
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A lactic acid dioxolane as a bio-based solvent for lithium-ion batteries: physicochemical and electrochemical investigations of lithium imide-based electrolytes†
Massimo
Melchiorre‡
 ab,
Khai Shin
Teoh‡
cd,
Juan Luis
Gómez Urbano
cd,
Francesco
Ruffo
*ae and
Andrea
Balducci
*cd
ab,
Khai Shin
Teoh‡
cd,
Juan Luis
Gómez Urbano
cd,
Francesco
Ruffo
*ae and
Andrea
Balducci
*cd
aDipartimento di Scienze Chimiche, Università degli Studi di Napoli Federico II, Complesso Universitario di Monte S. Angelo, Via Cintia 21, 80126, Napoli, Italy. E-mail: ruffo@unina.it
bISusChem S.r.l., Piazza Carità 32, 80134 Napoli, Italy
cInstitute for Technical Chemistry and Environmental Chemistry, Friedrich-Schiller University Jena, Philosophenweg 7a, 07743 Jena, Germany. E-mail: andrea.balducci@uni-jena.de
dCenter for Energy and Environmental Chemistry Jena (CEEC Jena). Friedrich-Schiller University Jena, Philosophenweg 7a, 07743 Jena, Germany
eConsorzio Interuniversitario di Reattività Chimica e Catalisi (CIRCC), Via Celso Ulpiani 27, 70126, Bari, Italy
First published on 2nd January 2025
Abstract
In this study we report for the first time the application of an emerging bio-based solvent derived from lactic acid, namely 5-methyl-1,3-dioxolane-4-one (LA-H,H), as an electrolyte component for lithium-ion batteries (LIBs). Electrolyte formulations consisting of this novel bio-solvent and imide conducting salts (i.e. lithium bis(trifluoromethanesulfonyl)imide, LiTFSI, and lithium bis(fluorosulfonyl)imide, LiFSI) and the additive vinylene carbonate (VC) are prepared and thoroughly evaluated. Resulting formulations demonstrate suitable transport properties (e.g., conductivity, viscosity) and considerably low flammability compared to standard electrolyte formulations. The compatibility of the novel imide-based electrolytes with benchmark active materials such as graphite (GR) and lithium iron phosphate (LFP) are explored. The results indicate that the use of LA-H,H-LiTFSI 1 M 5 wt% VC allows high electrochemical performance in terms of rate-capability and cycling stability for both the graphite (339 mA h g−1 at 1C) and the LFP (100 mA h g−1 at 1C) electrodes. The suitability of this novel electrolyte configuration was further demonstrated through the assembly of a lab-scale full-cell LIB showing remarkable rate capability and cycling stability. These results indicate that LA-H,H can be used as an electrolyte component for LIBs, and pave the way for its use as bio-based solvent in energy storage systems.
Green foundation1. This work introduces a novel bio-based and biodegradable solvent, namely 5-methyl-1,3-dioxolane-4-one (LA-H,H), for lithium-ion batteries. LA-H,H can be produced from biomass derived synthons, such as lactic acid, which reduces the burden associated with conventional electrolyte components, generally relying on non-renewable sources.2. The use of a bio-derived solvent along with imide-based salts overcomes some inherent limitations of conventional electrolytes, such as HF formation or room temperature flammability. Besides, the herein studied formulations demonstrate similar transport properties to those of conventional electrolytes produced from non-renewable sources and a wide voltage window. 3. The combination of this novel type of solvent with suitable bio-derived co-solvents and additives will be pursued with the aim of improving the overall sustainability, safety and performance of lithium-ion battery electrolytes. |
Introduction
One of the main challenges of this century is the energetic transition from fossil-based feedstocks to fully renewable energy sources (RESs).1 The recent escalation of extreme climatic events,2,3 combined with global warming due to greenhouse effects,4,5 clearly underlines the need to reduce our carbon footprint by moving from a linear to a circular economy.6 Within this framework, the energy storage systems (ESSs) have a key role as RESs are unstable and intermittent.7 Among them, lithium-ion batteries (LIBs) have become one of the most widely used energy storage devices8 thanks to their high energy density (up to 300 W h kg−1),9 long cycle life (thousands of cycles),10 low self-discharge rate (<2% per month),11 and high versatility, which makes this technology suitable for tailored design systems in both micro (e.g. electronics)12 and macro scales (e.g. battery packs and uninterruptible power supply systems).13The LIB energy storage mechanism relies on reversible intercalation/deintercalation of Li+ in the anode and cathode materials. This process is mediated by the electrolyte, which enables Li+ transport between the electrodes. Despite the effective storage performances being mainly defined by the cathode and anode properties (e.g. specific capacity of active materials, additives, binder, thickness, etc.), the electrolyte also is a key component of LIBs that should not be overlooked.14 Proper electrolytes should have good transport properties15 (e.g. high ionic conductivity, low viscosity, high lithium transfer number), high thermal and electrochemical stability range, and low risk and ecotoxicity profiles (e.g. low toxicity, bio-based and biodegradable natures).16
One of the best well-known liquid-electrolyte formulations is the so-called LP30,17 which consists of a 1 M solution of lithium hexafluorophosphate (LiPF6) conducting salt in a binary equal-weight mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC) solvents. Also, different additives are generally added to the formulation (e.g. vinylene carbonate – VC). Despite its good overall performances, EC/DMC-LiPF6 electrolytes have crucial concerns mostly related to safety issues18 (e.g. emission of HF by LiPF6 hydrolysis) and to the use of EC, which is mostly produced from toxic compounds (e.g. ethylene oxide) derived from non-renewable feedstocks (e.g. crude oil). These last aspects have driven the development of alternative liquid electrolytes intending to improve both their safety and sustainability profiles, without compromising the overall performance of the cell.19–21
In the field of bio-based solvents, dioxolanes (DOXs) of α-hydroxy acids (AHAs) family are emerging as alternative dipolar aprotic solvents. Among them, 5-methyl-1,3-dioxolane-4-one (LA-H,H) has recently found applications as dipolar aprotic reaction medium for cross-coupling and nucleophilic substitution reactions.22 Moreover, previous results have shown that LA-H,H overcomes other DOXs in energy storage applications thanks to its enhanced transport properties, enhanced ability to solvate electrolyte salts, and extended electrochemical voltage window. Specifically, a solution of 1 M TEMABF4 in LA-H,H achieved ionic conductivity values of 8.5 mS cm−1 at 20 °C, whereas formulations with LA-H,Me and LA-Me,Me displayed only 1.5 and 0.2 mS cm−1, respectively.23
Furthermore, LA-H,H proved to be biodegradable,24 and computational investigations pointed out that in general DOXs may have a non-toxic profile (non-mutagenic and non-carcinogenicity).22 These previous findings, along with its bio-derived origin and biocompatibility, position LA-H,H as a promising solvent alternative for application in LIBs.
The DOXs can be prepared by AHAs ketalization promoted by Brønsted acids (e.g. p-toluensulfonic acid – p-TsOH) in a multigram scale and with reasonable yield.22,25–27 Specifically, LA-H,H can be obtained by ketalization of lactic acid with a proper carbonyl precursor (e.g. paraformaldehyde, trioxane). The general reaction scheme for DOXs synthesis is reported in Scheme 1. Ketalization and acetalization reactions are generally well scalable, an example of this process is represented by solketal production starting from glycerol and acetone, which can be conveniently implemented at industrial level.28 Moreover, the mentioned precursors of LA-H,H are well-established bulk chemicals, economically viable, and widely exploited in the polymer industry.29–31 Those conditions are fundamental characteristics of an alternative solvent candidate.
 | ||
| Scheme 1 DOX synthesis scheme. In compound labels, the first two letters refer to the parent AHA (LA, lactic acid; iBu, α-hydroxyisobutyric acid; GA, glycolic acid), and the following letters refer to the substituent groups in position 2 (–H, –CH3). | ||
In this study, we explore the feasibility of using LA-H,H as solvent for LIBs in combination with two imide-based lithium salts: lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) and lithium bis(fluorosulfonyl)imide (LiFSI). These conductive salts have attracted high attention from academia and industry thanks to their advantageous features for energy storage applications, including high solubility, excellent associated conductivity, favorable Li+ transfer number and robust thermal and chemical stability.32
More in detail, the wide electrochemical stability of LiTFSI has enabled its use in various applications such as water-in-salt electrolytes for electrochemical double-layer capacitors (EDLCs) or lithium–sulfur (Li–S) batteries.33,34
However, although LiTFSI salt is widely known for its exceptional stability against spontaneous hydrolysis, its classification as a per- and polyfluoroalkyl substance raises environmental concerns.35,36 The presence of strong C–F bonds in LiTFSI contributes to its significant persistence in the environment, and therefore to its potential bioaccumulation and biomagnification. Thus, the industrial application of LiTFSI in LIBs needs to be strongly regulated and connected to a dedicated disposal industry, otherwise, their leakage in the environment can lead to global contamination (e.g. soil and surface water) associated with health hazards (e.g. liver injury).37,38 Besides, the high cost of LiTFSI has limited its large-scale use. Combining economic and environmental considerations with the chemical stability of this salt, its recycling and reuse can be effectively achieved through water-based processes. This kind of approach has recently been shown to be a promising sustainable solution.39 In contrast, LiFSI is more susceptible towards spontaneous hydrolysis due to its weaker F–S bonds.40,41 This reaction is notably pronounced under specific conditions (e.g., in basic, acidic environments or at elevated temperatures). Compared to LiPF6 salt, LiFSI exhibits considerably lower reactivity with water, and there is no clear evidence of HF formation at room temperature.42 From an economic and environmental perspective, LiFSI appears as a promising alternative salt due to its reduced price and lower fluorine content.43 The safety profile assessment of the imide-based salts should also consider their behavior in LIBs under extreme conditions (e.g., fires or explosions). In particular, potential thermal risks associated with these salts should not be overlooked, since their thermal degradation can be readily triggered under such conditions. This may eventually result in significant exothermic heat generation and the release of reactive radicals and hazardous anhydrides, such as SOx and NOx. It is worth noting that the thermal reaction mechanism of the salts is strongly influenced by the solvents used.44 Nevertheless, the intrinsic chemical stability of these imide-salts in mild conditions (e.g. ambient temperature, moisture) could also have a beneficial impact on the safety profile of logistic operations (e.g. storage, transport). Overall, for further development of LIBs with enhanced safety and sustainability, it is of paramount importance to comprehensively assess the chemical, environmental, and economic profiles of the salts used. While being aware of the potential drawbacks of using imide-based salts from the mentioned aspects, they are considered benchmark candidates for use with the bio-based LA-H,H solvent in the LIB investigations.
Herein, we report a deep examination about LA-H,H-based electrolytes prepared with both LiTFSI and LiFSI (1 M), especially focusing on their transport properties, thermal behavior, and electrochemical performances in combination with anode (graphite – GR) and cathode (lithium iron phosphate – LFP) benchmark active materials. The aim of this work is to explore a new sustainable battery solvent, not the sustainability of the used benchmark salts. From the investigation, the use of a film-forming additive as VC proved to be necessary since both electrolytes were unable to provide a stable solid–electrolyte interface (SEI). The best electrolyte candidate, LA-H,H-LiTFSI 1 M 5 wt% VC was successfully used to assemble a full LIB cell.
Results and discussion
Electrolyte characterization
As previously noted, LA-H,H is evaluated herein as a novel alternative solvent for application in LIBs. Besides its bio-derived origin, pristine LA-H,H exhibits several appealing solvent properties. A direct comparison with EC and DMC is collected in Table S1.† The LA-H,H bio-solvent was employed for preparing 1 M electrolyte solutions with LiTFSI or LiFSI conducting salts. First, the transport properties of the electrolyte formulations and the pristine solvent were evaluated. The viscosity was explored from −30 to 40 °C, while conductivity was investigated between −30 and 80 °C. Compared to the pure solvent LA-H,H, both electrolytes displayed higher viscosity values over the whole temperature range (Fig. 1a). Nevertheless, it is worth mentioning that the viscosity value at 20 °C for pristine LA-H,H is comparable to that of EC/DMC mixture (1.62 vs. 1.68 mPa s, respectively) as reported in Table S1.† From the comparison of the viscosity and conductivity values of the imide-based electrolytes (Fig. 1a and b), it can be appreciated that the transport properties of LA-H,H-LiFSI were slightly better than those of its LiTFSI counterpart. More in detail, the LiFSI-based electrolyte showed higher conductivity values over the whole temperature range, and lower viscosity between −30 and 0 °C. Overall, considering the results at 20 °C, LA-H,H-LiTFSI reached 4.66 mPa s and 4.85 mS cm−1, while LA-H,H-LiFSI achieved 4.33 mPa s and 6.22 mS cm−1. In both cases, the results are comparable with standard formulations, such as 1 M LiPF6 in EC/DMC 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 vol (η = 4.44 mPa s; σ = 10.7 mS cm−1), and other solvent mixtures with similar chemical nature like acetals (tetramethylglyoxal-TMG and tetraethylglyoxal-TEG mixture with carbonates: η = 5–11 mPa s−1; σ = 3–4 mS cm−1).45 The volumetric expansion coefficient of the pure LA-H,H solvent was also assessed (αv = 0.00101 °C−1) over 0–80 °C temperature range (Fig. S1†), which is comparable to EC:DMC mixture (0.0012 °C−1) and DMC and EC (details in Table S1†). Overall, a low expansion coefficient of solvents and electrolytes would contribute to mitigate the mechanical stress (e.g. overpressure) associated with temperature variations.46
:1 vol (η = 4.44 mPa s; σ = 10.7 mS cm−1), and other solvent mixtures with similar chemical nature like acetals (tetramethylglyoxal-TMG and tetraethylglyoxal-TEG mixture with carbonates: η = 5–11 mPa s−1; σ = 3–4 mS cm−1).45 The volumetric expansion coefficient of the pure LA-H,H solvent was also assessed (αv = 0.00101 °C−1) over 0–80 °C temperature range (Fig. S1†), which is comparable to EC:DMC mixture (0.0012 °C−1) and DMC and EC (details in Table S1†). Overall, a low expansion coefficient of solvents and electrolytes would contribute to mitigate the mechanical stress (e.g. overpressure) associated with temperature variations.46
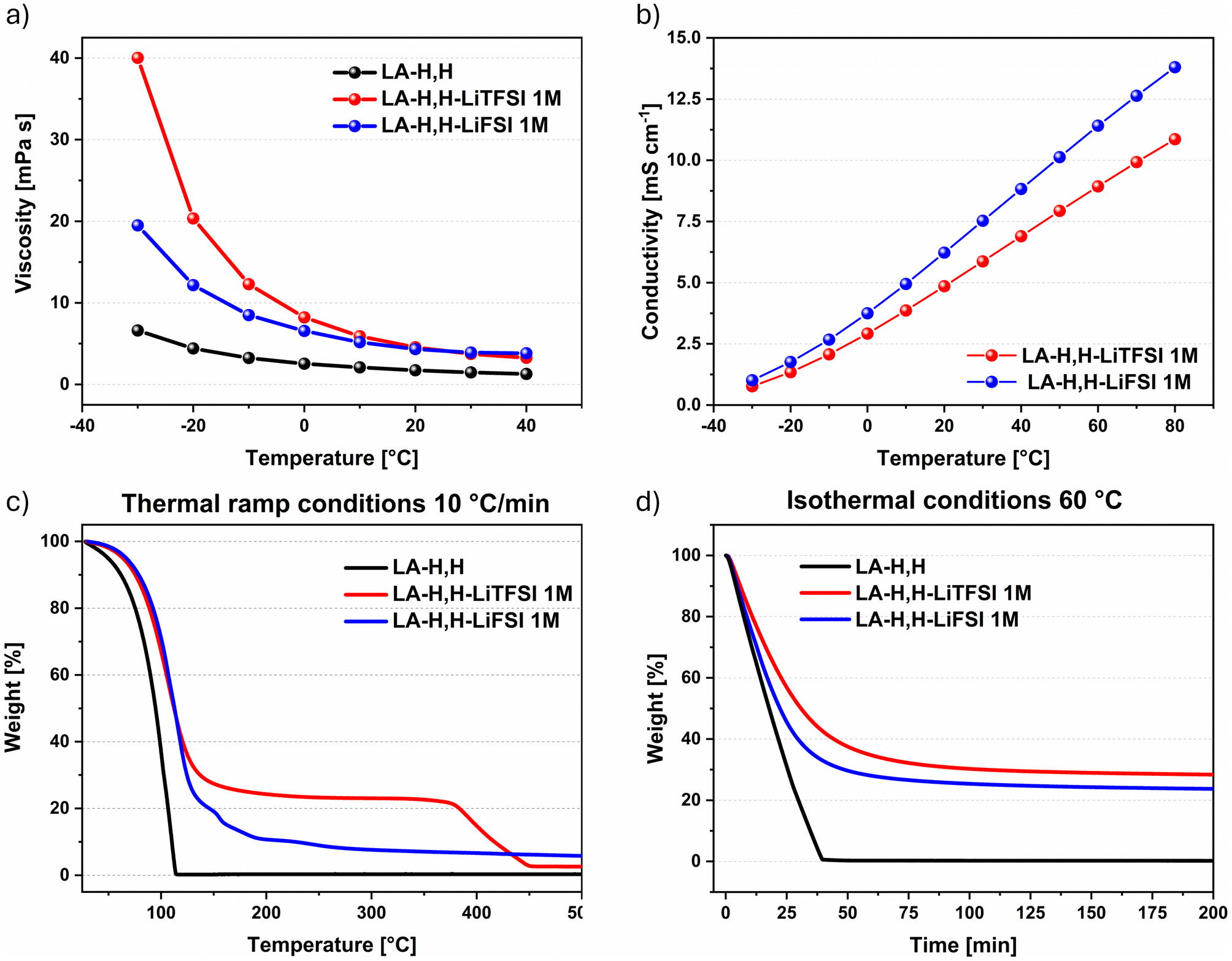 | ||
| Fig. 1 (a) Viscosity measured from −30 °C to 40 °C, and (b) conductivity measured from −30 °C to 80 °C, (c) dynamic TGA from 30 °C to 550 °C, and (d) isothermal TGA at 60 °C. | ||
At 20 °C, LA-H,H in LiTFSI and LiFSI electrolytes show a density value of 1.30 g cm−3 and 1.27 g cm−3 respectively (Fig. S2†). In order to get insights in the thermal stability of the herein studied samples, thermogravimetric investigations (Fig. 1c and d) were performed in both dynamic (10 °C min−1 thermal ramp) and isothermal modes (60 °C). Compared to the pure solvent, in both cases the presence of lithium salts tends to slow down the evaporation rate. This is reasonable due to the relation between the increase of the vapor pressure and the evaporation rate, caused by the establishment of specific solvent–solute interactions typical of colligative properties.47 Under dynamic investigation, LA-H,H-LiTFSI and LA-H,H-LiFSI reached respectively 90 wt% of their initial mass at 75 °C and 78 °C, while the pure solvent reached this threshold at 61 °C. Both imide salts showed decomposition temperature in agreement with literature data48,49 (LiTFSI above 300 °C, LiFSI 160 °C), and the residual masses in both profiles (LiTFSI 2.5 wt%, and LiFSI 5.5 wt%) are mainly related to residual decomposition products. In the isothermal conditions at 60 °C, LA-H,H-LiTFSI and LA-H,H-LiFSI reached 57 wt% and 46 wt% within 25 min, respectively, while LA-H,H reached 31 wt% within the same time. The evaporation rate vs. water in the isothermal condition (water evaporation curve not reported in Fig. 1d) was estimated after 5 min at 60 °C, and both electrolytes resulted in evaporating 30–32% slower than pure water. In all cases, the weight profiles were smooth, suggesting that the solvent evaporation was not affected by thermal degradation. The flash point of the pure solvent, LiTFSI- and LiFSI-based electrolytes resulted to be 60 °C, 61 °C and 60 °C respectively. These values are three times higher than those of conventional EC/DMC-LiPF6 systems (22–24 °C),50,51 which may contribute to improved handling and storage safety under standard conditions. Nevertheless, it is worth noting that the flash point alone is not a definitive indicator of the overall flammability of the cell due to the significantly higher temperatures reached in a real battery thermal runaway event.
Following, the electrochemical stability windows of the LA-H,H-based electrolytes (Fig. S3†) were explored using a platinum (Pt) disc as working electrode. Both electrolytes showed a lower limit of 0.06 V vs. Li+/Li, while LA-H,H-LiTFSI showed a slightly higher upper limit (5.63 V vs. Li+/Li) compared to LA-H,H-LiFSI (5.43 V vs. Li+/Li). Overall, a comparable ESW of 5.4–5.6 ΔV was achieved by the two LA-H-H-based electrolytes. To validate the ESW results, a different working electrode – glassy carbon (GC) was used instead of platinum.
For the LA-H,H-LiTFSI electrolyte, a reasonable ESW of 4.9 ΔV was demonstrated. In both cases, the obtained ESW ranges are enough also to consider their application in other energy storage devices (e.g. 1,3-dioxolane is used in Li–S batteries),52 and as reaction media for organic electrosynthesis (e.g. problematic dipolar aprotic solvents like THF and DMA are commonly used).53
Electrochemical and thermal stability results, combined with transport properties (collected in Table 1), make these electrolytes suitable candidates for their investigation in LIBs devices.
| Parameter | Units | Electrolyte | |
|---|---|---|---|
| LA-H,H-LiTFSI 1 M | LA-H,H-LiFSI 1 M | ||
| a Values measured at 20 °C. | |||
| Viscositya | [mPa s] | 4.66 | 4.33 |
| Conductivitya | [mS cm−1] | 4.85 | 6.22 |
| Densitya | [g cm−3] | 1.30 | 1.27 |
| Flash point | [°C] | 61 | 60 |
| E Cathodic | [V vs. Ag] | −2.99 | −2.99 |
| [V vs. Li+/Li] | 0.06 | 0.06 | |
| E Anodic | [V vs. Ag] | 2.58 | 2.38 |
| [V vs. Li+/Li] | 5.63 | 5.43 | |
Half-cell: graphite electrodes
The electrochemical stability of the two electrolytes was first assessed without additives in a half-cell 2-electrode configuration versus metallic lithium using GR as the working electrode (Fig. S4a and b†). In both cases, the electrolytes were not stable during the first lithium intercalation process, showing a plateau at 1.0–0.8 V vs. Li+/Li, characteristic of solvent co-intercalation and graphite exfoliation process.54 As a result, the following cycles recorded at 0.1C displayed a low capacity that was further reduced upon cycling. This could be related to the strong solvation shell created between the LA-H,H molecules and the Li+ ion, as typically observed also for similar carbonate solvents like propylene carbonate.55 To overcome this issue, VC was included in the electrolyte formulations to stabilize the graphite surface during the first cycles through the formation of a stable solid electrolyte interface (SEI).56–58 In the beginning, VC was added to the electrolytes up to 2 wt%, but the solvent co-intercalation effect happened similarly in the case of LiFSI (Fig. S4c†). On the other hand, the LA-H,H-LiTFSI 2 wt% VC showed a more promising potential profile for the first cycle at 0.1C (Fig. S4d†), showing the typical intercalation potential profile, albeit with a very low coulombic efficiency (Fig. S4e†). However, the subsequent cycles marked the GR electrode instability during the C-rate test (Fig. S4f†). Therefore, the amount of additive was raised to 5 wt%, resulting in a reversible intercalation process for the LA-H,H LiTFSI 1 M electrolyte as shown by the galvanostatic charge/discharge profile of the first cycle at 0.05C (Fig. 2a). This is corroborated by the appearance of three characteristic plateaus at 0.20, 0.12, and 0.08 V vs. Li+/Li, corresponding to the different lithium intercalation stages into graphite. Furtherly, the superimposable profiles at 0.1C at the beginning (in red) and at the end (in green) of the rate capability test (Fig. 2b) highlight the ability to reversibly intercalate lithium ions after applying current densities up to 2C. The cell with LA-H,H-LiFSI 1 M 5 wt% VC electrolyte experienced a sharp specific capacity fade after cycling at 2C, proving to be unstable within the rate capability test (Fig. 2c, potential profiles in Fig. S5a and b†). To prove a beneficial impact on the LiFSI electrolyte stability, the VC amount was increased to 10 wt%. As shown in Fig. S6a and b,† the performance measured in both the rate capability and long-cycling tests considerably improved. However, in this case, the amount of additive necessary to stabilize the electrode/electrolyte is relatively high. Further investigations may be directed to its use in combination with other co-solvents, but this aspect is outside the scope of this work.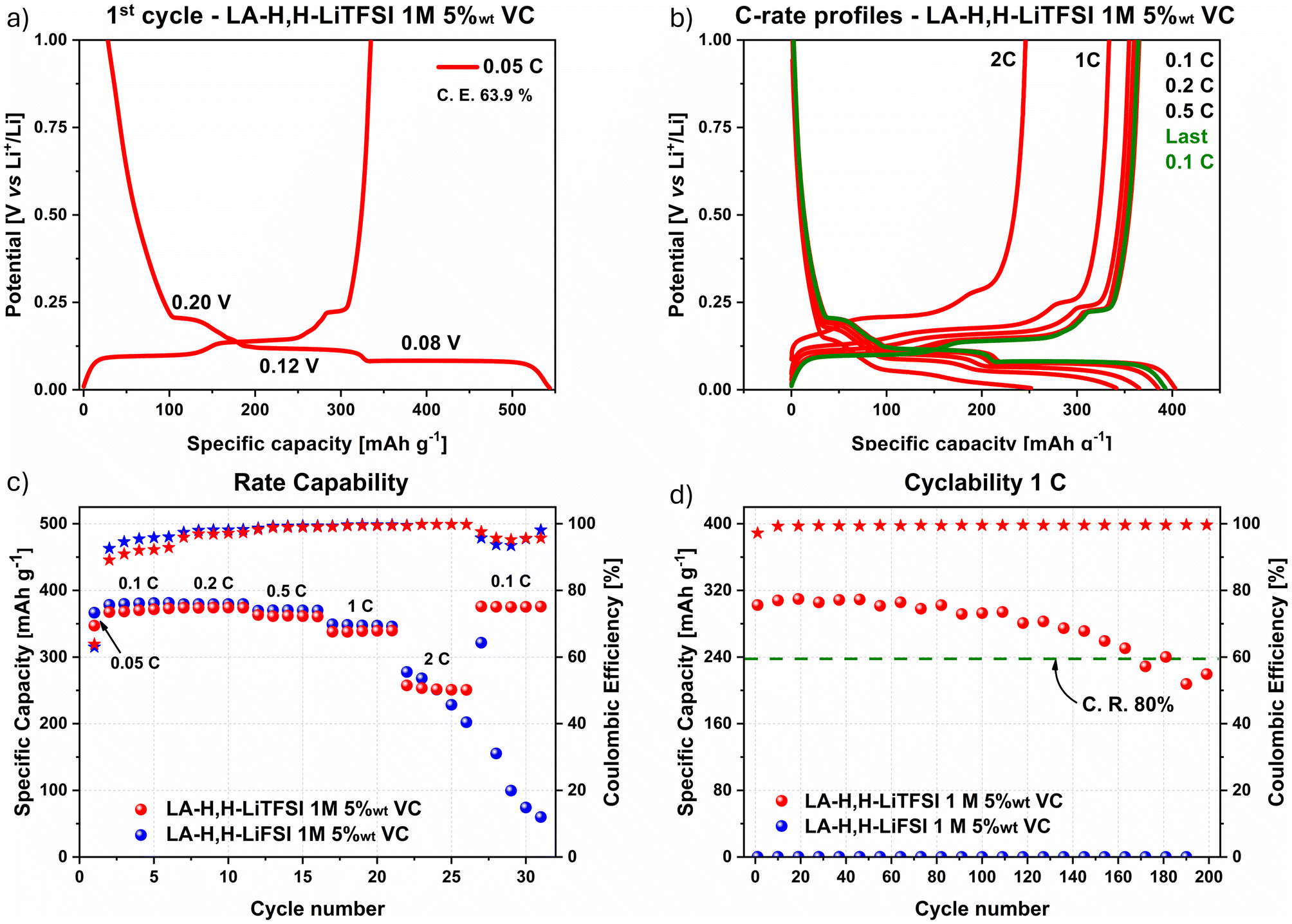 | ||
| Fig. 2 GR galvanostatic charge/discharge profile with LA-H,H-LiTFSI 1 M 5 wt% VC of (a) 1st cycle at 0.05C, and (b) profiles at different current densities over capability test. (c) Results from rate capability (5 cycles per each current density), and (d) cyclability (200 cycles at 1C). C. E.: coulombic Efficiency. C. R.: capacity retention. | ||
Considering LA-H,H-LiTFSI 1 M 5 wt% VC, the low coulombic Efficiency (C.E. 64%) obtained during the first cycle at 0.05C was probably related to irreversible processes involved in the formation of the SEI. Noteworthy, at low current density it almost reached the theoretical specific capacity (370 mA h g−1), while at the high current density of 2C, it was able to retain around 195–200 mA h g−1 (Fig. 2c), comparable to the results obtained with EC/DMC-LiPF6 1 M 2 wt% VC obtained in a precedent investigation20 (220–230 mA h g−1). In the cyclability test operated with a current density of 1C (Fig. 2d), LA-H,H-LiTFSI 1 M 5 wt% VC reached 160 cycles while retaining 80% of its initial capacity and very stable C.E. values within 99.2–99.6%.
