
Halogen-bond-assisted radical remote difunctionalization of bicyclo[1.1.1]butane skeletons†
Hui
Liu
,
Zhenda
Fu
,
Xingwei
Li
* and
Songjie
Yu
 *
*
Institute of Frontier Chemistry, School of Chemistry and Chemical Engineering, Shandong University, Qingdao, 266237, China. E-mail: lixw@snnu.edu.cn; yusongjie23@sdu.edu.cn
First published on 15th November 2024
Abstract
Transition-metal-free radical remote difunctionalization of bicyclo[1.1.1]butane skeletons in both two- and three-component fashions is presented. The reactions proceed via halogen-bond-assisted polyfluoroalkyl radical addition to newly designed 1-vinylbicyclo[1.1.1]pentanes, followed by strain-release-driven C–C bond cleavage to generate a strained cyclobutylmethyl radical. In the two-component reaction, iodine atom transfer to the resulting cyclobutylmethyl radical with polyfluoroiodides forms a broad array of strained 1,6-polyfluorocarboiodinated products, while boron atom transfer with bis(catecholato)diboron releases various strained 1,6-polyfluorocarboborylated products in the three-component reaction. This redox-neutral reaction features mild conditions, ease of operation, high atom economy, functional group tolerance, and a broad substrate scope, and offers a practical and sustainable approach for the synthesis of a range of challenging polyfluoroalkylated cyclobutane skeletons containing iodine and boron as versatile transformation handles for further useful derivatizations.
Introduction
Strained cyclobutane scaffolds are widely found in a large number of pharmaceuticals and polycyclic natural products.1 The inherent stereochemistry and unique chemical space of the strained cyclobutane structure allow the compounds to exhibit a variety of unique physicochemical properties and biological activities.2 On the other hand, fluorine is an indispensable tool in medicinal discovery, as fluorine can significantly improve a molecule's potency and permeability, modulate its pKa and lipophilicity, and control its conformation.3 Owing to these fascinating characteristics, an increasing number of molecules in which cyclobutane is the rigid core scaffold and fluorine is the pharmacokinetic mediator, have been reported recently with several previously unexplored biological activities (Scheme 1a).4 Although these bioactive fluorocyclobutane skeletons have been developed, current synthetic processes mostly suffer from tedious procedures, poor atom economy, and low generality, thus blocking their further medicinal applications.5 In this context, developing a general functional-group-tolerant and easily operated strategy for the sustainable construction of such scaffolds would be highly desirable.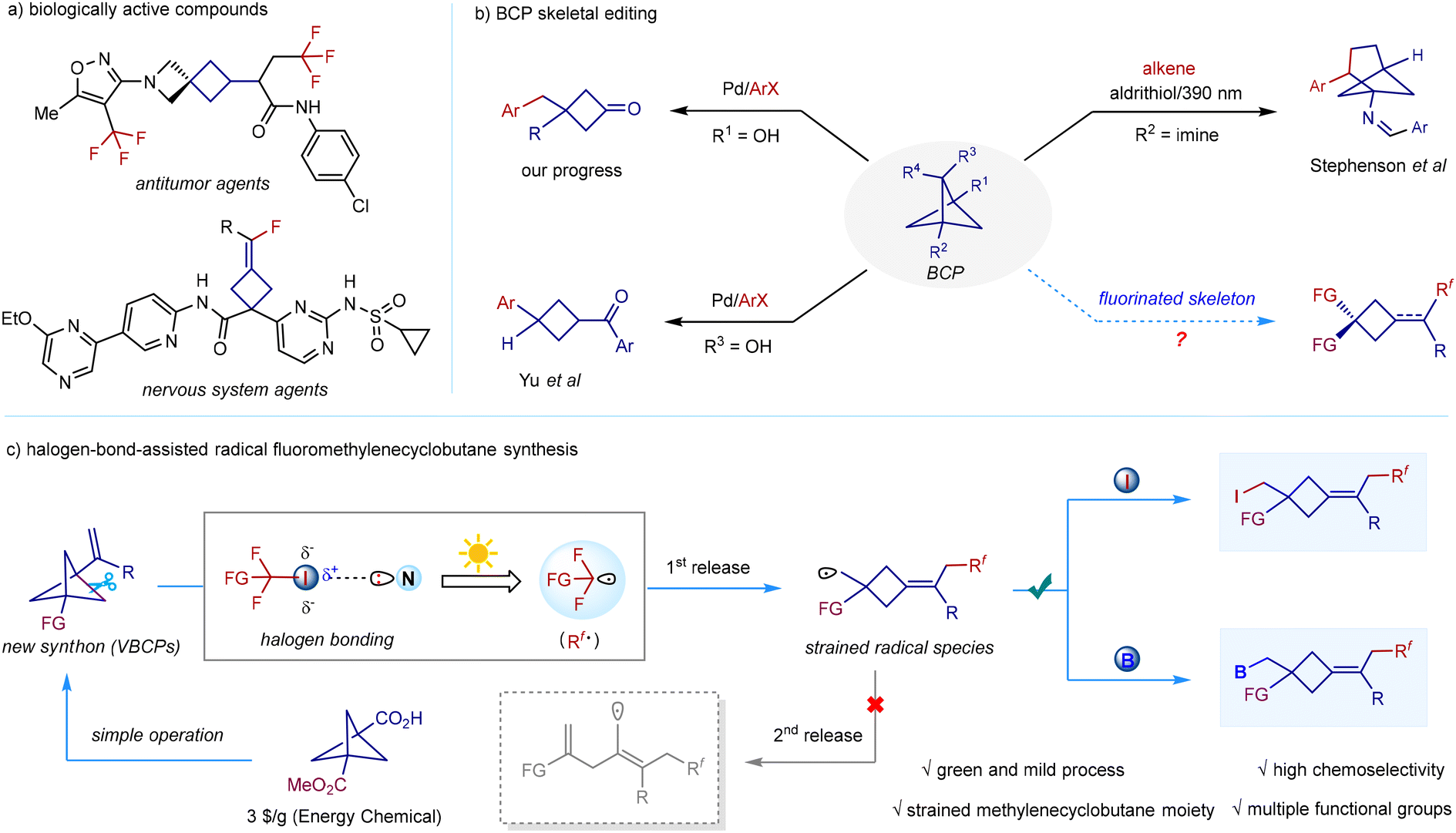 | ||
| Scheme 1 (a) Related biologically active compounds. (b) BCP skeletal editing. (c) Halogen-bond-assisted radical fluoromethylenecyclobutane synthesis. | ||
Functionalized bicyclo[1.1.1]pentanes (BCPs) are excellent bioisosteres of phenyl rings in pharmaceutical evolution,6 and their synthetic processes have attracted substantial attention over the past decade.7 Compared to the well-studied BCP synthesis, catalytic BCP transformations,8 especially chemoselective BCP skeletal editing, lag largely behind. As a matter of fact, investigating the chemical reactivity of the highly strained BCP skeleton is of great importance because this would not only reveal the metabolic pathways of BCP-derived drugs, but also provide access to more challenging new scaffolds. Thus, both these significant aspects have driven synthetic chemists to firmly place the exploration of BCP skeletal editing on their agenda (Scheme 1b). In 2021, Stephenson reported an elegant photochemical conversion of bicyclo[1.1.1]pentan-1-amines to bicyclo[3.1.1]heptan-1-amines via formal [4 + 2]cycloaddition with activated alkenes.9 In 2022, our group developed the first palladium-catalyzed chemoselective C–C activation of strained bicyclo[1.1.1]pentan-1-ols and their coupling with various organohalides, offering an expedient approach to a wide range of multi-functionalized cyclobutanone or β,γ-enone skeletons.10 Very recently, Yu's group also disclosed a palladium-catalysed stereospecific C–C arylation of bicyclo[1.1.1]pentan-2-ols to afford various cis-1,3-difunctionalized cyclobutanes.11 Despite the progress in catalytic BCP skeletal editing, these valuable methods commonly require a transition metal catalyst, a ligand, and a stoichiometric amount of a base, or otherwise they suffer from problematic reaction selectivity. To our knowledge, sustainable chemoselective skeletal editing of BCPs to access drug-involved fluorocyclobutane skeletons is unknown.
