga2O3 (829 ppm, see Table S5†) facilitated the complete glycolysis of PEF, resulting in a yield of 75.7% bis(hydroxyethyl)-2,5-furandicarboxylate (BHEFDC) and 24.3% BHEFDC dimer.
The de-polymerization product was further processed by cooling and filtering twice, resulting in the separation of two distinct solids: solid I and solid II. In particular, upon completion of the glycolysis reaction, the mixture was rapidly cooled to quench the reaction. The resulting precipitate was stored at −10 °C for a period of time, then filtered and washed with deionized water to obtain solid I, and solid I can be completely dissolved in DMSO (Fig. S7 in the ESI†). The filtrate was then concentrated using a rotary evaporator and stored at −10 °C, and the subsequent precipitate was filtered and washed to obtain solid II. In addition, the absence of 1H NMR signals corresponding to PEF, coupled with the appearance of new singlet peaks at δ 4.60 ppm and 4.14 ppm attributed to the methylene protons of the terminal group derived from ethylene glycol (Fig. 4A), indicates complete de-polymerization of PEF. The signal in the 1H NMR spectrum at δ 4.74–4.80 ppm corresponds to the chemical shifts of four protons associated with the two methylene groups located at the center of the 2,5-furandicarboxylate dimer, indicating that solid I is composed of oligomers. The average degree of solid I was determined to be 2, based on the peak area in the 1H NMR spectrum (Fig. 4A). In contrast, the 1H NMR peak distribution of solid II resembles that of solid I. However, based on NMR calculations, the average degree of polymerization for solid II is found to range between 1 and 2. The glycolysis products of PEF were further evaluated in the ESI, with HPLC-MS results presented in Fig. S8 and S9, and summarized in Table S4 in the ESI.† While this work primarily focuses on developing a new strategy for in situ closed-loop recovery of PEF, the recovery of BHEFDC following glycolysis was also achieved through simple refrigeration and filtration procedures. These findings further underscore the exceptional efficiency of γ-Ga2O3 with defects in the in situ glycolysis of PEF.
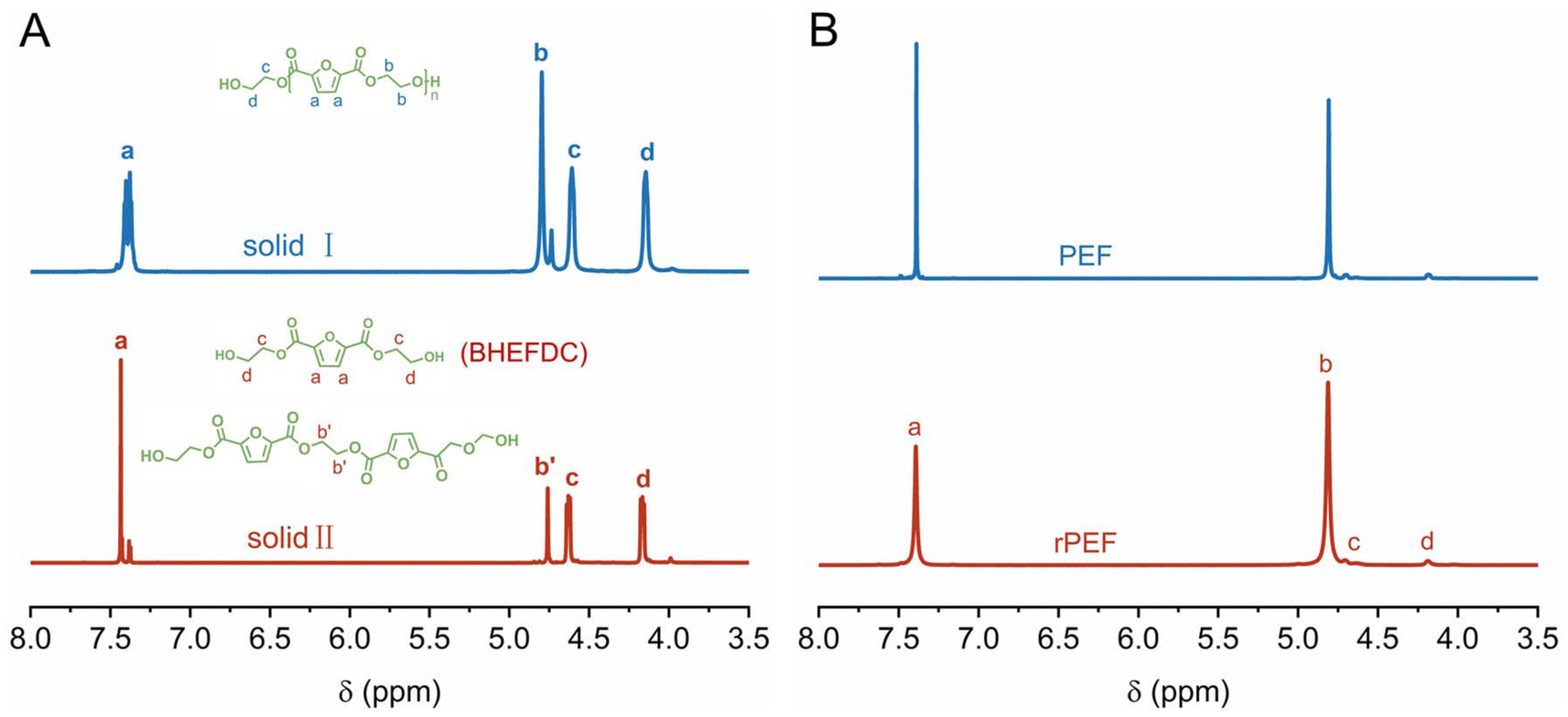 | ||
| Fig. 4 (A) 1H NMR spectra of PEF glycolytic products of solid I and solid II and (B) 1H NMR spectra of the rPEF and PEF. | ||