Half-cell: lithium-iron-phosphate electrodes
Investigation with LFP cathode materials using both imide-based electrolytes was directly conducted using 5 wt% VC as the lowest amount suitable to achieve proper stability with GE, at least in combination with LiTFSI as conductive salt. In this case, both electrolytes achieved a reversible lithiation/delithiation process as shown by the galvanostatic charge/discharge profiles of the first cycle at 0.05C (Fig. 3a and S7a†), and by the representative cycles at different current densities from the rate capability test (Fig. 3b and S7b†). The 0.05C and 0.1C profiles clearly showed the typical delithiation plateaus at ≈3.45 V vs. Li+/Li,59,60 which are related to the two-phase transition comprising the release of Li+ ions in the electrolyte and the contextual oxidation of Fe(II) to Fe(III). Additionally, in this case, the characteristic profile at 0.1C after the rate capability test (in green and orange in Fig. 3b and Fig. S7b† respectively) highlighted that current densities up to 5C may be applied without compromising the active material crystalline lattice.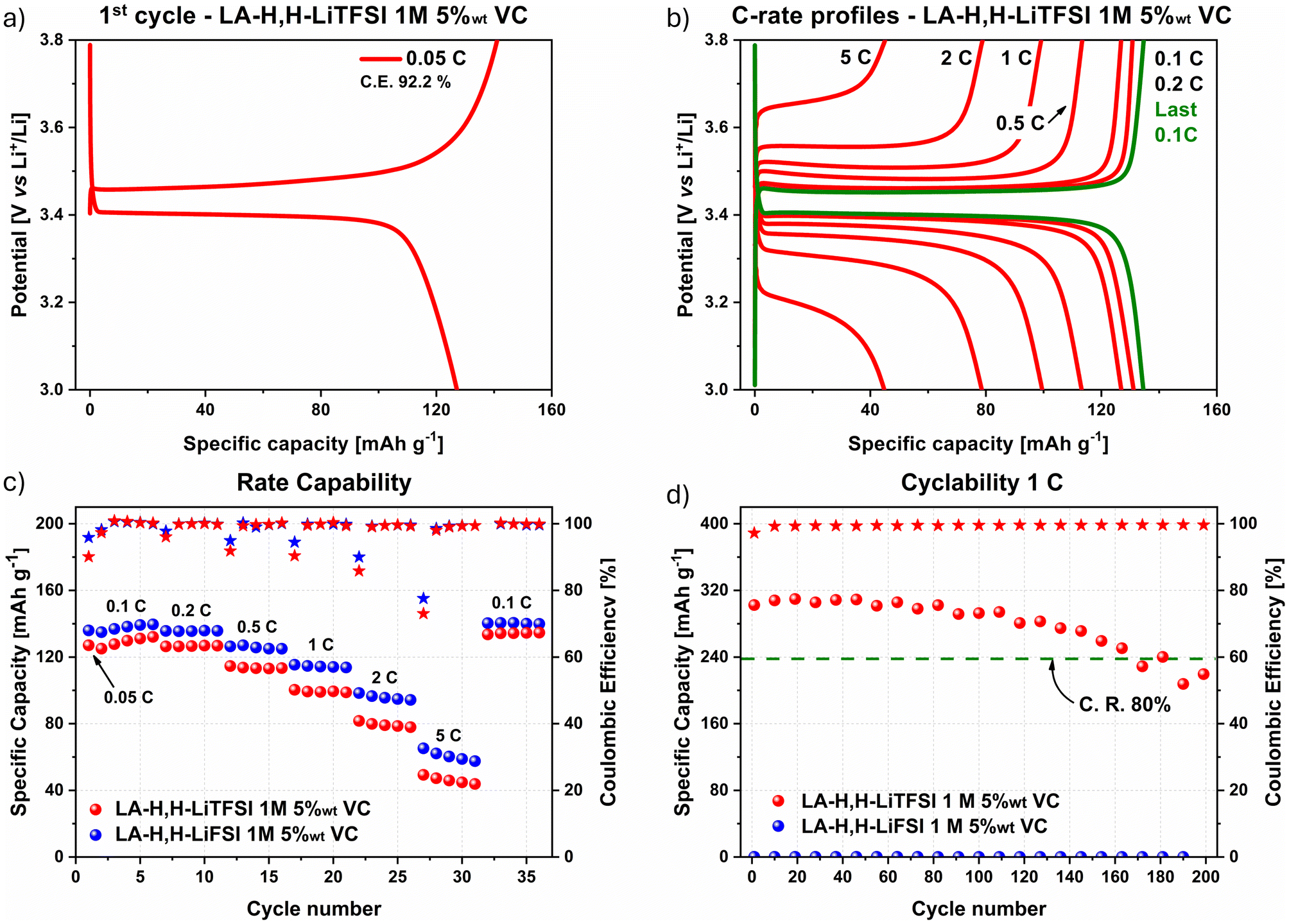 | ||
| Fig. 3 LFP galvanostatic charge/discharge profile with LA-H,H-LiTFSI 1 M 5 wt% VC of (a) 1st cycle at 0.05C, and (b) profiles at different current densities over capability test. (c) Results from rate capability (5 cycles per each current density), and (d) cyclability (200 cycles at 1C). C. E.: coulombic Efficiency. C. R.: capacity retention. | ||
Rate capability and cyclability tests are shown in Fig. 3c and d for both LiTFSI (in red) and LiFSI (in blue) imide-containing cells. In the first cycle at 0.05C, a C.E. of 90.1% and 95.8% were achieved with LA-H,H-LiTFSI 1 M 5 wt% VC and LA-H,H-LiFSI 1 M 5 wt% VC, respectively. In the rate capability test, the C.E. increased within the first cycles at 0.1C and remained stable within 99.0–99.9% over the following cycles. The LiFSI-based electrolyte overperforms its LiTFSI counterpart in terms of capacity in the whole range of C-rates applied, achieving 140 mA h g−1 at 0.1C and ≈60 mA h g−1 at 5C, while the LiTFSI-based electrolyte delivered 134 mA h g−1 and ≈45 mA h g−1, respectively. Considering the theoretical capacity of LFP active material (170 mA h g−1), both electrolytes gained roughly 80% of the available capacity. However, this loss is mainly related to the quality of the used active material since comparable results were achieved also with LP30 (≈140 mA h g−1 at 0.1C) as benchmark electrolyte (Fig. S8†). As shown by a long cycling test at 1C (Fig. 3d), both electrolytes retained more than 80% of their initial capacity after 200 cycles with comparable specific capacity (LiTFSI 106–85 mA h g−1; LiFSI 112–90 mA h g−1). It is worth highlighting that both electrolytes displayed excellent C.E. values (above 99.5%) along cycling. Overall, the slightly higher performance achieved with LiFSI can be mainly ascribed to its better transport properties (viscosity and conductivity) as described in the previous section. However, considering its incompatibility with GR (cf.Fig. 2c), the LiTFSI-electrolyte 5 wt% VC was chosen for further investigation in a full-cell setup.
Full-cell
Considering the promising results obtained from graphite and LFP half-cells using LiTFSI in LA-H,H-LiTFSI 1 M 5 wt% VC, its performance in a LIB full-cell configuration was evaluated. Prior to full-cell assembly, the graphite anode was precycled and lithiated to mitigate irreversible lithium loss on graphite during the initial galvanostatic cycles of the full-cell (further details can be found in the Experimental section). The galvanostatic profiles of the LIB at different rate capabilities are shown in Fig. S9a† (1C: 170 mA g−1). Specific capacity values delivered at corresponding rates considering the total active mass of both electrodes are represented in Fig. 4a. The lab-scale LIB demonstrates a remarkable rate capability, delivering ca. 85 mA h g−1 at 0.1C and still retaining a specific capacity above 60 mA h g−1 at 2C.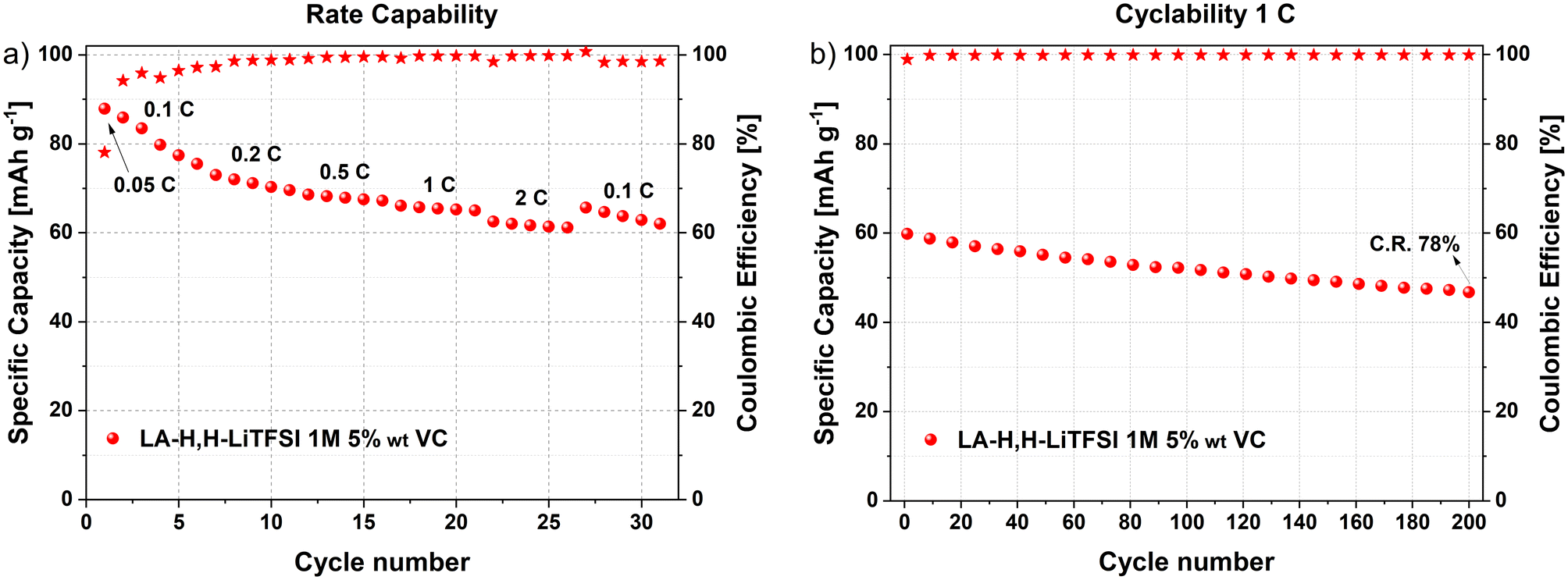 | ||
| Fig. 4 LFP/graphite LIB full-cell with LA-H,H-LiTFSI 1 M 5 wt% VC. (a) Results from rate capability (5 cycles per current density), and (b) cyclability (200 cycles at 1C). C. R.: capacity retention. | ||
It is worth noting that, despite showing an initial C.E. of ca. 80% in the first cycle at 0.05C, the reversibility of the cell increased in the following cycles, reaching >96% at 0.1C and >99.0% at the subsequent rates. Following the rate capability, a long-term cycling test at 1C was performed (Fig. 4b). The device demonstrated suitable cycling stability, retaining 78% of its initial capacity after 200 charge/discharge cycles, coupled with a constant C.E. of 99.8%. The voltage profiles of the LIB full-cell and the potential profiles of its respective electrodes (LFP and graphite) after 100 cycles at 1C are displayed in Fig. S9b.† No evident signs of ageing were observed from the LFP/graphite potential curves, displaying their respective characteristic profiles for lithium intercalation.
These results indicate that the novel bio-based solvent (in combination with LiTFSI and VC) enables efficient lithium intercalation in the LIB full-cell. It is worth mentioning that further cell optimization in terms of mass balancing and pre-forming steps will be required to increase the electrochemical features of the cell. Nevertheless, the suitable performance of the herein assembled lab-scale LIB demonstrates the suitability for the application of bio-based LA-H,H solvent in energy storage applications.
Experimental
Chemicals and materials
The salts LiTFSI (Solvionic, 99.9%), LiFSI (Solvionic, 99.9%), and VC (Thermo Scientific Chemicals, 98%) were directly used without further purifications. The LA-H,H solvent was prepared from lactic acid (Sigma-Aldrich, ∼90%) and paraformaldehyde (Sigma Aldrich, for synthesis), using p-toluenesulfonic acid (Sigma Aldrich, ACS reagent, ≥98.5%) as a catalyst and petroleum ether bp 40–60 °C(Sigma Aldrich) as solvent. Synthetic details for DOXs synthesis are already reported elsewhere.22 Briefly, the reaction was performed at reflux under vigorous magnetic stirring for 24 h using a Dean–Stark apparatus to remove water from the reaction mixture. Then the crude mixture was cooled in an ice bath and treated with Na2CO3 for 30 min. The reaction crude was filtered, and the volatile solvent evaporated under reduced pressure. The product was then purified by vacuum distillation (8 mbar) at 36–38 °C (yield 60%, purity <99%) and characterized through Nuclear Magnetic Resonance (NMR) with a Bruker Avance Ultrashield 400 (Bruker Corporation) operating at the proton frequency of 400 MHz. The following abbreviations have been adopted, s, singlet; d, doublet; q, quartet. Product structure was found in agreement with literature data:22,251H NMR (400 MHz, CDCl3) δ 5.51 (s, 1H), 5.38 (s, 1H), 4.27 (q, J = 6.8, 1H), 1.47 (d, J = 6.8, 3H). 13C NMR (101 MHz, CDCl3) δ 173.62, 93.93, 69.63, 15.82. Product purity was evaluated by 1H NMR analysis. An example of two 1H NMR spectra related to different LA-H,H distillation portions (head and core fractions) used to evaluate the sample purity are depicted in Fig. S10.† Further details concerning contaminants and side products identified during this investigation are included in Paragraph S1.†Before being used, LA-H,H was dried over 3 Å molecular sieves (sieves loading 10 wt%) for at least 48 h at room temperature inside an argon-filled glove box. The water content was confirmed to be below 10 ppm through Karl-Fischer titration (C20 Coulometric KF Titrator, Mettler Toledo). All electrolytes and cells (Swagelok) were prepared and assembled in a dry atmosphere using an argon-filled glove box (MasterLAB, MBraun).
Physicochemical characterization
The viscosities of the electrolytes were determined by a Modular Compact Rheometer MCR 102 (Anton Paar RheoCompass) using a plate-plate geometry at a shear rate of 1000 s−1, and temperatures were controlled in a range from −30 to 40 °C by a built-in Peltier element. Ionic conductivity measurements were measured using a Modulab XM ECS (Ametek SI) potentiostat while the temperature was controlled by a climatic chamber (BINDER) from −30 to 80 °C.Conductivity results were obtained using a two parallel platinized-platinum electrodes cell with a known cell constant, as detailed in previous literature.61 Density was measured using a density meter DMA 4100 M (Anton Paar) in the range of 0–80 °C. Thermogravimetric analyses (TGA) were performed using a PerkinElmer STA 6000 instrument under a nitrogen atmosphere (purge rate 20 mL min−1; gas pressure of 2.2 bar). Dynamic measurements were conducted using a heating rate of 10 °C min−1 from 30 to 550 °C, while isothermal measurements were performed at 60 °C for 24 h. Flash points were measured, according to the EN ISO 3679 standard, using a flash point tester NPV 310 model (Normalab) and a rapid equilibrium closed cup method.
Electrode and cell preparation
The following active materials, binders, and additives have been used for the electrode manufacture: graphite – GR (TIMREX SFG 6, TIMCAL); carbon black Super P C65 – CB (C ENERGY, Imerys); sodium carboxymethylcellulose – NaCMC (CRT 2000 GA, Walocel); lithium iron phosphate, LiFePO4 (Südchemie); styrene–butadiene rubber – SBR (Nanografil); activated carbon – AC (Norit Supra 50); polytetrafluoroethylene – PTFE (60 wt% aqueous dispersion, Sigma-Aldrich).Slurries were prepared using a ball mill (Mini-Mill, Laval Lab; 7 spheres, 5 min, oscillation frequency 50 Hz) to achieve a homogeneous dispersion. Graphite slurries were prepared using a mass ratio of 90:5:5 respectively with graphite (GR), binder (NaCMC), additive (CB), and water as a dispersant (≈3 mL g−1vs. total powders mass). LFP slurries were prepared using two binders (NaCMC and SBR) with the following mass ratio 90:5:3.75:1.25 respectively of LiFePO4, CB, NaCMC, and SBR. Graphite and LFP slurries were evenly cast respectively on copper and etched aluminium foils (etching process 1 min at 60 °C in a 5 wt/v% KOH solution), and subsequently dried at room temperature overnight. Then, electrode discs were punched (∅ 12 mm) and further dried overnight under vacuum at 65 °C. The electrode mass loading was adjusted through the wet thickness of the coater. The graphite electrode active masses were 1.1–1.7 mg cm−2, whereas the LFP electrodes weighed 2.5–3.6 mg cm−2. Swagelok-type cells were assembled with proper electrodes and a glass fibre separator (Whatman GF/D, ∅ 13 mm).
Self-standing oversized active carbon electrodes were prepared with the following mass ratio 85:10:5 respectively of AC, CB, and PTFE as the binder. A premix of carbon powders was prepared and placed into a beaker, then the PTFE binder was consecutively added. The mixture was kept under mechanical stirring at 60 °C using ethanol (Sigma Aldrich, 96%) as dispersant solvent until obtaining a smooth paste. The mixture was then placed on a glass plate, evenly spread, and punched (∅ 12 mm). Electrode discs were dried first at room temperature for 12 h and subsequently overnight under vacuum at 65 °C.
Electrochemical measurements
The electrochemical stability window (ESW) was evaluated using a 3-electrodes cell setup with a Pt disc as a working electrode, an Ag wire as a quasi-reference electrode, and a self-standing oversized AC (Norit) as the counter electrode. For the sake of comparison, the ESW measurements were also performed using the same configuration but with a glassy carbon electrode as the working electrode. Linear sweep voltammetry (LSV) measurements were carried out at a scan rate of 1 mV s−1 in two different cells to determine the respective anodic and cathodic stability limits (current threshold of ±0.5 mA cm−2).Graphite and LFP electrodes were investigated in a two-electrode half-cell configuration, using a lithium disc simultaneously as a counter and reference electrode. For graphite electrodes, galvanostatic charge/discharge measurements were performed between 0.005–2.0 V vs. Li+/Li at various C rates (1C: 372 mA g−1), followed by long-term cycling at 1C within the same potential range. Analogous measurements were performed on LFP electrodes within 3.0–3.8 V vs. Li+/Li (1C: 170 mA g−1). The specific capacity and current densities reported are calculated on the basis of active material mass of the electrode.
Prior to LIB full-cell assembly, the graphite electrodes were subjected to a pre-formation step in a separate cell against metallic lithium. The pre-formation protocol consisted in one initial cycle at 0.05C and 10 cycles at 0.1C between 0.005–2.0 V vs. Li+/Li followed by a last discharge at 0.1C to 150 mV. The LIB full cells were then assembled using the as-preformed graphite electrode as counter electrode, a LFP electrode as working electrode, and metallic lithium as reference electrode. An electrode active mass ratio of 3:1 (LFP to graphite) was used for the full-cell assembly. Galvanostatic charge/discharge measurements were performed at different C-rates (1C: 170 mA g−1), within the cell voltage of 2.0–3.9 V vs. Li+/Li. The measurements were followed by long-term cycling tests at 1C. The specific capacity values in the full-cell were calculated based on the total mass of LFP and graphite active materials.
All the electrochemical measurements were conducted using a BioLogic VMP 3 or MPG 2 and an Arbin LBT21084 multichannel potentiostatic–galvanostatic system.
Conclusions
In this study, we report for the first time the use of the bio-based solvent 5-methyl-1,3-dioxolan-4-one as a component of electrolytes for LIBs. Among the investigated electrolytes, the LA-H,H-LiTFSi 1 M 5 wt% VC exhibits suitable physicochemical properties and large electrochemical stability. Its use enables good rate capability and cycling stability in both graphite (≈339 mA h g−1 at 1C) and LFP half-cells (≈100 mA h g−1 at 1C). Furthermore, we show that LFP/graphite LIB full-cell containing this innovative electrolyte displays outstanding rate capability and high cycling stability, retaining 78% of its initial capacity after 200 galvanostatic cycles at 1C.It is important to remark that this bio-based solvent is biodegradable, it can be prepared using rather cheap chemicals and, moreover, the chemical structure of the class of 1,3-dioxolan-4-ones compounds could be tailored by proper choice of substituents, achieving a potential panel of solvents with tunable properties.
Taking these points into account, LA-H,H can be certainly considered a promising solvent for LIB and, more in general, for energy storage devices.
Author contributions
Investigation, data curation, formal analysis, writing – original draft M.M. and K.S.T (lead). Investigation, supervision, writing – original draft J.L.G.U. (supporting). Conceptualization, funding acquisition, supervision, writing – review & editing F.R. and A.B (lead).Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. M. and F. R. want to acknowledge the financial support from the Ministero dell'Università e della Ricerca provided through the “LABSolve” project (Progetti di Ricerca di Rilevante Interesse Nazionale – Bando 2022 Prot. 20229P7PPM). J. L. G. U. and A. B. want to acknowledge the financial support of the European Union's Horizon Europe transport program under the project SiGNE (grant agreement no 101069738). K. S. T. and A. B. would like to thank the financial support from Deutsche Forschungsgemeinschaft (DFG) for project BA4956/24-1 (Forschungsgruppe FuncHeal FOR5301).References
- EU-Commission , European Green Deal – Research & innovation call, Publications Office of the European Union, 2021.
- M. Beniston and D. B. Stephenson, Glob. Planet. Change, 2004, 44, 1–9 CrossRef.
- A. T. D. Perera, V. M. Nik, D. Chen, J.-L. Scartezzini and T. Hong, Nat. Energy, 2020, 5, 150–159 CrossRef.
- W. F. Lamb, T. Wiedmann, J. Pongratz, R. Andrew, M. Crippa, J. G. J. Olivier, D. Wiedenhofer, G. Mattioli, A. A. Khourdajie, J. House, S. Pachauri, M. Figueroa, Y. Saheb, R. Slade, K. Hubacek, L. Sun, S. K. Ribeiro, S. Khennas, S. de la Rue du Can, L. Chapungu, S. J. Davis, I. Bashmakov, H. Dai, S. Dhakal, X. Tan, Y. Geng, B. Gu and J. Minx, Environ. Res. Lett., 2021, 16, 073005 CrossRef CAS.
- K. O. Yoro and M. O. Daramola, in Advances in Carbon Capture, ed. M. R. Rahimpour, M. Farsi and M. A. Makarem, Woodhead Publishing, 2020, pp. 3–28, DOI:10.1016/B978-0-12-819657-1.00001-3.
- R. Sanguino, A. Barroso, S. Fernández-Rodríguez and M. I. Sánchez-Hernández, Environ. Sci. Pollut. Res., 2020, 27, 1–7 CrossRef PubMed.
- W. Wang, B. Yuan, Q. Sun and R. Wennersten, J. Energy Storage, 2022, 52, 104812 CrossRef.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2020, 19, 1151–1163 CrossRef CAS PubMed.
- M. R. Palacín and A. de Guibert, Science, 2016, 351, 1253292 CrossRef PubMed.
- N. A.-Z. R-Smith, M. Moertelmaier, G. Gramse, M. Kasper, M. Ragulskis, A. Groebmeyer, M. Jurjovec, E. Brorein, B. Zollo and F. Kienberger, Energy Rep., 2023, 10, 3394–3401 CrossRef.
- B. Hu and X. Wang, J. Micromech. Microeng., 2021, 31, 114002 CrossRef CAS.
- T. Chen, Y. Jin, H. Lv, A. Yang, M. Liu, B. Chen, Y. Xie and Q. Chen, Trans. Tianjin Univ., 2020, 26, 208–217 CrossRef.
- R. Marom, S. F. Amalraj, N. Leifer, D. Jacob and D. Aurbach, J. Mater. Chem., 2011, 21, 9938–9954 RSC.
- P. Zhou, X. Zhang, Y. Xiang and K. Liu, Nano Res., 2023, 16, 8055–8071 CrossRef CAS.
- J. Xing, S. Bliznakov, L. Bonville, M. Oljaca and R. Maric, Electrochem. Energy Rev., 2022, 5, 14 CrossRef CAS.
- S. Kainat, J. Anwer, A. Hamid, N. Gull and S. M. Khan, Mater. Chem. Phys., 2024, 313, 128796 CrossRef CAS.
- P. V. Chombo and Y. Laoonual, J. Power Sources, 2020, 478, 228649 CrossRef CAS.
- M. Armand, P. Axmann, D. Bresser, M. Copley, K. Edström, C. Ekberg, D. Guyomard, B. Lestriez, P. Novák, M. Petranikova, W. Porcher, S. Trabesinger, M. Wohlfahrt-Mehrens and H. Zhang, J. Power Sources, 2020, 479, 228708 CrossRef CAS.
- K. S. Teoh, M. Melchiorre, S. Darlami Magar, M. Hermesdorf, D. Leistenschneider, M. Oschatz, F. Ruffo, J. L. Gómez Urbano and A. Balducci, Adv. Mater., 2024, 36, 2310056 CrossRef CAS PubMed.
- K. S. Teoh, M. Melchiorre, S. Darlami Magar, C. Leibing, F. Ruffo, J. L. Gómez-Urbano and A. Balducci, Small, 2024, 2407850 Search PubMed.
- M. Melchiorre, P. H. M. Budzelaar, M. E. Cucciolito, R. Esposito, E. Santagata and F. Ruffo, Green Chem., 2023, 25, 2790–2799 RSC.
- M. Melchiorre, R. Esposito, M. Di Serio, G. Abbate, A. Lampasi, A. Balducci and F. Ruffo, Energies, 2021, 14, 4250 CrossRef CAS.
- M. Melchiorre, C. Melchiorre, M. Moracci, P. I. Somma, M. Markiewicz, S. Stolte, P. Cerruti, F. Ruffo and A. Carpentieri, Soc. Sci. Res. Netw. (SSRN), 2024 DOI:10.2139/ssrn.4932208.
- S. A. Cairns, A. Schultheiss and M. P. Shaver, Polym. Chem., 2017, 8, 2990–2996 RSC.
- H. S. Masahiko Okada and M. Atsumi, Macromolecules, 1984, 17, 1840–1843 CrossRef.
- T. Miyagawa, F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1861–1865 CrossRef CAS.
- J. A. Vannucci, M. N. Gatti, N. Cardaci and N. N. Nichio, Renewable Energy, 2022, 190, 540–547 CrossRef CAS.
- M. Singhvi and D. Gokhale, RSC Adv., 2013, 3, 13558–13568 RSC.
- N.-A. A. B. Taib, M. R. Rahman, D. Huda, K. K. Kuok, S. Hamdan, M. K. B. Bakri, M. R. M. B. Julaihi and A. Khan, Polym. Bull., 2023, 80, 1179–1213 CrossRef CAS.
- A. Dorieh, M. Farajollah Pour, S. Ghafari Movahed, A. Pizzi, P. Pouresmaeel Selakjani, M. Valizadeh Kiamahalleh, H. Hatefnia, M. H. Shahavi and R. Aghaei, Prog. Org. Coat., 2022, 165, 106768 CrossRef CAS.
- J. Xing, S. Bliznakov, L. Bonville, M. Oljaca and R. Maric, Electrochem. Energy Rev., 2022, 5, 14 CrossRef CAS.
- A. Manthiram, Y. Fu, S. H. Chung, C. Zu and Y. S. Su, Chem. Rev., 2014, 114, 11751–11787 CrossRef CAS PubMed.
- Y. Zhang, N. H. C. Lewis, J. Mars, G. Wan, N. J. Weadock, C. J. Takacs, M. R. Lukatskaya, H. G. Steinruck, M. F. Toney, A. Tokmakoff and E. J. Maginn, J. Phys. Chem. B, 2021, 125, 4501–4513 CrossRef CAS PubMed.
- S. Di Muzio, A. Paolone and S. Brutti, J. Electrochem. Soc., 2021, 168, 100514 CrossRef.
- S. F. Lux, L. Terborg, O. Hachmöller, T. Placke, H. W. Meyer, S. Passerini, M. Winter and S. Nowak, J. Electrochem. Soc., 2013, 160, A1694–A1700 CrossRef CAS.
- M. Sands, X. Zhang, A. Gal, M. Laws, M. Spinella, Z. M. Erdogan and J. Irudayaraj, Environ. Int., 2024, 185, 108556 CrossRef CAS PubMed.
- J. L. Guelfo, P. L. Ferguson, J. Beck, M. Chernick, A. Doria-Manzur, P. W. Faught, T. Flug, E. P. Gray, N. Jayasundara, D. R. U. Knappe, A. S. Joyce, P. Meng and M. Shojaei, Nat. Commun., 2024, 15, 5548 CrossRef CAS PubMed.
- K. S. Teoh, M. Melchiorre, S. Darlami Magar, C. Leibing, F. Ruffo, J. L. Gomez-Urbano and A. Balducci, Small, 2024, e2407850, DOI:10.1002/smll.202407850.
- Z. Li, L. Wang, X. Huang and X. He, Adv. Funct. Mater., 2024, 34, 2408319 CrossRef CAS.
- D. Reber, R. Figi, R.-S. Kühnel and C. Battaglia, Electrochim. Acta, 2019, 321, 134644 CrossRef CAS.
- H.-B. Han, S.-S. Zhou, D.-J. Zhang, S.-W. Feng, L.-F. Li, K. Liu, W.-F. Feng, J. Nie, H. Li and X.-J. Huang, J. Power Sources, 2011, 196, 3623–3632 CrossRef CAS.
- Y. Cai, H. Zhang, Y. Cao, Q. Wang, B. Cao, Z. Zhou, F. Lv, W. Song, D. Duo and L. Yu, J. Power Sources, 2022, 535, 231481 CrossRef CAS.
- L. Zhao, A. Inoishi and S. Okada, J. Power Sources Adv., 2021, 12, 100079 CrossRef CAS.
- L. Köps, C. Leibing, L. H. Hess and A. Balducci, J. Electrochem. Soc., 2021, 168, 010513 CrossRef.
- Y. Shen, S. Wang, H. Li, K. Wang and K. Jiang, J. Energy Storage, 2023, 64, 107164 CrossRef.
- D. Mackay and I. van Wesenbeeck, Environ. Sci. Technol., 2014, 48, 10259–10263 CrossRef CAS PubMed.
- M. Kerner, N. Plylahan, J. Scheers and P. Johansson, RSC Adv., 2016, 6, 23327–23334 RSC.
- M. Kerner, N. Plylahan, J. Scheers and P. Johansson, Phys. Chem. Chem. Phys., 2015, 17, 19569–19581 RSC.
- A. Tsurumaki, M. Agostini, R. Poiana, L. Lombardo, E. Lufrano, C. Simari, A. Matic, I. Nicotera, S. Panero and M. A. Navarra, Electrochim. Acta, 2019, 316, 1–7 CrossRef CAS.
- G. G. Eshetu, S. Grugeon, S. Laruelle, S. Boyanov, A. Lecocq, J.-P. Bertrand and G. Marlair, Phys. Chem. Chem. Phys., 2013, 15, 9145–9155 RSC.
- G. Liu, Q. Sun, Q. Li, J. Zhang and J. Ming, Energy Fuels, 2021, 35, 10405–10427 CrossRef CAS.
- C. Ma, P. Fang, Z.-R. Liu, S.-S. Xu, K. Xu, X. Cheng, A. Lei, H.-C. Xu, C. Zeng and T.-S. Mei, Sci. Bull., 2021, 66, 2412–2429 CrossRef CAS PubMed.
- G. C. Chung, H. J. Kim, S. I. Yu, S. H. Jun, J. w. Choi and M. H. Kim, J. Electrochem. Soc., 2000, 147, 4391 CrossRef CAS.
- S.-K. Jeong, M. Inaba, Y. Iriyama, T. Abe and Z. Ogumi, J. Power Sources, 2008, 175, 540–546 CrossRef CAS.
- L. El Ouatani, R. Dedryvère, C. Siret, P. Biensan, S. Reynaud, P. Iratçabal and D. Gonbeau, J. Electrochem. Soc., 2009, 156, A103 CrossRef CAS.
- A. Lewandowski, A. Swiderska-Mocek and L. Waliszewski, J. Solid State Electrochem., 2012, 16, 3391–3397 CrossRef CAS.
- S.-c. Kinoshita, M. Kotato, Y. Sakata, M. Ue, Y. Watanabe, H. Morimoto and S.-i. Tobishima, J. Power Sources, 2008, 183, 755–760 CrossRef CAS.
- L. Yang, W. You, X. Zhao, H. Guo, X. Li, J. Zhang, Y. Wang and R. Che, Nanoscale, 2019, 11, 17557–17562 RSC.
- F. Yu, L. Zhang, Y. Li, Y. An, M. Zhu and B. Dai, RSC Adv., 2014, 4, 54576–54602 RSC.
- L. H. Hess and A. Balducci, ChemSusChem, 2018, 11, 1919–1926 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc05476h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |