
Multitasking rhodium-catalyzed remote C(sp3)–H functionalization reactions of acyclic dienes to yield benzene-fused heterocycles†
Yuta
Sato
a,
Momoko
Nagafuchi
a,
Masaharu
Takatsuki
a,
Tsuyoshi
Matsuzaki
b,
Takeyuki
Suzuki
 b,
Makoto
Sako
a and
Mitsuhiro
Arisawa
*a
b,
Makoto
Sako
a and
Mitsuhiro
Arisawa
*a
aGraduate School of Pharmaceutical Sciences, Osaka University, 1-6, Yamada-oka, Suita, Osaka 565-0871, Japan. E-mail: arisaw@phs.osaka-u.ac.jp
bComprehensive Analysis Center, SANKEN, Osaka University, 8-1, Mihogaoka, Ibaraki, Osaka 567-0047, Japan
First published on 8th November 2024
Abstract
Remote C(sp3)–H bond functionalization reactions are environmentally benign methods for not only acyclic molecules but also cyclic molecules. However, products in previous reports have been limited to aliphatic five-membered ring structures. Herein, we developed an unprecedented multitasking rhodium-catalyzed reaction that can synthesize benzene-fused heterocyclic compounds, 2,3-disubstituted dihydrobenzofurans, through a remote C(sp3)–H bond functionalization reaction (chain-walking/cycloisomerization/hydroboration reaction). This catalytic system promotes a twice-occurring chain-walking/functionalization set in one pot, thus allowing the synthesis of dihydrobenzofurans with various side chain lengths at the 2- and 3-positions.
Introduction
Carbon–hydrogen (C–H) bonds are one of the most fundamental building blocks in organic molecules, and the site-selective functionalization of C–H bonds using transition metal catalysis has attracted the interest of many researchers.1,2 Particularly challenging is the functionalization of C(sp3)–H bonds due to the low acidity of alkane C(sp3)–H bonds and the low coordination ability of these bonds to metals. One approach to this challenge is a chain-walking/functionalization reaction using a multitasking catalyst.3 Multitasking catalysts are catalysts that promote multiple reactions successively. Multitasking catalysts are useful both synthetically and environmentally, because they can significantly change the structures of compounds in one pot. In this method, the typically inert C(sp3)–H bond is functionalized after the alkene is isomerized and the double bond is moved along the carbon chain. Various reports have detailed the remote alkylations,4 arylations,5 aminations,6 borylations,7 carbonylations,8etc., using palladium, nickel, rhodium, iridium, cobalt, iron, etc., with alkene chain-walking. However, most of these reports involve introducing functional groups onto alkyl chains through hydro-functionalization reactions, and examples, combining alkene chain-walking with reactions constructing cyclic structures (Fig. 1a), such as diene cycloisomerization reactions, are limited to cases using palladium catalysts, as reported by Kochi, Kakiuchi and co-workers (Fig. 1b).9 They reported palladium-catalyzed chain-walking/cycloisomerization reactions of 1,n-dienes using 1,10-phenanthroline9a,b or bioxazoline ligands.9c However, in these reports, the products were limited to aliphatic five-membered ring compounds, and there were no examples of applying chain-walking/cycloisomerization in the synthesis of benzene-fused heterocyclic compounds, such as 2,3-dihydrobenzofuran, although benzene-fused heterocyclic compounds are key skeletons in medicinal research. Also, in previous examples using palladium catalysts, the substituents on alkenes were usually limited to hydrogen or methyl groups, and the functional group tolerance of these methods has been unclear. Additionally, in the above cycloisomerization of dienes, the control of the position of the double bond after the reaction is a challenge (Fig. 1b).9d For example, if an alkene on the side chain is isomerized after the cycloisomerization of a 1,6-diene to form a five-membered ring compound, and it moves to the inside of the ring, the substituent on the side chain becomes a simple alkyl chain and subsequent chemical conversion is difficult. On the other hand, if isomerization of the alkene to the inside of the ring could be suppressed, further modification of the side chain in a one-pot process, such as hydroboration of the alkene moiety, would be possible.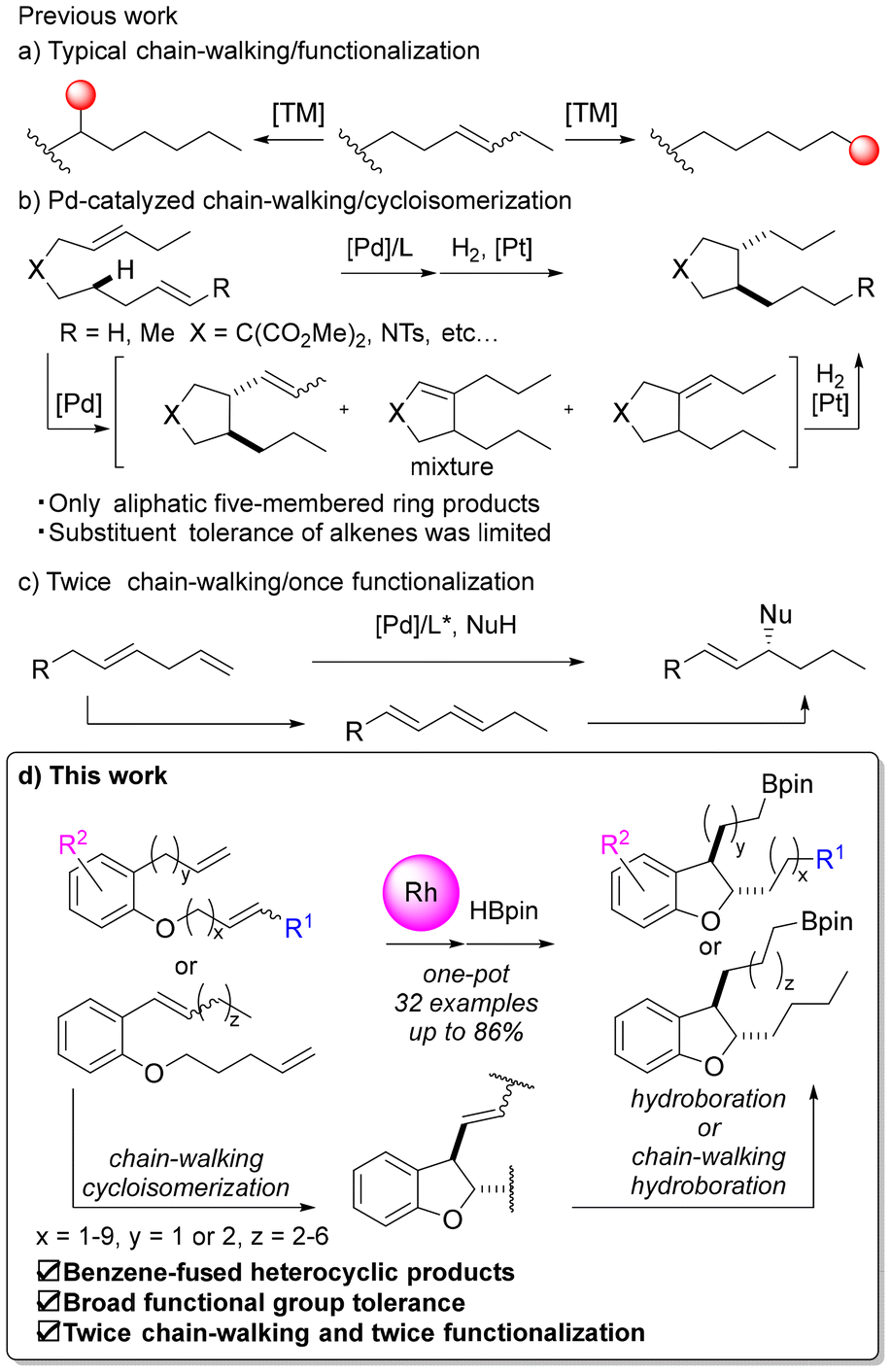 | ||
| Fig. 1 Chain-walking/functionalization reactions and this work. | ||
Furthermore, there have been several reported cases of multiple functionalization or chain-walking progressions, such as functionalization/chain-walking/functionalization,10 chain-walking/chain-walking/functionalization11 and chain-walking/functionalization/chain-walking.12 For example, He et al. reported allylation following chain-walking of dienes (Fig. 1c).11 However, there were no reported examples of reactions in which the chain-walking/functionalization set was repeated twice in one pot.
Therefore, in this study, we developed sustainable catalytic reactions with the following three features: (1) chain-walking and cycloisomerization of the internal alkene proceeds to yield 2,3-dihydrobenzofuran; (2) second chain-walking and subsequent hydroboration occur; and (3) hydroboration of the alkene on the side chain can be promoted while avoiding isomerization to the internal ring (Fig. 1d).
Results and discussion
We have previously developed a tandem catalytic reaction involving isomerization of cyclopropanes, isomerization of allyl ethers, cycloisomerization of dienes, and aromatization of 2,3-dihydrobenzofurans, resulting in the generation of benzofurans.13 As shown in Table 1, we started to optimize the reaction conditions to selectively obtain dihydrobenzofuran 2a using an internal alkene 1a with an n-butyl group as a substrate. First, we stirred a p-xylene solution (0.1 M) containing the substrate (0.12 mmol), 10 mol% Wilkinson's catalyst, and 10 mol% silver trifluoromethanesulfonate at 120 °C for 24 h (entry 1, best conditions mentioned in a previous report13). However, the cyclized compounds 2a and 2a′ were not obtained.|
|
||||
|---|---|---|---|---|
| Entry | [Rh] | Ligand | Temp (°C) | 2a/2a′a (%) |
| a NMR yield (1,3,5-trimethoxybenzene was used as an internal standard). b The reaction time was 14 h. c Isolated yield. | ||||
| 1 | RhCl(PPh3)3 | — | 120 | 0/0 |
| 2 | [Rh(C2H4)2Cl]2 | L1 | 120 | 14/63 |
| 3 | [Rh(C2H4)2Cl]2 | L2 | 120 | 0/0 |
| 4 | [Rh(C2H4)2Cl]2 | L3 | 120 | 69/26 |
| 5 | [Rh(C2H4)2Cl]2 | L3 | 100 | 75/17 |
| 6 | [Rh(C2H4)2Cl]2 | L3 | 110 | 82/17 |
| 7 | [Rh(C2H4)2Cl]2 | L4 | 110 | 0/0 |
| 8 | [Rh(C2H4)2Cl]2 | L5 | 110 | 17/0 |
| 9 | [Rh(C2H4)2Cl]2 | L6 | 110 | 69/20 |
| 10 | [Rh(C2H4)2Cl]2 | L7 | 110 | 60/7 |
| 11b | [Rh(C2H4)2Cl]2 | L3 | 110 | 82c/15c |
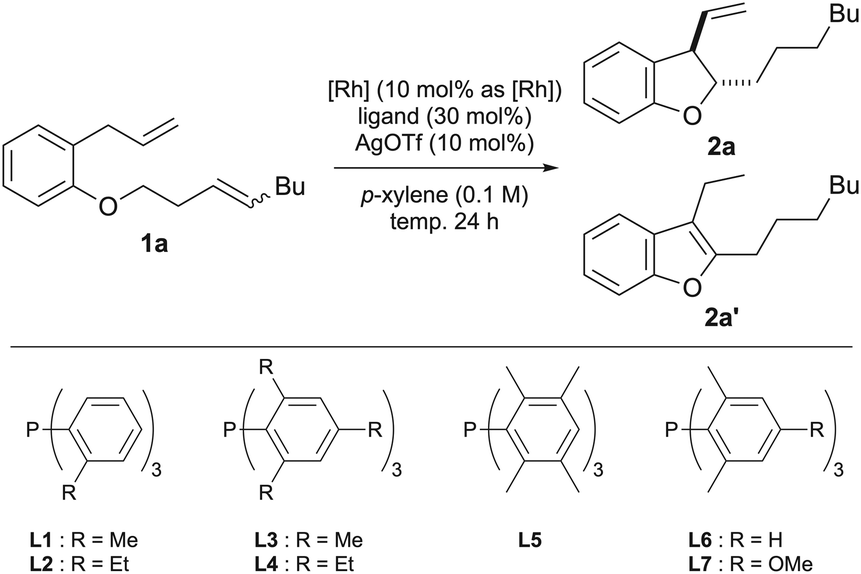
Therefore, the rhodium catalyst was changed to [Rh(C2H4)2Cl]2 and the effect of the ligand was investigated. Using L1 as the ligand, which is bulkier than triphenylphosphine, the desired dihydrobenzofuran 2a and benzofuran 2a′ were obtained in 14% and 63% yields, respectively (entry 2). When L2 with an ethyl group as a substituent at the ortho position on the benzene ring was used, cycloisomerization did not proceed (entry 3). In big contrast, the use of L3 with a methyl group at 2, 4 and 6-positions on the benzene ring gave 2a in 69% yield and 2a′ in 26% yield, reversing the selectivity of the products (entry 4). Therefore, we investigated the reaction temperature with L3 (entries 5 and 6) and obtained 2a in a high yield (82%) when the reaction was conducted at 110 °C. When L4, which has ethyl groups at positions 2, 4 and 6 on the benzene ring, was used, isomerization of the alkene proceeded, however, no cyclized product was obtained (entry 7). Subsequently, ligands with methyl groups at the 2, 3, 5 and 6 positions of the benzene ring were examined, but the yield of cyclized products decreased (entry 8). These results indicate that bulky ligands are effective in inhibiting aromatization; however, too much crowding of the metal center reduces the efficiency of cyclization itself. One possible reason why a moderately bulky ligand was effective is that the rotation of the alkyl chain of the intermediate, which is caused by ring formation by carbometalation or insertion of the alkene moiety of 2 into the rhodium hydride, is suppressed by the steric repulsion between the bulky ligand and the side chain at the dihydrobenzofuran 2-position (Fig. 2).