Given the excellent performance of γ-Ga2O3 with defects in catalyzing both PEF polymerization and glycolysis, we utilized it for the in situ re-polymerization recycling of PEF to its original polymer form (rPEF). We conducted the glycolysis reaction of ground PEF (2 g) with EG (8 g) in a three-necked round-bottomed flask. Without isolating the main intermediate glycolysis product, bis(hydroxyethyl)-2,5-furandicarboxylate (BHEFDC), and removing the γ-Ga2O3 catalyst, we utilized the unpurified reaction mixture and the residual of γ-Ga2O3 to catalyze the polymerization of BHEFDC to rPEF. The re-polymerized product was identified as PEF, exhibiting a promising Mn of 43 kg mol−1 (Fig. 4B and Table S6 in the ESI†). Additionally, the thermal properties of rPEF show no degradation and are nearly identical to those of the original PEF polymer (Fig. S11 and Table S4 in the ESI†). The efficacy of this innovative in situ closed-loop recycling method is demonstrated by its seamless transitions from polymerization to de-polymerization and subsequent re-polymerization.
Given that PET is a major plastic present in waste streams and subject to recycling, the ability to efficiently separate and recycle PEF from PET is crucial to prevent cross-contamination of either recycling stream. Equal amounts of commercial PET and PEF synthesized using γ-Ga2O3-6 were mixed and subjected to glycolysis under the conditions of 180 °C and atmospheric pressure, for 2 h. Notably, PEF can be depolymerized completely to its monomer, while PET is retained as large white particles. The PET granules were collected, rinsed with deionized water and anhydrous ethanol, and then dried under vacuum at 60 °C for 12 h (Scheme 1). Post-reaction gravimetric measurements indicated no significant mass change in PET, confirming its resistance to de-polymerization by the γ-Ga2O3-6 catalyst under the specified glycolysis conditions. In contrast, PEF was completely depolymerized, as evidenced by the total disappearance of PEF particles from the reaction mixture. The distinct reactivity difference between PEF and PET under identical glycolysis conditions highlights the selective catalytic efficiency of γ-Ga2O3 with defects for the PEF glycolysis reaction. The unique catalytic properties of oxygen vacancy-deficient γ-Ga2O3 offer a viable solution to enhance the sustainability of complex polymer system recycling processes.
 | ||
| Scheme 1 Glycolysis process of PEF catalyzed by nano γ-Ga2O3 with defects and a commercial PET polyester mixture. | ||
Density functional theory (DFT) calculation on the glycolysis reaction of PEF
To investigate the glycolysis pathway of PEF in the presence of the γ-Ga2O3 catalyst with oxygen-vacancy defects, we constructed a model of nano-Ga2O3 surfaces with oxygen-vacancy defects using DFT calculations (Fig. S12 in the ESI†). Ethane-1,2-diyl bis(furan-2-carboxylate) (EDBFC) was used as model molecules to simplify the calculation process (Fig. S13 in the ESI†). As depicted in Fig. 5, EDBFC initially approaches the catalyst surface to form *C12H10O6 (steps A1 to B1), with an adsorption energy of −42.49 kJ mol−1. Subsequently, *C2H6O2 forms on the catalyst surface through the interaction with ethylene glycol (steps B1 to C1), with an adsorption energy of −52.12 kJ mol−1. During steps C1 to D1, a nucleophilic substitution reaction occurs on the surface of the nano-catalyst with defects, reinforced by the coordination between the strongly acidic Ga3+ ions and the carbonyl oxygen of the ester bond in the EDBFC. This facilitates the nucleophilic attack of EG on the carbonyl carbon, forming a tetrahedral transition state (D1) from TS 1. The departure of Ga facilitates the protonation of the carbonyl group in TS 2, leading to the formation of 2-hydroxyethyl carboxyfuran (steps D1 to E1). The energy barrier for the formation of TS 1 is 146.56 kJ mol−1, while that for TS 2 is 110.53 kJ mol−1, indicating that the formation of TS 1 is the rate-determining step of the reaction. Ga3+ ions located at the oxygen vacancy defects on the nano-sized Ga2O3 surfaces serve as the active sites for catalyzing the glycolysis reaction of PEF. The formation of these active sites is further enhanced by electrostatic interactions between the Ga3+ ions and the oxygen-deficient sites. This conclusion is derived from a detailed analysis of the electronic structure and bonding configurations altered by the presence of these ions on the nanostructured material surfaces. The intentionally created oxygen vacancy defects on the Ga2O3 surfaces provide a unique environment where gallium ions are more readily available to interact with reactant molecules, creating a favorable microenvironment for the adsorption and activation of PEF molecules, which enhances the affinity of Ga3+ to PEF glycolytic substrates, facilitating the chemical transformations required for the breakdown of PEF into simpler compounds. The same mechanism explains the pathway of Ga2O3 in catalyzing the glycolysis of PET (see Fig. S14 and S15 in the ESI†) and its high activity in the catalytic polymerization of PEF (see Fig. S16 and S17 in the ESI†). Finally, based on DFT calculations, it was found that the adsorption energy of H2O on Ga2O3 in the reaction system is 0.302 eV, indicating that the likelihood of adsorption is low and confirming that the interaction between H2O and Ga is sufficiently weak, implying that water will not poison the reaction medium. Additionally, the adsorption energies of Ga2O3 with oxygen in furan, hydroxyl oxygen in EG, and carbonyl oxygen are −0.148 eV, −0.305 eV, and −0.583 eV, respectively (see Table S7 in the ESI†), indicating that carbonyl oxygen has the strongest affinity. These results further support our proposed mechanism, suggesting that the CO–Ga interaction is the most favorable.
