
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sensor-integrated gut-on-a-chip for monitoring senescence-mediated changes in the intestinal barrier†
Konstanze
Brandauer
 a,
Alexandra
Lorenz
a,
Silvia
Schobesberger
a,
Patrick
Schuller
a,
Martin
Frauenlob
a,
Sarah
Spitz
ab and
Peter
Ertl
*ab
a,
Alexandra
Lorenz
a,
Silvia
Schobesberger
a,
Patrick
Schuller
a,
Martin
Frauenlob
a,
Sarah
Spitz
ab and
Peter
Ertl
*ab
aTU Wien, Faculty of Technical Chemistry, Getreidemarkt 9, 1060 Vienna, Austria. E-mail: peter.ertl@tuwien.ac.at
bAustrian Cluster for Tissue Regeneration, Vienna, Austria
First published on 20th February 2025
Abstract
The incidence of inflammatory bowel disease among the elderly has significantly risen in recent years, posing a growing socioeconomic burden to aging societies. Moreover, non-gastrointestinal diseases, also prevalent in this demographic, have been linked to intestinal barrier dysfunction, thus highlighting the importance of investigating aged-mediated changes within the human gut. While gastrointestinal pathology often involves an impaired gut barrier, the impact of aging on the human gastrointestinal barrier function remains unclear. To explore the effect of senescence, a key hallmark of aging, on gut barrier integrity, we established and evaluated an in vitro gut-on-a-chip model tailored to investigate barrier changes by the integration of an impedance sensor. Here, a microfluidic gut-on-a-chip system containing integrated membrane-based electrode microarrays is used to non-invasively monitor epithelial barrier formation and senescence-mediated changes in barrier integrity upon treating Caco-2 cells with 0.8 μg mL−1 doxorubicin (DXR), a chemotherapeutic which induces cell cycle arrest. Results of our microfluidic human gut model reveal a DXR-mediated increase in impedance and cell hypertrophy as well as overexpression of p21, and CCL2, indicative of a senescent phenotype. Combined with the integrated electrodes, monitoring ∼57% of the cultivation area in situ and non-invasively, the developed chip-based senescent-gut model is ideally suited to study age-related malfunctions in barrier integrity.
Introduction
The worldwide incidence of inflammatory bowel disease in people over 60 years is about 6–14/100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 per year and is still rising among the elderly, creating a significant socioeconomic burden.1 Additionally, recent evidence suggests that non-gastrointestinal disorders prevalent among the aging population, such as Parkinson's disease and rheumatoid arthritis, originate in the gastrointestinal tract (GIT) and can be associated with an impaired barrier.2–4 However, the effect of aging on the gastrointestinal barrier still remains unclear.5–7 While numerous animal in vivo studies have demonstrated that aging increases intestinal permeability, encouraging bacterial components to penetrate through the gut barrier,8–10 human studies remain inconclusive. To illustrate, a large-scale study by Krueger et al. encompassing over 300 human patient samples from the GIT revealed no age-mediated change in tissue resistance,5 thus highlighting potential interspecies differences between humans and animals.11 It is also important to note that various factors, including bacterial diversity, pathology, and immune dysfunction, contribute to barrier disruption, rendering the identification of “healthy” aging-related alterations in the GIT challenging.12,13 Determining barrier changes that originate exclusively from aged intestinal cells in vivo is particularly difficult because of the wide range of factors that can influence barrier permeability. Alternative approaches to investigate age-associated changes within the intestinal barrier are advanced in vitro cell-based systems, which promise a better understanding of the relationship between age and (non-)gastrointestinal diseases in older individuals by uncoupling complex cellular processes.
000 per year and is still rising among the elderly, creating a significant socioeconomic burden.1 Additionally, recent evidence suggests that non-gastrointestinal disorders prevalent among the aging population, such as Parkinson's disease and rheumatoid arthritis, originate in the gastrointestinal tract (GIT) and can be associated with an impaired barrier.2–4 However, the effect of aging on the gastrointestinal barrier still remains unclear.5–7 While numerous animal in vivo studies have demonstrated that aging increases intestinal permeability, encouraging bacterial components to penetrate through the gut barrier,8–10 human studies remain inconclusive. To illustrate, a large-scale study by Krueger et al. encompassing over 300 human patient samples from the GIT revealed no age-mediated change in tissue resistance,5 thus highlighting potential interspecies differences between humans and animals.11 It is also important to note that various factors, including bacterial diversity, pathology, and immune dysfunction, contribute to barrier disruption, rendering the identification of “healthy” aging-related alterations in the GIT challenging.12,13 Determining barrier changes that originate exclusively from aged intestinal cells in vivo is particularly difficult because of the wide range of factors that can influence barrier permeability. Alternative approaches to investigate age-associated changes within the intestinal barrier are advanced in vitro cell-based systems, which promise a better understanding of the relationship between age and (non-)gastrointestinal diseases in older individuals by uncoupling complex cellular processes.
While traditional 2D cultivation techniques have been instrumental in expanding our knowledge of various pathologies over decades,14,15 the generation of more physiologically representative models, so-called organ-on-a-chips capable of mimicking functional units of the human body, have increasingly become the cell culture system of choice.16 These organ-on-a-chip systems have been shown to accurately emulate the physiologically relevant microenvironment of the GIT by incorporating factors such as shear stress, exerted by cyclic strain or fluid flow, and nutrient and oxygen gradients, facilitated by the cellular assembly in a villus structure.16–21 Another notable advantage of organ-on-a-chip platforms is the possibility to integrate complementary sensing strategies, enabling non-invasive, in situ, and real-time monitoring of cellular functions.22 For monitoring an impaired barrier, an important parameter is the barrier integrity, which can be analyzed based on the transepithelial electrical resistance (TEER) using e.g. traditional chopstick electrodes or impedance sensors. In both approaches, the formed cell layer's barrier function is evaluated non-invasively by measuring the electrical resistance across the cell layer.23,24 However, in contrast to planar impedance sensors, changes in the positioning of the chopstick electrodes lead to resistance inaccuracies. Additionally, the chopsticks must be handled with care when inserted into the test well to prevent cellular damage and furthermore, the non-uniformity of the electric field throughout the cell layer usually results in a methodical overestimation of TEER.24–26 We have recently reported the development of a microfluidic cell-barrier analysis system containing embedded membrane-based electrode microarrays,27–29 which eliminate most limitations of TEER measurements, including resistance inaccuracies and non-uniformity of the electric field throughout the cell layer,24–26 thus allowing precise detection and continuous monitoring of cell attachment,30 differentiation,31 migration,32 proliferation,33 inflammation,34 and invasion processes.35
While cellular phenotypes of healthy intestinal cell barriers are studied in detail, little is known about the impact of age on cell barrier function in vitro. Cellular senescence, a key hallmark of aging, describes a permanent state of cell cycle arrest accompanied by a range of phenotypical changes.12,36 Deciphering the function of senescent cells in the GIT is, therefore, crucial for understanding the intricate relationship between aging, gut homeostasis, and barrier integrity. General senescence-mediated changes include increased expression of the cyclin-dependent kinase inhibitors p16 and p21 since they are responsible for senescence-associated cell cycle arrest. Furthermore, the chemokine CCL2 is part of the senescence-associated secretory phenotype and serves as another marker for senescent cells.36–38 Besides, the lysosomal activity and consequently the senescence-associated β-galactosidase (SA-β-gal) activity increases,36 and alterations in cellular morphology and size have also been linked to senescence-related modifications.39 While in an aged state, intestinal epithelial cells typically overexpress the pore-forming protein claudin-2, no significant changes are observed in the tight junction adapter protein ZO1.40
In this study, we developed a membrane-based impedance sensor tailored for a gut-on-a-chip platform to dynamically monitor the barrier integrity of a senescent gut model. Compared to a previous study,27 the system was optimized to streamline the fabrication, improve handling, and align the apical cultivation area with the dimensions of a 24-well Transwell (TW) setup, often used to investigate gut models. First, the redesigned electrodes were characterized, demonstrating stable, reproducible, and robust measurements in a humidified environment. To validate their cellular applicability, the biocompatibility for Caco-2 cells was assessed, and the sensor's ability to monitor Caco-2 barrier formation over 7 days was successfully confirmed by comparing FITC dextran diffusion assays with impedance spectroscopy. Physiological features of the gut model, including aminopeptidase activity, mucus production, ZO1 expression, and microvilli formation, were evaluated to ensure its functional relevance.
To assess the sensor's capability to measure barrier alterations induced by senescence, Caco-2 cells were treated with a low dose of doxorubicin (DXR) for 6 days, a well-established senescence inducer.41–44 Cellular senescence was confirmed by expressing senescence-associated genes (p21 and CCL2) analyzed via qPCR. These experiments highlight the sensor's ability to detect senescence-mediated changes in real-time, non-invasively, and continuously across more than half of the cultivation area.
In contrast to standard TW systems and chopstick electrodes, this sensor-integrated microfluidic platform provides superior spatial and temporal resolution. The combination of organ-on-a-chip technology, integrated impedance sensors, and senescent gut models offers an advanced tool to investigate age-related gastrointestinal processes over extended cultivation time, facilitating a deeper understanding of the physiological and pathological changes in elderly individuals.