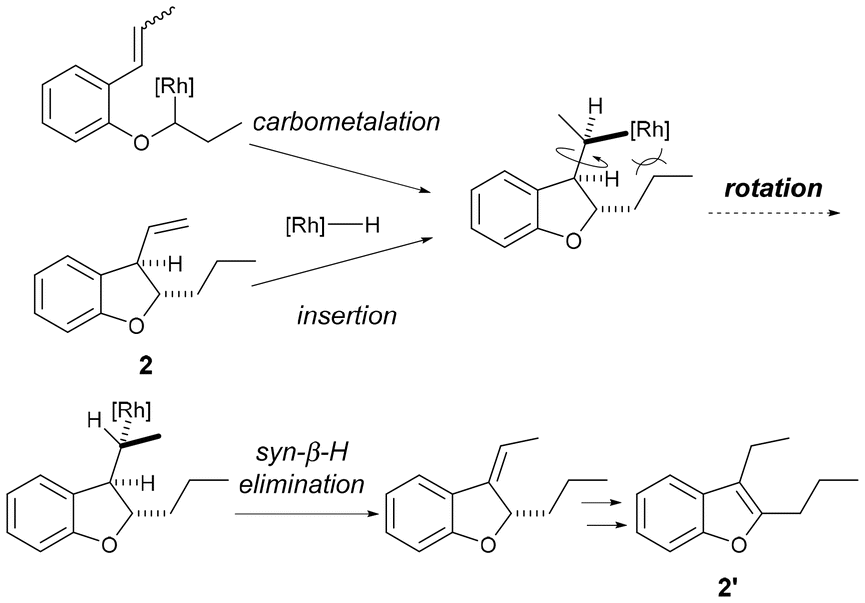 | ||
| Fig. 2 Ligand effects to yield 2 and 2′. | ||
This might have prevented the 2,3-dihydrobenzofuran from taking a conformation that would allow syn-β-hydrogen elimination of the hydrogen at the 3-position to proceed, thereby suppressing aromatization to 2′. To further investigate the electronic effects of substituents on the benzene ring of the ligand, we examined the substituent at the para-position of the benzene ring. When L6, which does not have an electron-donating methyl group, was used, the selectivity of the product decreased, and when L7, which has a more electron-donating methoxy group, was used, the yield of the cyclized product decreased. These results indicate that L3 was the optimal ligand. Finally, the reaction time was examined, and it was found that the yield was maintained even if the reaction was stopped after 14 hours (entry 11). Therefore, the conditions of entry 11, in which 5 mol% [Rh(C2H4)2Cl]2, 30 mol% L3, and 10 mol% silver trifluoromethanesulfonate were added and stirred at 110 °C in p-xylene for 14 hours, were determined to be optimal conditions. The effects of substituents on the ligand were compared using DFT calculations, but the calculations did not provide data to supplement the experimental results. Details of the calculations are described in the ESI.†
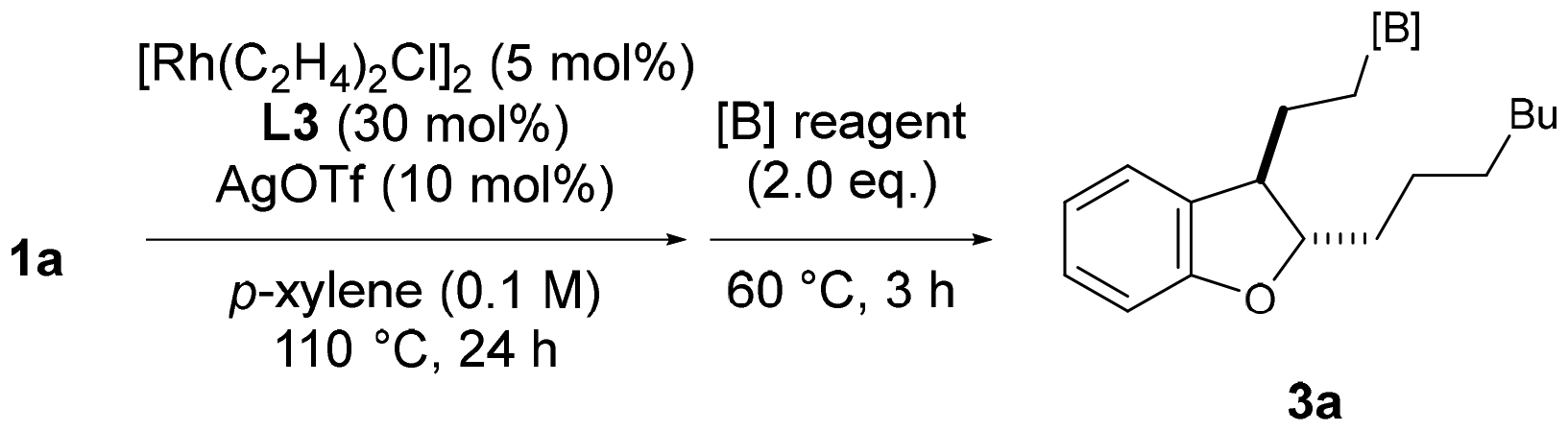
When 1a was subjected to the reaction conditions reported by Kochi and Kakiuchi9a using a palladium phenanthroline complex, the total yield of the cyclized product was 17%, indicating that the rhodium catalyst system is suitable for obtaining dihydrobenzofuran. One-pot hydroboration was then attempted by adding two equivalents of borylating agent after stirring under optimal conditions. First, pinacol borane was added, yielding 81% of 3a (Table 2, entry 1). On the other hand, when bis(pinacolato)diboron or bis(neopentylglycolato)diboron was used, no borylated product was obtained (entries 2 and 3). Therefore, we decided to investigate the substrate scope of the chain-walking/cycloisomerization/hydroboration reaction using pinacol borane as the borylating agent.
First, as shown in Scheme 1, the effects of carbon chain length and substituents on the alkenyl ether were investigated. As a result of examining the carbon chain length of the terminal alkene, it was confirmed that long distance chain-walking proceeds, although a slight decrease in yield is observed as the carbon chain length increases (3b–3f). In addition, the introduction of an n-pentyl group at the end of the long alkene chain resulted in lower yields of the corresponding cyclized products compared to that of the unsubstituted substrates (3g and 3h). From this result, we hypothesized that the chain-walking of the internal alkene proceeds not only in the direction of the ether oxygen but also in the direction of the alkyl chain terminus, resulting in a dispersion of the alkene position and thus a decrease in the yield. We then examined 1i and 1j, which have a cyclohexane ring or a benzene ring at the alkyl end. These substrates resulted in lower yields of the cyclized compound when the reaction was conducted at 110 °C. When the reaction was carried out at 120 °C, the corresponding products were obtained in yields of 70% and 41%, respectively (3i and 3j). The corresponding dihydrobenzofuran 3k was also obtained from 1k, which has a conjugated alkene with the benzene ring. Substrates 1l to 1q, which have substituents such as esters, acetals, sulfonamides, and imides at the terminus, were used to further explore the functional group tolerance of this reaction. These substrates were subjected to the above optimal reaction conditions at 120 °C. The corresponding product 3l was obtained in good yield from 1l with an ester at the end. On the other hand, in the reaction of 1m with the α,β-unsaturated ester, the catalyst was deactivated before the cycloisomerization, and only isomerization of the alkene proceeded. This may be because the α,β-unsaturated ester is more stable, more electron deficient, and less reactive. In the study of 1n with acetal at the end, the corresponding dihydrobenzofuran 3n was obtained in a good yield despite its long chain. Product 3o was also obtained in a moderate yield from 1o having a sulfonamide at the end. From 1p, in which the terminal amine was protected with a Boc group and a Ts group, the product 3p was obtained in a good yield with the Boc group deprotected (the yield was calculated after oxidation of the boronate ester moiety into OH). The corresponding product 3q was also obtained in a good yield from 1q with a phthalimide substituted at the terminal end. These results indicate that our developed reaction can synthesize dihydrobenzofurans with different functional groups at the ends of the side chains at positions 2 and 3.
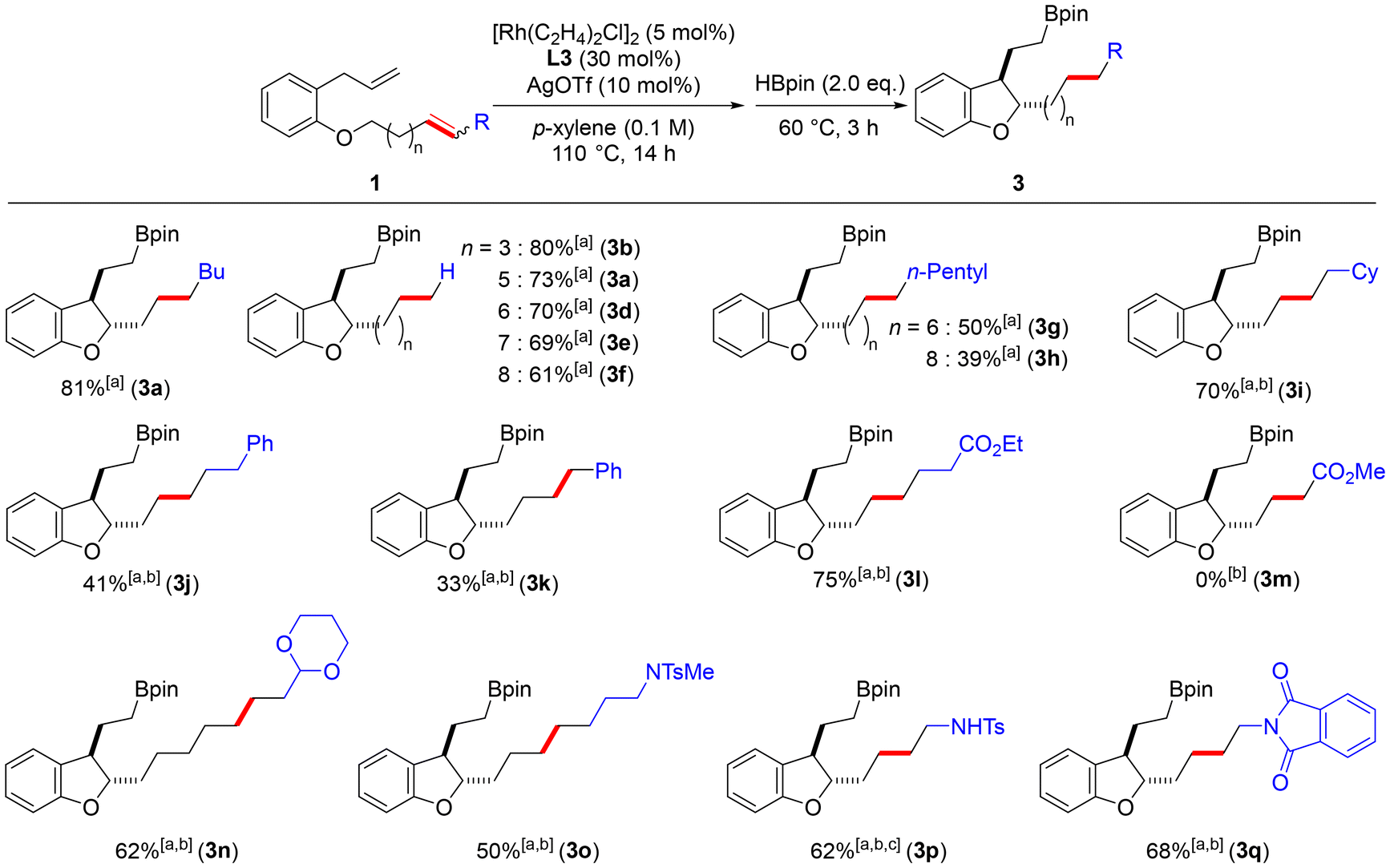 | ||
Scheme 1 Substrate scope (length of the alkenyl ethers and substituents of the alkene). a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Isolated yields. bThe first step at 120 °C. cFrom 1p (R = CH2NTsBoc). Yield was calculated after oxidation of the Bpin moiety into OH. Isolated yields. bThe first step at 120 °C. cFrom 1p (R = CH2NTsBoc). Yield was calculated after oxidation of the Bpin moiety into OH. | ||
Since it was found that the reaction tolerates a variety of substituents on the alkenyl ether, the effects of substituents on the benzene ring were subsequently investigated. Dihydrobenzofurans with substituents at positions 4 to 7 were synthesized by introducing substituents into the benzene ring of substrates having esters at the ends of ether side chains (Scheme 2). Dihydrobenzofurans bearing electron-withdrawing fluoro or chloro groups and electron-donating methoxy groups at positions 4 to 7 were obtained in moderate to good yields. In particular, dihydrobenzofurans with substituents at the 4-position were obtained in good yields (3r–3t). We believe that this is because the substituent at position 4 sterically prevents the alkene moiety of dihydrobenzofuran 2 from re-coordinating to rhodium, thereby suppressing aromatization. Furthermore, dihydrobenzofuran 3x with an ester at position 5 was obtained in a good yield from 1x. These results show that this reaction also tolerates various substituents on the benzene ring.
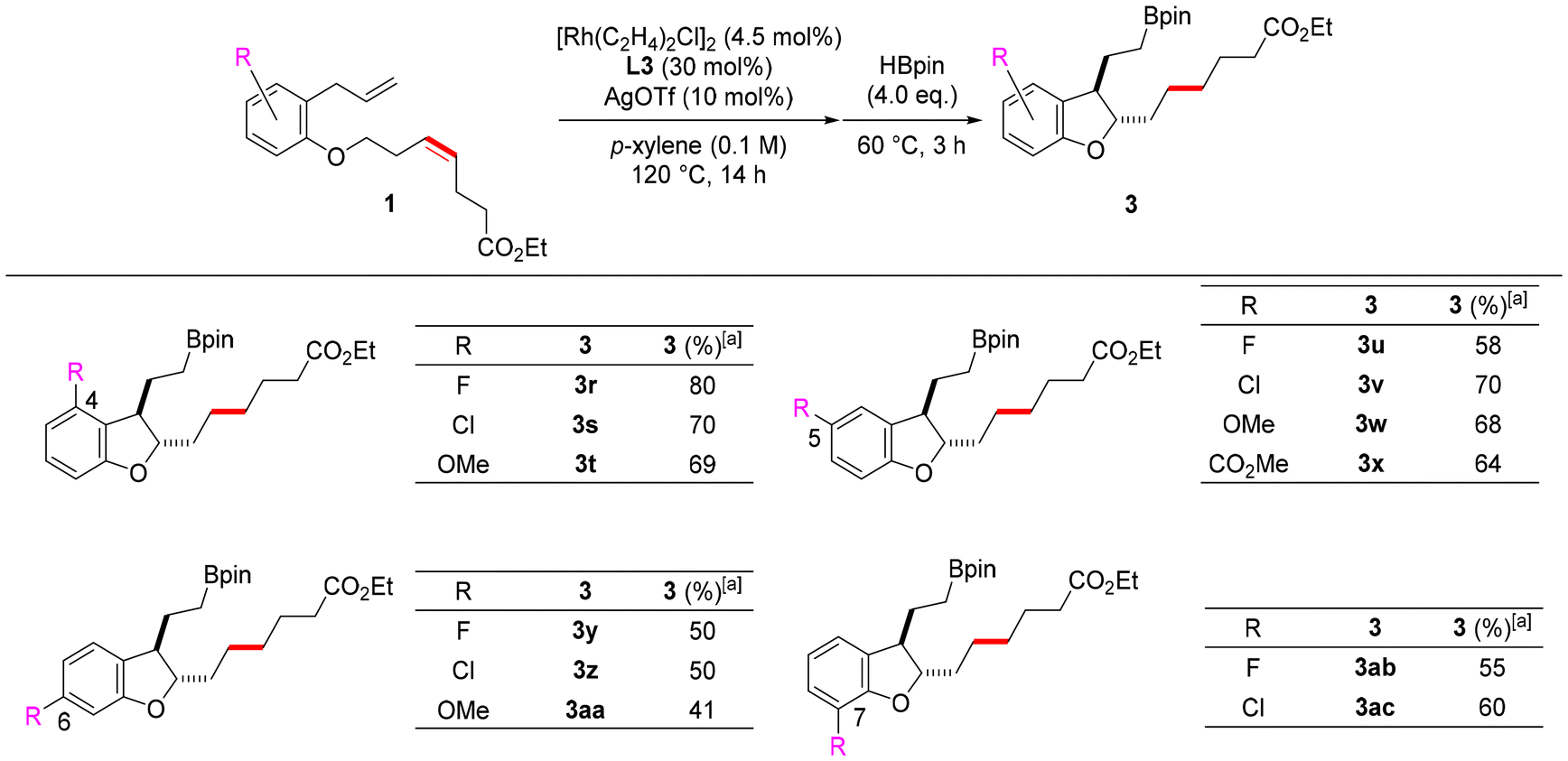 | ||
| Scheme 2 Substrate scope (substituents of the benzene ring). aIsolated yields. | ||
The length of the side chain at the ortho position of the alkoxy group on the benzene ring was then changed to examine whether chain-walking to the end of the side chain and subsequent hydroboration proceeded after cycloisomerization. When the substrate 1ad, whose carbon chain length was increased by one carbon, was used, chain-walking to the interior at two sites, cycloisomerization, chain-walking to the alkyl chain end, and subsequent hydroboration proceeded smoothly, and the desired 3ad was obtained in a high yield (86%, Scheme 3). The carbon chain length at which chain-walking/hydroboration proceeds after cycloisomerization was further investigated using substrates 1ae–1ag. As a result, 3ae and 3af were obtained in good yields from 1ae and 1af with internal alkenes substituted with n-propyl and n-butyl groups, respectively. The corresponding dihydrobenzofuran 3ag was also obtained in a moderate yield from 1ag having an n-heptyl group as a substituent of the alkene. These results indicate that this catalyst system also promotes chain-walking in both the internal and external directions of the alkyl chain and can introduce boronic esters at the ends of the side chains of various dihydrobenzofurans with different carbon lengths of the 3-position side chain. This reaction is the first example in which the chain-walking/functionalization set proceeds twice in one-pot.
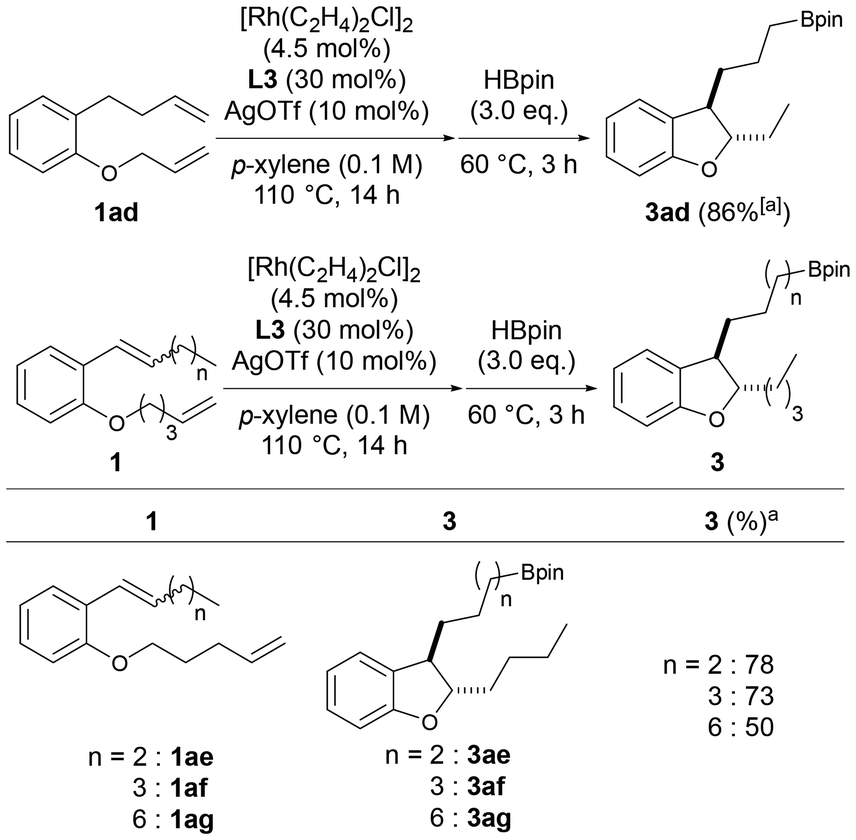 | ||
| Scheme 3 Substrate scope (length of the alkenyl side chain). aIsolated yields. | ||
To confirm the utility of the products obtained in this reaction, chemical transformations of the products were investigated (Scheme 4). First, we performed the conversion of 3l, which has an ester at the end of the 2-position side chain. Oxidation of the boronic ester moiety using sodium peroxoborate tetrahydrate was examined, and the corresponding alcohol 4 was obtained in a good yield (90%). The ester moiety at the 2-position side chain end can also be converted to alcohol by diisobutylaluminum hydride, and 5 was obtained in a good yield. Also, the boronic acid ester moiety at the 3-position side chain end of dihydrobenzofuran 3af, which was obtained by dual chain-walking/functionalization, could be subjected to Suzuki–Miyaura coupling conditions to introduce an aryl group (6). These results indicate that each of the 2- and 3-position side chain ends of the dihydrobenzofuran obtained by this reaction can be converted.
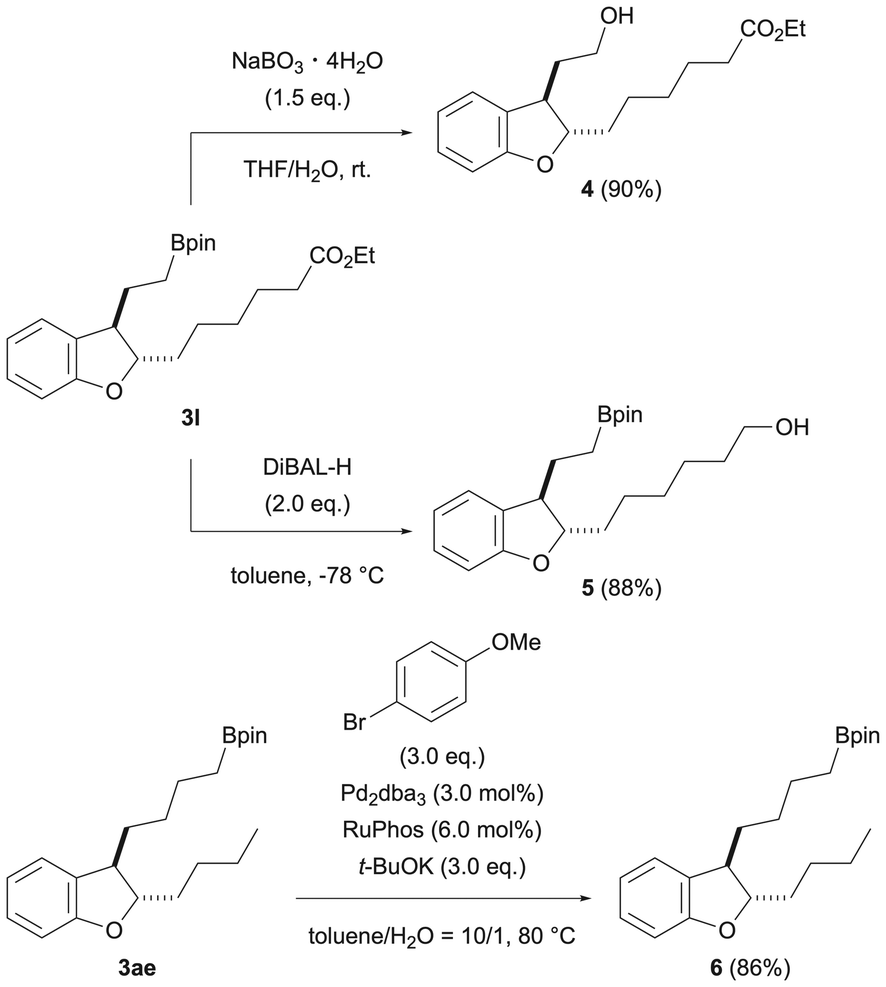 | ||
| Scheme 4 Chemical transformations. | ||
Finally, we considered the reaction mechanism of this reaction. Chain-walking mechanisms for alkenes include a 1,2-hydride shift mechanism involving repeated insertion of an alkene into a metal hydride and β-hydride elimination, an inner-sphere-type 1,3-hydride shift mechanism via the activation of C–H bonds adjacent to double bonds to form a π-allyl intermediate, and an outer-sphere-type 1,3-hydride shift mechanism where the ligand acts as a base.14
To investigate the chain-walking mechanism within the continuous reaction, we conducted deuterium labelling experiments. Substrates with deuterium-labelled terminal alkenes and internal alkenes were synthesized15 and subjected to the optimal conditions. The deuterium incorporation ratio of the products was calculated by adding chloroform-d as an internal standard to the chloroform solution of the products and measuring by 2H-NMR.
First, as shown in Fig. 3a, we subjected 1ah-d2, in which the terminal alkene was deuterated to 56%, to the conditions of the present catalytic reaction. The resulting product 2ah-d2 was found to have deuterium introduced at all positions except the oxygen α position. From these results, it is suggested that isomerization of the alkene in this reaction is reversible, proceeding through a 1,2-hydride shift mechanism involving repeated insertion of the alkene into a rhodium hydride and β-hydride elimination. Furthermore, when attempting the reaction with the 80% deuterium-labelled internal alkene 1ai-d1, we observed that the hydrogen in the terminal methyl group of the 2-position side chain of the resulting dihydrobenzofuran 2ai-d1 was exchanged with deuterium at 3% of the 12 protons in the six-carbon methylene chain, 4% of the terminal proton in the vinyl group at the 3-position side chain, 3% of the internal protons, and 2% of the protons at the 3-position (Fig. 3b). From these results, it was confirmed that chain-walking of the internal alkene occurs not only in the direction of the ether oxygen on the alkyl chain but also in the terminal direction. In all of these experiments, the introduction of deuterium into the α position of the ether oxygen was not observed. Therefore, it is assumed that the subsequent cycloisomerization, leading to vinyl ether, proceeds rapidly.
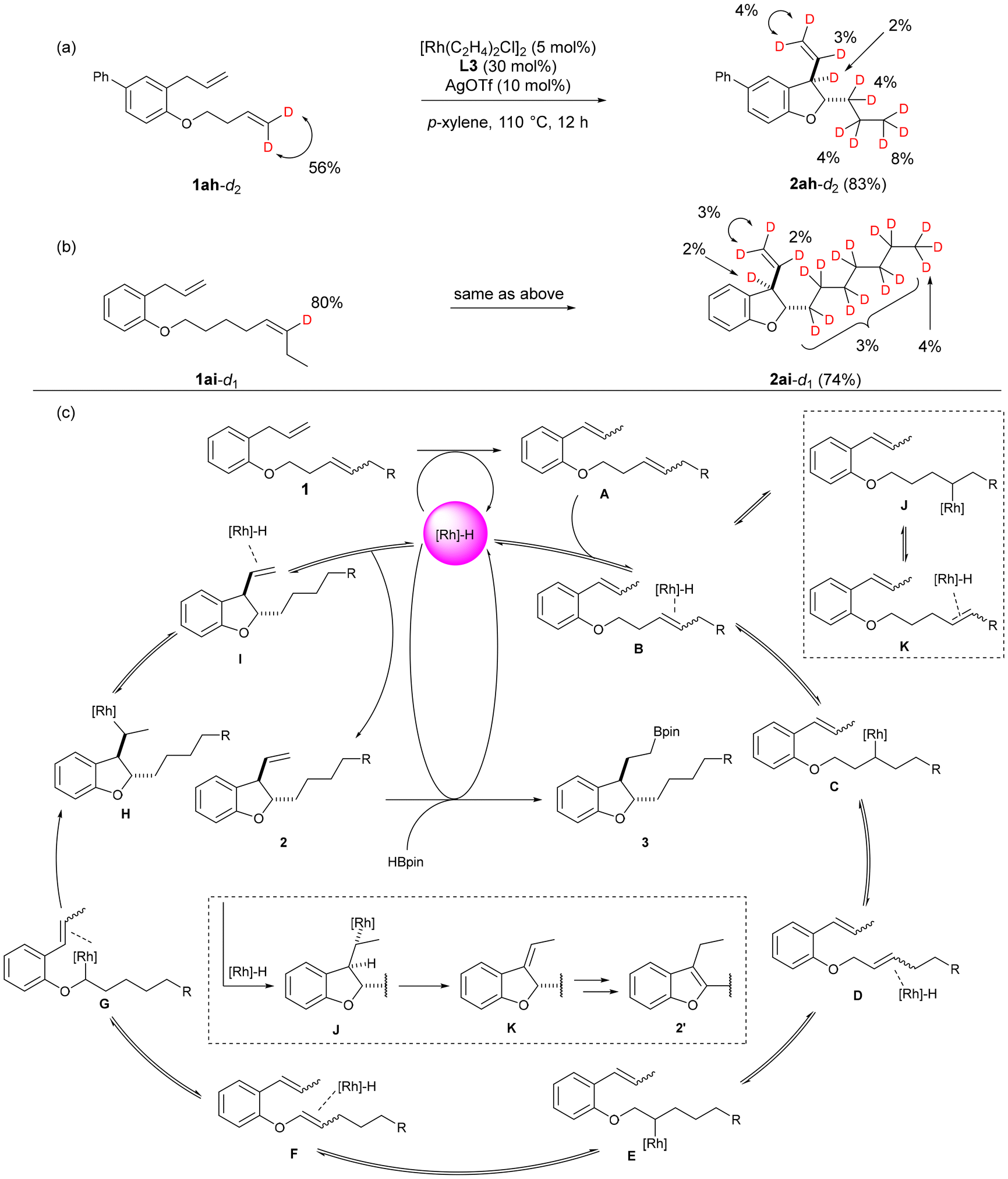 | ||
| Fig. 3 (a and b) Deuterium-labeled experiments. (c) A proposed reaction mechanism. | ||
Based on the results of the deuterium labelling experiments, we propose a plausible reaction mechanism for this reaction (Fig. 3c). First, the double bond of the allyl group of substrate 1 coordinates to the generated rhodium hydride in the system, and isomerization proceeds to form A. Subsequently, after the internal alkene on the ether side chain of A coordinates to the rhodium hydride, the alkene insertion leads to the formation of C, followed by the progression of β-hydride elimination to generate D. The insertion of the alkene and β-hydride elimination are repeated until the double bond reaches the α position of the oxygen. When this occurs, an alkyl rhodium intermediate G is generated.
Furthermore, G is converted to H through carbometalation, and subsequent β-hydride elimination leads to the formation of 2,3-dihydrobenzofuran 2. It is also suggested that isomerization of the alkene toward the end of the side chain, leading to intermediate K, occurs from B. However, due to the reversibility of the alkene isomerization, it is expected that, over time, the reaction will converge to form a cyclized product 2. Finally, the alkylrhodium species formed by the insertion of the terminal alkene of 2 into rhodium hydride reacts with pinacolborane or the alkene reacts with borylrhodium species to form 3.
Conclusions
In summary, we developed a multitasking Rh-catalyzed continuous reaction involving chain-walking, cycloisomerization, and hydroboration, leading to 2,3-disubstituted 2,3-dihydrobenzofurans. Through optimization, it was discovered that the use of a moderately bulky trimesitylphosphine ligand (L3) allows effective chain-walking and cycloisomerization while suppressing aromatization, enabling one-pot hydroboration. This reaction represents the first synthesis of benzene-fused heterocyclic compounds using chain-walking as an indirect method for C(sp3)–H bond functionalization. Furthermore, this catalytic reaction is the first example of a one-pot process involving two rounds of a chain-walking/functionalization set. Additionally, this reaction tolerates various substituents on the alkenyl ether and benzene ring, leading to the synthesis of polysubstituted 2,3-dihydrobenzofurans with different functional groups not only at the ends of the side chains at 2- and 3-positions,16 but also at 4–7 positions. Moreover, the obtained products are amenable for further chemical transformations. We believe that this reaction is a useful reaction that can solve these existing problems in chemistry. The results of deuterium labelling experiments suggest that the isomerization in this reaction is reversible and likely proceeds through a 1,2-hydride shift mechanism. Furthermore, the isomerization of the internal alkene was confirmed to proceed in both directions along the ether side chain.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest.Acknowledgements
This work was supported in parts by the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant Number 23H02608 (M. A.), the Grant-in-Aid from JST, Japan (CREST Grant Number JP JPMJCR20R1), the Naito Foundation (M. A.), The Sumitomo Foundation (M. A.), The Nippon Foundation – Osaka University Project for Infectious Disease Prevention (M. A.) and the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under Grant Number JP24ama121054 (M. A.). The computation was performed at the Research Center for Computational Science, Okazaki, Japan (project 24-IMS-C213).References
- Selected recent reviews of directed C–H functionalization reactions. (a) T. Dalton, T. Faber and F. Glorius, ACS Cent. Sci., 2021, 7, 245–261 CrossRef CAS PubMed; (b) R. Bisht, C. Haldar, M. M. M. Hassan, M. E. Hoque, J. Chaturvedi and B. Chattopadhyay, Chem. Soc. Rev., 2022, 51, 5042–5100 RSC; (c) J. H. Docherty, T. M. Lister, G. Mcarthur, M. T. Findlay, P. Domingo-Legarda, J. Kenyon, S. Choudhary and I. Larrosa, Chem. Rev., 2023, 123, 7692–7760 CrossRef CAS PubMed.
- Selected examples of nondirected C–H functionalization reactions. (a) J. Hao, X. Guo, S. He, Z. Xu, L. Chen, Z. Li, B. Song, J. Zuo, Z. Lin and W. Yang, Nat. Commun., 2021, 12, 1–11 CrossRef; (b) R. Rocaboy, D. Dailler, F. Zellweger, M. Neuburger, C. Salomé, E. Clot and O. Baudoin, Angew. Chem., Int. Ed., 2018, 57, 12131–12135 CrossRef CAS; (c) B. Tan, L. Bai, P. Ding, J. Liu, Y. Wang and X. Luan, Angew. Chem., Int. Ed., 2019, 58, 1474–1478 CrossRef CAS PubMed; (d) S. Le Cai, Y. Li, C. Yang, J. Sheng and X. S. Wang, ACS Catal., 2019, 9, 10299–10304 CrossRef; (e) J. L. Han, Y. Qin, C. W. Ju and D. Zhao, Angew. Chem., Int. Ed., 2020, 59, 6555–6560 CrossRef CAS; (f) R. Rocaboy, I. Anastasiou and O. Baudoin, Angew. Chem., Int. Ed., 2019, 58, 14625–14628 CrossRef CAS; (g) G. Zhang, M. Y. Li, W. B. Ye, Z. T. He, C. G. Feng and G. Q. Lin, Org. Lett., 2021, 23, 2948–2953 CrossRef CAS; (h) T. Čarný, R. Rocaboy, A. Clemenceau and O. Baudoin, Angew. Chem., Int. Ed., 2020, 59, 18980–18984 CrossRef; (i) A. Clemenceau, P. Thesmar, M. Gicquel, A. Le Flohic and O. Baudoin, J. Am. Chem. Soc., 2020, 142, 15355–15361 CrossRef CAS PubMed; (j) P. A. Provencher, J. F. Hoskin, J. J. Wong, X. Chen, J. Q. Yu, K. N. Houk and E. J. Sorensen, J. Am. Chem. Soc., 2021, 143, 20035–20041 CrossRef CAS PubMed; (k) T. Miyakoshi, N. E. Niggli and O. Baudoin, Angew. Chem., Int. Ed., 2022, 61, 1–8 CrossRef.
- (a) F. X. Felpin, J. Coste, C. Zakri and E. Fouquet, Chem. – Eur. J., 2009, 15, 7238–7245 CrossRef CAS; (b) G. You, Z. X. Chang, J. Yan, C. Xia, F. R. Li and H. S. Li, Org. Chem. Front., 2021, 8, 39–45 RSC; (c) X. Miao, C. Fischmeister, C. Bruneau and P. H. Dixneuf, ChemSusChem, 2009, 2, 542–545 CrossRef CAS; (d) S. Fustero, J. Mirõ, M. Sánchez-Rosellõ and C. Del Pozo, Chem. – Eur. J., 2014, 20, 14126–14131 CrossRef CAS.
- (a) F. Zhou, J. Zhu, Y. Zhang and S. Zhu, Angew. Chem., Int. Ed., 2018, 57, 4058–4062 CrossRef CAS; (b) F. Zhou, Y. Zhang, X. Xu and S. Zhu, Angew. Chem., Int. Ed., 2019, 58, 1754–1758 CrossRef CAS.
- (a) Y. He, Y. Cai and S. Zhu, J. Am. Chem. Soc., 2017, 139, 1061–1064 CrossRef CAS; (b) Y. He, C. Liu, L. Yu and S. Zhu, Angew. Chem., Int. Ed., 2020, 59, 9186–9191 CrossRef CAS PubMed; (c) A. J. Borah and Z. Shi, J. Am. Chem. Soc., 2018, 140, 6062–6066 CrossRef CAS PubMed.
- (a) J. Xiao, Y. He, F. Ye and S. Zhu, Chem, 2018, 4, 1645–1657 CrossRef CAS; (b) Y. Zhang, J. He, P. Song, Y. Wang and S. Zhu, CCS Chem., 2021, 3, 2259 CrossRef CAS; (c) C. Han, Z. Fu, S. Guo, X. Fang, A. Lin and H. Yao, ACS Catal., 2019, 9, 4196–4202 CrossRef CAS.
- (a) N. G. Léonard, W. N. Palmer, M. R. Friedfeld, M. J. Bezdek and P. J. Chirik, ACS Catal., 2019, 9, 9034–9044 CrossRef; (b) M. Hu and S. Ge, Nat. Commun., 2020, 11, 1–10 CrossRef; (c) J. Li, S. Qu and W. Zhao, Angew. Chem., Int. Ed., 2020, 59, 2360–2364 CrossRef CAS; (d) M. Zhang, Z. Liu and W. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202215455 CrossRef CAS PubMed.
- (a) F. Juliá-Hernández, T. Moragas, J. Cornella and R. Martin, Nature, 2017, 545, 84–88 CrossRef; (b) J. He, P. Song, X. Xu, S. Zhu and Y. Wang, ACS Catal., 2019, 9, 3253–3259 CrossRef CAS.
- (a) T. Kochi, T. Hamasaki, Y. Aoyama, J. Kawasaki and F. Kakiuchi, J. Am. Chem. Soc., 2012, 134, 16544–16547 CrossRef CAS PubMed; (b) T. Hamasaki, Y. Aoyama, J. Kawasaki, F. Kakiuchi and T. Kochi, J. Am. Chem. Soc., 2015, 137, 16163–16171 CrossRef CAS PubMed; (c) M. Shigekane, T. Arai, M. Tamura, T. Uchida, F. Kakiuchi and T. Kochi, Tetrahedron Lett., 2023, 114, 154292 CrossRef CAS; (d) T. Hamasaki, PhD thesis, Keio University, JP, 2015.
- (a) S. Kanno, F. Kakiuchi and T. Kochi, J. Am. Chem. Soc., 2021, 143, 19275–19281 CrossRef CAS PubMed; (b) S. Kanno, F. Kakiuchi and T. Kochi, J. Org. Chem., 2023, 88, 2621–2630 CrossRef CAS PubMed; (c) D. Zhu, Z. Jiao, Y. R. Chi, T. P. Gonçalves, K. W. Huang and J. S. Zhou, Angew. Chem., Int. Ed., 2020, 59, 5341–5345 CrossRef CAS PubMed.
- X. X. Chen, H. Luo, Y. W. Chen, Y. Liu and Z. T. He, Angew. Chem., Int. Ed., 2023, 62, e202307628 CrossRef CAS.
- S. Singh, J. Bruffaerts, A. Vasseur and I. Marek, Nat. Commun., 2017, 8, 1–10 CrossRef.
- Y. Sato, T. Matsuzaki, T. Takehara, M. Sako, T. Suzuki and M. Arisawa, Chem. Commun., 2022, 58, 415–418 RSC.
- A. Vasseur, J. Bruffaerts and I. Marek, Nat. Chem., 2016, 8, 209–219 CrossRef CAS PubMed.
- M. Yamamoto, K. Oshima and S. Matsubara, Chem. Lett., 2004, 33, 846–847 CrossRef CAS.
- Selected examples of the synthesis of trans-2,3-dihydrobenzofurans with alkyl substituents at the 2- and 3-positions. (a) Y. Liu, A. Lu, K. Hu, Y. Wang, H. Song, Z. Zhou and C. Tang, Eur. J. Org. Chem., 2013, 4836–4843 CrossRef CAS; (b) A. B. Dapkekar, C. Sreenivasulu, D. R. Kishiore and G. Satyanarayana, Asian J. Org. Chem., 2022, 11, e202200012 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc04927f |
| This journal is © The Royal Society of Chemistry 2025 |