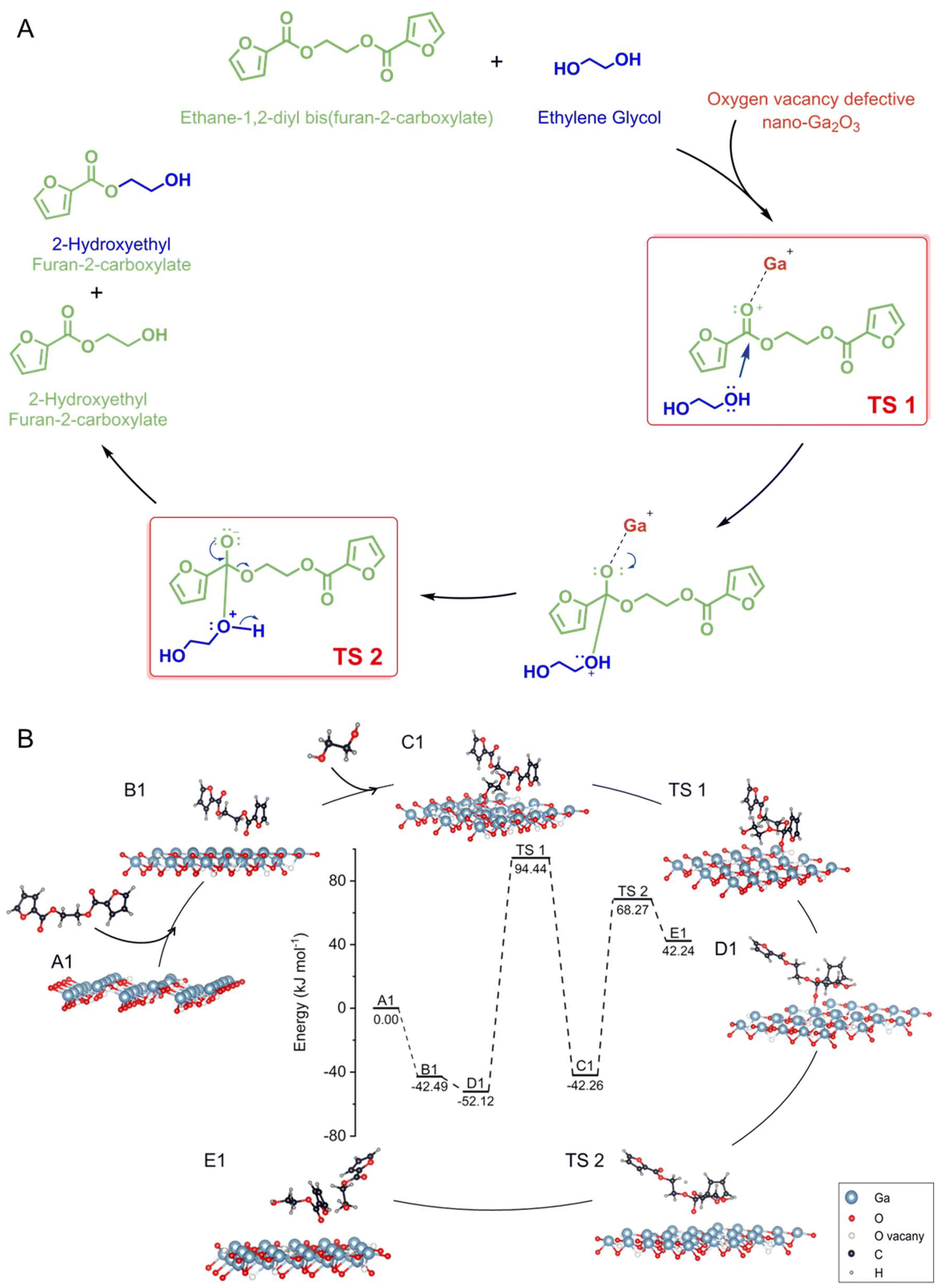 | ||
| Fig. 5 (A) Modelled activation mechanism for the glycolysis of PEF, using ethane-1,2-diyl bis(furan-2-carboxylate) to mimic PEF, ethylene glycol as a nucleophile, and nano-Ga2O3 with oxygen-vacancy defects as the catalyst, yielding 2-hydroxyethyl carboxyfuran as products; (B) energy curve and reaction structure formula of the ethane-1,2-diyl bis(furan-2-carboxylate) glycolysis reaction pathway on the nano-Ga2O3 surface with oxygen-vacancy defects. | ||
Conclusion
In this study, we have demonstrated that Ga2O3 monodisperse nanoparticles with oxygen vacancy defects γ- can facilitate an innovative in situ closed-loop chemical recycling process for bio-based PEF. This process achieves a seamless transition from catalytic polymerization to de-polymerization and re-polymerization. Modulating the oxygen vacancy density of γ-Ga2O3 enables highly active, controllable polymerization reactions, resulting in satisfactory PEF appearance. We designed an in situ-assisted glycolysis reaction using γ-Ga2O3 nanoparticles, achieving 100% conversion of PEF into monomers that meet the purity standards required for polymer synthesis. This process eliminates the need for additional specific catalysts or energy-intensive purification steps. Furthermore, the removal of γ-Ga2O3 is unnecessary during the transition from PEF to rPEF. The integration of this strategy with the non-toxic γ-Ga2O3 green catalyst aligns seamlessly with circular plastics economy goals and exhibits potential applicability to other polyesters. Notably, these nanoparticles can selectively catalyze the glycolysis of PEF in mixed plastics streams, paving new pathways for catalyst design and sustainable polymer recycling. This advancement significantly promotes circular economy within the polymer industry.Author contributions
Shaowei Wu: conceptualization, data curation, formal analysis, investigation, methodology, validation, visualization, writing – original draft, and writing – review and editing. Lu Li: funding acquisition, resources, supervision, and writing – review and editing. Lei Song: investigation, visualization, and writing – review and editing. Guannan Zhou: investigation and writing – review and editing. Lixin Liu: validation, and writing – review and editing. Hailan Kang: visualization and writing – review and editing. Rui Wang: conceptualization, funding acquisition, project administration, resources, supervision, and writing – review and editing. Guangyuan Zhou: funding acquisition, project administration, and resources.Data availability
All data generated or analyzed during this study are included in this published article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Key Research and Development Programme of China (2022YFC2105800), the Science and Technology Programme of Liaoning Province (No. 2022JH24/10200024) and the Medical-Industrial Joint Innovation Fund (No. DMU-1 & DICP UN202210).References
- F. Vidal, E. R. van der Marel, R. W. F. Kerr, C. McElroy, N. Schroeder, C. Mitchell, G. Rosetto, T. T. D. Chen, R. M. Bailey, C. Hepburn, C. Redgwell and C. K. Williams, Nature, 2024, 626, 45–57 CrossRef CAS.