Recently, transition metal-, and photocatalyst-mediated fluorocarbon-centred radical addition to alkenes has become an efficient method for alkene functionalization.12 Inspired by the well-established alkene fluorofunctionalization and successful conversion of BCPs to functionalized cyclobutanes, we reasoned that 1-vinylbicyclo[1.1.1]pentanes (VBCPs), which can be easily derived from commercially available 3-(methoxycarbonyl)bicyclo[1.1.1]pentane-1-carboxylic acid, would be promising starting materials to forge strained fluorocyclobutanes via the challenging fluorocarbon-centred radical addition, followed by a controllable partial-strain-release process (Scheme 1c). However, as a notable issue, the second strain-release process of the generated strained cyclobutylmethyl radical13 prior to the radical functionalization poses a great challenge. To avoid the double strain-release event, a sustainable reaction system that accommodates mild fluorocarbon-centred radical initiation and dynamically favourable atom transfer to the strained cyclobutylmethyl radical is required. Polyfluoroalkyliodides, a type of easily available fluoroalkylating reagent,14 are prone to undergo a linear halogen-bond interaction between the σ-hole of the iodine atom and a Lewis base,15 which leads to the lengthening of the carbon–iodine bond. Upon light excitation, this halogen-bonding complex steadily generates a fluorocarbon-centred radical through an intramolecular electron transfer event between the Lewis base and the CF–I σ* antibonding orbital in the absence of metals and photocatalysts, thus providing a more environmentally respectful alternative.16 Consequently, our proposal is aimed at leveraging the mild photochemical excitation of halogen-bonding complexes generated from easily available polyfluoroalkyliodides and Lewis bases to readily release reactive polyfluoroalkyl radicals, as well as the rapid atom (iodine or boron) transfer to the strained cyclobutylmethyl radical. Herein, we report our findings on the halogen-bond-assisted radical remote difunctionalization of 1-vinylbicyclo[1.1.1]pentanes in two- and three-component fashions, delivering various remote polyfluorocarboiodinated and polyfluorocarboborylated cyclobutane skeletons in which iodine or boron serves as a synthetically useful handle for further transformations.
Results and discussion
To develop this approach for remote iodopolyfluoroalkylation, VBCP 1 and polyfluoroalkyliodide 2 were chosen as representative substrates for optimization of the reaction conditions, as summarized in Table 1. Considering that the choice of Lewis bases significantly impacts formation of the halogen-bonding complex, we initiated these studies utilizing DBU as an initiator in MeCN solvent under blue LED irradiation. The desired fluorocyclobutane 3 was obtained in 95% yield (Table 1, entry 1). The employment of other radical initiators such as DABCO, AIBN, and Ph3P did not provide better efficiencies toward the formation of the desired product (entries 2–4). Several other solvents including THF, DMF, MeOH, and DCM were examined using DBU as the optimal initiator, but no improvement was achieved (entries 5–8). Although decreasing the amount of DBU to 10 mol% gave the desired product in 97% yield, 3 mol% DBU as an initiator afforded 3 in a reduced yield (entries 9 and 10). When the reaction was performed in the absence of DBU, a quite low reaction efficiency was observed (entry 11). The photochemical nature of this transformation was confirmed by performing the reaction in the absence of photoirradiation (entry 12). A thermally induced strategy showed low efficiency toward the formation of the fluoroalkyl radical, giving the desired product only in 13% yield (entry 13). The reaction was completely inhibited under air conditions (entry 14).|
|
||
|---|---|---|
| Entry | Variations from conditions | Yield/%a |
| Conditions: 1 (0.1 mmol), 2 (0.1 mmol), initiator (3–20 mol%), solvent (1 mL), blue LEDs (455 nm, 10 W), room temperature, nitrogen atmosphere, 12 h.a 1H NMR yield with 1,3,5-trimethoxybenzene as an internal standard.b Isolated yield. | ||
| 1 | None | 95(95)b |
| 2 | DABCO as an initiator | 88 |
| 3 | AIBN as an initiator | 50 |
| 4 | Ph3P as an initiator | 87 |
| 5 | THF as a solvent | 88 |
| 6 | DMF as a solvent | 93 |
| 7 | MeOH as a solvent | 50 |
| 8 | DCM as a solvent | 90 |
| 9 | 10 mol% DBU | 97(95)b |
| 10 | 3 mol% DBU | 71 |
| 11 | No initiator | 20 |
| 12 | No light | <5 |
| 13 | 100 °C instead of light | 13 |
| 14 | Under air | <5 |
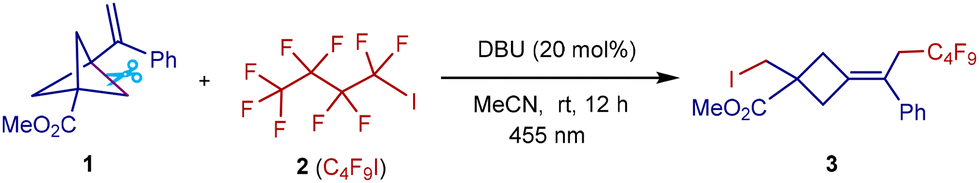
With the optimized conditions in hand, we first investigated the VBCP substrate scope using C4F9I as the bifunctional radical precursor (Scheme 2). The iodofluoroalkylation features good functional group tolerance, as installing various electron-rich (Me, tBu, and OMe) and electron-deficient (F, Cl, Ph, and naphthalene) groups at different positions on the benzene rings of VBCP skeletons was well tolerated (3–16). Interestingly, an electron-rich heteroaryl group was also compatible with the reactive polyfluoroalkyl radical, as the VBCP skeleton bearing a thiophene group was successfully converted to the desired product in 71% yield (17). In addition to aromatic groups, aliphatic groups such as methyl (18), ethyl (19), butyl (20), and benzyl (21) groups were tolerated as well, giving the desired fluorocyclobutanes in 78–90% yields. As expected, this protocol is amenable to use of other VBCP esters, including ethyl (22), tert-butyl (23), 1-adamantanethyl (24), benzyl (25–27) and 4-pyridinemethyl (28) esters. Apart from VBCP esters, ketone- and Weinreb amide-derived VBCPs (29–30), which provide extra electrophilic handles for further downstream modifications, were all compatible with the system, giving the corresponding poly-substituted cyclobutanes in high yields. Replacement of a strong electron-withdrawing ester group by a phenyl group in the VBCP skeleton is feasible, affording product 31 in 91% yield. The method was also applicable to several natural product-derived VBCPs, like valine (32), borneol (33), cholesterol (34), estrone (35), and tocopherol (36), thus offering a practical protocol for the post-modification of bioactive compounds.