Results and discussion
Initial characterization of the integrated porous membrane-based impedance sensor
A key technological feature of our microfluidic intestinal barrier-on-a-chip system is the integration of interdigitated gold electrodes (Scheme 1A) located on top of the embedded porous PET membrane to continuously monitor cell attachment, proliferation, differentiation, and gut integrity. The electrodes comprise a pair of 7 interdigitated gold fingers, covering ∼57% of the apical cultivation channel and thus enabling a broad detection area. Additionally, direct contact of the electrodes with epithelial cells ensures reliable in situ measurements. The PDMS-based microfluidic platform (Scheme 1B and C) is engineered to facilitate the polarization of the epithelial cell layer using an integrated porous membrane, which is essential for replicating the in vivo microenvironment of the apical and basolateral compartments in the human gut (Scheme 1D). The apical cell culture area (∼0.35 cm2) closely matches that of conventional 24-well TW systems (∼0.34 cm2), enabling direct comparisons between the 2 setups. The device, which is the size of a microscope slide, includes 3 replicates. Detailed technical drawings of the different layers and electrode layouts can be found in Fig. S1A–F.†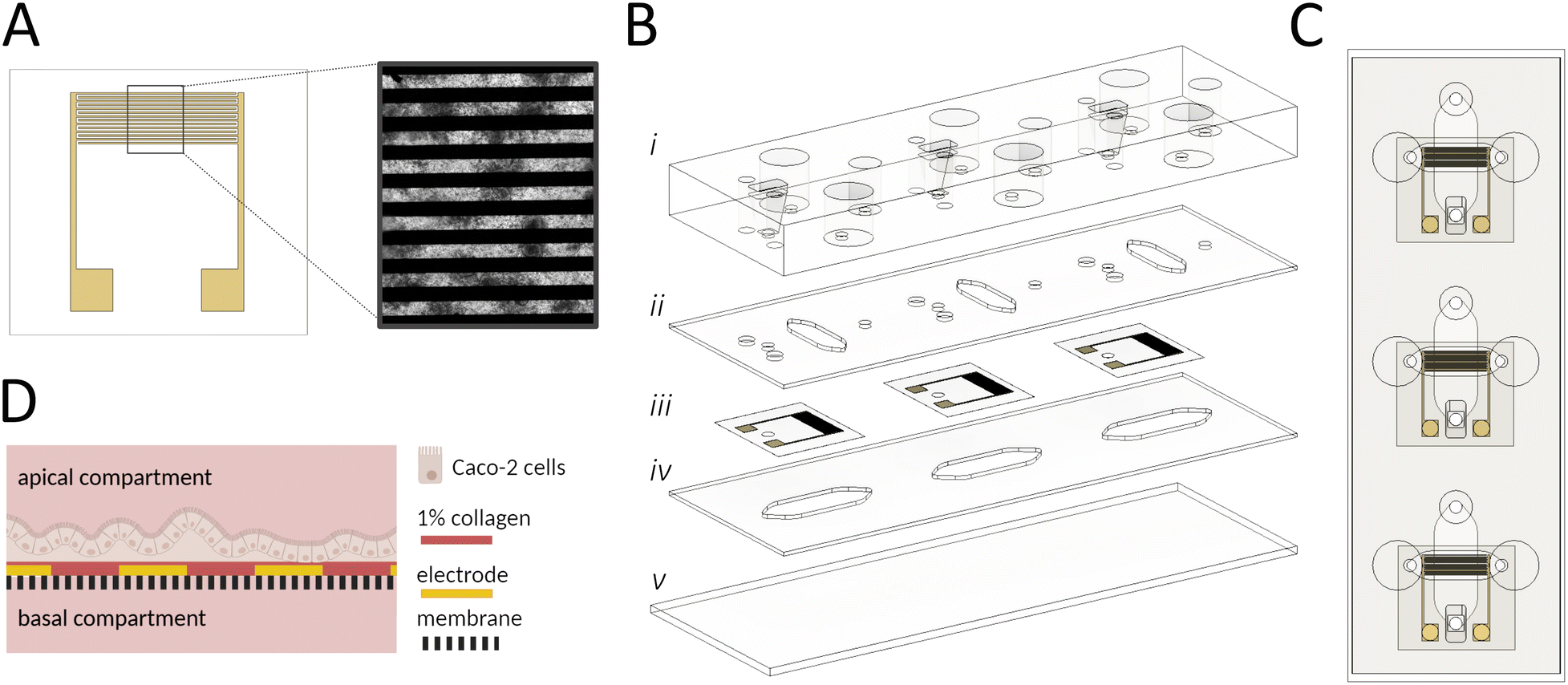 | ||
| Scheme 1 (A) The electrode design features a pair of 7 interdigitated gold rods; the microscope image (Caco-2 cells) demonstrates the optical accessibility of the device. (B) Exploded view of the sensor-integrated gut-on-a-chip system comprising (i) a cast PDMS layer providing a lid and medium reservoir, (ii) an apical channel, consisting of a 500 μm PDMS sheet that accommodates the intestinal cells, (iii) 3 gold-electrodes onto a porous PET membrane, (iv) a 500 μm PDMS layer, serving as a basal compartment and (v) a microscope slide. (C) Top view of the platform emphasizes the 3 individual cultivation channels and the electrode (yellow) position aligned to the apical channel. (D) Schematic cross-section of the apical and basal compartment with cells. The porous membrane is represented by the dashed line, gold rods are depicted in yellow, and rat tail collagen type I coating is illustrated in red. The illustration is created with https://BioRender.com. | ||
In the first step, the functionality of the interdigitated porous membrane-based gold electrodes was tested in the absence and presence of surface coatings using impedance spectroscopy. To assess the effect of surface modification on measured resistance, membranes were coated with 1% collagen type 1, a well-known promoter of cellular adherence and epithelial differentiation.45 As shown in Fig. 1A, collagen coating resulted in larger semicircles in the Nyquist plot and significantly higher impedance values in the frequency range of ∼0.1–400 Hz. These initial experiments, evidenced by impedance alterations in both Nyquist and Bode plots (Fig. 1A and B), demonstrate electrode functionality and the impact of surface modification in the lower frequency range.
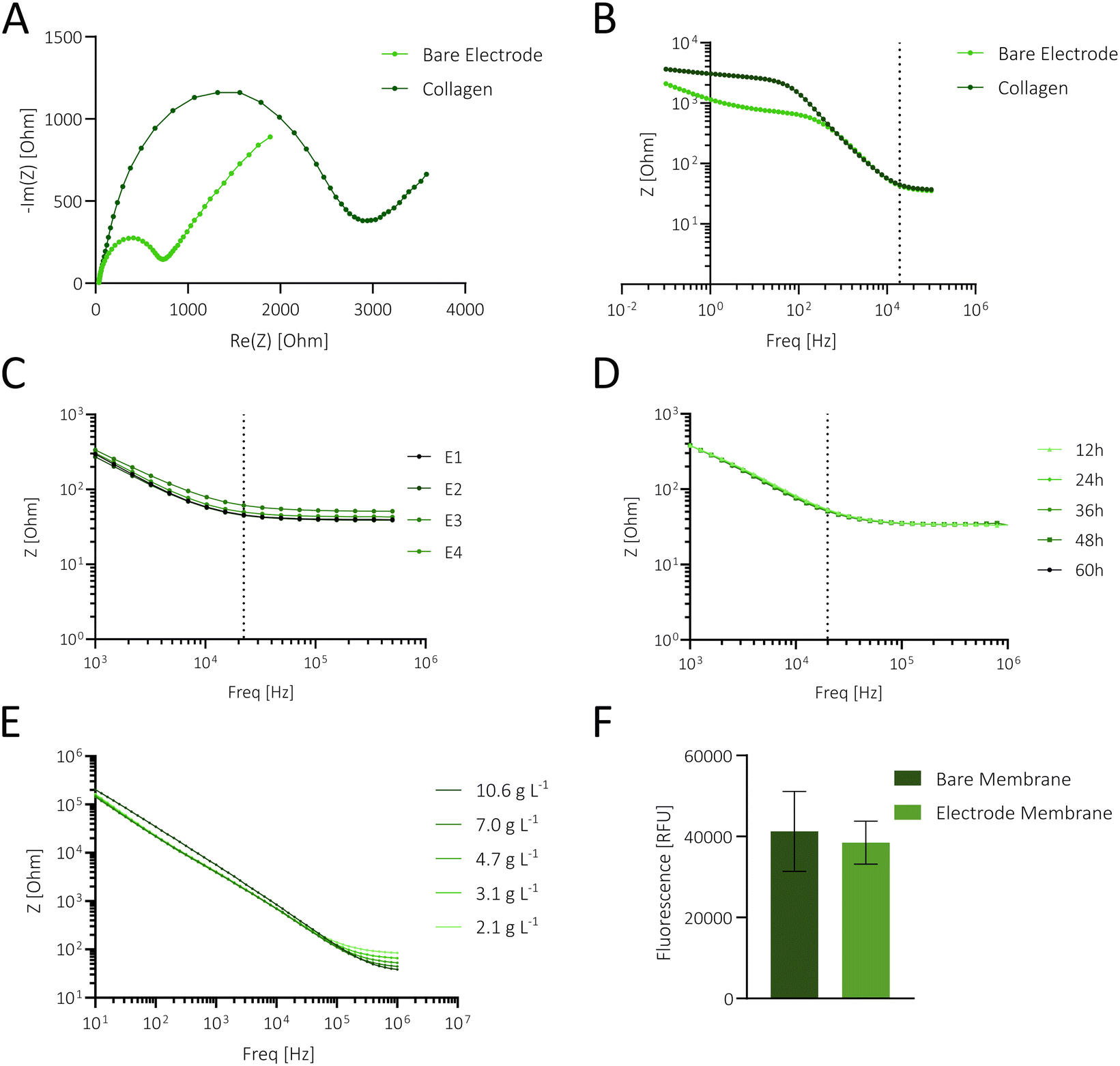 | ||
| Fig. 1 (A and B) Nyquist (A) and Bode plots (B) showing impedance measurements from an electrode before and after coating with 1% collagen type I. Impedance was measured in a 5 mM ferri-ferrocyanide solution from 0.1 Hz–100 kHz. (C) Bode plot depicting 4 different electrodes and repeated measurements in complete cell culture medium (n = 20), revealing electrode stability over 20 measurements (E = electrode). (D) Bode plot showing continuous measurements of medium from 1 kHz–1 MHz at different time points, revealing no significant impedance changes over time, thereby confirming the capability for long-term, stable measurements without signal variation at 20 kHz. (E) Impedance measurements from 1 Hz–1 MHz of diluted salt concentration (10.6–2.1 g L−1). (F) Relative fluorescence units (RFU) of a FITC–dextran solution sampled in the basal layer diffused through a bare or a gold electrode membrane, unveiling no pore blockage. Statistical significance was determined by unpaired t-test (n = 3; P = 0.688, ns is not shown). | ||
To assess the stability, reproducibility, and robustness of the applied biosensing system, the following experiments focused on determining electrode performance in the integrated gut-on-a-chip system in the presence of cell culture medium. Impedance measurements (n = 20) of 4 sensors, as shown in Fig. 1C, exhibited a relative standard deviation (RSD) in electrical resistance of 1.15 ± 0.91% (0.54 ± 0.40 Ohm) at 20 kHz, highlighting the stability and reproducibility of the measurement technique.
Subsequently, the robustness of the porous membrane-based sensors was tested by continuous impedance monitoring inside an incubator system (37 °C, 100% humidity, and 5% CO2) over a period of 60 h. As shown in Fig. 1D, the impedance measurements at the different time points are overlapping, and a detailed analysis of impedance-time traces (Fig. S2A†) at 20 kHz revealed a minor shift of 2.15 ± 0.7% (n = 3), confirming the robustness of the measurement setup in a humidified environment. Furthermore, batch-to-batch variations were assessed using bioimpedance sensors fabricated from 3 batches, each coated with collagen. Signal variations at 20 kHz yielded an RSD of 4.19% (Fig. S2B,†n = 9), demonstrating the reproducibility of the fabrication process.
To evaluate the influence of ionic concentration changes on sensor signals, in the next set of experiments, decreasing salt concentrations (10.6–2.1 g L−1) were measured using a frequency range from 1 MHz–1 Hz. In order to neglect the impact of temperature on the impedance value, reagents were prewarmed at RT. Results are shown in Fig. 1E, where higher salt content significantly decreased impedance values at higher frequencies (above 200 kHz up to 1 MHz), while just a neglectable change was observed at frequencies between 10–100 kHz. This confirms that variations in salt concentrations do not significantly influence barrier measurements at 20 kHz. To assess if the membrane-integrated sensor enables unaltered diffusion of nutrients from the basolateral compartment, an essential requirement for mimicking epithelial cell polarization in vitro, fluorescence diffusion studies were conducted. No significant difference between the sampled medium from the bare and sputtered membranes was found (see also Fig. 1F), confirming that sensor-modified membranes treated with various solvents during the electrode deposition process are not subject to pore clogging. The initial characterization experiments of the membrane-based electrode without cells demonstrate reliable, reproducible, and stable performance in a humidified environment. This setup facilitates continuous monitoring while supporting cellular differentiation via the membrane's porous structure.
Sensor validation in a gut model resembling key gastrointestinal features
First, the biocompatibility of the sensor-integrated porous membranes was tested using the human epithelial Caco-2 cell line. Experiments set out to investigate potentially harmful effects mediated by incomplete removal of cytotoxic resist components applied during the image reversal photolithography approach. A calcein-AM and ethidium bromide staining after 48 h cultivation of Caco-2 cells on the sensor-integrated porous membrane confirmed metabolic activity across the entire membrane area and only small amounts of death cells (see Fig. 2A). Additionally, a PrestoBlue™ Assay did not reveal significant changes in metabolic activity and viability (Fig. S2C†).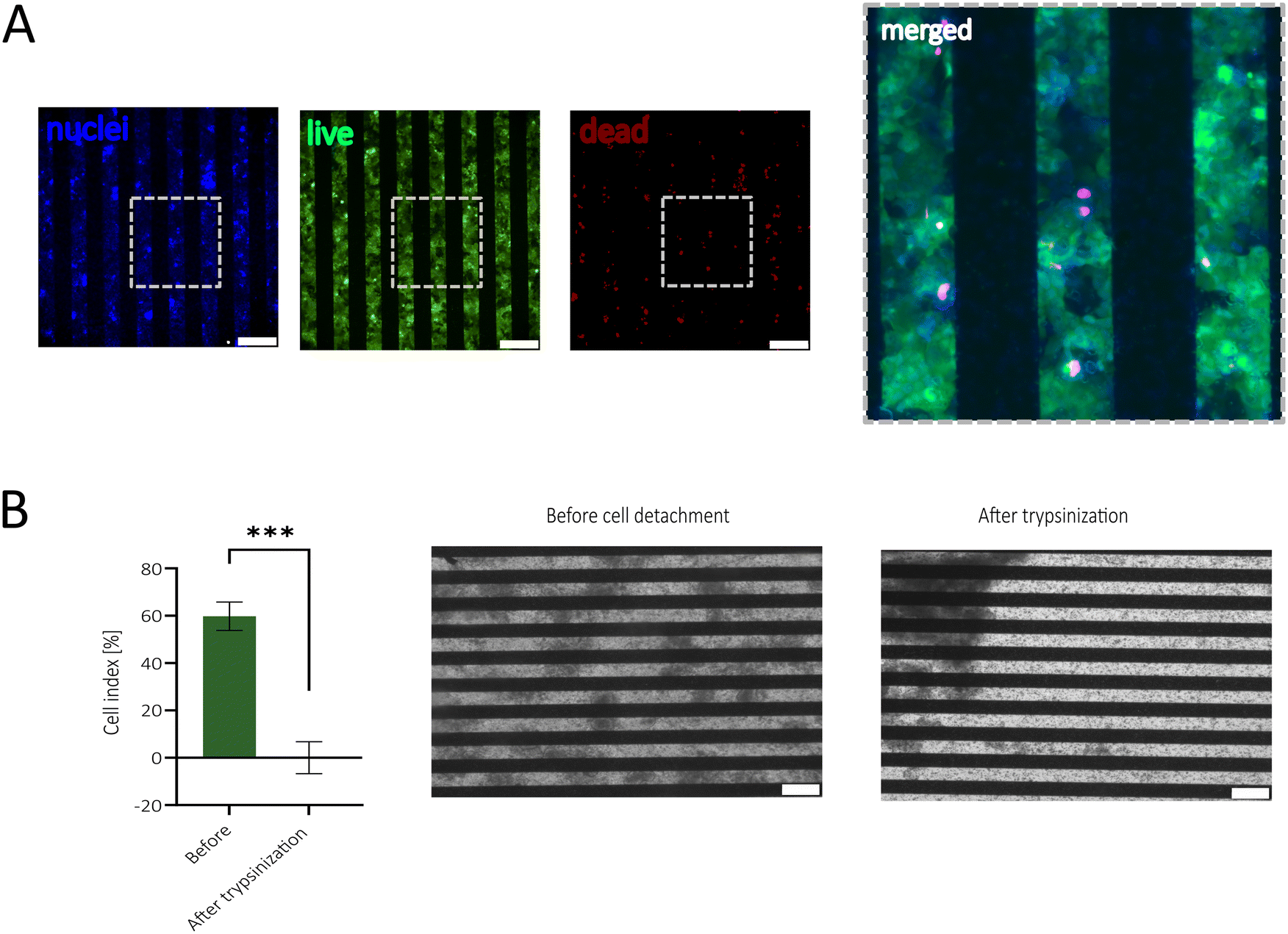 | ||
| Fig. 2 (A) Viability staining with Hoechst (nuclei), calcein-AM (live) and ethidium bromide (dead) conducted after 48 h of cultivation on an electrode membrane. The images depict spatially viable cells (green) and nuclei (blue) adjacent to the gold rods of the electrodes. A few dead (red) cells are visible scattered across the membrane. On the right-hand side, a zoom of the merged images is shown. (B) The cell index (CI) at 20 kHz (left) and phase contrast images (right) before and after the disruption of the cellular barrier, demonstrating no detachment of the sputtered gold after cell cultivation. Statistical differences were analyzed with a paired t-test (n = 6; P = 0.0002). All images were taken with a 10× magnification and scale bars are 200 μm. | ||
In the subsequent experiments, impedance measurements were conducted before and after cell detachment to assess whether epithelial cellular barriers affect electrode performance and maintain stable impedance values throughout extended cultivation periods (14 days). Here, the cell adhesion and barrier formation are described by the cell index (CI), introduced by Pan et al.,46 which is given by the ratio between the impedance background signal and the impedance signal coming from the cells. As depicted in Fig. 2B, long-term cultivation up to day 14 reached a CI of around 60% and after cell detachment, the signal went back to the original CI of 0.6%, demonstrating that the electrodes are suited to investigate cell coverage and that cellular adherence to the electrode does not influence electrode performance.
In a final electrode validation step, impedance-time traces were recorded every 24 h for 7 days (Fig. 3A), revealing a notable increase in impedance in the high-frequency range (1–100 kHz, Fig. 3A, upper right detail), thus indicating successful barrier formation. In contrast, at lower frequencies (<1 kHz) the capacitance becomes more dominant in the impedance values. Therefore, the slight decline in impedance at lower frequencies (Fig. 3A, bottom left detail) can be attributed to increased cell capacitance due to microvillus formation, leading to a larger surface area and the accumulation of extracellular matrix proteins on the surface of the electrodes.47–50
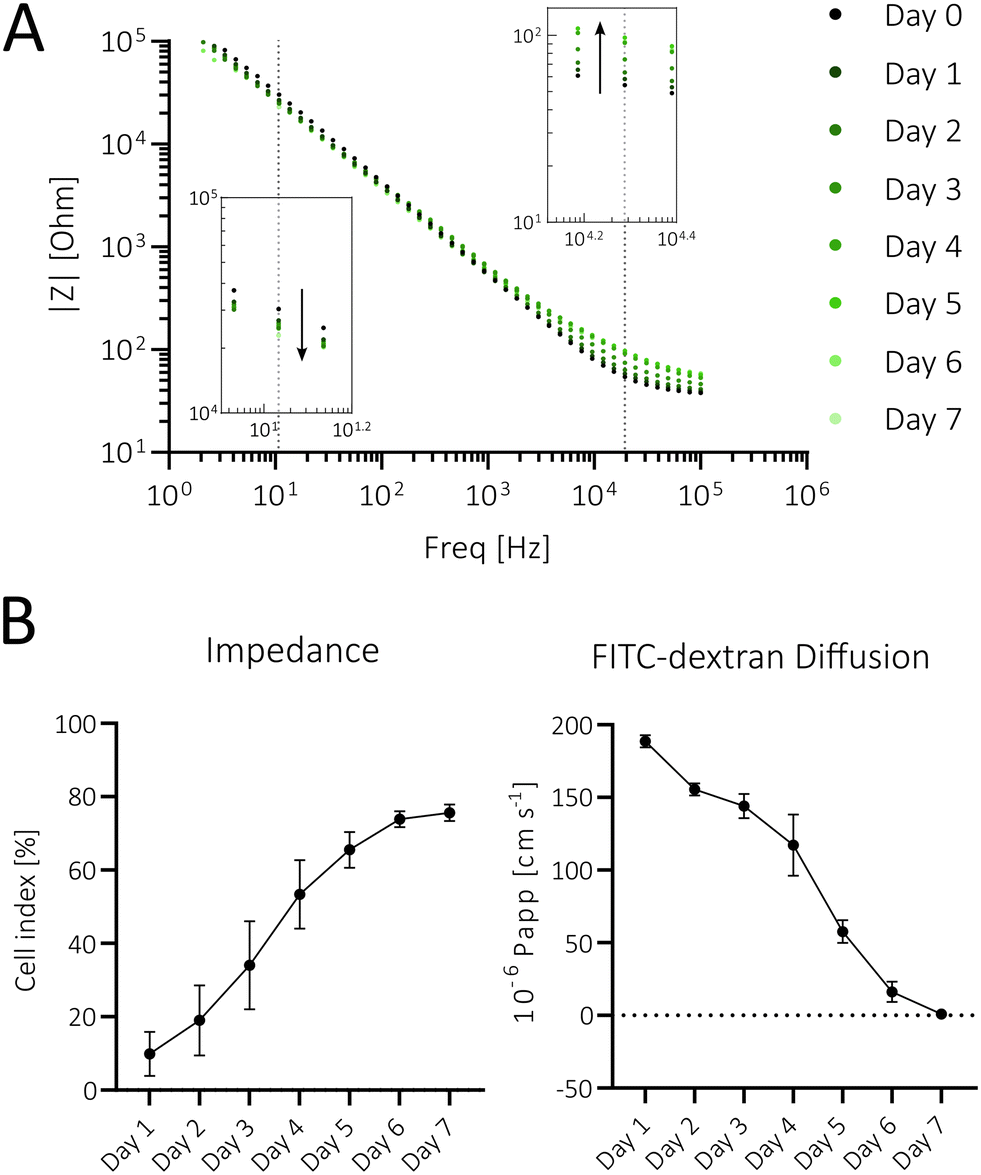 | ||
| Fig. 3 (A) Bode plot of a representative channel over 7 days, displaying the highest impedance increase at 20 kHz (upper right detail, dotted line) and decrease at 10 Hz (bottom left detail; dotted line). (B) The cell index (CI) at 20 kHz normalized to electrode membranes prior to cell seeding (left; n = 9) and apparent permeability (Papp; right, n = 6) over time. | ||
An additional frequency analysis showed the highest impedance changes occurring at ∼20 kHz, where CI exponentially increased and plateaued after 7 days (see Fig. 3B left; n = 9), thus indicating increased cell proliferation and barrier formation. To verify the electrode's functionality in terms of barrier formation, a FITC–dextran assay, the gold standard to study barrier permeability, was employed.51–53 Results shown in Fig. 3B right reveal an apparent permeability (Papp) of approx. 200 × 10−6 cm s−1 at day 1 that rapidly decreases over 7 days, inversely correlating to the CI increase observed over the same time period. On day 7, Caco-2 barriers displayed a Papp of 0.88 × 10−6 cm s−1, comparable to that of human intestinal tissue samples.54 This demonstrates the sensor's functionality in monitoring the barrier formation and establishing a tight barrier model. In order to show the application versatility for other intestinal in vitro models, impedance measurements of a direct epithelial co-culture (HT29-MTX with Caco-2) and an indirect endo- and epithelial co-culture (HUVECs with Caco-2) were successfully conducted (Fig. S3A and B†).
In order to prove the physiological relevance of the simplified gut model cultured in our sensor-integrated platform, the epithelial functionality, mucus secretion, ZO1 production, and microvilli formation were investigated. The intestinal functionality of the epithelial Caco-2 cells was assessed over time by measuring the specific activity of the apical brush border enzyme aminopeptidase.16 After just 7 days of cultivation without flow, aminopeptidase activity reached 11.08 nmol min−1 cm−2 (see Fig. 4A), indicating the differentiation of Caco-2 cells into functional enterocytes. The observed aminopeptidase activity value is comparable to a published gut-on-a-chip system (e.g. Jing et al. 2020) where Caco-2 cells were co-cultivated with endothelial cells and exposed to peristaltic shear forces.55 Furthermore, on-chip cultures showed a ∼3.5-fold increase in enzyme activity compared to TW cultures after 7 days. Notably, alcian blue staining on day 7 displayed a pronounced dark blue mucus layer on the apical side of the on-chip culture, suggesting a robust mucus formation, whereas TW cultures showed only a faint blue staining (Fig. 4B). More importantly, the Caco-2 cells in the on-chip model formed a complex 3D villi-like structure, visualized by the change in light transmission and areas out of focus, whereas the TW cultures primarily exhibited monolayers with occasional domes. Mucus staining was particularly intensified on the 3D regions on-chip, indicating enhanced differentiation of goblet-like cells and high secretion of mucus in vitro. Additionally, after 7 days of on-chip cultivation, cells expressed the tight-junction protein ZO1, as shown in Fig. 4C, which is essential for preserving cell polarity and controlling paracellular permeability as well as cell-to-cell adhesions.56 The fluorescence image (Fig. 4C) emphasizes the 3D structure of the barrier based on the darker out-of-focus areas. The microvilli structure in the intestine is crucial for nutrient absorption, secretion, and mechanotransduction and, therefore, has to be considered in an intestinal in vitro model.57 To confirm the brush border structure formation, scanning electron microscopy (SEM) images were captured on day 7. At 5000× magnification, microvilli were observed across the entire apical surface (Fig. 4D), with some microvilli appearing upright while others were flattened. Mucus residues were also visible (white arrows). A higher magnification image was taken, showing a cell with densely packed microvilli (Fig. 4D zoom) comparable to structures seen in other gut-on-a-chip models.58,59
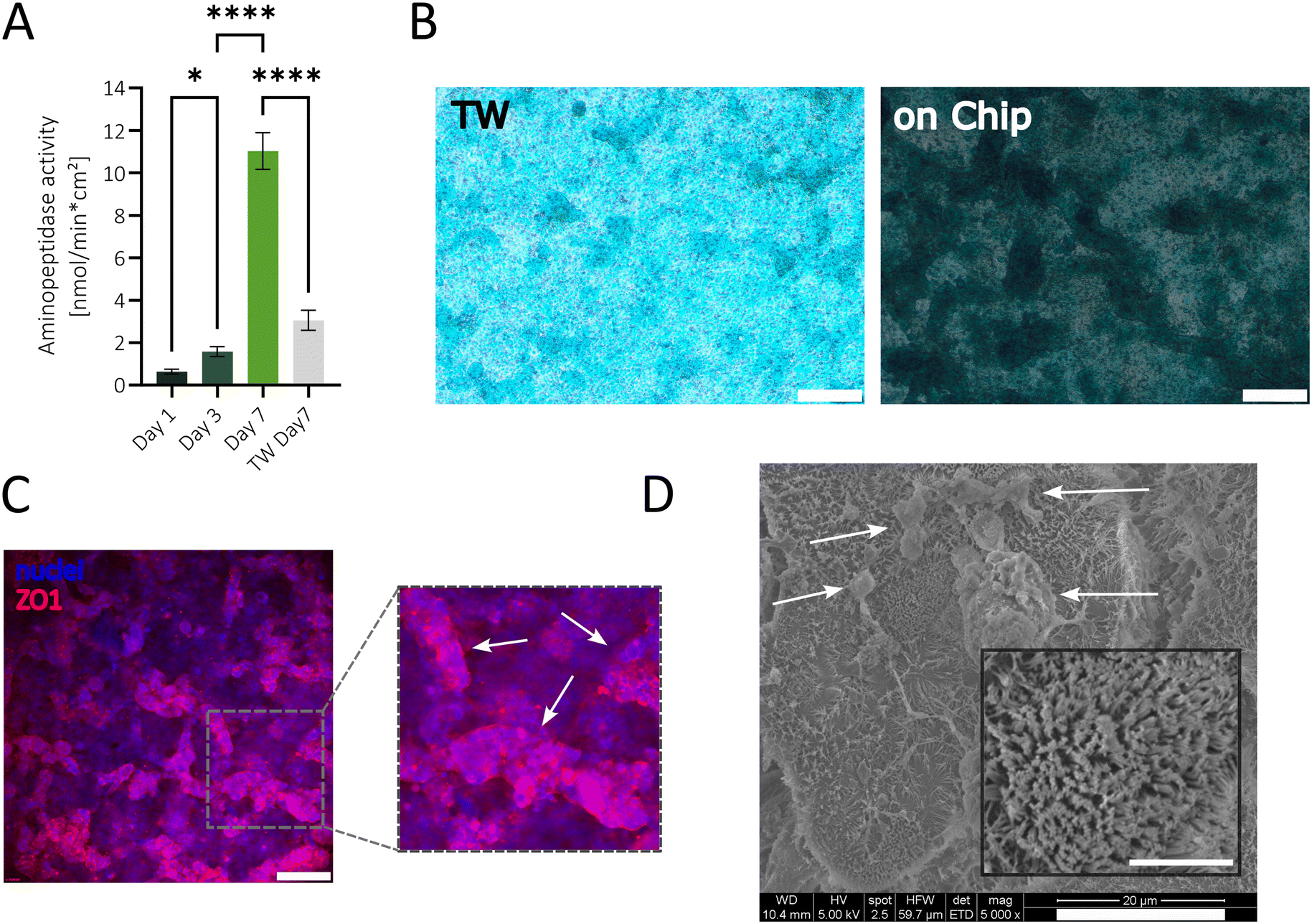 | ||
| Fig. 4 (A) Aminopeptidase activity analysis over 7 days on-chip to assess intestinal functionality, revealing a significant increase over time and enzyme activity comparison between Transwell (TW) and on-chip cultures on day 7, showing reduced enzyme activity in TW culture relative to on-chip culture. Statistical significance was calculated using an ordinary one-way ANOVA with a Turkey's multiple comparison test (n = 6, *P = 0.0176, ****P < 0.0001). (B) Alcian blue staining of the acidic mucopolysaccharides and morphological observations in TW system (left) and in the gut-on-a-chip platform (right) after 7 days, indicating an increased mucus production and pronounced 3D structures of the cells cultivated on-chip. Images were taken with a color camera and a 4× magnification. Scale bars are 200 μm. (C) Fluorescence images showing DAPI (blue) and ZO1 (red) staining after 7 days of cultivation, illustrating the expression of the adaptor protein of tight junctions across the membrane. The image was taken with a 10× magnification, and the scale bar corresponds to 200 μm. White arrows highlight focused cell areas, indicating villi-like 3D structures. (D) Scanning electron microscopy image with 5k× magnification (scale bar 20 μm) with a zoom (40k× magnification, scale bar 3 μm), showing microvilli on the cell surface and mucus residues (white arrows). | ||
To summarize, this easy-to-use and streamlined sensor-integrated gut-on-a-chip platform enables in situ and continuous monitoring of barrier formation and integrity of various cell culture applications. This platform offers a more physiologically relevant in vitro Caco-2 model than traditional TW systems since the on-chip model reveals enterocytic and goblet cell differentiation, microvilli formation and 3D assembly within just 7 days. This accelerated development may be attributed to nutrient gradients established within the microfluidic system due to the chamber dimensions and generated shear force during medium exchange.
Sensor application for monitoring barrier integrity in a senescent gut model
To test the capability of our sensor-integrated gut-on-a-chip platform for monitoring senescence-mediated changes in human intestinal cells, we exposed epithelial cells to DXR, which is a well-known inducer for cellular senescence by intercalating with the DNA and inhibiting DNA repair mechanisms and thus results in an irreversible cell cycle arrest.41,60 Furthermore, the anthracycline drug triggers the generation of reactive oxygen species, contributing to human aging.61,62Initial optimization studies were conducted to determine the optimal concentration of DXR (0.1–0.8 μg mL−1) capable of inducing cellular senescence while preserving high cellular viability. To confirm increased lysosomal activity in epithelial cells a SA-β-gal assay was conducted after 5 days of DXR exposure. Representative images are shown in Fig. S4A.† Results of the study (Fig. S4B†) revealed that in the presence of the DXR concentrations, 0.4 and 0.8 μg mL−1, 22% and 28% SA-β-gal-positive cells were detected, respectively, while no significant difference in the relative viability between 0.4 and 0.8 μg mL−1 -treated cultures was observed (Fig. S4C†). Consequently, in order to generate a more pronounced senescent phenotype, 0.8 μg mL−1 DXR was selected for all subsequent experiments.
Bioimpedance-time traces (20 kHz) of the treated intestinal barrier model are shown in Fig. 5A. The analysis revealed an increase in the change (%) of the CI in comparison to before (incubation, day 0) the treatment with 0.8 μg mL−1 DXR, featuring a significant signal shift following day 3 of DXR incubation (*P = 0.0390). This unexpected increase in barrier integrity was also confirmed using standard TW cell culture systems and chopstick electrodes (Fig. S4D†) with a cell density and cultivation area comparable to that of the microfluidic system. Microscopic examinations shown in Fig. 5B point to a hypertrophic cell state (white arrows), indicative of a senescent phenotype.44,63 Therefore, a more detailed size distribution analysis was utilized to further confirm that DXR-treated cells were predominantly bigger than the control, where the arithmetic mean of the controls was 13.5 μm, while that of the treated cells was 14.2 μm. Moreover, untreated cells did not attain a size greater than 25 μm, whereas the treated cells reached a size of up to 29 μm (Fig. 5C). The cellular exposure to DXR leads to a cell cycle arrest, stopping the proliferation and subsequently the cells gain in size.64,65 The general increase in cell size may have contributed to the above observed increased impedance signal by reducing the number of cell–cell connections per area and intercellular space.66 Gene expression analysis using qPCR, shown in Fig. 5D, reveals that treated cells did not overexpress the tight junction marker ZO1. Instead, the pore-forming protein claudin-2, associated with decreased barrier integrity, was slightly overexpressed.67 Increased cell sizes and unaltered ZO1 expression underline the indications of an increased impedance due to the phenotypical and morphological transition into cellular hypertrophy. The qPCR analysis further revealed a significant increase in p21 and CCL2 expression as well as a slight increase in claudin-2 expression upon treatment with DXR, a marker commonly upregulated in aged intestinal cells7,36,37,40 thus pointing at the establishment of a senescent intestinal in vitro model.
 | ||
| Fig. 5 (A) The cell index (CI) increases after adding 0.8 μg mL−1 DXR. Statistical analyses were conducted using mixed effect analysis (n = 8, *P = 0.0187) and unpaired t-tests at different time points (n = 8, *P < 0.0332). (B) Phase contrast images of cells on-chip treated (or non-treated) with DXR for 6 days. Hypertrophic cells are marked with a white arrow. Images were taken with a 20× magnification, and the scale bar is 50 μm. (C) Size distribution of treated and non-treated cells reveals increased size after a 6 day DXR treatment. The cell number of DXR-treated cells was normalized to the total number of the control. Nonlinear fitting was performed to determine the mean average. (D) Gene expression of markers responsible for barrier integrity and senescence after 6 days of DXR exposure, visualized as fold change (n = 3, **P = 0.0031, ****P < 0.0001, ns are not shown). Statistical analyses were conducted using a two-way ANOVA and a Sidak's multiple comparison test. | ||
Conclusions
The current study demonstrates the applicability of a gut-on-chip system for real-time, in situ, and label-free monitoring of cellular barrier function and morphological changes with high stability, reproducibility, and robustness. It is important to highlight that the integrated membrane-based impedance sensor allows for intestinal differentiation and polarization due to its porous feature, and it covers more than half of the entire cell culture area within the microfluidic system and, therefore, provides accurate information on cell–substrate interactions directly at the membrane interface. Additionally, compared to traditional chopstick electrodes, the impedance sensor offers higher sensitivity and eliminates positioning errors due to its fixed placement within the chip. The employed layer-by-layer fabrication process further allows for the straightforward generation of intestinal barriers using apical and basal compartments. As a result, gut epithelial cells are able to differentiate into functional enterocytes, produce a thick layer of mucus and microvilli, and form villi-like structures, thus mimicking important aspects of the human GIT already within one week of culture. Cells also developed the tight-junction protein ZO1 across the membrane, which is responsible for cell–cell adhesions and crucial for maintaining cell polarity and regulating paracellular permeability.56 Moreover, treatment with 0.8 μg mL−1 DXR induced senescence-associated phenotypes, including increased lysosomal activity, cell hypertrophy, and overexpression of the cyclin-dependent kinase inhibitor p21 and the chemokine CCL2. To the best of our knowledge, no publication has thus far shown the link between the senescence of gastrointestinal cells and an increased barrier integrity in vitro. Overall, the presented work addresses the need for novel aged gastrointestinal in vitro models to investigate the interplay of non-gastrointestinal diseases, such as Parkinson's and rheumatic disease, with the gut in the future.Material and methods
Gut-on-a-chip platform with integrated gold electrodes
For the fabrication of the gold electrodes, PET membranes (2000M12/580M303/47; it4ip S.A.) with 3 μm in pore size, a porosity of 8.00 × 105 pores cm−2 and 9 μm membrane thickness were used. The overall fabrication steps of the gold electrodes are illustrated in Fig. S5.† First, the membranes were washed for 15 min in distilled water (diH2O), followed by an isopropanol wash. After that, the membranes were dried on a heating plate at 120 °C to ensure isopropanol evaporation. Then, the cleaned PET membranes were reversibly glued to microscope slides with a low molecular polyvinylalcohol (PVA) solution (40 mg mL−1; 363170, Sigma-Aldrich). For this, the glasses were plasma-activated, and PVA solution was spin-coated on the slides at 800 rpm for 20 s. The membranes were immediately placed on the objective glasses, and to ensure gradual drying without causing wrinkles, they were heated from 70 °C to 150 °C. Then the photolithography resist LOR3A was spin-coated at 1000 rpm for 30 s and soft-baked at 150 °C for 180 s. The second resist LNR-003 (MicroChemicals) was spin-coated at 3000 rpm for 30 s followed by a soft-baking step at 100 °C for 90 s. The electrode geometry was transferred onto the samples via a UV light exposure of 80 mJ cm−2 using a photomask. The membranes were baked at 100 °C for 90 s, followed by a second UV exposure with 200 mJ cm−2 without the photomask. Then, the samples were developed for 45 s using the organic solvent solution AZ726 MIF (MicroChemicals) and rinsed with diH2O. An 80 nm gold layer was sputtered with a sputtering rate of 1.05 nm s−1. In order to remove the excess gold and the resist, the samples were soaked in N-methyl pyrrolidone. In the end, the membranes were released from the microscope slide by rinsing with diH2O and dried at 60 °C prior to integration. The polydimethylsiloxane (PDMS, Sylgard, Dow Corning)-based microdevice consists of a basolateral, an apical, and a top layer, serving as medium reservoirs and a lid. The fabrication steps are illustrated in Fig. S6.† The apical and basolateral channels, made of 500 μm PDMS foils, were adhered to the adhesives ARcare® 90106NB (Adhesives Research® Ireland, Ltd) before being cut by xurography to avoid alignment errors. A mold for the top layer was 3D printed with a Formlabs Form3B printer using Biomed Clear V1 Resin. Since the resolution of this printer did not result in complete transparency, an ARcare® 8259 was glued into the mold as an optical window for cellular examination under the microscope later. PDMS casting involved mixing the prepolymer and curing agent in a 10:1 volumetric ratio, followed by degassing in an exicator for 30 min. After curing for 6 h in a 60 °C oven, the cast PDMS was removed from the mold. The top layer, basolateral layer, apical layer, and microscope slide were plasma-activated, followed by bonding the apical layer to the top layer and the basolateral layer to the microscope slide. The resulting two components were then incubated at 80 °C for at least 4 h to achieve permanent bonding. To complete the membrane-based gut-on-a-chip system, the adhesive liners of the two components were removed, and the porous gold electrodes were sandwiched between them.
Cell culture on- and off-chip
In order to develop an in vitro gut model, mimicking aspects of the GIT, the colorectal adenocarcinoma cell line, Caco-2 (ATCC), was utilized. Standard cell culture reagents were purchased from Sigma-Aldrich. All cell culture experiments were carried out in a laminar flow hood at room temperature (RT) under sterile conditions. Media and other cell culture reagents were prewarmed at 37 °C or RT before usage. Caco-2 cells were maintained in minimum essential medium with Earl's salts (MEM; M0325) supplemented with 10% fetal bovine serum (FBS; F9665) and 1% antibiotics (AB; A5955) at 37 °C in a 5% CO2 humidified atmosphere. The medium was changed every 2 or 3 days. In order to ensure biological repeatability, cells with a passage number between 26–34 were split at 80–90% confluency and experiments were conducted using cells with a viability above 90%, as confirmed by Trypan Blue staining.In preparation for the cell culture experiment on-chip, the microfluidic devices were sterilized by wiping and priming the channels and reservoirs with 70% ethanol, drying the platform in a 60 °C oven, and subsequently exposing the platform to UV for 30 min. In order to facilitate cellular adherence and differentiation, membranes within the chip and in TW systems (662630, Greiner) were coated with 1% collagen type 1 (C3867) in a humidified incubator for 1 h followed by flushing with complete media. After reaching 90% confluency, cells were seeded onto the membranes at 1.0 × 105 cells cm−2 density.
To demonstrate the system's versatility, we established both a direct co-culture of Caco-2 and HT29-MTX-E12 cells (7:3 ratio) and an indirect co-culture of Caco-2 and HUVEC/TERT within the chips (Fig. S3A and B†). HT29-MTX-E12 cells (passage 35) were cultured following the previously described method, while HUVECs (passage 12) were maintained in EGM2 (C-22111, PromoCell) and seeded at a density of 0.2 × 105 cells cm−2 onto the collagen-coated membrane of the basolateral compartment 1 day prior to Caco-2 seeding.
Viability assay for toxicity assessment of electrodes and doxorubicin
The cell viability was evaluated after 48 h of cultivation in the microfluidic platform to assess the toxicity of the gold electrode membranes by using the PrestoBlue™ cell viability reagent (A13262; Invitrogen). Thereby, the stock solution was diluted 1:10 with complete media and added to the apical and the basolateral channels. After 1 h of incubation, 100 μL of the apical channel was sampled, and the fluorescence emission (at 590 nm) was measured in a plate reader (multimode plate reader, EnSpire 2300, Perkin Elmer). The fluorescence background was corrected by subtracting the control wells without any cells from all values. For visualization and spatial resolution of vital cells, Caco-2 cells were stained with 0.02 μM Hoechst, 2 μM calcein AM, and 4 μM ethidium bromide in cell culture media. The staining solution was incubated for 20 min and imaged at 10x magnification under a fluorescence microscope. Additionally, a PrestoBlue™ cell viability assay was performed to assess the toxicity of different DXR concentrations (0.1–0.8 μg mL−1). Caco-2 cells were cultivated in a standard 48-plate until they formed a monolayer. Subsequently, the cells were treated with DXR for 2 days, followed by the PrestoBlue™ assay.
Aminopeptidase assay to evaluate enterocytic differentiation
Using L-alanine-4-nitroanilide hydrochloride (A4N; Sigma-Aldrich) as a substrate, the specific activity of the apical brush border aminopeptidase enzyme expressed by differentiated human intestinal Caco-2 cell monolayers was utilized to assess human intestinal epithelial cell functionality. In order to analyze the differentiation into functional enterocytes, an adapted version of the aminopeptidase assay developed by Ferruza et al.68 was applied on days 1, 3, and 7. Briefly, a buffer with 10 mM Tris-HCl/150 mM NaCl was adjusted to pH 8 and mixed in a ratio of 1:30 with PBS to create a collection buffer. This collection buffer was used to prepare the reaction buffer containing 5 mM of the substrate L-alanine-p-nitro-anilide (L-Ala-NA), freshly made before usage. The apical and the basolateral layer of the microfluidic platform were rinsed twice with PBS. Subsequently, the reaction buffer was added to the apical channel, while the basolateral channel was filled with PBS. The reaction buffer was incubated for 20 min at 37 °C. Meanwhile, a standard curve of the hydrolysis product, p-nitroanilide (p-NA), was prepared in the linear range of 0.1–1.2 mM. After the incubation period, the solutions from both channels were sampled and transferred to a 96-well plate on ice. Finally, the absorbance of the p-NA was measured at 405 nm in a plate reader (multimode plate reader, EnSpire 2300, Perkin Elmer). For data analysis, absorbance values were subtracted by blanks and converted to concentrations by referencing to the standard curve. The data was plotted as a function of time and cell culture area (nmol min−1 cm−2).
Alcian blue staining to evaluate mucus production
To visualize acidic mucopolysaccharides, cells in the microfluidic and TW systems were fixed with 4% paraformaldehyde (PFA) for 20 min. The mucus was then acidified by incubating with 3% acetic acid for 3 min at RT. After rinsing with distilled water, the mucopolysaccharides were stained using an alcian blue staining solution (1431.1, Roth) for 30 min. Finally, the cells were washed with PBS until the supernatant became clear.Immunocytochemistry of tight-junction marker ZO1
Another vital aspect of intestinal epithelial cells is the tight junction formation. Therefore, the cells were stained after 1, 3, and 7 days with antibodies against ZO1, an intercellular membrane protein involved in the tight junction formation. For the staining, cells were rinsed with PBS and fixed with 4% PFA overnight at 4 °C. Afterward, the cells were rinsed with PBS containing Ca2+ and Mg2+ (PBS+), followed by a 15 min permeabilization step using 0.2% Triton X-100 in PBS+. In order to avoid unspecific antibody bindings, the samples were covered with a blocking solution containing 1% BSA in PBS+ for 2 h. The primary antibody against ZO1 (rabbit polyclonal antibody; 21773-1-AP, Proteintech®) was diluted at 1:200 and incubated on the cells overnight at 4 °C. The next day, the cells were washed twice with PBS+ and stained with a 1:1000 secondary antibody solution (goat anti-rabbit 555; A-32732; Invitrogen) for 2 h at RT. Subsequently, the cells were rinsed with PBS+ and counterstained for 1 h with DAPI (2 mg mL−1) 1:1000 diluted in PBS+. In the end, the cells were washed once with PBS+. Images were acquired using a fluorescence microscope (IX83, Olympus) and processed with ImageJ.
Scanning electron microscopy (SEM) to capture microvilli on the cell surface
To visualize microvilli on the apical side of Caco-2 cells, samples from day 7 were fixed in 2.5% glutaraldehyde for 2.5 h at RT. Membranes containing the cells were then cut and placed into a 48-well plate for serial dehydration in ethanol (50%, 60%, 70%, 80%, 90%, and 100%), followed by drying with 50% and 98.5% hexamethyldisilazane (AB111174, abcr GmbH) for 10 min each. The samples were gold sputtered and imaged with SEM (Quanta 200, FEI Company).Senescence-associated β-galactosidase assay to determine cell cycle arrest after adding doxorubicin
In order to induce senescence, cells were exposed to different DXR concentrations and stained with a senescence-associated β-galactosidase assay. For this, Caco-2 cells were cultivated in T25 culture flasks for 2 days to minimize cellular stress from passaging. They were then exposed to 0.1, 0.4, and 0.8 μg mL−1 DXR for 5 days. Subsequently, the cells were seeded in a 48-well plate in low density to facilitate manual cell counting. At the end of the DXR exposure, the chromogenic senescence-associated β-galactosidase assay from Debacq-Chainiaux et al. was employed.69 Briefly, cells were washed twice with PBS and fixed with 2% PFA and 0.2% glutaraldehyde for 5 min at RT. After another 2 washes with PBS, the cells were incubated with a staining solution containing 40 mM citric acid/Na phosphate buffer, 5 mM K4[F(CN)6]3H2O, 5 mM K3[F(CN)6], 150 mM sodium chloride, 2 mM magnesium chloride and 1 mg mL−1 X-gal. Following an 8 h incubation at 37 °C, the cells were washed and dried with methanol. Cells were examined under a light microscope with brightfield for the presence of blue precipitate. Representative images with a 40× magnification were taken, and positive cells were manually counted. The proportion of senescence-positive cells was normalized to the total cell count.RNA isolation, cDNA synthesis and qPCR to analyze senescence marker
In order to analyze the expression of senescence markers, total RNA was extracted from DXR-treated and untreated cells using innuPREP RNA Mini Kit 2.0 (845-KS-2040010, Innuscreen GmbH). After the isolation, the concentration of the extracted RNA was determined with a spectrophotometer DS-11 (Denovix) and diluted with nuclease-free water to 44 ng μL−1. Subsequently, 22 ng μL−1 RNA was transcribed using the high-capacity cDNA reverse transcription kit (4368814, Thermo Fisher Scientific) by following the manufacturer's instructions. ForqPCR, 20 μL reaction mixtures were prepared, comprising 2 μL of cDNA (4 ng), specific primer pairs (listed in Table S1†), 10 μL of Power Track SYBR Green Master Mix (A46012, Thermo Fisher Scientific), and nuclease-free water. Thermocycling conditions included 40 cycles at 95 °C for 5 s, 60 °C for 30 s, preceded by an initial polymerase activation step lasting for 2 min. Melting curve analysis, conducted between 60 and 95 °C, confirmed the specificity of amplification (Fig. S7†). Data analysis was performed using the Linegene real-time qPCR software. GAPDH served as a housekeeping gene and was used to calculate fold change in 2ΔΔCt.Size analysis of doxorubicin-treated cells
In order to determine cellular hypertrophy after the 6 day exposure to DXR, the cells were trypsinized and mechanically detached from the membrane. 10 μL cell suspension was then utilized to analyze the size distribution using the CellDrop™ cell counter (DeNovix). The number of cells per size was normalized to the total cell count of the untreated cells.Diffusion assay to analyze the intestinal barrier permeability
In order to evaluate the permeability of the cellular barrier, a diffusion assay was conducted using FITC–dextran (3–5 kDa, Sigma-Aldrich). This assay was performed daily throughout the cultivation period in the microfluidic platform. Cell culture medium containing 0.1 mg mL−1 fluorescence tracers was added to the apical channel and incubated for 15, 30, or 45 min. Subsequently, the entire solution from the basolateral layer was sampled and measured using a plate reader (multimode plate reader, EnSpire 2300, Perkin Elmer). By employing a calibration curve, the FITC–dextran concentration in the basal channel was determined, allowing for the calculation of the apparent permeability (Papp) using the following equation:Here, Papp describes the diffusion of the tracer through the barrier model in cm s−1; dCb represents the change in the FITC–dextran concentration in the basal channel; Vb denotes the sampled volume of the basal channel; dt is the duration of the diffusion; A stands for the diffusion area and C0 is the initial concentration of the FITC–dextran solution.
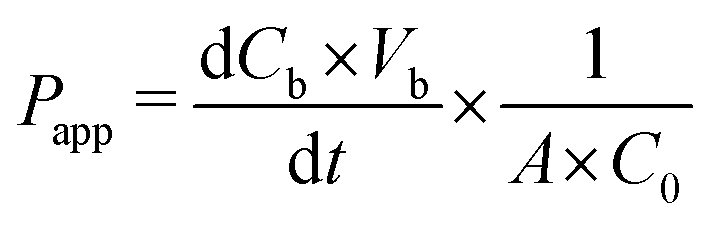
TEER measurement of senescent cells
To assess the TEER of the DXR-treated cells, chopstick electrodes from EVOM (STX-4 EVOM™, WPI) were used. Cells were cultivated on 24-well ThinCerts® membranes coated with 1% collagen until they reached a 70–80% confluency. Subsequently, cells were treated with 0.8 μg mL−1 DXR, and TEER was measured on days 2, 4, and 6 days of treatment. Changes in the TEER values were graphically visualized in a graph over time.Impedance measurement to detect barrier alterations and morphological changes
In order to monitor the barrier formation of the intestinal cells inside the gut-on-a-chip platform, the impedance was recorded daily. For the measurement using the in-house fabricated electrodes, the inlets and outlets of the microfluidic platforms were sealed with cell culture tape (236366PK, Thermo Fisher) and connected to a potentiostat (VMP3, Bio-Logic) via the contact pads of each electrode. The two-electrode electrochemical impedance sensing measurements were performed with a sinus amplitude of 10 mV, scanning from a maximum of 1 MHz to 0.1 Hz. In order to analyze the barrier formation over time, the cell index (CI), introduced by Pan et al.,46 was calculated at approximately 20 kHz using impedance signal of blanks without cells (Zblank) and the following equation: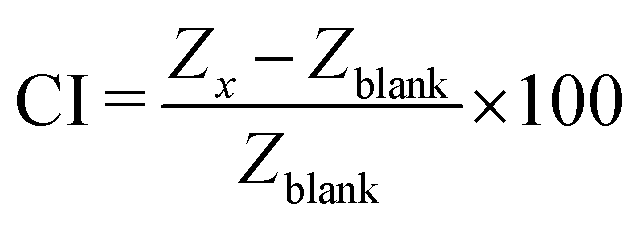
Statistical analysis
Statistical analysis and data visualization were conducted using the biostatistics program GraphPad Prism 10. For the assessment of statistical significance, Student's t-tests, one-way and two-way ANOVA, and Tukey's, Dunnett's, or Sidak's multiple comparison tests were performed. Normality was determined using either a Shapiro–Wilk or D'Agostino & Pearson test. The presence of single outliers was detected using a Grubb's test. Significance levels were denoted as follows: 0.1234 (ns.), 0.0332 (*), 0.0021 (**), 0.0002 (***), <0.0001 (****).Data availability
The data supporting this article have been included in the ESI.†Author contributions
Conceptualization: KB, SSp, methodology & investigation: KB, AL, SSp, material: KB, AL, PS, software and visualization: KB, analysis: KB, AL, writing and draft preparation: KB, writing – review and editing: all authors, supervision: SSp, SiS, MF, funding acquisition: PE.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors would like to thank Mario Rothbauer (TU Wien and Medical University of Vienna) for his advice, Elisabeth Eitenberger (TU Wien) for the SEM imaging, and Verena Stadlbauer from the Research Center Wels for providing us with the adenocarcinoma cell line Caco-2. Graphical illustrations were generated using the online software BioRender. The study was supported by the ENDOTARGET project (HORIZON-HLTH-2022-STAYHLTH-02-01/101095084). The authors acknowledge TU Wien Bibliothek for financial support through its Open Access Funding Program.References
- S. Taleban, J. F. Colombel, M. J. Mohler and M. J. Fain, J. Crohns Colitis, 2015, 9(6), 507–515, DOI:10.1093/ecco-jcc/jjv059
.
- R. A. Liddle, Brain Res., 2018, 1693, 201–206, DOI:10.1016/j.brainres.2018.01.010
- J. Parantainen, G. Barreto, R. Koivuniemi, H. Kautiainen, D. Nordström, E. Moilanen, M. Hämäläinen, M. Leirisalo-Repo, K. Nurmi and K. K. Eklund, Arthritis Res. Ther., 2022, 24(1), 256, DOI:10.1186/s13075-022-02946-z
- C. B. Forsyth, K. M. Shannon, J. H. Kordower, R. M. Voigt, M. Shaikh, J. A. Jaglin, J. D. Estes, H. B. Dodiya and A. Keshavarzian, PLoS One, 2011, 6(12), e28032, DOI:10.1371/journal.pone.0028032
- D. Krueger, K. Michel, F. Zeller, I. E. Demir, G. O. Ceyhan, J. Slotta-Huspenina and M. Schemann, J. Physiol., 2016, 594(2), 357–372, DOI:10.1113/JP271493
- A. Palmer, S. Epton, E. Crawley, M. Straface, L. Gammon, M. M. Edgar, Y. Xu, S. Elahi, J. Chin-Aleong, J. E. Martin, C. L. Bishop, C. H. Knowles and G. J. Sanger, Front. Neurosci., 2021, 15, 747067, DOI:10.3389/fnins.2021.747067
- A. R. Parrish, Tissue Barriers, 2017, 5(4), e1343172, DOI:10.1080/21688370.2017.1343172
- N. A. Mabbott, A. Kobayashi, A. Sehgal, B. M. Bradford, M. Pattison and D. S. Donaldson, Biogerontology, 2014, 16, 133–145, DOI:10.1007/S10522-014-9498-Z
- A. L. Man, N. Gicheva and C. Nicoletti, Cell. Immunol., 2014, 289(1–2), 112–118, DOI:10.1016/J.CELLIMM.2014.04.001
- L. Tran and B. Greenwood-Van Meerveld, J. Gerontol., Ser. A, 2013, 68(9), 1045–1056, DOI:10.1093/GERONA/GLT106
- M. Lucchetti, K. Oluwasegun Aina, L. Grandmougin, C. Jäger, P. Pérez Escriva, E. Letellier, A. S. Mosig and P. Wilmes, Adv. Healthcare Mater., 2024, 13(20), 2303943, DOI:10.1002/adhm.202303943
- J. L. Boyajian, M. Ghebretatios, S. Schaly, P. Islam and S. Prakash, Nutrients, 2021, 13(12), 4550, DOI:10.3390/nu13124550
- Y. Chen, S. E. Rudolph, B. N. Longo, F. Pace, T. T. Roh, R. Condruti, M. Gee, P. I. Watnick and D. L. Kaplan, Adv. Healthcare Mater., 2022, 11(16), 2200447, DOI:10.1002/adhm.202200447
- P. Zhuang, A. X. Sun, J. An, C. K. Chua and S. Y. Chew, Biomaterials, 2018, 154, 113–133, DOI:10.1016/j.biomaterials.2017.10.002
- N. S. Bhise, J. Ribas, V. Manoharan, Y. S. Zhang, A. Polini, S. Massa, M. R. Dokmeci and A. Khademhosseini, J. Controlled Release, 2014, 190, 82–93, DOI:10.1016/J.JCONREL.2014.05.004
- H. J. Kim, D. Huh, G. Hamilton and D. E. Ingber, Lab Chip, 2012, 12(12), 2165–2174, 10.1039/c2lc40074j
- S. R. Adam Kratz, G. Höll, P. Schuller, P. Ertl and M. Rothbauer, Biosensors, 2019, 9(3), 110, DOI:10.3390/BIOS9030110
- M. Lindner, A. Laporte, S. Block, L. Elomaa and M. Weinhart, Cells, 2021, 10(8), 2062, DOI:10.3390/cells10082062
- R. Kim and J. H. Sung, Adv. Healthcare Mater., 2024, 13(21), 2302777, DOI:10.1002/adhm.202302777
- L. F. Lorenzo-Martín, T. Hübscher, A. D. Bowler, N. Broguiere, J. Langer, L. Tillard, M. Nikolaev, F. Radtke and M. P. Lutolf, Nature, 2024, 629(8011), 450–457, DOI:10.1038/s41586-024-07330-2
- M. Fujii, M. Matano, K. Toshimitsu, S. Nishikori, S. Sugimoto and T. Sato, Cell Stem Cell, 2018, 23(6), 787–793.e6, DOI:10.1016/j.stem.2018.11.016
- S. Spitz, S. Schobesberger, K. Brandauer and P. Ertl, Bioeng. Transl. Med., 2023, 9(3), e10604, DOI:10.1002/BTM2.10604
- N. Ashammakhi, R. Nasiri, N. R. de Barros, P. Tebon, J. Thakor, M. Goudie, A. Shamloo, M. G. Martin and A. Khademhosseni, Biomaterials, 2020, 255, 120196, DOI:10.1016/j.biomaterials.2020.120196
- B. Srinivasan, A. R. Kolli, M. B. Esch, H. E. Abaci, M. L. Shuler and J. J. Hickman, J. Lab. Autom., 2015, 20(2), 107–126, DOI:10.1177/2211068214561025
- J. P. Vigh, A. Kincses, B. Ozgür, F. R. Walter, A. R. Santa-Maria, S. Valkai, M. Vastag, W. Neuhaus, B. Brodin, A. Dér and M. A. Deli, Micromachines, 2021, 12(6), 685, DOI:10.3390/mi12060685
- S. Palma-Florez, A. López-Canosa, F. Moralez-Zavala, O. Castaño, M. J. Kogan, J. Samitier, A. Lagunas and M. Mir, J. Nanobiotechnol., 2023, 21(1), 115, DOI:10.1186/s12951-023-01798-2
- P. Schuller, M. Rothbauer, S. R. A. Kratz, G. Höll, P. Taus, M. Schinnerl, J. Genser, N. Bastus, O. H. Moriones, V. Puntes, B. Huppertz, M. Siwetz, H. Wanzenböck and P. Ertl, Sens. Actuators, B, 2020, 312, 127946, DOI:10.1016/j.snb.2020.127946
- P. Schuller, M. Rothbauer, C. Eilenberger, S. R. A. Kratz, G. Höll, P. Taus, M. Schinnerl, J. Genser, P. Ertl and H. Wanzenboeck, MethodsX, 2019, 6, 2606–2613, DOI:10.1016/j.mex.2019.10.038
- M. G. Vasconez Martinez, E. I. Reihs, H. M. Stuetz, A. Hafner, K. Brandauer, F. Selinger, P. Schuller, N. Bastus, V. Puntes, J. Frank, W. Tomischko, M. Frauenlob, P. Ertl, C. Resch, G. Bauer, G. Povoden and M. Rothbauer, Biosensors, 2024, 14(2), 107, DOI:10.3390/bios14020107
- I. H. Heijink, S. M. Brandenburg, J. A. Noordhoek, D. S. Postma, D. J. Slebos and A. J. M. Van Oosterhout, Eur. Respir. J., 2010, 35(4), 894–903, DOI:10.1183/09031936.00065809
- T. Gerasimenko, S. Nikulin, G. Zakharova, A. Poloznikov, V. Petrov, A. Baranova and A. Tonevitsky, Front. Bioeng. Biotechnol., 2020, 7, 474, DOI:10.3389/fbioe.2019.00474
- C. R. Keese, J. Wegener, S. R. Walker and I. Giaever, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(6), 1554–1559, DOI:10.1073/PNAS.0307588100
- M. C. Lundien, K. A. Mohammed, N. Nasreen, R. S. Tepper, J. A. Hardwick, K. L. Sanders, R. D. Van Horn and V. B. Antony, J. Clin. Immunol., 2002, 22(3), 144–152, DOI:10.1023/A:1015420029430
- A. Migliorini, M. L. Angelotti, S. R. Mulay, O. O. Kulkarni, J. Demleitner, A. Dietrich, C. Sagrinati, L. Ballerini, A. Peired, S. J. Shankland, H. Liapis, P. Romagnani and H. J. Anders, Am. J. Pathol., 2013, 183(2), 431–440, DOI:10.1016/J.AJPATH.2013.04.017
- K. Benson, S. Cramer and H. J. Galla, Fluids Barriers CNS, 2013, 10(1), 1–11, DOI:10.1186/2045-8118-10-5
- N. Kudlova, J. B. De Sanctis and M. Hajduch, Int. J. Mol. Sci., 2022, 23(8), 4168, DOI:10.3390/ijms23084168
- M. Takasugi, Y. Yoshida, E. Hara and N. Ohtani, FEBS J., 2023, 290(5), 1348–1361, DOI:10.1111/febs.16381
- W. Lei, L. Jia, Z. Wang, Z. Liang, Z. Aizhen, Y. Liu, Y. Tian, L. Zhao, Y. Chen, G. Shi, Z. Yang, Y. Yang and X. Xu, Ageing Res. Rev., 2023, 87, 101900, DOI:10.1016/J.ARR.2023.101900
- R. Sharma, Probiotics Antimicrob. Proteins, 2022, 14(4), 648–663, DOI:10.1007/s12602-021-09903-3
- H. Zheng, C. Zhang, Q. Wang, S. Feng, Y. Fang and S. Zhang, Front. Immunol., 2022, 13, 1029948, DOI:10.3389/fimmu.2022.1029948
- K. Aoki, T. Sugasawa, Y. Yoshida, Y. Kawakami and K. Takekoshi, Int. J. Anal. Bio-Sci., 2022, 10(3), 60–66 CAS
- M. Biraud, J. Cortes, P. Cray, G. Kunzmann, J. Mohammed and C. M. Dekaney, bioRxiv, 2021, preprint, 2021–01, DOI:10.1101/2021.01.29.428764.
- K. Takaya and K. Kishi, Biogerontology, 2024, 25(4), 691–704, DOI:10.1007/s10522-024-10103-z
- B. P. Lohanathan, B. Rathinasamy, C. Y. Huang and V. P. Viswanadha, J. Biochem. Mol. Toxicol., 2022, 36(7), e23054, DOI:10.1002/JBT.23054
- M. D. Basson, G. Turowski and N. J. Emenaker, Experimental cell research, 1996, 225(2), 301–305, DOI:10.1006/excr.1996.0180
- Y. Pan, N. Hu, X. Wei, L. Gong, B. Zhang, H. Wan and P. Wang, Biosens. Bioelectron., 2019, 130, 344–351, DOI:10.1016/J.BIOS.2018.09.046
- C. M. Didier, J. F. Orrico, O. S. C. Torres, J. M. Castro, A. Baksh and S. Rajaraman, Microsyst. Nanoeng., 2023, 9(1), 22, DOI:10.1038/s41378-023-00488-1
- R. C. Huiszoon, S. Subramanian, P. Ramiah Rajasekaran, L. A. Beardslee, W. E. Bentley and R. Ghodssi, IEEE Trans. Biomed. Eng., 2019, 66(5), 1337–1345, DOI:10.1109/TBME.2018.2872896
- S. Subramanian, E. I. Tolstaya, T. E. Winkler, W. E. Bentley and R. Ghodssi, ACS Appl. Mater. Interfaces, 2017, 9(37), 31362–31371, DOI:10.1021/acsami.7b04828
- O. Y. F. Henry, R. Villenave, M. J. Cronce, W. D. Leineweber, M. A. Benz and D. E. Ingber, Lab Chip, 2017, 17(13), 2264–2271, 10.1039/c7lc00155j
- R. Freire, L. Ingano, G. Serena, M. Cetinbas, A. Anselmo, A. Sapone, R. I. Sadreyev, A. Fasano and S. Senger, Sci. Rep., 2019, 9(1), 1–15, DOI:10.1038/s41598-019-43426-w
- F. Dabbagh, H. Schroten and C. Schwerk, Pharmaceutics, 2022, 14(8), 1729, DOI:10.3390/pharmaceutics14081729
- S. Ladel, P. Schlossbauer, J. Flamm, H. Luksch, B. Mizaikoff and K. Schindowski, Pharmaceutics, 2019, 11(8), 367, DOI:10.3390/pharmaceutics11080367
- J. Westerhout, E. Van De Steeg, D. Grossouw, E. E. Zeijdner, C. A. M. Krul, M. Verwei and H. M. Wortelboer, Eur. J. Pharm. Sci., 2014, 63, 167–177, DOI:10.1016/J.EJPS.2014.07.003
- B. Jing, Z. A. Wang, C. Zhang, Q. Deng, J. Wei, Y. Luo, X. Zhang, J. Li and Y. Du, Front. Bioeng. Biotechnol., 2020, 8, 272, DOI:10.3389/fbioe.2020.00272
- M. S. Balda and K. Matter, EMBO J., 2000, 19(9), 2024–2033, DOI:10.1093/EMBOJ/19.9.2024
- B. N. G. Sajay, C. S. Yin and Q. Ramadan, J. Micromech. Microeng., 2017, 27(12), 124004, DOI:10.1088/1361-6439/AA96BD
- Y. Guo, Y. Xie and J. Qin, Biotechnol. J., 2024, 19(2), 2300390, DOI:10.1002/BIOT.202300390
- S. Jalili-Firoozinezhad, R. Prantil-Baun, A. Jiang, R. Potla, T. Mammoto, J. C. Weaver, T. C. Ferrante, H. J. Kim, J. M. S. Cabral, O. Levy and D. E. Ingber, Cell Death Dis., 2018, 9(2), 223, DOI:10.1038/s41419-018-0304-8
- R. Wang, L. Sun, S. Xia, H. Wu, Y. Ma, S. Zhan, G. Zhang, X. Zhang, T. Shi and W. Chen, Cell Death Dis., 2021, 12(5), 1–17, DOI:10.1038/s41419-021-03736-2
- P. Davalli, T. Mitic, A. Caporali, A. Lauriola and D. D'Arca, Oxid. Med. Cell. Longevity, 2016, 2016(1), 3565127, DOI:10.1155/2016/3565127
- F. Kullenberg, O. Degerstedt, C. Calitz, N. Pavlović, D. Balgoma, J. Gråsjö, E. Sjögren, M. Hedeland, F. Heindryckx and H. Lennernäs, Cells, 2021, 10(7), 1717, DOI:10.3390/CELLS10071717/S1
- P. Singh Rawat, A. Jaiswal, A. Khurana, J. Singh Bhatti and U. Navik, Biomed. Pharmacother., 2021, 139, 111708, DOI:10.1016/j.biopha.2021.111708
- K. W. Lanks and J. M. Lehman, Cancer Res., 1990, 50(15), 4776–4778 CAS
- S. Mouli, G. Nanayakkara, A. Alalasmari, H. Eldoumani, X. Fu, A. Berlin, M. Lohani, B. Nie, R. D. Arnold, A. Kavazis, F. Smith, R. Beyers, T. Denney, M. Dhanasekaran, J. Zhong, J. Quindry and R. Amin, Am. J. Physiol., 2015, 309(5), H844–H859, DOI:10.1152/AJPHEART.00182.2015
- M. Busch, G. Bredeck, A. A. M. Kämpfer and R. P. F. Schins, Environ. Res., 2021, 193, 110536, DOI:10.1016/J.ENVRES.2020.110536
- S. Venugopal, S. Anwer and K. Szászi, Int. J. Mol. Sci., 2019, 20(22), 5655, DOI:10.3390/IJMS20225655
- S. Ferruzza, C. Rossi, M. L. Scarino and Y. Sambuy, Toxicol. In Vitro, 2012, 26(8), 1247–1251, DOI:10.1016/j.tiv.2011.11.007
- F. Debacq-Chainiaux, J. D. Erusalimsky, J. Campisi and O. Toussaint, Nat. Protoc., 2009, 4(12), 1798–1806, DOI:10.1038/nprot.2009.191
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lc00896k |
| This journal is © The Royal Society of Chemistry 2025 |