- C. Shi, E. C. Quinn, W. T. Diment and E. Y. X. Chen, Chem. Rev., 2024, 124, 4393–4478 CrossRef CAS.
- Q. Dong, A. D. Lele, X. Zhao, S. Li, S. Cheng, Y. Wang, M. Cui, M. Guo, A. H. Brozena, Y. Lin, T. Li, L. Xu, A. Qi, I. G. Kevrekidis, J. Mei, X. Pan, D. Liu, Y. Ju and L. Hu, Nature, 2023, 616, 488–494 CrossRef CAS.
- S. Zhang, Q. Hu, Y. Zhang, H. Guo, Y. Wu, M. Sun, X. Zhu, J. Zhang, S. Gong, P. Liu and Z. Niu, Nat. Sci., 2023, 6, 965–973 Search PubMed.
- F. Wu, Y. Wang, Y. Zhao, M. Tang, W. Zeng, Y. Wang, X. Chang, J. Xiang, B. Han and Z. Liu, Sci. Adv., 2023, 9, eade7971 CrossRef CAS PubMed.
- X. Qu, G. Zhou, R. Wang, B. Yuan, M. Jiang and J. Tang, Green Chem., 2021, 23, 1871–1882 RSC.
- S. Westhues, J. Idel and J. Klankermayer, Sci. Adv., 2018, 4, eaat9669 CrossRef CAS PubMed.
- L. Monsigny, J.-C. Berthet and T. Cantat, ACS Sustainable Chem. Eng., 2018, 6, 10481–10488 CrossRef CAS.
- Q. Wang, X. Yao, Y. Geng, Q. Zhou, X. Lu and S. Zhang, Green Chem., 2015, 17, 2473–2479 RSC.
- L. T. J. Korley, T. H. Epps, B. A. Helms and A. J. Ryan, Science, 2021, 373, 66–69 CrossRef CAS PubMed.
- A. Kulkarni, G. Quintens and L. M. Pitet, Macromolecules, 2023, 56, 1747–1758 CrossRef CAS.
- M. D. de Dios Caputto, R. Navarro, J. L. Valentín and Á. Marcos-Fernández, J. Polym. Sci., 2022, 60, 3269–3283 CrossRef CAS.
- P. D. Goring and R. D. Priestley, JACS Au, 2023, 3, 2609–2611 CrossRef CAS PubMed.
- W. Zeng, Y. Zhao, F. Zhang, R. Li, M. Tang, X. Chang, Y. Wang, F. Wu, B. Han and Z. Liu, Nat. Commun., 2024, 15, 160 CrossRef PubMed.
- M. Hong and E. Y. X. Chen, Green Chem., 2017, 19, 3692–3706 RSC.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
- D. E. Fagnani, J. L. Tami, G. Copley, M. N. Clemons, Y. D. Y. L. Getzler and A. J. McNeil, ACS Macro Lett., 2021, 10, 41–53 CrossRef CAS PubMed.
- F. M. Lamberti, L. A. Román-Ramírez and J. Wood, J. Polym. Environ., 2020, 28, 2551–2571 CrossRef CAS.
- M. Rabnawaz, I. Wyman, R. Auras and S. Cheng, Green Chem., 2017, 19, 4737–4753 RSC.
- R. Nisticò, Polym. Test., 2020, 90, 106707 CrossRef.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y. X. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef CAS.
- L. Cederholm, P. Olsén, M. Hakkarainen and K. Odelius, Polym. Chem., 2023, 14, 3270–3276 RSC.
- V. Tournier, S. Duquesne, F. Guillamot, H. Cramail, D. Taton, A. Marty and I. André, Chem. Rev., 2023, 123, 5612–5701 CrossRef CAS PubMed.
- M. R. B. Mermet-Guyennet, J. Gianfelice de Castro, H. S. Varol, M. Habibi, B. Hosseinkhani, N. Martzel, R. Sprik, M. M. Denn, A. Zaccone, S. H. Parekh and D. Bonn, Polymer, 2015, 73, 170–173 CrossRef CAS.
- S. K. Burgess, J. E. Leisen, B. E. Kraftschik, C. R. Mubarak, R. M. Kriegel and W. J. Koros, Macromolecules, 2014, 47, 1383–1391 CrossRef CAS.
- K. Loos, R. Zhang, I. Pereira, B. Agostinho, H. Hu, D. Maniar, N. Sbirrazzuoli, A. J. D. Silvestre, N. Guigo and A. F. Sousa, Front. Chem., 2020, 8, 585 CrossRef CAS PubMed.
- J.-G. Rosenboom, D. K. Hohl, P. Fleckenstein, G. Storti and M. Morbidelli, Nat. Commun., 2018, 9, 2701 CrossRef.
- A. F. Sousa, R. Patrício, Z. Terzopoulou, D. N. Bikiaris, T. Stern, J. Wenger, K. Loos, N. Lotti, V. Siracusa, A. Szymczyk, S. Paszkiewicz, K. S. Triantafyllidis, A. Zamboulis, M. S. Nikolic, P. Spasojevic, S. Thiyagarajan, D. S. van Es and N. Guigo, Green Chem., 2021, 23, 8795–8820 RSC.
- G. Z. Papageorgiou, V. Tsanaktsis and D. N. Bikiaris, Phys. Chem. Chem. Phys., 2014, 16, 7946–7958 RSC.
- A. Pellis, K. Haernvall, C. M. Pichler, G. Ghazaryan, R. Breinbauer and G. M. Guebitz, J. Biotechnol., 2016, 235, 47–53 CrossRef CAS PubMed.
- S. Thiyagarajan, W. Vogelzang, R. J. I. Knoop, A. E. Frissen, J. van Haveren and D. S. van Es, Green Chem., 2014, 16, 1957–1966 RSC.
- Z. Terzopoulou, E. Karakatsianopoulou, N. Kasmi, M. Majdoub, G. Z. Papageorgiou and D. N. Bikiaris, J. Anal. Appl. Pyrolysis, 2017, 126, 357–370 CrossRef CAS.
- L. Papadopoulos, A. Zamboulis, N. Kasmi, M. Wahbi, C. Nannou, D. A. Lambropoulou, M. Kostoglou, G. Z. Papageorgiou and D. N. Bikiaris, Green Chem., 2021, 23, 2507–2524 RSC.
- M. B. Banella, J. Bonucci, M. Vannini, P. Marchese, C. Lorenzetti and A. Celli, Ind. Eng. Chem. Res., 2019, 58, 8955–8962 CrossRef CAS.
- G. J. Gruter, L. Sipos and M. A. Dam, Comb. Chem. High Throughput Screening, 2012, 15, 180–188 CrossRef CAS PubMed.
- Z. Terzopoulou, E. Karakatsianopoulou, N. Kasmi, V. Tsanaktsis, N. Nikolaidis, M. Kostoglou, G. Z. Papageorgiou, D. A. Lambropoulou and D. N. Bikiaris, Polym. Chem., 2017, 8, 6895–6908 RSC.
- I. Shigemoto, T. Kawakami, H. Taiko and M. Okumura, Polymer, 2011, 52, 3443–3450 CrossRef CAS.
- C. Alberti, K. Matthiesen, M. Wehrmeister, S. Bycinskij and S. Enthaler, Chem. Sel., 2021, 6, 7972–7975 CAS.
- E. Gabirondo, B. Melendez-Rodriguez, C. Arnal, J. M. Lagaron, A. Martínez de Ilarduya, H. Sardon and S. Torres-Giner, Polym. Chem., 2021, 12, 1571–1580 RSC.