 | ||
Scheme 2 VBCP substrate scope. Conditions: 1 (0.1 mmol), 2 (0.1 mmol), DBU (10 mol%), MeCN (1 mL), blue LEDs (455 nm, 10 W), room temperature, nitrogen atmosphere, 12 h. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 36 h. 36 h. | ||
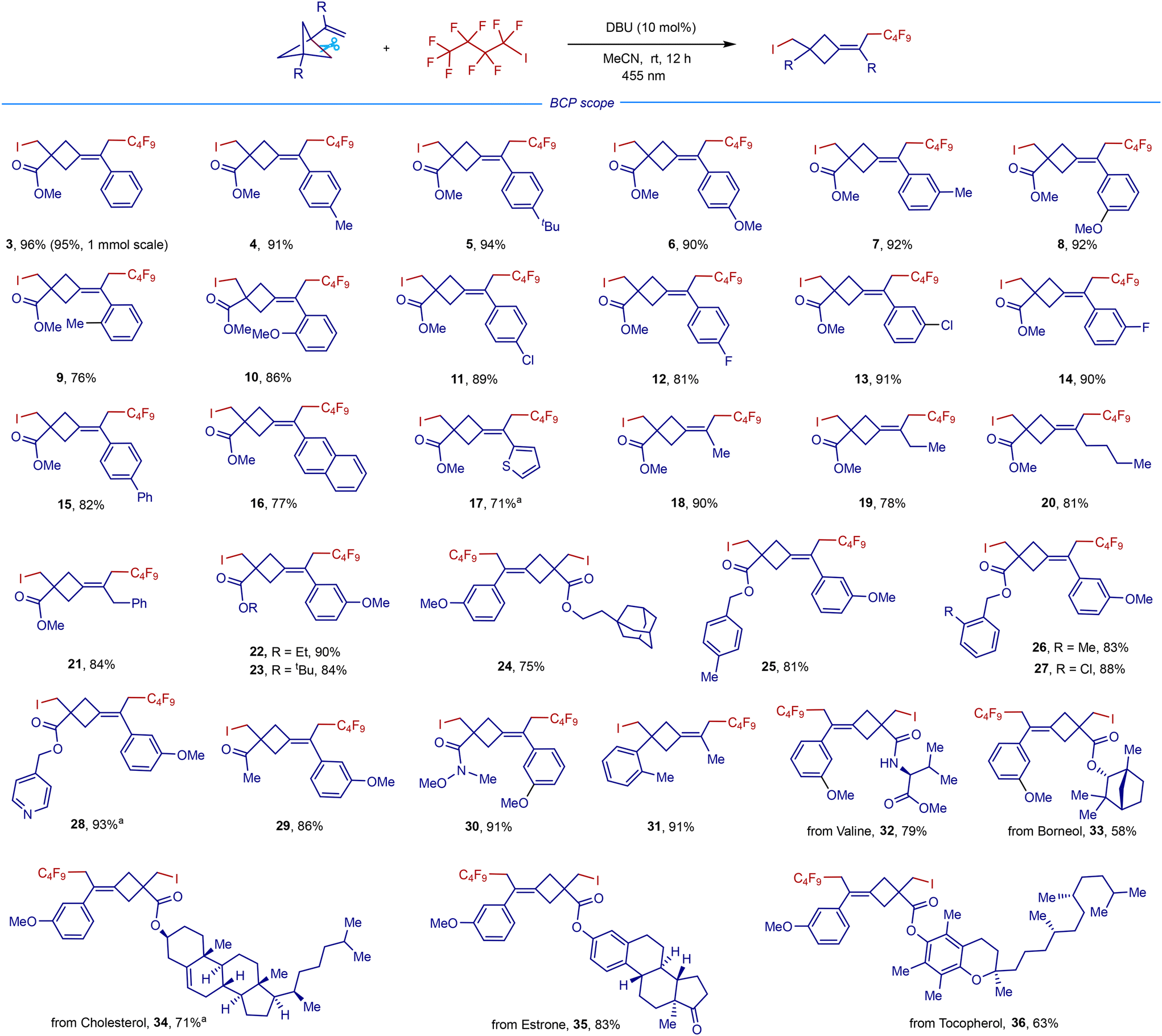
The scope and generality of this remote difunctionalization in terms of polyfluoroalkyliodides with representative VBCP 1e as a radical acceptor are summarized in Scheme 3. When using perfluoroalkyliodides containing CF3, C3F7, C6F13, C8F17, and C10F21 groups as radical precursors, the corresponding products 37–41 were obtained in high yields (71–95%). A good yield for the difluorocarboiodination was also achieved with ethyl difluoroiodoacetate as a substrate (42). Inspired by this result, we then examined the reaction with iododifluoromethyl ketones, which are easily available via difluoromethylene formal insertion into the C–H bonds of aldehydes.14b To our delight, a wide range of iododifluoromethyl aryl ketones could serve as efficient radical precursors to afford the corresponding difluoromethyl ketone-derived cyclobutanes in good yields (43–54). The benzene ring bearing either electron-donating groups such as methyl (44 and 46), tert-butyl (45), and methylthiol (47), or electron-withdrawing groups such as phenyl (48), naphthalene (49), chloro (50), and ester (51) undergoes the desired transformations smoothly, affording the corresponding products in 64–90% yields. Remarkably, the reactions with iododifluoromethyl ketones equipped with oxygen-, sulfur-, or nitrogen-containing heteroaromatic groups also gave the corresponding products 52–54 in good to excellent yields.
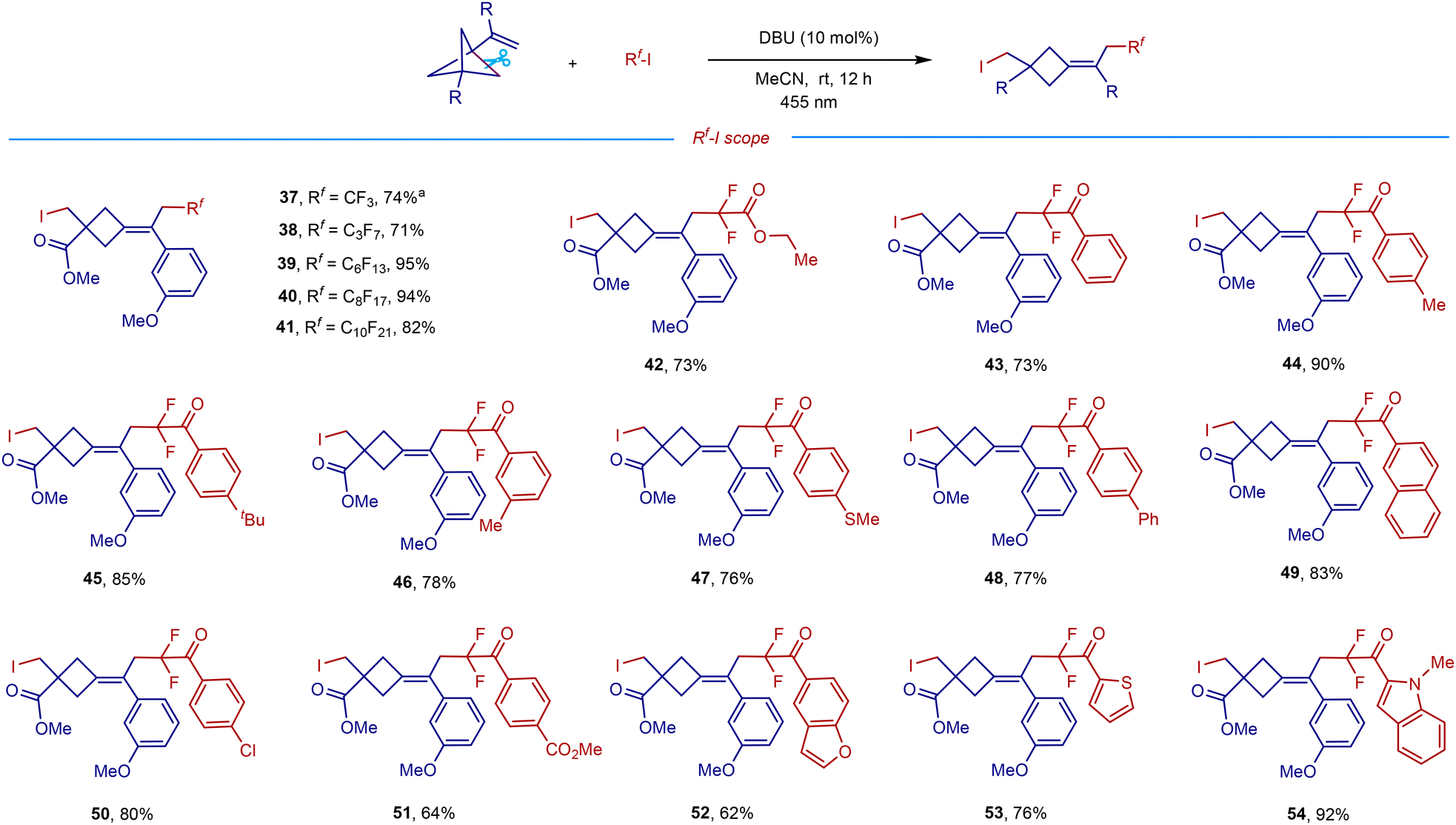 | ||
| Scheme 3 Polyfluoroalkyliodide scope. Conditions: VBCP (0.1 mmol), polyfluoroalkyliodide (0.1 mmol), DBU (10 mol%), MeCN (1 mL), blue LEDs (455 nm, 10 W), room temperature, nitrogen atmosphere, 12 h. aUsing 1.5 equiv. of 2 (0.15 mmol). | ||
As commonly used nucleophilic precursors, boronic esters are highly valuable synthetic handles to be transformed into a wide range of useful functional groups,17 and their transformation is complementary to the reactivity of the electrophilic organoiodides.18 Therefore, we tried to broaden access to useful polyfluorocarboborylated cyclobutane scaffolds. Inspired by the radical fluorocarboborylation of alkenes disclosed by Studer and coworkers,19 we then explored the three-component remote fluorocarboborylation of VBCPs with perfluoroalkyliodides and B2Cat2 (Scheme 4). Adding B2Cat2 to our reaction system in DMF solvent could allow the fluorocarboborylation to proceed smoothly, demonstrating the broad generality of this photochemical system. The reaction is able to tolerate various polyfluoroalkyl radical precursors, delivering the corresponding products in 39–56% isolated yields (55–60).
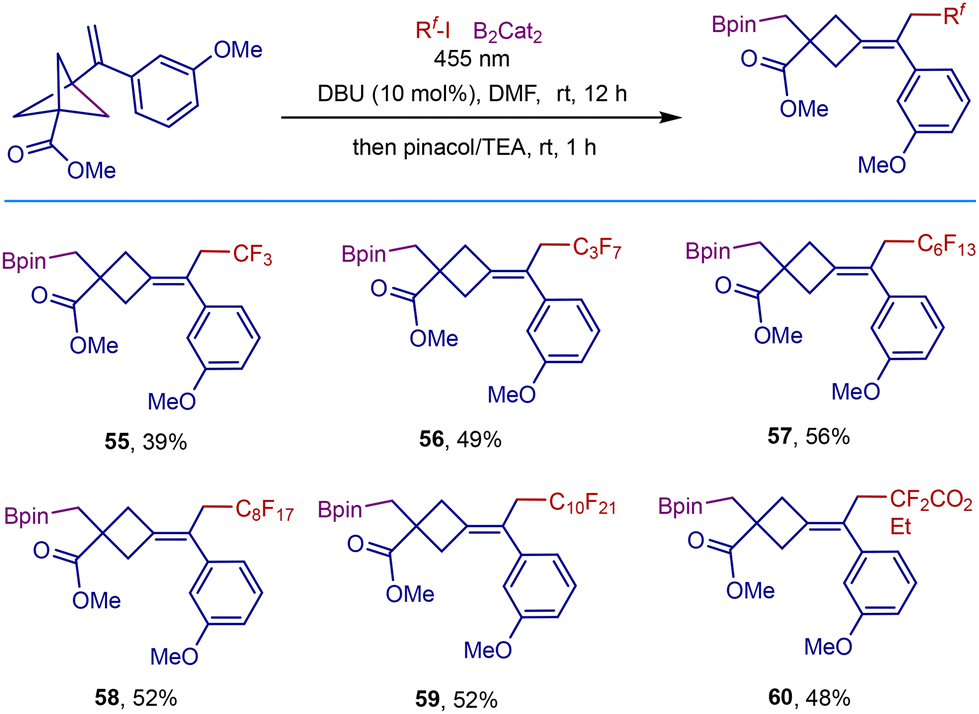 | ||
| Scheme 4 Three-component remote perfluorocarboborylation. Conditions: VBCP (0.1 mmol), polyfluoroalkyliodide (0.1 mmol), B2Cat2 (0.3 mmol), DBU (10 mol%), DMF (1 mL), blue LEDs (455 nm, 10 W), room temperature, nitrogen atmosphere, 12 h, then treatment with pinacol (0.9 mmol) in TEA (1 mL). | ||
Recognizing the importance of this strategy and its products, we explored the potential applications of our methodology (Scheme 5). The one-pot fluorocarboborylation followed by boron oxidation, could form the hydroxylated scaffold 61 in 55% yield. The boronic ester group of compound 58 could be converted to a vinyl group (62) in 64% yield through Zweifel olefination. Next, several divergent transformations of the iodine group were performed. Azidation of iodide 8 afforded the corresponding compound 63 in 92% yield. The resulting azide group could forge 1,2,3-triazole on the cyclobutane scaffold (64) with high efficiency via the classic click reaction. The reductive deiodination and nucleophilic substitution with imidazole gave the desired hydroxyl (65) or imidazolyl (66) derivatives in good yields.
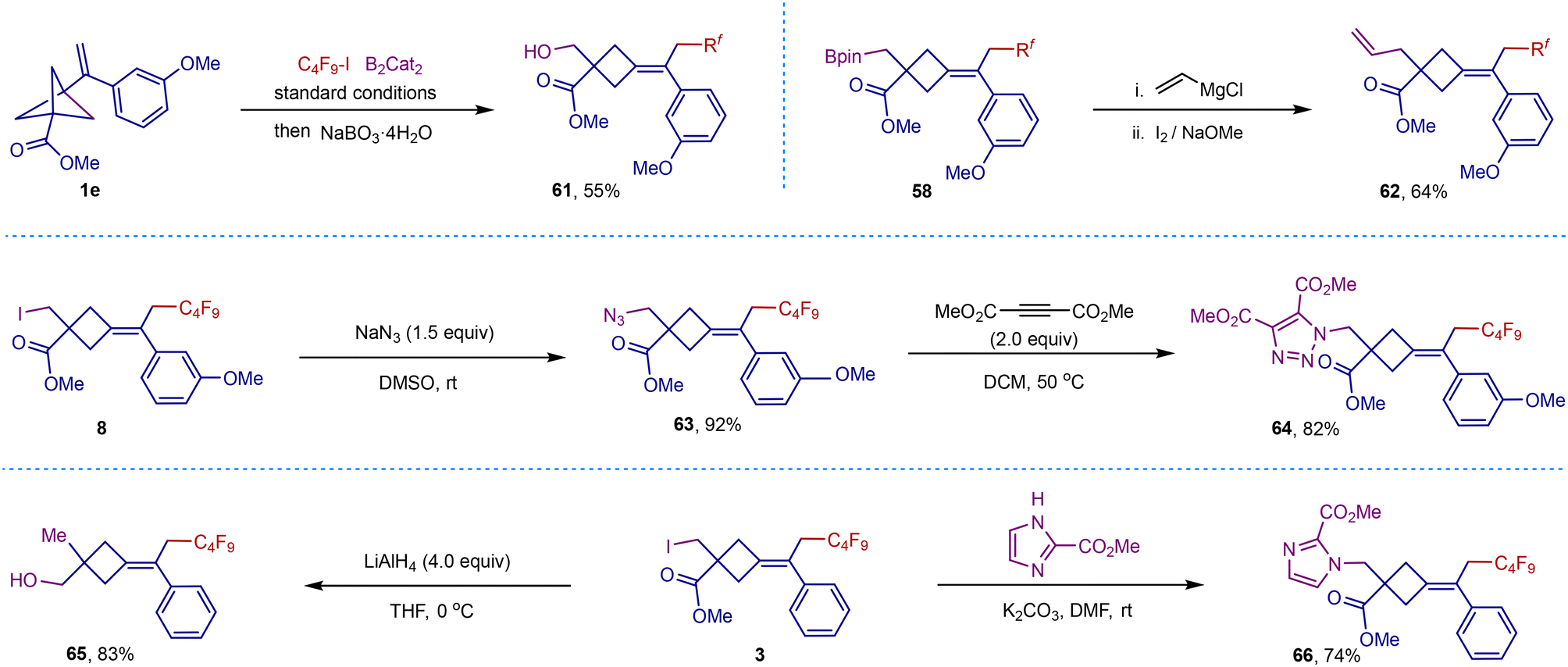 | ||
| Scheme 5 Product transformation. | ||
The fact that both light and DBU as initiators are required in the reaction indicates that these transformations are likely radical in nature. We therefore carried out a radical-inhibiting experiment. The reaction was completely suppressed when TEMPO was added, suggesting the involvement of a radical process in this transformation (Scheme 6a). UV/vis measurements showed that VBCP, C4F9I, and DBU all have no absorption around the operating wavelength (455 nm) in the UV-visible absorption spectra, thus excluding the possibility of direct substrate excitation. However, a bathochromic shift was observed by UV/vis absorption spectroscopy for a 1:1 mixture of C4F9I and DBU, which is likely ascribed to the formation of a halogen-bonding complex between polyfluoroalkyliodide and DBU (Scheme 6b). Switching polyfluoroalkyliodide to polyfluoroalkylbromide gave no desired product due to the tough cleavage of the carbon–bromine bond (Scheme 6c). On the basis of the above observations and previous reports,20 we proposed the mechanism outlined in Scheme 6d. Initiation of the radical chain begins with the formation of the halogen-bonding complex between polyfluoroalkyliodide and DBU. Photoexcitation of the complex triggers an intramolecular single-electron transfer to form a polyfluoroalkyl radical, followed by radical addition to VBCP. Strain-release-driven C–C cleavage of the VBCP skeleton generates the strained cyclobutylmethyl radical. Then rapid iodine atom transfer releases the final product and regenerates the polyfluoroalkyl radical. The measured quantum yield was determined to be Φ > 15, which is consistent with the above discussed radical chain process.
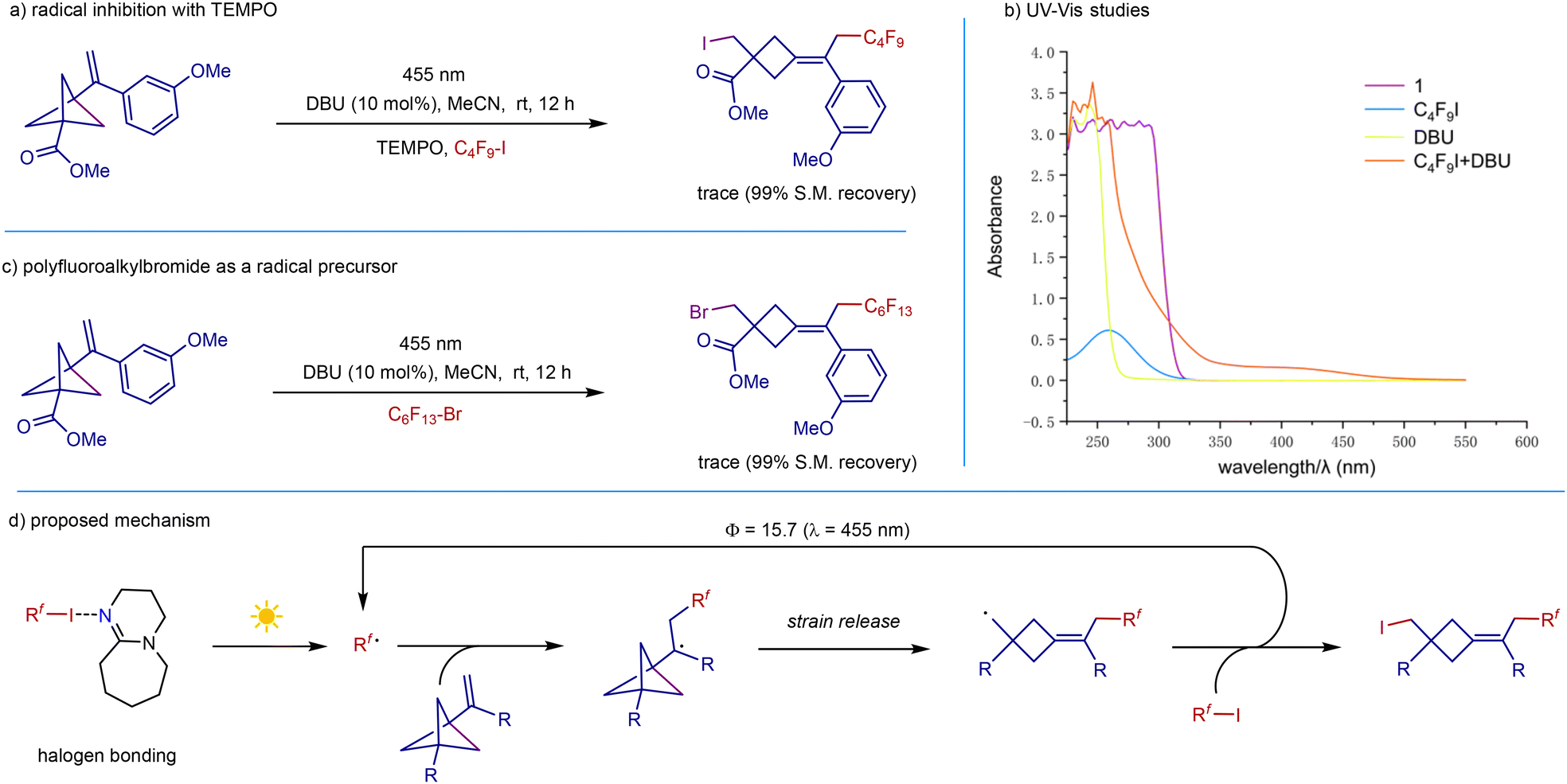 | ||
| Scheme 6 Mechanistic studies and a proposed reaction cycle. | ||
Conclusions
In summary, we have described a metal-free remote polyfluoroalkyliodination and polyfluoroborylation of well-tailored 1-vinylbicyclo[1.1.1]butane scaffolds initiated by halogen-bond-assisted polyfluoroalkyl radical addition. Both reactions proceeded under mild reaction conditions and showed broad substrate scopes in terms of 1-vinylbicyclo[1.1.1]butanes and polyfluoroalkyliodides, forging various highly functionalized fluorocyclobutanes containing iodine or boron as a synthetically useful handle. We anticipate that this green and efficient photochemical system will provide a new avenue for BCP transformations as well as fluorocyclobutane skeleton fabrications for research in chemistry and medicine.Author contributions
H. L. carried out the experiments. H. L., Z. F., X. L. and S. Y. analysed the data. S. Y. conceived the research and wrote the paper with input from all authors. S. Y. and X. L. directed the research.Data availability
Further details of the experimental procedure, NMR spectra, and HRMS data are available in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (No. 22371029) for financial support. S. Y. thanks the Taishan Scholar of Shandong Province (tsqnz20221108), and the Qilu Young Scholar of Shandong University for funding.References
- (a) Y.-Y. Fan, X.-H. Gao and J.-M. Yue, Sci. China: Chem., 2016, 59, 1126–1147 CrossRef CAS; (b) C. Hui, Y. Liu, M. Jiang and F. Wu, Trends Chem., 2022, 4, 677–681 CrossRef CAS; (c) J. Li, K. Gao, M. Bian and H. Ding, Org. Chem. Front., 2020, 7, 136–154 RSC; (d) V. M. Dembitsky, J. Nat. Med., 2007, 62, 1–33 CrossRef; (e) H. Zhao, Y. Lin, M. Jiang and B. Su, Chem. Sci., 2023, 14, 7897–7904 RSC; (f) V. M. Dembitsky, Phytomedicine, 2014, 21, 1559–1581 CrossRef CAS PubMed.
- (a) E. M. Carreira and T. C. Fessard, Chem. Rev., 2014, 114, 8257–8322 CrossRef CAS PubMed; (b) M. R. V. D. Kolk, M. A. C. H. Janssen, F. P. J. T. Rutjes and D. Blanco-Ania, ChemMedChem, 2022, 17, e202200020 CrossRef PubMed; (c) M. L. Wrobleski, G. A. Reichard, S. Paliwal, S. Shah, H. C. Tsui, R. A. Duffy, J. E. Lachowicz, C. A. Morgan, G. B. Varty and N. Y. Shih, Bioorg. Med. Chem. Lett., 2006, 16, 3859–3863 CrossRef CAS; (d) Y. Zheng, C. M. Tice and S. B. Singh, Bioorg. Med. Chem. Lett., 2014, 24, 3673–3682 CrossRef CAS.
- (a) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS; (b) Y. Yu, A. Liu, G. Dhawan, H. Mei and J. Han, Chin. Chem. Lett., 2021, 32, 3342–3354 CrossRef CAS; (c) Y. Zhu, J. Han, J. Wang, N. Shibata, M. Sodeoka, V. A. Soloshonok, J. A. S. Coelho and F. D. Toste, Chem. Rev., 2018, 118, 3887–3964 CrossRef CAS; (d) K. Mueller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef CAS; (e) J. Wang, M. Sánchez-Roselló, J. L. Aceña, D. C. Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2013, 114, 2432–2506 CrossRef PubMed.
- (a) L. D. Jennings, Z. Wawrzak, D. Amorose, R. S. Schwartz and D. B. Jordan, Bioorg. Med. Chem. Lett., 1999, 9, 2509–2514 CrossRef CAS PubMed; (b) H. Zhang, C. Cai and S. Zhu, CN patCN118359588, 2024 Search PubMed; (c) X. Wu, W. Deng, D. Lin, H. Mao, D. Chen, T. Zhao, J. Fu, N. Zhang, Z. D. Hang, Y. Xu and S. Zhao, CN patCN114685341, 2022 Search PubMed.
- (a) C. Hui, Z. Wang, Y. Xie and J. Liu, Green Synth. Catal., 2023, 4, 1–6 CrossRef; (b) V. L. Revil-Baudard and S. Z. Zard, Helv. Chim. Acta, 2021, 104, e2100106 CrossRef CAS; (c) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470–477 CrossRef CAS PubMed; (d) Y. Zhou, Z. Wu, J. Xu, Z. Zhang, H. Zheng and G. Zhu, Angew. Chem., Int. Ed., 2024, 63, e202405678 CrossRef CAS; (e) M. Silvi and V. K. Aggarwal, J. Am. Chem. Soc., 2019, 141, 9511–9515 CrossRef CAS PubMed; (f) X. Hu, W. Xu, Y. Liu and H. Guo, J. Org. Chem., 2023, 88, 2521–2534 CrossRef CAS PubMed.
- (a) A. F. Stepan, C. Subramanyam, I. V. Efremov, J. K. Dutra, T. J. O'Sullivan, K. J. DiRico, W. S. McDonald, A. Won, P. H. Dorff, C. E. Nolan, S. L. Becker, L. R. Pustilnik, D. R. Riddell, G. W. Kauffman, B. L. Kormos, L. Zhang, Y. Lu, S. H. Capetta, M. E. Green, K. Karki, E. Sibley, K. P. Atchison, A. J. Hallgren, C. E. Oborski, A. E. Robshaw, B. Sneed and C. J. O'Donnell, J. Med. Chem., 2012, 55, 3414–3424 CrossRef CAS PubMed; (b) N. D. Measom, K. D. Down, D. J. Hirst, C. Jamieson, E. S. Manas, V. K. Patel and D. O. Somers, ACS Med. Chem. Lett., 2017, 8, 43–48 CrossRef CAS; (c) P. K. Mykhailiuk, Org. Biomol. Chem., 2019, 17, 2839–2849 RSC.
- (a) W. Huang, S. Keess and G. Molander, Angew. Chem., Int. Ed., 2023, 62, e202302223 CrossRef CAS; (b) K. Schwrzer, H. Zipse, K. Karaghiosoff and P. Knochel, Angew. Chem., Int. Ed., 2020, 59, 20235–20241 CrossRef; (c) R. A. Shelp, A. Ciro, Y. Pu, R. R. Merchant, J. M. E. Hughes and P. J. Walsh, Chem. Sci., 2021, 12, 7066–7072 RSC; (d) M. Messner, S. I. Kozhushkov and D. A. Meijere, Eur. J. Org. Chem., 2000, 1137–1155 CrossRef CAS; (e) L. Yi, D. Kong, A. P. Kale, B. Maity, H. Yue, A. Gizatullin, R. Alshehri, R. Kancherla, L. Cavallo and M. Rueping, Angew. Chem., Int. Ed., 2024, e202411961 Search PubMed; (f) R. Gianatassio, J. M. Lopchuk, J. Wang, C.-M. Pan, L. R. Malins, L. Prieto, T. A. Brandt, M. R. Collins, G. M. Gallego, N. M. Sach, J. E. Spangler, H. Zhu, J. Zhu and P. S. Baran, Science, 2016, 351, 241–246 CrossRef CAS PubMed; (g) D. F. J. Caputo, C. Arroniz, A. B. Dürr, J. J. Mousseau, A. F. Stepan, S. J. Mansfield and E. A. Anderson, Chem. Sci., 2018, 9, 5295–5300 RSC; (h) R. A. Shelp and P. J. Walsh, Angew. Chem., Int. Ed., 2018, 57, 15857–15861 CrossRef CAS PubMed; (i) S. Yu, C. Jing, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2020, 59, 3917–3921 CrossRef CAS PubMed; (j) X. Zhang, R. T. Smith, C. Le, S. J. McCarver, B. T. Shireman, N. I. Carruthers and D. W. C. MacMillan, Nature, 2020, 580, 220–226 CrossRef CAS PubMed; (k) W. Dong, E. Yen-Pon, L. Li, A. Bhattacharjee, A. Jolit and G. A. Molander, Nat. Chem., 2022, 14, 1068–1077 CrossRef CAS PubMed.
