
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly efficient sulfurized Re–Ir catalysts for multiple N-methylation of ethylenediamine and its homologous series with CO2 and H2 in water solvent†
Min
Wang
a,
Mizuho
Yabushita
 *a,
Kazuki
Okuma
b,
Tomohiro
Shono
b,
Yoshinao
Nakagawa
a and
Keiichi
Tomishige
*ac
*a,
Kazuki
Okuma
b,
Tomohiro
Shono
b,
Yoshinao
Nakagawa
a and
Keiichi
Tomishige
*ac
aDepartment of Applied Chemistry, School of Engineering, Tohoku University, 6-6-07 Aoba, Aramaki, Aoba-ku, Sendai, Miyagi 980-8579, Japan. E-mail: m.yabushita@tohoku.ac.jp; tomishige@tohoku.ac.jp
bOrganic Materials Research Laboratory, Tosoh Corporation, Kaisei-cho 4560, Shunan, Yamaguchi 746-8501, Japan
cAdvanced Institute for Materials Research (WPI-AIMR), Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai, Miyagi 980-8577, Japan
First published on 13th May 2025
Abstract
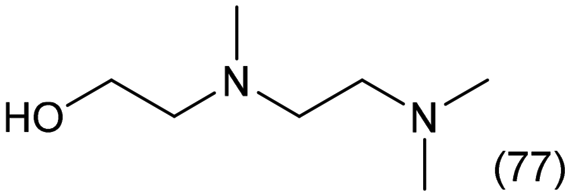
Sulfurized Re–Ir clusters on a carbon black support were demonstrated to be highly active catalysts for the quadruple N-methylation of ethylenediamine (EDA) with CO2 and H2 to yield N,N,N′,N′-tetramethylethylenediamine (TetraM-EDA). The catalytic activity of this catalyst was 6.4-fold higher than that of the previously reported sulfurized Re catalyst. This bimetallic catalyst was applicable to the multiple N-methylation of various diamines that consisted of C2/C3 units sandwiched between two amino groups, and their strong interaction arising from the chelating effect played a key role in the progress of N-methylation over the catalyst, as indicated by the kinetic study. Furthermore, the sulfurized Re–Ir species were mandatory for the N-methylation of EDA (namely, the hydrogenation step of N-formylated intermediates into N-methylated EDAs), and a lack of sulfurized species resulted in the formation of N-formylated compounds as major products instead of N-methylated ones. Detailed characterization suggested that the role of Ir species was to substantially reduce Re species. These functions offered not only high catalytic activity but also stability against air, the latter of which contributed to the suppression of oxidation and leaching of Re species after catalyst exposure to air.
 Mizuho Yabushita | Mizuho Yabushita received his Ph.D. from Hokkaido University in 2015 under Prof. Atsushi Fukuoka. He pursued his postdoctoral career in the groups of Prof. Alexander Katz at University of California, Berkeley and Prof. Fukuoka at Hokkaido University. He was promoted to an assistant professor of Tohoku University in Prof. Atsushi Muramatsu's group (2018–2020) and Prof. Keiichi Tomishige's group (since 2020). His current research interests are catalytic conversion of biomass and CO2 and design of porous materials. He is a member of Early Career Advisory Board of ChemSusChem (since 2023) and Early Career Editorial Board of Applied Catalysis A: General (since 2024). |
Green foundation1. This study offers a novel catalytic means of introducing four or more N-methyl groups to ethylenediamine and its homologous series using green reactants consisting of CO2 and H2 in water. This means is a promising alternative to classical organic chemistry approaches that rely on deleterious and hazardous stoichiometric reagents.2. The currently presented N-methylation proceeds well in the greenest solvent water, in stark contrast to previously reported catalytic N-methylation systems for aliphatic and aromatic monoamines operated in alkanes and tetrahydrofuran. 3. Further research will focus on the establishment of continuous flow systems for N-methylation of diamines as well as the replacement of precious metal components (i.e., Re and Ir) with abundant metals, both of which could lead to the industrialization of this process and contribute to the establishment of carbon neutrality. |
1. Introduction
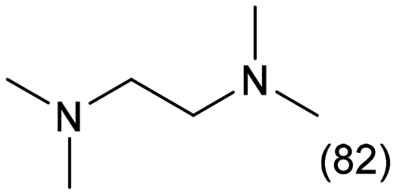
N,N,N′,N′-Tetramethylethylenediamine (TetraM-EDA) is a versatile compound, acting as a chelating agent and ligand for metal cations (particularly important for organo-lithium chemistry),1 biochemical reagent,2 crosslinking agent for epoxy resins,3 source of quaternary ammonium salts,4 polymerization initiator when combined with alkaline persulfate,5–7 and methylating agent under photoirradiation conditions.8 TetraM-EDA has also emerged as a promising green propellant fuel for replacing hydrazine-based fuels.9,10 Its global market size was 174.2 million USD in 2022 and is expected to grow up to 271.6 million USD by 2030.11 The classical synthetic method of TetraM-EDA, as filed in a patent by Bayer, involves an intermolecular reaction between 1,2-dichloroethane and dimethylamine (Fig. 1A).12 Another approach to synthesizing TetraM-EDA is the N-methylation of ethylenediamine (EDA), which is the simplest diamine, with methylating reagents. The classical N-methylation, known as Eschweiler–Clarke reaction, uses a mixture of formaldehyde and formic acid as sources of methyl groups (Fig. 1B).13–15 Iodomethane is also a well-known methylating agent, but its high reactivity has led to the over-functionalization of EDA to produce N,N,N,N′,N′,N′-hexamethylethylenediamine diiodide instead of TetraM-EDA.16 These traditional approaches for producing TetraM-EDA require stoichiometric reagents, and the development of greener catalytic processes, namely, using heterogeneous catalysts for introducing methyl groups into EDA, is thus highly desirable. | ||
| Fig. 1 Reported and current (non-)catalytic systems for synthesizing TetraM-EDA. | ||
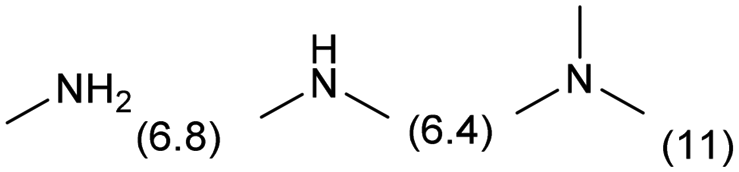
EDA exhibits both strong basicity (pKa = 9.98)17 and chelating effect toward metal cations. Upon the progress of step-by-step N-methylation of EDA toward TetraM-EDA, the basicity of amino groups increases from EDA (pKa = 9.98) to the intermediate compounds, N-methylethylenediamine (pKa = 10.14) and N,N′-dimethylethylenediamine (pKa = 10.17); meanwhile, the basicity of the end product TetraM-EDA (pKa = 9.28) is lower than that of EDA.17 The chelating effect of EDA and N-methylated EDAs for metal cations (e.g., Cu+) was reported to be reinforced upon increasing the number of N-methyl groups;18 such chelating effect should lead to the leaching of catalytically active species from heterogeneous catalysts. These facts posit that catalysts for quadruple N-methylation of EDA need to exhibit not only high activity for enabling multi-step N-functionalization but also high stability against EDA, intermediates, and TetraM-EDA. To date, various types of catalytic N-methylation of EDA have been reported. The reaction of EDA with methanol as a methylating agent with CuO–NiO/γ-Al2O3 in a fixed-bed flow reactor was reported by Chen et al. to produce TetraM-EDA in up to 92% yield at 453 K at 1.0 MPa H2 (Fig. 1C);19 yet, neither catalyst stability nor leaching of catalyst components was addressed in the report. The same research group employed the combination of RANEY® Ni catalyst and paraformaldehyde, the latter of which was a methylating agent, for the N-methylation of EDA and achieved 89% yield at 403 K (Fig. 1D).20 This Ni catalyst gradually lost its activity upon its reuse, but the reason for catalyst deactivation was not clarified. In a related work, the quadruple N-methylation of 1,6-diaminohexane was demonstrated by using a carbon-supported Pt catalyst and methanol.21 In addition to these thermocatalytic systems, photocatalytic quadruple N-methylation of EDA to TetraM-EDA was also reported; the ultraviolet-light irradiation conditions at room temperature with methanol and a TiO2-supported Pt–Pd bimetallic catalyst afforded 56% yield of TetraM-EDA (Fig. 1E).22
The above-mentioned methylating agents (i.e., methanol and paraformaldehyde) used for the catalytic N-methylation of EDA can be produced via hydrogenation of CO2 with H2;23–29 in this sense, direct N-methylation using CO2 and H2 as sources of N-methyl groups allows the shortening of overall processes and is thus attractive from the perspective of green chemistry. Also, the non-toxic, non-flammable, and ubiquitous nature of CO2 makes this gaseous compound a more ideal and safer methylating agent compared to classical and deleterious methylating agents such as iodomethane16,30 and diazomethane.31 The additional benefits of N-methylation using CO2 and H2 are the unnecessity of expensive reductants (e.g., hydrosilane)32 and generation of water as a sole co-product. Various homogeneous and heterogeneous catalysts have been reported for N-methylation of various amines with CO2 and H2 thus far, as comprehensively reviewed elsewhere.33–37 Typical target substrates were both aliphatic and aromatic primary monoamines, while the degree of their N-methylation (i.e., single N-methylation or double N-methylation) was dependent on catalysts and reaction conditions. Table S1† lists previously reported catalytic systems for double N-methylation of aliphatic/aromatic amines using CO2 and H2. The tripodal phosphine-ligated Ru(III) complexes were reported to be highly active homogeneous catalysts for the double N-methylation of aromatic and aliphatic amines.38,39 A Co(II) complex with a tripodal phosphine ligand and tin methanesulfonate was also reported for the double N-methylation of aniline and its derivatives.40 These findings have highlighted the great potential of metal complexes for catalytic N-methylation of amines; yet, due to the desire to improve the separability and reusability of catalysts, heterogeneous catalysts have been focused more on in this decade. The heterogeneous catalysts reported thus far are CuAlOx (i.e., CuOx nanoparticles deposited on AlOx),41 Pd/CuZrOx,42 and Au nanoparticles supported on γ-Al2O343 or metal–organic framework coating on SiO2 and Fe3O4.44 These catalysts exhibited a broad substrate scope including aromatic amines (i.e., aniline and its derivatives) as well as aliphatic amines. Meanwhile, no catalyst was reported for quadruple N-methylation of aliphatic primary diamines including EDA with CO2 and H2 regardless of the usefulness of their corresponding N-methylated products. Also, alkanes and tetrahydrofuran (THF) were typical solvents for N-methylation with CO2 and H2 (Table S1†), possibly due to the high solubility of each substrate in these solvents (i.e., aniline and aliphatic amines with a hydrophobic moiety such as a long alkyl chain, cycloalkyl group, and benzyl group). However, according to previously reported solvent guides, alkanes and THF were categorized into non-green solvents.45,46 Considering the environmental impact of solvents, the fact that water is always formed as a co-product in N-methylation with CO2 and H2, and the miscible nature of EDA, water is the most appropriate solvent. In this context, a catalytic system for quadruple N-methylation of EDA operable in water solvent needs to be developed.
We very recently found sulfurized Re species supported on carbon black (denoted as Re/C-BP) to be active for the quadruple N-methylation of EDA, with CO2 and H2 to produce TetraM-EDA in up to 84% yield in water solvent (Fig. 1F).47 The key finding for Re/C-BP was the high hydrogenation activity of sulfurized Re species that enabled the production of TetraM-EDA, while a lack of such sulfurized species yielded N-formylated EDAs as major products. In this catalytic system, water was demonstrated to be the most suitable reaction medium. However, the catalytic activity of Re/C-BP reflected by turnover frequency (TOF) on the basis of Re amount was not so high (i.e., 3.3 h−1, Table S1†) and thus needs to be improved. All the reaction orders with respect to EDA concentration, partial pressure of CO2, and that of H2 were positive (i.e., 0.5–0.7), indicating weak interaction between these three reactants and catalyst surfaces. To this end, the improvement of such interaction is necessary for increasing the coverage of catalyst surfaces with substrate molecules. Another issue of the Re/C-BP catalyst is the necessity of careful handling in an oxygen-free glove box in its repeated use; otherwise, oxophilic Re species readily undergo oxidation in air and are leached from the catalyst. Furthermore, the substrate scope of the sulfurized Re catalysts has not yet been investigated. In this context, this study aimed to develop a highly active and durable catalyst for the quadruple N-methylation of EDA by adding a second metal to Re/C-BP because such modification offers an opportunity to tune the catalytic performance including activity and stability.48–53 The substrate scope of the catalyst developed in this study was also investigated here.
2. Experimental
2.1. Reagents
All the reagents and gases used in this study for catalyst preparation and catalytic reactions were purchased from suppliers and used as received unless otherwise noted. The detailed information about each reagent and gas (e.g., supplier and specific surface area based on the Brunauer–Emmett–Teller (BET) equation (SBET)) is summarized in Table S2.† The main metal sources and catalyst support employed in this study were NH4ReO4 (Mitsuwa Chemical), Ir(NO3)4 (Furuya Metal), and carbon black (Black Pearls 2000, Cabot; denoted as C-BP; SBET = 1280 m2 g−1), respectively, based on our previous report47 and current catalyst screening (vide infra). In some cases, for desulfurization,54 C-BP was treated in a H2 flow (30 mL min−1) for 6 h at either 773 K or 973 K and used as a desulfurized support, which is denoted as C-BP(deS-773) or C-BP(deS-973), respectively.2.2. Catalyst preparation
Supported-(bi)metal catalysts (Re(–M)/support) were prepared by the typical (co-)impregnation method. A powdery support was impregnated with an aqueous solution containing metal precursor(s). The typical loading of Re was adjusted to 4 wt%. The molar ratios of M to Re were controlled by changing the amounts of metal precursors unless otherwise noted. After (co-)impregnation, drying on a hot plate at 363 K, and further drying in an oven at 383 K for 12 h, the resulting powder was obtained for catalytic tests and characterization.2.3. N-Methylation of EDA with CO2 and H2
Prior to N-methylation of EDA, each catalyst was pre-reduced in a H2 flow (30 mL min−1) at 773 K for 1 h. Without exposure to air, the reduced catalyst was transferred into an Ar-filled glove box. In the glove box, the reduced catalyst (0.10 g), EDA (0.24 g; 4 mmol), and water as a solvent (5.0 g) with a stirring bar were charged into a glass-lined stainless-steel autoclave (HIRO Company, inner volume 190 mL), which was then sealed. Note that water was previously found as the most appropriate solvent for the N-methylation of EDA in terms of reaction rate as well as a lack of undesired solvent-related side reactions.47 After purging with CO2, the autoclave was filled with 1.0 MPa CO2 at room temperature, followed by the introduction of 4.0 MPa H2 at room temperature. Subsequently, the reactor was heated in an aluminum block heater (ChemiChemi-200, Sibata Scientific Technology) to the designated temperature (typically 413 K, ramp time ca. 30 min) with magnetic stirring at 300 rpm. The inner temperature was monitored with a thermocouple inserted into the autoclave. In this study, the time when the inner temperature became the target one was defined as the beginning of reaction (i.e., 0 h). After a specific reaction time, the autoclave was cooled to room temperature in a water bath. The gaseous products in the autoclave were transferred into a plastic gas bag. The liquid mixture was collected with a washing solvent (50 g of a mixed solution consisting of distilled water (10 g) and ethanol (40 g)) and transferred into a vial. After the separation of liquid phase from the spent catalyst with a polytetrafluoroethylene syringe filter (0.20 μm mesh) and addition of 1,4-butanediol (or 2,3-butanediol) as an internal standard, the liquid phase was analyzed by using a gas chromatograph equipped with either a flame ionization detector (GC-FID; GC-2014, Shimadzu) or mass spectrometer (GC-MS; GCMS-QP2020NXC, Shimadzu, methane chemical ionization (CI)). The column for GC-FID and GC-MS was an InertCap for Amines capillary column (GL Sciences, ϕ0.32 mm × 30 m). The gaseous products were analyzed using the same GC-FID. The GC-MS data for identifying uncommercialized products (e.g., N-formylated compounds) are shown in Fig. S1–S3.†The conversion, yield, and carbon balance were calculated on the basis of the C2 unit originally present in the EDA molecule (i.e., CH2–CH2) because the formation of dimers and cracked compounds from EDAs and/or their N-methylated products was sometimes observed. The specific equations for calculating these values are eqn (1)–(3). Considering that EDA undergoes N-methylation at most four times, the degree of N-methylation (NMe) defined as shown in eqn (4) was also calculated in this study as one of the parameters that reflect the activity of the catalyst. Since EDA can undergo N-methylation at most four times, the upper limit of NMe is 400%. The abbreviations used in eqn (4) are as follows: M-EDA = N-methylethylenediamine; N,N-DM-EDA = N,N-dimethylethylenediamine; N,N′-DM-EDA = N,N′-dimethylethylenediamine; TriM-EDA = N,N,N′-trimethylethylenediamine; and TetraM-EDA = N,N,N′,N′-tetramethylethylenediamine.
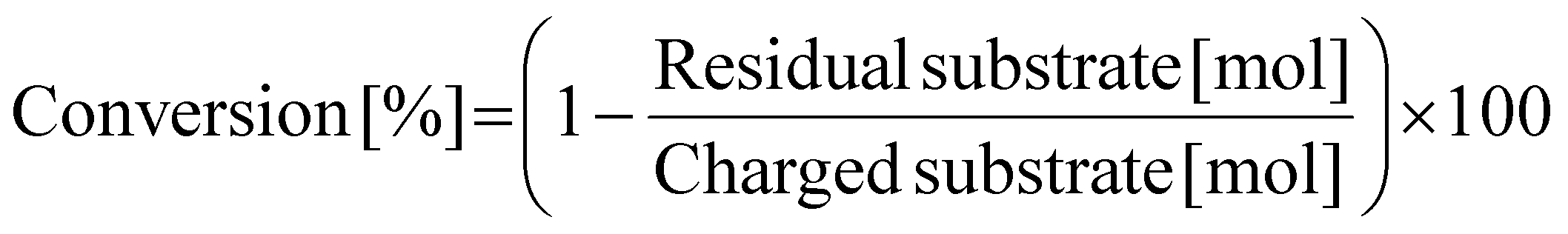 | (1) |
 | (2) |
 | (3) |
 | (4) |
The reusability of the catalyst was evaluated by repeating the reactions as follows. The spent catalyst was separated from the reaction mixture by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, washed with water and ethanol three times, and dried at room temperature for 12 h; these procedures were performed in air. After reduction in a H2 flow (30 mL min−1) at 773 K for 1 h, the resulting catalyst was charged into the reactor for the next run in the same manner as the 1st run (see Section 2.3). In this test, the reaction solution was analyzed by not only GC-FID (vide supra) but also inductively coupled plasma optical emission spectrometry (ICP-OES; iCAP6500, Thermo Fisher Scientific) in order to quantify the amount of metal species leached from the catalyst.
000 rpm, washed with water and ethanol three times, and dried at room temperature for 12 h; these procedures were performed in air. After reduction in a H2 flow (30 mL min−1) at 773 K for 1 h, the resulting catalyst was charged into the reactor for the next run in the same manner as the 1st run (see Section 2.3). In this test, the reaction solution was analyzed by not only GC-FID (vide supra) but also inductively coupled plasma optical emission spectrometry (ICP-OES; iCAP6500, Thermo Fisher Scientific) in order to quantify the amount of metal species leached from the catalyst.
2.4. Catalyst characterization
The X-ray diffraction (XRD) patterns of catalysts after the reduction in a H2 flow (30 mL min−1) at 773 K for 1 h and subsequent passivation at room temperature in a diluted O2 flow (2 vol% O2/Ar, 30 mL min−1) or after the reaction (vide supra) were obtained with a MiniFlex600 diffractometer (Rigaku, Cu Kα (0.154 nm) at 40 kV and 20 mA, scan speed 10° min−1). The specific surface area based on the Brunauer–Emmett–Teller (BET) equation was examined by N2 physisorption measurement at 77 K with an automated instrument (Gemini VII 2360, Micromeritics). X-ray fluorescence (XRF) spectroscopy (Bruker, S8 Tiger) was used to measure the actual loading amounts of metal species in the prepared catalysts. Prior to each measurement, the reduced and passivated catalyst (0.5 g) and internal standard of ZrO2 (0.1 g) were mixed well on a mortar. The high-angle annular dark field scanning transmission electron microscope (HAADF-STEM) images for the reduced and passivated catalysts were taken with a Titan3™ 60–300 Double Corrector microscope (Thermo Fisher Scientific) equipped with an energy-dispersive X-ray spectroscopy system (EDS; Super-X detector system, Thermo Fisher Scientific). The average particle size of supported metals was calculated from the HAADF-STEM images using the Sauter mean diameter (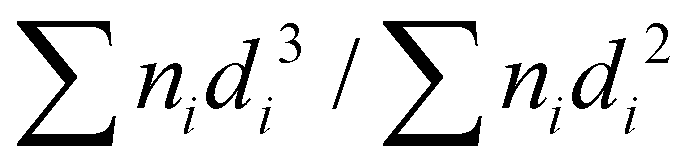 ; di = particle size, ni = number of particles with the size of di). A catalyst analyzer BELCAT II (MicrotracMRB) equipped with both a thermal conductivity detector (TCD) and a quadrupole mass spectrometer (Q-MS; BELMASS, MicrotracMRB) was employed to carry out temperature-programmed reduction with H2 (H2-TPR) in a 10 vol% H2/Ar flow (30 mL min−1) with a ramp rate of 10 K min−1. For the elucidation of dispersion of supported metal species, CO chemisorption was performed with the same analyzer. Prior to measurement, the catalysts were reduced in a H2 flow (30 mL min−1) at 773 K for 1 h, and then CO pulses were introduced at 323 K until the metal surfaces were saturated by CO. The amount of adsorbed CO was assumed to be equal to that of the surface metallic Re and/or Ir atoms (i.e., dispersion (DCO) [%] = CO [mol]/(Ir [mol] + Re [mol])).
; di = particle size, ni = number of particles with the size of di). A catalyst analyzer BELCAT II (MicrotracMRB) equipped with both a thermal conductivity detector (TCD) and a quadrupole mass spectrometer (Q-MS; BELMASS, MicrotracMRB) was employed to carry out temperature-programmed reduction with H2 (H2-TPR) in a 10 vol% H2/Ar flow (30 mL min−1) with a ramp rate of 10 K min−1. For the elucidation of dispersion of supported metal species, CO chemisorption was performed with the same analyzer. Prior to measurement, the catalysts were reduced in a H2 flow (30 mL min−1) at 773 K for 1 h, and then CO pulses were introduced at 323 K until the metal surfaces were saturated by CO. The amount of adsorbed CO was assumed to be equal to that of the surface metallic Re and/or Ir atoms (i.e., dispersion (DCO) [%] = CO [mol]/(Ir [mol] + Re [mol])).
X-ray absorption spectroscopy (XAS) was performed in the transmission mode at the BL14B2 beamline of SPring-8 (proposal no. 2024A1643). Two ion chambers were filled with N2/Ar = 85/15 (vol/vol) and N2/Ar = 50/50 (vol/vol) for I0 and I1, respectively. Standard samples (Re powder, ReO2, ReO3, and NH4ReO4 for Re L3-edge; Ir powder and IrO2 for Ir L3-edge) were measured for both X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) analyses. The data were managed using REX2000 version 2.6 software (Rigaku). The average valence of Re and Ir was determined from the XANES spectra by comparing the white line area of each sample with those of authentic samples, consistent with previous reports.47,55–61 The k3-weighted and Fourier-transformed EXAFS oscillations were acquired from the raw data in the k range of 30–120 nm−1. The empirical phase shift and amplitude function of Re–Re, Re–O, Ir–Ir, and Ir–O bonds were extracted from the data of Re powder, NH4ReO4, Ir powder, and IrO2, respectively, consistent with our previous reports.57–59,62–64 For both Re L3-edge and Ir L3-edge EXAFS, the distinction of Re and Ir as backscattering atoms is quite difficult; therefore, the empirical phase shift and amplitude functions for Re–Re and Re–Ir were extracted from the data of Re powder, and those for Ir–Ir and Ir–Re were done so from the data of Ir powder. Therefore, in the results of curve fitting (vide infra), the bonds of Re–Ir for Re L3-edge EXAFS and Ir–Re for Ir L3-edge EXAFS are represented as Re–Re (or Re–Ir) and Ir–Ir (or Ir–Re), respectively.59,62,63 The FEFF 8.20 program was used to calculate the theoretical phase shift and amplitude function of the Re–S and Ir–S shells.
3. Results and discussion
3.1. Screening of second metals in the Re–M/C-BP catalyst and optimization of metal components in N-methylation of EDA
To improve the activity of Re/C-BP, which was previously found to be an effective catalyst for the quadruple N-methylation of EDA,47 this catalyst was modified with second metals (M) at the M/Re molar ratio of 0.5 and tested for the reaction (Table 1). The loading amount of Re in previously reported Re/C-BP was 5 wt%,47 while the current investigation employed 4 wt% of Re species in order to control the conversion level to be moderate and enable the clear examination of the effects of second metals. For almost all the reactions in this study, water was employed as a reaction solvent because water was the most suitable solvent for the N-methylation of EDA as found previously47 and also ranked as the greenest solvent.45,46 Under these conditions, EDA was not converted at all without a catalyst or with C-BP only (entries 1 and 2). Meanwhile, the 4 wt% Re/C-BP catalyst gave N-methylethylenediamine (abbreviated as M-EDA; 10% yield) as a major product, and the further N-methylated EDAs consisting of N,N-dimethylethylenediamine (N,N-DM-EDA; 6.5%), N,N′-dimethylethylenediamine (N,N′-DM-EDA; 0.2%), N,N,N′-trimethylethylenediamine (TriM-EDA; 1.7%), and N,N,N′,N′-tetramethylethylenediamine (TetraM-EDA, 4.5%) were also produced (entry 3). Along with these N-methylated EDAs, N-formylated EDAs, which are intermediates of N-methylation,47 were detected to some extent. The degree of N-methylation (NMe), which is an indicator of catalytic activity in this reaction (see also Section 2.3), was calculated to be 47%. The addition of Ir, Ru, and Pt to Re/C-BP altered the major product from M-EDA to TetraM-EDA and greatly enhanced the catalytic activity for N-methylation, as reflected by at least 2.8-fold higher NMe compared to Re/C-BP (entries 4–6); namely, the modification of Re/C-BP with Ir exhibited 3.5-fold higher NMe (164%, entry 4) than Re/C-BP (47%, entry 3). The other modifiers consisting of Rh, Au, Ag, and Mo slightly increased NMe (entries 7–10), and the degree of improvement of catalytic activity by these metals was obviously inferior to Ir. Therefore, among the second metals tested in this study, Ir was found to offer the highest catalytic activity.| Entry | Catalyst | Conv. [%] | Yield [%] | N Me [%] | Bal. [%] | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N-Methylated EDA | N-Formylated EDA | Othersc | ||||||||||||
| M-EDA | N,N-DM-EDA | N,N′-DM-EDA | TriM-EDA | TetraM-EDA | F-EDA | F-M-EDA | F-DM-EDA | F-TriM-EDA | ||||||
| a Reaction conditions: EDA 4.0 mmol; Re–M/C-BP (Re 4 wt%, M/Re = 0.5, reduced in H2 at 773 K) 0.10 g; water 5.0 g; CO2 1.0 MPa + H2 4.0 MPa (r.t.); 413 K; 16 h. b Abbreviations: M-EDA = N-methylethylenediamine; N,N-DM-EDA = N,N-dimethylethylenediamine; N,N′-DM-EDA = N,N′-dimethylethylenediamine; TriM-EDA = N,N,N′-trimethylethylenediamine; TetraM-EDA = N,N,N′,N′-tetramethylethylenediamine; F-EDA = N-(2-aminoethyl)formamide; F-M-EDA = N-(2-aminoethyl)-N-methylformamide; F-DM-EDA = N-(2-(dimethylamino)ethyl)formamide; F-TriM-EDA = N-(2-(dimethylamino)ethyl)-N-methylformamide. c Including piperazine, 2-imidazolidinone, methylamine, and their corresponding N-methylated compounds. d Degree of N-methylation calculated using eqn (4). e Without impregnation of any metal. | ||||||||||||||
| 1 | None | 0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0 | 100 |
| 2 | C-BPe | 0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0 | 100 |
| 3 | Re/C-BP | 39 | 10 | 6.5 | 0.2 | 1.7 | 4.5 | 4.3 | 1.9 | 0.3 | 0.3 | 1.3 | 47 | 92 |
| 4 | Re–Ir/C-BP | 70 | 4.5 | 9.9 | 0.0 | 1.9 | 34 | 6.8 | 2.3 | 1.6 | 0.8 | 2.2 | 164 | 94 |
| 5 | Re-Ru/C-BP | 63 | 8.5 | 10 | 0.0 | 2.2 | 26 | 4.6 | 1.7 | 0.9 | 0.9 | 1.1 | 138 | 93 |
| 6 | Re–Pt/C-BP | 56 | 7.8 | 11 | 0.1 | 2.2 | 24 | 3.8 | 1.7 | 1.0 | 0.7 | 1.0 | 132 | 97 |
| 7 | Re–Rh/C-BP | 45 | 9.0 | 9.2 | 0.0 | 1.4 | 11 | 4.0 | 1.5 | 0.6 | 0.4 | 0.6 | 77 | 93 |
| 8 | Re–Au/C-BP | 40 | 11 | 7.7 | 1.6 | 0.0 | 8.1 | 4.4 | 2.0 | 0.3 | 0.4 | 0.9 | 59 | 95 |
| 9 | Re–Ag/C-BP | 50 | 9.0 | 7.8 | 0.4 | 1.3 | 8.5 | 7.7 | 3.0 | 0.9 | 0.6 | 3.2 | 63 | 93 |
| 10 | Re–Mo/C-BP | 51 | 9.5 | 8.4 | 0.0 | 1.7 | 11 | 7.1 | 2.8 | 0.8 | 0.7 | 1.1 | 76 | 92 |
To clarify the improvement of catalytic activity by the addition of Ir, some control catalysts were tested for the N-methylation of EDA under the same conditions as above, as listed in Table 2. The monometallic catalyst, 4 wt% Ir/C-BP, provided 27% NMe (entry 3), the value of which was lower than those given by Re–Ir/C-BP (Re 4 wt%, Ir/Re = 0.5, 0.10 g) (164%, entry 1) as well as 4 wt% Re/C-BP (0.10 g) (47%, entry 2). The use of sole Ir was, therefore, not effective for the N-methylation of EDA. The physical mixture of 4 wt% Re/C-BP (0.10 g) and 2 wt% Ir/C-BP (0.10 g), which offered the identical amounts of Re and Ir to the case of Re–Ir/C-BP (Re 4 wt%, Ir/Re = 0.5, 0.10 g) in the reaction system, increased the NMe to 109% (entry 4). This NMe value was higher than those given by the monometallic catalysts, 4 wt% Re/C-BP and 4 wt% Ir/C-BP, but lower than that by the bimetallic catalyst Re–Ir/C-BP (Re 4 wt%, Ir/Re = 0.5). These results demonstrated that both Re and Ir need to be placed on the same surface from the beginning to achieve high catalytic activity for the N-methylation of EDA. The reason why the physical mixture exhibited better activity than each monometallic catalyst would be due to the transfer of metal species between the different surfaces during the reaction65 and/or migration of active species such as hydrogen (i.e., hydrogen spillover) between the two catalyst surfaces.66,67
| Entry | Catalyst | Conv. [%] | Yield [%] | N Me [%] | Bal. [%] | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N-Methylated EDA | N-Formylated EDA | Othersc | ||||||||||||
| M-EDA | N,N-DM-EDA | N,N′-DM-EDA | TriM-EDA | TetraM-EDA | F-EDA | F-M-EDA | F-DM-EDA | F-TriM-EDA | ||||||
| a Reaction conditions: EDA 4.0 mmol; catalyst (reduced in H2 at 773 K) Re 4 mg, Ir 2 mg; water 5.0 g; CO2 1.0 MPa + H2 4.0 MPa (r.t.); 413 K; 16 h. b Abbreviations: M-EDA = N-methylethylenediamine; N,N-DM-EDA = N,N-dimethylethylenediamine; N,N′-DM-EDA = N,N′-dimethylethylenediamine; TriM-EDA = N,N,N′-trimethylethylenediamine; TetraM-EDA = N,N,N′,N′-tetramethylethylenediamine; F-EDA = N-(2-aminoethyl)formamide; F-M-EDA = N-(2-aminoethyl)-N-methylformamide; F-DM-EDA = N-(2-(dimethylamino)ethyl)formamide; F-TriM-EDA = N-(2-(dimethylamino)ethyl)-N-methylformamide. c Including piperazine, 2-imidazolidinone, methylamine, and their corresponding N-methylated compounds. d Degree of N-methylation calculated using eqn (4). e Re 4 wt%, Ir/Re = 0.5, 0.10 g. f Re 4 wt%, 0.10 g. g Ir 4 wt%, 0.10 g. h Physical mixture of Re/C-BP (Re 4 wt%, 0.10 g) and Ir/C-BP (Ir 2 wt%, 0.10 g). | ||||||||||||||
| 1 | Re–Ir/C-BPe | 70 | 4.5 | 9.9 | 0.0 | 1.9 | 34 | 6.8 | 2.3 | 1.6 | 0.8 | 2.2 | 164 | 94 |
| 2 | Re/C-BPf | 39 | 10 | 6.5 | 0.2 | 1.7 | 4.5 | 4.3 | 1.9 | 0.3 | 0.3 | 1.3 | 47 | 92 |
| 3 | Ir/C-BPg | 36 | 3.9 | 5.4 | 0.0 | 0.5 | 2.7 | 14 | 2.4 | 0.8 | 0.0 | 1.0 | 27 | 94 |
| 4 | Re/C-BP + Ir/C-BPh | 54 | 7.5 | 11 | 0.0 | 4.0 | 17 | 7.6 | 2.5 | 1.1 | 0.7 | 3.0 | 109 | 100 |
The optimal molar ratio of Ir to Re was then investigated. Fig. 2A and Table S3† represent the dependence of catalytic performance of Re–Ir/C-BP on the Ir/Re molar ratio at the constant Re loading of 4 wt%. The conversion and NMe gradually increased upon the increase of Ir/Re ranging from 0 to 0.5, while the further increase of Ir/Re did not affect the catalytic performance. In the case of the monometallic Re/C-BP catalysts, we previously observed a similar trend of catalytic activity against the loading amount of Re;47 at the high Re loading, the content of S in C-BP could be insufficient for the formation of ReSx species that contribute to the high catalytic activity for the N-methylation of EDA. Likewise, in the current study, the quantification of Re, Ir, and S present in the Re–Ir/C-BP catalysts by XRF confirmed the decrease of S/(Re + Ir) upon the increase in the loading amount of Ir (Table S3†). This trend implied that the shortage of S species at the high loading amount of metals made the formation of active sulfurized species difficult. Therefore, neither conversion nor NMe in the N-methylation of EDA got increased at the Ir/Re molar ratio of 0.5 or higher. The same situation was also found when the Re amount was increased with maintaining the Ir/Re molar ratio of 0.5 (Fig. 2B and Table S4†). In the range of 2–4 wt% of Re loading, both conversion and NMe were higher at the higher Re loading. Yet, these values plateaued at the Re loading ranging from 4 to 8 wt%. This trend can be rationalized again by the content of S against the loading amounts of Re and Ir. Overall, the Re–Ir/C-BP catalyst with the Re loading and Ir/Re molar ratio of 4 wt% and 0.5, respectively, was determined to be the optimum catalyst and employed for further investigation.
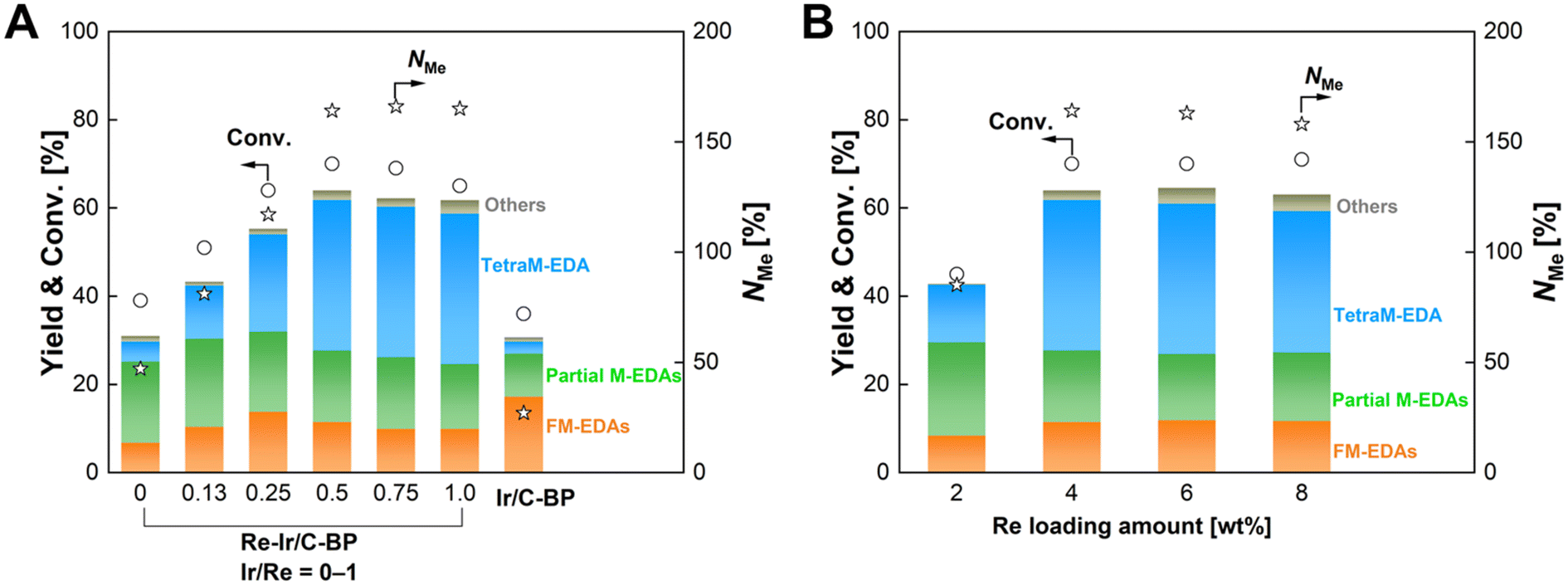 | ||
| Fig. 2 Effects of (A) Ir/Re molar ratio at the constant Re loading of 4 wt% and (B) Re loading at the constant Ir/Re ratio of 0.5 on the catalytic activity of Re–Ir/C-BP for the N-methylation of EDA. Reaction conditions: EDA 4.0 mmol; Re–Ir/C-BP (reduced in H2 at 773 K) or Ir/C-BP (Ir 4 wt%, reduced in H2 at 773 K) 0.10 g; water 5.0 g; CO2 1.0 MPa + H2 4.0 MPa (r.t.); 413 K; 16 h. In this figure, “Partial M-EDAs” consist of N-methylated EDAs possessing one to three methyl groups, and “FM-EDAs” are composed of N-formylated EDA and N-formylated partial M-EDAs. “Others” include piperazine, 2-imidazolidinone, methylamine, and their corresponding N-methylated compounds. The detailed data are summarized in Tables S3 and S4.† | ||
We previously found the sulfurized Re species, which were formed due to the presence of sulfur impurities in C-BP, to be the main contributor to the quadruple N-methylation of EDA into TetraM-EDA, while a lack of such species rather led to the N-formylation of EDA.47 The effect of sulfur content in the C-BP support was thus also elucidated in this work at the optimum Re loading of 4 wt% and Ir/Re molar ratio of 0.5. Table 3 shows the results of activity tests of Re–Ir on C-BP and desulfurized C-BP supports, the latter of which was prepared through desulfurization in H2 at either 773 K or 973 K. Note that the desulfurization treatment at 773 K and 973 K did not alter the specific surface area of the C-BP support. The lower S content was apt to decrease the conversion of EDA and NMe, the latter of which were reflected by the shift of the major products from N-methylated EDAs into N-formylated ones. Akin to the case of monometallic Re/C-BP, these data confirmed the importance of sulfurized species, whose characterization data are shown in Section 3.4, for the N-methylation of EDA.
| Entry | Catalyst | S BET [m2 g−1] | S/(Re + Ir)d | Conv. [%] | Yield [%] | N Me [%] | Bal. [%] | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N-Methylated EDA | N-Formylated EDA | Otherse | ||||||||||||||
| M-EDA | N,N-DM-EDA | N,N′-DM-EDA | TriM-EDA | TetraM-EDA | F-EDA | F-M-EDA | F-DM-EDA | F-TriM-EDA | ||||||||
| a Reaction conditions: EDA 4.0 mmol; Re–Ir/C-BP (Re 4 wt%, Ir/Re = 0.5, reduced in H2 at 773 K) 0.10 g; water 5.0 g; CO2 1.0 MPa + H2 4.0 MPa (r.t.); 413 K; 16 h. b Abbreviations: M-EDA = N-methylethylenediamine; N,N-DM-EDA = N,N-dimethylethylenediamine; N,N′-DM-EDA = N,N′-dimethylethylenediamine; TriM-EDA = N,N,N′-trimethylethylenediamine; TetraM-EDA = N,N,N′,N′-tetramethylethylenediamine; F-EDA = N-(2-aminoethyl)formamide; F-M-EDA = N-(2-aminoethyl)-N-methylformamide; F-DM-EDA = N-(2-(dimethylamino)ethyl)formamide; F-TriM-EDA = N-(2-(dimethylamino)ethyl)-N-methylformamide. c BET specific surface area. d Molar ratio of S to the sum of Re and Ir. e Including piperazine, 2-imidazolidinone, methylamine, and their corresponding N-methylated compounds. f Degree of N-methylation calculated using eqn (4). | ||||||||||||||||
| 1 | Re–Ir/C-BP | 1280 | 2.9 | 70 | 4.5 | 9.9 | 0.0 | 1.9 | 34 | 6.8 | 2.3 | 1.6 | 0.8 | 2.2 | 164 | 94 |
| 2 | Re–Ir/C-BP(deS-773) | 1260 | 0.6 | 60 | 5.0 | 11 | 0.0 | 1.1 | 17 | 13 | 3.1 | 2.2 | 0.7 | 0.8 | 97 | 95 |
| 3 | Re–Ir/C-BP(deS-973) | 1280 | 0.4 | 56 | 4.7 | 9.0 | 0.0 | 0.9 | 10 | 14 | 3.3 | 1.9 | 0.6 | 0.6 | 67 | 88 |
3.2. Optimization of reaction conditions with the Re–Ir/C-BP catalyst and its reusability in N-methylation of EDA
The effect of pressure of CO2 and H2 on the N-methylation of EDA was examined with the optimum catalyst of Re–Ir/C-BP (Re 4 wt%, Ir/Re molar ratio = 0.5). Initially, the ratio of pressure of CO2 to H2 (i.e., P(CO2)/P(H2)) was varied at their total pressure of 5.0 MPa (at room temperature), as summarized in Table S5.† Both the EDA conversion and NMe increased from the P(CO2)/P(H2) of 0.25 to 0.67 but got decreased upon the further increase of P(CO2)/P(H2) to 4.0. Such volcano-type dependence of reaction progress against P(CO2)/P(H2) could be connected to the involvement of equilibrium for the formation of possible intermediate adspecies derived from both CO2 and H2; for example, formate and formyl species were invoked as key intermediates in N-formylation/N-methylation of various amines.33–37 Although the optimum P(CO2)/P(H2) was 0.67 in terms of reaction progress, the excess dose of CO2 in catalytic systems rather decreases the efficiency of CO2 utilization. We, therefore, applied the P(CO2)/P(H2) of 0.25 in the following investigation. At the constant P(CO2)/P(H2) of 0.25, the total pressure was then altered in the range of 1.0–5.0 MPa (at room temperature). The EDA conversion and NMe both became higher at the higher total pressure. As discussed in Section 3.5, the reaction orders with respect to CO2 partial pressure and H2 partial pressure were 0.8 and 1.0, respectively; hence, the high-pressure conditions were apt to make the N-methylation of EDA faster.The reaction temperature varied from 373 K to 453 K with the Re–Ir/C-BP (Re 4 wt%, Ir/Re = 0.5) catalyst with the constant reaction time of 16 h to examine the effect of this parameter on the N-methylation of EDA, and the reaction data are shown in Fig. 3, Tables S6 and S7.† At 373 K, the dominant products were N-formylated EDAs. The higher reaction temperatures ranging from 393 K to 413 K gradually increased the conversion of EDA, yields of partial M-EDAs and TetraM-EDA, and NMe. Meanwhile, the even higher temperatures of 433 K and 453 K readily triggered undesired side reactions, and the detailed information about byproducts is listed in Table S7,† where the detected byproducts can be categorized into two groups. The first group consists of N,N,N′,N′′,N′′-pentamethyldiethylenetriamine, N-methylpiperazine, and N,N′-dimethylpiperazine along with methylamine, dimethylamine, and trimethylamine; these compounds could be produced via intermolecular condensation between two N-methylated EDA molecules and subsequent cyclization via the nucleophilic attack of the highly basic N-methylated amino moiety. The control experiment at 433 K and 453 K without the catalyst under CO2 1.0 MPa and H2 4.0 MPa (at room temperature) demonstrated that the formation of piperazine proceeded even without the catalyst. In contrast, the N-methylated compounds were not detected at all (Table S7†); therefore, even for the generation of N-methylated byproducts, the Re–Ir/C-BP catalyst was thus necessary. Another group includes 2-imidazolidinone, which was formed via non-reductive cyclization between EDA and CO2.68–72 Our control experiment at 433 K and 453 K posited that its formation proceeded regardless of the catalyst (Table S7†). In fact, the synthesis of 2-imidazolidinone from EDA and highly pressurized CO2 was previously demonstrated to proceed well at high temperatures in the absence of a catalyst.72–74 Consequently, too high reaction temperatures were unsuitable for the N-methylation of EDA into TetraM-EDA.
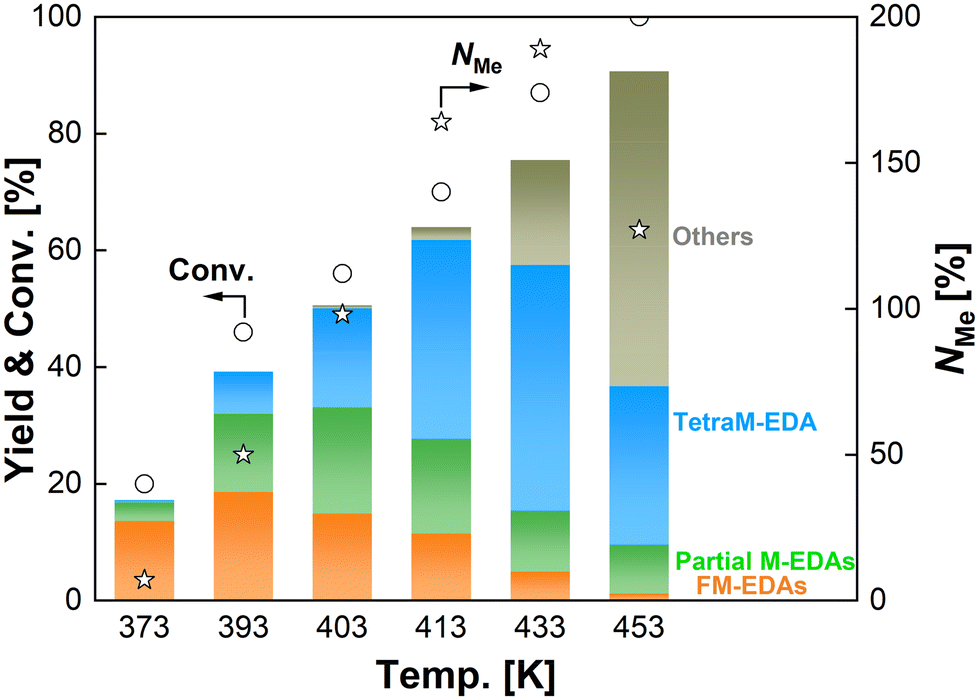 | ||
| Fig. 3 N-Methylation of EDA over Re–Ir/C-BP catalysts at different reaction temperatures. Reaction conditions: EDA 4.0 mmol; Re–Ir/C-BP (Re 4 wt%, Ir/Re = 0.5, reduced in H2 at 773 K) 0.10 g; water 5.0 g; CO2 1.0 MPa + H2 4.0 MPa (r.t.); 393–453 K; 16 h. In this figure, “Partial M-EDAs” consist of N-methylated EDAs possessing one to three methyl groups, and “FM-EDAs” are composed of N-formylated EDA and N-formylated partial M-EDAs. “Others” include piperazine, 2-imidazolidinone, methylamine, and their corresponding N-methylated compounds. The detailed data are summarized in Tables S6 and S7.† | ||
The time-course data were then acquired with the Re–Ir/C-BP (Re 4 wt%, Ir/Re = 0.5) catalyst at three reaction temperatures of 393 K, 403 K, and 413 K, the latter two of which are displayed in Fig. 4 (the detailed data including 393 K are listed in Tables S8–S10†). At the beginning of the reaction within 4 h at 403 K, the main product was N-(2-aminoethyl)formamide, followed by M-EDA as the secondary product (Table S8†). The extension of reaction time gradually shifted the major products from N-formylated EDAs (i.e., FM-EDAs in Fig. 4A) into N-methylated EDAs possessing one to four methyl groups (i.e., partial M-EDAs and TetraM-EDA in Fig. 4A). The observed trend was consistent with our previous report on the Re/C-BP catalyst,47 confirming that N-formylated EDAs were intermediates in the quadruple N-methylation of EDA into TetraM-EDA. After 120 h, the yield of TetraM-EDA reached 82%. Neither M-EDA nor N,N′-DM-EDA was detected at this reaction time, whereas N,N-DM-EDA and TriM-EDA were still observed since the further N-methylation of the latter two compounds was much slower than that of the former, as determined previously.47 The other detected byproducts were piperazine, methylamine, 2-imidazolidinone, and their corresponding N-methylated compounds with their total yield of 12%. The reaction conducted at 413 K reached a similarly high TetraM-EDA yield of 79% within a shorter reaction time of 96 h (Fig. 4B), compared to the case of 403 K, yet a slightly higher total yield of byproducts (18%) was observed. From the initial slope of this time course at 413 K, the TOF for introducing N-methyl groups based on the total mole of Re and Ir was calculated to be 21 h−1, which was 6.4-fold higher than the 3.3 h−1 for the previously reported Re/C-BP catalyst (Table S1†).47 Meanwhile, a high TetraM-EDA yield of 82% with the slight suppression of byproduct formation (9.7%) was achieved at 393 K, although a long reaction time of 120 h and three times higher amount of Re–Ir/C-BP (Re 4 wt%, Ir/Re = 0.5) catalyst relative to the cases of 403 K and 413 K were required (Table S10†). Due to the desire to shorten the reaction time as much as possible, the N-methylation of EDA in the following study was carried out at 413 K.
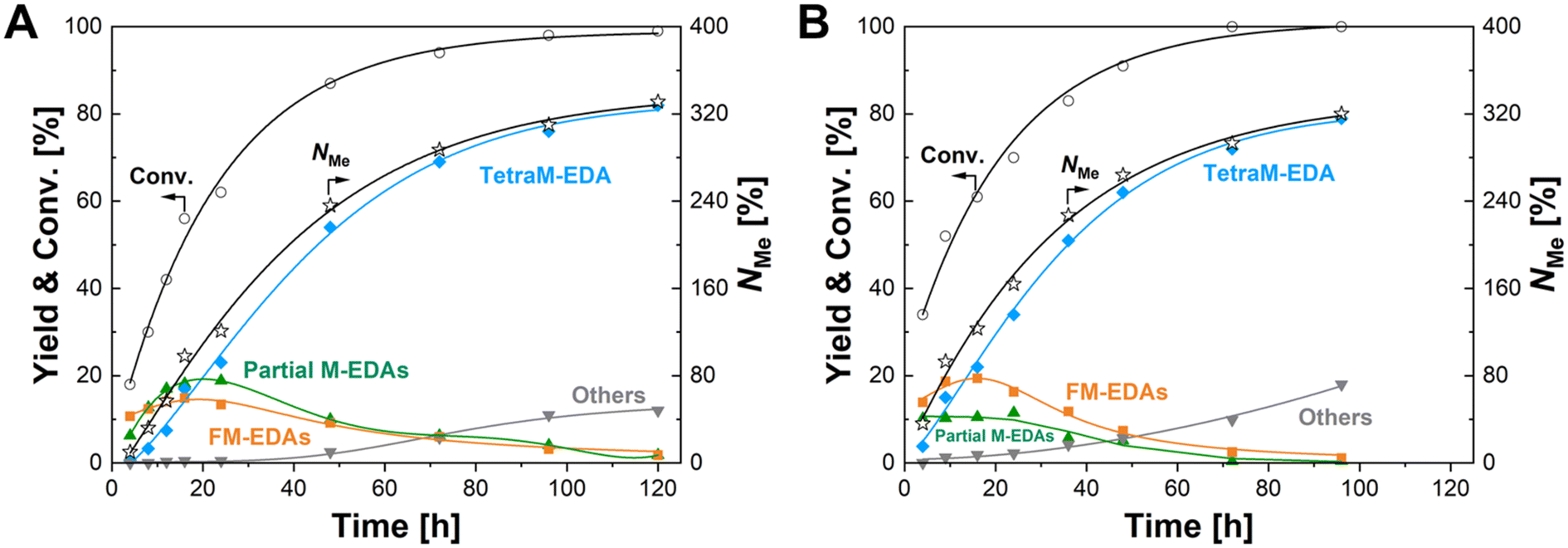 | ||
| Fig. 4 Time courses of N-methylation of EDA over the Re–Ir/C-BP catalyst at (A) 403 K and (B) 413 K. Reaction conditions: EDA 4.0 mmol; Re–Ir/C-BP (Re 4 wt%, Ir/Re = 0.5, reduced in H2 at 773 K) 0.10 g; water 5.0 g; CO2 1.0 MPa + H2 4.0 MPa (r.t.); 413 K; 4–120 h. In this figure, “Partial M-EDAs” consist of N-methylated EDAs possessing one to three methyl groups, and “FM-EDAs” are composed of N-formylated EDA and N-formylated partial M-EDAs. “Others” include piperazine, 2-imidazolidinone, methylamine, and their corresponding N-methylated compounds. The detailed data are summarized in Tables S8 and S9.† | ||
The reusability of the Re–Ir/C-BP (Re 4 wt%, Ir/Re = 0.5) catalyst was examined with the careful selection of reaction conditions to prevent both EDA conversion and NMe from reaching excessively high levels, thereby avoiding overestimation of its reusability.75 Besides, in this study, the spent catalyst was used for the next run after washing it with ethanol and water under air and reduction in H2 (see Section 2.3). The results of the reuse test are summarized in Fig. 5 and Table S11.† The Re–Ir/C-BP (Re 4 wt%, Ir/Re = 0.5) catalyst maintained its original activity for at least four reuses. In each run, the leaching of both Re and Ir was negligible (<0.1% against the amount of loaded metals, quantified by ICP-OES; Table S11†). This behavior is in stark contrast to the case of our previously reported monometallic Re/C-BP catalyst.47 The Re species in this monometallic catalyst were readily oxidized due to its high oxophilicity and >20% of Re was leached from the catalyst surfaces into water when being handled in air, leading to a loss of activity in its repeated use; therefore, this catalyst needed to be handled without exposure to air for its repeated usage. This difference between the bimetallic and monometallic catalysts confirmed that the modification of Re/C-BP with Ir not only enhanced the catalytic activity but also improved the stability against air. In addition to negligible leaching of Re and Ir, no observable aggregation of these metal species in the XRD patterns (Fig. S4†) also evidenced the good stability of the Re–Ir/C-BP catalyst in the N-methylation of EDA.
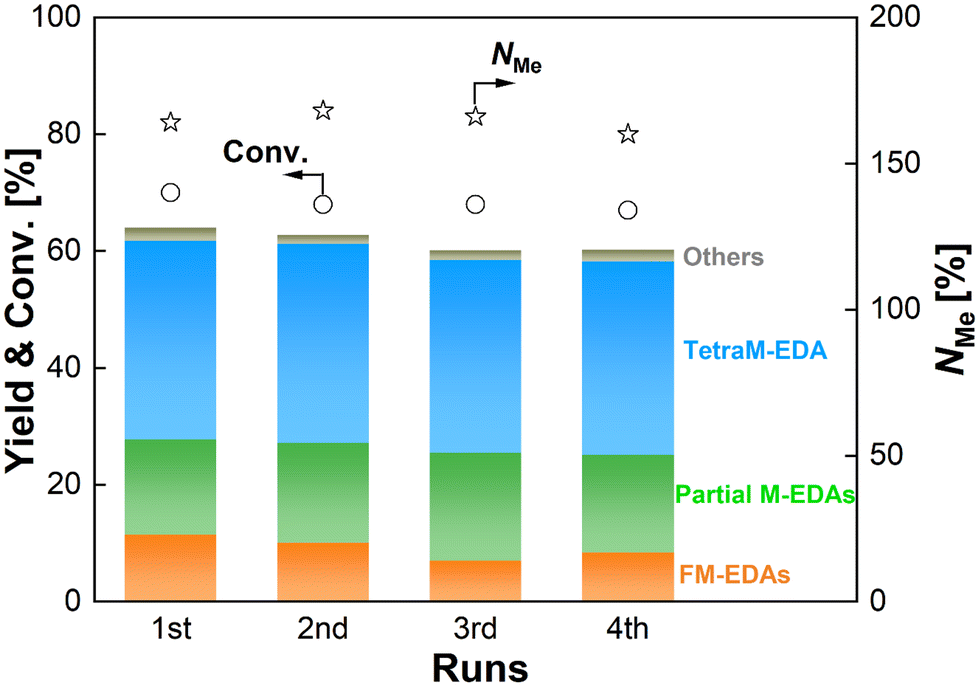 | ||
| Fig. 5 Reuse test of the Re–Ir/C-BP catalyst in the N-methylation of EDA. Reaction conditions: EDA 4.0 mmol; Re–Ir/C-BP (Re 4 wt%, Ir/Re = 0.5, reduced in H2 at 773 K) 0.10 g; water 5.0 g; CO2 1.0 MPa + H2 4.0 MPa (r.t.); 413 K; 16 h. Reuse procedure: After the reaction, the spent catalyst was separated by centrifugation and washed under air, followed by drying and reduction at 773 K in H2 for the next run. In this figure, “Partial M-EDAs” consist of N-methylated EDAs possessing one to three methyl groups, and “FM-EDAs” are composed of N-formylated EDA and N-formylated partial M-EDAs. “Others” include piperazine, 2-imidazolidinone, methylamine, and their corresponding N-methylated compounds. The detailed data are summarized in Table S11.† | ||
3.3. Substrate scope of the Re–Ir/C-BP catalyst in N-methylation
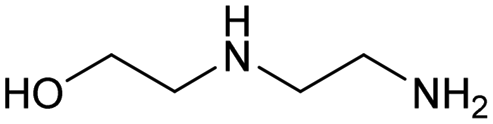
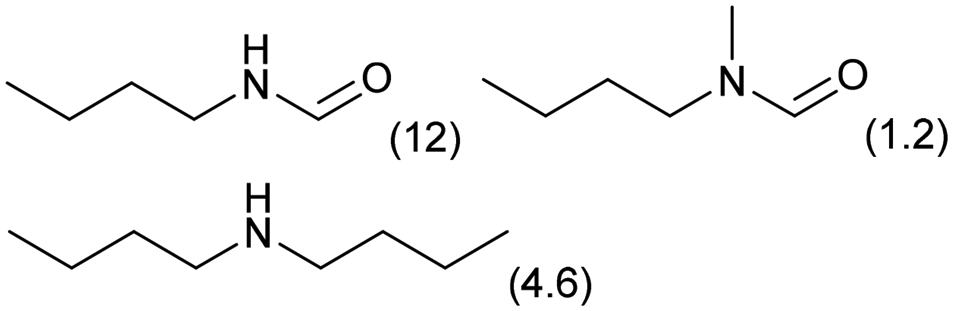
The applicability of the Re–Ir/C-BP (Re 4 wt%, Ir/Re = 0.5) catalyst was explored with a variety of amines including aliphatic diamines, aliphatic monoamines, and aromatic amine (i.e., aniline), as summarized in Table 4. EDA underwent the quadruple N-methylation as already discussed above (entry 1), and the same reaction proceeded well even in the presence of the methyl group (entry 2). Even though the methylene backbone between two amino groups was extended from two to three, the substrate successfully underwent N-methylation four times (entry 3). In stark contrast, the substrates with the methylene backbone between two amino groups consisting of four and six carbon atoms were hardly N-methylated, and only mono-N-methylated (and di-N-methylated) product(s) was detected (entries 4 and 5); instead, the major product was a mono-N-formylated compound. These data suggested the presence of an aliphatic C2 unit wedged between two amino groups in substrate molecules to be the prerequisite for their N-methylation over the Re–Ir/C-BP catalyst. Indeed, all the primary and secondary amino moieties in the triamines and tetramines in entries 6 and 7 were successfully N-methylated, respectively. Even in the presence of a hydroxyethyl group at one of the terminal amino groups, the substrate in entry 8 underwent triple N-methylation to yield the target product. These reaction results thus posited the aforementioned prerequisite for N-methylation over Re–Ir/C-BP. The aliphatic C2 (or C3) unit wedged between two amino groups is well-known to work as a chelating agent,76,77 while diamines consisting of longer methylene chains (≥C4) do not exhibit chelating effect.76 The strong interaction between diamines and catalytically active sites derived from chelating effect is thus assumed to be the key for the progress of N-methylation by Re–Ir/C-BP. Given that CO2 and H2 also need to be adsorbed and activated on the catalyst surface for the reaction progress, such strong interaction is assumed to be beneficial for the preferential adsorption and activation of the diamines with a C2 or C3 spacer over CO2 and H2. Indeed, the reaction order with respect to EDA concentration was determined to be zero, indicating the saturation of catalyst surfaces with EDA molecules, while those for CO2 and H2 pressure were positive (see Section 3.5). Due to a lack of such structure, the N-methylation of both aliphatic and aromatic mono-amines did not proceed well (entries 9–12). In an n-octane solvent, which and similar alkane solvents were typically used for N-methylation of both aliphatic and aromatic amines,41–44 the N-methylation of aniline proceeded slightly better than that in water, but the yield of the target product (i.e., N,N-dimethylaniline, 1.6%) was much lower than those given by reported catalysts (Table S1†).38–44 Altogether, the Re–Ir/C-BP (Re 4 wt%, Ir/Re = 0.5) catalyst exhibited an activity specific for aliphatic amines possessing a C2 (or C3) unit sandwiched between two amino groups.| Entry | Substrate | Catalyst [g] | Temp. [K] | Time [h] | Conv. [%] | Target product(s) (yield [%]) | Major side product(s) ([mmol]) |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: substrate 4.0 mmol for entry 1 or 2.0 mmol for entries 2–12; Re–Ir/C-BP (Re 4 wt%, Ir/Re = 0.5, reduced in H2 at 773 K); water 5.0 g; CO2 1.0 MPa + H2 4.0 MPa (r.t.). b n-Octane (5.0 g) was used as a solvent instead of H2O. | |||||||
| 1 |

|
0.1 | 403 | 120 | 100 |
|

|
| 2 |
|
0.3 | 393 | 192 | 100 |

|
|
| 3 |
|
0.3 | 393 | 192 | 100 |

|

|
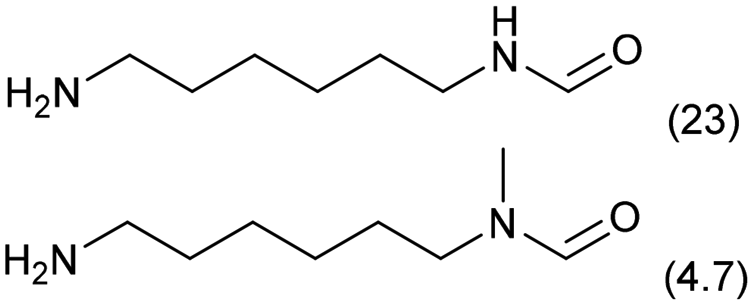
| 4 |
|
0.3 | 413 | 48 | 68 |
|
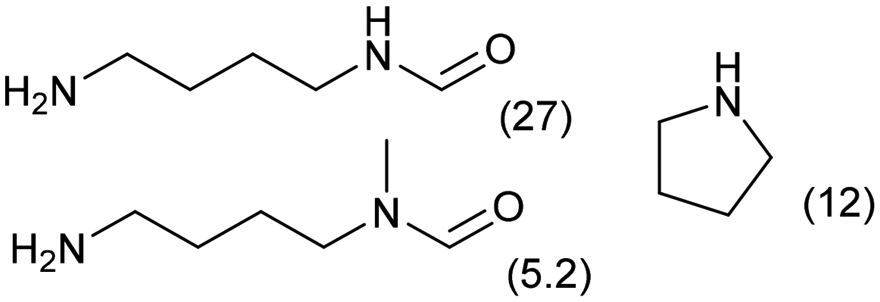
|
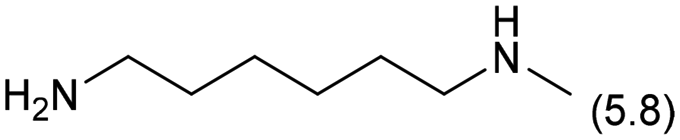
| 5 |

|
0.3 | 413 | 48 | 58 |
|
|
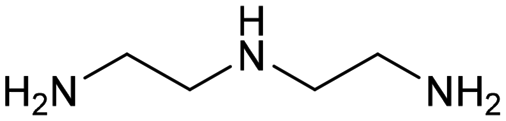
| 6 |
|
0.3 | 383 | 168 | 100 |
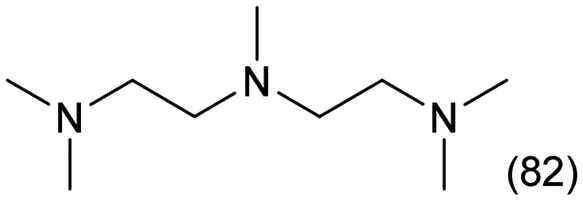
|
|
| 7 |

|
0.3 | 383 | 168 | 100 |
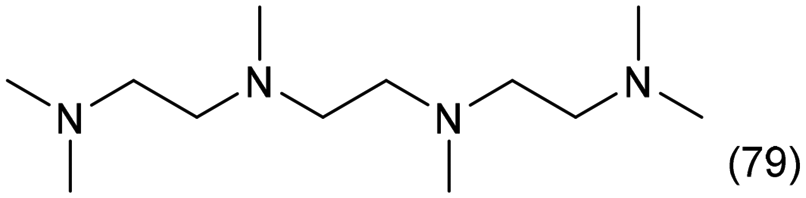
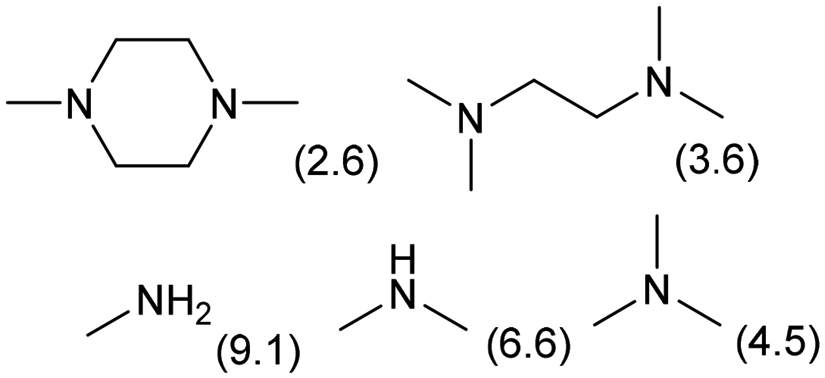
|
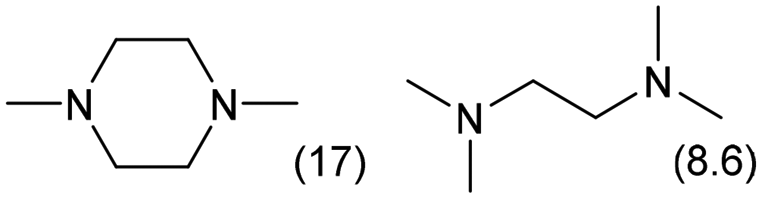
|
| 8 |
|
0.3 | 393 | 120 | 100 |
|
|
| 9 |
|
0.1 | 413 | 36 | 8.5 | — |
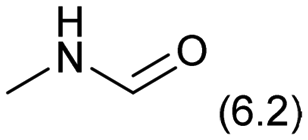
|
| 10 |
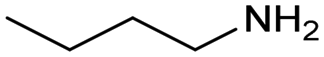
|
0.1 | 413 | 24 | 20 |
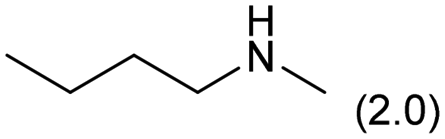
|
|
| 11 |

|
0.1 | 413 | 24 | 3.2 |
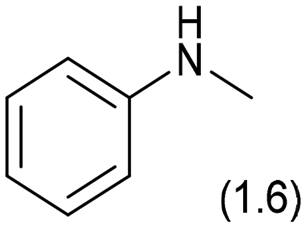
|
— |
| 12b |
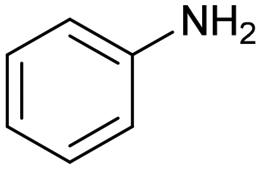
|
0.1 | 413 | 48 | 12 |
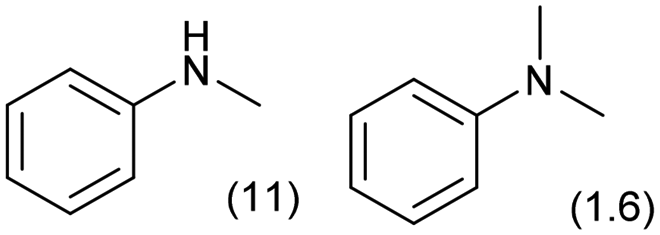
|
— |
3.4. Catalyst characterization
The Re–Ir/C-BP catalysts with different Ir/Re molar ratios were characterized by various techniques in order to reveal the active species for the N-methylation of EDA and its homologous series. Fig. 6 shows the HAADF-STEM images with EDS elemental mapping for the Re–Ir/C-BP catalysts and monometallic catalysts of Re/C-BP and Ir/C-BP after the reduction in H2 at 773 K and subsequent passivation. In all cases, the small and round-shaped particles were highly and uniformly dispersed on the C-BP support, which is consistent with our previously developed catalyst (5 wt% Re/C-BP).47 The distribution of particle size was shifted ever so slightly toward the larger particle side upon the increase in the Ir/Re ratio from 0.25 to 1.0 possibly because a higher total amount of Re and Ir species on the surfaces led to particle growth. Yet, the average particle size was still in the narrow range from 0.7 nm to 1.2 nm. The Re and Ir species seemed to be in close proximity to each other. Also, the HAADF-STEM image with EDS mapping for the optimum catalyst, Re–Ir/C-BP (Re 4 wt%, Ir/Re = 0.5), indicated that S atoms were homogeneously distributed in the entire catalyst, and their location was partially the same as those of Re and Ir (Fig. S5†), implying the formation of sulfurized species (vide infra). In the XRD patterns in Fig. S4,† no observation of Re- or Ir-derived peaks indicated the high dispersion of Re and Ir species, and this indication agreed well with the results of HAADF-STEM measurement. Another indication from the XRD patterns was no observable change of particle size of supported species due to a lack of Re- and Ir-derived peaks even after the four reuses. The preservation of particle size should be connected to the good reusability of the Re–Ir/C-BP catalyst (see Section 3.2).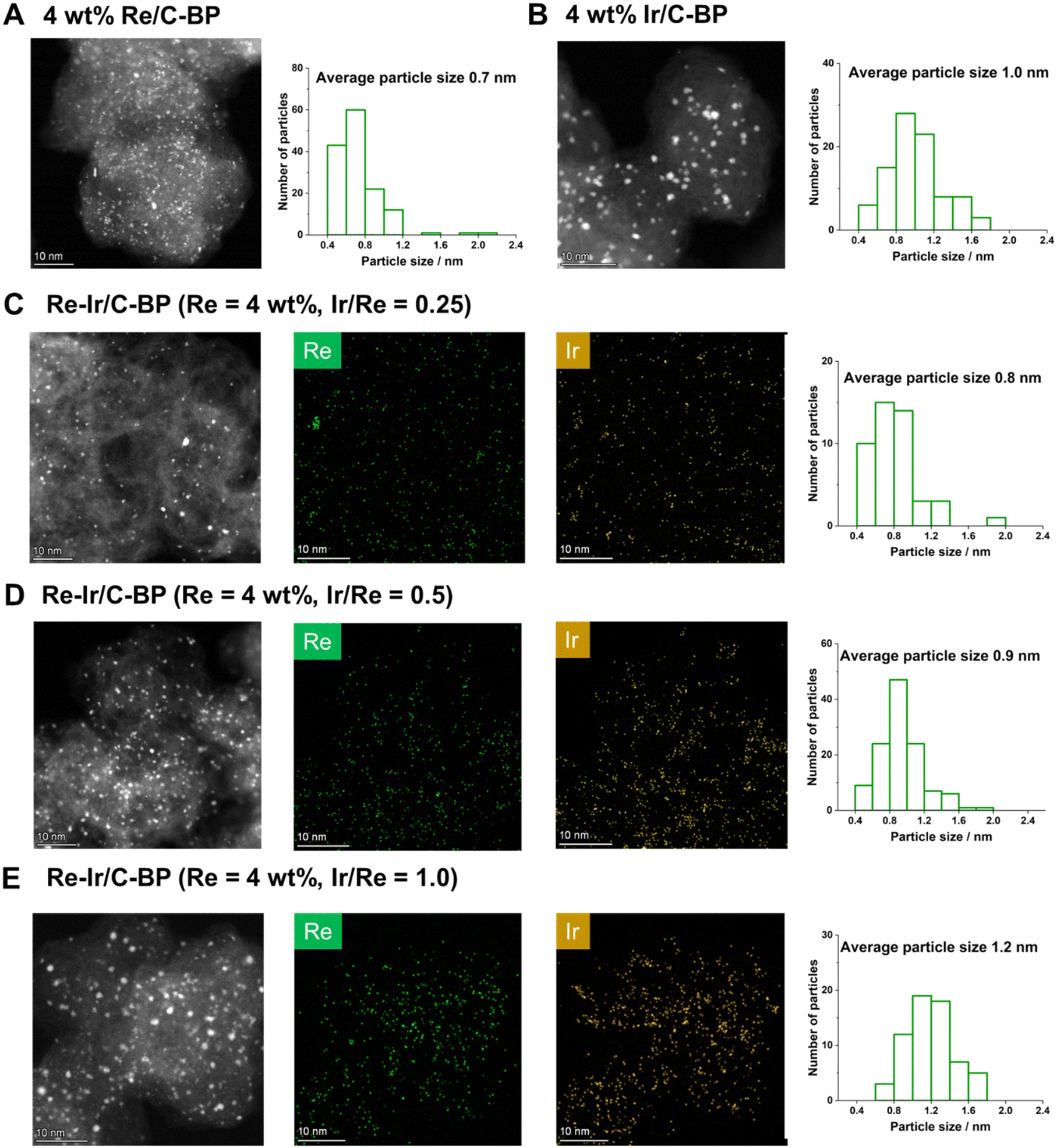 | ||
| Fig. 6 HAADF-STEM images and elemental mapping of catalysts after reduction (H2, 773 K) + passivation: (A) Re/C-BP (Re 4 wt%); (B) Ir/C-BP (Ir 4 wt%); (C) Re–Ir/C-BP (Re 4 wt%, Ir/Re = 0.25); (D) Re–Ir/C-BP (Re 4 wt%, Ir/Re = 0.5); (E) Re–Ir/C-BP (Re 4 wt%, Ir/Re = 1.0). | ||
The reducibility of Re and Ir species on C-BP was elucidated via H2-TPR measurement for C-BP impregnated with their precursors (Fig. S6 and Table S12†). In the H2-TPR profile for the monometallic Re species over C-BP, the main reduction peak appeared at 610 K. This observed profile was similar to that for previously reported 5 wt% Re/C-BP.47 The reduction temperature of Re species became lower at the higher Ir loading, suggesting that Ir helped the reduction of Re. Since all the catalysts tested in this study were reduced in H2 at 773 K, the reduction of supported Re and Ir species was expected to be completed. However, the estimation of oxidation states of Re and Ir from the H2-TPR profiles was unsuitable for catalysts with carbonaceous supports because of the possible interference from the reduction of the precursor by NH3 and/or functional groups of C-BP as well as generation of COx from C-BP via thermal degradation of oxygenated functionalities.78 To examine whether the top surfaces of Re and Ir particles worked as metallic sites, the CO uptake was measured after the reduction of each catalyst in H2 at 773 K, as listed in Table S13.† The CO uptake for 4 wt% Re/C-BP was determined to be 8.4 mmol g−1, and this value corresponded to the CO/Re molar ratio of 0.039 in spite of the quite small average particle size (i.e., 0.7 nm, vide supra). This contradiction indicated that a part of Re species on C-BP was oxidized; indeed, the Re species on C-BP bound to S as characterized previously.47 The molar ratio of CO/(Re + Ir) gradually increased upon the increase in the Ir/Re ratio from 0 to 0.5, while the further increase of Ir/Re ratio to 0.75 and 1.0 did not change the molar ratio of CO/(Re + Ir). Given that this observed trend between Ir/Re and CO/(Re + Ir) matched well with the dependence of catalytic activity for quadruple N-methylation of EDA on Ir/Re (Fig. 2), the enrichment of metallic sites that were able to interact with CO could be beneficial even for interacting with EDA and its homologous series. Such better interaction between Re–Ir/C-BP and EDA was indeed observed in the kinetic study (vide infra).
The series of Re–Ir/C-BP catalysts were further characterized by XAS after either reduction in H2 at 773 K or reaction. The values of average Re valence in the catalysts were determined from the white line intensity of the Re L3-edge XANES spectra in Fig. S7† and are summarized in Table 5. For the catalysts with the Ir/Re ratios between 0 and 0.5, the average valence of Re was in the narrow range from +2.1 to +2.4 (entries 6–8), which was also close to +2.1 for previously developed 5 wt% Re/C-BP.42 In the curve fitting analysis of the EXAFS data (Table 5 and Fig. S8†), the combination of three shells consisting of Re![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (or Re–O), Re–S, and Re–Re (or Re–Ir) fitted well. It should be noted that the employment of three shells for EXAFS analysis in this study is acceptable due to the satisfaction of the upper limit for the number of independent fitting parameters (Nind), which can be calculated from the equation of Nind ∼ 2ΔkΔR + 1 (Δk is the k range for Fourier transform and ΔR is the Fourier filtering range);79Nind in this study was calculated to be ∼12.5 (Δk = 90 nm−1 and ΔR = 0.2 nm; see Table 5). Due to the higher coordination numbers (CNs) for the latter two bonds than the former one, Re(–Ir)–Sx species were the major part in the catalysts. Indeed, the Re–Ir catalyst with the desulfurized support made the contribution of the Re–S shell minor (entry 11). Considering the higher catalytic activity of Re–Ir/C-BP compared to Re–Ir/C-BP(deS-973) (see Section 3.1), the presence of Re(–Ir)–Sx species was of great importance for the N-methylation of EDA. In the Re–Ir/C-BP catalyst with the Ir/Re molar ratio of 1.0, the Re valence was examined from the XANES spectrum to be +0.7 (entry 10), and its EXAFS data was fitted well using two shells Re–S and Re–Re (or Re–Ir). The high Ir content was assumed to enable the deeper reduction of Re species during the treatment in H2 at 773 K, which agreed well with the highest reducibility of this catalyst among all the Re–Ir/C-BP catalysts analyzed in H2-TPR (vide supra). Even in the case of Ir/Re of 0.5 (entry 9), the valence state of Re in the spent catalyst was +0.5, which was lower than that of the fresh catalyst (entry 8). This behavior was rationalized by the reduction of Re species during the N-methylation of EDA in the presence of H2 and was in contrast to the almost unchanged valence of Re species in 5 wt% Re/C-BP catalyst (+2.1 and +1.9 for the fresh and spent catalysts, respectively).47 This difference between the bimetallic and monometallic catalysts indicated that the co-presence of Ir species triggered the deeper reduction of Re species. Given that the Re–Ir/C-BP catalyst was reusable (see Section 3.2), the alteration of Re valence was not an issue for the catalytic activity, and rather the reduced species with sulfurized ones was suggested to be responsible for the higher activity of Re–Ir/C-BP than Re/C-BP.
O (or Re–O), Re–S, and Re–Re (or Re–Ir) fitted well. It should be noted that the employment of three shells for EXAFS analysis in this study is acceptable due to the satisfaction of the upper limit for the number of independent fitting parameters (Nind), which can be calculated from the equation of Nind ∼ 2ΔkΔR + 1 (Δk is the k range for Fourier transform and ΔR is the Fourier filtering range);79Nind in this study was calculated to be ∼12.5 (Δk = 90 nm−1 and ΔR = 0.2 nm; see Table 5). Due to the higher coordination numbers (CNs) for the latter two bonds than the former one, Re(–Ir)–Sx species were the major part in the catalysts. Indeed, the Re–Ir catalyst with the desulfurized support made the contribution of the Re–S shell minor (entry 11). Considering the higher catalytic activity of Re–Ir/C-BP compared to Re–Ir/C-BP(deS-973) (see Section 3.1), the presence of Re(–Ir)–Sx species was of great importance for the N-methylation of EDA. In the Re–Ir/C-BP catalyst with the Ir/Re molar ratio of 1.0, the Re valence was examined from the XANES spectrum to be +0.7 (entry 10), and its EXAFS data was fitted well using two shells Re–S and Re–Re (or Re–Ir). The high Ir content was assumed to enable the deeper reduction of Re species during the treatment in H2 at 773 K, which agreed well with the highest reducibility of this catalyst among all the Re–Ir/C-BP catalysts analyzed in H2-TPR (vide supra). Even in the case of Ir/Re of 0.5 (entry 9), the valence state of Re in the spent catalyst was +0.5, which was lower than that of the fresh catalyst (entry 8). This behavior was rationalized by the reduction of Re species during the N-methylation of EDA in the presence of H2 and was in contrast to the almost unchanged valence of Re species in 5 wt% Re/C-BP catalyst (+2.1 and +1.9 for the fresh and spent catalysts, respectively).47 This difference between the bimetallic and monometallic catalysts indicated that the co-presence of Ir species triggered the deeper reduction of Re species. Given that the Re–Ir/C-BP catalyst was reusable (see Section 3.2), the alteration of Re valence was not an issue for the catalytic activity, and rather the reduced species with sulfurized ones was suggested to be responsible for the higher activity of Re–Ir/C-BP than Re/C-BP.
| Entry | Sample | XANES | EXAFS | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Valenceb | Shell(s) | CNc | R [10−1 nm] | σ [10−1 nm] | ΔE0f [eV] | R f [%] | FF rangeh [nm–nm] | Valencei | ||
|
a EXAFS data are shown in Fig. S8.† For the curve fitting analyses, the raw data were k3-weighted and Fourier-transformed in the k range of 30–120 nm−1.
b Determined from the white line area intensity of XANES spectra (Fig. S7†).
c Coordination number.
d Bond distance.
e Debye–Waller factor.
f Difference in the origin of photoelectron energy between the reference and the sample.
g Residual factor.
h Fourier filtering range.
i Determined by EXAFS results: valence = CNReO × 7/4 + (CNRe–O + CNRe–S) × 4/6.
j Reduced in H2 at 773 K.
k Reaction conditions: EDA 4.0 mmol; Re–Ir/C-BP (Re 4 wt%, Ir/Re = 0.5, reduced in H2 at 773 K) 0.10 g; water 5.0 g; CO2 1.0 MPa + H2 4.0 MPa (r.t.); 413 K; 16 h.
l Valence state based on chemical composition.
m The values in parentheses are the results of fitting for the experimental EXAFS data of ReS2 using Re–S and Re–Re shells.
|
||||||||||
| 1 | Re powder | 0l | Re–Re | 12 | 2.76 | — | — | — | — | |
| 2 | ReO2 | +4l | Re–O | 2 | 1.94 | — | — | — | — | |
| Re–O | 4 | 2.11 | — | — | — | — | ||||
| 3 | ReS2 | +4l | Re–S | 6 (5.9)m | 2.38 (2.37)m | (0.070)m | (10.0)m | (1.0)m | (0.122–0.322)m | (+3.9)m |
| Re–Re | 3 (3.0)m | 2.76 (2.81)m | (0.060)m | (3.8)m | ||||||
| 4 | ReO3 | +6l | Re–O | 6 | 1.87 | — | — | — | — | |
| 5 | NH4ReO4 | +7l | ReO |
4 | 1.73 | — | — | — | — | |
| 6 | Re/C-BP (Re 4 wt%)j | +2.4 | ReO |
0.7 | 1.73 | 0.060 | –5.0 | 1.7 | 0.122–0.322 | +3.1 |
| Re–S | 2.8 | 2.36 | 0.075 | 10.0 | ||||||
| Re–Re | 2.0 | 2.66 | 0.060 | –3.9 | ||||||
| 7 | Re–Ir/C-BP (Re 4 wt%, Ir/Re = 0.13)j | +2.2 | ReO |
0.6 | 1.70 | 0.060 | –5.2 | 1.6 | 0.122–0.322 | +3.0 |
| Re–S | 2.9 | 2.36 | 0.070 | 10.0 | ||||||
| Re–Re (or Re–Ir) | 2.7 | 2.66 | 0.060 | –2.5 | ||||||
| Re–Ir/C-BP (Re 4 wt%, Ir/Re = 0.25)j | +2.2 | ReO |
0.4 | 1.71 | 0.060 | –6.4 | 1.9 | 0.122–0.322 | +2.4 | |
| Re–S | 2.6 | 2.35 | 0.085 | 10.0 | ||||||
| Re–Re (or Re–Ir) | 3.6 | 2.66 | 0.075 | –7.8 | ||||||
| 8 | Re–Ir/C-BP (Re 4 wt%, Ir/Re = 0.5)j | +2.1 | Re–O | 0.8 | 2.10 | 0.060 | 10.0 | 0.5 | 0.122–0.322 | +2.3 |
| Re–S | 2.6 | 2.36 | 0.060 | –6.5 | ||||||
| Re–Re (or Re–Ir) | 3.8 | 2.66 | 0.060 | 10.0 | ||||||
| 9 | Re–Ir/C-BP (Re 4 wt%, Ir/Re = 0.5, after reaction)k | +0.5 | Re–S | 2.3 | 2.36 | 0.060 | –7.1 | 0.7 | 0.122–0.322 | +1.6 |
| Re–Re (or Re–Ir) | 3.6 | 2.66 | 0.060 | 10.0 | ||||||
| 10 | Re–Ir/C-BP (Re 4 wt%, Ir/Re = 1.0)j | +0.7 | Re–S | 1.6 | 2.35 | 0.060 | –7.5 | 0.5 | 0.122–0.322 | +1.1 |
| Re–Re (or Re–Ir) | 4.9 | 2.66 | 0.060 | 10.0 | ||||||
| 11 | Re–Ir/C-BP(deS-973) (Re 4 wt%, Ir/Re = 0.5)j | +2.2 | Re–O | 1.5 | 2.11 | 0.060 | 9.8 | 0.7 | 0.122–0.322 | +1.6 |
| Re–S | 0.9 | 2.35 | 0.060 | –4.4 | ||||||
| Re–Re (or Re–Ir) | 7.2 | 2.67 | 0.098 | –7.6 | ||||||
The values of Ir average valence for the series of Re–Ir/C-BP catalysts were determined from the Ir L3-edge XANES spectra (Fig. S9†) in the same manner as the case of Re L3-edge XANES and are listed in Table 6. In the case of the monometallic catalyst 4 wt% Ir/C-BP, the average valence of Ir was +2.9, and this positive value originated from the formation of sulfurized species (i.e., IrSx), suggested by the curve fitting analysis of its EXAFS data (entry 3 in Table 6 and Fig. S10†). The presence of Re species diminished the average valence of Ir slightly to ca. +2 regardless of the Ir/Re ratios (entries 4–6), implying that the presence of Re led to the slightly deeper reduction of Ir species compared to the monometallic case. The curve fitting analysis of the EXAFS oscillations indicated that these positive values were mainly due to the presence of the Ir–S bond, while the major constituent was Ir–Ir, indicating that a part of Ir on these catalysts was sulfurized. The decrease in CN of the Ir–S bond was observed for the Re–Ir/C-BP (Ir/Re = 1.0) catalyst possibly because of the shortage of S originally present in C-BP against the total loading amount of Re and Ir (entry 8). The same reason was invoked for the decrease of CN of the Ir–S bond for the Re–Ir catalyst supported on the desulfurized C-BP (entry 9). In the case of the spent Re–Ir/C-BP (Ir/Re = 0.5) catalyst, the average valence of Ir did not change significantly, and the coordination environment was maintained (entry 7), which was connected to the good catalyst reusability.
| Entry | Sample | XANES | EXAFS | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Valenceb | Shell(s) | CNc | R [10−1 nm] | σ [10−1 nm] | ΔE0f [eV] | R f [%] | FF rangeh [nm–nm] | Valencei | ||
| a EXAFS data are shown in Fig. S10.† For the curve fitting analyses, the raw data were k3-weighted and Fourier-transformed in the k range of 30–120 nm−1. b Determined from the white line area intensity of XANES data (Fig. S9†). c Coordination number. d Bond distance. e Debye–Waller factor. f Difference in the origin of photoelectron energy between the reference and the sample. g Residual factor. h Fourier filtering range. i Determined by EXAFS results: valence = (CNIr–O + CNIr–S) × 4/6. j Reduced in H2 at 773 K. k Reaction conditions: EDA 4.0 mmol; Re–Ir/C-BP (Re 4 wt%, Ir/Re = 0.5, reduced in H2 at 773 K) 0.10 g; water 5.0 g; CO2 1.0 MPa + H2 4.0 MPa (r.t.); 413 K; 16 h. l Valence state based on chemical composition. | ||||||||||
| 1 | Ir powder | 0l | Ir–Ir | 12 | 2.76 | — | — | — | — | |
| 2 | IrO2 | +4l | Ir–O | 6 | 1.98 | — | — | — | — | |
| 3 | Ir/C-BP (Ir 4 wt%) | +2.9 | Ir–S | 1.5 | 2.28 | 0.060 | 10.0 | 0.1 | 0.141–0.346 | +1.0 |
| Ir–Ir | 7.2 | 2.73 | 0.093 | –5.1 | ||||||
| 4 | Re–Ir/C-BP (Re 4 wt%, Ir/Re = 0.13)j | +2.2 | Ir–O | 0.5 | 1.98 | 0.060 | 1.1 | 0.2 | 0.141–0.346 | +1.3 |
| Ir–S | 1.4 | 2.33 | 0.075 | 7.3 | ||||||
| Ir–Ir (or Ir–Re) | 6.5 | 2.77 | 0.100 | 0.6 | ||||||
| 5 | Re–Ir/C-BP (Re 4 wt%, Ir/Re = 0.25)j | +2.2 | Ir–O | 0.3 | 2.01 | 0.060 | –10.0 | 0.2 | 0.141–0.346 | +1.1 |
| Ir–S | 1.4 | 2.32 | 0.080 | 8.4 | ||||||
| Ir–Ir (or Ir–Re) | 9.1 | 2.75 | 0.100 | –1.6 | ||||||
| 6 | Re–Ir/C-BP (Re 4 wt%, Ir/Re = 0.5)j | +2.0 | Ir–S | 1.4 | 2.32 | 0.072 | 7.8 | 0.1 | 0.141–0.346 | +0.9 |
| Ir–Ir (or Ir–Re) | 8.9 | 2.71 | 0.100 | –7.5 | ||||||
| 7 | Re–Ir/C-BP (Re 4 wt%, Ir/Re = 0.5, after reaction)k | +2.6 | Ir–S | 1.3 | 2.34 | 0.063 | 10.0 | 0.8 | 0.141–0.346 | +0.9 |
| Ir–Ir (or Ir–Re) | 8.0 | 2.72 | 0.100 | –6.7 | ||||||
| 8 | Re–Ir/C-BP (Re 4 wt%, Ir/Re = 1.0)j | +1.8 | Ir–S | 0.8 | 2.27 | 0.073 | 2.6 | 0.2 | 0.141–0.346 | +0.5 |
| Ir–Ir (or Ir–Re) | 9.1 | 2.70 | 0.100 | –6.0 | ||||||
| 9 | Re–Ir/C-BP(deS-973) (Re 4 wt%, Ir/Re = 0.5)j | +1.7 | Ir–S | 0.8 | 2.27 | 0.060 | 9.9 | 0.3 | 0.141–0.346 | +0.5 |
| Ir–Ir (or Ir–Re) | 6.3 | 2.69 | 0.096 | –10.0 | ||||||
Altogether, the aforementioned characterization data demonstrated that in the optimal Re–Ir/C-BP (Re 4 wt%, Ir/Re = 0.5) catalyst, the particles consisting of Re and/or Ir with ca. 0.9 nm diameter were dispersed uniformly on the C-BP support. The Re species were more sulfurized, compared to the Ir species, yet not fully sulfurized; in other words, both metallic and sulfurized species were involved in these particles. Considering that S atoms were originally present in C-BP, the Re species were assumed to be in close proximity to the support surface and modified with the Ir species. The top surface of particles supported on C-BP was able to behave as metallic sites. Considering the reaction results in Tables 2 and 3, the steps of N-formylation and subsequent hydrogenation were assumed to be promoted mainly by the metallic and sulfurized Re species, respectively, while the Ir species behaved as a promotor for deep reduction of Re species.
3.5. Kinetic study on N-methylation of EDA over the Re–Ir/C-BP catalyst
Kinetic analysis of the N-methylation of EDA was conducted with the optimal catalyst, Re–Ir/C-BP (Re 4 wt%, Ir/Re = 0.5). The dependence of concentration of EDA, partial pressure of H2, and that of CO2 on the conversion rate of EDA was measured at 413 K, and the corresponding double logarithmic plots are drawn in Fig. 7 (the detailed reaction data are listed in Tables S14–S16†). The reaction order with respect to the EDA concentration was determined to be 0, indicating that the catalyst surfaces were saturated with EDA molecules. In contrast, a lack of Ir species on the catalyst surfaces (i.e., 5 wt% Re/C-BP) previously provided 0.5 of the reaction order with respect to the EDA concentration in the same reaction,47 suggesting the lower affinity of Re/C-BP for EDA compared to the Re–Ir/C-BP catalyst. As suggested by the CO chemisorption measurements (vide supra), the addition of Ir enriched the metallic sites, which could be favorable for interacting with the substrate molecules and result in the enhancement of catalytic activity. The reaction orders with respect to partial pressure of H2 and CO2 were evaluated to be 1.0 and 0.8, respectively, both of which were larger than the reported reaction orders over Re/C-BP (i.e., 0.5 for partial pressure of H2 and 0.6 for that of CO2).47 The increases of these reaction orders possibly arose from the competitive adsorption of EDA and gaseous compounds over the surfaces of the Re–Ir/C-BP catalyst. The key behavior of the Re–Ir/C-BP catalyst was thus to exhibit high affinity for EDA, as suggested from these kinetic results.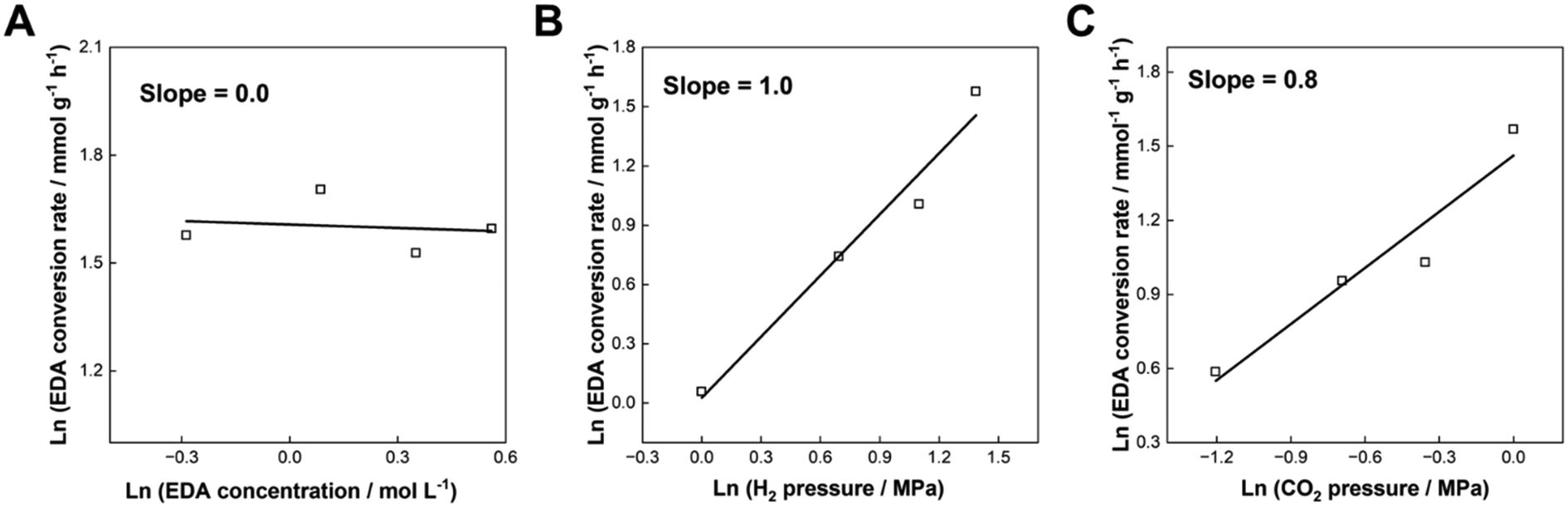 | ||
| Fig. 7 Double logarithmic plots of EDA conversion rate as a function of (A) concentration of EDA, (B) partial pressure of H2, and (C) partial pressure of CO2 in the N-methylation of EDA over Re–Ir/C-BP (Re 4 wt%, Ir/Re = 0.5). Reaction conditions: EDA 4.0–10.0 mmol for panel A or 4.0 mmol for panels B and C; Re–Ir/C-BP (Re 4 wt%, Ir/Re = 0.5, reduced in H2 at 773 K) 0.10 g; water 5.0 g; CO2 1.0 MPa + H2 4.0 MPa (r.t.) for panel A, CO2 1.0 MPa + H2 1.0–4.0 MPa + Ar (4-P(H2)) MPa (r.t.) for panel B, or CO2 0.3–1.0 MPa + H2 4.0 MPa + Ar (1-P(CO2)) MPa (r.t.) for panel A; 413 K; 0.5–6 h. The detailed data are summarized in Tables S14–S16.† | ||
4. Conclusions
Sulfurized Re–Ir bimetallic catalysts supported on carbon black (Re–Ir/C-BP) were developed as highly active and durable catalysts for the quadruple N-methylation of ethylenediamine (EDA) with CO2 and H2 as the sources of methyl groups. The optimum catalyst, Re–Ir/C-BP (Re 4 wt%, Ir/Re molar ratio = 0.5), exhibited 21 h−1 turnover frequency (TOF) for introducing N-methyl groups on the basis of the mole of Re and Ir, and this TOF was 6.4-fold higher compared to a previously developed sulfurized Re catalyst. The reaction operated under the optimized conditions afforded the target product of N,N,N′,N′-tetramethylethylenediamine (TetraM-EDA) in up to 82% yield. This catalyst was also effective for the multiple N-methylation of various aliphatic amines that possess the C2 (or C3) unit wedged between two amino groups. On the other hand, aliphatic monoamines, aniline, and diamines composed of ≥C4 methylene backbones wedged between two amino groups did not undergo N-methylation well. Such contrasting reactivity of substrates suggested that the strong interaction of the C2 (or C3) unit wedged between two amino groups (i.e., chelating effect) with the catalytically active sites was the prerequisite for the N-methylation over the Re–Ir/C-BP catalyst. The kinetic study on the N-methylation of EDA indeed indicated that the catalyst surfaces were readily saturated with the substrate molecules. The combination of various characterization techniques indicated the presence of highly dispersed sulfurized Re and Ir species in the same particles (ca. 0.9 nm), where the Re species were more sulfurized than the Ir species. The possible role of Ir was the deep reduction of Re species, which led to the suppression of Re leaching after the use of the Re–Ir/C-BP catalyst and its subsequent exposure to air. This feature of Re–Ir/C-BP offered better ease of handling compared to Re/C-BP, which lost >20% of Re species after the same treatment. The catalytic N-methylation system using CO2 and H2 operating in water could pave the way for a green approach to preparing N-methylated aliphatic amines.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was financially supported by a Grant-in-Aid for Specially Promoted Research and International Leading Research from the Japan Society for the Promotion of Science (JSPS KAKENHI; grant no. 23H05404 and 23K20034). M. W. acknowledges the financial support from the China Scholarship Council (CSC). We thank the Technical Division of School of Engineering, Tohoku University for their help with ICP-OES and XRF as well as Mr. Yuichiro Hayasaka (The Electron Microscopy Center, Tohoku University) for his support with STEM measurement.References
- D. B. Collum, Acc. Chem. Res., 1992, 25, 448–454 CrossRef CAS.
- A. Espinosa, M. Sohail, M. Habib, K. Naveed, M. Saleem, H. Rehman, I. Hussain, A. Munawar and S. Ahmad, Polyhedron, 2015, 90, 252–257 CrossRef CAS.
- I. Hamerton, B. J. Howlin and P. Jepson, Coord. Chem. Rev., 2002, 224, 67–85 CrossRef CAS.
- G. Li, F. Ma, J. Li, G. Chen, P. Yang, N. Yu, J. Chen, Y. Wang and X. Zhang, J. Dispersion Sci. Technol., 2024, 45, 1319–1327 CrossRef CAS.
- X. D. Feng, X. Q. Guo and K. Y. Qiu, Makromol. Chem., 1988, 189, 77–83 CrossRef CAS.
- K.-J. Yao and F.-H. Liu, J. Appl. Polym. Sci., 1995, 56, 9–15 CrossRef CAS.
- W.-J. Zhou, M. E. Wilson, M. J. Kurth, Y.-L. Hsieh, J. M. Krochta and C. F. Shoemaker, Macromolecules, 1997, 30, 7063–7068 CrossRef CAS.
- F. Liu, Z.-P. Ye, Y.-Z. Hu, J. Gao, L. Zheng, K. Chen, H.-Y. Xiang, X.-Q. Chen and H. Yang, J. Org. Chem., 2021, 86, 11905–11914 CrossRef CAS PubMed.
- Y. Wu, X. Kong, T. Yu, Z. Mai, S. Cao, Q. Yu, J. Liang, S. S. Nagaraja, S. M. Sarathy, Z. Huang and C. Tang, Combust. Flame, 2023, 248, 112584 CrossRef CAS.
- Q. Zhu, B. Liu, Z. Hu, S. Chen, Q. Xu and Z. Wang, Proc. Combust. Inst., 2024, 40, 105426 CrossRef CAS.
- Strategic Drivers and Barriers in Tetramethylethylenediamine (TEMED) Market 2025–2033, https://www.datainsightsmarket.com/reports/tetramethylethylenediamine-temed-1815447#, (accessed March 2025).
- K. Hammerstrom and G. Spielberger, US Pat, 4053516A, 1977 Search PubMed.
- W. Eschweiler, Ber. Dtsch. Chem. Ges., 1905, 38, 880–882 CrossRef.
- H. T. Clarke, H. B. Gillespie and S. Z. Weisshaus, J. Am. Chem. Soc., 1933, 55, 4571–4587 CrossRef CAS.
- D. Waldmüller, B. J. Kotsatos, M. A. Nichols and P. G. Williard, J. Am. Chem. Soc., 1997, 119, 5479–5480 CrossRef.
- C. Thomas, A. Milet, F. Peruch and B. Bibal, Polym. Chem., 2013, 4, 3491–3498 RSC.
- P. Paoletti, R. Barbucci, A. Vacca and A. Dei, J. Chem. Soc. A, 1971, 310–313 RSC.
- X. Wang and D.-S. Yang, J. Phys. Chem. A, 2006, 110, 7568–7576 CrossRef CAS PubMed.
- J.-M. Huang, L.-F. Xu, C. Qian and X.-Z. Chen, Chem. Pap., 2012, 66, 304–307 CAS.
- X. Ge, C. Luo, C. Qian, Z. Yu and X. Chen, RSC Adv., 2014, 4, 43195–43203 RSC.
- M. A. R. Jamil, A. S. Touchy, M. N. Rashed, K. W. Ting, S. M. A. H. Siddiki, T. Toyao, Z. Maeno and K. Shimizu, J. Catal., 2019, 371, 47–56 CrossRef CAS.
- Z. Lv, Z. Hong, C. Qian and S. Zhou, Catal. Sci. Technol., 2023, 13, 5058–5070 RSC.
- L. Zhang, Z.-J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2017, 56, 11326–11353 CrossRef CAS PubMed.
- W.-H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936–12973 CrossRef CAS PubMed.
- A. Modak, P. Bhanja, S. Dutta, B. Chowdhury and A. Bhaumik, Green Chem., 2020, 22, 4002–4033 RSC.
- X. Jiang, X. Nie, X. Guo, C. Song and J. G. Chen, Chem. Rev., 2020, 120, 7984–8034 CrossRef CAS PubMed.
- J. Hu, Y. Cai, J. Xie, D. Hou, L. Yu and D. Deng, Chem, 2024, 10, 1084–1117 CAS.
- K. Cheng, Y. Li, J. Kang, Q. Zhang and Y. Wang, Acc. Chem. Res., 2024, 57, 714–725 CrossRef CAS PubMed.
- N. Onishi and Y. Himeda, Acc. Chem. Res., 2024, 57, 2816–2825 CrossRef CAS PubMed.
- R. A. W. Johnstone and M. E. Rose, Tetrahedron, 1979, 35, 2169–2173 CrossRef CAS.
- H. Yang, B. Martin and B. Schenkel, Org. Process Res. Dev., 2018, 22, 446–456 CrossRef CAS.
- O. Jacquet, X. Frogneux, C. D. N. Gomes and T. Cantat, Chem. Sci., 2013, 4, 2127–2131 RSC.
- A. Tlili, E. Blondiaux, X. Frogneux and T. Cantat, Green Chem., 2015, 17, 157–168 Search PubMed.
- Y. Li, X. Cui, K. Dong, K. Junge and M. Beller, ACS Catal., 2017, 7, 1077–1086 CrossRef CAS.
- G. Naik, N. Sarki, V. Goyal, A. Narani and K. Natte, Asian J. Org. Chem., 2022, 11, e202200270 CrossRef CAS.
- I. Dutta, S. S. Gholap, M. M. Rahman, D. Tan, L. Zhang, S. U. Dighe and K.-W. Huang, Chem. – Asian J., 2024, 19, e202400497 CrossRef CAS PubMed.
- S. Lin, J. Liu and L. Ma, J. CO2 Util., 2024, 54, 101759 CrossRef.
- K. Beydoun, T. vom Stein, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2013, 52, 9554–9557 CrossRef CAS PubMed.
- Y. Li, I. Sorribes, T. Yan, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 12156–12160 CrossRef CAS PubMed.
- Z. Ke, Y. Zhao, R. Li, H. Wang, W. Zeng, M. Tang, B. Han and Z. Liu, Green Chem., 2021, 23, 9147–9153 RSC.
- X. Cui, X. Dai, Y. Zhang, Y. Deng and F. Shi, Chem. Sci., 2014, 5, 649–655 RSC.
- X. Cui, Y. Zhang, Y. Deng and F. Shi, Chem. Commun., 2014, 50, 13521–13524 RSC.
- X.-L. Du, G. Tang, H.-L. Bao, Z. Jiang, X.-H. Zhong, D. S. Su and J.-Q. Wang, ChemSusChem, 2015, 8, 3489–3496 CrossRef CAS PubMed.
- Z. Ran, J. Liu, M. A. Mushtaq, X. Shao, H. Liu, X. Du, S. Hou and S. Ji, Catal. Today, 2022, 405–406, 309–320 CrossRef CAS.
- P. G. Jessop, Green Chem., 2011, 13, 1391–1398 RSC.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC.
- M. Wang, D. Kanemaru, M. Yabushita, K. Okuma, T. Shono, Y. Nakagawa and K. Tomishige, ChemCatChem, 2025, 17, e202401803 CrossRef CAS.
- M. Sankar, N. Dimitratos, P. J. Miedziak, P. P. Wells, C. J. Kiely and G. J. Hutchings, Chem. Soc. Rev., 2012, 41, 8099–8139 RSC.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075–8098 RSC.
- K. Tomishige, Y. Nakagawa and M. Tamura, Green Chem., 2017, 19, 2876–2924 RSC.
- K. Tomishige, M. Yabushita, J. Cao and Y. Nakagawa, Green Chem., 2022, 24, 5652–5690 RSC.
- L. Liu and A. Corma, Chem. Rev., 2023, 123, 4855–4933 CrossRef CAS PubMed.
- L. Liu, B. Liu, Y. Nakagawa, S. Liu, L. Wang, M. Yabushita and K. Tomishige, Chin. J. Catal., 2024, 62, 1–31 CrossRef CAS.
- G. Zhang, Y. Du, Y. Zhang and Y. Xu, J. Ind. Eng. Chem., 2014, 20, 487–493 CrossRef CAS.
- M. Rønning, T. Gjervan, R. Prestvik, D. G. Nicholson and A. Holmen, J. Catal., 2001, 204, 292–304 CrossRef.
- Y. Ishida, T. Ebashi, S. Ito, T. Kubota, K. Kunimori and K. Tomishige, Chem. Commun., 2009, 5308–5310 RSC.
- Y. Takeda, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, ACS Catal., 2015, 5, 7034–7047 CrossRef CAS.
- S. Liu, Y. Okuyama, M. Tamura, Y. Nakagawa, A. Imai and K. Tomishige, Catal. Today, 2016, 269, 122–131 CrossRef CAS.
- L. Liu, T. Asano, Y. Nakagawa, M. Tamura, K. Okumura and K. Tomishige, ACS Catal., 2019, 9, 10913–10930 CrossRef CAS.
- M. Gu, L. Liu, Y. Nakagawa, C. Li, M. Tamura, Z. Shen, X. Zhou, Y. Zhang and K. Tomishige, ChemSusChem, 2021, 14, 642–654 CrossRef CAS PubMed.
- K. Yamaguchi, J. Cao, M. Betchaku, Y. Nakagawa, M. Tamura, A. Nakayama, M. Yabushita and K. Tomishige, ChemSusChem, 2022, 15, e202102663 CrossRef CAS PubMed.
- Y. Nakagawa, Y. Shinmi, S. Koso and K. Tomishige, J. Catal., 2010, 272, 191–194 CrossRef CAS.
- Y. Amada, Y. Shinmi, S. Koso, T. Kubota, Y. Nakagawa and K. Tomishige, Appl. Catal., B, 2011, 105, 117–127 CrossRef CAS.
- Y. Takeda, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, Catal. Sci. Technol., 2016, 6, 5668–5683 RSC.
- T. Wang, M. Tamura, Y. Nakagawa and K. Tomishige, ChemSusChem, 2019, 12, 3615–3626 CrossRef CAS PubMed.
- Y. Nakagawa, S. Tazawa, T. Wang, M. Tamura, N. Hiyoshi, K. Okumura and K. Tomishige, ACS Catal., 2018, 8, 584–595 CrossRef CAS.
- K. Yamaguchi, Y. Nakagawa, C. Li, M. Yabushita and K. Tomishige, ACS Catal., 2022, 12, 12582–12595 CrossRef CAS.
- M. Tamura, K. Noro, M. Honda, Y. Nakagawa and K. Tomishige, Green Chem., 2013, 15, 1567–1577 RSC.
- J. Peng, M. Yabushita, Y. Li, R. Fujii, M. Tamura, Y. Nakagawa and K. Tomishige, Appl. Catal., A, 2022, 643, 118747 CrossRef CAS.
- R. Fujii, M. Yabushita, D. Asada, M. Tamura, Y. Nakagawa, A. Takahashi, A. Nakayama and K. Tomishige, ACS Catal., 2023, 13, 1562–1573 CrossRef CAS.
- R. Fujii, M. Yabushita, Y. Li, Y. Nakagawa and K. Tomishige, ACS Catal., 2023, 13, 11041–11056 CrossRef CAS.
- M. Yabushita, R. Fujii, Y. Nakagawa and K. Tomishige, ChemCatChem, 2024, 16, e202301342 CrossRef CAS.
- B. M. Bhanage, S. Fujita, Y. Ikushima and M. Arai, Green Chem., 2003, 5, 340–342 RSC.
- C. Wu, H. Cheng, R. Liu, Q. Wang, Y. Hao, Y. Yu and F. Zhao, Green Chem., 2010, 12, 1811–1816 RSC.
- S. L. Scott, ACS Catal., 2018, 8, 8597–8599 CrossRef CAS.
- Y. Hojo, Y. Sugiura and H. Tanaka, J. Inorg. Nucl. Chem., 1977, 39, 715–720 CrossRef CAS.
- R. L. Fanshawe, A. Mobinikhaledi, C. R. Clark and A. G. Blackman, Inorg. Chim. Acta, 2000, 307, 27–32 CrossRef.
- J. L. Figueiredo, M. F. R. Pereira, M. M. A. Freitas and J. J. M. Órfão, Carbon, 1999, 37, 1379–1389 CrossRef CAS.
- E. A. Stern, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 9825–9827 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5gc01522g |
| This journal is © The Royal Society of Chemistry 2025 |