- B. Agostinho, A. J. D. Silvestre and A. F. Sousa, Green Chem., 2022, 24, 3115–3119 RSC.
- F. Kawai, Y. Furushima, N. Mochizuki, N. Muraki, M. Yamashita, A. Iida, R. Mamoto, T. Tosha, R. Iizuka and S. Kitajima, AMB Express, 2022, 12, 134 CrossRef CAS.
- H. P. Austin, M. D. Allen, B. S. Donohoe, N. A. Rorrer, F. L. Kearns, R. L. Silveira, B. C. Pollard, G. Dominick, R. Duman, K. El Omari, V. Mykhaylyk, A. Wagner, W. E. Michener, A. Amore, M. S. Skaf, M. F. Crowley, A. W. Thorne, C. W. Johnson, H. L. Woodcock, J. E. McGeehan and G. T. Beckham, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E4350–E4357 CrossRef CAS PubMed.
- S. A. Younis, E. Amdeha and R. A. El-Salamony, J. Environ. Chem. Eng., 2021, 9, 104619 CrossRef CAS.
- A. M. Deliormanlı, J. Mater. Sci.: Mater. Med., 2015, 26, 67 CrossRef.
- C. H. Nguyen, J. A. Field and R. Sierra-Alvarez, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2020, 55, 168–178 CrossRef CAS PubMed.
- A. A. Ismail, I. Abdelfattah, M. Faisal and A. Helal, J. Hazard. Mater., 2018, 342, 519–526 CrossRef CAS PubMed.
- C. H. Nguyen, C. Zeng, S. Boitano, J. A. Field and R. Sierra-Alvarez, Int. J. Toxicol., 2020, 39, 218–231 CrossRef CAS PubMed.
- R. Raphael, W. F. Aswathy and E. I. Anila, MRS Commun., 2024 DOI:10.1557/s43579-024-00647-z.
- X. Li, X. Wang, J. Luo, J. Jiang, K. Ding, L. Ye, Y. Xiong, D. Pang, H. Li, P. Yu, C. Kong, L. Ye, H. Zhang and W. Li, Mater. Today Commun., 2023, 35, 106118 CrossRef CAS.
- W. Xu, T. Peng, Y. Li, F. Xu, Y. Zhang, C. Zhao, M. Fang, S. Han, D. Zhu, P. Cao, W. Liu and Y. Lu, Nanomaterials, 2022, 12, 1125 CrossRef CAS PubMed.
- D. Wawrzynczyk, M. Nyk and M. Samoc, Chem. Phys., 2015, 456, 73–78 CrossRef CAS.
- Y. Xu, X. Wang, D. Yang, Z. Tang, M. Cao, H. Hu, L. Wu, L. Liu, J. McLeod, H. Lin, Y. Li, Y. Lifshitz, T.-K. Sham and Q. Zhang, ACS Catal., 2021, 11, 10159–10169 CrossRef CAS.
- Y. Zhang, S. Jiao, J. Zhang, S. Liu, D. Wang, S. Gao and J. Wang, CrystEngComm, 2022, 24, 1789–1794 RSC.
- G. Mu, Y. Zeng, Y. Zheng, Y. Cao, F. Liu, S. Liang, Y. Zhan and L. Jiang, Green Energy Environ., 2023, 8, 831–841 CrossRef CAS.
- L. Kong, J. Ma, C. Luan, W. Mi and Y. Lv, Thin Solid Films, 2012, 520, 4270–4274 CrossRef CAS.
- A. Janotti, J. B. Varley, P. Rinke, N. Umezawa, G. Kresse and C. G. Van de Walle, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 085212 CrossRef.
- B. J. Morgan and G. W. Watson, J. Phys. Chem. C, 2010, 114, 2321–2328 CrossRef CAS.
- X. Pan, M.-Q. Yang, X. Fu, N. Zhang and Y.-J. Xu, Nanoscale, 2013, 5, 3601–3614 RSC.
- A. Naldoni, M. Allieta, S. Santangelo, M. Marelli, F. Fabbri, S. Cappelli, C. L. Bianchi, R. Psaro and V. Dal Santo, J. Am. Chem. Soc., 2012, 134, 7600–7603 CrossRef CAS.
- J.-L. Chiang, B. K. Yadlapalli, M.-I. Chen and D.-S. Wuu, Nanomaterials, 2022, 12, 3601 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc03803g |
| This journal is © The Royal Society of Chemistry 2025 |