- (a) Z. J. Garlets, J. N. Sanders, H. Malik, C. Gampe, K. N. Houk and H. M. L. Davies, Nat. Catal., 2020, 3, 351–357 CrossRef CAS; (b) I. F. Yu, J. L. Manske, A. Diéguez-Vázquez, A. Misale, A. E. Pashenko, P. K. Mykhailiuk, S. V. Ryabukhin, D. M. Volochnyuk and J. F. Hartwig, Nat. Chem., 2023, 15, 685–693 CrossRef CAS.
- A. S. Harmata, T. E. Spiller, M. J. Sowden and C. R. J. Stephenson, J. Am. Chem. Soc., 2021, 143, 21223–21228 CrossRef CAS.
- (a) S. Yu, Y. Ai, L. Hu, G. Lu, C. Duan and Y. Ma, Angew. Chem., Int. Ed., 2022, 61, e202200052 CrossRef CAS; (b) Y. Geng, Y. Ma, R. Huang, X. Li and S. Yu, Green Chem., 2023, 25, 221–228 RSC; (c) Y. Ma, Y. Ai and S. Yu, Synlett, 2023, 359–363 CAS.
- Z. Fan, D. A. Strassfeld, H. S. Park, K. Wu and J. Q. Yu, Angew. Chem., Int. Ed., 2023, 62, e202303948 CrossRef CAS PubMed.
- (a) J. Yu, Z. Wu and C. Zhu, Angew. Chem., Int. Ed., 2018, 57, 17156–17160 CrossRef CAS; (b) M. Luo, S. Zhu, J. Yin, C. Yang, L. Guo and W. Xia, Org. Chem. Front., 2024, 11, 4748–4756 RSC; (c) Z. Ma, X. Wu and C. Zhu, Chem. Rec., 2023, 23, e202200221 CrossRef CAS; (d) L. Song, D.-M. Fu, L. Chen, Y.-X. Jiang, J.-H. Ye, L. Zhu, Y. Lan, Q. Fu and D.-G. Yu, Angew. Chem., Int. Ed., 2020, 59, 21121–21128 CrossRef CAS PubMed; (e) J. Yu, H. Zhang, X. Wu and C. Zhu, CCS Chem., 2021, 3, 1426–1434 Search PubMed; (f) T. Rawner, E. Lutsker, C. A. Kaiser and O. Reiser, ACS Catal., 2018, 8, 3950–3956 CrossRef CAS; (g) F. Lucio-Martínez and W. Chaładaj, Adv. Synth. Catal., 2023, 365, 2092–2125 CrossRef; (h) X. Geng, F. Lin, X. Wang and N. Jiao, Org. Lett., 2017, 19, 4738–4741 CrossRef CAS; (i) D.-T. Xie, H.-L. Chen, D. Wei, B.-Y. Wei, Z.-H. Li, J.-W. Zhang, W. Yu and B. Han, Angew. Chem., Int. Ed., 2022, 61, e202203398 CrossRef CAS; (j) J.-S. Lin, F.-L. Wang, X.-Y. Dong, W.-W. He, Y. Yuan, S. Chen and X.-Y. Liu, Nat. Commun., 2017, 8, 14841–14851 CrossRef PubMed; (k) S. Engl and O. Reiser, ACS Catal., 2020, 10, 9899–9906 CrossRef CAS; (l) C. Rosso, J. D. Williams, G. Filippini, M. Prato and C. O. Kappe, Org. Lett., 2019, 21, 5341–5345 CrossRef CAS PubMed; (m) S. Barata-Vallejo, M. V. Cooke and A. Postigo, ACS Catal., 2018, 8, 7287–7307 CrossRef CAS.
- (a) K. U. Ingold, B. Maillard and J. C. Walton, J. Chem. Soc., Perkin Trans. 2, 1981, 2, 970–974 RSC; (b) G. Sissengaliyeva, F. Dénès, V. Girbu, V. Kulcitki, E. Hofstetter and P. Renaud, Adv. Synth. Catal., 2023, 365, 2568–2576 CrossRef CAS; (c) G. Li, T. Wang, F. Fei, Y.-M. Su, Y. Li, Q. Lan and X.-S. Wang, Angew. Chem., Int. Ed., 2016, 55, 3491–3495 CrossRef CAS.
- (a) H.-P. Cao and Q.-Y. Chen, J. Fluorine Chem., 2007, 128, 1187–1190 CrossRef CAS; (b) A. Liu, C. Ni, Q. Xie and J. Hu, Angew. Chem., Int. Ed., 2023, 62, e202217088 CrossRef CAS PubMed.
- (a) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- (a) X. Tang and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 814–817 CrossRef CAS PubMed; (b) F. Sladojevich, E. McNeill, J. Börgel, S.-L. Zheng and T. Ritter, Angew. Chem., Int. Ed., 2015, 54, 3712–3716 CrossRef CAS PubMed; (c) L. Woźniak, J. J. Murphy and P. Melchiorre, J. Am. Chem. Soc., 2015, 137, 5678–5681 CrossRef PubMed; (d) Y. Wang, J. Wang, G.-X. Li, G. He and G. Chen, Org. Lett., 2017, 19, 1442–1445 CrossRef CAS PubMed; (e) E. Zhu, X.-X. Liu, A.-J. Wang, T. Mao, L. Zhao, X. Zhang and C.-Y. He, Chem. Commun., 2019, 55, 12259–12262 RSC.
- (a) C. Sandford and V. K. Aggarwal, Chem. Commun., 2017, 53, 5481–5494 RSC; (b) A. J. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412–443 RSC.
- (a) R. Lekkala, R. Lekkala, B. Moku, K. P. Rakesh and H.-L. Qin, Eur. J. Org. Chem., 2019, 2769–2806 CrossRef CAS; (b) S. B. Ankade, A. B. Shabade, V. Soni and B. Punji, ACS Catal., 2021, 11, 3268–3292 CrossRef CAS; (c) J. Gu, X. Wang, W. Xue and H. Gong, Org. Chem. Front., 2015, 2, 1411–1421 RSC.
- Y. Cheng, C. Mück-Lichtenfeld and A. Studer, J. Am. Chem. Soc., 2018, 140, 6221–6225 CrossRef CAS PubMed.
- (a) R. L. Sutar and S. M. Huber, ACS Catal., 2019, 9, 9622–9639 CrossRef CAS; (b) D. Jovanovic, M. P. Mohanan and S. M. Huber, Angew. Chem., Int. Ed., 2024, 63, e202404823 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc05166a |
| This journal is © The Royal Society of Chemistry 2025 |