
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into the role of transition and noble metals mediating photochemical vapor generation†
Ralph E.
Sturgeon
 *a,
Enea
Pagliano
a,
Gisele S.
Lopes
b,
Renato S. A.
Neto
b and
Jane K. S.
Brito
b
*a,
Enea
Pagliano
a,
Gisele S.
Lopes
b,
Renato S. A.
Neto
b and
Jane K. S.
Brito
b
aMetrology Research Center, National Research Council Canada, Inorganic Chemistry, Ottawa, Canada K1A 0R6. E-mail: Ralph.sturgeon@nrc-cnrc.gc.ca
bDepartamento de Química Analítica e Físico-Química, Centro de Ciencias, Campus do Pici, Universidade Federal do Ceara, Fortaleza, CE, Brazil
First published on 30th October 2024
Abstract
The recent expansion of the suite of elements amenable to photochemical vapor generation (PVG) is primarily linked to the addition of mg L−1 concentrations of selected transition metals (TMs) to the photolysis medium, principally Fe, Cd, Co, Ni and Cu. Their presence enhances synthesis yields of several analytical targets, particularly carbonylated species, in some cases by orders of magnitude. A consideration of curated analytical PVG literature reveals substantial inconsistencies with the current use of generalized ligand-to-metal charge transfer processes to mechanistically account for these so-called TM “sensitizer” effects via their enhancement in free radical populations participating in the PVG synthesis routes. In this study, a novel approach utilizes an independent window for evaluation of the effects of added TMs on radical production based on an examination of the altered concentration profiles of H2, CO, CH4 and CO2 generated in formic and acetic acid media, whose origins lie with the precursor free radicals responsible for the analytical PVG process. A photocatalytic mechanism induced by homogeneous co-generation of TM nanoparticles is proposed which more reasonably accounts for both the altered gas profiles and their notable selectivity evident with improved PVG efficiencies of specific analytes. A tutorial approach to the topic has been adopted in an effort to provide a balanced framework within which the various processes are comprehensively discussed with relevance to state-of-the-art PVG techniques and current literature.
 Ralph E. Sturgeon | Ralph Sturgeon obtained his PhD is 1977 and has been with the National Research Council of Canada, Ottawa since that time and is currently an Emeritus Researcher. His interests lie in trace element analysis, vapor generation, organometallic speciation and production of Certified Reference Materials with a focus on atomic and mass spectrometric detection techniques. The fundamentals of vapor generation techniques currently occupy his attention. |
 Enea Pagliano | Enea Pagliano completed his PhD studies in analytical chemistry at Scuola Normale Superiore of Pisa and since 2013 has been a research officer at the National Research Council of Canada. Enea is a team member of the Joint Committee for Traceability in Laboratory Medicine, a member of the IUPAC Subcommittee on Metrology in Chemistry, and an adjunct research professor at Carleton University. His research has focused on the development of primary analytical methods for metrological applications. |
 Gisele S. Lopes | Gisele S. Lopes is a Full Professor at Universidade Federal do Ceara, Ceara, Brazil. She obtained her PhD in analytical chemistry in 2002 from Universidade Federal de São Carlos, Sao Paulo, Brazil. Her interest lies in analytical chemistry, with emphasis on metrology, trace element analysis, sample preparation and chemometrics. |
 Renato S. A. Neto | Renato S. A. Neto is currently a PhD student in analytical chemistry at Universidade Federal do Ceara, Ceara, Brazil. He obtained his Master's and Bachelor's degrees from this institution in 2019 and 2017, respectively. His current interest focuses on trace element analysis. |
 Jane K. S. Brito | Jane K. S. Brito is currently a PhD student in analytical chemistry at Universidade Federal do Ceara, Ceara, Brazil. She obtained her Master's degree in 2021 from Universidade Federal do Ceara and her Bachelor's from Universidade Federal do Piauí, Piauí, Brazil. Her current interest is primarily trace element analysis. |
Introduction
Vapor generation techniques coupled with analytical atomic and mass spectrometry have been in continuous use for more than 50 years.1 High synthesis yields, improved sample introduction efficiency, inherent matrix separation and enhanced detection power sustain continued interest in both their analytical application and mechanistic understanding. Amongst these, photochemical vapor generation (PVG) is one of the most recent and actively pursued, primarily because of its simplicity, green chemistry, widest scope of application and interest in acquiring a more complete fundamental comprehension of this novel phenomenon; it is currently amenable for analytical use with some 25 elements, including transition and semi-metals as well as the halogens.The premise of PVG rests on a mechanistic model that the targeted analytes are converted to volatile species during UV irradiation of a (aqueous) medium (typically) containing low molecular weight carboxylic acids (R-COOH; primarily formic and/or acetic, although ethanol has been utilized).2–5 Emission of UV-C from low pressure Hg discharge sources gives rise to both homolysis of water (induced via 185 nm absorption) and photolysis of the carboxylate anion, yielding both aquated electrons (e(aq)−) as well as a suite of powerful reducing radicals (H˙, R˙, and CO2˙−). These, and their subsequent thermal reaction products, interact with ionic analytes to ultimately form their volatile free atoms, hydrides, carbonyls or alkylated derivatives, depending on the element and photochemical medium used.
The deleterious presence of concomitant metal cations (and anions such as Cl− and NO3−) in real samples has long been recognized as a primary shortcoming of PVG reactions, leading to interferences often ascribed to radical scavenging effects.2–4 Nevertheless, it is evident that the most recent advances in PVG have all accrued from use of mg L−1 concentrations of select transition metal ions (TMs) deliberately added to reaction media to enhance the yield of many PVG reactions, as summarized in a recent report by Hou and colleagues.6 Such systems are more appealing (and currently more interesting) than resorting to addition of typically heterogeneous semiconductor (SC) catalysts, such as TiO2,7–10 or the use of unique metal organic frameworks (MOFs) which have also improved PVG performance for several elements.11 A summary of applications of SC catalysts in PVG systems has been presented by Zou et al.12 but, despite interest, they have enjoyed only limited use.
Gao et al.13 first highlighted the unique impact of added TMs, noting the addition of a 5 mg L−1 concentration of Ni(II) increased the earlier optimized yield of photogenerated lead species 4100-fold. This study was rapidly followed by a succession of publications from many laboratories, which repeatedly confirmed similar efficacious effects, particularly in systems targeting the generation of carbonylated transition metals. For lack of a more informed mechanism of action, the role of these added TMs was putatively designated to be a “sensitizer effect” in early publications. However, the only known occurrence of true sensitization with an organic compound being used for application with PVG has been the use of anthranilic acid as a homogeneous photosensitizer for PVG speciation of mercury.14 This term should thus be discontinued until the mechanistic aspects of TM mediated reactions are more clearly understood. Additions of Cd(II),15,16 Co(II),17–22 Cu(II),23–25 Fe(II/III),26–31 Ni(II)13 and, to a lesser extent, V(IV/V)32 have been evaluated for both their individual performance characteristics as well as unique synergistic effects arising from their various combinations.6,33–36 Of equal interest is the sole report of in situ formation of nano-CdSe, which promoted reduction of Se(VI) to volatile SeH2via irradiation of a sample augmented with 10 mg L−1 Cd(II);37 this finding deserves more extensive study.
Whereas the general role of added TMs functioning in such a “homogeneous” or dissolved manner (as opposed to use of typical SCs or MOFs) is not completely understood at this time, enhanced PVG synthesis of the methylhalogens (XCH3) from dilute acetic acid containing added TMs23,25,38 appears to rest on a well-established foundation and likely occurs via either ˙CH3 capture of X˙ from its ligand-to-metal charge transfer (LMCT) cage arising from photolysis of an aquated TM-X complex (for I− and Br−),39–41 or by oxidative addition-reductive elimination reactions between ˙CH3 and the TM-X complex (for Cl− and F−). The latter is the most likely mechanism for methylfluoride generation since current photochemical reactors are unable to generate an intermediate charge transfer to solvent (CTTS) cage complex due to source output being limited to >185 nm.38,42 These scenarios, leading to catalytic cycles, are illustrated in Fig. 1 for the case of a Cu(II)-X complex.
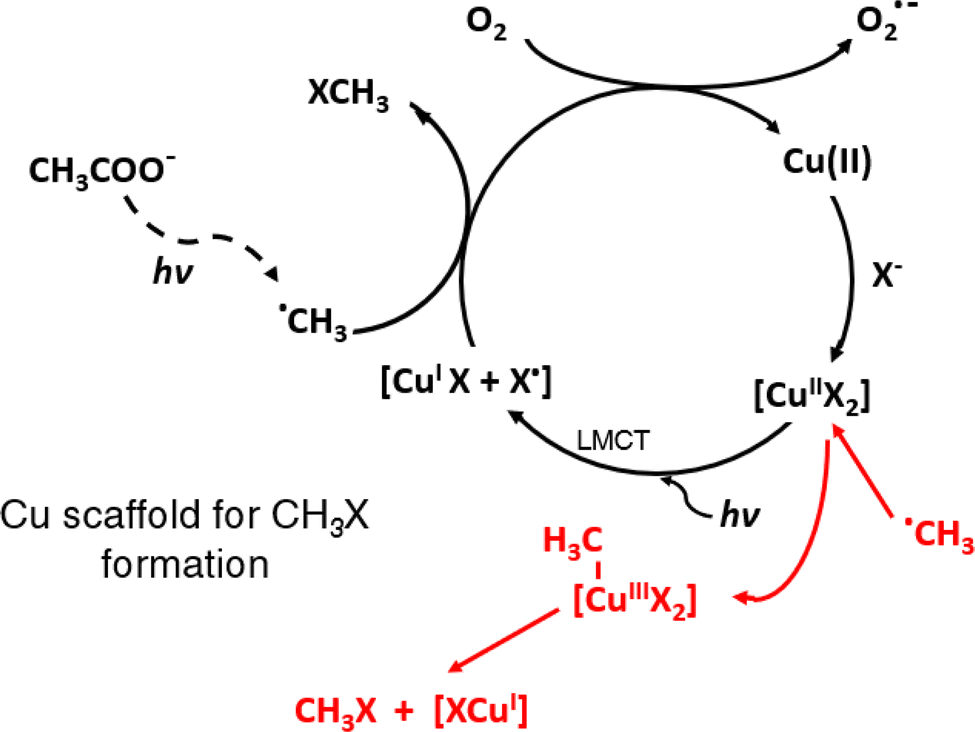 | ||
| Fig. 1 TM-assisted PVG of halogens using added Cu(II) as an example of aquo-metal halide complex formation followed by (i) ˙CH3 capture of halide atom (X˙) formed by a LMCT process or (ii, shown in red) oxidative addition of ˙CH3 to CuX2 complex followed by reductive elimination of CH3X and subsequent catalytic re-oxidation of Cu(I) by dissolved oxygen or other oxidants in the system. | ||
From the broader perspective of general application of TMs beyond the halogens, Sturgeon and colleagues initially proposed a framework for their action based on an augmented production of reducing radicals arising from the photolysis of TM–carboxylate complexes induced by (longer wavelength) LMCT processes.1,3,4 This, in principle, could enhance PVG reaction kinetics, leading to increased overall analyte generation efficiencies because a broader band of the emission output from the photochemical source could be utilized to produce additional needed radical partners. Such reactions are commonplace in the inorganic and environmental photochemical literature, occurring by exposure of aqueous solutions containing carboxylic acids to UV-B radiation, readily available from solar and low pressure mercury discharge sources.43,44 Additionally, direct evidence of such enhanced radical production in the presence of added TMs is supported by ESR spin trapping experiments which confirm elevated (but unquantified) levels of reducing radicals (CO2˙−, ˙CH3) with concurrently decreased concentrations of ˙OH in irradiated TM-doped PVG reaction media.6,20,32,45,46 These reports compliment similar such increases earlier noted in classical SC-mediated photochemical systems.47,48 As a consequence, the LMCT-based mechanism has proliferated, essentially unchallenged, and has become pervasive throughout more recent PVG literature.
The pronounced role of TMs mediating homogeneous PVG reactions likely stems from their d shell configurations which give rise to varied redox properties and multiple oxidation states to serve as scaffolds fostering interaction of reaction partners (cf.Fig. 1). However, an elementary qualitative meta-analysis of the common effects of added TMs published since the first report by Gao et al. in 2015 (ref. 13) readily reveals both trends and inconsistencies with this general LMCT model of action. Most evident is the overwhelming use of Cd, Cu, Fe, Ni and Co for this purpose. As noted above, these elements possess multiple oxidation states and can be readily reduced to their atomic form during PVG. Additionally, with the exception of the halogens, in every experimental system, an optimum concentration of the added TM is evident beyond which higher concentrations lead to reduced PVG yields (this is often ascribed to a “shadowing effect” arising from the filtering and consequent diminished effective intensity of the UV source by TM d–d transitions and charge transfer transitions of TM complexes). Noteworthy, however, is that apart from the halogens the optimal concentration of added TM varies significantly from one targeted analyte system to another, despite otherwise overall identical photoreactor conditions (i.e., same source, same irradiation conditions, ostensibly the same photochemical medium). PVG enhancement yields thus selectively vary considerably from one analyte to another, sometimes by factors of 10–103, despite similar (and often identical) TM concentrations being used, suggesting that there is a specific interaction between the analyte and the added TM. This is further reinforced by the observation that the relative effectiveness of a specific TM added to a given analyte system is often completely different in another analyte system (i.e., analyte specific effects).6 As well, the existence of combinations of co-added TMs, inducing synergistic effects on analyte response, is both surprising and highly variable; similar to their single element impacts. The most effective combinations of TMs and their relative concentrations beneficial to one analyte are typically unique from those of another analyte6,33,36 such that no universal combination/concentration can be a priori recommended. Apart from ESR data, these observations occur against a backdrop of increased radical production sometimes visually evident from changes in the rate of generation of stable molecular gases whose precursor species are radicals such as H˙ and ˙CH3 arising from the PVG process (as evidenced by enhanced gas bubble segmentation in the liquid outlet line from the photoreactor) when particular TMs are employed.
It appears clear that no single, simple mechanism of action of added TMs, such as the LMTC concept first promulgated by Sturgeon et al.,1,3,4 can be used to account for the diversity and inconsistency of these observations. As noted by Ford,49 “…it is clear that the successful use of inorganic photochemistry, whether in energy science, biomedicine, or other foreseen or unforeseen application, needs to be based on a sound understanding of the fundamental principles that define the mechanisms of these systems…”, further experimentation is required to shed light on this phenomenon if knowledgeable use of the phenomenon is to be used to its full advantage. In response to this, an investigation of the relative concentrations of stable small molecules (CO, H2, CO2 and CH4) generated during UV photolysis of acetic and formic acids, and the impact of added TMs on changes to their profiles was undertaken. As these gases are the products of radical precursors arising from direct photolytic and subsequent thermal reactions, this provides a novel independent approach into an examination of the effects of TM mediated PVG on radical production to complement those based on limited ESR detection of these primary radicals. This research is framed within the context of an overarching tutorial review of this subject matter to enhance the perspective of the newly generated results and their interpretation based on a new approach to the mechanism of action of added TMs founded on the production of TM nanoparticles (NPs) possessing intrinsic properties of high chemical and size related selectivity for photochemical reactions.
Experimental
Instrumentation
A 19 W thin-film flow through photoreactor (Beijing Titan Instrument Co. Ltd, Beijing, China) fitted with synthetic quartz conduits (internal volume 0.72 mL) directly embedded in the low pressure mercury discharge exposed aqueous samples to vacuum UV radiation of 185 nm, as described in earlier publications.13,50,51 Prepared liquid samples were continuously introduced into the photoreactor at 1–2 mL min−1, typical of those used throughout the published PVG literature, using a Gilson Minipuls 2 peristaltic pump (Mandel Scientific, Villiers, Le Bel, France). Effluent from the reactor was directed to a glass thin-film gas–liquid separator (GLS) repurposed from a model 2600 mercury analyzer (Tekran Instruments Corporation, Toronto, Canada).Liquid waste evacuation from the GLS was accomplished using a separate peristaltic pump. A flow of Ar passing through the GLS in a countercurrent direction to the admitted irradiated sample served to efficiently strip volatile gases from the liquid phase and direct them, via a 2 mm i.d. PTFE line, to a 15 cm long (4 mm i.d.) glass dryer tube packed with granular NaOH immobilized between plugs of glass wool. This unit served to remove moisture and acidic vapors. Fresh dryer units were used for each day of measurements. Gas flow was regulated with a calibrated rotameter (Matheson Tri-Gas Inc., Montgomeryville, USA) in line with a precision metering valve and verified for consistency (28 ± 1 mL min−1) at the beginning and end of each set of experiments via its rate of volumetric displacement of water. Following passage of the gas stream through the NaOH drying tube, it was directed to 10 mL glass headspace (HS) vials fitted with magnetic ring caps and PTFE/silicone septa seals via a short length of 1 mm i.d. tygon line terminating in a 22 gauge luer mounted stainless steel needle. The needle was used to manually pierce septa during gas sampling and was inserted to the bottom of the HS vial to ensure efficient flushing of the vial in tandem with a second such needle located just beneath the septum which served as a relief vent to the atmosphere. In this manner excess gas pressure was released to a short length of 2 mm i.d. tygon tubing to ensure a representative HS gas sample. This vent line terminated a few mm below the surface of a water reservoir to ensure no back diffusion of atmospheric gases into the vial occurred during flush/fill operations or after the inlet needle was withdrawn so as to guarantee that the fill pressure in each vial was reproducible and atmospheric, as evidenced by the cessation of bubbling after the input line was removed. Filled vials were transferred to a CTC Analytics PAL RSI autosampler (Zwingen, Switzerland) connected to a double-inlet Agilent 7890A GC system (Santa Clara, CA) equipped with both Agilent TCD and Agilent 7000 TripleQuad mass spectrometry detectors. Before analysis, each sample was incubated at 35 °C for 5 min. Thereafter, a headspace (HS) volume was sampled and injected onto the GC column (syringe held at 40 °C; 4 mm double gooseneck liner held at 200 °C). In all cases, an oven heating program for the chromatography comprised the following: 5 min at 40 °C then 20 °C min−1 ramp to 120 °C with a hold for 5 min (14 min run time), followed by rapid cooling in preparation for the next run. A schematic of the irradiation and gas collection system is presented in Fig. 2.
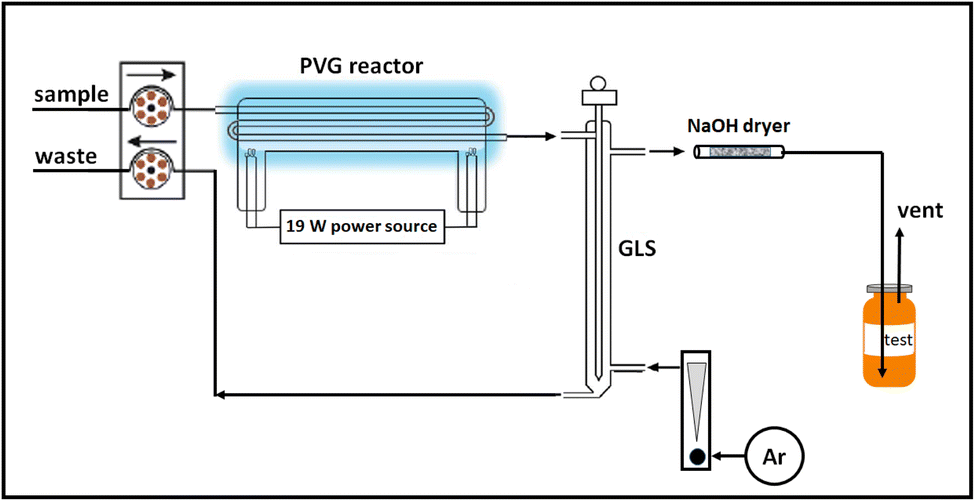 | ||
| Fig. 2 Schematic of photolysis and gas collection system. | ||
In an effort to prolong the lifetime and ensure reliability of the GC columns, the amount of water and carboxylic acid vapor transfer to the headspace vials was minimized with use of the short column NaOH dryer. More than 90% of acid vapor and water was eliminated with this approach, which unfortunately also significantly (∼92%) adsorbed the CO2 from the gas stream, necessitating its subsequent determination via independent analytical runs which bypassed the dryer column.
A Shimadzu GC-2010 Plus gas chromatograph equipped with a sensitive dielectric barrier discharge ionization detector (DIB) was used for the determination of methanol in irradiated and non-irradiated solutions of 5% (v/v) acetic acid. An identical apparatus and approach to PVG was used as described above. Solid phase microextraction of headspace vapor derived from 1 mL volumes of sample solutions placed in 7 mL glass HS vials sealed with PTFE/silicone septa was manually performed using a SUPELCO assembly comprising a fused silica fiber hosting an 85 μm poly-acrylate (PA) coating. Analyte separation was performed on a Shimadzu 30 m × 0.25 mm × 0.25 μm SH-Rtx-Wax column.
Reagents and materials
The following materials were all sourced from MilliporeSigma Canada Ltd (Oakville, ON): ACS reagent grade formic (>96%) and glacial acetic (>99.7%) acids, ACS reagent grade ammonium hydroxide (28.0–30.0% NH3 basis), ACS reagent grade (>99.0%) potassium nitrate, cobalt(II) acetate tetrahydrate (ACS reagent, ≥98.0%), copper(II) acetate monohydrate (ACS reagent, ≥98%), nickel(II) acetate tetrahydrate (98%) and cadmium acetate dihydrate (reagent grade 98%). Concentrated nitric acid was purified in-house by sub-boil distillation. High-purity (18 MΩ cm) water (DIW) was generated in a Milli-Q Advantage system (MilliporeSigma) fed with distilled water and was used for preparation of all test solutions. HPLC grade methanol, sourced from JT Baker (Philadelphia, USA), was used for preparation of calibration standards.A custom mixture of NIST traceable 1000 ppm (v/v) calibration gases (H2, CO2, CO and CH4) in a balance of 5 N Ar was ordered from Messer Canada Inc. (Mississauga, Ontario Canada).
Procedures
Stock solutions of minimum 5000 mg L−1 Cd(II), Co(II), Cu(II) and Ni(II) were prepared in DIW and serially diluted for use as TM additives to test solutions of acetic and formic acids, which were themselves prepared in large batches through volumetric dilution with DIW to yield concentrations in the range 1–20% (v/v). To reliably assess the impact of each experimental parameter, a minimum of 3 to 5 replicate samples were irradiated and gases released from the GLS were collected in separate HS vials during continuous supply of the test solutions to the photoreactor.The photolysis system was typically conditioned for 20–30 min to ensure the UV source was at a steady-state temperature as this impacts not only the primary source intensity,52 but also solution temperature which may have a minor effect on the GLS operation since gas solubility obeys an inverse temperature trend. It may also be noteworthy to point out that older lamps, which may have decreased spectral output despite running hotter, would compromise the reproducibility of the detected gas analytes amongst laboratories. During conditioning, DIW was continuously pumped through the system and all liquid and gas flow rates were set, stabilized and monitored before prepared samples were admitted for photolysis. Earlier experiments were conducted to pre-establish the optimal conditions for gas transfer flow rates and Ar back pressures with the aim of achieving efficient transfer and flushing of headspace vials within reasonably short time periods without excessive dilution of the released analyte gases. For this study, an Ar flow of 28 ± 1 mL min−1 was found effective, permitting vial flush times of typically 6 min to be used, resulting in a turn-over flushing of >15 vial volumes when real samples were processed.
The concentration ranges of TMs added to the formic and acetic acid media typically spanned reported values curated from the latest PVG literature so as to yield data/conclusions relevant to current PVG use.
Nitric acid was diluted 10-fold with DIW and used to occasionally clean the inner surfaces of the irradiated quartz lines in the PVG reactor to ensure removal of any reduced TM deposits (none visible) that may affect the photolysis processes. This was consecutively followed by several mL volumes of DIW, dilute liquid ammonia (NH3·H2O) and DIW to efficiently clean/condition the conduits.
For the analysis of H2, a 500 μL injection (25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 split ratio) of sample HS was performed and separation was obtained using an Agilent HP-PLOT Molesieve column (30 m × 0.32 mm × 25 μm, P/N 19091P-MS8) operated at constant N2 flow rate of 1.8 mL min−1. H2 was detected at 2.4 min by TCD (temperature: 220 °C, reference flow: 18 mL min−1 N2; makeup flow: 6 mL min−1 N2).
:1 split ratio) of sample HS was performed and separation was obtained using an Agilent HP-PLOT Molesieve column (30 m × 0.32 mm × 25 μm, P/N 19091P-MS8) operated at constant N2 flow rate of 1.8 mL min−1. H2 was detected at 2.4 min by TCD (temperature: 220 °C, reference flow: 18 mL min−1 N2; makeup flow: 6 mL min−1 N2).
For measurements of CH4 and CO, a 750 μL injection (15:1 split) of sample HS drawn from the purged 10 mL glass sampling vials was performed and separation was obtained on the HP-PLOT Molesieve column operated at constant He flow rate of 1.8 mL min−1. Both CH4 and CO were detected by TCD at 7.1 and 9.9 min, respectively (temperature: 220 °C, reference flow: 18 mL min−1 He; makeup flow: 6 mL min−1 He).
For the analysis of CO2 and light hydrocarbons (notably C2H6), a 250 μL injection (100:1 split ratio) of sample HS vial content was performed and separation was obtained on an Agilent DB-1701 column (30 m × 0.25 mm × 1 μm, P/N 122-0733) operated at constant He flow rate of 1.0 mL min−1 (transfer line temperature: 250 °C). The CO2 was detected at 1.5 min by mass spectrometry in SIM mode at m/z 44, 45 and 46, whereas m/z 25, 26, 27 and 30 were monitored for the presence of other potential hydrocarbons (i.e., C2H6).
For expediency, the following protocol was followed: multiple analyses for each target gas were completed in all experimental systems before proceeding to measurement of the next gas of interest. Thus, CO and CH4 were conveniently measured in each chromatographic run in all studied systems using a He carrier gas and TCD before repeating all experiments for detection of H2 with use of a N2 carrier gas. A full day for equilibration of the GC system was used when carrier gas was changed. Lastly, CO2 and C2H6 were determined by GC-MS in SIM mode with He carrier gas, as described above.
For each test solution prepared, gas products from 3–5 replicate irradiated samples were collected in separate HS vials for analysis, reflecting products generated during steady-state photolysis of each solution. Analysis of the sample HS in a given collection vial required ∼14 min to complete for each associated GC run. This led to the need for overnight operation for periods of up to 9 h; hence the need to pre-confirm the temporal stability of the HS gas samples over a period of up to 24 h. Irrespective of TCD or MS response, analyte peak area integration was used to quantify all species based on use of proprietary Agilent MassHunter software.
Concentrations of the analyte gases produced and collected in the HS vials were based on responses from a single customized NIST traceable high purity Ar multi-gas standard containing 1000 ppm (v/v) of each gas to calibrate the TCD and MS detection systems. This was achieved by direct transfer/flushing of each headspace vial in a flow of 150 mL min−1 of this calibration gas for typically 3 minutes in a manner identical to that described for the samples (including dryer column as appropriate). As analytical runs may last several hours, the stability of the sampled gases and repeatability of the detection systems were verified with an earlier set of studies as being fit-for-purpose (see Results and discussion). Additionally, replicate standards were prepared at the beginning of a set of experimental irradiations and two further replicates were freshly prepared at the end of such a sequence, which typically required 2–4 hours. This permitted a convenient assessment of any drift in instrument response or physical loss of sample components by diffusion/leakage during the measurement phase, as this required up to 9 hours to complete. Each HS vial was sampled only once as each needle puncture of the septum may open an avenue for potential accelerated escape of the analyte gases. Up to a 7% loss of response was incurred for both CH4 and CO if a second HS volume was withdrawn; this manifested as a 12% loss for H2. It was serendipitous that an earlier decision to employ a single 1000 ppm standard proved to provide a most useful reference signal that was within a factor of 2-5-fold of the range of all analyte HS gas concentrations sampled from the various PVG systems, making calibration of the detection systems over a wide concentration range unnecessary since TCD and MS are characterized as possessing several orders of magnitude linear range of operation.
For the determination of methanol, reference 5% (v/v) acetic acid solutions were irradiated at a flow rate of 2 mL min−1 (22 s IT) and 1.0 mL subsamples were drawn from the outlet of the flow-through photoreactor, transferred to HS vials and capped to await subsequent analysis by GC-DIB. A 20 min headspace extraction was performed while equilibrating the sample at 50 °C under magnetic stirring (500 rpm). SPME injections were manually undertaken in split mode (1:40) with the injector at 250 °C using He (99.999%, Messer Gases, Brazil) for sample transport and ionization at a flow rate of 1.3 mL min−1. The column was subjected to a multistep thermal program starting with an initial 40 °C ramped to 50 °C at 2 °C min−1 and held for 1 min; a rate of 12.5 °C min−1 to 75 °C with hold for 2 min and finally a ramp of 30 °C min−1 to a final temperature of 230 °C followed by cooling. The detector was maintained at 300 °C during the total analysis time of 16 min. Instrument calibration was performed using an external calibration curve prepared in the range 1.0–30.0 mg L−1 methanol (5 point) spiked to ultrapure Milli-Q water and SPME extracted and analyzed in a manner identical to that for the irradiated 5% (v/v) acetic acid solutions. Non-irradiated 5% (v/v) solutions of acetic acid served as the method blank to undertake any needed corrections to the data. Peak height response was used for characterization.
Safety considerations
The full range and identity of compounds produced during PVG is unknown. Standard safety precautions should be taken during all experiments and an adequate ventilation/exhaust system should be used.Results and discussion
Performance of sampling/detection systems
Fig. 3 shows the “long-term” temporal stability reflecting the combined contribution from collection (HS purging of vials), subsequent HS sampling of the 1000 ppm gas standard, as well as detector stability for each gas species over a period of 1–4 weeks of measurements. Individual error bars represent estimates of the standard deviations associated with 2–4 measurements taken during evaluation of impacts of individual experimental variables on the given days (note: repeatability precision is generally too small for CH4 and CO data and are hidden within the marker points in panel 3A). Panel 3A shows TCD detection of CH4 and CO in He carrier (within laboratory intermediate precision = 1.5% and 1.2%, n = 10 and 11, respectively) over the course of a one month study. Panel 3B summarizes the trends arising during more than a week of monitoring both TCD of H2 in N2 carrier (intermediate precision = 2.4%, n = 10), and GC-MS extracted chromatograms (SIM @ m/z 44) for CO2 in He carrier gas (intermediate precision = 6.4%, n = 10). These data provide assurance in both the repeatability of the performance of the sampling and detection systems during a given analytical run as well as their longer-term stability (intermediate precision) over time periods ranging from a week to more than a month of study, thereby instilling confidence in direct comparisons of data repeatedly obtained with various solutions subjected to various experimental variables.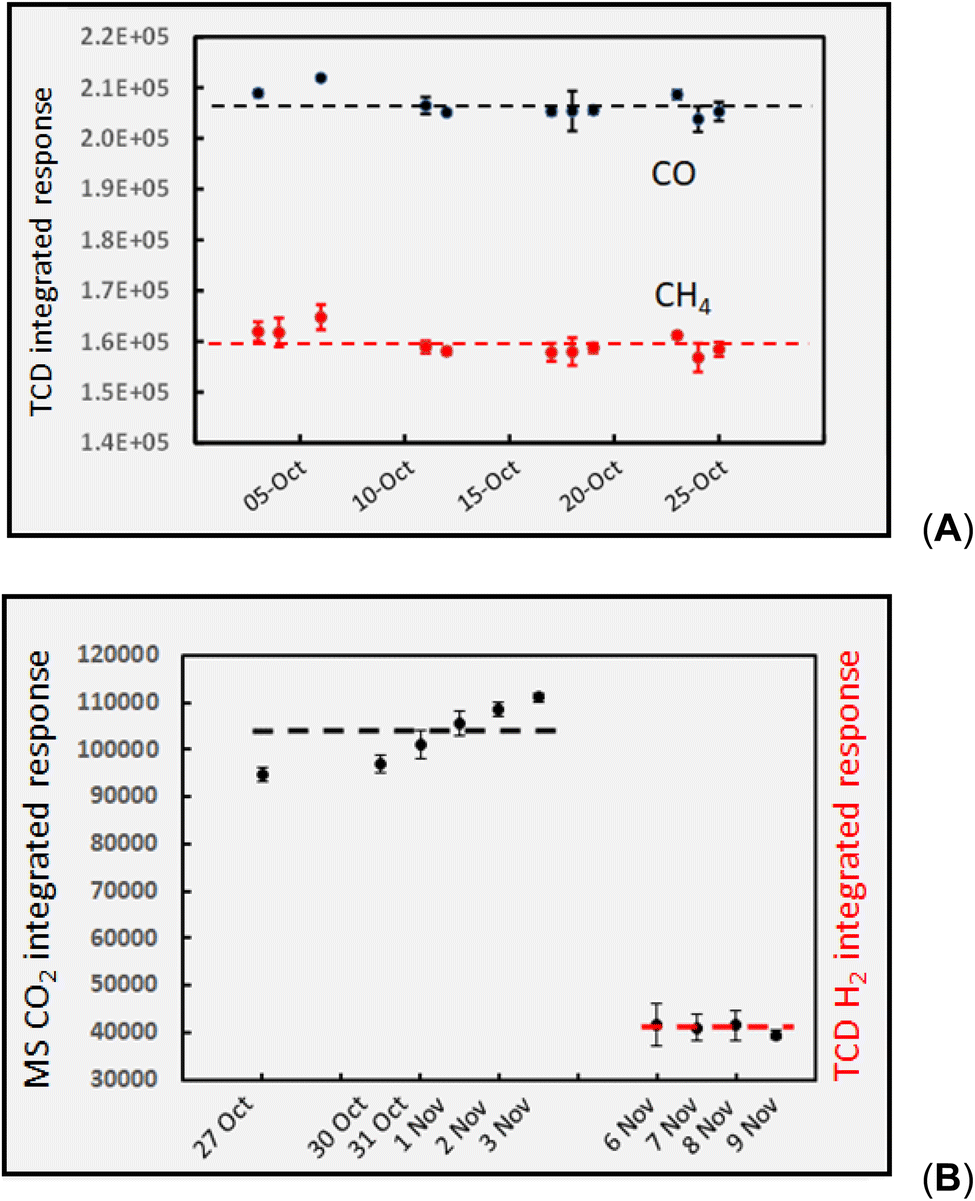 | ||
| Fig. 3 “Long-term” temporal stability (within lab intermediate precision) for detection of 1000 ppm calibration standards: (A) CO (upper) with intermediate SD = 1.2%, n = 10 and repeatability SD = 0.81%, n = 4; CH4 (lower, red) with intermediate SD 1.5%, n = 11 and repeatability SD = 0.76%, n = 4; (B) CO2 (upper) with intermediate SD = 6.4%, n = 10 and repeatability SD = 2.3%, n = 4; H2 (lower, red) with intermediate SD = 2.4% n = 10 and repeatability SD = 2.6%, n = 3. Ordinate displays integrated response characterizing chromatographic peaks. | ||
Baseline separation of the various peaks was achieved and no extraneous interferences encountered. With the exception of specific intensities, signals arising from the 1000 ppm standards were identical to those characterizing these gases present in the effluent from the GLS derived from photolysis of the formic and acetic acids, with/without added TMs. Hydrogen was the only analyte that suffered measurable temporal instability, as losses from the HS vials by diffusion amounted to 5% within 4 h and 20% after 24 h. Loss rates accruing over typical 6 h data collections were assumed linear and corrected for using changes in bracketed standard intensities. Use of the NaOH dryer has no discernible impact on response for CH4, CO, and H2, although it efficiently removes water and acidic vapors, thereby protecting the life of the GC column.
As a consequence of the overall stability of the experimental system, integrated detector response is consistently presented as the measurement metric throughout this study rather than normalized comparisons of responses which can misrepresent data and lead to ambiguous interpretations otherwise based on stand-alone snapshots of the system. Instrument response, generated under stable measurement/sampling conditions permits the reader to comprehensively overview all data and formulate consistent trends and comparisons for each analyte of interest as experimental conditions are varied.
Part 1: PVG in the absence of added TMs
The vacuum-UV photochemistry of water is well established53–59 and essentially summarized by the following homolysis and (to a lesser extent) charge-transfer-to-solvent reactions, respectively: | (1a) |
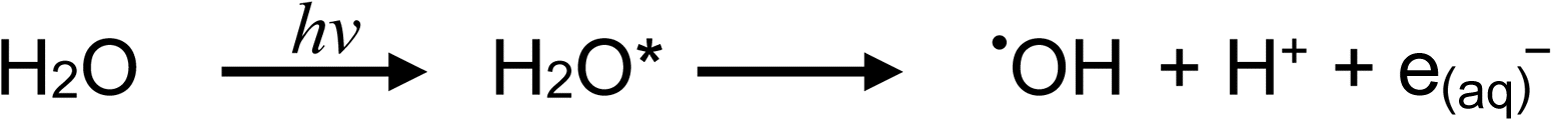 | (1b) |
Typical of low pressure mercury discharge lamps, the intensity of 184.9 nm photons emitted by the flow-through photoreactor used in this study is likely 40-fold lower than that of the primary 253.7 nm Hg resonance line.60 Any H˙, ˙OH and e(aq)− so formed provide primary radicals that participate in the overall photolysis of dilute aqueous solutions of formic acid.60,61 In the absence of radical scavengers (i.e., added carboxylic acids), diffusional rates of radical recombination likely inhibit significant free H2 liberation. Zechner and Getoff57 have demonstrated that only when formate is present at <10−4 mol L−1 [i.e., <4 × 10−4% (v/v)] do reactions (1a) and (b) proceed, as otherwise the 185 nm incident radiation is preferentially absorbed by formate. With increasing formate concentration, reaction (7) (see below) occurs with generation of H2. Recently, Hu et al.62 reported photochemical hydride generation of As(III) and As(V) from an aqueous mixture of 1 × 10−3 mol L−1 HCOO− containing 2 × 10−3 mol L−1 SO32−, noting ESR detection of minor amounts of H˙, indirect evidence of e(aq)−, but no mention of CO2˙–. Generation of AsH3 was attributed to the action of redox processes involving H˙ and e(aq)− derived from reaction (1) and photolysis of added HSO3−, suggesting that H˙ remains an active intermediate despite the recognized high pH of this reaction medium which would lead to rapid recombination of H˙ and ˙OH radicals. Direct generation of H˙ and e(aq)− by primary photolysis of the HCOO− in accordance with reactions (2d) and (2e) (below) was not considered in this study, although at a concentration of 1 × 10−3 mol L−1, absorption of 185 nm photons preferentially occurs by formate rather than water. More recently, Jeníková et al.63 explored the feasibility of PVG of Ru, Re and Ir from low concentrations of formic acid, noting that relatively efficient PVG processes occur during photolysis of even pure water if it is pre-saturated with CO. Thus, it was clear that efficient reduction of these elements was achieved due to available H˙ and e(aq)− radicals.
It is also expedient to review the UV-B and -C induced photolysis of formic and acetic acids, as well as briefly consider a few model LMCT reactions induced by added TMs in these acids. Reactions presented below summarize the pertinent photochemical processes that give rise to radical species which (apart from contributing to the generation of volatile metal and halogen species of analytical interest) are also the precursors to the permanent gases which are the principal subjects of this study.
Photolysis of formic acid (gas and condensed phases) has been studied for almost a century. Contrary to the belief of many organic photochemists, generation of CO occurs in addition to CO2, albeit not as the primary reaction.57,58,64–66 Highest relative CO/CO2 is achieved at lower irradiation wavelengths due to rapid dissociation of the excited trans form of HCOOH to yield CO and H2O. Several dissociation channels have also been identified by studies using 248 and 193 nm irradiation65 (e.g., reactions (2a)–(2e)), yielding the usual powerful reducing radicals (e(aq)−, CO2˙− and H˙) which participate in subsequent thermal events (e.g., reactions (3)–(12) and denoted throughout as dashed arrows), ultimately giving rise to detectable CO, CO2 and H2 and characterized by overall reactions (c) and (d):
 | (2a) |
 | (2b) |
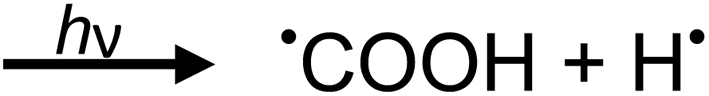 | (2c) |
 | (2d) |
 | (2e) |
 | (3) |
 | (4) |
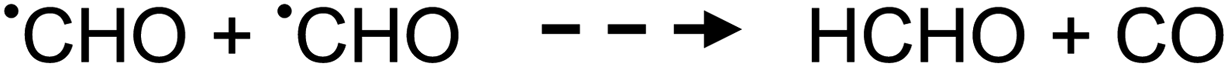 | (5) |
 | (6) |
 | (7) |
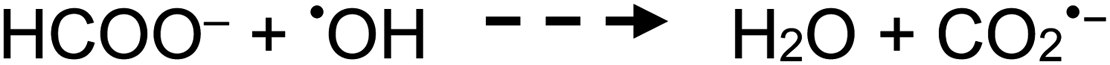 | (8) |
 | (9) |
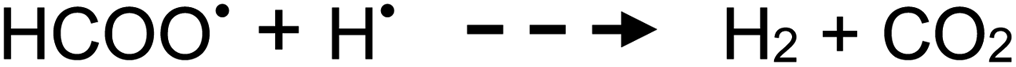 | (10) |
 | (11) |
 | (12) |
 | (c) |
 | (d) |
Photolysis of aqueous solutions of acetic acid have been equally long studied, wherein several routes arise and stable product yields are dependent on the degree of ionization of the acid.67–71 Due to the greater complexity of the system, reported stable reaction products have included CH4 and CO2, along with H2, methanol, formic acid and C2H6, summarized by generalized reaction e:
 | (13a) |
 | (13b) |
 | (13c) |
 | (13d) |
 | (13e) |
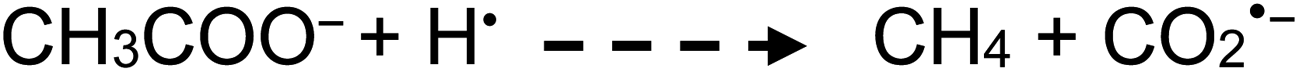 | (14) |
 | (15) |
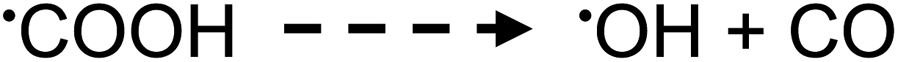 | (16) |
 | (17) |
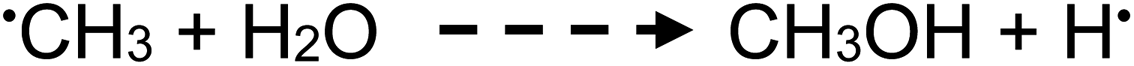 | (18) |
 | (19) |
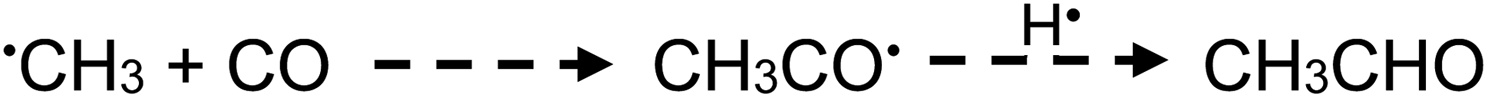 | (20) |
 | (21) |
 | (22) |
 | (23) |
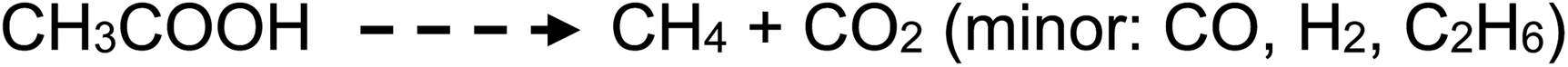 | (e) |
Note that no consideration has been given to the existence of stable dimers of both carboxylic acids in aqueous solutions72 which may initiate their own sets of radical based reactions.67
Addition of TMs to either acid results in the formation of TM-carboxylate complexes, which may exhibit not only d–d transitions of the metal center, but also photochemical (LMTC) reactions, as noted earlier. Typical examples are presented below for the case of iron and copper,42,44,60 wherein LMCT processes take place at significantly longer wavelengths outside the UV-C spectral window needed for homolysis of water and oxidation of formic/acetic acids. Thus, advantage may be taken of the intense UV-B spectral region emitted from low pressure mercury discharge sources, enhancing generation efficiencies of H˙ and ˙CH3 radicals as well as production of CO2:
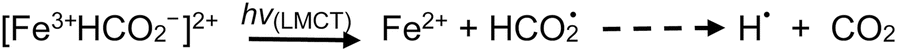 | (24) |
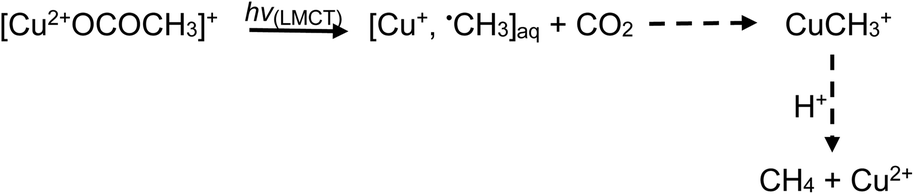 | (25) |
Apart from the above processes potentially directly influencing the sources and concentrations of radicals giving rise to CO, CO2, H2 and CH4 in TM-doped PVG systems, the added TMs may also be involved in secondary thermal radical catalytic redox reactions which further influence the relative concentrations of some gases, including, for example:50,73,74
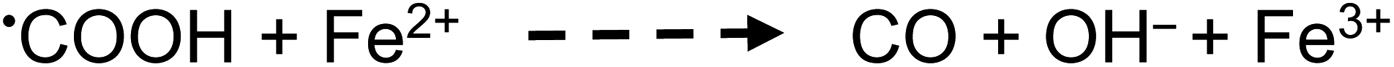 | (26) |
 | (27) |
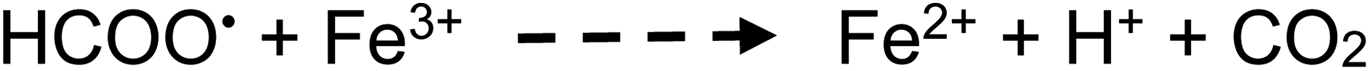 | (28) |
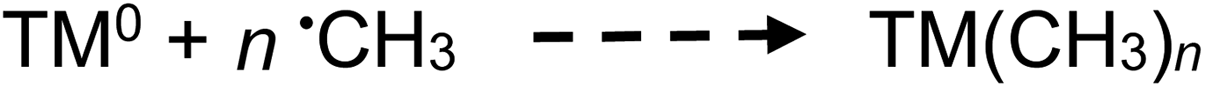 | (29) |
The extent to which added TMs perturb generation of radical intermediates should thus be reflected in relative changes in HS gas composition between irradiated TM-spiked and -unspiked PVG media. As noted earlier, this provides a new tool for probing mechanistic aspects of PVG which should not only complement results from earlier ESR studies, but also provide a broader profile, as H2 and CO become useful probes in addition to the usual ˙CH3, ˙OH and CO2˙− radical species reported with ESR, which primarily serve as indicators of the reductive potential of irradiated media.
Although the principal objective of this study was to utilize these results to establish comparative rates of production of the various gases as a function of experimental variables, use of Henry's Law and the known dilution factor arising from the GLS gas transfer process suggest that, at the least, semiquantitative concentrations of CO, CO2, CH4 and H2 may also be estimated. The solubility of CO2 in water presents the worst case scenario as its dissolution is the most favorable, as evident from its Henry's Law constant, typically an order of magnitude greater than that for the other gases due to its reaction to form H2CO3. All photolysis media used in this study have a pH < 3, providing favorable conditions for decreased solubility of CO2 as the reaction to form carbonic acid is suppressed. Thus, complete release of even CO2 can be expected.75,76 Irrespective, relative changes in the targeted gas concentrations in the TM-unspiked formic and acetic acid systems can be directly compared to those generated in the presence of added TMs to provide insight into the relevance of the LMCT model in accounting for changes in PVG efficiencies because all experimental variables are carefully replicated and any perturbation reflects only the impact of the added TM.
To appreciate the effects of the presence of added TMs, it is instructive to first consider the baseline or “endogenous” concentrations of the gases of interest that are photolytically generated in their absence and their response to changes in experimental variables typically undertaken during optimization of PVG efficiency, i.e., impact of the concentrations of the formic and acetic acids, sample irradiation time and use of mixtures of these acids.
To ensure the relevance of these experiments, a comprehensive examination of the PVG literature published since 2015 based on available use of thin-film flow through photoreactors was used to identify relevant concentration ranges of formic and acetic acids for study, typical concentrations of added TMs to be investigated as well as irradiation times. As such, observed trends can be correlated with those typical of published analytical PVG results wherein response from synthesis of targeted volatile metal species has been examined under similar experimental conditions.
Contamination introduced by the sampling process was validated by running DIW blanks through the fully powered PVG system. No response was detected from CO, CH4 and H2 while that for CO2 was consistently in the range 5–10% of the typical concentrations evolved from irradiated samples of 5% (v/v) formic and acetic acids, corresponding to 285 ± 50 ppm CO2 in the collected sample HS (averaged over 7 days of testing). Its source was never investigated in detail and although the DIW system was fitted with a UV oxidation unit, it is possible that it was inefficient and the “blank” CO2 measurement corresponded to oxidation of residual organic contaminants in the water, as the level appears too high to be the result of dissolution of atmospheric CO2 (nominally 420 ppm) over such short exposure times.
Hydrogen production via photolysis of water
Photolysis of pure water was investigated for evolution of H2, as this is the only gaseous species of interest potentially produced. In connection with the results of Zechner and Getoff,57 the impact of dilute solutions of formic acid serving as an H˙ radical scavenger (via reaction (7)) was also examined.Replicate GC-TCD analyses of HS H2 content derived from the photolysis of DIW revealed no quantifiable response above baseline noise. Homolysis reaction (1a) is likely rapidly reversible as initially a cage complex must be generated permitting subsequent diffusion limited recombination of the H˙ and ˙OH species. Interestingly, however, is that photolysis of a very dilute solution of 0.004% (v/v) formic acid generated measurable quantities of H2, far in excess of that expected based on the amounts released from subsequently explored solutions of 1000-fold higher concentrations. Thus, while photolysis of 5% HCOOH at a flow rate of 2 mL min−1 typically provided 380 ± 40 ppm H2 in the HS (see Table 1), that from 0.004% (v/v) formic acid produced 110 ± 10 ppm H2. The disproportionate synthesis of H2 in this very dilute medium suggests that either homolysis of water is efficient and reaction (7) serves to trap the generated H˙ before extensive radical recombination can occur to yield H2 (as with commonly used ESR spin trapping reagents), or that the penetration depth of 185 nm photons is enhanced by at least 100-fold as the HCOOH concentration is reduced from 5 to 0.004%. Irrespective, recent studies by Musil et al.63 which reported on the successful generation of several metal carbonyls during photolysis of only deionized water that was pre-saturated with CO provides evidence for the efficacy of reactions (1a) and (b) liberating reducing radicals that create free atoms of metals which subsequently coordinate with available CO to yield their volatile molecular carbonyls. Moreover, sufficient liberation of H˙ is evident in 0.004% formic acid to permit synthesis of volatile species of hydrides, such as Se.63
| CO | H2 | CH4 | CO2 | |
|---|---|---|---|---|
| a Intermediate precision expressed as standard deviation over duration of study encompassing (n) measurements. Note: data are HS concentrations and thus uncorrected for dilution arising from GLS Ar carrier gas flow of 28 mL min−1. | ||||
| Formic | 3760 ± 220(11) | 380 ± 40(7) | — | 4440 ± 310(13) |
| Acetic | 14 ± 4(9) | 240 ± 35(6) | 1180 ± 150(14) | 1700 ± 130(9) |
Gas production via photolysis of formic acid
A formic acid medium is the simplest to consider first, as only H2, CO and CO2 are of interest. All data are reported based on the volumetric concentrations of the carboxylic acids (rather than molarity) as this is the most common means of preparing such experimental PVG media and reporting their impact in the analytical literature. Note also that, unless specifically calculated, all concentration data reported for the various gases are uncorrected for the dilution effect imposed by the 28 mL min−1 flow of Ar transport gas used to flush the products from the GLS to the HS sample vials. | ||
| Fig. 4 Effect of formic acid concentration on analyte gas response (production) at 2 mL min−1 solution flow rate (IT = 22 s). (A) CO production, uncorrected HS [CO] = 3810 ± 930 ppm (repeatability SD, n = 2) at 5% (v/v) formic acid; (B) H2 response (production), uncorrected HS [H2] = 400 ± 60 ppm (repeatability SD, n = 3) at 5% (v/v) formic acid; (C) CO2 response (production), uncorrected HS [CO2] = 4290 ± 100 ppm (repeatability SD, n = 5) at 5% formic acid. Ordinate displays integrated response characterizing chromatographic peaks. | ||
Several points merit discussion of these data. It is noteworthy that the non-linear asymptotic characteristic of this curve mirrors those illustrating the impact of carboxylic acid concentration (in both HCOOH and CH3COOH systems) on analytical PVG yields for numerous elements.16,22,26,28,30,31,77–84 The downward curvature is frequently attributed to the decreased depth of penetration of the UV-C radiation into the bulk of the sample as the acid concentration is increased, thereby lowering the effective photon flux to the bulk or core of the flowing solution. In the case of formic acid (and similarly acetic), its significant molar absorption coefficient (34.9 M−1 cm−1 at 185 nm)85 results in a penetration depth (depth at which attenuation of source intensity of 185 nm radiation is reduced to 10%) of 240 μm into a 5% (v/v) medium (similar results hold for acetic acid as well). Unless turbulent mixing is achieved in the 2 mm diameter sample conduit, its central core receives significantly fewer photons; photolysis efficiency is expected to correspondingly diminish with increased [HCOOH].
Additionally, it has been reported that the rate of photolysis of HCOOH is dependent on its degree of ionization.59,70 Being a weak acid, increased HCOOH concentration corresponds to a decreased relative [HCOO−] and a potentially concurrent quadratic relationship between [HCOOH] and CO yield. Adams and Hart60 concluded that UV photolysis of HCOOH resulted in production of CO via reactions 3, 5, 6 and 11, involving ˙CHO and ˙COOH intermediates. Unless the [HCOOH] is <10−4 mol L−1 [i.e., <0.001% (v/v)], the contribution of subsequent thermal reactions involving H˙ and ˙OH (arising from homolysis of water, reaction (1)) with HCOO− (reactions (7) and (8)) to generate H2 and CO2˙− are insignificant due to the molar absorption coefficient for H2O being 103-fold smaller than that of HCOO− at 185 nm.57,85
Based on its low solubility, Henry's law predicts a negligible concentration of CO remains dissolved in the irradiated solution; furthermore, the thin-film geometry of the GLS serves to efficiently release it to the counter-current flow of 28 mL min−1 Ar. Reactions (3), (5), (6) and (11) serve to generate CO, but irrespective of the mechanism, reaction (c) shows that a 5% (v/v, ∼1.18 mol L−1) solution of HCOOH irradiated at a flow rate of 2 mL min−1 will quantitatively generate 2.4 mmol min−1 CO, equivalent to 53 mL min−1. When diluted in a flow of 28 mL min−1 Ar, the sampled HS mixture should contain 6.5 × 105 ppm CO. Based on the average determined HS CO concentration of 3760 ± 220 ppm (cf.Table 1), an apparent oxidation (photolysis) efficiency of only 0.6% is revealed. Considering the possible need for an ionized precursor59 and the relatively low flux of source photons at 185 nm, this estimate is perhaps not surprising. Due to the low degree of ionization (α) of HCOOH (i.e., α = 1.2% at 1.18 mol L−1; Ka = 1.78 × 10−4), a 5-fold increase in [HCOOH] yields a 2.2-fold increase in [HCOO−] and can quantitatively account for the experimentally observed rise in CO as the formic acid concentration is augmented from 2 to 10% (v/v), evident in Fig. 4A. This suggests that penetration of UV-C photons may not present such a significant impediment to photolysis in this flow-through reactor in this range of formic acid concentration. Although not shown in Fig. 4A, decreasing the sample flow rate to 1 mL min−1 (thereby increasing the irradiation time to 43 s), resulted in a 1.5-fold increase in CO from a 2% (v/v) HCOOH medium to yield a 3520 ± 190 ppm (repeatability SD: n = 3) CO headspace concentration. Note that when corrected for the rate of delivery of product at the exit of the GLS, this would result in an equivalent 3-fold increase in yield for a doubling of the irradiation time. Unaccounted for losses of primary generated CO via such processes as the water gas shift reaction are unlikely due to both the low temperature and low H2 partial pressure, as well as the absence of a catalyst.
An estimate of overall photolysis efficiency with respect to H2 production may be made based on reaction d. Using data averaged from all experiments based on a 22 s photolysis of 5% HCOOH (sample flow rate of 2 mL min; H2 = 380 ± 40 ppm, intermediate SD, n = 7, cf.Table 1) in the collected HS samples and calculations similar to those outlined above for CO, suggests an oxidation efficiency of 0.05%, an order of magnitude inferior to that based on CO data. It is clear that these reactions are not directly coupled (reactions (c) and (d)) and details of the quantum yields, excitation/dissociation and kinetics of processes leading to CO and H2 generation would have to be taken into account to arrive at more accurate estimates of reaction yields. Nevertheless, together they consistently suggest a low overall oxidation efficiency. Unless a complete analysis of all reaction products can be accomplished, differentiation of the chemical (thermal) interactions of the various radicals cannot be discerned.
Similar to CO (and not shown in Fig. 4B), H2 generation from 5% (v/v) HCOOH at a 1 mL min−1 sample flow rate (irradiation time of 43 s) was enhanced 1.9-fold to yield 720 ± 75 ppm (repeatability SD, n = 3) headspace concentration. This occurs despite the 2-fold reduction in the rate of delivery of H2 to the GLS, indicating that a doubling of irradiation time enhanced efficiency of H2 production by 4-fold. Similar correlations are evident in the analytical PVG literature wherein response from targeted (UV-stable) metal analytes frequently increases with irradiation time but typically not as dramatically as for these gases, indicating a potential loss mechanism which may be putatively assigned as a concurrent competitive UV photolytic decomposition of the (unstable) metal analyte species.
Over a measurement period of 10 days, comprising 9 discrete experiments, an average (blank corrected) concentration of 4440 ± 310 ppm CO2 (intermediate SD, n = 13; cf.Table 1) was determined in the sample HS comprising GLS effluent from photolysis of 5% (v/v) formic acid at 2 mL min−1 (IT = 22 s).
Gas production via photolysis of acetic acid
The majority of published literature concludes that CO2 and CH4 are the principle gaseous products of photolysis of acetic acid. However, other reported by-products have included C2H4, CH3OH and acetaldehyde, likely arising from secondary recombination reactions consuming ˙CH3 radicals. Several thermal radical reactions also lead to production of CO (reactions (16) and (21)) and H2 (reaction (23)), although they are expected to be of low yield. As is the case with HCOOH, at the concentration levels of interest to this study, direct photolysis of CH3COOH and CH3COO− likely dominate reactions as compared to those initiated by H˙ and ˙OH homolysis processes arising from eqn (1a) and (b) due to their higher molar absorptivity compared to that for DIW. Note that the equivalent molarity of the photolyzed solutions of formic and acetic differ by a factor of 1.4, reflecting the lower concentration of the neat acetic acid; this is expected to result in lower HS concentrations of the measured gases common to the two systems.:1 stoichiometry for oxidation of CH3COOH summarized by equation e, an efficiency of 0.5% can be estimated, somewhat higher than that calculated below from data based on production of CH4 in this system, but similar to that calculated for the formic acid system.
 | ||
| Fig. 5 Effect of acetic acid concentration on analyte gas response (generation) at 2 mL min−1 solution flow rate (IT = 22 s). (A) CO2 production, uncorrected HS [CO2] = 1730 ± 30 ppm (repeatability SD, n = 4) at 5% (v/v) acetic acid. (B) CO production, uncorrected HS [CO] = 14 ± 4 ppm (repeatability SD, n = 9) at 5% (v/v) acetic acid. (C) H2 production, uncorrected HS [H2] = 290 ± 40 ppm (repeatability SD, n = 3) at 5% (v/v) acetic acid. Ordinate displays integrated response characterizing chromatographic peaks. | ||
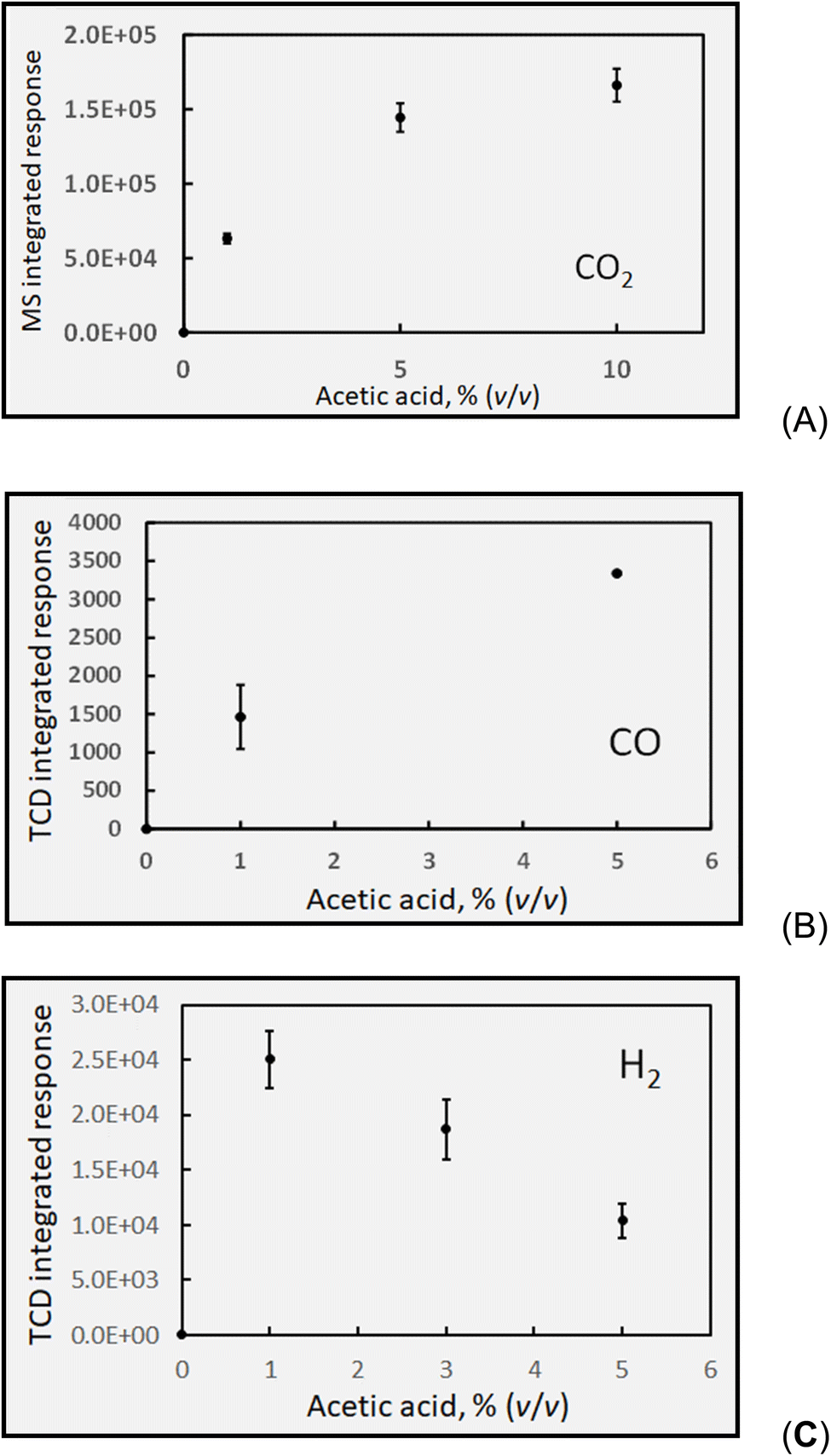
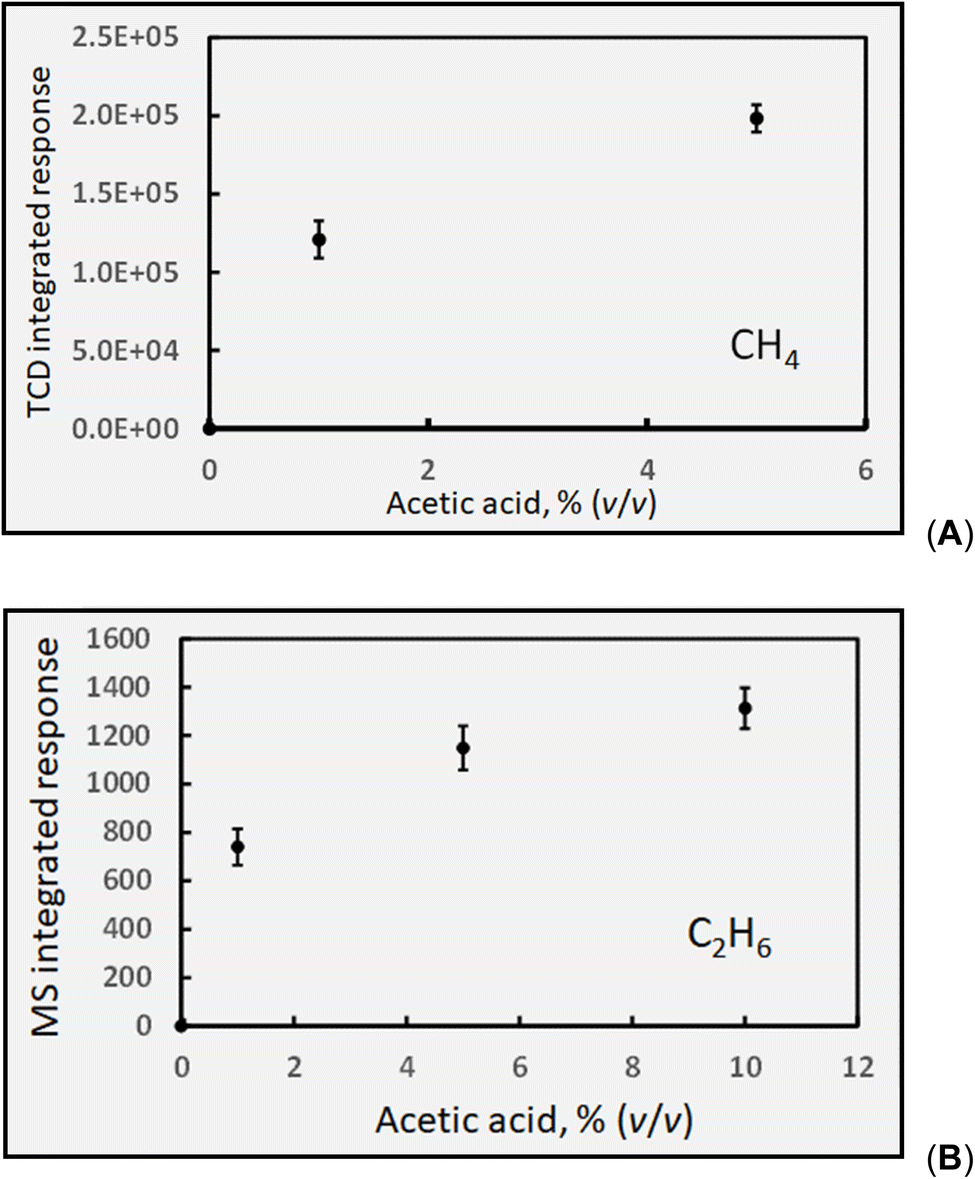 | ||
| Fig. 6 Effect of acetic acid concentration on response (generation) of: (A) HS CH4 at 2 mL min−1 solution flow rate (IT = 22 s). Uncorrected HS [CH4] = 1250 ± 60 ppm (repeatability SD, n = 3) for 5% (v/v) CH3COOH. (B) HS C2H6 response (generation) at 2 mL min−1 solution flow rate. Uncorrected HS [C2H6] generated in 5% acetic acid estimated as ∼30 ppm (error bars show repeatability SD: n = 3). Ordinate displays integrated response characterizing chromatographic peaks. | ||
The methyl radical is quite reactive and can function as both an oxidant and reductant, which may alter its perceived stoichiometry of net production as presented by overall reaction e. During analytical PVG of the halogens, its oxidizing character is evident in the capture of nucleophilic X˙ to yield CH3X. By contrast, it also serves as a strong reductant towards water, producing methanol, as illustrated by reaction (18), which will become more significant at higher concentrations of acetic acid. The general shape of the response curve is similar to that obtained with analytical PVG optimization experiments for elements synthesized as methylated species from an acetic acid medium.
Over the course of one month, comprising 7 discrete experiments, an average concentration of 1180 ± 150 ppm (intermediate SD, n = 14; cf.Table 1) CH4 was determined in the sample HS derived from the GLS effluent from photolysed 5% (v/v) acetic acid solutions. Based on this, and neglecting any potential losses of ˙CH3 precursor via other reactions, an estimate of the overall oxidation efficiency of CH3COOH at a flow rate of 2 mL min−1 in accordance with reaction (e) and calculated in the same manner as described earlier for CO in the formic acid system, yields a photolysis efficiency of 0.2% [note that a 5% (v/v) solution of CH3COOH corresponds to an 0.85 mol L−1 solution (and a smaller α of 0.4%)].
Loss of ˙CH3via reaction (18) can occur to yield methanol. To estimate the impact of this potential sink, the concentration of methanol produced in irradiated 5% acetic acid was determined by SPME-GC-DIB to be 3.7 ± 0.3 mg L−1 (reproducibility SD, n = 3). Assuming no methanol formed in the irradiated samples was lost through evaporation during the short collection phase, and that all methanol originated from photogenerated ˙CH3 reacting with water in accordance with reaction (18), the yield can be calculated to be 1.7 × 10−7 mol mL−1, equivalent to a synthesis efficiency of 0.014%. This may be contrasted to the earlier estimate of 0.2% generation efficiency ˙CH3 derived from the measured yield of CH4 in this medium (Table 1, characterizing reaction (e)) and to the conclusion that losses of CH4 through formation of methanol amount to less than 10%.
:H2 reportedly produced during photolysis of aqueous solutions of acetic acid was 1:17, an estimated concentration of ethane in the GLS effluent during photolysis of 5% (v/v) acetic acid yields 16 ppm. Similarly, Mozia et al.87 reported a C2H6:CH4 ratio of 0.03 for the photo-Kolbe oxidation of acetic acid solution in the presence of decorated TiO2, suggesting the concentration of ethane could be in the range of 30 ppm herein. It is clear that this species is characterized by low generation efficiency, despite the likely beneficial effects arising from the presence of photocatalysts in such media.87
Table 1 presents a summary of the HS concentrations of stable gas species generated during the photolysis of 5% (v/v) reference concentrations of both formic and acetic acids irradiated for 22 s in the flow-through photoreactor (solution flow = 2 mL min−1). As has been noted throughout, the detected HS concentrations are not corrected for volumetric dilution introduced by the carrier gas flow of 28 ± 1 mL min−1 Ar, but may be corrected as attempted for some earlier examples used for estimation of oxidation efficiency of the acids based on overall reaction stoichiometries (eqn (13a)–(13e)). Moreover, with a 1.4-fold higher molarity of formic acid, all other factors aside (relative α, solubility), relative concentrations of their one common gas, CO2, should theoretically be 40% higher in formic acid. Details of interaction of the source intensity and wavelength with the different acids need to be taken into account along with all subsequent thermal reactions involving the photolytically generated precursor radicals in order to present a clearer picture, but this is not possible within the scope of this study.
Photolysis of mixed acid media
A number of analytical PVG studies have reported enhanced synthesis yields of targeted metal species when using mixtures of formic and acetic acids.20,21,28,88 Hou and colleagues20 utilized spin-trapping ESR detection techniques to present evidence that more CO2˙− was generated in a mixed formic/acetic acid medium than in their individual solutions of the same concentration. In the case of Te and other semi-metals that give rise to methylated PVG analogs, it is possible that differences in the molar absorption coefficients for formic acid at lower wavelengths increase generation of CO2˙− to provide a more reducing medium for efficient metal ion reduction, although it is ˙CH3 available from acetic acid that drives final product formation. Additionally, CO can rapidly scavenge ˙OH to provide a yet more favorable population of H˙ reducing radicals:89 | (30) |
It is noteworthy, however, that use of mixtures of carboxylic acids does not universally endow analytical PVG with positive impacts. Musil and colleagues reported that PVG of both Mo30 and W16 provided superior generation efficiency of their metal carbonyls (as well as for those of Fe, Ni and Co) from solely formic acid as compared to a formic/acetic mixture. For transition metals which form primarily stable carbonyl species, this negative impact can be ascribed to the consumption of CO by ˙CH3 radicals to yield ˙CH3CO and subsequently CH3COCOCH3, CH3CHO and CH3COOH.8 As an example, the relative effects of a mixed acid medium comprising 5% (v/v) formic and 5% acetic (v/v) on production of CO and CH4 headspace gases are compared to those of their individual solutions in Fig. 7.
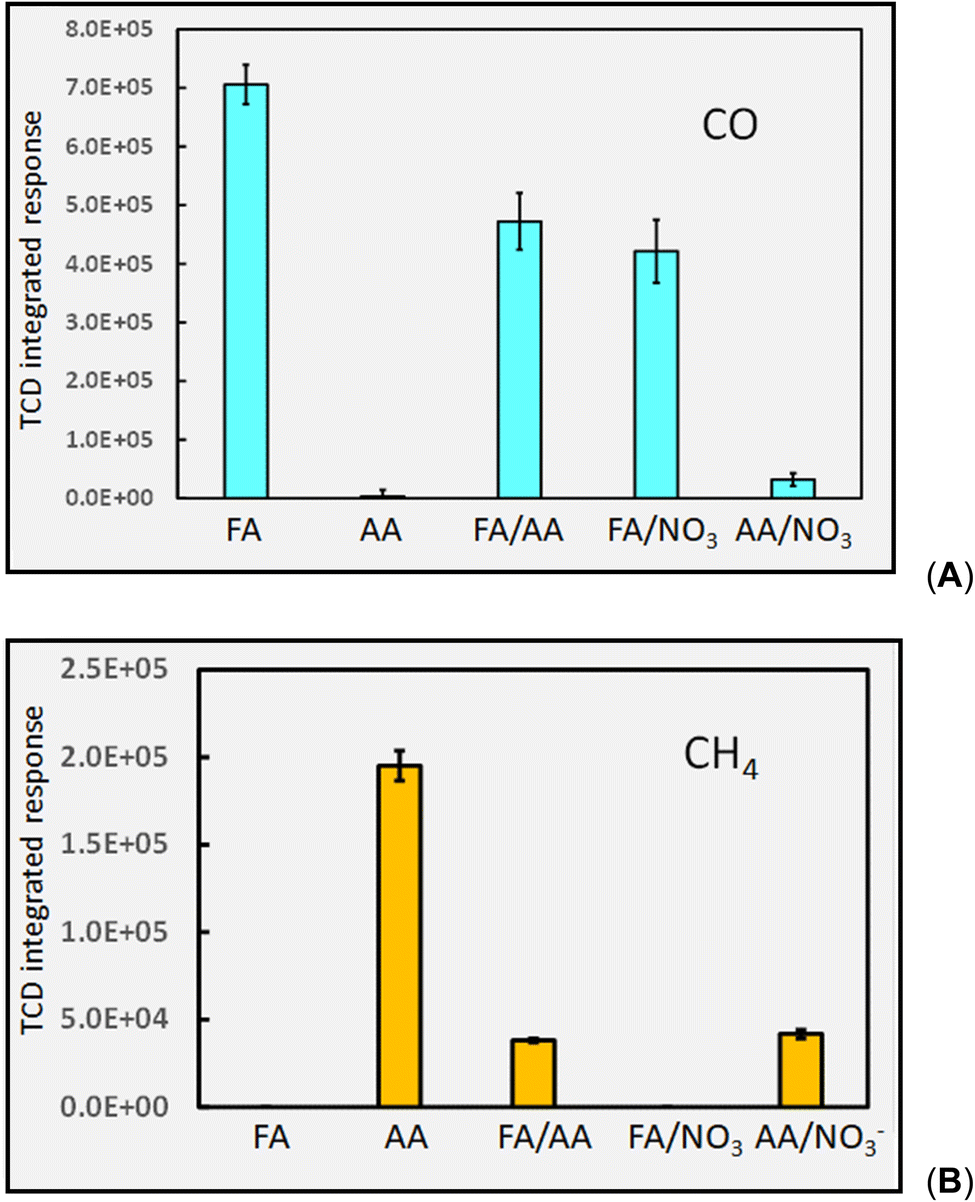 | ||
| Fig. 7 Impact of PVG medium composition on HS response from: (A) CO and (B) CH4. FA = 5% formic acid (v/v); AA = 5% acetic acid (v/v); FA/AA = mixture of 5% (v/v) each acid; FA/NO3− = 5% (v/v) formic acid containing 10 mM NO3−; AA/NO3− = 5% acetic acid (v/v) containing 10 mM NO3−. 2 mL min−1 solution irradiation (IT = 22 s). Repeatability SD, n = 3. Ordinate displays integrated response characterizing chromatographic peaks. | ||
Responses (repeatability SD, n = 3) to production of both CO in formic acid and CH4 in acetic acid (5% (v/v)) are in statistical agreement with those shown earlier in Fig. 4A and 6A respectively, illustrating the stability of the systems throughout this study. Importantly, addition of acetic acid to formic acid produces a significant depletion of CO in this medium whereas addition of formic acid to acetic acid results in a more deleterious effect on the generation of CH4 in the mixture. Both effects may be attributed to reaction 31.40
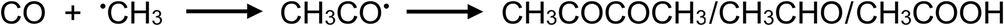 | (31) |
Impact of NO3− as a common interference on gas production
Fig. 7 also illustrates the significant effect addition of 10 mM NO3− (present as KNO3) into the generation medium has on decreasing CO and CH4 production by an order of magnitude, far more impactful that the effect of any other experimental variable examined. Dissolved O2, NO3−, NO2− and Cl− are recognized quenchers of radical based reactions due to their affinity for hydrated electrons and hydrogen atoms.90–95 | (32) |
 | (33) |
 | (34) |
 | (35) |
Suppression of CH4 generation in acetic acid containing added NO3− is also a consequence of the scavenging of H˙. Although all analytical PVG systems suffer to some extent from the presence of oxidants such as nitrate and nitrite (a product of photoreduction of NO3−),1 PVG yield of Se(IV) from formic and acetic acid media presents a particularly severe example.96,97
Fig. 8 quantifies the very severe interference (i.e., almost complete suppression) that the presence of 10 mM NO3− has on H2 production from 5% solutions of acetic and formic acids. Its seemingly more dramatic impact on the formic acid system may be a consequence of the more complex environment present in acetic acid. The consequences of thermal reactions involving radicals other than those of H˙ (as illustrated in reactions (32) and (34)) with the photolysis products of nitrate are unknown. A gas profile of the photolysis products of nitrate would likely shed more light on what is occurring in these systems to provide for a more holistic picture of events.
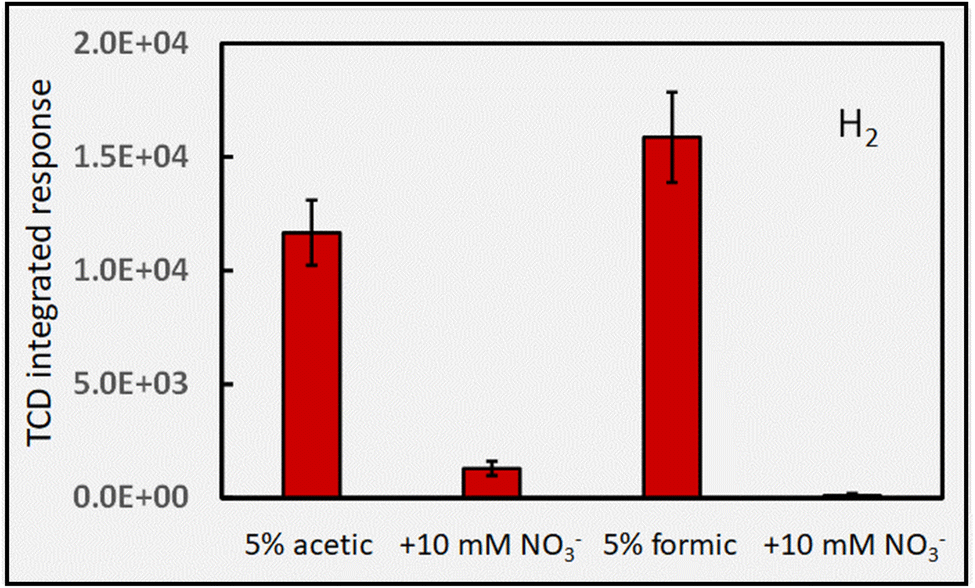 | ||
| Fig. 8 Impact of the presence of 10 mM NO3− on HS H2 response (generation) during photolysis of 5% (v/v) formic and acetic acids at 2 mL min−1 (IT = 22 s). Repeatability SD, n = 3. Ordinate displays integrated response characterizing chromatographic peaks. | ||
It is interesting that PVG of Se(IV) from formic acid generates a mixture of both SeH2 (∼70%) and SeCO (∼30%). However, in the presence of 10 mM NaNO3, formation of SeH2 is completely suppressed, with only SeCO being produced but at 3-fold its intensity in nitrate-free media.96 Based on the direct evidence presented in Fig. 8 and the potential reactions (32)–(34), it is abundantly clear that efficient scavenging of H˙ radicals is responsible for this. Note also that the extent of H2 generation from PVG media is typically insufficient to support atomization of hydrides in conventional heated quartz tube atomizers.96,98
Part 2: PVG in the presence of added TMs
Having gained fundamental insight into the influence of several experimental variables on the “baseline” photochemical production of stable small gas molecules from acetic and formic acid media, it is instructive to examine the impact of added transition metal ions (TMs). Their effects on precursor radical species that lead to H2, CO, CO2 and CH4 may provide enhanced understanding of the mechanism(s) of action of these metal ion-assisted processes. As noted earlier, this study was not intended to be comprehensive, but rather limited to an examination of use of Cd(II), Co(II), Cu(II) and Ni(II) added to 5% (v/v) solutions of acetic and formic acids in an effort to minimize the complexity and number of experiments needed to be undertaken. For consistency, test solutions were irradiated at flow rates of 2 mL min−1. An arbitrary but analytically efficacious concentration of each TM was identified from the more recent PVG literature related to use of flow-through photoreactors. Those perceived as having a significant influence on gas production were further examined by evaluation of their impact as a function of added concentration. It is acknowledged that further to their use with thin-film flow-through reactors, their beneficial influence has also recently been reported with use of standard (germicidal) low pressure mercury lamp sources limited to 254 nm output.26,31,38,83,99Amongst the terminologies employed in the PVG literature, the use of TMs to enhance synthesis yields of targeted analytical species has variously been described as inducing a “photoassisted” process due to the action of a “photocatalyst”, a “photoredox catalyst” or a “photosensitizer”. In view of the current paucity of mechanistic information, a more generalized description evoked by the phrase “TM-assisted” or “TM-mediated” is thus suggested for use herein, with a preference for the former terminology. By analogy, Kozlowski and Loon suggested that the most general term applicable to the entire field of TM-assisted reactions for organic synthesis be “photocatalysis”, since the mechanism of energy transfer is typically unknown.100
TM-mediated gas production from formic acid
000-fold enhancement in PVG yield of W(CO)6 elicited by its presence.16 Such impact is more congruent with a catalytic than LMCT effect. Whereas Cd(II) is beneficial for PVG of W, Co(II) is completely ineffective and Cu(II) is reportedly 1000-fold less effective than that induced by the presence of Cd(II). As another typical example, the 4100-fold increase in yield of PVG generated Pb from formic acid following the addition of 6 μg mL−1 Ni(II)13 can be realized despite the minor enhancement in [CO] produced with addition of 20 Ni(II) μg mL−1 to this medium, evident in Fig. 9. A further example is evident from the work of Hou et al., reporting that in the presence of 20 μg mL−1 of Co(II) or Cd(II), the PVG-ICP-MS response for Ru increased by 140- and 45-fold, respectively.34 From the perspective of the small relative changes in the concentration of available CO as a reaction partner, it is not possible to readily account for these effects. Although TM reactions such as illustrated in eqn (26) may influence production of CO, the effects of such increases and their specificity are difficult to rationalize based on such simple scenarios. However, if the CO concentration in solution is sufficiently in excess of the target analyte during PVG in the absence of added TM, then it will not be the rate limiting or critical reagent supporting synthesis. The impact of the presence of the TM on other gases, as proxies for radicals, must also be examined for a more holistic approach.
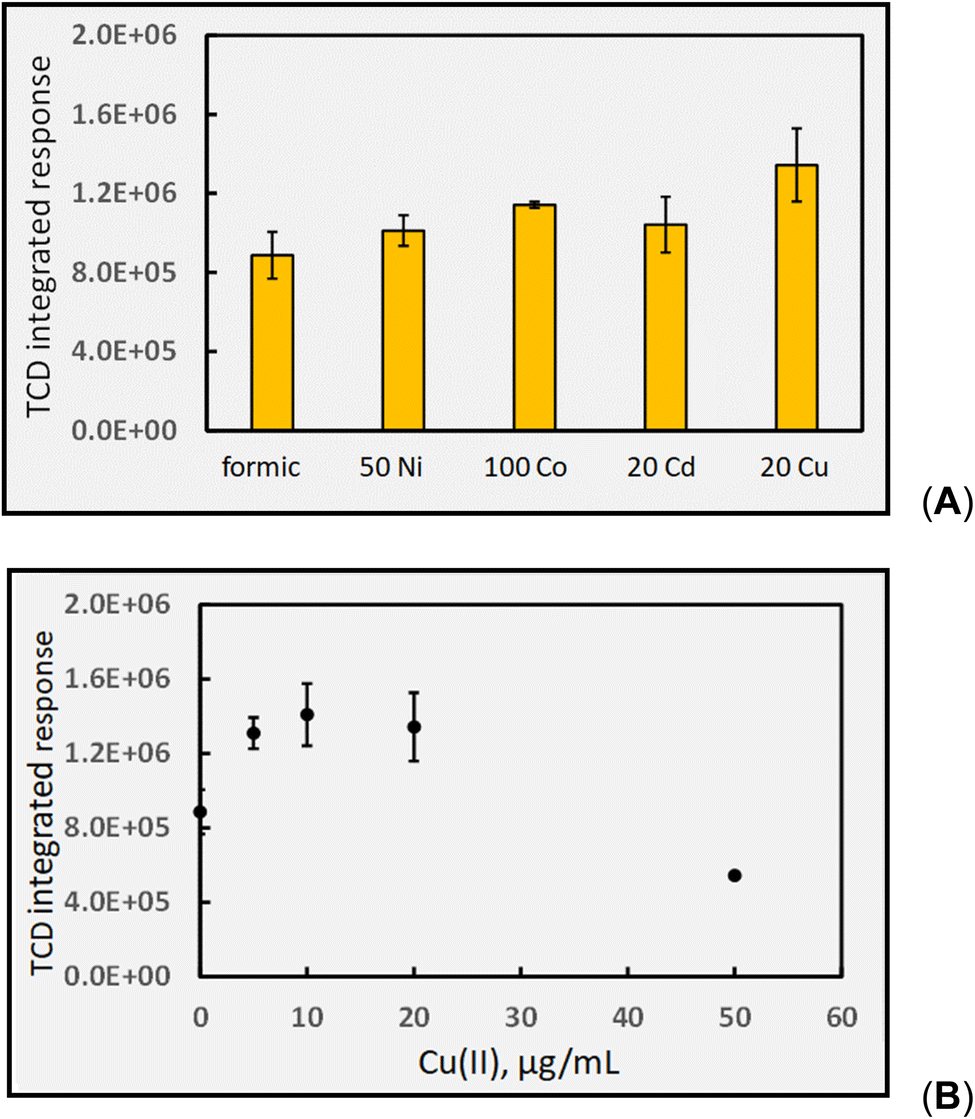 | ||
| Fig. 9 Impact of added TMs (TM concentrations given as μg mL−1) on HS CO response (generation). Photolysis of 5% (v/v) formic acid at a solution flow rate of 2 mL min−1 (IT = 22 s). Ordinate displays integrated response characterizing chromatographic peaks. (A) Uncorrected HS [CO] increases from 4290 ± 570 ppm to 6550 ± 900 ppm (repeatability SD, n = 3) in the presence of 20 μg mL−1 Cu(II). (B) Effect of increasing Cu(II) on HS CO response (generation). | ||
Selecting Cu(II) for further investigation, as it appears the most influential, Fig. 9B shows the effects of increasing concentrations of Cu(II) in the 5% formic acid medium on CO generation. In this example, the initial arbitrary selection of 20 μg mL−1 shown in Fig. 9A was serendipitously close to that proving optimal in this system. Noteworthy is the overall shape of the function in that a maximum is present, reflecting the typical performance metric reported with all analytical PVG systems wherein the PVG yield of the targeted volatile metallic analyte species follows a similar profile noted here for the effects of added TMs.13,16,20,24,27,32,34,35,82 The initial rate of increase with [Cu(II)] is short-ranged and one explanation frequently used to account for the decreasing efficiency at higher concentrations of added TMs is that their presence introduces an increasing shadowing effect whereby absorption of incident radiation due to TM d–d and charge transfer transitions of their carboxylate complex(es) inhibits primary photochemical reactions needed to generate the targeted analytical products. Unfortunately, it is rare that any additional absorption bands are identified in the UV spectra of TM adulterated PVG solutions and thus this argument is generally weak. Formation of Cu0 NPs at high concentrations could lead to a scavenging of free CO and release of volatile Cu–CO species. In such situations, of course, the presence of Cu(II) in test samples at this concentration may likely be considered an interferant in the PVG process.
Further to this point, and in connection with the earlier introduced reaction 29 involving consumption of methyl radicals in the presence of NPs, it is instructive to consider the following general reaction which should be taken into consideration for synthesis of TM-carbonyls, as it often presents a practical limit to the issue of selecting the optimal concentration of added TM to effect the largest impact on the desired analyte PVG reaction while minimizing adverse effects:
 | (36) |
High concentrations of TM carbonyl compounds can result in significant contamination of mass spectrometer detection systems as they are employed at 10's of ppm or higher concentrations. An equally important consideration, however, is the impact of co-generation of such compounds on the consumption of available CO, H2 or H˙ species. A semiquantitative estimate of this effect may be undertaken using PVG generation of Ni(CO)4 as an example. This is an analytically attractive process and occurs with high efficiency at trace concentrations. Assuming application of a 50 μg mL−1 concentration of Ni(II) as a PVG mediator, as used in this study, significant amounts of Ni(CO)4 would be generated. It is rather unlikely that this process continues with uniform high efficiency over such a broad concentration range, but if 50% is assumed, this would amount to a consumption of approximately 75 μL min−1 CO during a typical photolysis operation wherein only 320 μL min−1 CO is photolytically generated in 5% (v/v) formic acid containing no added TMs (cf. earlier discussions of overall oxidation efficiency). This may represent a significant sink for available CO. Similar calculations come into play considering hydrogen and methane. Very likely, PVG of TM carbonylated species occurs with significantly lower efficiencies at concentrations orders of magnitude higher than heretofore examined under analytical PVG concentrations. However, it is possible that actual generation of CO and H2via photolysis may be significantly higher (by up to 50%) in the Ni(II) and Co(II) systems studied here than is represented in Fig. 9 and 10 (below) due to its competitive uptake (apparent loss) as a TM carbonyl compound. In this connection, it would be of interest to determine residual TM content in the waste solutions of photolysed media.
 | ||
| Fig. 10 Impact of added TMs (TM concentrations given as μg mL−1) on HS H2 response (generation). Photolysis of 5% (v/v) formic acid at a solution flow rate of 2 mL min−1 (IT = 22 s). Ordinate displays integrated response characterizing chromatographic peaks. (A) Uncorrected HS [H2] increases from 370 ± 20 ppm to 2880 ± 80 ppm (repeatability SD, n = 3) in the presence of 20 μg mL−1 Cu(II). (B) Effect of increasing Cu(II) on HS H2 response. | ||
The issue arises as to what means, in an otherwise identical system, can the products of photolysis be so significantly altered with regard to CO or H2 for a given added TM ion; again, LMCT reactions are completely inconsistent in this regard.
Fig. 10B shows the impact of [Cu(II)] on H2 generation. As with CO generation, an optimum concentration is evident, suggesting a possible common origin. The relatively rapid drop in enhanced production of CO and H2 with a further increase in added Cu(II) cannot be accounted for by either spectral shadowing or invoking a LMCT model of enhanced radical production, despite interpretation of ESR evidence, which unfortunately has never been quantitatively investigated.
A further observation that is not possible to rationalize is the synergistic effect of co-addition of two TMs to the sample solution, a frequent observation in the more recent analytical PVG literature wherein combination of individual TMs provides for an enhanced synthesis of targeted analytes beyond the summed impact of either.6,33–36Fig. 11 illustrates the synergistic impact of the simultaneous presence of both Co(II) and Cd(II) in a 5% solution of formic acid wherein the yield of H2 is clearly greater than the sum of the impact of either TM alone. Obviously, if each TM-formate complex contributes to an enhanced production of radicals through their associated LMCT processes, it is difficult to account for a synergistic effect when both are present; the mechanism itself must be altered.
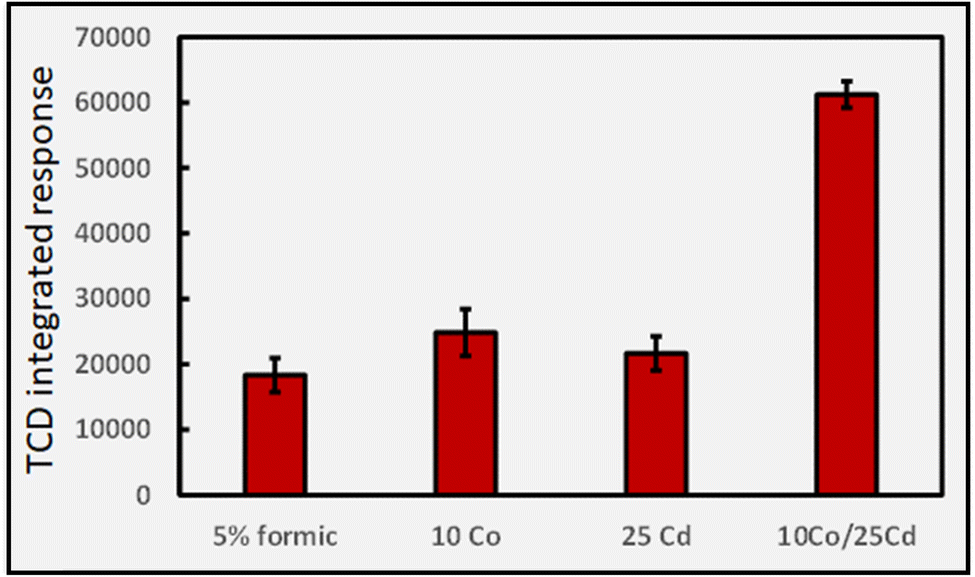 | ||
| Fig. 11 Synergistic effect of the simultaneous presence of two TMs (concentrations given as μg mL−1) on HS H2 response (generation) during photolysis of 5% (v/v) formic acid at a solution flow rate of 2 mL min−1 (IT = 22 s; repeatability SD, n = 3). Ordinate displays integrated response characterizing chromatographic peaks. | ||
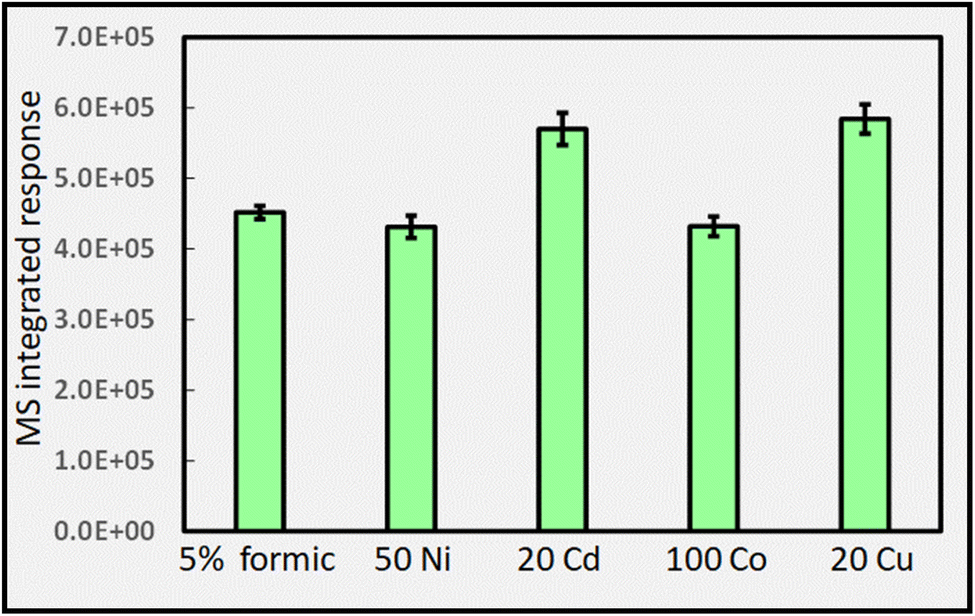 | ||
| Fig. 12 Impact of added TMs (concentrations given in μg mL−1) on HS CO2 response (generation) during photolysis of 5% (v/v) formic acid at a solution flow rate of 2 mL min−1 (IT = 22 s; repeatability SD, n = 3). Ordinate units are integrated response over chromatographic peak. | ||
TM-mediated gas production from acetic acid
 | ||
| Fig. 13 Impact of added TMs (concentrations given as μg mL−1) on HS CH4 response (generation) from photolysis of 5% (v/v) acetic acid at 2 mL min−1 (IT = 32 s; repeatability SD, n = 3). Ordinate displays integrated response characterizing chromatographic peaks. | ||
As Co(II) appears most beneficial for CH4 generation, the impact of varying its concentration was examined in the range 0–200 μg mL−1. An optimum was evident near 75 μg mL−1, beyond which response decreased, reaching 70% of maximum at 200 μg mL−1. This pattern produced by increasing TM concentrations remains consistent with all other gases generated in the formic acid system as well as being reflected by their impact on analyte targeted analytical PVG systems.
The use of a mixture of added Cd(II) and Co(II) (25 and 10 μg mL−1, respectively) on CH4 production did not lead to a synergistic effect, providing the same response as that from 25 μg mL−1 Cd(II) alone. In accordance with the possible existence of an ionic bimetallic-coupled photoredox catalytic cycle,44 such a TM mixture, demonstrated to occur for one analytical PVG system, should be viable in all, irrespective of the intended analyte target.
As earlier discussed in relation to Fig. 6B, photolysis of acetic acid generates C2H6 as a consequence of reaction 19. The impact of added Co(II) in the range 0–200 μg mL−1 on C2H6 generation was remarkable in that it provided a rather monotonic decrease in C2H6 response from 1080 ± 160 (repeatability SD: n = 4) to 690 ± 50 (n = 4). Clearly, the reaction is influenced by the presence of Co(II) but the mechanism of action is unclear since, as shown in Fig. 13, Co(II) enhances CH4 production (implying enhancements in precursor ˙CH3 production) but its presence apparently inhibits reaction (19).
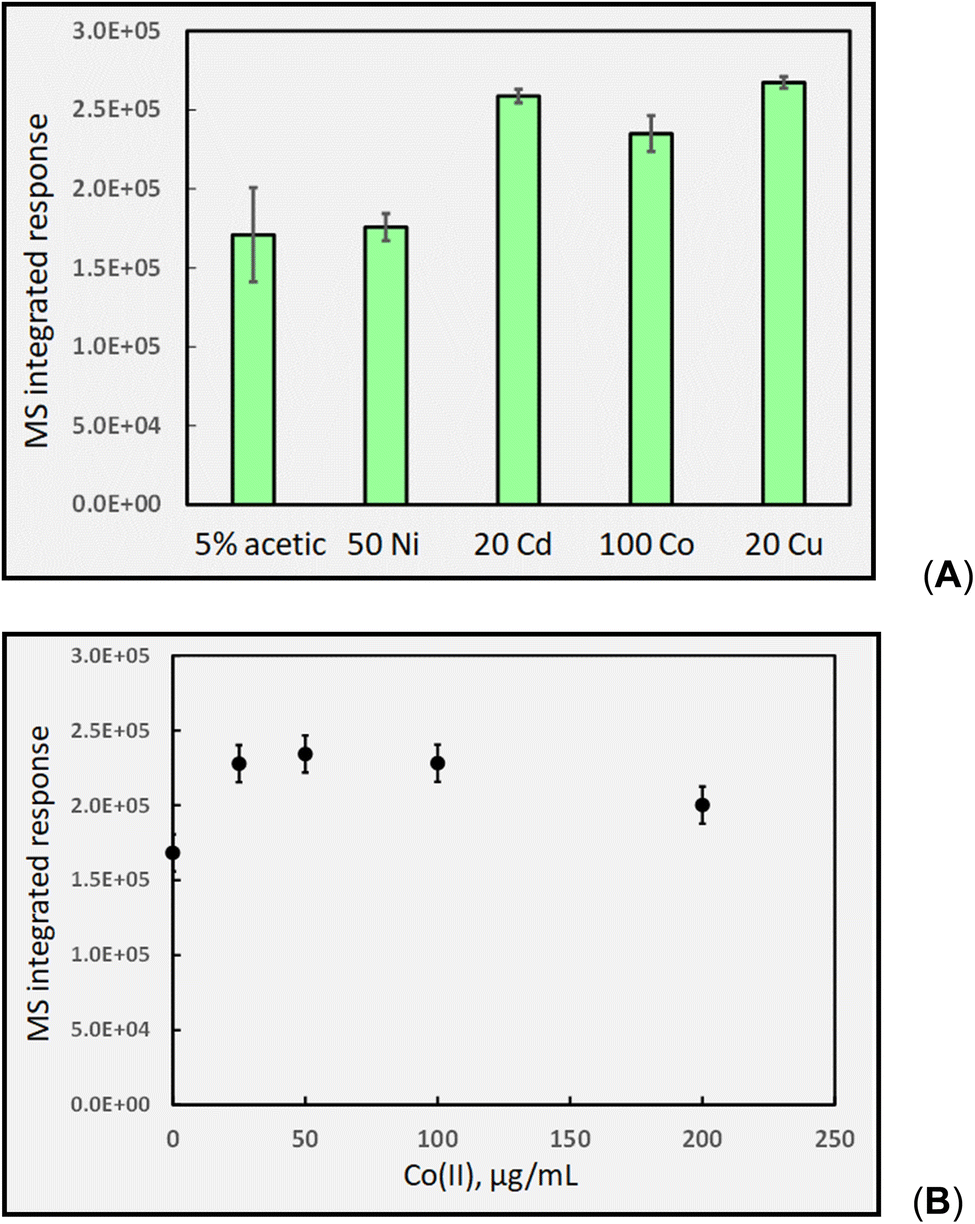 | ||
| Fig. 14 (A) impact of added TMs (concentrations given as μg mL−1) on response (generation) of HS CO2 during photolysis of 5% (v/v) acetic acid at 2 mL min−1 (IT = 32 s). Ordinate displays integrated response characterizing chromatographic peaks; (B) effect of added Co(II) on HS CO2 response (generation). Uncorrected HS [CO2] increases from 1670 ± 100 ppm to 2160 ± 40 ppm (repeatability SD, n = 4) in the presence of 100 μg mL−1 Co(II). | ||
Fig. 14B shows the typical impact of increasing Co(II) concentration on CO2 generation, once again illustrating the presence of a generic optimum that is also reflected in the analytical PVG literature for generation of volatile species of all targeted analytes, as discussed earlier.
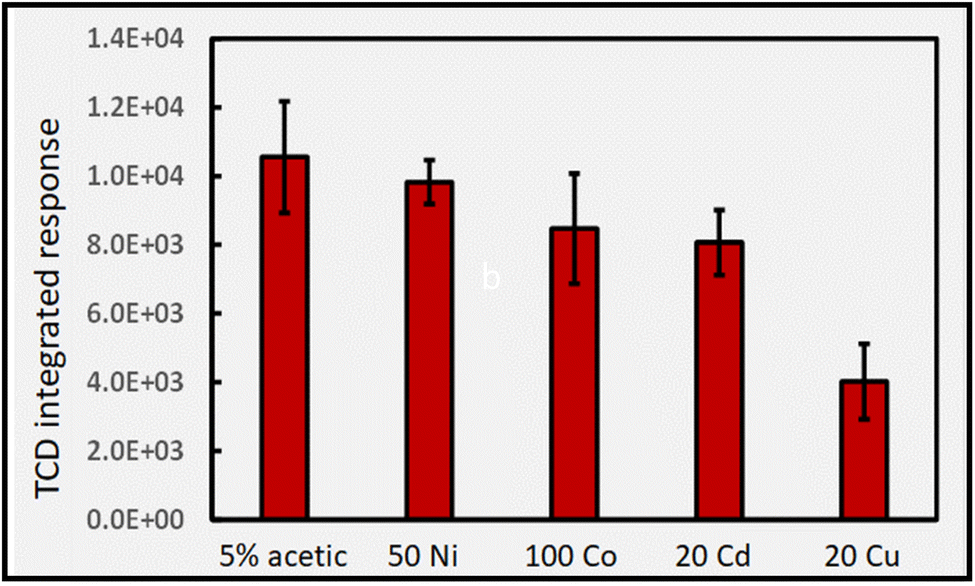 | ||
| Fig. 15 Impact of added TMs (concentrations given as μg mL−1) on response (generation) of H2 from photolysis of 5% (v/v) acetic acid at 2 mL min−1 (IT = 32 s; repeatability SD: n = 3). Uncorrected HS [H2] in 5% acetic acid is 270 ± 40 ppm. Ordinate displays integrated response characterizing chromatographic peaks. | ||
Although a synergistic effect on H2 production was very pronounced when a combination of Co(II) and Cd(II) was present in 5% formic acid (cf.Fig. 11) no such trend arises when the experiment is replicated using 5% acetic acid, highlighting the uniqueness of catalyst substrate/reactant interactions in changing from formic to acetic acid.
Part 3. Possible mechanisms of action of added TMs
A general model based on enhanced radical production in formic and acetic acids in the presence of added TMs is supported by direct ESR evidence (mostly limited to ˙CH3 and CO2˙− with a single report for H˙62). LMCT reactions involving TM-carboxylate complexes efficiently harvest longer wavelength photons from the photoreactors, potentially leading to more favorable PVG reaction kinetics. However, this study exposes several shortcomings with use of this model:(i) A limitation or saturation in the magnitude of the analytical effect as the concentration of TMs is increased as generally exhibited by an asymptotic impact profile that is not likely due to spectral shadowing (minimal effect in the UV-B region supporting LMCT processes) and generally fails to reveal evidence of any significant or distinctive TM-carboxylate LMCT bands in examined UV-Vis spectra of TM-spiked PVG solution media which could account for their action; note that PVG halide systems display broad concentration ranges over which added TMs do not result in decreased synthesis efficiencies, highlighting a distinctly different mechanism of action.
(ii) A specificity of effects unique to particular TM-analyte combinations is evident in the PVG literature; in accordance with a generalized LMCT model, a positive impact should be expected which transcends specific TM-analyte combinations as no perturbation to the photochemical system is made apart from the addition of a TM; such specificity cannot be accounted for by LMCT processes and appears equally evident in the impact of specific TMs affecting yields of stable gases which arise from radical precursors.
(iii) Synergistic interactions amongst added TMs cannot be accounted for based on LMCT reactions proportionally contributing to the population of “radical reagents”; a simple sum of the effects of each is expected due to their independence; as noted earlier, possible ionic bimetallic-coupled photoredox catalytic cycles44 may occur, but should remain effective irrespective of the coupled analyte target.
(iv) Inconsistent effects amongst the generation patterns of gases in the presence of the various TMs occur, a most dramatic example being a decrease in methane and hydrogen production in the presence of Cu, which cannot be accounted for by LMCT events.
(v) Catalytic redox cycle reactions, such as those proposed by Adams and Hart60 involving the presence of variable valence TMs (i.e., reactions (26)–(28)), may occur with photolytically generated radicals such as ˙COOH and HCOO˙ enhancing rates of generation of CO and CO2, but this effect (at least for Ni, Co, Cd and Cu) is quantitatively small (<30% change) for both CO (Fig. 9) and CO2 (Fig. 12) and any such increases in intermediate reducing radicals would not be expected to yield major enhancements in PVG analyte efficiencies.
An alternative model that may more comprehensively and consistently account for all of the above suggests intervention by homogeneous generation of (photo)catalytic nanoparticles (NPs) from the added TMs. Their formation during the PVG process cannot be disputed; they have been visually evident, as reported by the formation of pink-colored Se0 deposits when formic acid solutions containing mg L−1 concentrations of Se(IV) are subjected to PVG96 and, more recently, identified as being present in several PVG systems by TEM imaging techniques.24,33,37,101,102 A most recent example, presented in Fig. 16, shows islands or clusters comprising bimetallic Cd and Co NPs isolated from a mixture of 50 μg mL−1 Cu(II)/100 μg mL−1 Co(II) in 0.045% (v/v) formic acid following PVG. Individual NPs typically appear to be <5 nm in diameter within their 50 nm clusters.
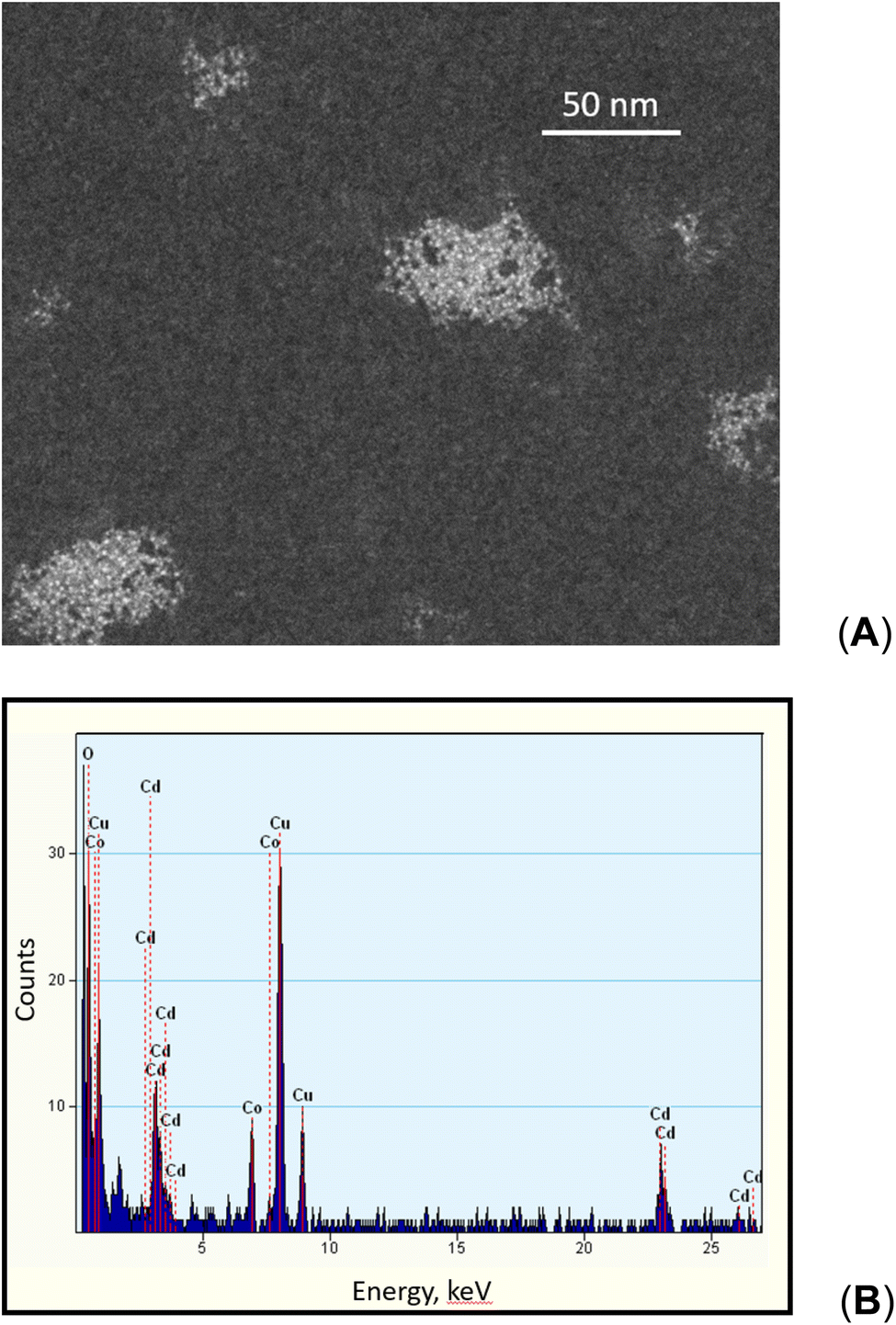 | ||
| Fig. 16 (A) TEM image of clusters of mixed Cd/Co comprising NPs of diameter < 5 nm synthesized during PVG of 0.045% (v/v) formic acid containing 50 μg mL−1 Cd(II)/100 μg mL−1 Co(II); (B) EDX profile of a cluster (images courtesy of M. Couillard, EME/NRC). | ||
Although single particle ICP-MS (SP-ICP-MS) would seem to be a convenient technique for detection of such NPs, no such reports have appeared, possibly because of insufficient detection power due to their small size (<10 nm) coupled with the elevated background arising from the high dissolved ionic fraction of 10's of mg L−1 added TMs.103,104 It would appear that, for the selected TMs used in this study (Ni, Co, Cd and Cu), current SP-ICP-MS detection approaches are not tenable (note that the 50 nm islands shown in Fig. 16A would be disaggregated prior to applying SP-ICP-MS due to commonly used sample preparation techniques).
Catalytic (thermal) processes can broadly be characterized as either associative or redox. With the former, one or more reactant molecules are adsorbed onto active surface sites in such an energetically favorable environment that intermediates form and products desorb. The latter redox mechanism engages changes in oxidation state of the catalytic material innate to their variable valence states, such as involvement of surface oxygen vacancies. In both cases, initial adsorption or surface interaction is inherent to the catalytic process and efficiencies are influenced by the Sabatier principle. In the context of PVG, which is driven by UV-C irradiation, it is evident that excited states of reactant molecules are already engaged to homogeneously produce the needed radicals that lead to analytical products in the absence of added TMs. Addition of TMs to this system may, in successful cases, result in production of TM NPs and photocatalytic effects associated with excitation of electron–hole pairs either within their conduction band (intraband excitation) or between the d-band and the conduction band (interband excitation) where they can reduce surface-adsorbed species or serve as oxidizing agents, allowing redox processes to occur at lower UV energies and with higher efficiency by utilizing longer wavelength source radiation.105 Most recent evidence favors the role of interband transitions.106 In this vein, it may be possible to imagine a scenario akin to electrophotocatalysis75 wherein the SC properties of the TM and/or transition metal oxide (TMO) NPs serve to establish the electrochemical potentials helping to drive reactions of surface adsorbed species.107
Such significant impacts of heterogeneous SC and MOF additives are already well-appreciated and utilized across many fields of photochemical research and application. Specifically, early PVG studies reported extensive use and performance enhancements arising from the presence of nano-TiO2 and noble metal decorated TiO2 systems.7,8,10,29,108 These structures facilitate rapid reactions on their surfaces to which the reaction partners have adsorbed, and make more efficient use of photogenerated electrons as a consequence of their electronic bandgaps, as detailed by Amal and colleagues in the case of Se(VI),109 to provide more energetically favorable routes for radical generation. ESR studies have corroborated enhanced photochemical yields of CO2˙− in formic acid47 and ˙CH3 in acetic acid48 in the presence of added SCs.
Zou et al.12,110 have summarized uses of not only conventional heterogeneous catalysts such as MOFs and TiO2, but also homogeneous “catalysts” based on TMs added to PVG systems. However, no mechanistic pathways were proffered by which the latter actually functioned or could be justified to be addressed as “catalysts”.
In contrast to their heterogeneous counterparts (primarily TiO2 to date), the size, shape and composition of metallic NPs significantly affects their catalytic performance; as they become smaller, new chemical and physical properties emerge as specific surface areas increase along with increases in reactive edge, step and corner sites. Below some critical particle size, changes also occur in their electronic structures which become more quantized, leading to discrete electronic states rather than those of a continuous spectrum in a macroscopic bulk metal.111–113 Correspondingly, their size influences not only their impact on reaction rates, but also product selectivity as they possess varying crystallographic structures which have implications for chemical reactivity. Exchange interactions of adsorbate electrons with conduction band electrons alter the NP surface electron density, affecting the adsorption of the surface-bound species as well as the wavelength of the localized absorption band (Localized Surface Plasmon Resonance, LSPR), potentially controlling synthesis products as well as selectivity of the adsorber.114 Decay of resonant surface plasmons may result in direct excitation of an electron to an unoccupied adsorbate acceptor state, weakening the chemical bonds within the adsorbate to induce dissociation of the adsorbed species. Even in TM NPs lacking strong plasmonic resonance due to their unfilled d-orbitals, interband excited electrons in these metal NPs can facilitate reactions at the NPs' surface via visible light excitation.106
The ubiquitous presence of dissolved oxygen (∼8 mg L−1) and its consequent photolysis by-products (e.g., O2˙− or  ) in PVG media suggests two potential roles. Firstly, the nascent TM atoms or their clusters may readily form TMO species (and binary TMOs with mixed TMs) with SC properties analogous to the well-known TiO2 discussed above.115 Such TMOs have also been actively utilized to promote catalytically enhanced redox reactions by nature of the variable valence properties of elements such as Cu, Ni, Co and Fe and numerous vacancy sites on their surfaces.116,117 Possible redox (electrochemical) reactions needed to reduce targeted analytical species during PVG could be catalytically enhanced in the presence of TMOs, aiding the homogeneous photoreduction reactions ascribed to H˙, R˙, and CO2˙−. Second, dissolved oxygen may play a direct role in a catalytic cycle wherein excited photogenerated surface electrons may react with O2 to create positively biased NP surface sites that foster adsorption of anionic partners (i.e., ionized carboxylate species) to further promote overall reactions.118
) in PVG media suggests two potential roles. Firstly, the nascent TM atoms or their clusters may readily form TMO species (and binary TMOs with mixed TMs) with SC properties analogous to the well-known TiO2 discussed above.115 Such TMOs have also been actively utilized to promote catalytically enhanced redox reactions by nature of the variable valence properties of elements such as Cu, Ni, Co and Fe and numerous vacancy sites on their surfaces.116,117 Possible redox (electrochemical) reactions needed to reduce targeted analytical species during PVG could be catalytically enhanced in the presence of TMOs, aiding the homogeneous photoreduction reactions ascribed to H˙, R˙, and CO2˙−. Second, dissolved oxygen may play a direct role in a catalytic cycle wherein excited photogenerated surface electrons may react with O2 to create positively biased NP surface sites that foster adsorption of anionic partners (i.e., ionized carboxylate species) to further promote overall reactions.118
Over the past few decades, numerous organic and inorganic reactions have been catalyzed using both colloidal (free) and supported TM and TMO nanocatalysts105,119–122 with a maximum in utilization and selectivity culminating in the extreme of (host matrix supported) single atom catalysts.123–125 Synthetic organic chemists are currently making significant use of the photochemistry of formate salts to perform the roles of reductant, carbonyl source and hydrogen atom transfer reagent in a variety of reactions.126 Correspondingly, it is not surprising that interest in their application to inorganic PVG systems has surfaced. With the above properties in mind, a model based on the impact of in situ photoreductive synthesis of TM NPs in PVG systems should be explored to more comprehensively rationalize observations reported in this study, as well as account for those in the recent PVG literature to a better degree than can be achieved using the current LMCT model.
It is clear that as NP catalysts exhibit significant size-dependent photoactivities,105 this property naturally gives rise to an optimum particle size distribution whose effects may vary, depending on the PVG analytical target, due to the need for simultaneously compatible surface interactions. Additionally, the spectral output of the photoreactor likely plays a role in determining the optimal NP size as there will be a trade-off between the impact of available photon intensity and the efficiency of its LSPR effect. In the flow-through photoreactor, time-dependent development of an optimal NP size distribution will be directly influenced by the initial concentration of the added TM. Whereas the precise mechanism and kinetics of formation of reduced atoms in solution and their agglomeration remains largely unknown for most systems, recent studies suggest that following initial reduction to atoms, small semiconducting clusters form which rapidly transition to plasmonic nanoparticles via agglomeration.127,128 An optimal TM solution concentration is thus to be expected and has been reported in every successful application of a TM-mediated PVG system. Decreasing performance characteristics at higher concentrations, often attributed to spectral shadowing effects, more likely manifests due to time dependent changes in NP size which alter the selective adsorption interactions of reactants onto the NPs. This is also mirrored in their influence on the relative yields of the photolysis gas products, as investigated in this study. It is expected that such TM concentration dependencies are unique and differ in range with different TMs and, by consequence because of their specificity of developed surface properties, also vary with the PVG analyte target; a TM most suitable for one PVG analyte may exhibit completely different performance characteristics with a different analyte, as attested to in the PVG literature.
Use of specific combinations of added TMs leading to unique synergistic impacts on PVG efficiencies is also readily rationalized by recognizing that bimetallic NPs can distinctively enhance catalytic properties for radical production as compared to those of their monometallic constituents.111,114,116 They would also be expected to exhibit diverse catalytic effects on different PVG analyte systems due to specific surface adsorption phenomena, consistent with observations in the PVG literature6,33–36,45 and in the general field of catalysis wherein small alterations to bi- or multi-metallic catalyst composition may significantly change desired performance.129,130
The addition of Cu(II) to formic acid significantly increases the yield of H2 (cf.Fig. 10) whereas the opposite occurs in the acetic acid system [decreasing both CH4 (Fig. 13) and H2 (Fig. 15)]. It has been reported that (photolytically generated) precursor radicals can be captured by suspensions of TM NPs in aqueous media, leading to their reduction and formation of long-lived NP-(CH3)x and NP-Hx surface species.73,74,131 Adsorption of H˙ onto the Cu0 NP surface fosters their desorption as H2 in a classical heterogeneous catalytic mechanism, thereby accounting for the enhanced H2 production. In acetic acid, additional reactions may occur due to the presence of ˙CH3 and a much smaller population of H˙ (possibly arising from photolysis of HCOOH impurity) to make this matter more complicated. Co-adsorption of ˙CH3 and H˙ would lead to production of a small increase in CH4 and a substantial decrease in H2 production due to the excess ˙CH3 in this system. Despite this, an overall decrease in CH4 generation may be arise due to reaction of adsorbed ˙CH3 with adjacent water molecules to produce CH3OH. Post photolysis thermal reactions of generated radicals in the acetic acid system are more varied than in formic acid.
In this adsorbed state, a variety of reactions, including those with adsorbed CO2, may proceed to yield acetic acid with a concurrent reduction in generation of CH4, H2 and C2H6.132 It is acknowledged that the cited publication (ref. 132) refers to a non-aqueous thermal reaction, but the principle reactions may still occur in the PVG system and would, moreover, be dependent on the TM NP composition and hence reflect the variable impact of the different TMs (and their combinations) examined in this study. Clearly, interpreting the impact of TMs via the LMCT model cannot account for any of the above observations.
In an effort to present a visual summary of the results, a chemometric analysis of the relative impact of the addition of each TM mediator on each measured gas in both acid media was attempted. Hierarchical Cluster Analysis (HCA), an unsupervised pattern recognition chemometric tool, was performed using MetaboAnalyst 6.0 software (freely available at https://www.metaboanalyst.ca/) to highlight any patterns that may be present. Uncorrected HS concentrations generated for each gas in the presence and absence of added TMs represented the sample input data. The gas concentrations were normalized, using auto-scaling, by which the data were centered on their mean divided by its standard deviation in each carboxylic acid. Such data pre-processing was necessary to ensure that all samples were accorded the same weight in subsequent treatment. After pre-processing, the (Euclidean) distances amongst the samples (original single point “clusters”) were calculated and interconnected using a Ward D linkage algorithm based on minimum variance when merging clusters. Fig. 17 summarizes the results wherein concentrations are reflected as colors (the deepest red representing highest while deepest blue reflects the lowest) and the horizontal and vertical lines comprise the dendrograms, i.e., summaries of the distance matrix. It is evident that the HCA treatment readily identifies which added TM has had the greatest impact on relative generation of a given gas compared to photolysis of the “unspiked” 5% (v/v) acid system. As examples, in a 5% (v/v) formic acid medium, FA Cu20 (i.e., 20 μg mL−1 Cu2+ added) revealed greatest impact on the generation of elevated concentrations of CO compared to its effect on the other gases; however, the highest concentration of H2 was obtained using FA Co100, and for CO2, FA Cd20 is unique. Turning to the 5% (v/v) acetic acid system, it is clear that greatest increase in concentration of CH4 occurs in the presence of 100 μg mL−1 added Co, i.e., AA Co100. Most significant is that overall, the impact of each added TM is unique for each gas in each acid and no clusters can be identified.
 | ||
| Fig. 17 HCA results depicted as a “heatmap” (Euclidean distances for clustering algorithm using Ward D methodology). AA: 5% (v/v) acetic acid; FA: 5% (v/v) formic acid; element and number designations represent added transition metal mediator to the PVG medium at the indicated concentration in μg mL−1. | ||
Conclusions and future studies
A complementary and novel approach to the usual direct observation of the impact of added TMs on the synthesis yields of PVG target analytes is presented here based on their effects on changes in the relative concentrations of generated H2, CO, CH4 and CO2 as stable end products of photolysis. Reducing radicals currently envisioned as responsible for analytical PVG processes (˙H, e(aq)−, ˙CH3, CO2˙−) are also precursors to these gases along with CO, permitting use of this new tool for such investigations. A photocatalytic mechanism mediated by homogeneous co-generation of TM/TMO nanostructures has been proposed which may more comprehensively account for their reported analytical effects than is possible via LMCT processes, particularly the notable selectivity accompanying improved PVG efficiencies of specific analytes.In contrast to frequent high and robust vapor generation efficiencies for numerous analytes, oxidation of formic and acetic acids (and corresponding radical production) appears to be very inefficient. Despite use of state-of-the-art flow-through photoreactors, overall oxidation does not exceed 1% under typical operating conditions. However, with the low concentrations of analytical targets (∼100 ng mL−1), generation of a 104-fold excess molar ratios of active radicals can readily account for high synthesis efficiencies of targeted metals and halogens.
The overall impact of added TMs likely derives from a combination of enhanced reaction rates arising from the in situ formation of (photo)catalytic TM/TMO structures coupled with an acknowledged background of LMCT reactions, both contributing to increased production of reducing radicals. However, very substantially increased synthesis yields of analytical targets amounting to more than an order of magnitude, and those exhibiting a synergistic response to specific combinations of added TMs, can more likely be attributed to the intervention of in situ generated catalytic surfaces.
Hou and colleagues37 noted that whereas CdSe, CdTe and CdS nanocatalysts were each able to catalyse the PVG generation of Se(CH3)2 from Se(VI), ZnS was distinctly inactive. This led to the conclusion that the metal centre of the SC catalyst (i.e., Cd) is key to successful use. This supports the high specificity of TM-NP-target analyte interactions. Clearly, significantly more work is required to investigate the impacts and actual mechanisms arising from addition of TMs to PVG systems. For comparison, the most common SC TMO in use, TiO2, should be examined for its influence on the profiles of generated stable gases. Building on the work of Amal et al.,109 and the purported mechanisms of action revealed for reduction of Se(VI), it would be of interest to determine in what manner this catalyst and its metal decorated forms perturb the stable gas profiles. Interestingly, these may be significantly altered in the presence of TiO2.71,133 Dey et al.133 reported that, in addition to the usual H2, CO and CO2 arising from photolysis of formic acid, a small yield of CH4 was also detected and putatively accounted for by chemical reduction of CO2. Although Gao and colleagues29 suggested that a synergistic interaction was evident for PVG of Te when iron was added to a mixture of acetic and formic acids containing TiO2, more likely TM NPs of reduced Fe were sequestered onto the existing catalyst surface to alter its Fermi level and provide for a more efficient reducing environment consistent with the characteristics of such a “decorated” catalyst.
The influence of solution pH was not examined in this study. Considering that pH is one of the most effective variables influencing not only the degree of ionization of the carboxylic acids, but the electrical charge characteristics of possible catalytic surfaces and thereby their affinity for adsorption of species, it may play a critical role governing the action of resultant TM-NPs and should be examined in detail as it may also influence the growth and lifetime of the NPs.
Potential losses of detected gaseous products of PVG may arise as a result of their precursor radical constituents participating in various competitive parallel thermal reactions. This was shown to be the case for methanol, which induced a small negative bias in detected CH4 concentrations in photolysed acetic acid and was implicated in potential loss of CO by formation of acetaldehyde during photolysis of formic acid. The concentration profiles of such water miscible products under various experimental conditions, notably in the presence of added TMs, need to be examined to more comprehensively expand the scope of this study to more rigorously account for any additional loss mechanisms for CO as well as CH4 and H2 proposed earlier in the presence of Cu0.
It would be of interest to contrast the results of this study with those obtained using a non-ozone generating low pressure photoreactor, precluding vacuum UV output and limited to wavelengths >254 nm, to ascertain whether the same trends are evident. Such a study is also interconnected with an evaluation of interference effects arising from TMs and whether any resulting NPs formed serve to enhance or mitigate interferences such as those due to nitrate/nitrite.
In this connection, these gas studies should be augmented with results generated from aerated and deoxygenated systems. This aspect is not currently controlled during PVG, despite being a potentially significant variable that may contribute to analytical PVG interference alleviation as well as alteration of the profile of gas production due to changes in the mechanisms of photolysis itself, as reported by Dey et al. during a study of TiO2 mediated photolysis of formic acid.133
Lastly, and likely of most immediate importance to the present discussion, more effort directed to detection and characterization of proposed TM/TMO species utilizing microscopic and optical techniques such as TEM, dynamic light scattering (DLS),134 asymmetric flow field-flow fractionation with multiangle light scattering (AF4-MALS)135 and (nano)particle tracking analysis (NTA)136 are needed in order to more convincingly underpin the foundations of this model. TEM imaging is potentially influenced by sample preparation techniques and is done off-line, typically via evaporation of a small volume of the photolysed sample onto a supporting grid, inviting the possibility of post-photolysis NP growth and nucleation (or even their dissolution) or interference from simple crystallization of the concomitant excess TM salt. DLS, on the other hand, can be readily configured for continuous on-line examination of the effluent from the GLS in real time, without operator intervention, providing particle size and distribution data in the range 1–1000 nm. NTA further provides information on particle size distribution in the range 10–1000 nm and number density of typically 106–109 cm−3. To place this latter metric into perspective, if an added 50 μg mL−1 Co(II) concentration is under investigation and assumed to be quantitatively transformed into 10 nm particles during photolysis, the resultant number density would be 1 × 1013 cm−3. Provided only 10−5% fraction was so formed, this would still be within the particle density range for NTA detection. As discussed earlier, although SP-ICP-MS may appear to be an ideal tool for such investigations, the current lack of any results supported by this technique may simply point to insufficient detection power if the responsible nanoparticles prove to typically be <10 nm in diameter.
Apart from the proposal that TM NPs are strongly implicated in the action of TM-mediated PVG, no description of an explicit mechanism by which this occurs has been advanced herein, aside from arguments suggesting that the analytical target ion or carboxylate complex is likely involved in order to account for the high specificity of effect with different TMs. Such mechanisms are beyond the scope of this work and need to be elucidated, but guidance may be drawn from the increasing literature devoted to metallaphotocatalysis of TMs under visible light to foster numerous organic reactions137,138 as well as a better understanding of the influence of metals used for catalyst decoration on available reactive electron species.139 In this connection, the influence of reaction wavelength requires attention, as initially investigated by Hou and colleagues for the limited case of PVG of Se wherein the attributes of a laser driven light source were highlighted as an ideal tool for this purpose.140
As with earlier complementary tutorials on PVG,3,4 this contribution bears a significant shortcoming in that it has neither proved nor disproved any proposed fundamental mechanisms. However, it aspires to provide a potentially more coherent framework with which to rationally approach the subject of TM-mediated PVG while hopefully enticing interested researchers and experts from complementary disciplines to conduct further investigations of many of the intriguing facets presented herein which will contribute to the continuing advancement of this field.
Data availability
Data supporting the experimental findings of this study are presented within the article; those mentioned by way of a simple summary statement are available from the corresponding author upon reasonable request.Author contributions
RES conceptualization, methodology, investigation, writing original draft, creation of original figures, data interpretation; EP methodology, investigation, data curation, review and editing; GSL methodology, investigation, data curation, review and editing; RSAN investigation; JKSB investigation.Conflicts of interest
The authors declare that there are no conflicts of interest associated with this manuscript.Acknowledgements
The authors are grateful to M. Couillard (Clean Energy Innovation Research Center/NRC) for kindly providing the TEM image of Cd/Co NP clusters (Fig. 16) and S. Musil (Institute of Analytical Chemistry of the Czech Academy of Sciences, Brno) for numerous helpful comments to this work.References
- Vapor Generation Techniques for Trace Element Analysis: Fundamental Aspects, ed A. D'Ulivo and R. E. Sturgeon, Elsevier, 2022 ISBN: 9780323858342 Search PubMed.
- R. E. Sturgeon and P. Grinberg, Some speculations on the mechanisms of photochemical vapor generation, J. Anal. At. Spectrom., 2012, 27, 222–231 RSC.
- R. E. Sturgeon, Photochemical vapor generation: a radical approach to analyte introduction for atomic spectrometry, J. Anal. At. Spectrom., 2017, 32, 2319–2340 RSC.
- D. Leonori and R. E. Sturgeon, A unified approach to mechanistic aspects of photochemical vapor generation, J. Anal. At. Spectrom., 2019, 34, 636–654 RSC.
- R. E. Sturgeon, Photo-sono-thermo-chemical vapor generation techniques, in Vapor Generation Techniques for Trace Element Analysis: Fundamental Aspects, ed A. D'Ulivo and R. E. Sturgeon, Elsevier, 2022 chapter 7 Search PubMed.
- Y. Zhen, H. Chen, M. Zhang, J. Hu and X. Hou, Cadmium and cobalt ions enhanced-photochemical vapor generation for determination of trace rhenium by ICP-MS, Appl. Spectrosc. Rev., 2022, 57, 318–337 CrossRef CAS.
- E. Kikuchi and H. Sakamoto, Kinetics of the reduction reaction of selenate ions by TiO2 photocatalyst, J. Electrochem. Soc., 2000, 147, 4589–4593 CrossRef CAS.
- Q. Wang, J. Liang, J. Qiu and B. Huang, Online pre-reduction of selenium(VI) with a newly designed UV/TiO2 photocatalysis reduction device, J. Anal. At. Spectrom., 2004, 19, 715–716 RSC.
- K. Chen, I. Hsu and Y.-C. Sun, Determination of methylmercury and inorganic mercury by coupling short-column ion chromatographic separation, on-line photocatalyst-assisted vapor generation, and inductively coupled plasma mass spectrometry, J. Chromatogr. A, 2009, 1216, 8933–8938 CrossRef CAS.
- C.-H. Lin, Y. Chen, Y.-A. Su, Y.-T. Luo, T.-T. Shih and Y.-C. Sun, Nanocomposite-coated microfluidic-based photocatalyst-assisted reduction device to couple high-performance liquid chromatography and inductively coupled plasma-mass spectrometry for online determination of inorganic arsenic species in natural water, Anal. Chem., 2017, 89, 5891–5899 CrossRef CAS.
- J. Jia, Z. Long, C. Zheng, X. Wu and X. Hou, Metal organic frameworks CAU-1 as new photocatalyst for photochemical vapor generation for analytical atomic spectrometry, J. Anal. At. Spectrom., 2015, 30, 339–342 RSC.
- Z. Zou, J. Hu, F. Xu, X. Hou and X. Jiang, Nanomaterials for photochemical vapor generation analytical atomic spectrometry, TrAC, Trends Anal. Chem., 2019, 114, 242–250 CrossRef CAS.
- Y. Gao, M. Xu, R. Sturgeon, Z. Mester, Z. Shi, R. Galea, P. Saull and L. Yang, Metal ion-assisted photochemical vapor generation for the determination of lead in environmental samples by MC-ICPMS, Anal. Chem., 2015, 87, 4495–4502 CrossRef CAS.
- G. Chen, B. Lai, N. Mei, J. Liu and X. Mao, Mercury speciation by differential photochemical vapor generation at UV-B vs. UV-C wavelength, Spectrochim. Acta, Part B, 2017, 137, 1–7 CrossRef CAS.
- J. Zhou, D. Deng, Y. Y. Su and Y. Lv, Determination of total inorganic arsenic in water samples by cadmium ion assisted photochemical vapor generation-atomic fluorescence spectrometry, Microchem. J., 2019, 146, 359–365 CrossRef CAS.
- J. Vyhnanovský, R. E. Sturgeon and S. Musil, Cadmium assisted photochemical vapor generation of tungsten for detection by ICPMS, Anal. Chem., 2019, 91, 13306–13312 CrossRef.
- Y. Yu, Q. Zhao, H. L. Bao, Q. Mou, Z. M. Shi, Y. L. Chen and Y. Gao, Determination of trace bismuth in environmental waters by ICP-MS with cobalt ion-assisted photochemical vapour generation, Geostand. Geoanal. Res., 2020, 44, 617–627 CrossRef CAS.
- Q. Mou, L. Dong, L. Xu, Z. L. Song, Y. Yu, E. H. Wang, Y. H. Zhao and Y. Gao, Integration of cobalt ion assisted Fenton digestion and photochemical vapor generation: a green method for rapid determination of trace cadmium in rice, J. Anal. At. Spectrom., 2021, 36, 1422–1430 RSC.
- Y. Yu, J. J. Hu, X. N. Zhao, J. C. Liu and Y. Gao, Photochemical vapor generation for germanium: synergistic effect from cobalt/chloride ions and air-liquid interfaces, Anal. Bioanal. Chem., 2022, 414, 5709–5717 CrossRef CAS PubMed.
- W. Zeng, J. Hu, H. J. Chen, Z. R. Zou, X. D. Hou and X. M. Jiang, Cobalt ion-enhanced photochemical vapor generation in a mixed acid medium for sensitive detection of tellurium(IV) by atomic fluorescence spectrometry, J. Anal. At. Spectrom., 2020, 35, 1405–1411 RSC.
- W. Zeng, Z. H. Hu, J. Luo, X. D. Hou and X. M. Jiang, Highly sensitive determination of trace antimony in water samples by cobalt ion enhanced photochemical vapor generation coupled with atomic fluorescence spectrometry or ICP-MS, Anal. Chim. Acta, 2022, 1191, 339361 CrossRef CAS PubMed.
- T. Xu, J. Hu and H. J. Chen, Transition metal ion Co(II)-assisted photochemical vapor generation of thallium for its sensitive determination by inductively coupled plasma mass spectrometry, Microchem. J., 2019, 149, 103972 CrossRef CAS.
- J. Hu, R. E. Sturgeon, K. Nadeau, X. D. Hou, C. Zheng and L. Yang, Copper ion assisted photochemical vapor generation of chlorine for its sensitive determination by sector field inductively coupled plasma mass spectrometry, Anal. Chem., 2018, 90, 4112–4118 CrossRef CAS PubMed.
- J. J. Hu, Y. Yu, Z. L. Xiao and Y. Gao, Photochemical vapor generation of zinc and gallium, Microchem. J., 2023, 193, 109178 CrossRef CAS.
- R. E. Sturgeon and E. Pagliano, Evidence for photochemical synthesis of fluoromethane, J. Anal. At. Spectrom., 2020, 35, 1720–1726 RSC.
- R. M. de Oliveira, D. L. G. Borges, P. Grinberg and R. E. Sturgeon, High-efficiency photoreductive vapor generation of osmium, J. Anal. At. Spectrom., 2021, 36, 2097–2106 RSC.
- Y. L. Wang, L. L. Lin, J. Liu, X. F. Mao, J. H. Wang and D. U. Qin, Ferric ion induced enhancement of ultraviolet vapour generation coupled with atomic fluorescence spectrometry for the determination of ultratrace inorganic arsenic in surface water, Analyst, 2016, 141, 1530–1536 RSC.
- Y. Yu, Y. Jia, Z. Shi, Y. Chen, S. Ni, R. Wang, Y. Tang and Y. Gao, Enhanced photochemical vapor generation for the determination of bismuth by inductively coupled plasma mass spectrometry, Anal. Chem., 2018, 90, 13557–13563 CrossRef CAS.
- H. Y. He, X. H. Peng, Y. Yu, Z. M. Shi, M. Xu, S. J. No and Y. Gao, Photochemical vapor generation of tellurium: synergistic effect from ferric ion and nano-TiO2, Anal. Chem., 2018, 90, 5737–5743 CrossRef CAS.
- J. Šoukal, R. E. Sturgeon and S. Musil, Efficient photochemical vapor generation of molybdenum for ICPMS detection, Anal. Chem., 2018, 90, 11688–11695 CrossRef.
- E. Nováková, K. Horová, V. Červený, J. Hraníček and S. Musil, UV photochemical vapor generation of Cd from a formic acid based medium: optimization, efficiency and interferences, J. Anal. At. Spectrom., 2020, 35, 1380–1388 RSC.
- L. Dong, H. J. Chen, Y. Y. Ning, Y. W. He, Y. Yu and Y. Gao, Vanadium species-assisted photochemical vapor generation for direct detection of trace tellurium, Anal. Chem., 2022, 94, 4770–4778 CrossRef CAS PubMed.
- X. Deng, L. Dong, H. Chen, W. Wang, Y. Yu and Y. Gao, Sensitive determination of arsenic by photochemical vapor generation with inductively coupled plasma mass spectrometry: synergistic effect from antimony and cadmium, Anal. Chem., 2024, 96, 652–660 CrossRef CAS PubMed.
- Q. Yang, H. Chen, J. Hu, K. Huang and X. Hou, Simultaneous detection of ruthenium and osmium by photochemical vapor generation-inductively coupled plasma-mass spectrometry, Anal. Chem., 2022, 94, 593–599 CrossRef CAS PubMed.
- S. Musil, J. R. Vyhnanovský and R. E. Sturgeon, Ultrasensitive detection of ruthenium by coupling cobalt and cadmium ion-assisted photochemical vapor generation to inductively coupled plasma mass spectrometry, Anal. Chem., 2021, 93, 16543–16551 CrossRef CAS.
- S. Musil, E. Jeníková, J. R. Vyhnanovský and R. E. Sturgeon, Highly efficient photochemical vapor generation for sensitive determination of iridium by inductively coupled plasma mass spectrometry, Anal. Chem., 2023, 95, 3694–3702 CrossRef CAS.
- F. Xu, Z. Zou, J. He, M. Li, K. Xu and X. Hou, In situ formation of nano-CdSe as a photocatalyst: cadmium ion-enhanced photochemical vapour generation directly from Se(VI), Chem. Commun., 2018, 54, 4874–4877 RSC.
- R. M. de Oliveira, D. L. G. Borges, P. Grinberg, Z. Mester and R. E. Sturgeon, Copper-ion assisted photochemical vapor generation of bromide and bromate, J. Anal. At. Spectrom., 2021, 36, 1235–1243 RSC.
- J. M. Carraher, O. Pestovsky and A. Bakac, Transition metal ion-assisted photochemical generation of alkyl halides and hydrocarbons from carboxylic acids, Dalton Trans., 2012, 41, 5974–5980 RSC.
- A. Bakac, Effects of halide ions and carbon monoxide on the reactions of iron(III) with alkyl radicals, Croat. Chem. Acta, 2001, 74, 633–640 CAS.
- R. Ge, Z. Xu, K. Yang and H. Ge, Transition-metal-catalyzed C(sp3)-H bond fluorination reactions, Chem Catal., 2024, 4, 101009 CrossRef CAS.
- G. Ferraudi, Photochemical generation of metastable methylcopper complexes. Oxidation-reduction of methyl radicals by copper complexes, Inorg. Chem., 1978, 17, 2506–2508 CrossRef CAS.
- A. W. Adamson, W. L. Waltz, E. Zinato, D. W. Watts, P. D. Fleischauer and R. D. Lindholm, Photochemistry of transition-metal coordination compounds, Chem. Rev., 1968, 68, 541–585 CrossRef CAS.
- P. Ciesla, P. Kocot, P. Mytych and Z. Stasicka, Homogeneous photocatalysis by transition metal complexes in the environment: a review, J. Mol. Catal. A: Chem., 2004, 224, 17–33 CrossRef CAS.
- J. Hu, H. Chen, X. Hou and X. Jiang, Cobalt and copper ions synergistically enhanced photochemical vapor generation of molybdenum: mechanism study and analysis of water samples, Anal. Chem., 2019, 91, 5938–5944 CrossRef CAS PubMed.
- J. Hu, C. Li, Y. Zhen, H. Chen, J. He and X. Hou, Current advances of chemical vapor generation in nontetrahydroborate media for analytical atomic spectrometry, Trends Anal. Chem., 2022, 155, 116677 CrossRef CAS.
- L. L. Perissinotti, M. A. Brusa and M. A. Grela, Yield of carboxyl anion radicals in their photocatalytic degradation of formate over TiO2 particles, Langmuir, 2001, 17, 8422–8427 CrossRef CAS.
- M. Kaise, H. Nagai, K. Tokuhashi and S. Kondo, Electron spin resonance studies of photocatalytic interface reactions of suspended M/TiO2 (M = Pt, Pd, Ir, Rh, Os or Ru) with alcohol and acetic acid in aqueous media, Langmuir, 1994, 10, 1345–1347 CrossRef CAS.
- P. C. Ford, From curiosity to applications. A personal perspective on inorganic photochemistry, Chem. Sci., 2016, 7, 2964–2986 RSC.
- T. Suzuki, R. E. Sturgeon, C. Zheng, A. Hioki, H. Tao and T. Nakazato, Influence of speciation on response from selenium to UV-photochemical vapor generation, Anal. Sci., 2012, 28, 807–811 CrossRef CAS PubMed.
- R. E. Sturgeon, Quantitation of bromine by ICP-ToF-MS following photochemical vapor generation, Anal. Chem., 2015, 87, 3072–3079 CrossRef CAS.
- H. Coutinho de Jesus, P. Grinberg and R. E. Sturgeon, System optimization for determination of cobalt in biological samples by ICP-OES using photochemical vapor generation, J. Anal. At. Spectrom., 2016, 31, 1590–1604 RSC.
- G. V. Buxton and R. M. Sellers, The radiation chemistry of metal ions in aqueous solution, Coord. Chem. Rev., 1977, 22, 195–274 CrossRef CAS.
- M. G. Gonzalez, E. Oliveros, M. Worner and A. M. Braun, Vacuum-ultraviolet photolysis of aqueous reaction systems, J. Photochem. Photobiol., C, 2004, 5, 225–246 CrossRef CAS.
- J. A. Kloepfer, V. H. Vilchiz, V. A. Lenchenkov, A. C. Germaine and S. E. Bradforth, The ejection distribution of solvated electrons generated by the one-photon photodetachment of aqueous I− and two photon ionization of the solvent, J. Chem. Phys., 2000, 113, 6288–6307 CrossRef CAS.
- K. Zoschke, H. Börnick and E. Worch, Vacuum-UV radiation at 185 nm in water treatment – a review, Water Res., 2014, 52, 131–145 CrossRef CAS.
- J. Zechner and N. Getoff, Photoproduction of hydrated electrons from formate ions at 184.9 nm, Int. J. Radiat. Phys. Chem., 1974, 6, 215–218 CrossRef CAS.
- G. Imoberdorf and M. Mohseni, Modeling and experimental evaluation of vacuum-UV photoreactors for water treatment, Chem. Eng. Sci., 2011, 66, 1159–1167 CrossRef CAS.
- G. V. Buxton and R. M. Sellers, Acid dissociation constant of the carboxyl radical. Pulse radiolysis studies of aqueous solutions of formic acid and sodium formate, J. Chem. Soc., Faraday Trans. 1, 1973, 69, 555–559 RSC.
- G. E. Adams and E. J. Hart, Radiolysis and photolysis of aqueous formic acid. Carbon monoxide formation, J. Am. Chem. Soc., 1962, 84, 3994–3999 CrossRef CAS.
- E. J. Hart, Mechanism of the γ-ray induced oxidation of formic acid in aqueous solution, J. Am. Chem. Soc., 1951, 73, 68–73 CrossRef CAS.
- J. Hu, C. Li, H. Chen and X. Hou, Photochemical hydride generation in sulfite medium: sample introduction into an inductively coupled plasma mass spectrometer for the determination of ultratrace inorganic arsenic, Anal. Chem., 2023, 95, 10498–10503 CrossRef CAS.
- E. Jeníková, J. Vyhnanovský, K. Hašlová, R. E. Sturgeon and S. Musil, Efficient photochemical vapor generation from low concentration formic acid media, Anal. Chem., 2024, 96, 1241–1250 CrossRef PubMed.
- A. J. Allmand and L. Reeve, The photochemical decomposition of aqueous formic acid solutions, J. Chem. Soc., 1926, 129, 2852–2863 Search PubMed.
- H. Su, Y. He and F. Konga, Photodissociation of formic acid, J. Chem. Phys., 2000, 113, 1891–1897 CAS.
- J. Thøgersen, S. K. Jensen, O. Christiansen and S. R. Keiding, Fast photodynamics of aqueous formic acid, J. Phys. Chem. A, 2004, 108, 7483–7489 Search PubMed.
- M. Burton, The photolysis of acetic acid, J. Am. Chem. Soc., 1936, 58, 692 CAS.
- P. Ausloos and E. W. R. Steacie, The vapor phase photolysis of acetic acid, Can. J. Chem., 1955, 33, 1530–1535 CAS.
- J. L. Wilkerson and W. A. Guillory, Condensed phase photochemistry of acetic acid in the vacuum UV, J. Photochem., 1977, 7, 261 CrossRef.
- L. J. Mittal, J. P. Mittal and E. Hayon, Photo-induced decarboxylation of aliphatic acids and esters in solution. Dependence upon state of protonation of the carboxyl group, J. Phys. Chem., 1973, 77, 1482–1487 CrossRef CAS.
- S. Hamid, I. Ivanova, T. H. Jeon, R. Dillert, W. Choi and D. W. Bahnemann, Photocatalytic conversion of acetate into molecular hydrogen and hydrocarbons over Pt/TiO2: pH dependent formation of Kolbe and Hofer-Moest products, J. Catal., 2017, 349, 128–135 CrossRef CAS.
- R. Chelli, R. Righini and S. Califano, Structure of liquid formic acid investigated by first principle and classical molecular dynamics simulations, J. Phys. Chem. B, 2005, 109, 17006–17013 CrossRef CAS PubMed.
- R. Bar-Ziv, I. Zilbermann, T. Zidki, G. Yardeni, V. Shevchenko and D. Meyerstein, Coating Pt0 nanoparticles with methyl groups: the reaction between methyl radicals and Pt0 NPs suspended in aqueous solutions, Chem.–Eur. J., 2012, 18, 6733–6736 CrossRef CAS PubMed.
- R. Bar-Ziv, I. Zilbermann, O. Oster-Golberg, T. Zidki, G. Yardeni, H. Cohen and D. Meyerstein, On the lifetime of the transients (NP)-(CH3)n (NP=Ag0, Au0, TiO2 nanoparticles) formed in the reactions between methyl radicals and nanoparticles suspended in aqueous solutions, Chem.–Eur. J., 2012, 18, 4699–4705 CAS.
- M. König, J. Vaes, E. Klemm and D. Pant, Solvents and Supporting Electrolytes in the Electrocatalytic Reduction of CO2, iScience, 2019, 19, 135–160 Search PubMed.
- R. Sander, Compilation of Henry's law constants (version 4.0) for water as solvent, Atmos. Chem. Phys., 2015, 15, 4399–4981 CAS.
- Y. Gao, R. E. Sturgeon, Z. Mester, X. Hou, C. Zheng and L. Yang, Direct determination of trace antimony in natural waters by photochemical vapor generation ICPMS: method optimization and comparison of quantitation strategies, Anal. Chem., 2015, 87, 7996–8004 CAS.
- R. M. de Oliveira and D. L. G. Borges, UV photochemical vapor generation of noble metals (Au, Ir, Pd, Pt and Rh): a feasibility study using inductively coupled plasma mass spectrometry and seawater as a test matrix, J. Anal. At. Spectrom., 2018, 33, 1700–1706 Search PubMed.
- P. Grinberg and R. E. Sturgeon, Photochemical vapor generation of iodine for detection by ICP-MS, J. Anal. At. Spectrom., 2009, 24, 508–514 CAS.
- C. Zheng, R. E. Sturgeon and X. Hou, UV photochemical vapor generation and in situ preconcentration for determination of ultra-trace nickel by flow injection graphite furnace atomic absorption spectrometry, J. Anal. At. Spectrom., 2009, 24, 1452–1458 CAS.
- H. Duan, Z. Gong and S. Yang, Online photochemical vapour generation of inorganic tin for inductively coupled plasma mass spectrometric detection, J. Anal. At. Spectrom., 2015, 30, 410–416 CAS.
- L. Dong, W. Wang, Y. Ning, X. Deng and Y. Gao, Detection of trace antimony by vanadium (IV) ion assisted photochemical vapor generation with inductively coupled plasma mass spectrometry measurement, Anal. Chim. Acta, 2023, 1251, 341006 CrossRef CAS PubMed.
- E. Jeníková, E. Nováková, J. Hraníček and S. Musil, Ultra-sensitive speciation analysis of tellurium by manganese and iron assisted photochemical vapor generation coupled to ICP-MS/MS, Anal. Chim. Acta, 2022, 1201, 339634 CrossRef PubMed.
- S. Li, Y. Gao, Y. Yu, H. He, X. Hu, S. Ni, Z. Shi, X. Peng and R. Liu, Direct determination of trace lead in seawater by inductively coupled plasma mass spectrometry after photochemical vapor generation, At. Spectrosc., 2017, 38, 37–43 CrossRef CAS.
- J. L. Weeks, G. M. A. C. Meaburn and S. Gordon, Absorption coefficients of liquid water and aqueous solutions in the far ultraviolet, Radiat. Res., 1963, 19, 559–567 CrossRef CAS.
- I. Janik and G. N. R. Tripathi, The nature of the CO2− radical anion in water, J. Chem. Phys., 2016, 144, 154307 CrossRef.
- S. Mozia, A. Heciak and A. W. Morawski, Photocatalytic acetic acid decomposition leading to the production of hydrocarbons and hydrogen on Fe-modified TiO2, Catal. Today, 2011, 161, 189–195 CrossRef CAS.
- Y. Jia, Q. Mou, Y. Yu, Z. Shi, Y. Huang, S. Ni, R. Wang and Y. Gao, Reduction of interferences using Fe-containing metal–organic frameworks for matrix separation and enhanced photochemical vapor generation of trace bismuth, Anal. Chem., 2019, 91, 5217–5224 CrossRef CAS.
- G. B. Porter and S. W. Benson, The reaction of carbon monoxide with free radicals, J. Am. Chem. Soc., 1953, 75, 2773–2774 CrossRef CAS.
- H. A. Schwarz and A. J. Salzman, The radiation chemistry of aqueous nitrite solutions: the hydrogen peroxide yield, Radiat. Res., 1958, 9, 502–508 CrossRef CAS.
- A. J. Elliot and F. C. Sopchyshyn, The radiolysis at room temperature and 118 oC of aqueous solutions containing sodium nitrate and either sodium formate or 2-propanol, Can. J. Chem., 1983, 61, 1578–1582 CrossRef CAS.
- G. L. Hug, D. M. Camaioni and I. Carmichael, EPR detection of HNO2− in the radiolysis of aqueous nitrite and quantum chemical calculation of its stability and hyperfine parameters, J. Phys. Chem. A, 2004, 108, 6599–6604 CrossRef CAS.
- J. Jortner, M. Ottolenghi and G. Stein, On the photochemistry of aqueous solutions of chloride, bromide and iodide ions, J. Phys. Chem., 1964, 68, 247–255 CrossRef CAS.
- U. Shuali, M. Ottolenghi, J. Rabani and Z. Yelin, On the photochemistry of aqueous nitrate solutions excited in the 195 nm band, J. Phys. Chem., 1969, 73, 3445–3451 CrossRef CAS.
- M. Daniels, R. V. Meyers and E. V. Belardo, Photochemistry of the aqueous nitrate system. I. Excitation in the 300-m.mu. band, J. Phys. Chem., 1968, 72, 389–399 CrossRef CAS.
- X. Guo, R. E. Sturgeon, Z. Mester and G. J. Gardner, UV Vapor generation for determination of Se by heated quartz tube AAS, Anal. Chem., 2003, 75, 2092–2099 CrossRef CAS.
- G. S. Lopes, R. E. Sturgeon, P. Grinberg and E. Pagliano, Evaluation of approaches to the abatement of nitrate interference with photochemical vapor generation, J. Anal. At. Spectrom., 2017, 32, 2378–2390 RSC.
- M. Rybínová, V. Červený, J. Hraníček and P. Rychlovský, UV-photochemical vapor generation with quartz furnace atomic absorption spectrometry for simple and sensitive determination of selenium in dietary supplements, Microchem. J., 2016, 124, 584–593 CrossRef.
- J. Vyhnanovský, D. Yildiz, B. Stadlerov and S. Musil, Efficient photochemical vapor generation of bismuth using a coiled teflon reactor: Effect of metal sensitizers and analytical performance with flame-in-gas-shield atomizer and atomic fluorescence spectrometry, Microchem. J., 2021, 164, 105997 CrossRef.
- M. Kozlowski and T. Yoon, Editorial for the special issue on photocatalysis, J. Org. Chem., 2016, 81, 6895–6897 CrossRef CAS.
- H. Bao, X. Peng, Z. Song, Y. Ning, Y. Yu and Y. Gao, Natural mineral assisted photochemical vapor generation for determination of trace inorganic arsenic by inductively coupled plasma mass spectrometry, Microchem. J., 2021, 170, 106689 CrossRef CAS.
- R. E. Sturgeon, Unpublished Results; Presented at the 16th Rio Symposium on Atomic Spectrometry, Bento Goncalves, Brazil, November 28-30, 2023 Search PubMed.
- F. Laborda, A. C. Gimenez-Ingalaturre, E. Bolea and J. R. Castillo, About detectability and limits of detection in single particle inductively coupled plasma mass spectrometry, Spectrochim. Acta, Part B, 2020, 169, 105883 CrossRef CAS.
- S. Lee, X. Bi, R. B. Reed, J. F. Ranville, P. Herckes and P. Westerhoff, Nanoparticle size detection limits by single particle ICP-MS for 40 elements, Environ. Sci. Technol., 2014, 48, 10291–10300 CrossRef CAS.
- L. Liu, X. Zhang, L. Yang, L. Ren, D. Wang and J. Ye, Metal nanoparticles induced photocatalysis, Natl. Sci. Rev., 2017, 4, 761–780 CrossRef CAS.
- J. Zhao, S. C. Nguyen, R. Ye, B. Ye, H. Weller, G. A. Somorjai, A. P. Alivisatos and F. D. Toste, A comparison of photocatalytic activities of gold nanoparticles following plasmonic and interband excitation and a strategy for harnessing interband hot carriers for solution phase photocatalysis, ACS Cent. Sci., 2017, 3, 482–488 CrossRef CAS PubMed.
- H. Huang, K. A. Steiniger and T. H. Lambert, Electrophotocatalysis: combining light and electricity to catalyze reactions, J. Am. Chem. Soc., 2022, 144, 12567–12583 CrossRef CAS.
- L. Zhang, M. Zhang, X. Guo, X. Liu, P. Kang and X. Chen, Sorption characteristics and separation of tellurium ions from aqueous solutions using nano-TiO2, Talanta, 2010, 83, 344–350 CrossRef CAS PubMed.
- V. Nguyen, D. Beydoun and R. Amal, Photocatalytic reduction of selenite and selenate using TiO2 photocatalyst, J. Photochem. Photobiol., A, 2005, 171, 113–120 CAS.
- Z. Zou, Y. Zhen, C. Zheng and X. Hou, “Catalysts in photochemical vapor generation” in Vapor Generation Techniques for Trace Element Analysis: Fundamental Aspects, ed A. D'Ulivo and R. E. Sturgeon, Elsevier, 2022, Chapter 8 Search PubMed.
- K. An and G. A. Somorjai, Nanocatalysis I: Synthesis of metal and bimetallic nanoparticles and porous oxides and their catalytic reaction studies, Catal. Lett., 2015, 145, 233–248 CAS.
- P. V. Kamat, Photophysical, photochemical and photocatalytic aspects of metal nanoparticles, J. Phys. Chem. B, 2002, 106, 7729–7744 CrossRef CAS.
- J. A. Adekoya, K. O. Ogunniran, T. O. Siyanbola, E. O. Dare and N. Revaprasadu, “Band structure, morphology, functionality, and size dependent properties of metal nanoparticles”, in Noble and Precious Metals - Properties, Nanoscale Effects and Applications, M. Seehra and A. D. Bristow (Eds), Chapter 2, IntechOpen, 2018, pp. 15 - 42. ISBN: 978-1-78923-293-6 Search PubMed.
- P. Han, W. Martens, E. R. Waclawik, S. Sarina and H. Zhu, Metal nanoparticle photocatalysts: synthesis, characterization and application, Part. Part. Syst. Charact., 2018, 35, 1700489 CrossRef.
- A. Krishnan, A. Swarnalal, D. Das, M. Krishnan, V. S. Saji and S. M. A. Shibli, A review on transition metal oxides based photocatalysts for degradation of synthetic organic pollutants, J. Environ. Sci., 2024, 139, 389–417 CrossRef CAS.
- H. Xu, Q.-Y. Wang, M. Jiang and S. Li, Application of valence-variable transition-metal-oxide-based nanomaterials in electrochemical analysis: A review, Anal. Chim. Acta, 2024, 1295, 342270 CrossRef CAS.
- A. M. Abu-Dief, Development of metal oxide nanoparticles as semiconductors, Nanomater. Nanotechnol., 2020, 1, 5–10 Search PubMed.
- K. Roy, C. K. Sarkar and C. K. Ghosh, Photocatalytic activity of biogenic silver nanoparticles synthesized using potato (Solanum tuberosum) infusion, Spectrochim. Acta, Part A, 2015, 146, 286–291 CrossRef CAS.
- R. Narayanan and M. A. El-Sayed, Catalysis with transition metal nanoparticles in colloidal solution: nanoparticle shape dependence and stability, J. Phys. Chem. B, 2005, 109, 12663–12676 CrossRef CAS PubMed.
- A. A. Ponce and K. J. Klabunde, Chemical and catalytic activity of copper nanoparticles prepared via metal vapor synthesis, J. Mol. Catal. A: Chem., 2005, 225, 1–6 CrossRef CAS.
- M. A. El-Sayed, J. Yu, G. Liu and M. Jaroniec, Non-noble plasmonic metal-based photocatalysts, Chem. Rev., 2022, 122, 10484–10537 CrossRef PubMed.
- J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, The merger of transition metal and photocatalysis, Nat. Rev. Chem., 2017, 1, 1–17 CrossRef.
- F. Kraushofer and G. S. Parkinson, Single-atom catalysis: insights from model systems, Chem. Rev., 2022, 122, 14911–14939 CrossRef CAS.
- S. K. Kaiser, Z. Chen, D. F. Akl, S. Mitchell and J. Pérez-Ramírez, Single-atom catalysts across the Periodic Table, Chem. Rev., 2020, 120, 11703–11809 CrossRef CAS.
- L. Xiangdong, W. Jia, N. Chao, X. Yongjie and L. Fuwei, Manipulating the structure of metal at atomic level to enhance the catalytic performance, Chem Catal., 2024, 4, 100810 CrossRef.
- Y. Huang, J. Hou, L. Zhan, Q. Zhang, W. Tang and B. Li, Photoredox activation of formate salts: hydrocarboxylation of alkenes via carboxyl group transfer, ACS Catal., 2021, 11, 15004–15012 CrossRef CAS.
- M. Biegel, T. Schikarski, P. C. Lopez, L. Gromotka, C. Lübbert, A. Völkl, C. Damm, J. Walter and W. Peukert, Efficient quenching sheds light on early stages of gold nanoparticle formation, RSC Adv., 2023, 13, 18001–18013 RSC.
- K. Cao, J. Biskupek, C. T. Stoppiello, R. L. McSweeney, T. W. Chamberlain, Z. Liu, K. Suenaga, S. T. Skowron, E. Besley, A. N. Khlobystov and U. Kaiser, Atomic mechanism of metal crystal nucleus formation in a single-walled carbon nanotube, Nat. Chem., 2020, 12, 921–928 CrossRef CAS PubMed.
- B. Huang, Y. Liu, H. Kobayashi, Z. Tan, T. Yamamato, T. Toriyama, S. Matsumura, S. Kawaguchi, Y. Kubota, H. Zheng and H. Kitagawa, CuxRu1−x catalysts for carbon neutralization with CH4 or CO production, Chem Catal., 2023, 3, 100705 CrossRef CAS.
- T. S. Rodrigues, A. G. M. da Silva and P. H. C. Camargo, Nanocatalysis by noble metal nanoparticles: controlled synthesis for the optimization and understanding of activities, J. Mater. Chem. A, 2019, 7, 5857–5874 RSC.
- J. Ma, X. Tan, Q. Zhang, Y. Wang, J. Zhang and L. Wang, Exploring the size effect of Pt nanoparticles on the photocatalytic nonoxidative coupling of methane, ACS Catal., 2021, 11, 3352–3360 CrossRef CAS.
- R. Zhang, L. Song and B. Wang, The interaction mechanism of CO2 with CH3 and H on Cu {111} surface in synthesis of acetic acid from CH4/CO2: A DFT study, Appl. Catal., A, 2012, 443–444, 50–58 CrossRef CAS.
- G. R. Dey, K. N. R. Nair and K. K. Pushpa, Photolysis studies on HCOOH and HCOO− in presence of TiO2 photocatalyst as suspension in aqueous medium, J. Nat. Gas Chem., 2009, 18, 50–54 CrossRef CAS.
- P. J. Wyatt, Differential light scattering and the measurement of molecules and nanoparticles: A review, Anal. Chim. Acta: X, 2021, 7–8, 100070 CAS.
- H. Kato, A. Nakamura, K. Takahashi and S. Kinugasa, Accurate size and size-distribution determination of polystyrene latex nanoparticles in aqueous medium using dynamic light scattering and asymmetrical flow field flow fractionation with multi-angle light scattering, Nanomaterials, 2012, 2, 15–30 CrossRef CAS PubMed.
- Y. Matsuura, N. Ouchi, A. Nakamura and H. Kato, Determination of an accurate size distribution of nanoparticles using particle tracking analysis corrected for the adverse effect of random Brownian motion, Phys. Chem. Chem. Phys., 2018, 20, 17839–17846 RSC.
- W.-M. Cheng and R. Shang, Transition metal-catalyzed organic reactions under visible light: recent developments and future perspectives, ACS Catal., 2020, 10, 9170–9196 CAS.
- S. De Kreijger, F. Glaser and L. Troian-Gautier, From photons to reactions: key concepts in photoredox catalysis, Chem Catal., 2024 DOI:10.1016/j.checat.2024.101110.
- H. Sato and T. Sugimoto, Direct operando identification of reactive electron species driving photocatalytic hydrogen evolution on metal-loaded oxides, J. Am. Chem. Soc., 2024, 146, 24800–24807 CrossRef CAS PubMed.
- Z. Zou, Y. Tian, W. Zeng, X. Hou and X. Jiang, Effect of variable ultraviolet wavelength and intensity on photochemical vapor generation of trace selenium detected by atomic fluorescence spectrometry, Microchem. J., 2018, 140, 189–195 CAS.
Footnote |
| † Presented at the 16th Rio Symposium on Atomic Spectrometry, Bento Gonçalves, Brazil, November 28–30, 2023. |
| This journal is © The Royal Society of Chemistry 2025 |