
Rapid automated production of tubular 3D intestine-on-a-chip with diverse cell types using coaxial bioprinting†
Heeju
Song
a,
Yeonjin
Hong
 a and
Hyungseok
Lee
*ab
a and
Hyungseok
Lee
*ab
aDepartment of Smart Health Science and Technology, Kangwon National University, Chuncheon, Republic of Korea. E-mail: ahl@kangwon.ac.kr; Tel: +82 33 250 6309
bDepartment of Mechanical and Biomedical, Mechatronics Engineering, Kangwon National University, 1, Kangwondaehak-gil, Chuncheon-si, Gangwon-do, Republic of Korea
First published on 3rd December 2024
Abstract
Despite considerable animal sacrifices and investments, drug development often falters in clinical trials due to species differences. To address this issue, specific in vitro models, such as organ-on-a-chip technology using human cells in microfluidic devices, are recognized as promising alternatives. Among the various organs, the human small intestine plays a pivotal role in drug development, particularly in the assessment of digestion and nutrient absorption. However, current intestine-on-a-chip devices struggle to accurately replicate the complex 3D tubular structures of the human small intestine, particularly when it comes to integrating a variety of cell types effectively. This limitation is primarily due to conventional fabrication methods, such as soft lithography and replica molding. In this research, we introduce a novel coaxial bioprinting method to construct 3D tubular structures that closely emulate the organization and functionality of the small intestine with multiple cell types. To ensure stable production of these small intestine-like tubular structures, we analyzed the rheological properties of bioinks to select the most suitable materials for coaxial bioprinting technology. Additionally, we conducted biological assessments to validate the gene expression patterns and functional attributes of the 3D intestine-on-a-chip. Our 3D intestine-on-a-chip, which faithfully replicates intestinal functions and organization, demonstrates clear superiority in both structure and biological function compared to the conventional 2D model. This innovative approach holds significant promise for a wide range of future applications.
1. Introduction
In traditional drug development, the reliance on animal testing has raised concerns due to significant physiological differences between animals and humans,1 potentially making these models unreliable in predicting treatment outcomes.2 Controlled and systematic animal experiments may depict the effects of drugs in an overly favorable light, as they often exclude many variables associated with the complex environment of the human body.3 Ethical concerns and recognition of interspecies distinctions have led some nations to ban animal testing for cosmetics and raw ingredients.4 Recently, the FDA approved legislation to replace animal testing with cell-based experiments in the drug approval process,5 sparking increased interest in developing alternative methods for testing. This shift is expected to drive greater research and funding into in vitro models using human cells as substitutes for animal testing.The digestive system encompasses organs responsible for ingestion and excretion.6 Within this system, the gastrointestinal tract includes a series of interconnected organs forming a continuous tube that serves as the pathway for food from the mouth to the anus. The human small intestine, situated between the stomach and large intestine, exhibits distinct internal layers, including the intestinal lumen, mucosa, submucosa, muscularis propria, and serosa. The intestinal lumen serves as the conduit for ingested substances, while the mucosal layer features specialized villi responsible for nutrient absorption. Nutrients are transported through the villi via blood vessels and lacteals within the submucosal layer. The muscularis propria and serosal layers contain specific muscle types that facilitate peristalsis and provide tissue protection, respectively. The widespread use of oral medications, attributable to their ease of manufacture and distribution,7 along with patients' adherence to medical prescriptions,8 underscores the critical importance of understanding drug absorption mechanisms in the small intestine. Consequently, developing an ideal in vitro intestinal model is imperative for advancing drug delivery optimization.
Two-dimensional (2D) transwell models have long been regarded as the predominant method for investigating intestinal physiology and function as conventional intestinal in vitro models. These models allow cells to be cultured under different conditions in two chambers separated by a porous membrane, enabling the analysis of the migratory properties of cells. Culturing Caco-2, an intestinal epithelial cell line, on transwell models yields outcomes that closely mimic the structure and function of native small intestinal epithelium.9 The function of the small intestine can be analyzed by applying reagents to the upper chamber where Caco-2 are cultured, followed by the analysis of samples from the lower chamber. Owing to their simplicity and ease of use, 2D transwell models are widely used at present as in vitro models. Considerable efforts have been made to improve these models by stacking different types of small intestinal cells in a single layer to mimic the structural characteristics of the small intestine or co-culturing different types of cells in a single layer.10,11 However, transwell models still have inherent limitations in the functional expression of Caco-2 cells owing to the constraints in accurately mimicking the native environment.12
Organ-on-a-chip is a three-dimensional (3D) platform fabrication technique incorporating microfluidic systems, enabling the culture of tissue-specific cells within an environment that mimics the conditions encountered in the human body.13 Microchannel platforms offer precise size control in laboratory settings, facilitate rapid fluid mixing, and provide economic advantages through minimal reagent usage.14 For example, the intestine-on-a-chip (IOC) features flexible silicon chambers positioned on both sides of a microchannel with a porous membrane in the center.15 Although the IOC successfully emulates the 2D microenvironment of the small intestine, its structural configuration deviates from that of the actual small intestine, which typically exhibits a long cylindrical shape with a hollow center. This aspect highlights a limitation in its design. To date, numerous in vitro intestinal models have been developed. However, none have successfully replicated the long, cylindrical structure of the actual small intestine, crucial for mimicking the 3D biological environment. Therefore, in this study, we engineered a 3D tubular structure designed to replicate the cylindrical morphology of the small intestine using extrusion-based coaxial bioprinting. To recreate a 3D environment, we attached intestinal epithelial cells to the inner surface of this biocompatible tubular structure to establish a more authentic intestinal epithelium. We also explored the integration of small intestinal smooth muscle cells and vascular endothelial cells into the structure using bioprinting technology. This innovative approach not only replicated the structural characteristics of the actual small intestine but also enabled the use of various cell types within the IOC system, thereby enhancing the model's functionality and paving the way for future applications in drug testing.
2. Materials and methods
2.1. Cell culture
Caco-2, a human colon adenocarcinoma-derived intestinal epithelial cell line (HTB-37; ATCC, USA), were cultured for 5 to 15 passages. Initially, the cells were seeded at a density of 2 × 106 cells in 150 mm cell culture dishes (20150; SPL, Republic of Korea). The cell culture medium used was Dulbecco's modified Eagle's medium (DMEM) containing 4.5 g L−1 glucose, L-glutamine, and sodium pyruvate (10-013-CV; Corning, USA), supplemented with 10% (v/v) fetal bovine serum (FBS; 35-015-CV; Corning, USA) and 1% (v/v) penicillin (30-002-CI; Corning, USA). The medium was changed every other day.The hybrid cell line EA.hy926, resulting from the fusion of human umbilical vein endothelial cells (HUVECs) and A549, a thioguanine-resistant human lung carcinoma cell line, served as the selected vascular endothelial cell line in this study due to its distinctive endothelial characteristics. EA.hy926 were sourced from ATCC, USA, with accession number CRL-2922. The culture conditions and medium composition for EA.hy926 were identical to those used for Caco-2.
Human small intestine smooth muscle cells (HSISMC), sourced from Innoprot, Spain, under the trade name P10751, are primary cells isolated from healthy human small intestine. These cells were cultured in a smooth muscle cell medium (SMCM, with growth supplements, P60125; Innoprot, Spain). The cells from passages two to five were seeded at a density of 2 × 106 cells in 150 mm cell-culture dishes, and the medium was refreshed every other day.
When the cells reached confluence at 37 °C in a humidified atmosphere with 5% CO2 (BB15 CO2 Incubator, 51023121; Thermo Fisher, USA), a two-step detachment process was used. Initially, the cells underwent two rinses with 1× PBS (SM-P02-100, Solmate; GeneAll Biotechnology, Republic of Korea). The cells were then subjected to trypsin treatment (25300054, Gibco; Thermo Fisher, USA) for 2 min to facilitate detachment from the culture dish. The resulting cell suspension was transferred to a 50 mL conical tube (352070, Falcon; Corning, USA) containing 5 mL of the medium to neutralize the trypsin. The remaining cells in the dishes were incubated for an additional 2 min to ensure thorough collection. To ensure the collection of all remaining cells, 5 mL of the medium was spread across the dishes multiple times. The harvested cells were then transferred to the conical tube to the greatest extent possible. Finally, the collected cells were centrifuged at 800 RPM for 6 min in a centrifuge (1248R; Labogene, Republic of Korea) maintained at 4 °C.
2.2. Preparation of bioinks
A 3% (w/v) gelatin bioink was formulated using Caco-2 culture medium as the solvent. Porcine skin-derived gelatin powder (G2625, Sigma-Aldrich; Merck, Germany) was dissolved by heating it in a 37 °C water bath for 1 h. During this process, the gelatin bioink was periodically agitated by gently oscillating it vertically to prevent the powder from solidifying inside the conical tube. As the gelatin bioink maintains a liquid state at 37 °C, the cell-containing gelatin bioink was created by directly adding the required amount to the cell pellet, achieving a cell density of 7 × 106 cells per mL.Collagen sponge (derived from porcine skin, type I; PC-001; Dalim Tissen, Republic of Korea) was introduced into a solution of 17 mM acetic acid (1006-3705; Daejung Chemical & Metals, Republic of Korea) and stirred at 330 RPM for 6 h, forming a 1.5% (w/v) stock solution. Subsequently, the collagen stock solution was prepared proportionally to achieve a 1% (w/v) collagen bioink. This process included the addition of 10× PBS to one-tenth of the final solution volume and 1 M sodium hydroxide (S2018; Biosesang, Republic of Korea) to 1% (v/v) of the final solution volume for pH neutralization. The pH was adjusted in the range of 7.2–7.4 using minimal quantities of 1 M acetic acid and 1 M sodium hydroxide. The remaining solution was supplemented with DMEM, constituting four-fifths of the final solution volume. Lastly, cell pellets, along with the medium accounting for one-fifth of the final solution volume, were introduced to produce a 1% (w/v) collagen bioink containing cells. For the middle layer, a 1% collagen bioink with a cell density of EA.hy926 at 2 × 106 cells per mL.
A 4% (w/v) alginate bioink was prepared by dissolving alginic acid sodium salt powder (71238, Sigma-Aldrich; Merck, Germany) in 1× PBS. The mixture was stirred using a heating magnetic stirrer (SP88857105, Thermo Scientific; Themo Fisher, USA) at 140 °C and 440 RPM for 2 h, with a room temperature of 20 °C. The 4% (w/v) alginate bioink was mixed with a 1% (w/v) collagen bioink containing cells in a 1:1 ratio, resulting in a 2% (w/v) alginate–0.5% (w/v) collagen bioink with a cell density of HSISMC at 4 × 106 cells per mL.
2.3. Testing of rheological properties
The printability of the bioinks and stability of the fabricated structures were assessed using a rheometer (MCR92; Anton Paar, Austria). Initially, the storage and loss moduli of the gelatin bioinks at different concentrations were measured at a frequency of 1 Hz across ten temperature intervals ranging from the refrigeration temperature of 4 °C to the physiological body temperature of 37 °C. Gelatin samples, 1 mm thick, were prepared in 35 mm Petri dishes (353001, Falcon; Corning, USA) and stored in a 4 °C refrigerator for at least 6 h prior to testing.To evaluate the printability of collagen-based bioinks in the context of pre-crosslinking treatments, we analyzed the storage and loss moduli for 1% collagen bioink during both heating and cooling phases. Similar to the gelatin bioinks, these experiments were conducted at a frequency of 1 Hz, and the resulting data were presented in terms of tan(δ), representing the ratio of the loss modulus to the storage modulus. Moreover, after establishing suitable preparation conditions for automated production, we scrutinized viscosity curves for collagen-based bioinks characterized by lower melting points. This examination aimed to affirm that the pre-crosslinking treatment rendered the collagen-based bioink attainable in a printable gel state, ensuring its suitability for use in the printing process.
The storage moduli of both alginate–collagen and collagen scaffolds, cultured in the incubator for 12 h, were determined to assess their structural stability. Sample preparation involved casting 1.5-mm-thick layers of alginate–collagen bioinks and collagen bioinks in 35 mm Petri dishes. The samples were subjected to several processing steps: 15 min in a 37 °C incubator, 10 min in a 10 °C cooling environment, 10 min in a 2% (w/v) calcium chloride solution (1098, Duksan Pure Chemicals, Republic of Korea), 5 min in 1× PBS, followed by a thorough wash with 1× PBS, and subsequent replenishment with fresh media. The storage moduli of these samples were measured under consistent shear strain conditions of 0.5% and angular frequencies ranging from 1 to 100 rad s−1.
2.4. Extrusion-based coaxial bioprinting
Coaxial nozzles enable distinct materials to flow autonomously through separate nozzles with a shared center, ultimately merging into a unified strand-like structure at the nozzle tip. The number of overlapping layers corresponds to the number of nozzles. In this study, a triple-layer coaxial nozzle, specifically a triaxial needle (Ramé-hart, USA), consisting of 21G-17G-13G, was used for the printing process. The extrusion-based 3D bioprinter used was sourced from EDmic Bio (Republic of Korea). Based on the results related to rheological properties, 2% (w/v) alginate–0.5% (w/v) collagen and 1% (w/v) collagen bioinks were incubated at 37 °C for 15 min to ensure printability. Simultaneously, 3% (w/v) gelatin bioink was stored at 4 °C to transition into a gel state. All bioinks were then loaded into the cylinders of the 3D bioprinter, which was maintained at 10 °C. After waiting for 10 min to allow for temperature adjustment, different pressure conditions were applied: 30 kPa for the alginate–collagen bioink, 20 kPa for the collagen bioink, and 100 kPa for the gelatin bioink, to print tubular structures. After treatment with 2% (w/v) calcium chloride for 10 min, warm culture media caused the gelatin bioink to transition into a liquid state, facilitating the formation of a hollow channel and activating Caco-2 (Video S1†). Simultaneously, the collagen-based bioinks became cross-linked.2.5. Fabrication of testing in vitro platforms
The tubular structure, emulating the small intestine, was printed within a chip platform. The IOC platform, obtained from EDmicBio Inc. (Republic of Korea), is a polystyrene-based culture system designed to optimize media and reagent usage, while facilitating convenient handling and analysis. Different pressure conditions were applied to alginate–collagen bioink, collagen bioink, and gelatin bioink, at 30 kPa, 20 kPa, and 100 kPa, respectively. The tubular structure was secured using autoclaved 10% (w/v) low melting point agarose (AR1022; Biosesang, Republic of Korea). After a two-day attachment period for Caco-2, both ends of the structures were trimmed to allow perfusion with fresh medium. The luminal sides were filled with medium for Caco-2 and EA.hy926, while the basolateral side received medium for HSISMC. Media changes were performed every two days.For comparison, 12-well plate-sized transwell models (37012; SPL, Republic of Korea) were used as the control group. The seeding conditions involved using 500 μL of medium containing 1.25 × 105 cells of Caco-2 in the upper compartment and 1 mL of cell-free medium in the lower compartment. The medium in both compartments was changed every other day in equal volumes. In the dynamic culture system, a rocker (SHRK04DG; Ohaus, USA) was used for cultivation. For the dynamic culture models, a rocker was used to implement flow conditions of 4 °C and 10 RPM. These conditions were chosen to ensure even coverage on both sides of the culture and prevent detachment of the Caco-2 cells from the surface.
2.6. Cell labeling
Cell trackers were used to confirm the co-culture of the three cell types within the tubular structure. Caco-2 cells in the luminal area were labeled with CellTracker CM-DiI Dye (C7001, Invitrogen; Thermo Fisher, USA), while EA.hy926 cells in the inner layer were labeled with CellTracker Blue CMAC Dye (C2110, Invitrogen; Thermo Fisher, USA). HSISMC cells in the outer layer were labeled with CellTracker Green CMFDA Dye (C2925, Invitrogen; Thermo Fisher, USA). The 3D z-stack images were analyzed using a confocal microscope (AX; Nikon, Japan). The excitation wavelengths for the lasers were 385 nm, 470 nm, and 525 nm, which resulted in EA.hy926 appearing blue, HSISMC appearing green, and Caco-2 appearing yellow. All cells were incubated with the dyes for 30 minutes, using a ratio of 1 μL of dye per 1 mL of culture medium.2.7. Cell viability assessment
Living cells were labeled with green fluorescence using calcein AM, while dead cells were marked with a red fluorescent signal induced by ethidium homodimer (EthD-1). Cell staining was performed using a commercially available kit, the LIVE/DEAD viability/cytotoxicity kit for mammalian cells (L3224, Invitrogen; Thermo Fisher, USA). The tubular structures containing Caco-2, EA.hy926, and HSISMC were cultured for a period of 7 d, with cell densities of each cell type adjusted to 2 × 106 cells per mL for clear fluorescent imaging. The stained cells were subsequently observed and analyzed using a fluorescence microscope (Eclipse TS2, Nikon, Japan).2.8. Immunocytochemistry (ICC) assay
Immunofluorescence is a crucial technique used for visualizing the spatial distribution of specific proteins within cells, thereby confirming the presence or absence of target proteins in a given construct through confocal microscopy. For cell fixation in this study, IOC and transwell models, cultured for seven days, were prepared. Initially, they were rinsed twice with 1× PBS and subsequently treated with a 4% paraformaldehyde solution (BPP-9004, T&I; BioPrince, Republic of Korea) for 15 min at room temperature. Following the removal of the paraformaldehyde solution, the models underwent two additional washes with 1× PBS. Subsequent treatment with 0.2% (v/v) Triton X-100 (TR1020-500-00; Biosesang, Republic of Korea) for 15 min at room temperature was performed, followed by two more washes with 1× PBS. Next, the models were subjected to a blocking step using a 2% (w/v) fixative solution (Blocker BSA, 37525; Thermo Fisher, USA) for 1 h at room temperature, followed by another round of washing with 1× PBS. Subsequently, the models were stored in a refrigerator at −4 °C. Experimental procedures encompassed a sequence of steps as follows: a 6 h incubation with primary antibodies, followed by a 12 h period in fresh 1× PBS, and concluding with a 6 h incubation involving secondary antibodies and additional fluorescent dyes. Detailed information regarding the types of reagents and their dilution ratios can be found in Table 1.| Type | Product name | Brand | Catalog number | Dilution |
|---|---|---|---|---|
| 1st | ZO-1 monoclonal antibody (ZO1-1A12), Alexa Fluor™ 594 | Invitrogen | 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 194 194 |
1:200 |
| 1st | Villin polyclonal antibody | Invitrogen | PA5-29078 | 1:200 |
| 1st | MUC2 polyclonal antibody | Invitrogen | PA5-103083 | 1:200 |
| 1st | Lysozyme polyclonal antibody | Invitrogen | PA5-16668 | 1:100 |
| 2nd | Goat anti-rabbit IgG (H + L), superclonal recombinant secondary antibody, Alexa Fluor 488 | Invitrogen | A27034 | 1:1000 |
| Dye | DAPI (4′,6-diamidino-2-phenylindole, dihydrochloride) | Invitrogen | D1306 | 1:500 |
| Dye | Alexa Fluor™ 488 phalloidin | Invitrogen | A12379 | 1:400 |
2.9. Gene expressions with qRT-PCR
Quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR) is a precise technique used for the real-time measurement and analysis of specific gene expression levels. To evaluate the presence and expression of the genes in both models after 7 d of culture, RNA extraction from the models was carried out using the RiboEX reagent (301-002; GeneAll Biotechnology, Republic of Korea), following the manufacturer's guidelines. Reverse transcription was performed using the HyperScript RT Master Mix (601-710; GeneAll Biotechnology, Republic of Korea) on 1–5 μg of RNA to convert it into complementary DNA (cDNA). During the analysis of the cDNA samples using the qRT-PCR kit (TB Green Premix Ex TaqTM, RR420A; Takara, Japan), a three-step PCR technique was used instead of the two-step PCR technique recommended in the guide. This modification was made to enhance amplification efficiency on the Thermal Cycler Dice® Real Time System III (TP950; Takara, Japan). All results were normalized with respect to the cellular reference gene GAPDH, employing the ddCT method provided in the manufacturer's software (Takara). The primer sequences used in this analysis (Table 2) were procured from Bioneer, Republic of Korea.| Gene | Target | Primer type | Primers (5′-3′) | Genebank accession |
|---|---|---|---|---|
| GAPDH | Housekeeping gene | Forward | TGGAAGGACTCATGACCACAG | NM_001357943.2 |
| Reverse | TCCACCACTGACACGTTGG | |||
| CYP3A4 | Metabolism | Forward | CCCACAAAGCTCTGTCCGAT | NM_001202855.3 |
| Reverse | TATCATAGGTGGGTGGTGCC | |||
| CD31 | Vascular endothelial cells | Forward | GTCCCTGATGCCGTGGAAA | NM_000442.5 |
| Reverse | GGAGCAGGGCAGGTTCATAA | |||
| ACTA2 | Smooth muscle cells | Forward | AGCGCAAATACTCTGTCTGG | NM_001406464.1 |
| Reverse | CAGAGAGGAGCAGGAAAGTGT | |||
| CTNND1 | Cell morphology | Forward | CTGGTAAGAGAAGTGAGTGGTG | NM_001085469.2 |
| Reverse | CTAAAGTGAGAGGGGGCAATAC | |||
| VIL1 | Microvilli found on the villous top | Forward | GCCTCGATGGAAGCAACAAA | NM_007127.3 |
| Reverse | CGGTGAGAAAATGAGACCCTAC | |||
| LYZ | Protective enzyme secreted in crypts | Forward | GCCAAATGGGAGAGTGGTTAC | NM_000239.3 |
| Reverse | CCTGGGGTTTTGCCATCATTAC | |||
| TJP1 | Barrier function | Forward | GGAGGGTGAAGTGAAGACAATG | NM_001330239.4 |
| Reverse | CTGCTGGTTAGTATGTCTGTGG | |||
| MUC2 | Mucus secretion | Forward | GCCCTCTAACAACTACTCCTCT | NM_002457.5 |
| Reverse | GGTTTTCCAGAATCCAGCCAG |
2.10. Staining of mucus layer
Alcian blue (B8438, Sigma-Aldrich; Merck, Germany) is a cationic dye used for staining mucin-like acidic polysaccharides within tissues. To prepare the models for staining, they were initially fixed using a 4% formaldehyde solution for 30 min at room temperature. This fixation step was crucial for preserving the structural integrity of the cells. Following fixation, the models were immersed in an Alcian blue solution for a duration of 2 h at room temperature. Careful and thorough washing was performed to ensure complete removal of any residual dye solution. Finally, visual examination was conducted under a microscope (Eclipse TS2; Nikon, Japan) to estimate the quantity of mucus secretion by assessing the degree of staining.2.11. Permeability test
Intestinal permeability was assessed using a 4 kDa fluorescent dye (4 kDa FITC-Dextran, FD4, Sigma-Aldrich; Merck, Germany). The reagent was prepared by dissolving the fluorescent dye in phenol red-free DMEM (A1443001, Gibco; Thermo Fisher, USA) to prevent any potential influence of the solvent on fluorescence measurements. A total volume of 600 μL of the fluorescent dye was introduced into the section corresponding to the intestinal lumen, located in the upper chamber of the transwell model and side chambers of the IOC. Concurrently, 1.2 mL of phenol red-free DMEM was added to the basolateral section, representing the external portion of the intestine. All models were incubated on the rocker for 5 min to ensure even distribution of the reagent and subsequently examined using a fluorescent microscope.2.12. Drug screening tests
Acetaminophen (APAP, A7085, Sigma-Aldrich; Merck, Germany) was dissolved in a dimethyl sulfoxide (DMSO, D2650, Sigma-Aldrich; Merck, Germany) solution to create a 1 M APAP stock solution, which was subsequently diluted in the culture medium to achieve concentrations of 5 mM, 10 mM, and 20 mM. To evaluate the effects of APAP, each APAP solution was introduced into the luminal sides of the IOC, cultured for 7 d under dynamic conditions. To assess the toxicity induced by APAP, the LIVE/DEAD Viability/Cytotoxicity Kit was used to stain and distinguish between live and dead cells.2.13. Image analysis
Image data were analyzed using the open-source image processing software, ImageJ (National Institutes of Health, USA). To determine villous heights in the models, cell construct lengths were measured in accordance with the provided scale bar. Additionally, ImageJ was used to quantify transmittance resulting from Alcian blue staining, based on RGB values. For transwell models, the brightest field devoid of cells served as the blank reference, while the darkest field displaying differentiated villi was used as the analyzed reference. For the IOC, the blank reference was defined as the alginate–collagen layer, while the inner channel containing Caco-2 was designated as the analyzed reference. Additionally, to assess the permeability and dispersion of fluorescent particles, the green values within the image data were observed and quantified using ImageJ.2.14. Statistical analysis
The data are presented as mean ± standard deviations. Statistical analysis of the variables was conducted using Student's t-test. Results with p-values greater than 0.05 were considered non-significant (ns), those with p ≤ 0.05 were denoted with a single asterisk (*), those with p ≤ 0.01 were marked with two asterisks (**), and those with p ≤ 0.001 were indicated with three asterisks (***), signifying increasing levels of statistical significance.3. Results and discussion
3.1. Identification of suitable bioinks for automated production
The fabrication process for the IOC is illustrated in Fig. 1, providing a detailed overview of the 3D extrusion-based coaxial bioprinting technique used to shape tubular structures that mimic the morphology of the small intestine. In the illustration, the exterior part is represented in purple, signifying the presence of alginate–collagen bioink containing HSISMC, while the interior part is depicted in pink, indicating the use of collagen bioink containing EA.hy926. The yellow portion represents the gelatin bioink containing Caco-2.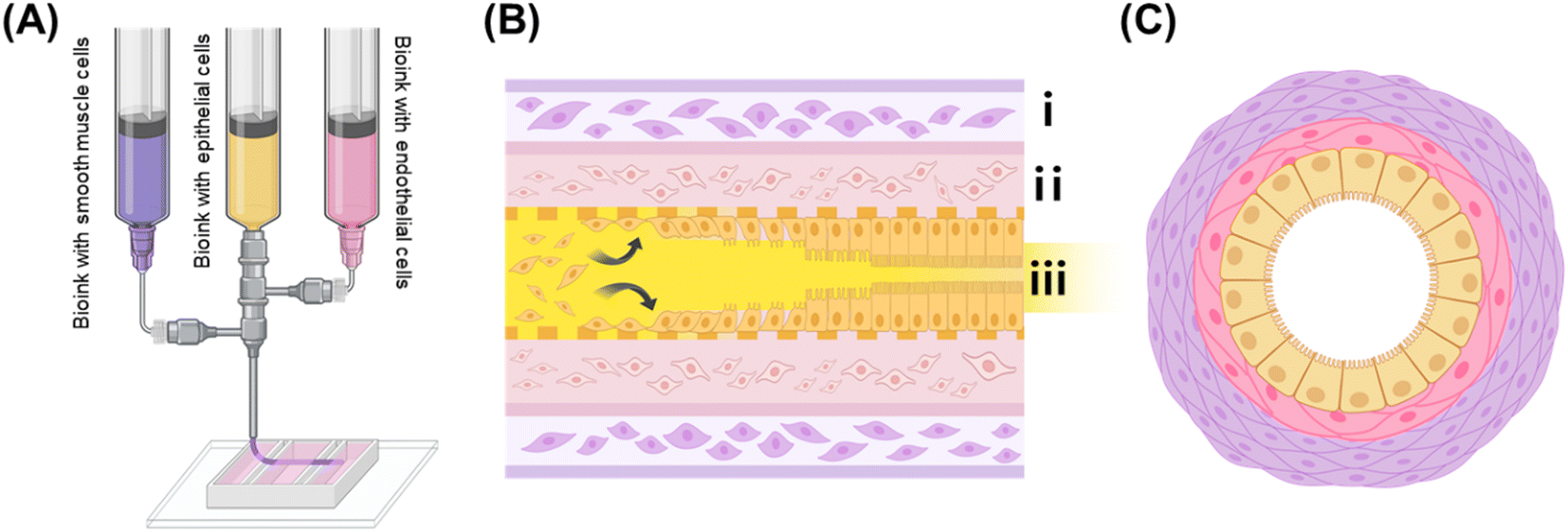 | ||
| Fig. 1 (A) Printing procedure over the chip platform. (B) (i) Indicates the presence of HSISMC within the alginate–collagen layer, (ii) denotes the incorporation of EA.hy926 within the collagen layer, and (iii) illustrates the attachment of Caco-2 within the gelatin bioink after the removal of gelatin as a sacrificial material. (C) Representation of the tubular structure produced in this research. Created using https://BioRender.com. | ||
In coaxial nozzle bioprinting, F-127 is frequently chosen as a material due to its favorable properties. However, it has certain limitations, particularly when it comes to encapsulating cells. One major issue is that F-127 can negatively affect cell viability, making it less ideal for applications where cell survival is critical.16 Additionally, even when used in bioprinting without cells, F-127 remains in a gel-like state at the typical cell culture temperature of 37 °C. This characteristic requires an extra step to remove the material after printing,17 which adds complexity to the overall process. Contrarily, the choice of gelatin bioink as a sacrificial material ensures uniform cell encapsulation and creates passageways without negatively impacting the Caco-2 until they adhere to the inner structure (Fig. 1(B)). Notably, gelatin bioinks can exhibit variations in viscosity due to cooling and heating cycles, potentially affecting printing conditions.18 Consequently, our experimental design adheres to a specific cooling cycle, accounting for the process of initially forming the cell suspension in a liquid state and subsequently allowing it to solidify at 4 °C. Thus, this study aimed to fabricate the tubular structure presented in Fig. 1(C).
To determine the optimal gelatin concentration for forming the inner layer of the structure, we analyzed the rheological properties of gelatin bioinks at concentrations of 1%, 2%, and 3% (w/v) and evaluated their response to temperature changes (Fig. 2(A)). Maintaining the gelatin bioink in an optimal gel state is crucial to ensure consistent ejection pressure while avoiding extremely low temperatures or high pressures that might damage the cells. A 2% gelatin concentration could maintain the gel state at 10 °C while ensuring stability at room temperature (20 °C). However, we selected a 3% gelatin concentration for its enhanced ability to manage the effects of cell density on printability and bioink properties.19,20
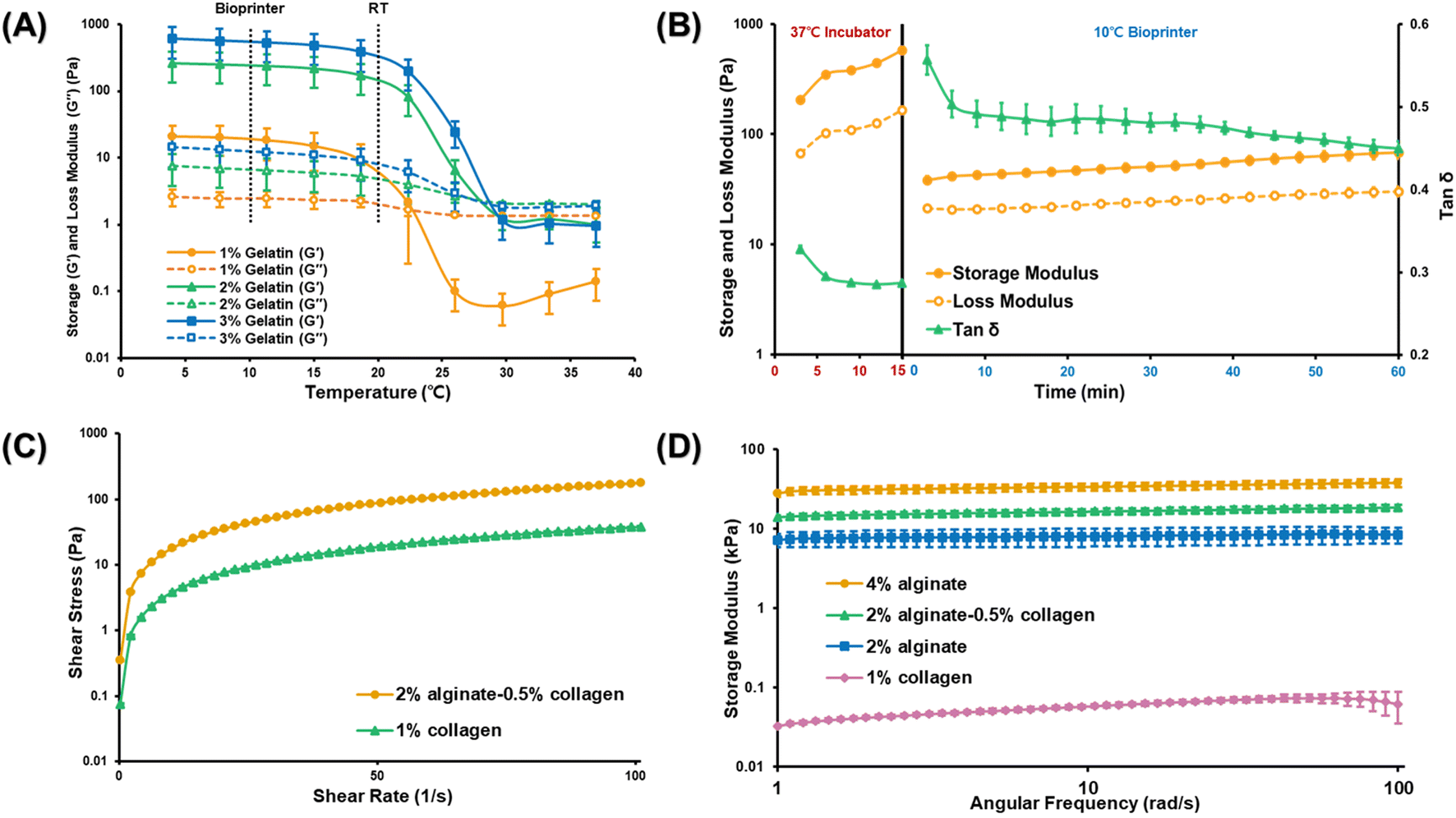 | ||
| Fig. 2 (A) Rheological properties of the gelatin bioinks at different concentrations and temperatures. (B) Rheological properties of the collagen bioink subjected to temperature treatment. The results demonstrate that the pre-crosslinked collagen bioink can be used without additional support material. (C) Viscosity curves for the melting point of the alginate–collagen and collagen bioinks. (D) Storage modulus values, indicating the stability of the long-term culture systems. | ||
For the middle vascular layer of the intestine structure, collagen bioink was selected. This choice was grounded in two main reasons. Ensuring stable adhesion of the Caco-2 cells to the inner surface, and providing a stable environment for maintaining vascular cells. Uncrosslinked collagen bioinks are generally weak and unsuitable for 3D bioprinting without additional support materials. These bioinks solidify when exposed to temperatures above 37 °C, and it is typically recommended to crosslink them for at least 30 min to achieve optimal results. In this study, we explored whether partial crosslinking could improve the printability of collagen bioinks compared with the standard use of uncrosslinked collagen bioinks in the bioprinting process. To achieve this, the bioinks were exposed to a temperature of 37 °C for 15 min and then to 10 °C for an additional 60 min. The aim was to assess if these conditions could positively influence the printability of the collagen bioinks. Fig. 2(B) shows how the storage modulus, loss modulus, and the ratio of these values, tan(δ), change over time at different temperatures. A tan(δ) value greater than 1 indicates a liquid state, while a value closer to 0 indicates a solid state. From these observations, we determined that the best printing condition is when tan(δ) is in the range of 0.4–0.6, representing a scenario where the gelatin bioink in the central layer is supported by a thin, stable layer. Fig. 2(B) indicates that the collagen bioink, after being crosslinked at 37 °C for 15 min, achieves a stable tan(δ) value at 10 °C after approximately 10 min, suggesting that these conditions are effective for stable printing.
To establish the muscle layer that supports the structural integrity of the small intestine, a combination of alginate and collagen bioinks was used. Alginate bioinks are commonly used in coaxial nozzle printing techniques owing to their rapid and straightforward gelation when exposed to calcium chloride.21,22 Based on previous recommendations for optimal printing conditions, a combination of 4% (w/v) alginate bioink and 2% (w/v) calcium chloride was used as the crosslinker for the alginate–collagen bioink in this study.21 Furthermore, to mitigate the risk of cell death associated with high calcium chloride concentration or extended crosslinking duration,16 we established a crosslinking period for the 2% calcium chloride solution, ensuring it did not exceed 15 min. Given the acidic pH of calcium chloride solutions based on 1× PBS, which could be detrimental to the cells, DMEM was used as the solvent for calcium chloride preparation. Consequently, we opted to employ DMEM as the solvent for calcium chloride preparation.
While alginate and calcium chloride are commonly favored in coaxial 3D extrusion bioprinting owing to their convenient sol–gel transition properties,23 alginate is not conducive to long-term cell culture within 3D constructs due to its lack of cell-binding sites.24 To enhance cell viability and create a more faithful tissue mimic, we incorporated collagen bioink in conjunction with alginate bioink. The 4% alginate bioink was combined with 1% collagen bioink in a 1:1 ratio for effective formulation.
To demonstrate the consistent printability of the collagen-based bioinks (collagen bioink and collagen–alginate bioink) under specific conditions, we assessed their viscosity profiles at low melting points. The relationship between shear stress and shear rate was examined after crosslinking at 37 °C for 15 min and stabilization at 10 °C for 10 min, consistent with the conditions set before initiating the printing process (Fig. 2(C)). Both alginate–collagen bioink and collagen bioink exhibited pseudoplastic behavior, characterized by a decreasing rate of shear stress as shear rate increased. This behavior indicates that they are shear-thinning fluids, where viscosity diminishes as shear rate escalates. Such a rheological profile is highly desirable for extrusion-based bioprinting processes.
After confirming the printability of the bioinks, we evaluated their mechanical stability—a critical prerequisite for the safe and sustained cultivation of cells. Notably, increased stiffness and density do not necessarily yield superior outcomes, as excessive rigidity can negatively impact biological functions such as cell proliferation and differentiation, potentially resulting in unexpected cell behavior within the tissue.25,26 Additionally, it is essential to consider any changes in stiffness over the culture period, as rapid degradation of the material stiffness may lead to structural collapse and loss of support for the cells.27 As illustrated in Fig. 2(D), both the alginate and collagen bioinks exhibited a consistent storage modulus within an angular frequency range of 1–100 rad s−1, indicating that both bioinks retained their structural stability even after 12 h of incubation. The inclusion of collagen bioink did not compromise structural integrity, as evidenced by the stable storage modulus, which remained similar to that of the 4% alginate and 2% alginate scaffolds. Moreover, the collagen in the endothelial layer exhibited the necessary softness to facilitate the growth of endothelial cells, and the small pores did not hinder their capacity to form a vascular network (Fig. 2(D)).
3.2. Formation of small-intestine-like tubular structures
After the printing process, we observed that the gelatin layer in the inner section liquefied, allowing the cells to gain mobility and adhere to the walls using the chosen bioinks (Video S1†). This effect is likely due to the presence of numerous cell-binding functional groups in the collagen-based middle layer, facilitating strong adhesion of Caco-2 cells. Fig. 3(A) shows the intended shape of the intestinal tubular structure in this study. To confirm that this shape was achieved using a combination of epithelial, endothelial, and smooth muscle cells, we captured microscopic images of the tubular structures after a 7-day co-culture, as shown in (B). As a result, we verified that all three cell types could successfully adhere and co-culture within the model. In addition to 2D microscopic images, we obtained 3D confocal images immediately after printing through cell labeling (Fig. S1†), which confirmed that the epithelial, vascular, and intestinal muscle cells formed distinct layers, establishing a 3D, perfusable structure directly post-printing.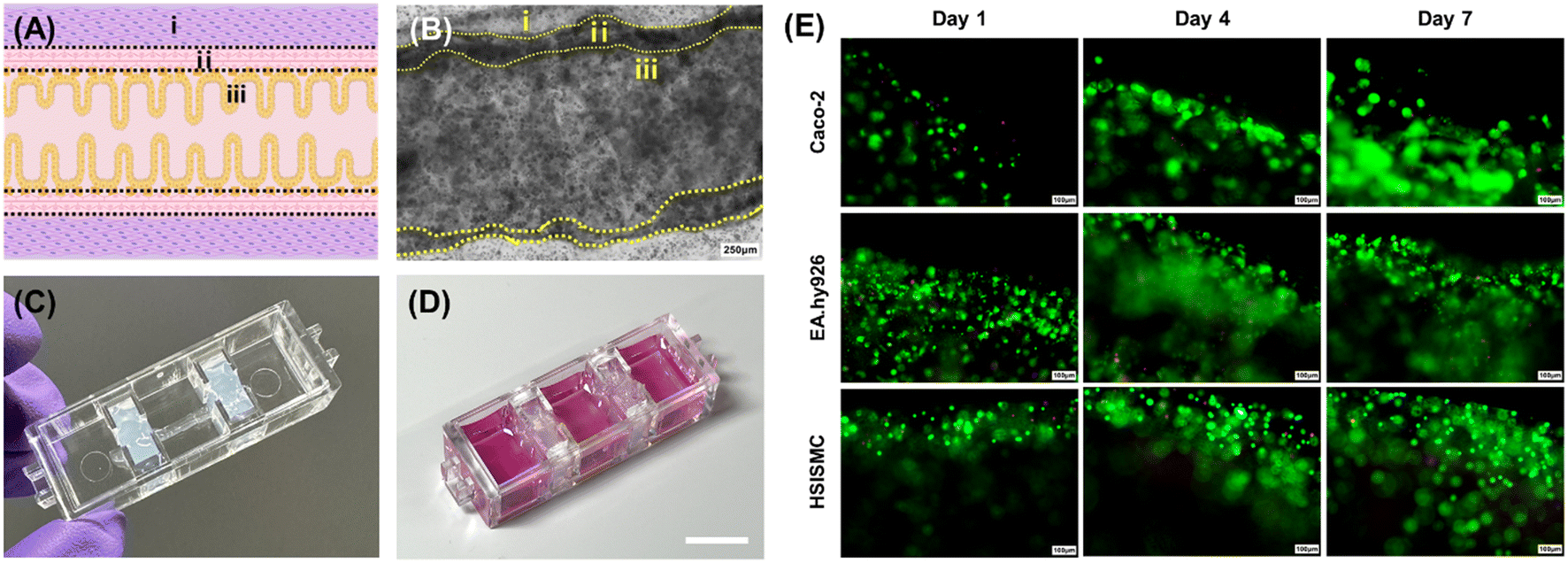 | ||
| Fig. 3 (A) Intended tubular structure resembling the small intestine. (i) Alginate–collagen layer containing HSISMC, (ii) collagen layer with EA.hy926, and (iii) attached Caco-2 forming the epithelium, mimicking the small intestine (the internal lumen of the small intestine). (B) Microscopic images of the tubular structure with Caco-2 cultured for 7 d (scale bar: 250 μm). Final small intestine-on-a-chip (IOC) created on the chip platform using a bioprinting technique (C) without medium, and (D) with medium (scale bar: 1 cm). (E) Live/dead assay to assess the viability of Caco-2, EA.hy926, and HSISMC. Live and dead cells were labeled with green and red fluorescence, respectively (scale bar: 100 μm). The figure was created using https://BioRender.com. | ||
Furthermore, Fig. 3(C) illustrates the bioprinted and fabricated tubular small intestine structure onto the chip platform, generating the final intestine-on-a-chip (IOC) model. We stained the Caco-2 within the tubular structures in the IOC using calcein AM and ethidium homodimer, as depicted in Fig. 3(D). Our findings demonstrated that epithelial cells actively proliferated and successfully adhered to the collagen endothelial layer. The significantly low number of dead cells observed by day 7 of culture strongly suggests that the gelatin bioink effectively shaped the hollow channel without inducing cytotoxicity.
The same experimental approach was employed for both endothelial cells and smooth muscle cells, with these cells integrated into the collagen and alginate–collagen layers, respectively. As illustrated in Fig. 3(D), both cell types exhibited robust growth in their designated layers, with no significant cell death observed. These results underscore the effectiveness of co-culturing smooth muscle cells, endothelial cells, and epithelial cells within the tubular structures, demonstrating that the system is capable of replicating the physiological environment of the small intestine and supporting normal cell functionality.
3.3. Formation of villi and crypts
The numerous villi within the epithelial layer exhibit a finger-like structure that enhances the surface area, facilitating increased nutrient absorption.28 Therefore, accurately replicating the villi is crucial for developing effective in vitro intestinal models, as it ensures the model's functionality in nutrient absorption and overall intestinal health. Dynamic culture systems significantly contribute to enabling cells cultured within a model to mimic natural growth mechanisms.29 In experiments aimed at inducing villi differentiation in intestinal epithelial cells, the applied shear stress has been identified as a pivotal factor.30 Thus, in this study, we investigated whether villi could be formed under dynamic culture conditions using rockers, which represent simpler, cleaner, and more accessible cultivation apparatus. To evaluate the effectiveness of villi formation in the dynamic culture environment of the IOC, we divided the experimental groups into three categories: 2D transwell dynamic, IOC static, and IOC dynamic (Fig. 4(A)). Immunostaining was performed to visualize filamentous actin (F-actin), a structural protein present in the small intestine and a key component of the cellular cytoskeleton, to assess villi formation. Confocal microscopy was then used to capture images of the stained cells (Fig. 4(B)). Both the IOC cultured under static and dynamic conditions exhibited the accumulation of Caco-2, forming well-defined villi. As depicted in Fig. 4(C), the villi in the IOC, exceeding a height of 100 μm, displayed significant growth regardless of the specific culture conditions. In contrast, the monolayer thickness of the epithelial cell in the transwell model was less than 50 μm.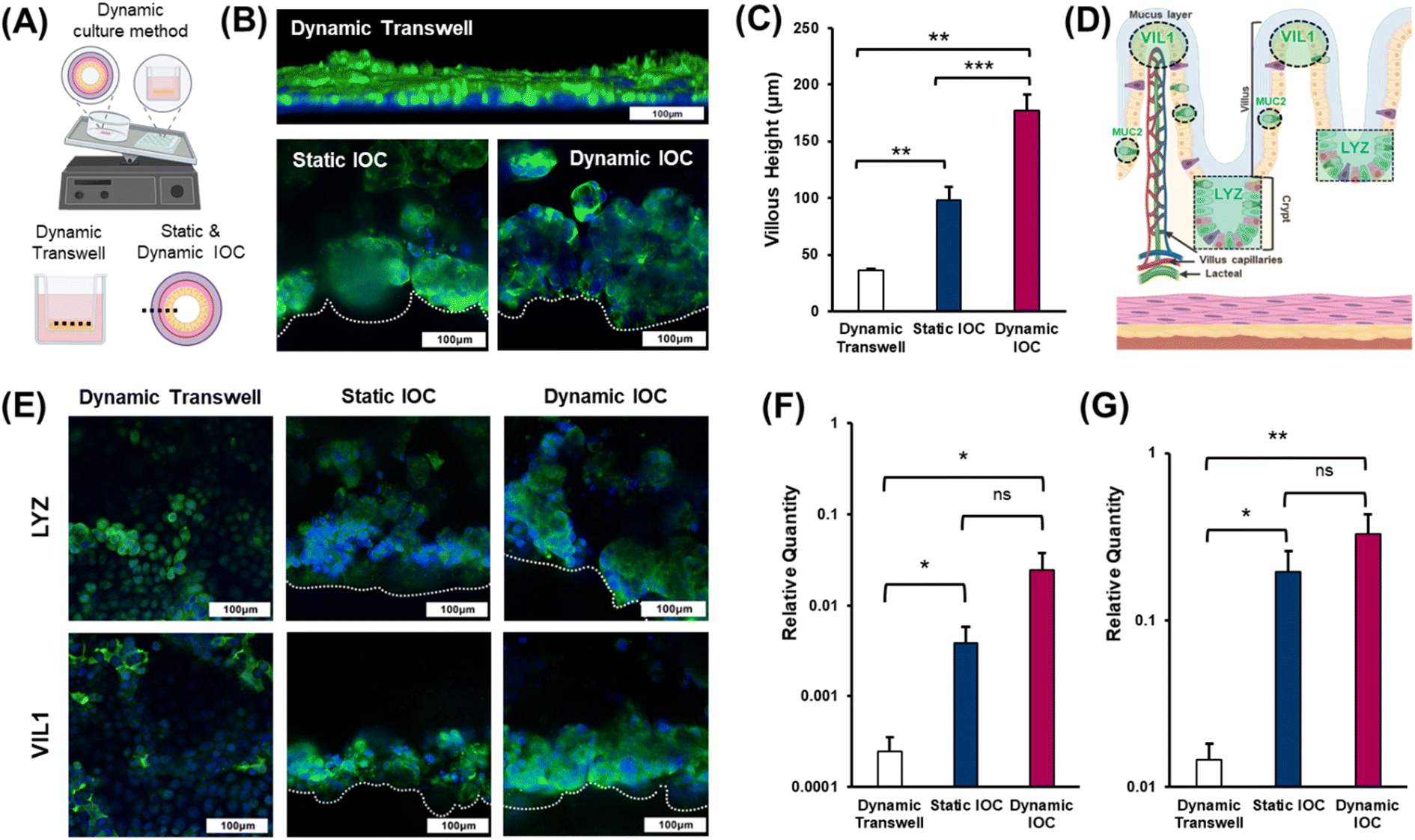 | ||
| Fig. 4 (A) Dynamic culture methods for Transwell and intestine-on-a-chip (IOC), and viewpoints on Transwell vs. static/dynamic IOC (dotted line represents the immunostaining viewpoint). (B) Cell morphology in various cell culture systems, examined by immunostaining F-actin in the models. F-actin was visualized as green fluorescence, and cell nuclei, stained with DAPI, appeared as blue fluorescence (scale bar: 100 μm). (C) Villous heights in all three types of models were measured using ImageJ. (D) Cellular and structural organization of the human small intestine, including the secretion of LYZ and presence of VIL1. (E) LYZ and VIL1 were detected by immunostaining, appearing as green fluorescence, while nuclei were stained with DAPI, appearing as blue fluorescence (scale bar: 100 μm). Relative expressions of (F) LYZ and (G) VIL1, determined through qRT-PCR experiments. The figure was created with https://BioRender.com. | ||
Lysozyme (LYZ) is an enzyme that plays a crucial role in the innate immune system, defending against bacterial infections.31 Within the villi, LYZ is primarily secreted at the crypts and plays a crucial role in protecting the epithelial cells. It is evenly distributed throughout both the mucus layer and crypts, ensuring comprehensive protection for the entire epithelial layer. Microvilli, situated on the apical side of the villi, serve to increase the surface area of the cell membrane, enhancing the functionality of the villi.32 The brush border, composed of numerous microvilli, is positioned closest to the intestinal lumen and plays a pivotal role in nutrient absorption. Villin-1 (VIL1), a cytoskeletal protein found in microvilli, contributes to structural support and amplifies the surface area for nutrient absorption (Fig. 4(D)). The results of villi height measurements across the different models (Fig. 4(C)) demonstrated a certain pattern pertaining to the effectiveness of villi formation that was consistent with the levels of LYZ and VIL1 staining observed in each model (Fig. 4(E)). Specifically, the staining intensities of both LYZ and VIL1 increased progressively from the dynamic transwell model to the static IOC model, and these values were the highest in the dynamic IOC model. These findings confirm that the dynamic IOC model not only supports the formation of well-structured villi but also replicates the key functionalities of crypts and villi most effectively. Therefore, the dynamic IOC model was validated as the superior platform for mimicking the structural and functional characteristics of the small intestine. The relative gene expression levels of LYZ and VIL1, determined through qRT-PCR, further supported these observations, showing that the dynamic conditions in the IOC model led to the tallest villi and most pronounced crypt formation (Fig. 4(F) and (G)).
3.4. Biological characterizations related to functionalities
The luminal surface of the small intestine is exposed to numerous external pathogens and potential threats, including mechanical injury, chemical irritants, intestinal bacteria, and ingested pathogens. To protect the epithelial cells from these risks and prevent cellular dehydration by trapping water, a protective mucus layer must be present. This mucus layer is primarily composed of Mucin2 (MUC2), a glycoprotein secreted within the small intestine.33 Additionally, the intestinal barrier function is crucial for regulating the selective passage of ions and molecules across compartments with distinct biological structures.34 Maintaining the integrity of this barrier is essential for small intestine homeostasis, and the expression of zonula occludens-1 (ZO-1), a tight junction protein, serves as an indicator to assess the functionality of the models.We assessed the presence of MUC2 in both the transwell and IOC models to evaluate their ability for Mucin2 secretion. As shown in Fig. 5(A) and (B), dynamic transwell models exhibited limited MUC2 expression, whereas the IOC demonstrated more pronounced and positive MUC2 expression. Based on these findings, the presence of well-defined villi and crypts in the IOC model appears to significantly enhance MUC2 expression and secretion, indicating that cell interactions within the IOC structure play a key role in epithelial function. Additionally, previous studies support that the presence of endothelial cells, smooth muscle cells, and epithelial cells enhances epithelial functionality, suggesting a synergistic effect on the barrier integrity and immune responses of epithelial layers.35 Given that flow rates considerably affect the morphology of the intestinal barrier,36 it is essential to highlight the distinction in ZO-1 expression levels between the dynamic transwell model and static IOC. Despite the exposure to dynamic conditions in the transwell models, the static IOC exhibited superior ZO-1 expression levels, as illustrated in Fig. 5(A) and (C). These findings suggest that the formation of villi through the creation of a 3D tubular structure (Video S2†) is associated with improved barrier function in small intestinal in vitro models. In this context, the IOC cultured under dynamic conditions demonstrated the most favorable outcomes among the three models.
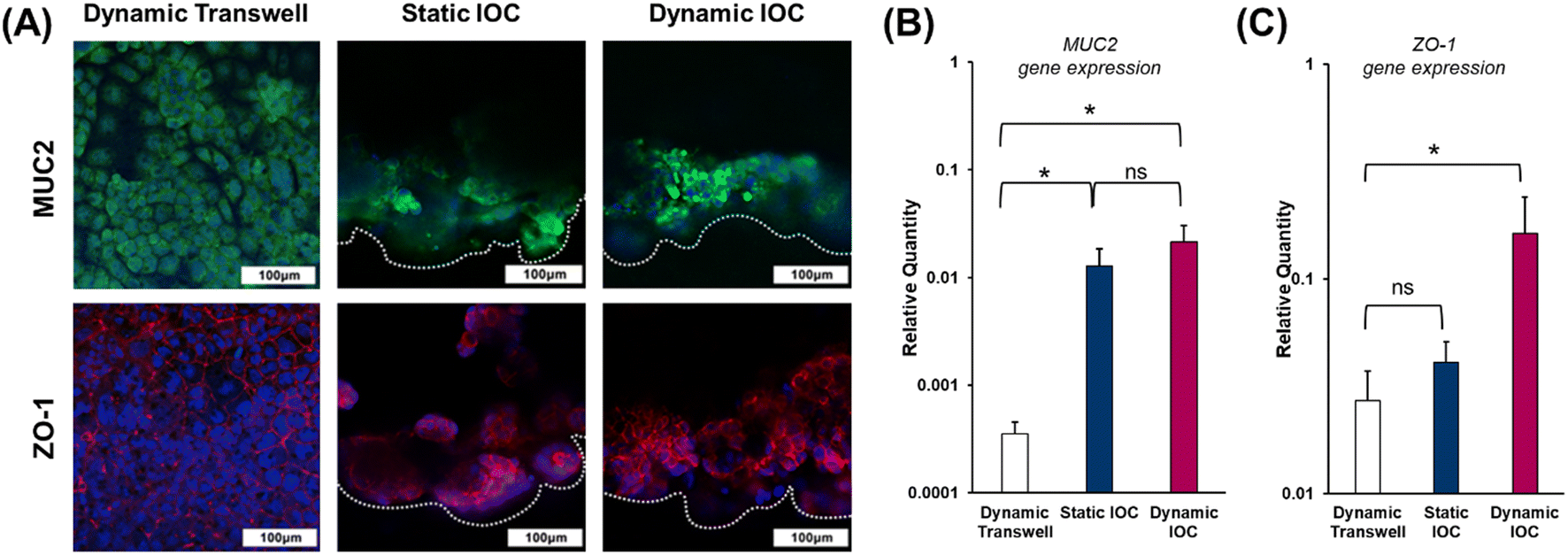 | ||
| Fig. 5 Evaluation of MUC2 protein secretion of the mucus layer and ZO-1 for intestinal barrier integrity. (A) Immunostaining of MUC2 and ZO-1, with MUC2 detected in green and ZO-1 in red. Nuclei stained with DAPI (blue) (images were obtained from the same regions as in the schematic shown in Fig. 3., scale bar: 100 μm). Relative expressions of (B) MUC2 and (C) ZO-1, determined through qRT-PCR experiments. | ||
3.5. Evaluation of functionalities
To evaluate the intestinal functionalities of the in vitro models, we conducted Alcian blue staining assays for MUC2 secretion and permeability tests for barrier function after 7 d of cultivation. Alcian blue staining was used to assess the presence and thickness of the mucus layer (Fig. 6(A)). The intensity of the staining in the microscopic images reflects the thickness of the mucus layer, with the dynamic IOC showing a thicker mucus layer compared with the other groups. To quantify the mucus layer thickness, we used ImageJ to analyze grayscale values for transmittance comparison (Fig. 6(B)). ImageJ analysis revealed that the dynamic IOC had the lowest transmittance values, indicating a higher level of MUC2 secretion, which resulted in a thicker mucus layer. Consequently, these results confirm that the dynamic IOC exhibits the highest level of MUC2 secretion among the models tested.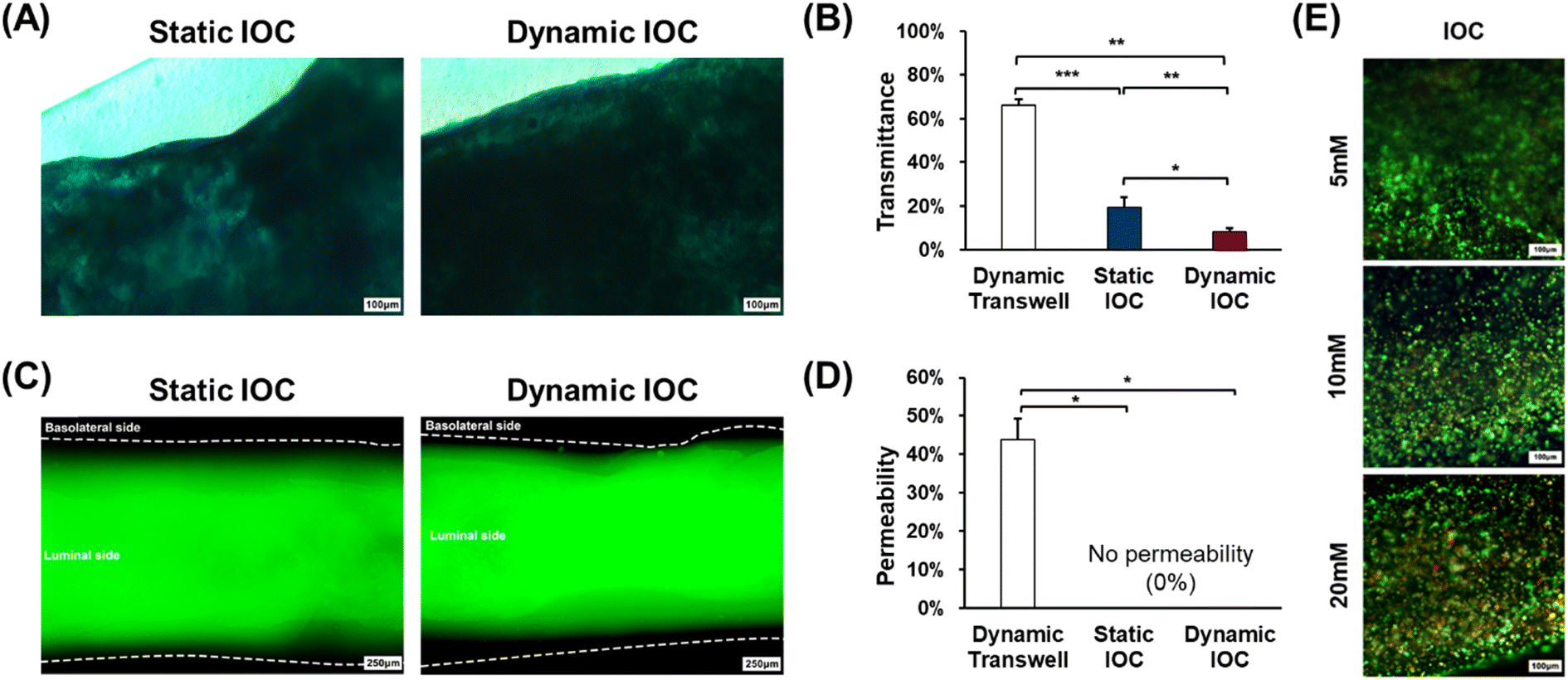 | ||
| Fig. 6 (A) Alcian blue staining assay to assess mucus secretions (scale bar: 100 μm). Darker coloration indicates a thicker mucus layer. Quantification of (B) transmittance, reflecting the secretion of the mucus layer, performed using ImageJ. (C) Permeability tests conducted using a 4 kDa FITC-dextran solution and visualized under a fluorescent microscope (scale bar: 100 μm). Quantification of (D) permeability, indicative of barrier integrity, performed using ImageJ. (E) Live/dead assay to evaluate cell viability in the IOC following APAP treatment. Living and dead cells emit green and red fluorescence, respectively (scale bar: 100 μm). | ||
In the permeability test, a low molecular weight fluorescent dye with a molecular weight of 4 kDa was applied to the luminal side of IOC models. This molecular weight is adequately small to traverse the pericellular pathway, allowing for permeability experiments with more stringent criteria compared with the 40 kDa threshold, which just passes through the intercellular space. Fluorescent molecules rapidly passed through the porous membrane in the dynamic transwell model within only 5 min of treatment. In contrast, both the static and dynamic IOC models effectively prevented the passage of the fluorescent dye to the basolateral side, as illustrated in Fig. 6(C). To quantify permeability, we analyzed the intensity of the green fluorescence on the basolateral side across the three models using ImageJ software (Fig. 6(D)). This analysis revealed that the IOC models exhibited a lower level of fluorescence on the basolateral side compared with the dynamic transwell model, indicating better barrier function. These results confirm that the IOC models, cultured for 7 d, maintain robust intestinal barrier integrity, effectively preventing the passage of fluorescent molecules and demonstrating their capability to accurately replicate the intestinal barrier function.
Acetaminophen (APAP) is a commonly used over-the-counter analgesic and antipyretic.37 However, it has been reported to potentially harm the intestinal epithelium and pose a risk of liver toxicity.38,39 In light of these findings, we conducted drug metabolism tests using APAP concentrations of 5 mM, 10 mM, and 20 mM. Fig. 6(E) shows images depicting live and dead cells within the IOC. The number of dead cells within the IOC increased with rising concentrations of APAP. These observations suggest that the IOC effectively metabolized the drug, reinforcing its suitability as an intestinal in vitro model for drug delivery system studies.
4. Conclusion
This study was aimed at developing an IOC with a 3D tubular structure that closely mimics the human small intestine, utilizing bioprinting technology. This research was driven by the need for an improved in vitro model of the small intestine, given the limitations of existing models. Our fabricated tubular structures, emulating the small intestine, consisted of epithelial cells adhering to the inner wall, closely resembling the natural small intestine. These structures featured multiple layers, with interior and exterior layers, facilitating the incorporation of vascular endothelial cells and intestinal smooth muscle cells to replicate the composition of the small intestine within each layer. The IOC developed in this study outperformed conventional transwell models, even without dynamic culture conditions. This underscores the importance of achieving distinct villi and crypts in establishing an effective small intestinal in vitro model. Thus, the proposed IOC emerges as an ideal model. In conclusion, the proposed IOC represents a significant advancement in achieving structural resemblance to the small intestine compared with existing in vitro models. Furthermore, its innovative co-culture with intestinal cell types opens new avenues for exploring previously unknown mechanisms of the small intestine.Data availability
The data cannot be made publicly available upon publication because they are owned by a third party and the terms of use prevent public distribution. The data that support the findings of this study are available upon reasonable request from the authors.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by a National Research Foundation of Korea (NRF) grants funded by the Korea government (MSIT) (No. 2020R1C1C1011147 and RS-2024-00423107).References
- G. A. Van Norman, JACC: Basic Transl. Sci., 2019, 4, 845–854 Search PubMed.
- A. Nair, M. A. Morsy and S. Jacob, Drug Dev. Res., 2018, 79, 373–382 CrossRef CAS.
- S. H. Richter, Lab. Anim., 2017, 46, 343–349 CrossRef.
- M. Cassotta, J. J. Bartnicka, F. Pistollato, S. Parvatam, T. Weber, V. D'Alessandro, L. F. Bastos and S. Coecke, Eng. Life Sci., 2022, 22, 564–583 CrossRef CAS.
- E. Y. Adashi, D. P. O'Mahony and I. G. Cohen, Am. J. Med., 2023, 136, 853–854 CrossRef CAS PubMed.
- G. J. Tortora and B. H. Derrickson, Principles of anatomy and physiology, John Wiley & Sons, 2018 Search PubMed.
- S. V. Sastry, J. R. Nyshadham and J. A. Fix, Pharm. Sci. Technol. Today, 2000, 3, 138–145 CrossRef CAS PubMed.
- A. Paes, A. Bakker and C. Soe-Agnie, Pharm. World Sci., 1998, 20, 73–77 CrossRef CAS PubMed.
- M. Schweinlin, S. Wilhelm, I. Schwedhelm, J. Hansmann, R. Rietscher, C. Jurowich, H. Walles and M. Metzger, Tissue Eng., Part C, 2016, 22, 873–883 CrossRef CAS.
- F. Leonard, E.-M. Collnot and C.-M. Lehr, Mol. Pharmaceutics, 2010, 7, 2103–2119 CrossRef CAS PubMed.
- F. Antunes, F. Andrade, F. Araújo, D. Ferreira and B. Sarmento, Eur. J. Pharm. Biopharm., 2013, 83, 427–435 CrossRef CAS PubMed.
- S.-M. Jung and S. Kim, Front. Microbiol., 2022, 12, 767038 CrossRef.
- C. M. Leung, P. De Haan, K. Ronaldson-Bouchard, G.-A. Kim, J. Ko, H. S. Rho, Z. Chen, P. Habibovic, N. L. Jeon and S. Takayama, Nat. Rev. Methods Primers, 2022, 2, 33 CrossRef CAS.
- Z. Li, B. Zhang, D. Dang, X. Yang, W. Yang and W. Liang, Sens. Actuators, A, 2022, 344, 113757 CrossRef CAS.
- H. J. Kim, D. Huh, G. Hamilton and D. E. Ingber, Lab Chip, 2012, 12, 2165–2174 RSC.
- N. Cao, X. Chen and D. Schreyer, Int. Scholarly Res. Not., 2012, 2012, 516461 Search PubMed.
- G. Gao, J. H. Lee, J. Jang, D. H. Lee, J. S. Kong, B. S. Kim, Y. J. Choi, W. B. Jang, Y. J. Hong and S. M. Kwon, Adv. Funct. Mater., 2017, 27, 1700798 CrossRef.
- S. A. Irvine, A. Agrawal, B. H. Lee, H. Y. Chua, K. Y. Low, B. C. Lau, M. Machluf and S. Venkatraman, Biomed. Microdevices, 2015, 17, 1–8 CrossRef CAS.
- N. Diamantides, C. Dugopolski, E. Blahut, S. Kennedy and L. J. Bonassar, Biofabrication, 2019, 11, 045016 CrossRef CAS PubMed.
- G. J. Gillispie, A. Han, M. Uzun-Per, J. Fisher, A. G. Mikos, M. K. K. Niazi, J. J. Yoo, S. J. Lee and A. Atala, Tissue Eng., Part A, 2020, 26, 1349–1358 CrossRef CAS PubMed.
- A. Kjar, B. McFarland, K. Mecham, N. Harward and Y. Huang, Bioact. Mater., 2021, 6, 460–471 CAS.
- T. S. Mohan, P. Datta, S. Nesaei, V. Ozbolat and I. T. Ozbolat, Prog. Biomed. Eng., 2022, 4, 022003 CrossRef.
- K. Y. Lee and D. J. Mooney, Prog. Polym. Sci., 2012, 37, 106–126 CrossRef CAS.
- T. Hu and A. C. Lo, Polymer, 2021, 13, 1852 CAS.
- F. Brandl, F. Sommer and A. Goepferich, Biomaterials, 2007, 28, 134–146 CrossRef CAS.
- K. Saha, A. J. Keung, E. F. Irwin, Y. Li, L. Little, D. V. Schaffer and K. E. Healy, Biophys. J., 2008, 95, 4426–4438 CrossRef CAS.
- L. Meinel, S. Hofmann, V. Karageorgiou, L. Zichner, R. Langer, D. Kaplan and G. Vunjak-Novakovic, Biotechnol. Bioeng., 2004, 88, 379–391 CrossRef CAS PubMed.
- J. DeSesso and C. Jacobson, Food Chem. Toxicol., 2001, 39, 209–228 CrossRef CAS PubMed.
- C. K. Byun, K. Abi-Samra, Y. K. Cho and S. Takayama, Electrophoresis, 2014, 35, 245–257 CrossRef CAS.
- W. Shin, C. D. Hinojosa, D. E. Ingber and H. J. Kim, iScience, 2019, 15, 391–406 CrossRef CAS.
- N. Gassler, World J. Gastrointest. Pathophysiol., 2017, 8, 150 Search PubMed.
- L. G. Van Der Flier and H. Clevers, Annu. Rev. Physiol., 2009, 71, 241–260 CrossRef CAS PubMed.
- N. W. Toribara, J. R. Gum, P. J. Culhane, R. E. Lagace, J. W. Hicks, G. M. Petersen and Y. S. Kim, J. Clin. Invest., 1991, 88, 1005–1013 CrossRef CAS PubMed.
- K. R. Groschwitz and S. P. Hogan, J. Allergy Clin. Immunol., 2009, 124, 3–20 CrossRef CAS PubMed.
- C. J. Childs, H. M. Poling, K. Chen, Y.-H. Tsai, A. Wu, C. W. Sweet, A. Vallie, M. K. Eiken, S. Huang, R. Schreiner, Z. Xiao, A. S. Conchola, M. F. Anderman, E. M. Holloway, A. Singh, R. Giger, M. M. Mahe, K. D. Walton, C. Loebel, M. A. Helmrath, S. Rafii and J. R. Spence, bioRxiv, 2023, preprint, DOI:10.1101/2023.11.06.565830.
- K. Kulthong, L. Duivenvoorde, B. Z. Mizera, D. Rijkers, G. Ten Dam, G. Oegema, T. Puzyn, H. Bouwmeester and M. van der Zande, RSC Adv., 2018, 8, 32440–32453 RSC.
- M. J. Hodgman and A. R. Garrard, Crit. Care Clin., 2012, 28, 499–516 CrossRef.
- C. Schäfer, K. R. Schröder, O. Höglinger, S. Tollabimazraehno and M. R. Lornejad-Schäfer, Cell. Physiol. Biochem., 2013, 32, 431–447 CrossRef PubMed.
- G. G. Graham, M. J. Davies, R. O. Day, A. Mohamudally and K. F. Scott, Inflammopharmacology, 2013, 21, 201–232 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lc00731j |
| This journal is © The Royal Society of Chemistry 2025 |