
Detecting telomerase activity at the single-cell level using a CRISPR-Cas12a-based chip†
Yateng
Jiang‡
a,
Yanping
Wang‡
 b,
Wen
Luo
a,
Xiaowei
Luan
a,
Zhibin
Zhang
a,
Yongchun
Pan
a,
Bangshun
He
*c,
Yanfeng
Gao
*b and
Yujun
Song
*a
b,
Wen
Luo
a,
Xiaowei
Luan
a,
Zhibin
Zhang
a,
Yongchun
Pan
a,
Bangshun
He
*c,
Yanfeng
Gao
*b and
Yujun
Song
*a
aCollege of Engineering and Applied Sciences, State Key Laboratory of Analytical Chemistry for Life Science, Nanjing University, Nanjing 210023, China. E-mail: ysong@nju.edu.cn
bSchool of Medical Imaging, Wannan Medical College, Wuhu 241002, China. E-mail: gaoyanfeng@wnmc.edu.cn
cDepartment of Laboratory Medicine, Nanjing First Hospital, Nanjing Medical University, Nanjing 210006, China. E-mail: bhe@njmu.edu.cn
First published on 18th November 2024
Abstract
The intimate association between telomerase activity and cancer has driven the exploration of diverse methodologies for its precise detection. However, detecting telomerase activity at the single-cell level remains a significant challenge. Herein, we present a MOF–DNA barcode-amplified CRISPR-Cas12a strategy integrated with a single-cell microfluidic chip for ultrasensitive detection of telomerase activity. DNA-functionalized UiO-66 nanoparticles act as signal transducers, effectively converting telomerase activity into DNA activation strands, which subsequently trigger the trans-cleavage activity of CRISPR-Cas12a. This amplification-based assay could be integrated with a microfluidic chip to enable highly sensitive detection of telomerase activity at the single-cell level, offering promising advancements in early cancer diagnosis.
1 Introduction
Telomerase, as a ribose reverse transcriptase, plays a critical role in the enzymatic synthesis of telomeric repeats (TTAGGG)n at chromosomal termini.1 In normal cellular dynamics, mitotic events are typically associated with a gradual reduction in telomere length. Conversely, cancer cells exhibit a distinctive ability to sustain the telomere length due to the overexpression of telomerase,2 thereby facilitating continuous cell growth and proliferation.3,4 Consequently, meticulous and precise detection of telomerase activity has emerged as an indispensable factor in the domains of cancer diagnostics, therapeutic interventions, and continuous monitoring.5Currently, the prevailing method for telomerase detection relies on the polymerase chain reaction (PCR)-based telomeric repeat amplification protocol (TRAP).6 However, the limitations of the TRAP, including time consumption and false positive signals induced from the PCR, restrict their application, especially in remote settings.7 This necessitates the development of a PCR-free assay, prompting the exploration of alternative methodologies, such as surface Raman enhancement,8 fluorescence techniques,9 and nanomaterial-assisted detection.10 Unfortunately, these approaches often compromise single-cell sensitivity or are hindered by protracted protocols, thereby impeding their broad applicability.
Recently, the clustered regularly interspaced short palindromic repeats (CRISPR)-Cas12a system has emerged as a compelling alternative due to its procedural simplicity, rapid execution, and cost-effectiveness.11 Moreover, it can be employed for both the quantitative detection of non-nucleic acids (e.g., ATP and Na+)12 and the rapid, sensitive detection of nucleic acids (e.g., SARS-CoV-2 RNA and DNA),13 underscoring its potential as a robust tool for telomerase detection.14–16 However, the CRISPR-based detection of single-cell telomerase activity requires a highly sensitive signal transduction strategy and an effective telomerase isolation approach, which is still challenging.
Bio-barcode assays,17 renowned for their efficacy in signal conversion and amplification, have been combined with nanomaterials to detect prostate antigens, achieving a performance six orders of magnitude superior to that of the traditional ELISA method.18 To fully exploit the advantages of bio-barcode technology, constructing barcode nanocarriers is essential.19–21 Among them, metal–organic frameworks (MOFs) emerge as exemplary nanocarriers, distinguished by their high porosity and structural tunability,22 which facilitate substantial nucleic acid modifications and enable their integration with CRISPR-Cas12a for ultrasensitive detection.23 Furthermore, microfluidic chips have significantly enhanced single-cell manipulation, serving as potent tools for isolating and extracting telomerase from individual cells. Significantly, this discovery suggests the potential to integrate the MOF–DNA bio-barcode-amplified CRISPR-Cas12a system with a microfluidic chip, offering a viable pathway for highly sensitive detection of telomerase activity.
In this work, a MOF–DNA bio-barcode-amplified CRISPR-Cas12a system allowing dual amplification was proposed for detecting telomerase activity, and when combined with a radial microfluidic chip enabling efficient single-cell capture and isolation, the platform was capable of highly sensitive telomerase activity detection at the single-cell level (Scheme 1). Specifically, the oligonucleotide sequences that act as telomerase-binding substrates (TSs) were conjugated to functionalized magnetic silica nanoparticles (MSNPs) and could be elongated by the extracted telomerase to form MSNP–TS extension probes. Then, the MSNP–TS extension probes were hybridized with UiO-66 nanoparticles functionalized with strands complementary to the telomerase-elongated sequence and CRISPR-Cas12a activation strands (UiO-66 probes) via complementary base pairing. Upon the introduction of phosphate, UiO-66 decomposed, and the activation strands were released, thus activating the CRISPR-Cas12a system and resulting in the trans-cleavage of the FQ reporter (single-strand DNA (ssDNA) sequences labeled with fluorescein phosphoramidite (FAM) and benzothiazole quencher (BQ) at both ends), to generate a fluorescence signal that is indicative of telomerase activity. Specifically, it is triggered by crRNA–target DNA hybridization, facilitated by PAM binding for dsDNA target recognition and cleavage, and maintained by releasing the PAM-distal DNA fragment, allowing Cas12a to cleave non-target ssDNA.24 The constructed telomerase activity assay platform demonstrated rapid and effective detection of telomerase activity at the single-cell level. The dual-amplification effect, including the binding of UiO-66 containing activation strands onto the MSNP–TS extension probes and the trans-cleavage of the FQ reporter via UiO-66-released activation strands, endows the platform with comparable detection time and specificity, as well as superior sensitivity compared with other single-cell telomerase detection methods reported in the literature.25–28 Furthermore, the general applicability of the platform was validated in a variety of cancer cells exhibiting higher telomerase activity. The results conclusively substantiated the platform's versatility and underscored its potential for extensive applications in clinical cancer diagnosis.
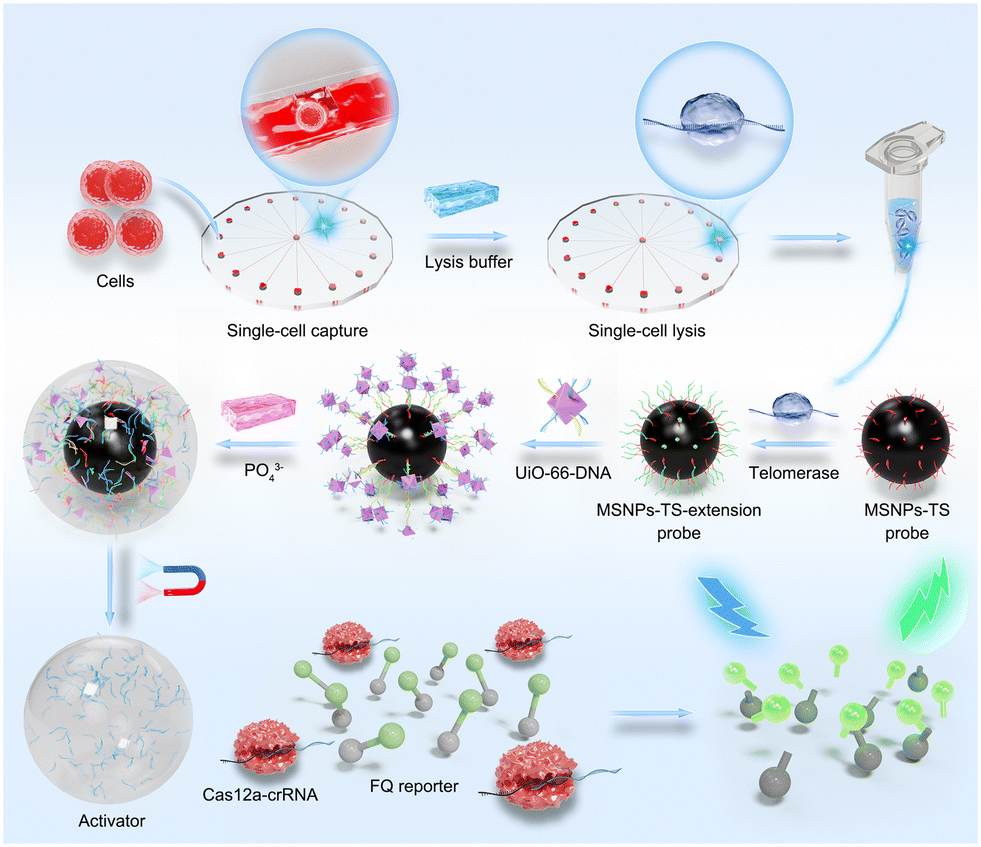 | ||
| Scheme 1 Schematic illustration of single-cell telomerase activity detection through a radial microfluidic chip integrated with MOF–DNA bio-barcode-amplified CRISPR-Cas12a technology. | ||
2 Materials and methods
2.1 Preparation of UiO-66
UiO-66 was synthesized by a solvothermal reaction with minor modifications.29 ZrOCl2·8H2O (21 mg, 0.066 mmol) was dissolved adequately in 1 mL of N,N-dimethylformamide (DMF). In another separate vial, 1,4-benzenedicarboxylic acid (50 mg, 0.3 mmol) was thoroughly dissolved in 3 mL of DMF and heated to 90 °C. A ZrOCl2 solution and 1.5 mL of acetic acid were then added, followed by an 8 h reaction at 90 °C, resulting in the formation of UiO-66 nanoparticles.2.2 DNA functionalization of UiO-66
The activation strands, the complementary strands to the telomerase-elongated sequence and UiO-66 nanoparticles were mixed in HEPES buffer at a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:100, to control the ratio of the two nucleic acids on UiO-66. The specific maximum number of activation strands and TS complementary strands on a single UiO-66 nanoparticle is approximately 2.4 × 10−9 pmol. The amalgamated solution was incubated overnight on a shaker at room temperature. To optimize DNA loading, NaCl was introduced to the HEPES buffer to a final concentration of 100 mM. Subsequently, excess DNA was eliminated through centrifugation (7000 rpm, 5 min), and the resulting precipitate was washed multiple times with the buffer. After a blocking step with 1% BSA, the prepared material was then stored for subsequent use.
:10:100, to control the ratio of the two nucleic acids on UiO-66. The specific maximum number of activation strands and TS complementary strands on a single UiO-66 nanoparticle is approximately 2.4 × 10−9 pmol. The amalgamated solution was incubated overnight on a shaker at room temperature. To optimize DNA loading, NaCl was introduced to the HEPES buffer to a final concentration of 100 mM. Subsequently, excess DNA was eliminated through centrifugation (7000 rpm, 5 min), and the resulting precipitate was washed multiple times with the buffer. After a blocking step with 1% BSA, the prepared material was then stored for subsequent use.
2.3 Preparation and modification of MSNPs
First, Fe3O4 nanoparticles were obtained by the solvothermal method.30 Briefly, 2.73 g of FeCl3·6H2O and 7.22 g of anhydrous sodium acetate were added to 80 mL of ethylene glycol with magnetic stirring. After full dissolution, 1 g of polyethylene glycol (PEG) was added, and the solution was transferred to a Teflon-lined stainless-steel autoclave (100 mL), heated for 12 h at 200 °C in an oven and then naturally cooled. The resulting precipitate was washed several times with water and ethanol. For the preparation of MSNPs, 120 mg of Fe3O4 was fully dissolved in a mixture of 25 mL of water and 80 mL of ethanol by sonication. The sequential addition of 2 mL of ammonia and 2 mL of tetraethyl orthosilicate, along with sonication at approximately 20 °C, was followed by overnight mechanical stirring. The resulting solution was washed three times with ethanol and water before drying. To introduce amino groups to the MSNPs, the MSNPs (50 mg) were thoroughly mixed with methanol by sonication, followed by the addition of 100 μL of APTES and continued sonication for 20 min. The mixture was shaken overnight and washed three times with ethanol. Via the glutaraldehyde cross-linking method,31 TS was conjugated to the surface of MSNPs–NH2. Specifically, glutaraldehyde (30% v/v) was added to the solution of MSNP–NH2 (1 mg mL−1), which was then washed 5 times with PBS (10 mM, pH 7.4) after 1 h of shaking at room temperature. Afterwards, 1 μM TS was added to the above solution, followed by incubation overnight at room temperature. The free TSs were separated using a magnet and stored in PBS for further applications in biomedical research. Prior to the cross-linking reaction between the UiO-66–DNA nanoprobes and the MSNP–DNA nanoprobes, the MSNP–DNA nanoprobes were thoroughly washed five times with HEPES buffer to eliminate any residual PBS buffer solution. This step is crucial in preventing the potential disintegration of the UiO-66–DNA nanoparticles due to residual phosphate ions, thereby ensuring the accuracy and integrity of the experimental results.2.4 Detection of telomerase
Following the amalgamation of the UiO-66 probes and MSNP–TS extension probes, the hybridization reaction system was incubated at 37 °C for 1 h. The mixture was then washed three times with HEPES buffer and ultrapure water (DNase/RNase-free, sterile) using a magnet. Subsequently, 10 μL of phosphate solution (8 mM) was added, and the mixture was subjected to a 30 min agitation period to liberate the activation strands from UiO-66, which were then disengaged from the MSNPs. The supernatants were separated using a magnetic field and utilized for fluorescence analysis based on the CRISPR-Cas12a system. The CRISPR-Cas12a system (100 μL) incorporated the above supernatants (including the activation strands), 20 nM Cas12a nuclease, 250 nM CRISPR RNA (crRNA, which is transcribed and processed from the CRISPR locus to yield mature RNAs, each encoding a distinct spacer sequence32) and 100 nM FQ reporter in 1× NEB 2.1 buffer. Specifically, the crRNA strand was chosen to be highly complementary to the target DNA strand (the activation strand) to ensure precise targeting and effective system activation. The incubation spanned 1 h at 37 °C under shaded conditions for fluorescence spectra measurement in a quartz cuvette, with an excitation wavelength of 480 nm and an emission wavelength of 520 nm.2.5 Design and fabrication of the microfluidic chip
Initially, a 10 μL min−1 flow of 1× PBS was introduced into the radial microfluidic chip, where each microchannel is capable of capturing a single cell for single-cell analysis. Specifically, a cell suspension with a density of 1 × 107 cells per mL was introduced into the microfluidic chip at a flow rate of 10 μL min−1. As the cell suspension passed through the microchannels, the integrated hook-shaped structures within the channels efficiently separated and captured individual cells, driven by the inertial forces and interactions between cells. These microscale hook structures were optimized on the basis of the size, shape, and physical properties of single cells. Once a single cell was captured, HEPES buffer was introduced to flush away excess cells, leaving only the captured single cell. After rinsing with the buffer, a mixture of NP-40 and 1% PMSF was introduced into the chip to lyse the captured cells at room temperature. This mixture ensured thorough cell lysis while preserving the integrity of the intracellular components, particularly the telomerase extracts of interest.The microfluidic chip used for single-cell telomerase activity analysis was meticulously designed using AutoCAD software and fabricated using traditional soft lithography techniques.33,34 The chip was molded from polydimethylsiloxane (PDMS, Sylgard 184, Dow Corning). Specifically, the precursor and curing agent were thoroughly mixed at a 10:1 ratio (base:curing agent, w/w), stirred for 10 min, and then poured into the mold. After degassing in a vacuum chamber for 2 h, the material was cured at 70 °C for 3 h. The resulting PDMS chip was carefully peeled off from the mold, and inlet and outlet holes were drilled. The PDMS chip was then irreversibly bonded to a clean glass substrate through oxygen plasma treatment, followed by incubation at 80 °C for 15 min to ensure strong adhesion.
3 Results and discussion
3.1 Characterization of DNA-modified UiO-66
MSNPs have exhibited unique potential in bio-separation35 and diagnostics36 due to their exceptional magnetic response properties and surface chemical modifications.37 In accordance with a previously reported method,30 ∼300 nm Fe3O4 was synthesized via a solvothermal method (Fig. S1†). Subsequently, the surface of the silica shell was chemically modified, resulting in a unique core–shell structure with a diameter of 1 μm (Fig. S2†). The functionalization of the amino groups on the MSNPs was confirmed by a significant change in the zeta potential of the particles after modification (Fig. S3†). By employing the glutaraldehyde cross-linking method, TSs with terminal amino modifications were successfully grafted onto the surface of MSNPs, which was extended through repetitive sequences catalyzed by the extracted telomerase to generate MSNP–TS extension probes.UiO-66 nanoparticles were synthesized via a solvothermal method,29 and their size was modulated by the addition of acetic acid.38 To determine the size and morphology, the UiO-66 nanoparticles were examined by transmission electron microscopy (TEM) and scanning electron microscopy (SEM) (Fig. 1b and S4†). The corresponding micrographs revealed that homogeneous distributions of UiO-66 nanoparticles with octahedral shapes of ∼88 nm were synthesized (Fig. S4†). Due to the abundant modification sites on the surface, DNA with a terminal phosphate could be attached to UiO-66 nanoparticles (Fig. 1a), through the unsaturated metal sites of the MOF via the site occupancy effect.39 Previous research using a ligand strut modification approach demonstrated that UiO-66 nanoparticles could accommodate an average DNA loading capacity of 17 ± 6 pmol cm−2, which was approximately double that of the reporter.40 Consequently, the UiO-66-to-DNA ratio was set at 1:10, enabling a higher loading capacity of nucleic acids onto UiO-66 nanoparticles to amplify the fluorescence signal.23
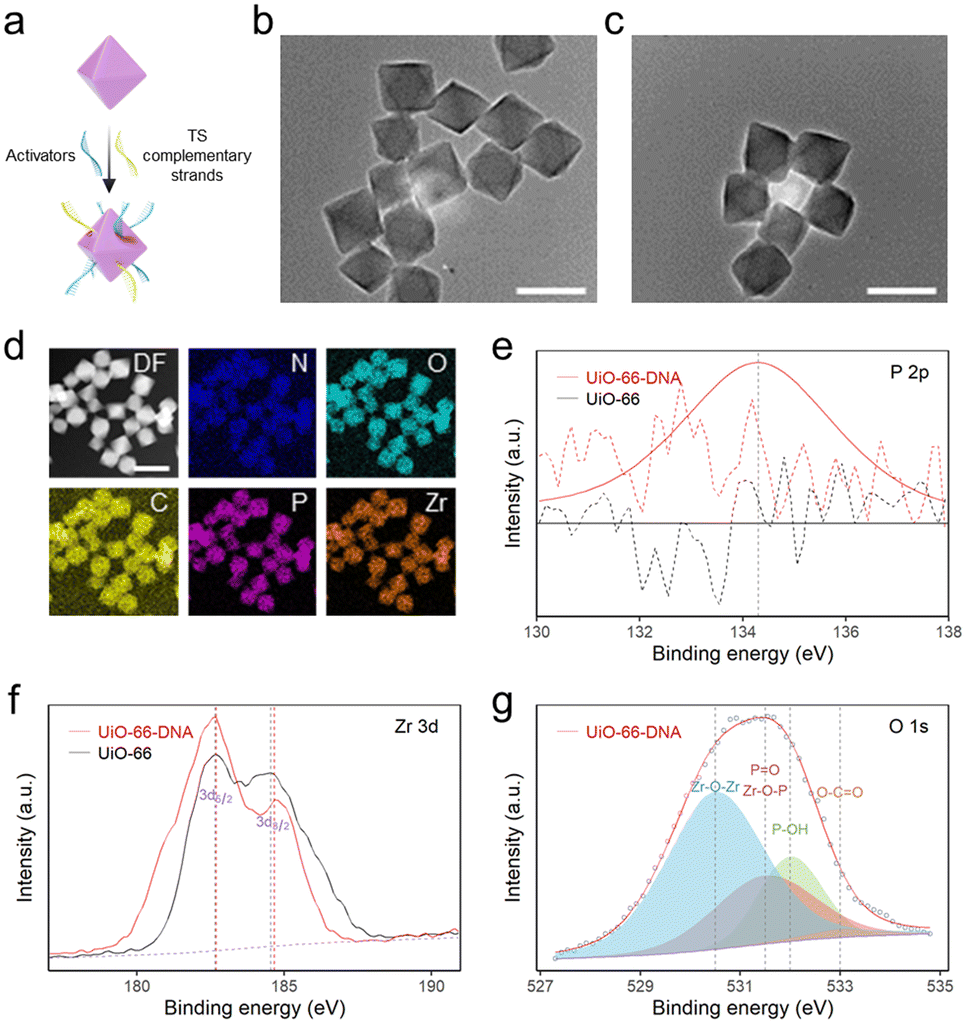 | ||
| Fig. 1 Characterization of DNA-functionalized UiO-66. (a) Schematic illustration of the functionalization of UiO-66 with DNA. (b) TEM image of UiO-66. Scale bar: 100 nm. (c) TEM image of UiO-66–DNA. Scale bar: 100 nm. (d) STEM image and elemental mappings of UiO-66–DNA. The scale bar denotes 100 nm. (e)–(g) High-resolution XPS spectra of UiO-66 and UiO-66–DNA, focusing on the P 2p, Zr 3d, and O 1s regions. | ||
To study the effect of DNA modification on UiO-66, elemental mapping was performed. The images displayed a uniform dispersion of Zr, O and P, suggesting that DNA was successfully modified onto the MOF surface (Fig. 1d). Additionally, the attachment of DNA on UiO-66 was analyzed by high-resolution X-ray photoelectron spectroscopy (XPS) (Fig. 1e–g). The existence of a distinct P 2p peak in UiO-66–DNA, in contrast to that in UiO-66, was readily apparent. The Zr 3d peak of UiO-66–DNA slightly redshifted. The upward shift in the binding energy may be a consequence of the replacement of carboxylic groups coordinated with Zr–O clusters by phosphoric groups. In contrast to the Zr atom, the P atom is more electronegative, resulting in a decrease in the electron cloud density surrounding the Zr atom and an increase in the binding energy of internal electrons.41 Through deconvolution, the O 1s spectrum was resolved into four distinct peaks. Notably, the presence of a peak at 531.5 eV could be interpreted as evidence for the formation of Zr–O–P bonds, further supporting the binding of DNA to the MOF.42 Additionally, the successful attachment of DNA on the MOF surface was further confirmed by the zeta potentials and Fourier transform infrared (FTIR) spectra of UiO-66–DNA (Fig. S5 and S6†).
3.2 Process of DNA release from UiO-66
In our previous study, we found that DNA modified on UiO-66 could be released in the presence of phosphate ions.23 By virtue of the site-occupancy effect, single-stranded DNA could be modified on UiO-66 through phosphate groups binding to unoccupied Zr sites.40 DNA modification had little effect on the morphology of UiO-66 (Fig. 1c). However, upon the introduction of phosphate ions, the phosphate ions effectively displaced the oligonucleotide phosphates, instead of binding to the Zr sites. This displacement was driven by the stronger binding energy of the phosphate ions toward the Zr sites than that of the terminal phosphate groups on the DNA.39,43 Notably, an excessive concentration of phosphate ions poses a risk to the structural integrity of the MOF, potentially leading to its dissociation. The terminal phosphate-modified oligonucleotides interact with the unsaturated metal sites (CUSs) on the surface of UiO-66. This interaction results in the stable conjugation of phosphate-functionalized DNA to UiO-66 through the formation of Zr–O–P bonds. However, as the concentration of free PO43− ions increases, these ions are more likely to bind to the Zr sites on the MOF than to the DNA molecules. This is because the binding energy of PO43− to the Zr centers is greater than that of the oligonucleotide phosphates. This displacement process reduces the binding energy between UiO-66 and DNA, causing the DNA to be released from the surface of the UiO-66 nanoparticles (Fig. S7–S9†), ultimately leading to the dissociation of the UiO-66–DNA complex (Fig. 2a). To validate these findings, a comprehensive analysis utilizing TEM was performed on UiO-66 nanoparticles subjected to treatment with 8 mM phosphate at various time intervals (Fig. 2b). The emergence of discernible small pores on the surface of the UiO-66 nanoparticles within a relatively short cleavage period of 5 min indicated the initiation of dissociation. After a duration of 30 min, substantial dissociation of the UiO-66 frameworks was observed, with only a few residues remaining. These observations provided preliminary evidence substantiating the feasibility of DNA release from the MOF structure.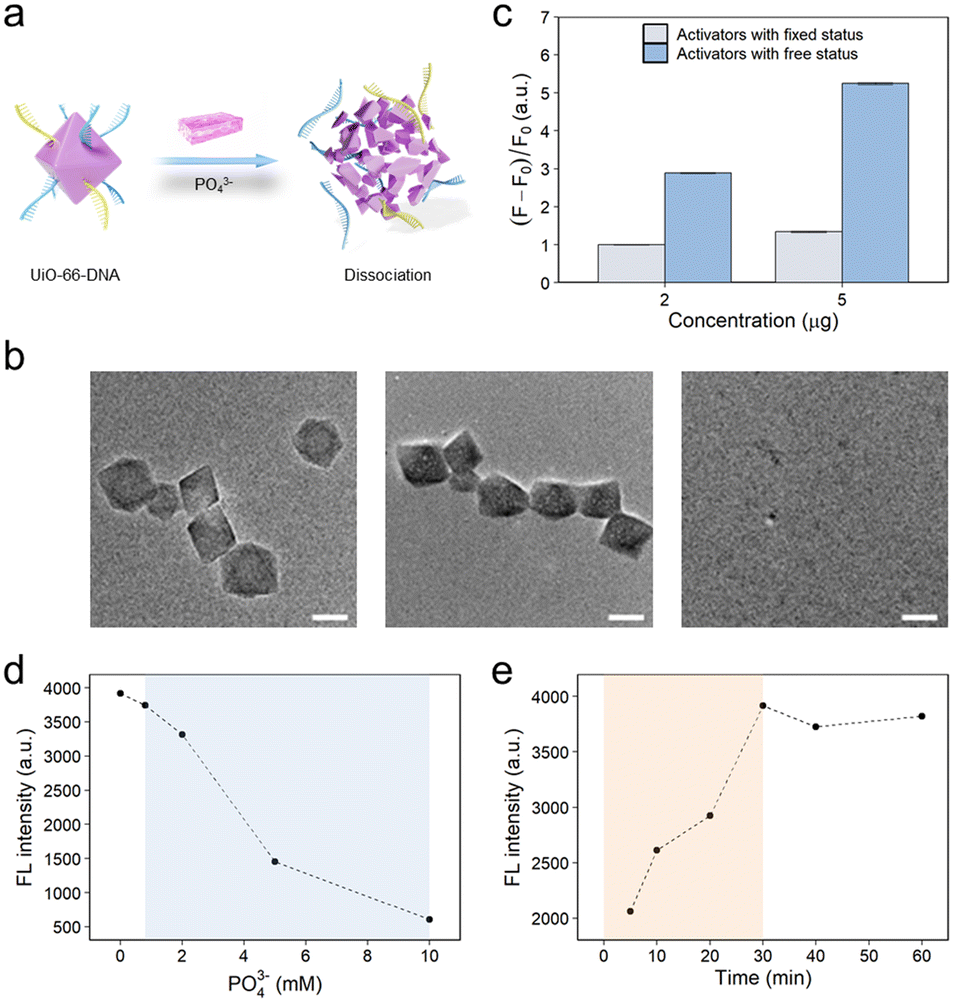 | ||
| Fig. 2 Characterization of DNA release from UiO-66–DNA in response to phosphate ions. (a) Schematic illustration showing the release of DNA from UiO-66–DNA upon exposure to phosphate ions. (b) TEM images of UiO-66–DNA treated with 8 mM phosphate at various time intervals (0, 5, and 30 min, from left to right). Scale bar: 50 nm. (c) Fluorescence intensity analysis in the CRISPR-Cas12a reaction system, comparing the fluorescence activators in different states: activators bound to UiO-66, and activators released from UiO-66. F0 represents the fluorescence intensity at 520 nm in the absence of the telomerase, and F represents the fluorescence intensity in the presence of telomerase. Data are presented as mean ± SD from three independent experiments. (d) Effects of various phosphate concentrations on Cas12a protein activity. (e) Time-dependent ssDNA release from UiO-66–DNA upon treatment with 8 mM phosphate. | ||
In the CRISPR-Cas12a system, the final fluorescence signal was found to increase proportionally with the amount of added activation strands (Fig. S10†). Furthermore, the free activation strands exhibited greater ability to induce trans-cleavage of the Cas protein than the activation strands immobilized on the surface of the MOF. This discernible dissimilarity resulted in a marked amplification of the ultimate fluorescence signal, as illustrated in Fig. 2c. The rationale behind this disparity could be attributed to the spatial site barrier engendered by the DNA moieties affixed to the UiO-66 nanoparticles, thereby impeding the effective activation of the Cas protein by DNA targeting. The experimental parameters were rigorously optimized to enhance the final fluorescence signal. Notably, excess phosphate could detrimentally impede the activity of the Cas protein. The results suggested that the activity of the Cas protein was impeded when the phosphate concentration within the CRISPR-Cas12a system (100 μL) was calibrated to 0.8 mM (Fig. 2d). Additionally, the impact of the cleavage time of the UiO-66 nanoparticles on the ultimate fluorescence signal was also investigated (Fig. 2e).
Within a span of 30 min, the UiO-66 nanoparticles had essentially underwent dissociation, liberating the activation strands. Remarkably, these results aligned with the previously reported literature23,44 as well as the TEM image presented in Fig. 2b.
3.3 Quantitative analysis of telomerase activity
To perform the quantitative analysis, telomerase extracted from a human lung cancer cell line (A549 cells) or a mouse melanoma cell line (B16 cells) was diluted based on predetermined cell counts. Telomerase catalytically added repeated DNA sequences to the ends of TSs, resulting in the successful hybridization between the two probes via complementary base pairing (Scheme 1). As demonstrated in the SEM image (Fig. S11†), the surface of the MSNP–TS extension probes was extensively covered by a substantial number of UiO-66–DNA nanoprobes, which contained activation strands and strands complementary to the telomerase elongation sequence on the surfaces. After the introduction of phosphate, the activation strands were released for fluorescence detection by the activation of trans-cleavage activity (Fig. 3a). As illustrated in Fig. 3b and c, the final fluorescence signal increased with increasing number of cells, suggesting a concomitant increase in telomerase activity. Moreover, as shown in Fig. S12,† the fluorescence signal demonstrated a linear correlation with the number of cells within the range of 10 to 1000. The limit of detection, which was calculated according to the IUPAC definition, revealed the ability to detect telomerase activity at the single-cell level.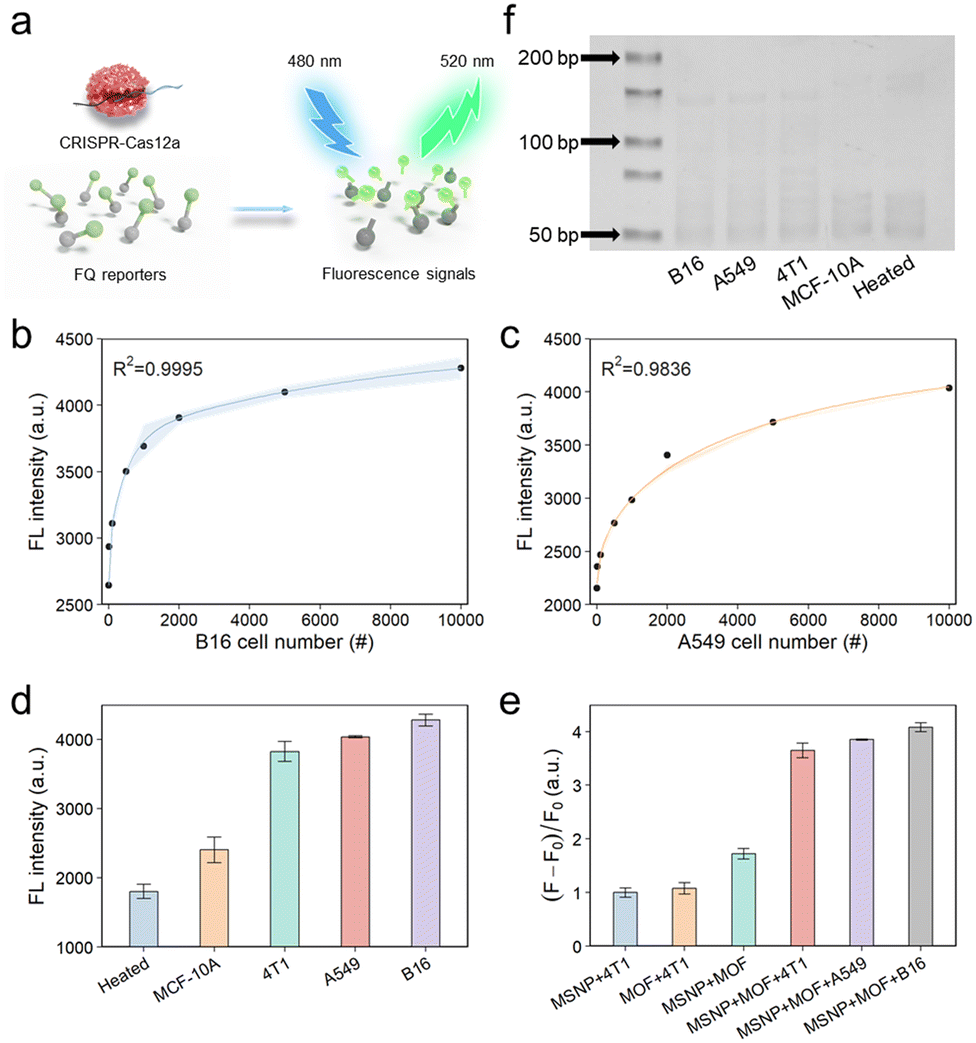 | ||
| Fig. 3 Sensitivity and selectivity analysis of the telomerase detection platform. (a) trans-Cleavage activity of Cas12a used for the amplification of the fluorescence detection signal. (b) Calibration curves for detecting telomerase activity in various numbers of B16 cells. (c) Calibration curves for detecting telomerase activity in various numbers of A549 cells. (d) Detection of telomerase activity in the CRISPR-Cas12a system across different types of cells, with 10000 cells used in each case. (e) Conditions for measuring the fluorescence intensity at 520 nm. Data are presented as mean ± SD of the triplicate samples. (f) Comparison of telomerase activity across different cell lines using the conventional TRAP assay. | ||
3.4 Sensitivity and selectivity of the detection platform
To investigate the specificity of the detection platform, various telomerase extracts were prepared, including those from a mouse breast carcinoma cell line (4T1 cells), a human lung cancer cell line (A549 cells), a mouse melanoma cell line (B16 cells), and a normal human breast epithelial cell line (MCF-10A cells). First, various cell line extracts were detected (Fig. 3d), and the results illustrated that the fluorescence intensity of the malignant tumor cell groups (4T1, A549, and B16 cells) surpassed those of the normal cell group (MCF-10A cells) and the heated negative control group (telomerase extracted from the cancer cells was heated at 100 °C for 10 min). This difference could be attributed to the inhibition of telomerase activity in normal cells and the deactivation of telomerase under elevated temperature conditions.45 Conversely, tumor cells exhibited increased telomerase activity.46 Next, we assessed the specificity of each detection probe. As shown in Fig. 3e, the observed fluorescence signals in the experimental results markedly exceeded those in their respective control groups, indicating the exceptional selectivity of the probes in detecting telomerase activity. In parallel, telomerase activity was also evaluated by the conventional TRAP method (Fig. 3f and S13†), revealing that the results were consistent with those obtained using the above methods. However, it is imperative to acknowledge that the telomerase activity assessed via the TRAP method is inherently semi-quantitative, exhibiting sensitivity inferior to that of our meticulously designed platform. These findings support the successful construction of an exceptionally sensitive and universally applicable framework for the precise detection of telomerase activity.3.5 On-chip single cell capture test for detection of telomerase activity
To further improve the performance of the developed telomerase activity detection platform at the single-cell level, we implemented a radial microfluidic chip (Fig. 4a) enabling single-cell capture and isolation for telomerase extraction. This radial circular microfluidic chip was prepared using conventional soft lithography and rapid polydimethylsiloxane (PDMS) molding techniques.33 As depicted in Fig. 4b, this microfluidic chip consisted of one inlet, 16 channels and 16 outlets. Upon introduction into the inlet, the cells within each channel were captured. Moreover, the continuous flow enabled the separation of cell doublets or clusters. Each channel was equipped with a customized hook-like trap, as depicted in Fig. 4c, designed to accommodate cell sizes ranging from 20 to 30 μm. This consideration takes into account the inherent flexibility of cells, ensuring tight capture (Fig. 4d and S14†). After the capture efficiency was optimized, a flow rate of 10 μL min−1 and a cell concentration of 107 cells per mL were selected. Moreover, to authenticate the viability of the captured cells rather than dead cells or cell fragments, A549 cells used as model cells were stained with a calcium xanthophyll dye. The successful capture of single live cells was visually confirmed through the manifestation of green fluorescence, as exemplified in Fig. 4e. This demonstrated the exceptional efficacy of the microfluidic chip in capturing individual live cells.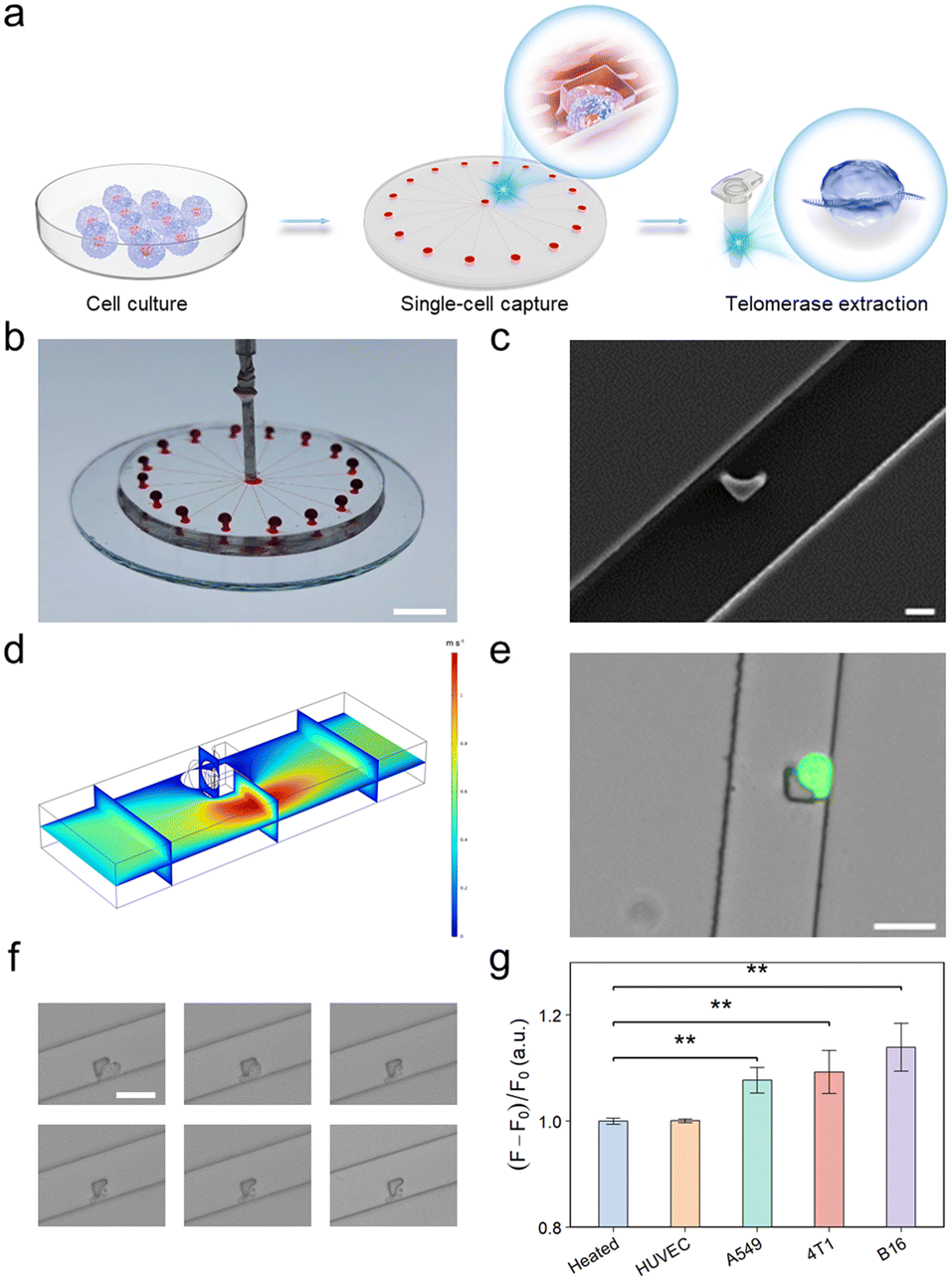 | ||
| Fig. 4 Microfluidic chip with single-cell capture capability enabling sensitive detection of telomerase at the single-cell level. (a) Schematic diagram illustrating the workflow for single-cell capture and telomerase extraction. (b) Photograph of a microfluidic chip filled with dye to aid visualization. Scale bar: 0.5 cm. (c) SEM image of the hook-shaped trap structure within the microfluidic chip. Scale bar: 10 μm. (d) Simulation of the fluid flow field within the microchannel after single-cell capture. (e) Merged microscopy images showing the capture of a single A549 cell stained with calcium xanthophyll. (f) Optical micrographs of single-cell lysates at different time intervals (0, 5, 10, 20, 30, and 60 min). Scale bars: 50 μm. (g) Detection of telomerase activity of different single cells via the CRISPR-Cas12a system. Statistical analysis was performed using a two-tailed Student's t-test. *P < 0.05, **P < 0.01. | ||
Afterwards, to optimize the conditions of single-cell lysis for telomerase extraction, a series of bright-field plots of A549 cell lysis over various durations were analyzed (Fig. 4f). Notably, discernible changes in the cell membrane, characterized by crumpling, were observed within the initial 2 min, and the complete lysis of the cells was achieved within 30 min. This observation provided compelling evidence supporting the successful extraction of telomerase. Following the successful extraction of telomerase from individual cells, the subsequent detection process was carried out according to the established method. The results are graphically presented in Fig. 4g, showing a significant difference in fluorescence intensity between the single malignant tumor cell groups and the negative control groups. This differential fluorescence intensity provided robust confirmation that the devised single-cell capture microfluidic chip, in conjunction with the integrated MOF–DNA bio-barcode-amplified CRISPR-Cas12a technology, was adept at detecting telomerase activity at the single-cell level.
4 Conclusions
In summary, we developed an innovative MOF–DNA bio-barcode-amplified CRISPR-Cas12a technology assisted by a microfluidic chip for single-cell telomerase activity analysis. The utilization of UiO-66 as a signal transducer plays a pivotal role in primary signal amplification through the binding and release of substantial DNA bar-codes. Subsequent self-signal amplification by the CRISPR-Cas12a system achieved secondary signal amplification through the efficient cleavage of the FQ reporter. When integrated with a single-cell capture microfluidic chip, the approach achieved high sensitivity at the single-cell level. The assay strategy, when applied to various malignant tumor cells, normal cells, and heated-negative control groups, demonstrated the versatility and selectivity of the platform in detecting telomerase activity. Compared with the conventional TRAP assay and other documented single-cell detection techniques, this method efficiently facilitates the detection of telomerase activity in individual cells within 3 h. This study introduces a novel approach for assessing telomerase activity at the single-cell level, thereby broadening the potential applications of the platform in the field of cancer diagnosis.The human normal breast cell line (MCF-10A cells), human lung cancer cell line (A549 cells), mouse melanoma cell line (B16 cells) and mouse breast carcinoma cell line (4T1 cells) were obtained from the Institute of Chinese Academy of Medical Sciences (Shanghai, China).
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Yateng Jiang: conceptualization, data curation, investigation, methodology, and writing – original draft. Yanping Wang: investigation, visualization, funding acquisition, and writing – review & editing. Wen Luo: investigation, methodology, and writing – review & editing. Xiaowei Luan: writing – review & editing. Zhibin Zhang: writing – review & editing. Bangshun He: supervision and writing – review & editing. Yanfeng Gao: conceptualization, data curation, formal analysis, investigation, methodology, funding acquisition, project administration, supervision, and writing – review & editing. Yujun Song: supervision, funding acquisition, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (22477056 and 82472379), the National Key Research and Development Program of China (2019YFA0709200), the Jiangsu Provincial Key Research and Development Program (BE2021373), the Natural Science Foundation of Anhui Province (2408085QH276), the Scientific Research Foundation of Education Department of Anhui Province of China (2024AH051909 and 2024AH051911), the Fundamental Research Funds for the Central Universities (2024300315), the State Key Laboratory of Analytical Chemistry for Life Science (5431ZZXM2304), and the Program for Innovative Talents and Entrepreneur in Jiangsu.References
- E. Martínez-Balsalobre, J. García-Castillo, D. García-Moreno, E. Naranjo-Sánchez, M. Fernández-Lajarín, M. A. Blasco, F. Alcaraz-Pérez, V. Mulero and M. L. Cayuela, Nat. Commun., 2023, 14, 5912 CrossRef PubMed.
- W. C. Hahn, S. A. Stewart, M. W. Brooks, S. G. York, E. Eaton, A. Kurachi, R. L. Beijersbergen, J. H. M. Knoll, M. Meyerson and R. A. Weinberg, Nat. Med., 1999, 5, 1164–1170 CrossRef CAS PubMed.
- R. Qian, L. Ding and H. Ju, J. Am. Chem. Soc., 2013, 135, 13282–13285 CrossRef CAS PubMed.
- Y. Pan, X. Luan, Y. Gao, F. Zeng, X. Wang, D. Zhou, W. Li, Y. Wang, B. He and Y. Song, ACS Nano, 2023, 17, 4515–4525 CrossRef CAS PubMed.
- L. Sun, Q. Zhao, X. Liu, Y. Pan, Y. Gao, J. Yang, Y. Wang and Y. Song, Chem. Commun., 2020, 56, 6969–6972 RSC.
- E. Savoysky, K.-i. Akamatsu, M. Tsuchiya and T. Yamazaki, Nucleic Acids Res., 1996, 24, 1175–1176 CrossRef CAS PubMed.
- X. Zhou and D. Xing, Chem. Soc. Rev., 2012, 41, 4643–4656 RSC.
- T. Zheng, E. Feng, Z. Wang, X. Gong and Y. Tian, ACS Appl. Mater. Interfaces, 2017, 9, 36596–36605 CrossRef CAS PubMed.
- Z. Zhang, E. Sharon, R. Freeman, X. Liu and I. Willner, Anal. Chem., 2012, 84, 4789–4797 CrossRef CAS PubMed.
- S. Huang, Q. Zhang, H. Yao, W. Wang, J.-R. Zhang and J.-J. Zhu, ACS Sens., 2020, 5, 1149–1157 CrossRef CAS PubMed.
- X. Ding, K. Yin, Z. Li, R. V. Lalla, E. Ballesteros, M. M. Sfeir and C. Liu, Nat. Commun., 2020, 11, 4711 CrossRef CAS PubMed.
- Y. Xiong, J. Zhang, Z. Yang, Q. Mou, Y. Ma, Y. Xiong and Y. Lu, J. Am. Chem. Soc., 2019, 142, 207–213 CrossRef PubMed.
- M. Hu, R. Liu, Z. Qiu, F. Cao, T. Tian, Y. Lu, Y. Jiang and X. Zhou, Angew. Chem., Int. Ed., 2023, 62, e202300663 CrossRef CAS PubMed.
- H. Wang, S. Wang, H. Wang, Y. Liang and Z. Li, Talanta, 2023, 253, 123999 CrossRef CAS.
- M. Cheng, E. Xiong, T. Tian, D. Zhu, H.-q. Ju and X. Zhou, Biosens. Bioelectron., 2021, 172, 112749 CrossRef CAS PubMed.
- X. Chen, G. Cao, J. Zhang, Y. Deng, X. Luo, M. Yang, D. Huo and C. Hou, Sens. Actuators, B, 2021, 333, 129553 CrossRef CAS.
- S. Zhi, X. Zhang, J. Zhang, X.-y. Wang and S. Bi, ACS Nano, 2023, 17, 13533–13544 CrossRef CAS PubMed.
- J.-M. Nam, C. S. Thaxton and C. A. Mirkin, Science, 2003, 301, 1884–1886 CrossRef CAS.
- N. Boehnke and P. T. Hammond, JACS Au, 2021, 2, 12–21 CrossRef PubMed.
- S. Chandra and T. Hu, ACS Nano, 2022, 16, 9985–9993 CrossRef CAS.
- S. Das, C. J. Lyon and T. Hu, ACS Nano, 2024, 18, 9784–9797 CrossRef CAS PubMed.
- S. M. Moosavi, A. Nandy, K. M. Jablonka, D. Ongari, J. P. Janet, P. G. Boyd, Y. Lee, B. Smit and H. J. Kulik, Nat. Commun., 2020, 11, 4068 CrossRef CAS PubMed.
- Q. Zhao, B. Pan, W. Long, Y. Pan, D. Zhou, X. Luan, B. He, Y. Wang and Y. Song, Nano Lett., 2022, 22, 9714–9722 CrossRef CAS.
- D. C. Swarts and M. Jinek, Mol. Cell, 2019, 73, 589–600 CrossRef CAS PubMed.
- H. Wang, H. Wang, C. Liu, X. Duan and Z. Li, Chem. Sci., 2016, 7, 4945–4950 RSC.
- Y. Gao, J. Xu, B. Li and Y. Jin, Biosens. Bioelectron., 2016, 81, 415–422 CrossRef CAS.
- Y. Gao, J. Xu, B. Li and Y. Jin, ACS Appl. Mater. Interfaces, 2016, 8, 13707–13713 CrossRef CAS.
- C.-c. Li, J. Hu, M. Lu and C.-y. Zhang, Biosens. Bioelectron., 2018, 122, 51–57 CrossRef CAS PubMed.
- H. Wu, Y. S. Chua, V. Krungleviciute, M. Tyagi, P. Chen, T. Yildirim and W. Zhou, J. Am. Chem. Soc., 2013, 135, 10525–10532 CrossRef CAS PubMed.
- V. Mamaeva, J. M. Rosenholm, L. T. Bate-Eya, L. Bergman, E. Peuhu, A. Duchanoy, L. E. Fortelius, S. Landor, D. M. Toivola and M. Lindén, et al. , Mol. Ther., 2011, 19, 1538–1546 CrossRef CAS PubMed.
- K. Adamiak and A. Sionkowska, Int. J. Biol. Macromol., 2020, 161, 550–560 CrossRef CAS PubMed.
- S. R. Rananaware, E. K. Vesco, G. M. Shoemaker, S. S. Anekar, L. S. W. Sandoval, K. S. Meister, N. C. Macaluso, L. T. Nguyen and P. K. Jain, Nat. Commun., 2023, 14, 5409 CrossRef CAS PubMed.
- Y. Gao, Y. Wang, B. He, Y. Pan, D. Zhou, M. Xiong and Y. Song, Angew. Chem., Int. Ed., 2023, 62, e202302000 CrossRef PubMed.
- B. Ning, S. Chandra, J. Rosen, E. Multala, M. Argrave, L. Pierson, I. Trinh, B. Simone, M. D. Escarra and S. Drury, et al. , ACS Nano, 2023, 17, 1206–1216 CrossRef CAS PubMed.
- S. Noreen, A. Maqbool, I. Maqbool, A. Shafique, M. M. Khan, Y. Junejo, B. Ahmed, M. Anwar, A. Majeed and M. Abbas, et al. , Mater. Chem. Phys., 2022, 285, 126132 CrossRef CAS.
- B. Fortuni, T. Inose, M. Ricci, Y. Fujita, I. Van Zundert, A. Masuhara, E. Fron, H. Mizuno, L. Latterini and S. Rocha, et al. , Sci. Rep., 2019, 9, 2666 CrossRef PubMed.
- Z. Li, J. C. Barnes, A. Bosoy, J. F. Stoddart and J. I. Zink, Chem. Soc. Rev., 2012, 41, 2590–2605 RSC.
- Y. Gu, D. Xie, Y. Ma, W. Qin, H. Zhang, G. Wang, Y. Zhang and H. Zhao, ACS Appl. Mater. Interfaces, 2017, 9, 32151–32160 CrossRef CAS PubMed.
- K. Yu, T. Wei, Z. Li, J. Li, Z. Wang and Z. Dai, J. Am. Chem. Soc., 2020, 142, 21267–21271 CrossRef CAS PubMed.
- S. Wang, C. M. McGuirk, M. B. Ross, S. Wang, P. Chen, H. Xing, Y. Liu and C. A. Mirkin, J. Am. Chem. Soc., 2017, 139, 9827–9830 CrossRef CAS PubMed.
- Y.-M. Zheng, L. Yu and J. P. Chen, J. Colloid Interface Sci., 2012, 367, 362–369 CrossRef CAS PubMed.
- J. Yang, Y. Dai, X. Zhu, Z. Wang, Y. Li, Q. Zhuang, J. Shi and J. Gu, J. Mater. Chem. A, 2015, 3, 7445–7452 RSC.
- Z. Wang, Y. Fu, Z. Kang, X. Liu, N. Chen, Q. Wang, Y. Tu, L. Wang, S. Song and D. Ling, et al. , J. Am. Chem. Soc., 2017, 139, 15784–15791 CrossRef CAS PubMed.
- W. Long, J. Yang, Q. Zhao, Y. Pan, X. Luan, B. He, X. Han, Y. Wang and Y. Song, Anal. Chem., 2023, 95, 1618–1626 CAS.
- N. W. Kim, M. A. Piatyszek, K. R. Prowse, C. B. Harley, M. D. West, P. L. C. Ho, G. M. Coviello, W. E. Wright, S. L. Weinrich and J. W. Shay, Science, 1994, 266, 2011–2015 CrossRef CAS PubMed.
- R. L. Flynn, K. E. Cox, M. Jeitany, H. Wakimoto, A. R. Bryll, N. J. Ganem, F. Bersani, J. R. Pineda, M. L. Suvà and C. H. Benes, et al. , Science, 2015, 347, 273–277 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lc00619d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |