
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBio-oil derived polyesteramides as water-degradable replacements for polyethylene†
Yohei
Yoshinaka
and
Stephen A.
Miller
 *
*
The George and Josephine Butler Polymer Research Laboratory, Department of Chemistry, University of Florida Gainesville, Florida 32611-7200, USA. E-mail: miller@chem.ufl.edu; Fax: +1-352-392-9741; Tel: +1-352-392-7773
First published on 5th February 2025
Abstract
The seed oil from Brassica carinata is a promising source for biobased aviation fuel. Interestingly, the seeds contain considerable sinapic acid and erucic acid, both of which can be utilized for bioplastic synthesis. From the latter, we report herein the synthesis of biobased and water-degradable polyesteramides (PEAs) via N,N′-bis(2-hydroxyethyl)brassylamide (BHEBA). This diol was prepared from ethanolamine and brassylic acid, a C13 oxidation product of the C22 erucic acid, which makes up 42% of the fatty acids present in this non-GMO (non-genetically modified) Ethiopian mustard seed oil. After optimization of conditions, BHEBA was polymerized with aliphatic diacids to obtain the designed PEAs with high purified yields (77–88%) and good molecular weights (Mn = 7000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 Da). The melting temperatures of these PEAs ranged from 130–139 °C, values comparable to those of several grades of polyethylene. Compared to shorter diacids, the brassylic acid incorporated into PEA structures improves hydrophobicity, and mechanical performance was not compromised after a daylong exposure to water. Furthermore, a 12-month PEA degradation study revealed significant hydrolytic degradation (at least 37% loss in Mn) under all the conditions studied: pH 2, pH 5, seawater, and deionized water. Their degradability was further evaluated under high-temperature conditions compared to several commercial plastics, establishing their superior degradability in seawater and deionized water. Chemical recyclability of PEA was demonstrated through facile aminolysis with ethanolamine to regenerate the BHEBA monomer in 84% yield. While further mechanical property improvement would be ideal, the results substantiate the high potential of brassylic acid-based polyesteramides to be eco-friendly replacements for some petroleum-derived commodity plastics, especially polyethylene.
700 Da). The melting temperatures of these PEAs ranged from 130–139 °C, values comparable to those of several grades of polyethylene. Compared to shorter diacids, the brassylic acid incorporated into PEA structures improves hydrophobicity, and mechanical performance was not compromised after a daylong exposure to water. Furthermore, a 12-month PEA degradation study revealed significant hydrolytic degradation (at least 37% loss in Mn) under all the conditions studied: pH 2, pH 5, seawater, and deionized water. Their degradability was further evaluated under high-temperature conditions compared to several commercial plastics, establishing their superior degradability in seawater and deionized water. Chemical recyclability of PEA was demonstrated through facile aminolysis with ethanolamine to regenerate the BHEBA monomer in 84% yield. While further mechanical property improvement would be ideal, the results substantiate the high potential of brassylic acid-based polyesteramides to be eco-friendly replacements for some petroleum-derived commodity plastics, especially polyethylene.
Green foundation1. We have introduced Brassica carinata seed oil as a scalable source of brassylic acid and used this building block for the synthesis of novel polyesteramides with properties indicating their potential as new, eco-friendly replacements for some commodity plastics, including polyethylene.2. The polyesteramides described possess high melting temperatures (Tm = 130–139 °C) comparable to that of polyethylene. Additionally, the brassylic acid component conferred hydrophobicity to the polyesteramides, resulting in maintained mechanical property performance after short-term exposure to water. Nonetheless, these hydrophobic polyesteramides exhibited water-degradability with more than 37% molecular weight loss after one year. 3. Further increasing polyesteramide molecular weight is desirable because that can improve mechanical properties and expand the potential applications. Moreover, a detailed study of polymer biodegradability—and toxicity of the released molecules—could further enhance the significance of this project. |
Introduction
The development of polymer science has provided human conveniences which will never be willingly abandoned. However, this development has been accompanied by unforeseen problems, notably stemming from the boundless production of commodity plastics. These issues include fossil fuel depletion,1,2 carbon dioxide production,3 and environmental persistence.4 While polymer stability can be a great advantage for certain applications, it is an unnecessary property in many cases given that 40% of plastics are produced for single use.5 Because of such poor degradability, 79% of post-consumer plastic waste is accumulated in landfills.6 Furthermore, plastic waste is often discarded inappropriately and remains in the open environment semi-permanently. It is estimated that 2–5% of all plastic waste enters the ocean and negatively affects the marine environment.7,8 Additionally, this waste eventually turns into microplastics that can easily disperse and pollute our environment directly or indirectly.9–11 While plastics provide us vast conveniences, they also cause many serious problems that should be addressed without delay.Of course, science can address such problems and considerable research effort has been directed toward next generation eco-friendly plastics,12–16 which can be biobased, degradable, recyclable, or some combination thereof. Biobased plastics are derived from biobased feedstocks such as lignin, carbohydrates, terpenes, and fatty acids. Degradable plastics decompose into benign, low molecular weight molecules under specific conditions. While several bioplastics are still under commercial development, they are expected to address the aforementioned problems, as their global production capacity increases from 2.2 billion kg per year (in 2023) to an estimated 7.4 billion kg per year in five years. However, current bioplastic production is less than 1% of total plastic production, meaning conventional commodity plastics still dominate the market.17 In order for eco-friendly plastics to penetrate the current market, their drawbacks must be remedied—including relatively poor polymer properties and high production costs compared to conventional commodity plastics.18
One potential solution stems from Brassica carinata (carinata), a seed crop grown commercially on four continents.19–22 While the main application of this seed crop is for bio-jet fuel, it also provides building blocks that can be utilized for bioplastic synthesis (Fig. 1).19,23 The limited land for growing biobased feedstocks is one common problem with bioplastic production;24 but carinata is a hardy species that can be grown off-cycle on otherwise unused land. Accordingly, it can be grown as a winter/cover crop in warm climates, such as the southeast United States. Furthermore, since carinata seed oil is inedible (too high molecular weight fatty acid profile), there is diminished concern about its effect on the food supply chain, which is problematic for edible biobased resources, including starch and common vegetable oils.25,26
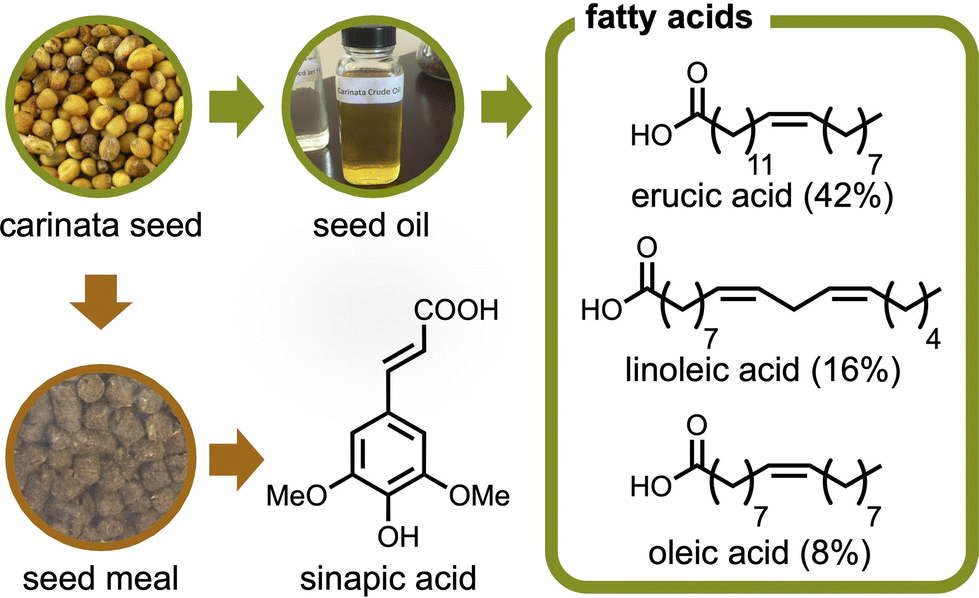 | ||
| Fig. 1 Chemicals available from carinata seed. Percentages shown describe the composition of each fatty acid in the seed oil, as reported by Warwick, et al.28 | ||
As an example of carinata-based plastics, our group previously studied the extraction of its seed meal to yield sinapic acid and its derivative was polymerized to give biobased polyesters with commercially applicable glass transition temperatures (Tg) ranging from 41 to 118 °C.27 Beyond sinapic acid, carinata provides several fatty acids like other seed crops. But interestingly, the primary component is erucic acid (42%), a fatty acid not common in other mustard species.28 This C22 cis-Δ13 mono-unsaturated fatty acid is readily oxidized to afford brassylic acid, an aliphatic carboxylic diacid possessing thirteen carbons.29 Targeted production quantities for sustainable aviation fuel (132.5 billion liters per year by 2050)30 has the potential to markedly increase the availability of brassylic acid, while lowering its cost. Although the brassylic acid market has exceeded 60 million USD per year, its production is much less than other diacids and its primary application is in the fragrance industry.31 The polymers derived from brassylic acid (C13) are much less studied compared to those from common aliphatic carboxylic diacids with shorter hydrocarbon chains, such as succinic acid (C4), adipic acid (C6), and sebacic acid (C10).
Still, several researchers reported polymers derived from brassylic acid with promising properties,32–34 establishing it as a viable biobased resource and providing additional value to carinata crop production. In order for brassylic acid-derived polymers to compete with extant commodity plastics, further improvement of polymer properties is desirable. One possible approach is to incorporate amide functional groups to create biobased aliphatic polyamides (nylons). The introduction of an amide group can induce chain–chain hydrogen bonding, significantly improving polymer properties such as melting temperature (Tm), glass transition temperature (Tg), and toughness, as originally reported by Carothers.35 Although a few nylons derived from brassylic acid have already been reported and exhibited good polymer properties,36,37 their high crystallinity made them very chemically stable and less degradable, which is not ideal for many single-use plastic applications.
To improve environmental degradation aptitude, we envisioned incorporating an ester functional group into the nylon structure and thus, creating aliphatic polyesteramides (PEAs) from brassylic acid. The ester group is a pervasive main-chain polymer linkage and, in several instances, its degradation can occur in the presence of water via hydrolysis.38–40 Although the polymer properties may be diminished by introducing ester functional groups, the remaining amide functional groups of the PEAs should counterbalance this effect. Thus, PEAs should possess better polymer properties than conventional polyesters and better degradation behavior than conventional polyamides. There are already several examples of aliphatic PEAs with good thermal properties, sufficient mechanical performance, biodegradability, hydrolytic degradability, and low gas permeability.41–50 In particular, Guégan and colleagues reported a series of PEAs derived from carboxylic diacids and diols containing amide groups prepared by amidation of carboxylic diacids with ethanolamine.41 This approach provides good guidance in that the starting materials are carboxylic diacids and ethanolamine generally (or potentially) available from biobased resources.51,52 Additionally, their obtained aliphatic PEAs showed admirable melting temperatures (Tm = 122–158 °C), comparable to those of some commodity plastics such as high density polyethylene.
To our knowledge, however, PEAs derived from brassylic acid have not been reported. It should be emphasized that the long aliphatic hydrocarbon segment of brassylic acid ([CH2]11) should render a less hydrophilic PEA, which has advantages for short-term dimensional stability in the event of water exposure. This conjecture is partially supported by the research of Smith and colleagues, who reported that polyamides with longer aliphatic chains absorbed less water over time.53 This is advantageous in some applications because the polymer performance is often compromised when absorbed water functions as a plasticizer. This plasticizing effect is especially concerning for polyamides because of the hydrophilicity accompanying the secondary amide functional group.54–56 While brassylic acid incorporation may improve hydrophobicity and expand potential applications, it may also slow down or arrest hydrolytic degradation. The net result cannot yet be predicted because the impact of hydrophilicity on the hydrolytic degradability of aliphatic PEAs is not well studied.
In this report, we demonstrate the synthesis of a series of PEAs from various aliphatic carboxylic diacids and N,N′-bis(2-hydroxyethyl)brassylamide (BHEBA) that was prepared from brassylic acid in two steps (Scheme 1). The properties of these polymers were studied, including their thermal properties and mechanical performance under wet conditions. The long-term water degradation behavior of PEA 13,6 is thoroughly investigated at room temperature in four different aqueous media: pH 2, pH 5, deionized (DI) water, and seawater. The water degradability of PEAs is directly compared to that of commodity plastics under high-temperature conditions. Additionally, the chemical recyclability of PEA 13,13 was investigated via aminolysis with ethanolamine.
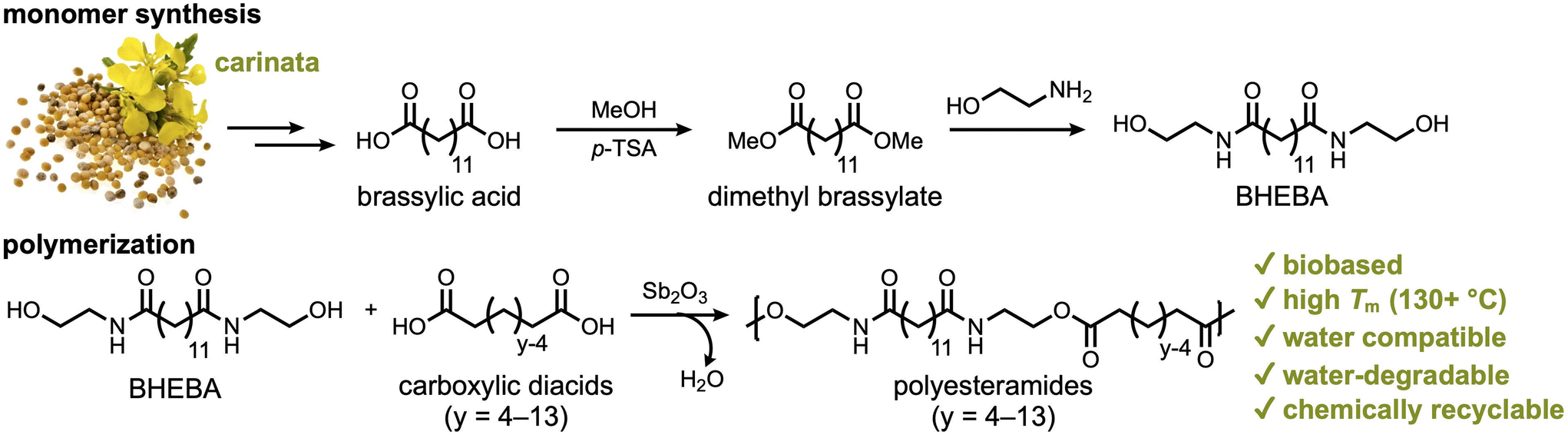 | ||
| Scheme 1 Synthesis of a series of polyesteramides (PEAs) from brassylic acid, ethanolamine, and carboxylic diacids of varying length. While carinata is a potential source of brassylic acid, the current industrial production of this diacid employs erucic acid from other species,57,58 such as crambe (Crambe abyssinica) and high erucic acid rapeseed (Brassica napus). | ||
Results and discussion
The synthesized polyesteramides (PEAs) are described here as “PEA x,y” where x is the carbon number of the carboxylic diacid used to prepare the bis-amidediol monomer and y is the carbon number of the carboxylic diacid used for the polymerization (Scheme 2). In all cases, the bis-amidediol monomer is prepared from ethanolamine. The experimental details are summarized in the ESI.† | ||
| Scheme 2 General representation of PEAs (polyesteramides) synthesized in this study. The number of carbons between amide N atoms is given by x. The number of carbons between ester O atoms is given by y. | ||
Polymerization optimization
The polymerization conditions were optimized by synthesizing PEA 13,6 from N,N′-bis(2-hydroxyethyl)brassylamide (BHEBA) and adipic acid, employing antimony oxide (Sb2O3) as the catalyst. The conditions and polymer characterization data are summarized in Table S3.† With a maximum polymerization temperature of 240 °C,38–40 intractable polymers were obtained, presumably because of cross-linking via N-carbonylation with adipic acid and/or polymer ester bonds. With a maximum polymerization temperature of 200 °C, the synthesized polymers were tractable as demonstrated by their dissolution in hexafluoroisopropanol (HFIP) or trifluoroacetic acid (TFA). Although good molecular weight values were obtained (Mn = 13900 Da, Mw = 69400 Da), the dispersity was broad (Đ = 4.98) and the melting temperature (Tm = 116 °C) was lower than in other trials. Optimal polymer properties were obtained when the polymerization temperature ramped from 160 to 180 °C over 8 hours, with vacuum applied during the latter half (Table 1, entry 1). The structure of PEA 13,6 was confirmed by 1H NMR analysis (Fig. S24†). Because of observed adipic acid sublimation, a slight excess of adipic acid (1.05 equivalents) was tested. This resulted in slightly higher Mn and Mw values, but a greater dispersity (Table 1, entry 2). Hence, 1.0 equivalent of adipic acid was deemed optimal. Subsequent entries of Table 1 show catalyst screening results and indicate that Sb2O3 is the preferred catalyst (Table 1, entry 1).
| Entry | Catalyst (2 mol%) | Equivalents of adipic acidb | M n (Da) | M w (Da) | Đ |
|---|---|---|---|---|---|
| a The polymerization system was initially heated to 160 °C under a nitrogen atmosphere for 4 hours, followed by dynamic vacuum at the same temperature for 2 hours. Then, the temperature was raised to 180 °C for 2 hours. b Molar equivalents of adipic acid versus BHEBA. c Measured by gel permeation chromatography (GPC) using sodium trifluoroacetic acid (NaTFA, 20 mM) dissolved in hexafluoroisopropanol (HFIP) as the mobile phase. The columns were kept at 40 °C and polymethylmethacrylate (PMMA) standards were employed. | |||||
| 1 | Sb2O3 | 1.00 | 10500 |
31200 |
3.0 |
| 2 | Sb2O3 | 1.05 | 11100 |
36400 |
3.3 |
| 3 | p-TSA | 1.00 | 5800 | 23000 |
4.0 |
| 4 | Zn(OAc)2 | 1.00 | 6100 | 17600 |
2.9 |
| 5 | K2CO3 | 1.00 | 3700 | 9000 | 2.4 |
| 6 | H2SO4 | 1.00 | 5000 | 16600 |
3.3 |
| 7 | Sn(Oct)2 | 1.00 | 8200 | 24600 |
3.0 |
| 8 | Ti(OBu)4 | 1.00 | 9600 | 27800 |
2.9 |
| 9 | No catalyst | 1.00 | 4500 | 12500 |
2.8 |
Synthesis of a polyesteramide series
With the optimized conditions, a PEA series was synthesized, wherein the carboxylic diacid varied from C4 to C13 (PEA 13,y with y = 4–13); the polymers were obtained with 77–88% post-precipitation yield (Table 2). For comparison, PEA 6,6 was synthesized under the same conditions. While most PEAs had good molecular weights, lower molecular weight products were initially obtained with succinic acid (C4), glutaric acid (C5) and brassylic acid (C13): PEA 13,4 (Mn = 1000 Da); PEA 13,5 (Mn = 6000 Da); and PEA 13,13 (Mn = 6300 Da), respectively. Further optimization provided improved results for PEA 13,13 (Mn = 8600 Da) by using 4 mol% of Sb2O3 and 1.05 equivalents of brassylic acid, although the dispersity broadened somewhat (3.3 to 3.8). While this approach also improved the Mn of PEA 13,5 (Mn = 7400 Da), it did not work for PEA 13,4. The very limited Mn values for PEA 13,4 can be explained by competitive chain back-biting, wherein a secondary amide nitrogen initially attacks the carboxylic acid terminus or an ester group already within the main-chain, forming an 8-membered ring (Fig. S2A†), which can then isomerize, exergonically (ΔG453 = −12.5 kcal mol−1), to N-hydroxyethylsuccinimide (Fig. S2F†), which we have confirmed by 1H NMR (Fig. S33†). Höcker and colleagues have reported that the same thermodynamically favorable by-product competes with chain extension (Fig. S2E†).59,60 Our computational results suggest that cyclization to N-hydroxyethylsuccinimide is rather exergonic (Fig. S2D, A and F,† ΔG453 = −8.7 kcal mol−1) compared to chain extension (Fischer esterification, Fig. S2D,† ΔG453 = +7.1 kcal mol−1) at our typical polymerization temperature of 180 °C, where possible 5-membered ring volatilization further signals low molecular weight. Because of this apparent side reaction and our inability to obtain decent molecular weights for PEA 13,4, this polymer is excluded from discussion in the following section.|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Polymer | Yieldb (%) | M n (Da) | M w (Da) | Đ | T d5 (°C) | T g (°C) | T m (°C) |
| a With 1.0 equivalent carboxylic diacid and 2 mol% Sb2O3 as catalyst, the polymerization system was initially heated to 160 °C under a nitrogen atmosphere for 4 hours, followed by dynamic vacuum at the same temperature for 2 hours. Then, the temperature was raised to 180 °C for 2 hours. b Reported as a mass yield following polymer precipitation, except for entry 1 which preceded polymer precipitation. c Measured by GPC using NaTFA (20 mM) dissolved in HFIP as the mobile phase. The columns were kept at 40 °C and PMMA standards were employed. d 5% mass loss degradation temperature as measured by thermogravimetric analysis (TGA) with a 10 °C min−1 heating rate under nitrogen flow. e Measured by differential scanning calorimetry (DSC) with a 10 °C min−1 heating rate under nitrogen flow. The 3rd cycle thermogram was used for the analysis. f T g values were not determined from the obtained thermograms. g 1.05 equivalents of carboxylic diacid and 4 mol% Sb2O3 were used. | ||||||||
| 1 | PEA 13,4 | 82 | 1000 | 1700 | 1.8 | 275 | N.D.f | 91 |
| 2g | PEA 13,5 | 77 | 7400 | 22200 |
3.0 | 284 | 0 | 139 |
| 3 | PEA 13,6 | 78 | 10500 |
31200 |
3.0 | 285 | 0 | 137 |
| 4 | PEA 13,7 | 78 | 9600 | 30800 |
3.2 | 303 | −1 | 136 |
| 5 | PEA 13,8 | 83 | 7900 | 23800 |
3.0 | 306 | 2 | 137 |
| 6 | PEA 13,9 | 83 | 10700 |
34300 |
3.2 | 306 | 3 | 136 |
| 7 | PEA 13,10 | 88 | 9700 | 31600 |
3.3 | 309 | N.D.f | 130 |
| 8 | PEA 13,11 | 81 | 8100 | 24700 |
3.0 | 303 | N.D.f | 135 |
| 9 | PEA 13,12 | 83 | 7000 | 21800 |
3.1 | 307 | N.D.f | 132 |
| 10g | PEA 13,13 | 84 | 8600 | 32700 |
3.8 | 309 | N.D.f | 135 |
| 11 | PEA 6,6 | 81 | 12200 |
36000 |
2.9 | 290 | 9 | 130 |

Thermal properties of polyesteramides
The thermal properties of the PEAs in Table 2 were studied by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). Their 5% degradation temperatures (Td5, 284–309 °C) were high enough for practical applications and substantially above their melting temperatures (Tm, 130–139 °C), which is preferred for melt processing. The glass transition temperatures (Tg) of these polymers were either not clearly observed or around 0 °C—substantially lower than that of nylon 6,6 (44 °C) or nylon 12 (50 °C),61,62 but decidedly higher than that of polycaprolactone (−60 °C).63Interestingly, their Tm values fell within a small range of 130–139 °C. Still, a slight decreasing trend was observed among the even y carbon series (137, 137, 130, 132 °C) and the odd y carbon series (139, 136, 136, 135, 135 °C) (Fig. S4†)—in line with the presumption that additional methylene groups diminish crystallinity by diluting the interchain amide hydrogen bonding. In this regard, PEAs behave more like polyamides than polyesters since the latter generally exhibit increasing melting temperatures with an increasing number methylene groups per repeat unit.54,64–67 Nevertheless, these counterbalancing effects for polyesters and for polyamides may explain the narrow Tm range observed for the polyesteramides of Table 2. It should be noted that similar trends were observed with related, aliphatic PEAs as their methylene composition was varied.49,50 Additionally, the small Tm range of these PEAs is probably a consequence of employing the long BHEBA monomer, which diminishes the length effect of the diacid employed. For comparison, the Tm range of the nylon x,6 series (x = 2–12) is 315–204 °C,54 but that of the nylon x,13 series (x = 3–11, from various diamines and brassylic acid) is much narrower at 191–176 °C.36
With little surprise, the Tm values of PEAs in this study are much lower than those of related nylons such as nylon 6,6 (Tm = 265 °C).61 However, the Tm values are still comparable to those of some commodity plastics, including low-density polyethylene (LDPE, Tm ∼ 108 °C)68 and high-density polyethylene (HDPE, Tm ∼ 134 °C).66 The thermal properties of PEAs were further analyzed by comparing them with consumer grade LDPE and HDPE via DSC. Table 3 summarizes the melting and crystallization temperatures as well as the enthalpies of melt (ΔHm) and crystallization (ΔHc) of PEA 13,6, PEA 13,13, LDPE, and HDPE (additional PEA data are summarized in Table S7†). A laboratory solvent bottle (LDPE) and a milk jug (HDPE) were analyzed as representative commercial articles. A polymer's crystallization rate is often evaluated by the gap between its melting temperature and crystallization temperature (ΔT = Tm − Tc) and this was around 30 °C for PEA 13,6 and PEA 13,13. In comparison, both LDPE and HDPE exhibited ΔT around 15 °C. This implies that our PEAs cannot crystallize as fast as polyethylene, but ΔT = 30 °C is still considered small.41,69 Also, the ΔT values for the PEA 13,y series ranged from 23 to 34 °C (Table S7†), meaning some were more comparable to polyethylene. Within the PEA 13,y series, the ΔHm and ΔHc values generally increased with increasing length of the aliphatic diacid employed. The ΔHm ranged from 37 J g−1 (PEA 13,6) to 64 J g−1 (PEA 13,12), achieving 55% of the value for LDPE and 30% of the value for HDPE.
| Entry | Polymer | T m (°C) | T c (°C) | T m − Tc (°C) | ΔHm (J g−1) | ΔHc (J g−1) |
|---|---|---|---|---|---|---|
| a Measured by DSC with a 10 °C min−1 heating rate under nitrogen flow. The 3rd cycle thermogram was used for the analysis. b LDPE from a laboratory solvent bottle. c HDPE from a milk jug. | ||||||
| 1 | PEA 13,6 | 137 | 105 | 32 | 37 | 46 |
| 2 | PEA 13,13 | 135 | 106 | 29 | 52 | 57 |
| 3 | LDPEb | 112 | 99 | 13 | 117 | 121 |
| 4 | HDPEc | 135 | 120 | 15 | 213 | 215 |
Mechanical properties of polyesteramides
Polymer mechanical testing was performed with samples prepared by cutting a film manually along the mold (Fig. S5†). In order to minimize the inconsistency of this method, multiple specimens (9–12) were tested and Table 4 summarizes the average of the top five samples for each polymer. Still, the mechanical data presented here should be considered relative since inadvertent defects in the narrow section of the specimens can result in premature deformation and breakage events (see Fig. S6†). The primary purpose of this study is to understand the effect of water exposure on the mechanical performance, which is described in the next section.| Entry | Polymer | M n (Da) | σ max (MPa) | ε (%) | E (MPa) |
|---|---|---|---|---|---|
| a Determined by a tensile test with rate = 1.5 mm min−1. b The number average molecular weight of the crude products, measured by GPC using NaTFA (20 mM) dissolved in HFIP as the mobile phase. The columns were kept at 40 °C and PMMA standards were employed. c Maximum stress. d Strain at complete break. e Young's modulus, calculated from the slope from 0% to 1% of strain. f Sample brittleness precluded data acquisition. g Values summarized from the literature.70–76 | |||||
| 1 | PEA 13,5 | 5500 | N.D.f | N.D.f | N.D.f |
| 2 | PEA 13,6 | 9600 | 23.1 ± 0.9 | 65 ± 4 | 192 ± 19 |
| 3 | PEA 13,7 | 8100 | 23.2 ± 0.9 | 82 ± 14 | 215 ± 17 |
| 4 | PEA 13,8 | 7800 | 29.6 ± 2.3 | 52 ± 8 | 280 ± 31 |
| 5 | PEA 13,9 | 9000 | 26.6 ± 2.1 | 40 ± 5 | 218 ± 12 |
| 6 | PEA 13,10 | 11300 |
29.2 ± 0.9 | 33 ± 3 | 260 ± 13 |
| 7 | PEA 13,11 | 8100 | 28.4 ± 2.4 | 117 ± 18 | 277 ± 35 |
| 8 | PEA 13,12 | 8000 | 31.3 ± 1.8 | 76 ± 27 | 302 ± 34 |
| 9 | PEA 13,13 | 7500 | 28.2 ± 2.6 | 57 ± 15 | 298 ± 45 |
| 10 | PEA 6,6 | 14000 |
26.2 ± 1.8 | 131 ± 31 | 221 ± 24 |
| 11 | LDPEg | 70k–120k | 12–20 | 281–950 | 138–200 |
| 12 | HDPEg | 20k–3000k | 20–30 | 180–870 | 745–1683 |
Unfortunately, specimens could not be prepared for PEA 13,5 because of poor mechanical properties (brittleness). In contrast, good maximum stress (σmax) values were observed for the other PEAs, ranging from 23.1–31.3 MPa. These σmax values were comparable to or better than values reported for HDPE and LDPE (reported as stress at break).70 Although the trend is imperfect, the σmax values tend to be larger as hydrocarbon chain length increases. This may seem counterintuitive, considering a dilution of amide bond density, but comports with the increased heats of melt (Table S8†) and thus, increased crystallinity, postulated for this PEA series. While a similar increasing trend for Young's modulus was observed (E = 192–302 MPa), the strain values (ε) varied broadly and no discernable trend was identified.
For comparison, the mechanical properties of PEA 6,6 were also studied with our protocol. PEA 6,6 showed maximum stress (σmax) and Young's modulus (E) comparable to the PEA 13,y series, but it showed a much higher strain (ε). We can ascribe this, partially, to a higher molecular weight (Mn = 14000 Da) of PEA 6,6, an effect reported in another class of polyesteramides.42 Additionally, the lower crystallinity of PEA 6,6 (ΔHm = 27 J g−1) implies a greater amorphous region, which generally improves structural flexibility. While our PEAs showed σmax and Young's modulus comparable to the reported values of commercial LDPE, the strain (ε) was markedly higher for the polyethylenes.70–74 While some of this advantage is attributable to additives in the commercial materials and imprecise sample preparation of our PEAs, further improvements in the PEA 13,y series are clearly necessary before these polyesteramides can mimic polyethylenes across all important parameters.
An important lever for improving the strain of PEAs is polymer molecular weight. While our Mn values near 10000 Da are useful for ascertaining thermal properties (Tm and Tg), they may be insufficient to reveal the full potential of PEA mechanical properties. For example, the ideal mechanical properties of commercial nylon 6,6 are not reached until Mn exceeds 20000 Da. Given that high viscosity halted the stirring for most of our polymerizations, PEA molecular weight would assuredly increase by more efficient stirring in sophisticated reactors.
Water absorption of the polyesteramides
While polyamides tend to absorb water because of their hydrophilic amide functionality, the brassylic acid-derived PEAs of this study may behave differently because of the less hydrophilic ester functionality and long, hydrophobic methylene sequences ([CH2]11) in the main-chain. To investigate this possibility, we initially studied water absorption of PEA 13,6, PEA 13,13, PEA 6,6, and nylon 6,6 by storing samples in deionized (DI) water for 24 hours. The amount of absorbed water was determined by measuring the mass loss upon heating the samples at 100 °C for 2 hours via TGA. As predicted, minimal water loss was found for PEA 13,6 (2.4%) and PEA 13,13 (1.0%). In contrast, more than double the water loss was observed for PEA 6,6 (5.4%) and nylon 6,6 (5.8%) (Fig. 2A).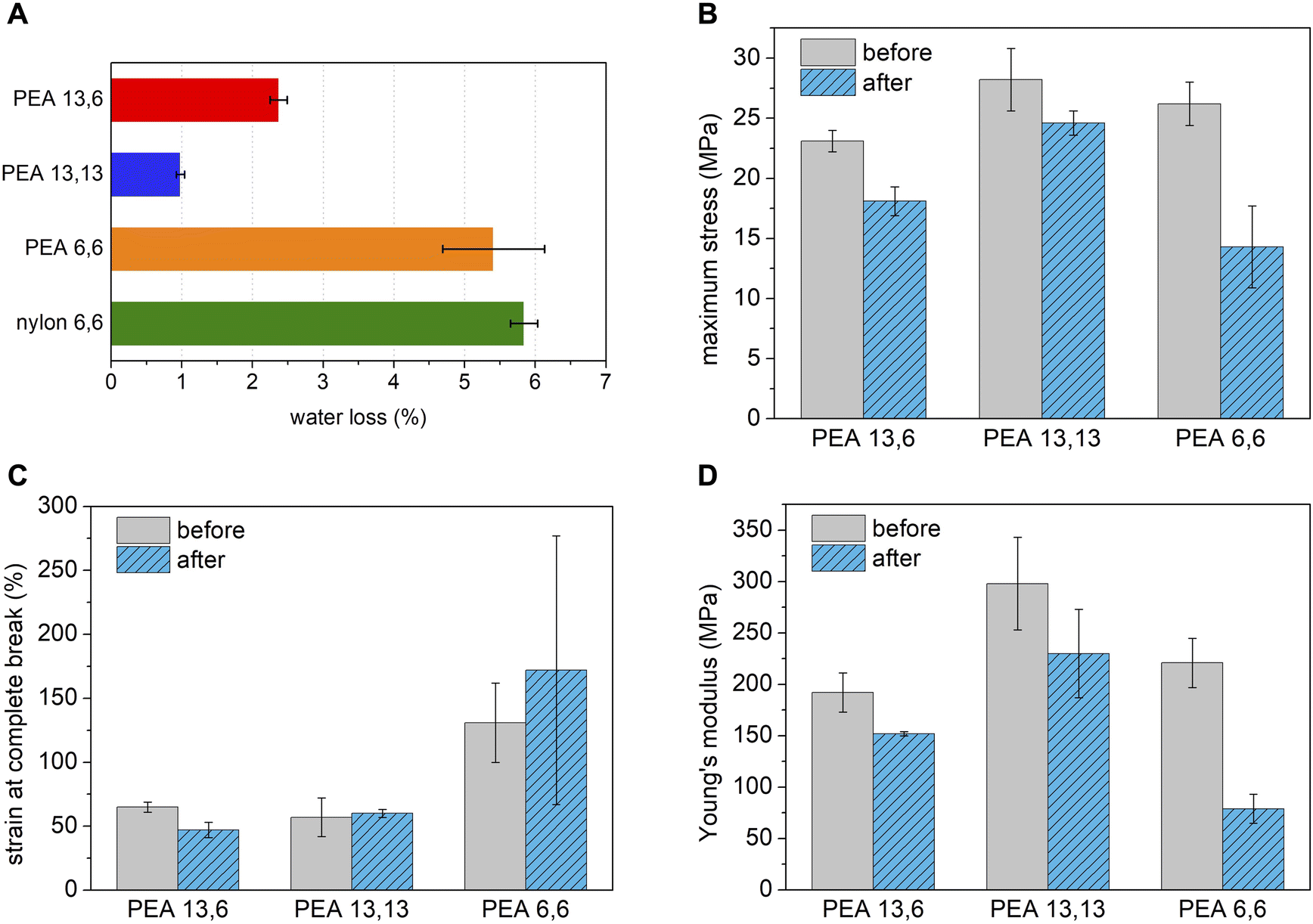 | ||
| Fig. 2 (A) Amount of DI water absorbed in 24 hours by PEAs and nylon 6,6, as determined by mass loss at 100 °C over 2 hours (TGA). The maximum stress (B), percent strain at break (C), and Young's modulus (D) of PEAs before and after storing samples in DI water for 24 hours. | ||
In order to further support the superior water compatibility of our polyesteramides, PEA 13,6, PEA 13,13, and PEA 6,6 were stored in DI water for 24 hours and their mechanical properties were compared to their original values (Fig. 2B–D). As expected, the mechanical performance of PEA 13,6 and PEA 13,13 were largely maintained. In contrast, significant mechanical property changes were observed for PEA 6,6, suggesting that the long methylene sequences of the incorporated brassylic acid are responsible for repelling water and maintaining mechanical performance. Based on these results, PEA 6,6 is better suited for dry applications, while PEA 13,6 and PEA 13,13 can tolerate water exposure, at least for short intervals, which is sufficient for a greater variety of single-use applications.
Hydrolytic degradation of polyesteramide
The water-degradability of PEA 13,6 was studied by agitating polymer samples in four different aqueous media—pH 2, pH 5, DI water, and seawater—on an orbital shaker and monitoring their molecular weight for 12 months. As shown in Fig. 3, 80% molecular weight (Mn) loss was observed in the pH 2 solution, while approximately 40% Mn loss was observed in the other three media. The fastest degradation in the pH 2 medium is accounted for by acid-catalyzed ester hydrolysis, as commonly reported for aliphatic polyesters.77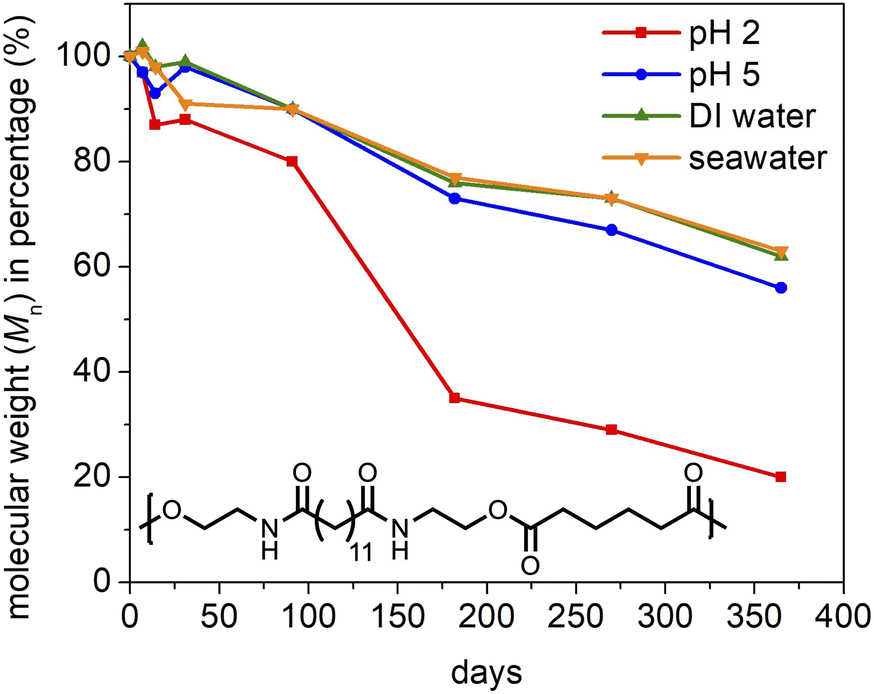 | ||
| Fig. 3 Hydrolytic degradation study of PEA 13,6 in four different aqueous media: pH 2, pH 5, DI water, and seawater. The samples were agitated on an orbital shaker at room temperature for specific durations (1 week, 2 weeks, 1 month, 3 months, 6 months, 9 months, and 12 months). The water-degradability was evaluated by the change in the number average molecular weight compared to the initial value. | ||
The results for each medium were extrapolated (linearly, via the last four data points) to estimate the time required for complete hydrolysis of PEA 13,6: 14 months at pH 2; 27 months at pH 5; 33 months in DI water; and 34 months in seawater (Fig. S14†). Despite the simplicity of this estimate, the trends suggest that PEA 13,6 will degrade under environmentally relevant conditions markedly faster than the vast majority of commercial polymers, such as polyethylene, polypropylene, polyethylene terephthalate (thousands of years),4 and nylon 6,6 (one hundred years).78 We speculate that the amide and ester functionalities work symbiotically to effect polymer degradation. The amides improve polymer hydrophilicity while the esters function as weak links in the main-chain as they are kinetically more prone to hydrolysis than the amides.38 Ester hydrolysis was confirmed by 1H NMR analysis of the filtered residue collected after PEA 13,6 was stirred in DI water for over three years. Signature peaks for BHEBA were identified (Fig. S15†). Additionally, given the effect of crystallinity on polymer degradation rates,79 the lower hydrogen bond density and lower crystallinity of PEA 13,6 compared to nylon 6,6 further explain the hydrolytic aptitude of PEA 13,6. It should be noted that without added water, PEA 13,6 stored in a vial under ambient conditions showed essentially no molecular weight loss after one year (Fig. S102†).
High-temperature hydrolytic degradation of polyesteramides
To better understand the degradability of polyesteramides, PEA 13,6 and PEA 13,13 were subjected to high-temperature degradation conditions by agitation (shaker) in DI water or seawater at 60 °C for up to 6 months. High temperatures can simulate the passage of time and can, thus, replace longer experiments. For comparison, several other polymers were included in this study: PEA 6,6 was chosen to assess the effect of methylene sequence length; polylactic acid (PLA) was tested as a representative commercial bioplastic; polyethylene terephthalate (PET) was considered as a persistent commodity polyester; and nylon 6,6 was selected as a persistent commodity polyamide.Similar high-temperature hydrolysis results were obtained for DI water and seawater, as shown in Fig. 4. Although PEA 13,6 and PEA 13,13 were mostly degraded in 3 months, the molecular weight loss of PEA 6,6 stagnated, and its molecular weight remained around 40% of its original value. Because of its greater hydrophilicity, we expected the PEA 6,6 to degrade faster than the PEA 13,y polymers. One explanation is that PEA 6,6 did hydrolyze faster, but our analysis method—which analyzed the Mn of the solid portion only (isolated from the water soluble monomers and oligomers by filtration)—biased our results. This effect probably contributed to the initial increase in Mn observed for PEA 6,6—which seems to be mostly attributable to truncation of the lower molecular weight fraction, according to GPC comparison of the initial and 7 day samples (Fig. S18†). Another explanation is that the sample's initial molecular weight is a factor more important than the concentration of hydrophilic functional groups (amides). Nonetheless, all PEA polymers were mostly or fully hydrolzyed after 6 months at 60 °C. This implies that the hydrophobic brassylate segment of the PEA 13,y polymers does not significantly slow their hydrolysis, at least when Mn is around 8000–10000 Da.
 | ||
| Fig. 4 High-temperature hydrolytic degradation study of PEA 13,6, PEA 6,6, PEA 13,13, PLA, PET, and nylon 6,6 in DI water (A) or seawater (B). Vials were agitated in a water bath shaker held at 60 °C for specific durations: 1 week, 2 weeks, 1 month, 2 months, 3 months, and 6 months. The water-degradability was evaluated by the change in the number average molecular weight compared to the initial value. | ||
Fig. 4 reveals that PLA exhibits the fastest degradation. This makes sense because the PLA samples employed were amorphous, with a Tg near 55 °C,80 whereas the other polymers tested all had Tm values considerably higher than the 60 °C temperature of the experiment. As expected, minimal Mn loss was measured for PET or nylon 6,6, in agreement with their established environmental persistence.
Chemical recyclability of PEA 13,13
Among the PEAs of this study, we consider PEA 13,13 to be the strongest candidate for the next generation of eco-friendly plastics for several reasons. First, it is built only from ethanolamine and brassylic acid, meaning it can be fully biobased. Second, it is hydrophobic and maintains polymer performance under wet conditions, which expands its application range. Third, it is ultimately water-degradable under longer-term, environmentally-relevant conditions. This is an attractive property considering the current fate of most plastics.11 However, society has stated goals of increased polymer recyclability81 and PEA 13,13 meshes very well with those aspirations. Because PEA 13,13 is only made from ethanolamine and brassylic acid, we envisioned that the straightforward aminolysis of PEA 13,13 with ethanolamine should depolymerize PEA 13,13 back into BHEBA. Accordingly, pure BHEBA was obtained with 84% yield in just 2 hours via aminolysis with 5 equivalents of ethanolamine, followed by recrystallization from ethanol (Fig. 5A). As concluded by 1H NMR analysis (Fig. 5B), the recovered depolymerization product was BHEBA identical to the original monomer synthesized from brassylic acid. This reconstituted BHEBA was repolymerized with brassylic acid and the resulting PEA 13,13 showed properties very similar to those of our original PEA 13,13 (Table S13†)—thereby demonstrating the chemical recyclability of PEA 13,13. | ||
| Fig. 5 (A) Chemical recycling of PEA 13,13 via aminolytic depolymerization and esterification polymerization. (B) 1H NMR (in DMSO-d6) comparison of BHEBA synthesized from brassylic acid (top) and BHEBA obtained via aminolytic depolymerization of PEA 13,13 (bottom). | ||
Production of PEA 13,13 from Brassica carinata
According to George and colleagues,19 the seed yield of carinata is typically 2800 kg ha−1 (in the southeastern United States) and the seed oil content is 40%. This production of about 1120 kg ha−1 of seed oil does not exceed that of palm oil, but compares favorably to that of rapeseed and excels most other vegetable oils.82,83 Since erucic acid constitutes 42% of this seed oil,28 about 470 kg ha−1 of erucic acid can be obtained, which converts to 339 kg ha−1 of brassylic acid through oxidation, using the 72% yield reported by Carlson and Sohns.29 This quantity can afford 361 kg ha−1 of crude PEA 13,13, using the 96.5% yield found in the present study. Wright and colleagues reported that around 4.9 million hectares of cropland remains fallow during the winter season in the southeast USA and has the potential to grow carinata as a winter crop.84 Maximally, 1.77 billion kg of PEA 13,13 can be produced from this idle land, which could replace about 1.5% of the worldwide market for polyethylene (116 billion kg in 2024, combined for LDPE and HDPE).85 This fraction could increase by an order of magnitude by expanding cropland on the six continents where carinata has been successfully grown.22 This fraction could further increase through chemical recycling of post-consumer PEA 13,13—a strategy not currently feasible for polyethylene.Conclusions
We have synthesized novel polyesteramides (PEAs) from a promising and expanding bio-oil crop, Brassica carinata (carinata). The primary interest in this seed oil is its conversion to sustainable aviation fuel, especially of the jet A type.86–88 But, many other applications are envisioned, including biobased plastic production via carinata-based brassylic acid, which can add value to this new industry.Herein, the bis-amidation of dimethyl brassylate with ethanolamine yielded the diol, N,N′-bis(2-hydroxyethyl)brassylamide (BHEBA), which was polymerized with linear aliphatic carboxylic diacids of various carbon number (y = 4–13). The PEA 13,y polymers were obtained in good yields and with acceptable molecular weights in most cases. Polymer melting temperatures (130–139 °C) were comparable to or greater than those of some commodity plastics such as HDPE and LDPE, and our preliminary mechanical testing revealed the potential for many practical applications, although further improvement is needed for a perfect polyethylene replacement. Importantly, our PEAs were less hydrophilic than other PEAs or polyamides and generally retained their mechanical properties after a daylong exposure to water, indicating their short-term water compatibility. Although slower hydrolytic degradation was implied by the polymer hydrophobicity, a yearlong hydrolysis study demonstrated that PEA 13,6 is quite water-degradable in a reasonable timescale under environmentally-relevant conditions. In seawater, for example, the molecular weight (Mn) of PEA 13,6 diminished to 63% of its original value over 365 days at room temperature. The degradation behaviors of PEA 13,13, PEA 13,6, and PEA 6,6 were further examined under accelerated conditions by agitation in a water bath heated to 60 °C for 6 months. While commercial PET and nylon 6,6 exhibited minimal degradation, the PEAs experienced considerable ester hydrolysis. Additionally, the chemical recyclability of PEA 13,13 was demonstrated by its aminolysis with ethanolamine, affording the pure, original monomer (BHEBA) in good yield (84%)—a process that converts all ester functional groups to the amide functional group already present.
The thermomechanical properties of these novel PEAs, along with their propensity for water-degradation, marks them as interesting candidates for replacing incumbent commercial polymers, especially LDPE. Note that the market price of erucic acid is $4.74 USD per kg89 and that of brassylic acid is $5.60 USD per kg90—both of which are expected to trend downward substantially with the expansion of carinata-based aviation fuel. Additional work is needed to fully evaluate the commercial viability of brassylic acid-based PEAs, including increasing polymer molecular weight, studying polymer/oligomer biodegradability, testing degradation product toxicity, and studying PEA film gas permeability.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was supported by the United States Department of Agriculture – National Institute of Food and Agriculture (USDA-NIFA) Bioenergy-Coordinated Agricultural Projects grant no. 2016-11231 (Southeast Partnership for Advanced Renewables from Carinata, SPARC) and the University of Florida.References
- N. Abas, A. Kalair and N. Khan, Review of fossil fuels and future energy technologies, Futures, 2015, 69, 31–49, DOI:10.1016/j.futures.2015.03.003.
- S. Shafiee and E. Topal, When will fossil fuel reserves be diminished?, Energy Policy, 2009, 37, 181–189, DOI:10.1016/j.enpol.2008.08.016.
- M. Höök and X. Tang, Depletion of fossil fuels and anthropogenic climate change-A review, Energy Policy, 2013, 52, 797–809, DOI:10.1016/j.enpol.2012.10.046.
- A. Chamas, H. Moon, J. Zheng, Y. Qiu, T. Tabassum, J. H. Jang, M. Abu-Omar, S. L. Scott and S. Suh, Degradation Rates of Plastics in the Environment, ACS Sustainable Chem. Eng., 2020, 8, 3494–3511, DOI:10.1021/acssuschemeng.9b06635.
- “Solving Plastic Pollution Through Accountability” from World Wildlife Fund. https://www.worldwildlife.org/publications/solving-plastic-pollution-through-accountability (accessed October 2024).
- R. Geyer, J. R. Jambeck and K. L. Law, Production, use, and fate of all plastics ever made, Sci. Adv., 2017, 3, e1700782, DOI:10.1126/sciadv.1700782.
- J. R. Jambeck, R. Geyer, C. Wilcox, T. R. Siegler, M. Perryman, A. Andrayan and K. L. Law, Plastic waste inputs from land into the ocean, Science, 2015, 347, 768–771, DOI:10.1126/science.1260352.
- J. G. B. Derraik, The pollution of the marine environment by plastic debris: a review, Mar. Pollut. Bull., 2002, 44, 842–852, DOI:10.1016/S0025-326X(02)00220-5.
- I. Wojnowska-Baryła, K. Bernat and M. Zaborowska, Plastic Waste Degradation in Landfill Conditions: The Problem with Microplastics, and Their Direct and Indirect Environmental Effects, Int. J. Environ. Res. Public Health, 2022, 19, 13223, DOI:10.3390/ijerph192013223.
- M. Cole, P. Lindeque, C. Halsband and T. S. Galloway, Microplastics as contaminants in the marine environment: A review, Mar. Pollut. Bull., 2011, 62, 2588–2597, DOI:10.1016/j.marpolbul.2011.09.025.
- P. G. C. Nayanathara Thathsarani Pilapitiya and A. S. Ratnayake, The world of plastic waste: A review, Cleaner Mater., 2024, 11, 100220, DOI:10.1016/j.clema.2024.100220.
- F. M. Haque, J. S. A. Ishibashi, C. A. L. Lidston, H. Shao, F. S. Bates, A. B. Chang, G. W. Coates, C. J. Cramer, P. J. Dauenhauer, W. R. Dichtel, C. J. Ellison, E. A. Gormong, L. S. Hamachi, T. R. Hoye, M. Jin, J. A. Kalow, H. J. Kim, G. Kumar, C. J. LaSalle, S. Liffland, B. M. Lipinski, Y. Pang, R. Parveen, X. Peng, Y. Popowski, E. A. Prebihalo, Y. Reddi, T. M. Reineke, D. T. Sheppard, J. L. Swartz, W. B. Tolman, B. Vlaisavljevich, J. Wissinger, S. Xu and M. A. Hillmyer, Defining the Macromolecules of Tomorrow through Synergistic Sustainable Polymer Research, Chem. Rev., 2022, 122, 6322–6373, DOI:10.1021/acs.chemrev.1c00173.
- Y. D. Y. L. Getzler and R. T. Mathers, Sustainable Polymers: Our Evolving Understanding, Acc. Chem. Res., 2022, 55, 1869–1878, DOI:10.1021/acs.accounts.2c00194.
- Y. Zhu, C. Romain and C. K. Williams, Sustainable polymers from renewable resources, Nature, 2016, 540, 354–362, DOI:10.1038/nature21001.
- B. von Vacano, H. Mangold, G. W. M. Vandermeulen, G. Battagliarin, M. Hofmann, J. Bean and A. Künkel, Sustainable Design of Structural and Functional Polymers for a Circular Economy, Angew. Chem., Int. Ed., 2023, 62, e202210823, DOI:10.1002/anie.202210823.
- S. A. Miller, Sustainable Polymers: Opportunities for the Next Decade, ACS Macro Lett., 2013, 2, 550–554, DOI:10.1021/mz400207g.
- “Bioplastics market development update 2023” from European Bioplastics. https://www.european-bioplastics.org/market/ (accessed October 2024).
- L. Avérous and E. Pollet, Biodegradable Polymers, in Environmental Silicate Nano-Biocomposites, ed. L. Avérous and E. Pollet, Springer, London, 1st edn, 2012, ch. 2, pp. 13–39. DOI:10.1007/978-1-4471-4108-2.
- S. George, R. Seepaul, D. Geller, P. Dwivedi, N. DiLorenzo, R. Altman, E. Coppola, S. A. Miller, R. Bennett, G. Johnston, L. Streit, S. Csonka, J. Field, J. Marois, D. Wright, I. M. Small and G. P. Philippidis, A regional inter-disciplinary partnership focusing on the development of a carinata-centered bioeconomy, GCB Bioenergy, 2021, 13, 1018–1029, DOI:10.1111/gcbb.12828.
- SPARC – Southeast Partnership for Advanced Renewables from Carinata. https://sparc-cap.org/ (accessed October 2024).
- “Nuseed Carinata” from Nuseed. https://nuseed.com/carinata/ (accessed October 2024).
- R. Seepaul, S. Kumar, J. E. Iboyi, M. Bashyal, T. L. Stansly, R. Bennett, K. J. Boote, M. J. Mulvaney, I. M. Small, S. George and D. L. Wright, Brassica carinata: Biology and agronomy as a biofuel crop, GCB Bioenergy, 2021, 13, 582–599, DOI:10.1111/gcbb.12804.
- D. C. Taylor, K. C. Falk, C. D. Palmer, J. Hammerlindl, V. Babic, E. Mietkiewska, A. Jadhav, E.-F. Marillia, T. Francis, T. Hoffman, E. M. Giblin, V. Katavic and W. A. Keller, Brassica carinata – a new molecular farming platform for delivering bio-industrial oil feedstocks: case studies of genetic modifications to improve very long-chain fatty acid and oil content in seeds, Biofuels, Bioprod. Biorefin., 2010, 4, 538–561, DOI:10.1002/bbb.231.
- J. Brizga, K. Hubacek and K. Feng, The Unintended Side Effects of Bioplastics: Carbon, Land, and Water Footprints, One Earth, 2020, 3, 45–53, DOI:10.1016/j.oneear.2020.06.016.
- R. Mülhaupt, Green Polymer Chemistry and Bio-based Plastics: Dreams and Reality, Macromol. Chem. Phys., 2013, 214, 159–174, DOI:10.1002/macp.201200439.
- A. Muscat, E. M. de Olde, I. J. M. de Boer and R. Ripoll-Bosch, The battle for biomass: A systematic review of food-feed-fuel competition, Global Food Secur., 2020, 25, 100330, DOI:10.1016/j.gfs.2019.100330.
- S. Shen, Extraction and Polymerization of Bioaromatics from Megacrops, PhD Thesis, University of Florida, 2019. https://ufdc.ufl.edu/ufe0055730/00001 Search PubMed.
- S. I. Warwick, R. K. Gugel, T. McDonald and K. C. Falk, Genetic variation of Ethiopian mustard (Brassica carinata A. Braun) germplasm in western Canada, Genet. Resour. Crop Evol., 2006, 53, 297–312, DOI:10.1007/s10722-004-6108-y.
- K. D. Carlson and V. E. Sohns, Brassylic Acid: Chemical Intermediate from High-Erucic Oils, Ind. Eng. Chem. Prod. Res. Dev., 1977, 16, 95–101, DOI:10.1021/i360061a020.
- P. Dwivedi, Sustainable aviation fuel production from Brassica carinata in the Southern United States, GBC Bioenergy, 2021, 13, 1854–1858, DOI:10.1111/gcbb.12900.
- Acumen Research and Consulting, Brassylic Acid Market Size to Reach USD 100.5 Million by 2032 growing at 4.7% CAGR. July 28, 2024. https://www.acumenresearchandconsulting.com/press-releases/brassylic-acid-market (accessed October 2024).
- L. Zhou, L. Wu, P. Qin and B.-G. Li, Synthesis and properties of long chain polyesters from biobased 1,5-pentanediol and aliphatic α,ω-diacids with 10–16 carbon atoms, Polym. Degrad. Stab., 2021, 187, 109546, DOI:10.1016/j.polymdegradstab.2021.109546.
- Y. Tachibana, T. Tsutsuba, K. Kageyama and K. Kasuya, Biodegradability of poly(butylene n-alkylenedionate)s composed of long-methylene chains as alternative polymers to polyethylene, Polym. Degrad. Stab., 2021, 190, 109650, DOI:10.1016/j.polymdegradstab.2021.109650.
- J. Fernάndez, H. Amestoy, H. Sardon, M. Aguirre, A. L. Varga and J.-R. Sarasua, Effect of molecular weight on the physical properties of poly(ethylene brassylate) homopolymers, J. Mech. Behav. Biomed. Mater., 2016, 64, 209–219, DOI:10.1016/j.jmbbm.2016.07.031.
- W. H. Carothers, in Collected Papers of Wallace Hume Carothers on High Polymeric Substance, ed. H. F. Mark and G. S. Whitby, Interscience Publishers, New York, 1940. https://books.google.com/books/about/Collected_Papers_of_Wallace_Hume_Carothe.html?id=TdogAAAAMAAJ Search PubMed.
- X. Cui, D. Yan and D. Xiao, Synthesis and characterization of novel polyamides based on tridecanedioic acid: Nylons 3 13, 5 13, 6 13, 7 13, 9, 13, 10 13, 11 13, e-Polymers, 2004, 13, 7–15, DOI:10.1515/epoly.2004.4.1.784.
- L.-H. Wang, F. J. B. Calleja, T. Kanamoto and R. S. Porter, The characterization and properties of nylon 13,13, Polymer, 1993, 34, 4688–4691, DOI:10.1016/0032-3861(93)90702-C.
- P. Qi, H.-L. Chen, H. T. H. Nguyen, C.-C. Lin and S. A. Miller, Synthesis of biodegradable polylactam esters from itaconic acid, Green Chem., 2016, 18, 4170–4175, 10.1039/C6GC01081D.
- O. Nsengiyumva and S. A. Miller, Synthesis, characterization, and water-degradation of biorenewable polyesters derived from natural camphoric acid, Green Chem., 2019, 21, 973–978, 10.1039/C8GC03990A.
- Y.-K. Su, G. N. Short and S. A. Miller, Renewable and water degradable polyimide-esters from citric acid, Green Chem., 2023, 25, 6200–6206, 10.1039/D3GC01779F.
- T. H. N. Nguyen, F. Balligand, A. Bormann, V. Bennevault and P. Guégan, Synthesis of new biobased linear poly(ester amide)s, Eur. Polym. J., 2019, 121, 109314, DOI:10.1016/j.eurpolymj.2019.109314.
- Y.-J. Jang, L. Sangroniz and M. A. Hillmyer, Ductile gas barrier poly(ester-amide)s derived from glycolide, Polym. Chem., 2022, 13, 3882–3891, 10.1039/D2PY00479H.
- N. Paredes, A. Rodríguez-Galán and J. Puiggalí, Synthesis and Characterization of a Family of Biodegradable Poly(ester amide)s Derived from Glycine, J. Polym. Sci., Part A: Polym. Chem., 1997, 36, 1271–1282, DOI:10.1002/(SICI)1099-0518(199806)36:8<1271::AID-POLA10>3.0.CO;2-3.
- J. Montané, E. Armelin, L. Asín, A. Rodríguez-Galán and J. Puiggalí, Comparative Degradation Data of Polyesters and Related Poly(ester amide)s Derived from 1,4-Butanediol, Sebacic Acid, and α-Amino Acids, J. Appl. Polym. Sci., 2001, 85, 1815–1824, DOI:10.1002/app.10379.
- P. A. M. Lips, R. Broos, M. J. M. van Heeringen, P. J. Dijkstra and J. Feijen, Incorporation of different crystallizable amide blocks in segmented poly(ester amide)s, Polymer, 2005, 46, 7834–7842, DOI:10.1016/j.polymer.2005.07.009.
- D. M. Guptill, B. S. Chinta, T. Kaicharla, S. Xu and T. R. Hoye, β-Methyl-δ-valerolactone-containing thermoplastic poly(ester-amide)s: synthesis, mechanical properties, and degradation behavior, Polym. Chem., 2021, 12, 1310–1316, 10.1039/D1PY00040C.
- S. K. Murase, L. Franco, L. J. del Valle and J. Puiggalí, Synthesis and Characterization of Poly(ester amides)s with a Variable Ratio of Branched Odd Diamine Units, J. Appl. Polym. Sci., 2014, 131, 40102, DOI:10.1002/app.40102.
- C. Rader, C. Weder and R. Marti, Biobased Polyester-Amide/Cellulose Nanocrystal Nanocomposites for Food Packaging, Macromol. Mater. Eng., 2021, 306, 2000668, DOI:10.1002/mame.202000668.
- A. Borriello, L. Nicolais and S. J. Huang, Poly(amide-ester)s Derived from Dicarboxylic Acid and Aminoalcohol, J. Appl. Polym. Sci., 2005, 95, 362–368, DOI:10.1002/app.21270.
- P. Rizzarelli, M. Cirica, G. Pastorelli, C. Puglisi and G. Valenti, Aliphatic poly(ester amide)s from sebacic acid and aminoalcohols of different chain length: Synthesis, characterization and soil burial degradation, Polym. Degrad. Stab., 2015, 121, 90–99, DOI:10.1016/j.polymdegradstab.2015.08.010.
- J. Brazdil and C. C. Ma, Process for making biobased products from sugars, WO Pat, WO2020028521A1, 2020 Search PubMed.
- M. Foti, R. Médici and H. Ruijssenaars, Biological production of monoethanolamine by engineered Pseudomonas putida, S12, J. Biotechnol., 2013, 167, 344–349, DOI:10.1016/j.jbiotec.2013.07.013.
- N. Ehrenstein, S. Dellsperger, C. Kocher, N. Stutzmann, C. Weder and P. Smith, New polyamides with long alkane segments: nylon 6.24 and 6.34, Polymer, 2000, 41, 3531–3539, DOI:10.1016/S0032-3861(99)00590-X.
- F. Stempfle, P. Ortmann and S. Mecking, Long-Chain Aliphatic Polymers To Bridge the Gap between Semicrystalline Polyolefins and Traditional Polycondensates, Chem. Rev., 2016, 116, 4597–4641, DOI:10.1021/acs.chemrev.5b00705.
- A. Touris, A. Turcios, E. Mintz, S. R. Pulugurtha, P. Thor, M. Jolly and U. Jalgaonkar, Effect of molecular weight and hydration on the tensile properties of polyamide 12, Results Mater., 2020, 8, 100149, DOI:10.1016/j.rinma.2020.100149.
- L. P. Razumovskii, V. S. Markin and G. Y. Zaikov, Sorption of water by aliphatic polyamides. Review, Polym. Sci. U.S.S.R., 1985, 27, 751–768, DOI:10.1016/0032-3950(85)90411-3.
- B. Cornils and P. Lappe, Dicarboxylic Acids, Aliphatic, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley, New Jersey, 2014. DOI:10.1002/14356007.a08_523.pub3.
- R. W. Johnson, C. M. Pollock and R. R. Cantrell, Dicarboxylic Acids, in Kirk–Othmer Encyclopedia of Chemical Technology, Wiley, New Jersey, 2010. DOI:10.1002/0471238961.0409030110150814.a01.pub2.
- T. Fey, M. Hölscher, H. Keul and H. Höcker, Alternating poly(ester amide)s from succinic anhydride and α,ω-amino alcohols: synthesis and thermal characterization, Polym. Int., 2003, 52, 1625–1632, DOI:10.1002/pi.1354.
- T. Fey, H. Keul and H. Höcker, Interconversion of Alternating Poly(ester amide)s and Cyclic Ester Amides from Adipic Anhydride and α,ω-Amino Alcohols, Macromol. Chem. Phys., 2003, 204, 591–599, DOI:10.1002/macp.200390021.
- S.-P. Rwei, Y.-C. Tseng, K.-C. Chiu, S.-M. Chang and Y.-M. Chen, The crystallization kinetics of Nylon 6/6T and Nylon 66/6T copolymers, Thermochim. Acta, 2013, 555, 37–45, DOI:10.1016/j.tca.2012.12.026.
- N. Ma, W. Liu, L. Ma, S. He, H. Liu, Z. Zhang, A. Sun, M. Huang and C. Zhu, Crystal transition and thermal behavior of Nylon 12, e-Polymers, 2020, 20, 346–352, DOI:10.1515/epoly-2020-0039.
- Y. Tokiwa, B. P. Calabia, C. U. Ugwu and S. Aiba, Biodegradability of Plastics, Int. J. Mol. Sci., 2009, 10, 3722–3742, DOI:10.3390/ijms10093722.
- C. Berti, A. Celli, P. Marchese, E. Marianucci, G. Barbiroli and F. D. Credico, The effect of aliphatic chain length on thermal properties of poly(alkylene dicarboxylate)s, e-Polymers, 2007, 7, 1–18, DOI:10.1515/epoly.2007.7.1.658.
- S. F. Marxsen, M. Häußler, M. Eck, S. Mecking and R. G. Alamo, Effect of CH2 run length on the crystallization kinetics of sustainable long-spaced aliphatic polyesters, Polymer, 2023, 282, 126181, DOI:10.1016/j.polymer.2023.126181.
- P. Ortmann and S. Mecking, Long-Spaced Aliphatic Polyesters, Macromolecules, 2013, 46, 7213–7218, DOI:10.1021/ma401305u.
- K. Nomura and N. W. B. Awang, Synthesis of Bio-Based Aliphatic Polyesters from Plant Oils by Efficient Molecular Catalysis: A Selected Survey from Recent Reports, ACS Sustainable Chem. Eng., 2021, 9, 5486–5505, DOI:10.1021/acssuschemeng.1c00493.
- M. M. Zagho, M. A. A. AlMaadeed and K. Majeed, Thermal Properties of TiO2NP/CNT/LDPE Hybrid Nanocomposite Films, Polymers, 2018, 10, 1270, DOI:10.3390/polym10111270.
- P. J. M. Serrano, B. A. van de Werff and R. J. Gaymans, Alternating polyesteramides based on 1,4-butylene terephthalamide: 3. Alternating polyesteramides based on mixtures of linear diols (4NTm,p), Polymer, 1998, 39, 83–92, DOI:10.1016/S0032-3861(97)00232-2.
- F. D. Stefano, M. Baur, C. D. Rosa and S. Mecking, Keto-Polyethylenes with Controlled Crystallinity and Materials Properties from Catalytic Ethylene-CO-Norbornene Terpolymerization, Macromolecules, 2024, 57, 1072–1079, DOI:10.1021/acs.macromol.3c02309.
- Y.-J. Jang, S. Nguyen and M. A. Hillmyer, Chemically Recyclable Linear and Branched Polyethylenes Synthesized from Stoichiometrically Self-Balanced Telechelic Polyethylenes, J. Am. Chem. Soc., 2024, 146, 4771–4782, DOI:10.1021/jacs.3c12660.
- X.-L. Li, R. W. Clarke, J.-Y. Jiang, T.-Q. Xu and E. Y.-X. Chen, A circular polyester platform based on simple gem-disubstituted valerolactones, Nat. Chem., 2023, 15, 278–285, DOI:10.1038/s41557-022-01077-x.
- S. Paszkiewicz, A. Szymczyk, D. Pawlikowska, J. Subocz, M. Zenker and R. Masztak, Electrically and Thermally Conductive Low Density Polyethylene-Based Nanocomposites Reinforced by MWCNT or Hybrid MWCNT/Graphene Nanoplatelets with Improved Thermo-Oxidative Stability, Nanomaterials, 2018, 8, 264, DOI:10.3390/nano8040264.
- M. Kojima, X. Wang, L. O. P. Go, R. Makino, Y. Matsumoto, D. Shimoyama, M. M. Abdellatif, J. Kadota, S. Higashi, H. Hirano and K. Nomura, Synthesis of High Molecular Weight Biobased Aliphatic Polyesters Exhibiting Tensile Properties Beyond Polyethylene, ACS Macro Lett., 2023, 12, 1403–1408, DOI:10.1021/acsmacrolett.3c00481.
- N. Maraschin, Polyethylene, Low Density, in Kirk–Othmer Encyclopedia of Chemical Technology, Wiley, New Jersey, 2005. DOI:10.1002/0471238961.12152316050219.a01.pub2.
- E. Benham and M. McDaniel, Polyethylene, High Density, in Kirk–Othmer Encyclopedia of Chemical Technology, Wiley, New Jersey, 2005. DOI:10.1002/0471238961.0809070811091919.a01.pub2.
- J. H. Jung, M. Ree and H. Kim, Acid- and base-catalyzed hydrolyses of aliphatic polycarbonates and polyesters, Catal. Today, 2006, 115, 283–287, DOI:10.1016/j.cattod.2006.02.060.
- R. Berstein and K. T. Gillen, Nylon 6.6 accelerating aging studies: II. Long-term thermal-oxidative and hydrolysis results, Polym. Degrad. Stab., 2010, 95, 1471–1479, DOI:10.1016/j.polymdegradstab.2010.06.018.
- K. Min, J. D. Cuiffi and R. T. Mathers, Ranking environmental degradation trends of plastic marine debris based on physical properties and molecular structure, Nat. Commun., 2020, 11, 727, DOI:10.1038/s41467-020-14538-z.
- G. L. Baker, E. B. Vogel and M. R. Smith, Glass Transitions in Polylactides, Polym. Rev., 2008, 48, 64–84, DOI:10.1080/15583720701834208.
- U.S. Plastics Pact. https://usplasticspact.org/ (accessed October 2024).
- T. D. Alcock, D. E. Salt, P. Wilson and S. J. Ramsden, More sustainable vegetable oil: Balancing productivity with carbon storage opportunities, Sci. Total Environ., 2022, 829, 154539, DOI:10.1016/j.scitotenv.2022.154539.
- “Vegetable oil yields per hectare worldwide as of 2020, by crop plant” from Statista. https://www.statista.com/statistics/1263201/vegetable-oil-yields-per-hectare-worldwide-by-type-globally/#:~:text=Global%20vegetable%20oil%20yields%20per%20hectare%20worldwide%202020%2C%20by%20type&text=Palm%20oil%20produces%20more%20oil,0.4%20metric%20tons%20per%20hectare (accessed October 2024).
- R. Seepaul, I. M. Small, P. Devkota, H. Y. Sintim, M. J. Mulvaney, S. George, R. G. Leon, S. V. Paula-Moraes, I. L. Esquivel, R. Bennett, A. Pokrzywinski, D. Geller, J. J. Marois and D. L. Wright, Carinata, the Sustainable Crop for a Bio-based Economy: Production Recommendations for the Southeastern United States: SS-AGR-384/AG389, 8/2023, EDIS, 2023, 4, 1–15, DOI:10.32473/edis-ag389-2023.
- “Market volume of polyethylene worldwide from 2015 to 2022, with a forecast for 2023 to 2030” from Statista. https://www.statista.com/statistics/1245162/polyethylene-market-volume-worldwide/ (accessed October 2024).
- “WORLD FIRST US-AUSTRALIA BIOFUEL FLIGHT TAKES OFF” from Qantus. https://www.qantasnewsroom.com.au/media-releases/world-first-us-australia-biofuel-flight-takes-off/ (accessed October 2024).
- C. Zhao, G. Colson, B. Karali and G. P. Philippidis, Drop-in ready jet biofuel from carinata: A real options analysis of processing plant investments, GCB Bioenergy, 2021, 13, 1624–1635, DOI:10.1111/gcbb.12873.
- M. J. Watson, P. G. Machado, A. V. da Silva, Y. Saltar, C. O. Ribeiro, C. A. O. Nascimento and A. W. Dowling, Sustainable aviation fuel technologies, costs, emissions, policies, and markets: A critical review, J. Cleaner Prod., 2024, 449, 141472, DOI:10.1016/j.jclepro.2024.141472.
- “Erucic Acid prices increase in Europe amidst strong demand outpacing supply” from ChemAnalyst. https://www.chemanalyst.com/NewsAndDeals/NewsDetails/erucic-acid-prices-increase-in-europe-amidst-strong-demand-outpacing-supply-27793 (accessed October 2024).
- Flag Chemical Industry Co., Ltd. Alibaba.com. https://www.alibaba.com/product-detail/High-quality-Brassylic-acid-1-11_1601271629129.html (accessed October 2024).
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, complete polymer characterization data, polymer degradation data, and computational methods. See DOI: https://doi.org/10.1039/d4gc05490c |
| This journal is © The Royal Society of Chemistry 2025 |