
Transition metal/photocatalyst-free synthesis of geminal diamines via a sandwich-like photoactive donor–acceptor–donor complex†
Ziyi
Xu‡
a,
Ziyang
Chen‡
a,
Shuyang
Liu‡
 a,
Jian
Gao
a,
Jinglan
Lei
a,
Min
Li
*a,
Yongqiang
Zhang
a,
Ziyu
Gan
a,
Limei
Yu
*a,
Shu-Xin
Liu
*b and
Yunhe
Jin
*a
a,
Jian
Gao
a,
Jinglan
Lei
a,
Min
Li
*a,
Yongqiang
Zhang
a,
Ziyu
Gan
a,
Limei
Yu
*a,
Shu-Xin
Liu
*b and
Yunhe
Jin
*a
aState Key Laboratory of Fine Chemicals, School of Chemistry, Dalian University of Technology, Dalian, 116024, China. E-mail: nulilimin@dlut.edu.cn; ochem@dlut.edu.cn; jinyh18@dlut.edu.cn
bDepartment of Nephrology, Dalian Municipal Central Hospital affiliated with Dalian University of Technology, Dalian Key Laboratory of Intelligent Blood Purification, No. 826 Xinan Road, Dalian, 116033, China. E-mail: shuxinliu@dlut.edu.cn
First published on 3rd April 2025
Abstract
The geminal diamine is an important molecular scaffold widely found in natural products, bioactive compounds, and pharmaceuticals. Nonetheless, the construction of this motif suffers from high temperature, excessive oxidants, and the utilization of transition metals. Herein, we present a transition metal/photocatalyst-free strategy for the synthesis of geminal diamines via photoinduced amidation of C(sp3)–H bonds. This reaction features high atom economy, broad substrate applicability, and good functional group tolerance. An unusual sandwich-like ternary donor–acceptor–donor complex resulting from π–π stacking and base action is proposed to be the key photosensitive species.
Green foundation1. Different from the traditional photoredox or EDA system, an unusual sandwich-like photosensitive ternary DAD complex is proposed to be the key species resulting from π–π stacking, electrostatic interaction, and base action. This method will provide a convenient and competitive approach for geminal diamine synthesis and provide new inspiration for green reaction construction.2. Previously, the construction of geminal diamines suffered from high temperatures, excessive oxidants, the utilization of transition metals, and unstable starting materials. Fully avoiding these issues, we herein present a novel transition metal/photocatalyst-free strategy for this valuable synthesis. 3. We think our work is green and atom-economic enough with only a low-cost inorganic base needed and a molecule of methanol being emitted. |
Introduction
Geminal diamines, as important molecular scaffolds, are widely found in natural products, bioactive compounds, and pharmaceuticals that are usually of enormous application value.1 In the past few decades, the synthesis of such compounds has usually been achieved by thermal-driven synthesis and transition metal catalysis (Scheme 1a).2 For example, Fu and coworkers reported a copper-catalyzed geminal diamine synthesis utilizing amide as a substrate and TBHP as an oxidant;2b with a similar strategy, Zhao and coworkers took advantage of the nucleophilicity of the lone-pair electrons on the N atom in benzonitrile to replace the amide as the N source.2d Although these methods provide feasible access to geminal diamine compounds, several problems such as high temperature and excessive oxidant addition still exist, limiting further application of this strategy to some extent.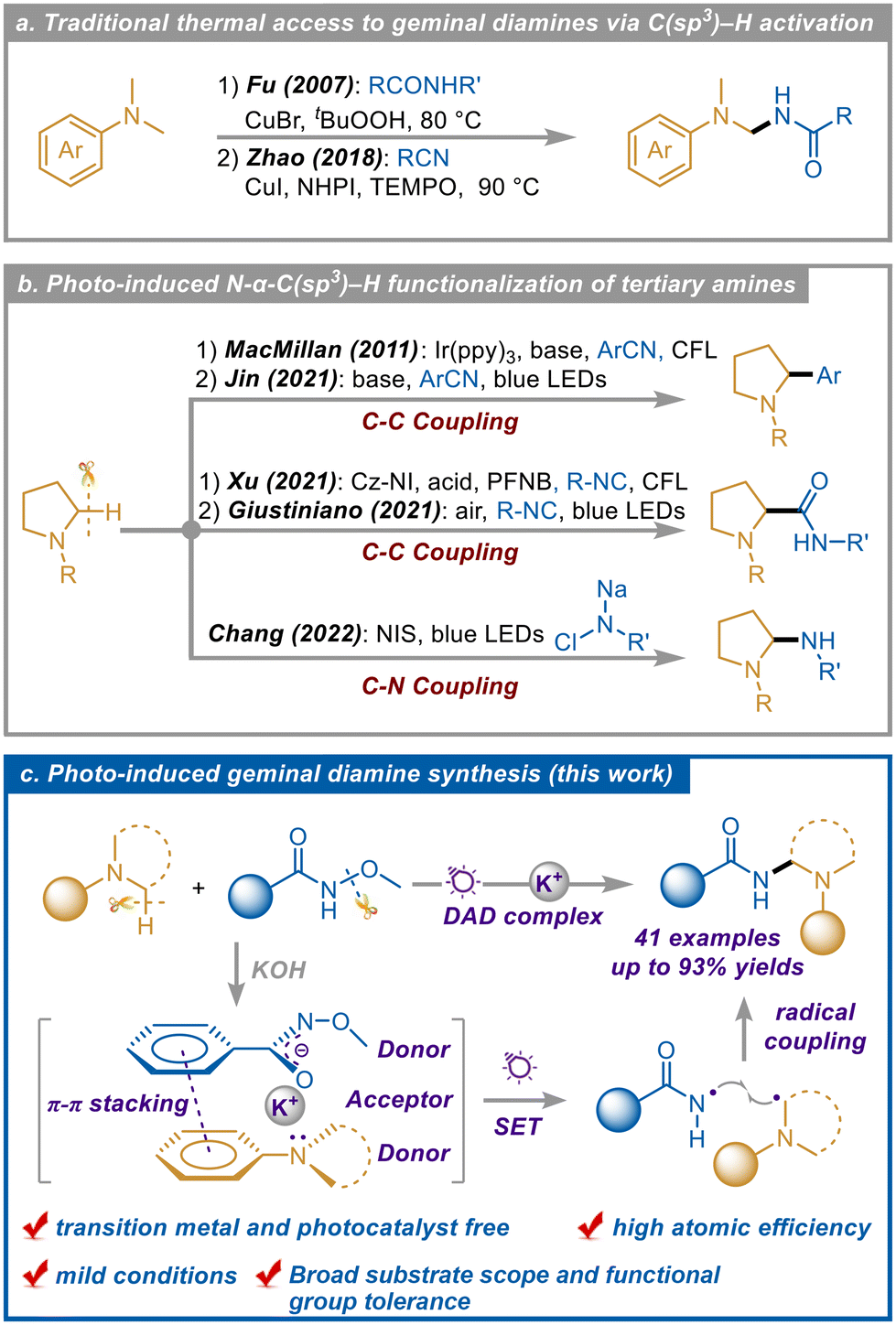 | ||
| Scheme 1 Research progress on the synthesis of geminal diamines involving tertiary amines. | ||
In the recent decade, photocatalysis has emerged as an environmentally friendly, sustainable, and unique methodology for the construction of various compounds.3 In photocatalytic systems, the reactions are always initiated by single electron transfer (SET) processes induced by photoexcitation of photosensitizers or photosensitive species, generating a range of active radical intermediates and promoting the progression of diverse reactions.4 As a significant part of this field, electron donor–acceptor (EDA) photochemistry, which generally involves an in situ photosensitive complex consisting of an electron donor and an acceptor, has experienced growth as a selective and durable radical generation approach via intra-complex SET, eliminating the requirement for transition metals or dyes.5 Tertiary amines are a kind of important active unit and electron donor, and their α-C–H functionalization has received extensive attention (Scheme 1b).6 In 2011, Macmillan et al. successfully arylated the tertiary amine α-C(sp3)–H bonds using Ir as a catalyst and benzonitrile compounds as substrates.7 Later, the synthesis of compounds with similar structures was achieved by Jin et al. through an EDA process.8 In 2021, amidation of the tertiary amine α-C(sp3)–H bond was conducted by the Ugi-type reaction of tertiary amine and isonitrile which was achieved by Xu9 through the traditional photocatalytic process and by Giustiniano6f through the EDA process. According to the above reaction systems, the EDA reaction pathway exhibits some advantages including simple conditions, operation convenience, and direct substrate activation. However, the essential matching between the electron donor and the acceptor for forming a stable photosensitive EDA complex always limits the rapid development of this chemistry. Moreover, most of these works focus on the construction of C–C bonds, which is not practical for the synthesis of geminal diamines. Impressively in 2022, the controllable amination of the tertiary amine α-C(sp3)–H bonds was achieved by the Chang group via a cyclic imine salt intermediate in situ.10 The system underwent a series of polar addition, elimination, photoinduced homolysis, hydrogen atom transfer, and halogen atom transfer processes, using somewhat unstable N-chloro-N-sodio-carbamates as amination reagents. Accordingly, considerable space and demand still exist for the facile and mild access to valuable geminal diamines with abundant-sourced and stable materials.
Based on previous works11 and our previous findings,12N-alkoxy benzamides can be applied as redox-neutral and atom-efficient amidation reagents for C(sp3)–H bonds via metal nitrene intermediates. However, additional photocatalysts or transition metal catalysts were essential in these systems. Employing an EDA pathway instead of these catalytic systems has not been reported due to the inadequate electron-deficient characteristics for N-alkoxy benzamides as an electron acceptor. In our design, a strong base may transform the N-alkoxy benzamide into an anion that can act as an electron donor instead, and this anion, the corresponding alkali metal cation, and another donor are proposed to form a tertiary photoactive complex for novel photo-transformation. Herein, we developed a photocatalyst/transition metal-free and efficient strategy to achieve photoinduced tertiary amine α-C(sp3)–H activation and C–N coupling based on the radicals generated from the photoexcited intra-SET process of a sandwich-like electron donor–acceptor–donor (DAD) ternary photoactive complex, and geminal diamines were successfully constructed via radical coupling (Scheme 1c). The protocol exhibited mild and green conditions, high atom utilization, wide substrate range, and good functional group tolerance.
Results and discussion
According to our previously established system,12a we started the investigation for the model reaction of N-phenylpyrrolidine (1a) and N-methoxybenzamide (1b) with fac-Ir(ppy)3 and CuCl as catalysts, Cs2CO3 as the base and MeCN as the solvent under irradiation with 405 nm LEDs (Table 1, entry 1). Satisfyingly, the desired geminal diamine product 1c was successfully obtained in a 27% yield. To our surprise, removing either or both catalysts did not markedly affect the yield of 1c, indicating the possible formation of a photosensitive species that was similar to the EDA complex in this reaction system (entries 2–4). Various bases including KOtBu, KOH, LiOH, NaOH, K3PO4, and DIPEA were tested then (entries 4–11), suggesting that strong bases with potassium ions significantly boosted the production of 1c with KOH appearing to be the best choice (entry 6). Further increasing the equivalents of KOH provided an enhanced yield of 76% (entry 7). Different types of solvents were also examined, showing acetonitrile to still be the best one (entries 12–15). We were pleased to find that as the illumination wavelength of the LEDs changed (entries 16–18), the yield of 1c finally reached its highest point of 92% at 395 nm (entry 17). Additionally, variations in leaving groups exhibited no positive effects (Table S1,† entries 19–21), and only N-ethoxybenzamide delivered the desired product in a comparable yield when the reaction time was long enough. Control experiments showed that the reaction gave a decreased yield when it was exposed to water or air (entries 22 and 23), and the reaction would not occur without light as an energy source (entry 24).| a Reaction conditions: 1a (0.1 mmol), 1b (3 equiv.), base (2 equiv.), and solvent (2 mL) under N2 and irradiation from 30 W LEDs for 48 h. b Isolated yield. c 1 equiv. of the base was added. d 0.2 mol% of fac-Ir(ppy)3 and 20 mol% of CuCl was added. e 0.2 mol% of fac-Ir(ppy)3 was added. f 20 mol% of CuCl was added. |
|---|

|
With the optimized reaction conditions in hand, the scope of tertiary amines was investigated first (Scheme 2). To our delight, kinds of N-aryl pyrrolidines with electron-donating groups including methyl (2c), tert-butyl (3c), methoxyl (4c and 9c), and methylthio (5c) were all suitable substrates, delivering the corresponding gem-diamines in good yields. Compounds containing trifluoromethyl are beneficial for drug discovery and the development of new materials. Significantly, trifluoromethyl-substituted N-aryl pyrrolidines at para- and meta-positions also provided the corresponding products smoothly (7c and 8c). When naphthyl pyrrolidine was employed as a substrate, 10c was obtained in an excellent yield, possibly due to the enlarged conjugation system and the promoted light absorption capacity. Furthermore, we were pleased to find that dimethyl arylamines with various functional groups were also effective candidates (11c–17c), and the reaction efficiency was generally higher than that of N-aryl pyrrolidines. As vital synthons, halogen-substituted N-aryl pyrrolidines and dimethyl arylamines were all demonstrated to be compatible substrates, offering a good chance of further derivatization (6c and 15c–17c).
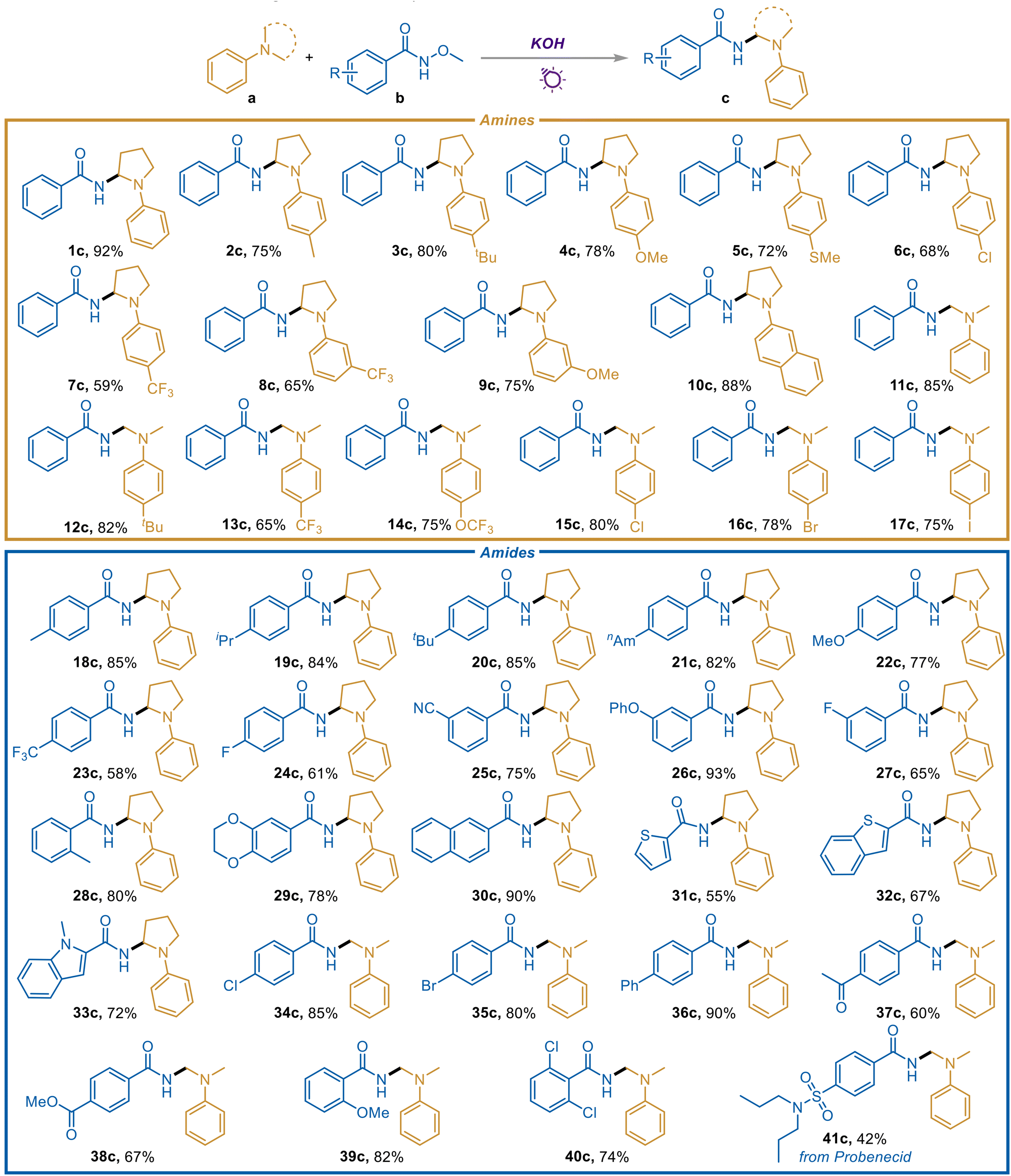 | ||
| Scheme 2 Substrate scope. Reaction conditions: amines (a, 0.2 mmol, 1 equiv.), amides (b, 3 equiv.), KOH (2 equiv.), MeCN (2 mL) under N2 and irradiation from 30 W 395 nm LEDs for 48 h. | ||
Subsequently, diverse amides were examined to extend the application scope (Scheme 2). It was gratifying to observe that a variety of electron-donating group substituted aromatic amides including alkyl (18c–21c and 28c) and ether (22c, 26c, 29c, and 39c) modified ones can furnish the corresponding gem-diamines in significant yields. The substituted position showed no obvious influence (18c and 28c, 24c and 27c). Besides, heterocyclic compounds are often considered to be toxic to metal catalysts, but in our transition metal-free system, the heteroaryl amides reacted smoothly and offered the heteroaromatic-containing geminal diamines effectively (31c–33c). Notably, halogen-substituted amides (24c, 27c, 34c, 35c, and 40c) were readily converted into the corresponding gem-diamines efficiently, rendering a wide platform for further derivatization. Even as an electron donor, the amide substrates are still gratifyingly tolerant to various electron-withdrawing groups including trifluoromethyl (23c), cyano (25c), acetyl (37c), and ester (38c), and the desired gem-diamines were obtained in good yields. An amide derived from Probenecid, which is primarily used in treating gout and hyperuricemia,13 was also tested, and the drug motif was successfully introduced to the α-amido tertiary amine structure (41c), facilitating the development of this method in further drug discovery.
To explore the reaction mechanism of this catalyst-free system, several mechanistic experiments were conducted. When stoichiometric radical quenchers including 2,2,6,6-tetramethyl-1-piperinedinyloxy (TEMPO), 2,6-di-tert-butyl-4-methylphenol (BHT), and 1,1-diphenyl-ethylene were respectively added to the reaction system, the yields of 3a were slashed dramatically, and the corresponding radical adducts were observed in High-Resolution Mass Spectrometry (HRMS) analyses, indicating that the reaction underwent a radical pathway (Scheme 3a, for details see the ESI†). To further investigate the photosensitive species inside the simple system, UV-visible absorption spectroscopy was performed on different combinations of the components. As displayed in Scheme 3b (for details see the ESI†), no obvious absorption was observed in the visible range for either 1a or 1b, or a simple mixture of 1a and 1b. The addition of KOH would slightly enhance the absorption of 1a or 1b, but the outcomes were still weak in the range over 390 nm. Exhilaratingly, a ternary mixture of 1a, 1b, and KOH emerged with strong absorption at values over 380 nm, indicating that a ternary complex of 1a, 1b, and K+ with the ability to absorb light successfully formed in situ. A distinct color change could also be observed in the real photograph. In addition, electron spin-resonance (ESR) experiments were carried out to further distinguish the radical category (Scheme 1c, for details see the ESI†). The ESR spectrum of a mixture of N,N-dimethyl-aniline (11a) and N-methoxybenzamide (1b) in MeCN with 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as a radical spin trap under dark conditions showed no signal of a trapped radical. After irradiation for 30 s, an obvious sextet signal (g = 2.0051, AN = 14.53 mT, AH = 21.35 mT) was observed, indicating the appearance of a carbon-centered radical14 (Scheme 3d). Furthermore, the key factors for the formation process of the ternary photosensitive complex were studied by substrate control experiments (Scheme 3d & e). When aliphatic tertiary amine 1-methyl pyrrolidine (42a), secondary amide tert-butyl pyrrolidine-1-carboxylate (43a), or aliphatic amide N-methoxy-3-phenylpropanamide (44b) was respectively employed as the substrate instead, no desired product was observed, implying that the π–π stacking interaction between the aromatic rings of the two substrates was vital for the formation of the photosensitive species. A tertiary amide N-methoxy-N-methylbenzamide (45b) without a reactive hydrogen was also tried and no product was provided, demonstrating the importance of the in situ deprotonation of 1b with KOH for the formation of the photosensitive complex. Moreover, the competition reaction of 3a with 46a and 22b with 37b showed that the assembling of the DAD complex favored aromatics with a denser electron density. These results further demonstrated that both substrate a and b acted as electron donors in the in situ formed complex.
 | ||
| Scheme 3 Mechanistic studies. (a) Control experiments with TEMPO, BHT, and 1,1-diphenylethylene. (b) UV-vis absorption spectrum. (c) Electron spin-resonance (ESR) spectrum. (d) Control experiments with different types of substrates. (e) Competition experiments. | ||
Based on the above results and our research experience,12a a plausible reaction mechanism for the present transition metal/photocatalyst-free synthesis of geminal diamines is proposed in Scheme 4. Initially, KOH deprotonates 1b (pKa 8.88)15 to form the amide anion I, transforming 1b from a relatively electron-deficient amide structure to an electron-rich conjugated anionic structure. Consequently, the electron-donor anion I, the electron-acceptor cation K+, and another electron-donor tertiary amine 1a can form a sandwich-like ternary photosensitive DAD complex in situ via π–π stacking and electrostatic interaction. The photoactive complex II flips to its excited state III under light irradiation, followed by an intra-SET process to generate the tertiary amine radical cation IV and the amide radical anion VI. With the assistance of base, the N-α-carbon radical V is delivered by eliminating the N-α-proton of IV (confirmed by EPR). Meanwhile, removing the methoxide anion as a leaving group from VI provides the nitrogen radical VII. Finally, a dual-radical coupling reaction between V and VII results in the desired gem-diamine 1c.
 | ||
| Scheme 4 Proposed reaction pathway. | ||
Conclusions
Overall, we herein develop a transition metal/photocatalyst-free method for the synthesis of valuable geminal diamines under extremely mild and simple conditions with high atom economy and good functional group compatibility. Different from the traditional EDA system, an unusual sandwich-like photosensitive ternary DAD complex is proposed to be the key species resulting from π–π stacking, electrostatic interaction, and base action. We believe that this method will provide an alternative and competitive approach for geminal diamine synthesis and also give some new inspiration for the activation and construction of special structures without photosensitizers.Author contributions
Y.J. supervised the project. Y.J., M.L., S.-X.L., and L.Y. guided the project. Y.J., Z.X., Z.C., and S.L. conceived and designed the study. All authors performed and analyzed the experiments. Z.X., Z.C., S.L., and Y.J. wrote and revised the paper.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the support from the Natural Science Foundation of Liaoning Province (2024-MSLH-080), the Fundamental Research Funds for the Central Universities (DUT24BK058), and the Applied Basic Research Project of Liaoning Province, China (2023JH2/101300091).References
- (a) Y. Nishimura, W. Wang, S. Kondo, T. Aoyagi and H. Umezawa, J. Am. Chem. Soc., 1988, 110, 7249–7250 CrossRef CAS; (b) H. Mitsunuma, M. Shibasaki, M. Kanai and S. Matsunaga, Angew. Chem., Int. Ed., 2012, 51, 5217–5221 CrossRef CAS PubMed.
- (a) V. V. Zhdankin, M. McSherry, B. Mismash, J. T. Bolz, J. K. Woodward, R. M. Arbit and S. Erickson, Tetrahedron Lett., 1997, 38, 21–24 CrossRef CAS; (b) Y. Zhang, H. Fu, Y. Jiang and Y. Zhao, Org. Lett., 2007, 9, 3813–3816 CrossRef CAS PubMed; (c) X. Liu, Y. Zhang, L. Wang, H. Fu, Y. Jiang and Y. Zhao, J. Org. Chem., 2008, 73, 6207–6212 CrossRef CAS PubMed; (d) B. Lin, S. Shi, Y. Cui, Y. Liu, G. Tang and Y. Zhao, Org. Chem. Front., 2018, 5, 2860–2863 RSC; (e) S. K. Singh, N. Chandna and N. Jain, Org. Lett., 2017, 19, 1322–1325 CrossRef CAS PubMed; (f) Q. Xia and W. Chen, J. Org. Chem., 2012, 77, 9366–9373 CrossRef CAS PubMed; (g) T. Song, Y. Luo, K. Wang, B. Wang, Q. Yuan and W. Zhang, ACS Catal., 2023, 13, 4409–4420 CrossRef CAS.
- (a) C. R. Stephenson, T. P. Yoon and D. W. MacMillan, Visible light photocatalysis in organic chemistry, John Wiley & Sons, 2018 CrossRef; (b) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed; (c) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed; (d) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS PubMed; (e) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (f) H.-T. Ji, K.-L. Wang, W.-T. Ouyang, Q.-X. Luo, H.-X. Li and W.-M. He, Green Chem., 2023, 25, 7983–7987 RSC; (g) J. Liu, L. Guo, Z. Chen, Y. Guo, W. Zhang, X. Peng, Z. Wang and Y.-F. Zeng, Chem. Commun., 2024, 60, 3413–3416 RSC.
- (a) I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS PubMed; (b) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed; (c) L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed; (d) P. Xiang, K. Sun, S. Wang, X. Chen, L. Qu and B. Yu, Chin. Chem. Lett., 2022, 33, 5074–5079 CrossRef CAS; (e) S. Dutta, J. E. Erchinger, F. Strieth-Kalthoff, R. Kleinmans and F. Glorius, Chem. Soc. Rev., 2024, 53, 1068–1089 RSC.
- (a) C. G. S. Lima, T. de M. Lima, M. Duarte, I. D. Jurberg and M. W. Paixão, ACS Catal., 2016, 6, 1389–1407 CrossRef CAS; (b) J. Wu, L. He, A. Noble and V. K. Aggarwal, J. Am. Chem. Soc., 2018, 140, 10700–10704 CrossRef CAS PubMed; (c) M.-C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Science, 2019, 363, 1429–1434 CrossRef CAS PubMed; (d) D. Leifert and A. Studer, Angew. Chem., Int. Ed., 2020, 59, 74–108 CrossRef CAS PubMed; (e) G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS PubMed; (f) A. K. Wortman and C. R. J. Stephenson, Chem, 2023, 9, 2390–2415 CrossRef CAS PubMed; (g) M. Sharique, B. Matsuo, A. Granados, S. Kim, M. Arshad, H. Oh, V. E. Wu, M. Huang, A. Csakai, L. A. Marcaurelle and G. A. Molander, Chem. Sci., 2023, 14, 14193–14199 RSC; (h) L. van Dalsen, R. E. Brown, J. A. Rossi-Ashton and D. J. Procter, Angew. Chem., Int. Ed., 2023, 62, e202303104 CrossRef CAS PubMed; (i) S. M. Treacy, D. R. Vaz, S. Noman, C. Tard and T. Rovis, Chem. Sci., 2023, 14, 1569–1574 RSC; (j) G. Guo, X. Li, J. Ma, Y. Shi, J. Lv and D. Yang, Chin. Chem. Lett., 2024, 35, 110024 CrossRef CAS; (k) M.-H. Zhou, J. Jiang and W.-M. He, Chin. Chem. Lett., 2025, 36, 110446 CrossRef CAS; (l) J. D. Lasso, D. J. Castillo-Pazos, J. M. Salgado, C. Ruchlin, L. Lefebvre, D. Farajat, D. F. Perepichka and C.-J. Li, J. Am. Chem. Soc., 2024, 146, 2583–2592 CrossRef CAS PubMed; (m) T. Yan, J. Yang, K. Yan, Z. Wang, B. Li and J. Wen, Angew. Chem., Int. Ed., 2024, 63, e202405186 CrossRef CAS PubMed; (n) X.-L. Huang, D.-L. Zhang, Q. Li, Z.-B. Xie, Z.-G. Le and Z.-Q. Zhu, Org. Lett., 2024, 26, 3727–3732 CrossRef CAS PubMed.
- (a) Y. Zhang, H. Peng, M. Zhang, Y. Cheng and C. Zhu, Chem. Commun., 2011, 47, 2354–2356 RSC; (b) F. Alonso, A. Arroyo, I. Martín-García and Y. Moglie, Adv. Synth. Catal., 2015, 357, 3549–3561 CrossRef CAS; (c) L. Niu, S. Wang, J. Liu, H. Yi, X.-A. Liang, T. Liu and A. Lei, Chem. Commun., 2018, 54, 1659–1662 RSC; (d) B. T. Matsuo, J. T. M. Correia and M. W. Paixão, Org. Lett., 2020, 22, 7891–7896 CrossRef CAS PubMed; (e) C. Russo, J. Amato, G. C. Tron and M. Giustiniano, J. Org. Chem., 2021, 86, 18117–18127 CrossRef CAS PubMed; (f) C. Russo, G. Graziani, R. Cannalire, G. C. Tron and M. Giustiniano, Green Chem., 2022, 24, 3993–4003 RSC; (g) X. Li, H. Zhao, L. Meng, J. Dong, C. Yang, Z. Wei, X. Wang, J. Xu and B. Fan, J. Org. Chem., 2023, 88, 5300–5310 CrossRef CAS PubMed.
- A. McNally, C. K. Prier and D. W. C. MacMillan, Science, 2011, 334, 1114–1117 CrossRef CAS PubMed.
- C. Xu, F.-Q. Shen, G. Feng and J. Jin, Org. Lett., 2021, 23, 3913–3918 CrossRef CAS PubMed.
- M.-J. Yi, H.-X. Zhang, T.-F. Xiao, J.-H. Zhang, Z.-T. Feng, L.-P. Wei, G.-Q. Xu and P.-F. Xu, ACS Catal., 2021, 11, 3466–3472 CrossRef CAS.
- W. Lee, D. Kim, S. Seo and S. Chang, Angew. Chem., Int. Ed., 2022, 61, e202202971 CrossRef CAS PubMed.
- (a) Z. Zhang, Y. Deng, M. Hou, X. Lai, M. Guan, F. Zhang, R. Qi and G. Qiu, Chem. Commun., 2022, 58, 13644–13647 RSC; (b) S. Xie, S. Tang, M. Hou, W. Xie, M. Guan, T. Bai, L. He and G. Qiu, Org. Lett., 2024, 26, 11134–11139 CrossRef CAS PubMed.
- (a) J. Lei, M. Li, Q. Zhang, S. Liu, H. Li, L. Shi, W.-F. Jiang, C. Duan and Y. Jin, Org. Lett., 2023, 25, 2300–2305 CrossRef CAS PubMed; (b) Z. Gan, J. Chen, H. Wang, Z. Xue, Z. Chen, Y. Zhang, L. Wang, H. Zi, S. Liu, L. Shi and Y. Jin, Org. Lett., 2024, 26, 7751–7756 CrossRef CAS PubMed; (c) R. Cui, Q. Liao, Y. Zhao, L. Wang, Y. Zhang, S. Liu, Z. Gan, Y. Chen, Y. Shi, L. Shi, M. Li and Y. Jin, Org. Lett., 2024, 26, 8222–8227 CrossRef CAS PubMed; (d) Y. Zhang, D. Fu, Z. Chen, R. Cui, W. He, H. Wang, J. Chen, Y. Chen, S.-J. Li, Y. Lan, C. Duan and Y. Jin, Nat. Commun., 2024, 15, 8619 CrossRef CAS PubMed.
- S. E. McKinney, H. M. Peck, J. M. Bochey, B. B. Byham, G. S. Schuchardt and K. H. Beyer, J. Pharmacol. Exp. Ther., 1951, 102, 208–214 CrossRef CAS PubMed.
- G. R. Buettner, Free Radical Biol. Med., 1987, 3, 259–303 CrossRef CAS PubMed.
- J. E. Bartmess, in PATAI'S Chemistry of Functional Groups, 2010, DOI:10.1002/9780470682531.pat0506.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5gc00426h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |