
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An electrochemical sensor integrated lab-on-a-CD system for phenylketonuria diagnostics†
Ipek
Akyilmaz
a,
Dilan
Celebi-Birand
 a,
Naim Yagiz
Demir
a,
Deniz
Bas
b,
Caglar
Elbuken
cd and
Memed
Duman
*a
a,
Naim Yagiz
Demir
a,
Deniz
Bas
b,
Caglar
Elbuken
cd and
Memed
Duman
*a
aHacettepe University, Institute of Science, Nanotechnology and Nanomedicine Division, Ankara, Turkey. E-mail: memi@hacettepe.edu.tr
bDepartment of Food Engineering, Faculty of Engineering, Cankiri Karatekin University, Cankiri, Turkey
cFaculty of Biochemistry and Molecular Medicine, University of Oulu, Oulu, Finland
dVTT Technical Research Centre of Finland Ltd., Finland
First published on 31st December 2024
Abstract
Phenylketonuria (PKU) is characterized by an autosomal recessive mutation in the phenylalanine hydroxylase (PAH) gene. Impaired PAH enzyme activity leads to the accumulation of phenylalanine (Phe) and its metabolites in the bloodstream, which disrupts the central nervous system and causes psychomotor retardation. Early diagnosis of PKU is essential for timely intervention. Moreover, continuous monitoring of blood Phe levels is indispensable for prognosis, requiring a robust and reliable monitoring system. This study presents an automated lab-on-a-CD-based system for early diagnosis and monitoring of PKU treatment. This miniaturised system contains CD-shaped disposable cartridges, a mini centrifuge, and an electrochemical sensing unit. Modified screen-printed gold electrodes were used for the electrochemical measurements in cartridges. Electrode modification was conducted by electrochemical graphene oxide reduction and deposition on the electrode surface, which increased the sensitivity of the measurement 1.5 fold. The system used amperometric detection to measure Phe in the blood through oxidation of NAD+ to NADH by the enzyme phenylalanine dehydrogenase. The limit of detection (LOD), limit of quantification (LOQ), and sensitivity of the system were 0.0524, 0.1587 mg dL−1 and 0.3338 μA mg−1 dL, respectively, within the 0–20 mg dL−1 measurement range (R2 = 0.9955). The performance of the lab-on-a-CD system was compared to the gold standard HPLC method. The accuracy was 83.1% for HPLC and 84.1% for the lab-on-a-CD system. In conclusion, this study successfully developed a portable diagnostic device for rapid (under 20 min), accurate and highly sensitive detection of Phe in whole blood.
Introduction
Phenylketonuria (PKU) is a metabolic disorder caused by the inability to process the amino acid phenylalanine due to an autosomal recessive mutation in the phenylalanine hydroxylase (PAH) gene. The protein product of this gene is an enzyme responsible for converting the amino acid phenylalanine (Phe) into tyrosine. Impaired PAH function leads to the accumulation of Phe and its metabolic byproducts (e.g., phenylpyruvic acid, phenyllactic acid, etc.) for people suffering from PKU. Elevated levels of Phe and phenyl derivatives in blood, urine and other body fluids disrupt the myelination in the central nervous system and lead to severe psychomotor retardation and other neurological complications such as seizures.1–3The prevalence of PKU varies across populations, with the highest incidence observed in low and middle-income countries. For instance, in Turkey 38 per 100.000 newborns are affected by PKU. In contrast, globally, the condition occurs at a rate of approximately 6 per 100.000 neonates, making PKU a widespread public health concern and burden for healthcare systems in underdeveloped and developing countries.4
Detecting PKU in newborns is crucial for immediate dietary management through a low Phe diet, which effectively prevents life-long cognitive impairment associated with the disorder. Despite advancements in the diagnosis of PKU, major improvements are required in close monitoring of blood Phe levels during follow-up care. A sensitive, robust, and low-cost system is required to confine Phe levels within the therapeutic range, to optimise treatment outcomes, and to eradicate the long-term consequences of PKU.
Commonly used methods in PKU screening are the Guthrie test,5 fluorometric assays,6 an enzyme-based colourimetric test,7 HPLC (for direct amino acid analysis) and tandem mass spectroscopy (MS/MS).5 These tests are usually conducted at well-equipped health centres and require technical infrastructure, advanced equipment and well-trained staff. Due to the inaccessibility of regular screen tests, cold storage and transportation of blood samples from the periphery to testing facilities increase costs. Therefore, it is important to develop an accessible, user-friendly, and low-cost blood Phe monitoring system.8
In today's healthcare landscape, particularly in developing countries where healthcare infrastructure is limited, there is an increasing demand for cost-effective, reliable and easy-to-use diagnostic solutions.9,10 As health organisations such as the National Institutes of Health (NIH) and the Bill and Melinda Gates Foundation have emphasized, point-of-care (POC) technologies are vital for timely diagnoses and treatments, especially in underserved areas.11
Centrifugal microfluidic disc technologies, also known as lab-on-a-CD (LoCD), represent a promising frontier in diagnostic applications, offering several advantages that make them ideal for POC settings.12–14 With their ability to perform complex analyses on small samples, LoCD devices alleviate the need for large, centralised laboratory infrastructures, making them favourable for remote or resource-limited environments. Their portability, low sample volume requirements, and rapid results hold significant potential for revolutionising the way diseases such as PKU are diagnosed and monitored, particularly in developing regions. The full potential of LoCD technology is yet to be realised; its impact could be transformative in addressing healthcare challenges globally.12
These compact devices integrate various components, including reservoirs, chambers, and microchannels to perform a variety of chemical and biochemical reactions. Fluids are moved within the disc using centrifugal force, eliminating the need for complex fluid handling systems.15 Thus the need for complex fluid interconnection and different types of pumps are eliminated from microfluidic devices.16 LoCD systems can be designed to be operated in an automated manner with minimal operator need. The only specific expertise required is for taking the sample from the patient and introducing it to the disposable disc. The rest of the measurement can be automated making LoCD valuable for medical and clinical applications in resource-limited settings.17
Recent studies have increasingly incorporated optical or electrochemical sensing modalities within microfluidic devices. These integrated systems have demonstrated the capability to deliver rapid and accurate analytical results. Among these, optical systems, often favoured for their accessibility and simple detector interfaces, have been widely adopted.18 However, the intricate alignment requirements of these systems have posed challenges for their implementation in POC settings. Conversely, electrochemical sensing is a promising alternative, offering advantages in miniaturization, cost-effectiveness, and portability. Furthermore, electrochemical methods have exhibited superior detection limits for biological analytes.19–21
Here, we aimed to develop a LoCD platform for early diagnosis and monitoring of PKU. The LoCD system developed in this study is an easy-to-use device that allows for rapid and accurate testing of PKU levels and is particularly useful for point-of-care testing. Moreover, the system utilising electrochemically reduced graphene oxide (ERGO)-modified gold electrodes is highly sensitive, and provides reliable results. Consequently, the system described in this study provides a valuable tool for identifying and effectively monitoring PKU, and it has the potential to be used to diagnose and manage other metabolic disorders.
Experimental
Materials and reagents
L-Phenylalanine (Phe, 98%), β-nicotinamide adenine dinucleotide sodium salt (NAD+, 95%), glycine, and hydrochloric acid (HCl), sodium hydroxide (NaOH), graphene oxide (GO) (4 mg mL−1, dispersion in H2O), sulfuric acid (H2SO4), potassium ferricyanide/ferrocyanide (K3/4Fe(CN)6), potassium chloride (KCl), sodium phosphate monobasic (Na2HPO4), and potassium phosphate monobasic (KH2PO4) were purchased from Sigma-Aldrich (Germany). Phenylalanine dehydrogenase from Rhodococcus sp. was acquired from ASA Spezialenzyme GmbH (Germany). Ethanol, used for electrode cleaning, was obtained from Isolab (Germany). Electrochemical reduction of GO was performed in a phosphate buffer (PB) solution, and reduction was characterised as described previously.22 β-Nicotinamide adenine dinucleotide, reduced disodium salt hydrate (NADH, grade I) was purchased from Roche (Germany), and NADH solutions were prepared in glycine buffer (pH 10.5). Human blood samples were collected from healthy individuals at Hacettepe University Hospital (Ankara, Turkey). All experiments were performed in compliance with the Guidelines on the Use of Human Biological Specimens of Hacettepe University and approved by the Ethics Committee of Hacettepe University. Informed consent was obtained for the collection of whole blood from human subjects. All reagents were freshly prepared using ultrapure water (18.2 M Ω cm−1) obtained from an Aqua Max-Ultra water purification system (South Korea).Apparatus
Microfluidic channels and chambers within the cartridges were designed utilizing SolidWorks 3D CAD Design software (USA). Polymethylmethacrylate (PMMA) plates were precisely cut to a thickness of 1 mm using AEON NOVA7 CO2 laser engraver and cutter (China) with 60% laser power and 15 mm s−1 speed. Three plates (top, middle and bottom) were assembled into cartridges using 3M 468 MP double-sided adhesive tapes (USA). Electrochemical measurements were conducted with a 910 PSTAT Mini potentiostat (Metrohm, Switzerland) by using the PSTAT software. Disposable screen-printed gold electrodes (SPE) (DRP 220 AT) were purchased from Metrohm Dropsens (Switzerland). Amperometric detection was performed at room temperature (25 °C). Blood plasma samples were centrifuged in a high-speed mini centrifuge (Isolab, Germany), and external electrochemical measurements were carried out. The centrifugation of blood within the fabricated cartridges was achieved using a custom-designed mini centrifugation module, containing a brushless DC motor (E-max Grand Turbo 2215/09; 12 V, 20k rpm) and hall effect sensor (Keyes) to modulate the rotational speed. Scanning electron microscopy (SEM) (Tescan, GAIA3+Oxford XMax 150 EDS, Czech Republic) was employed to characterise the surface morphology of the electrodes.Design, fabrication, and centrifugation of the CD cartridges
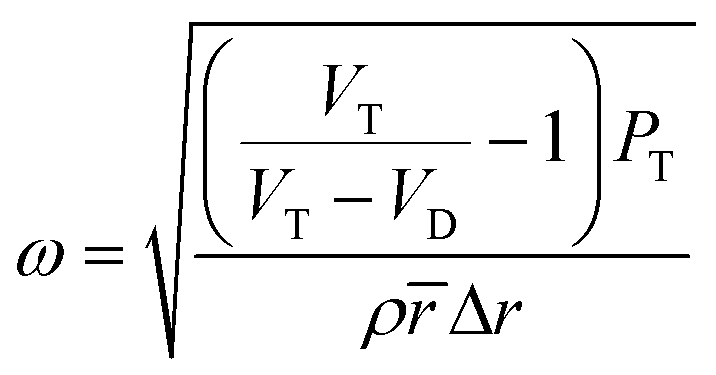 | (1) |
![[r with combining macron]](https://www.rsc.org/images/entities/i_char_0072_0304.gif) = 0.5 × (ro + ri), PT: atmospheric pressure, : mean radial position, VT: volume of gas trapped under atmospheric pressure, VD: volume of fluid flowing into the pneumatic chamber.
= 0.5 × (ro + ri), PT: atmospheric pressure, : mean radial position, VT: volume of gas trapped under atmospheric pressure, VD: volume of fluid flowing into the pneumatic chamber.
The angular frequency at which the disc should rotate to transfer the fluid in the pneumatic chamber was obtained by balancing the air and centrifugal pressures.24 The geometry of the siphon system within the cartridges was determined using eqn (1). The design ensured that the blood sample was subjected to high rotational speed as it passed through the sedimentation chamber while entering the analysis chamber at a lower rotational speed. The cartridge design and its dimensions are given in Fig. 1a and b, respectively.
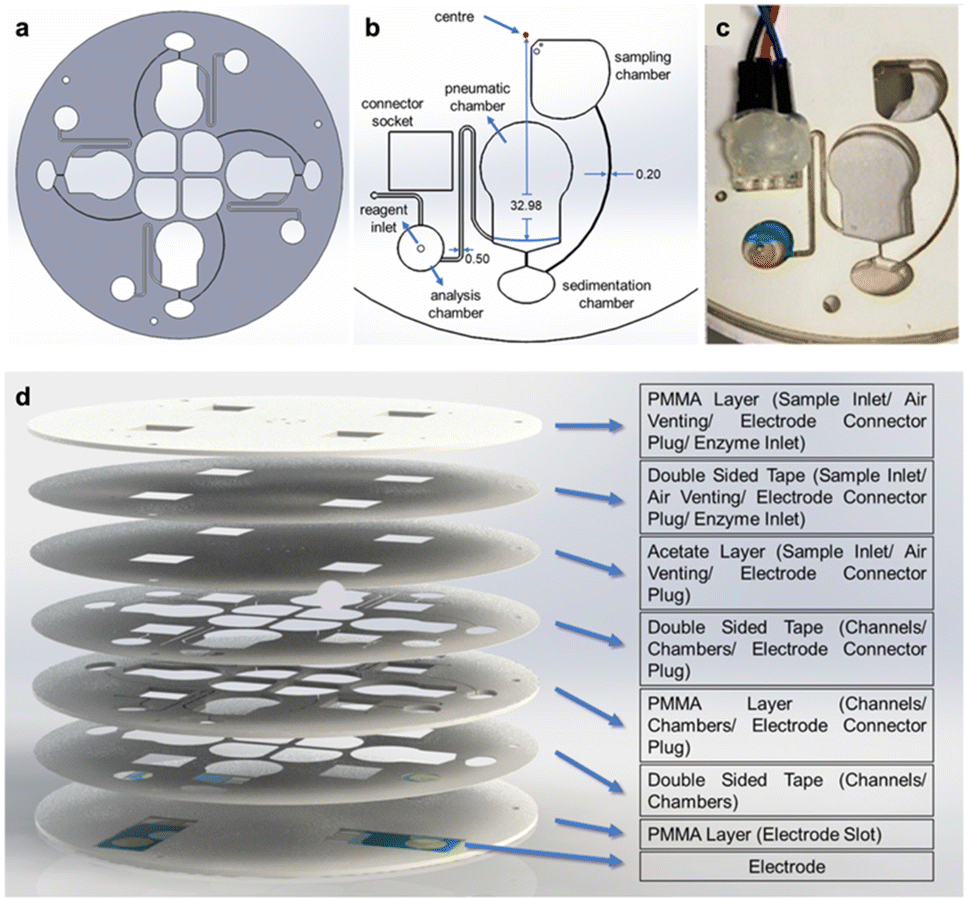 | ||
| Fig. 1 The design and the fabrication process of the cartridge used in the lab-on-a-CD system. The cartridge design (a), dimensions of the components (b), their installation on the CD (c), and layers of the disc (d) are illustrated and described. | ||
The connection adapter, designed for continuous contact between the pogo pins and electrodes, was created using SolidWorks software. This adapter was specifically designed for the measurement chamber (Fig. 1c). A connector socket was cut into the CD to allow for easy connection to the electrodes. Once the blood separation process is complete, the adapter with the pogo pins is inserted into the connector socket on the CD (Fig. 1c). This enables uninterrupted signal reception for the measurement process.
The analysis chamber (7.5 mm in diameter and 1 mm in height) was designed to accommodate approximately 45 μL of blood plasma. A small opening was created in the chamber to allow for the introduction of reagents after the measurement had stabilized. To prevent leaks during centrifugation, acetate paper was placed between the layers and secured with double-sided tape (Fig. 1d). After centrifugation, the acetate paper was punctured using an injector, and the reagents were added to the analysis chamber.
The electrodes were incorporated into the cartridge by using a laser cutting device to ablate a 500 μm deep groove in the bottom PMMA layer. The electrode was then positioned in this groove and aligned with the surface of the other layers to prevent any unevenness during the lamination process. This step was crucial to avoid leaks in the cartridge (Fig. 1d).
PMMA plates with a thickness of 1 mm and 0.13 mm double-sided transfer tape (3M, 468 MP) were cut using the previously specified laser settings. Once the electrodes were positioned in the analysis chamber, the screw holes and CD layers were aligned and laminated together. The assembly was then subjected to a 5 kg weight for 24 h to ensure leak-free adhesion of the double-sided tape.
Modification of the electrodes
Electrochemically reduced graphene oxide (ERGO) was prepared and deposited onto the surface using amperometry and cyclic voltammetry (CV). Before modification, the bare gold electrodes were cleaned with pure ethanol for 5 min and then rinsed with distilled water. Then, the electrodes underwent electrochemical cleaning using 0.5 M H2SO4 through 15 cycles of CV within a potential range of 0.0 to 1.6 V at a scanning rate of 100 mV s−1.Following electrode cleaning, graphene oxide (GO) reduction was achieved amperometrically by applying a constant potential of −1.2 V for 800 s. A solution containing 30 μg mL−1 of GO in 0.5 M NaNO3 solution at pH 4 was used.25 In CV, 1.5, 2, 2.5 mg mL−1 of GO were mixed with 0.1 M PB (70% v/v) at pH 6, and 40 μL of this mixture was then pipetted onto the electrode. GO reduction was performed through multiple cycles at 100 mV s−1 within a potential range of 0.1 to −1.5 V.26
Phe detection principle
Nicotinamide adenine dinucleotide (NAD+) and its reduced form, NADH, are essential coenzymes in dehydrogenase-catalysed enzymatic reactions. The nicotinamide nucleotide acts as an electron transfer mediator in biological systems. Detecting these compounds is vital for studying redox biological processes and identifying various enzyme substrates.27 The electrochemical system designed for phenylalanine (Phe) measurement is based on the following principle: first, the phenylalanine dehydrogenase (PheDH) enzyme produces phenylpyruvate and NADH in the presence of NAD+. | (2) |
| NADH → NAD+ + H+ + 2e | (3) |
Preparation of blood samples
Blood plasma samples were prepared following the manufacturer's instructions. The final phenylalanine (Phe) concentration in the samples was adjusted to 2, 4, 10, and 20 mg dL−1 by adding appropriate amounts of Phe solutions in deionised water. To adjust the plasma pH to 10.5, which is the optimal working pH for PheDH, the samples were diluted with 200 mM glycine buffer (pH 10.5) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 blood:buffer ratio.
:3 blood:buffer ratio.
Electrochemical detection of Phe
Amperometry was employed as the electrochemical measurement technique, with a potential of 0.65 V applied for 2000 s. The measurement of the sample started immediately following plasma separation. Once the measurement reached equilibrium, the reaction was initiated by adding reagents at various intervals between 600–800 s. Experiments were conducted using different reagents: PheDH alone, NAD+ alone (with PheDH added during sample preparation), and a mixture of PheDH and NAD+. The final concentrations in the solution were adjusted to 1.6 U mL−1 for PheDH, and 2.5 mM for NAD+. The detailed description of the design and optimisation of the device are specified in our patent (WIPO patent no. WO2023121632A2; Turkish patent no. TR2021020698A2).Results and discussion
Electrode modification and characterisation
Carbon-based nanomaterials, as working electrode modifications, can enhance electrochemical signals in analytical instruments.29,30 Graphene oxide (GO) is a widely used material for developing electrochemical biosensors.31 In our previous study, we demonstrated that electrochemically reducing GO-deposited gold electrodes improves phenylalanine sensing in the blood.22 In this study, amperometry and cyclic voltammetry (CV) were used to modify the gold electrode surface with electrochemically reduced graphene oxide (ERGO). A 1200 μM NADH solution was measured using CV on both modified gold electrodes and bare gold electrodes within a potential range of 0 to 1 V at a scanning speed of 100 mV s−1. A single cycle of measurement was performed to compare the current values. The results showed that electrochemical reduction using CV produced higher current values at lower potentials (Fig. 2a) due to enhanced specific surface area and increased electrochemical conductance. Therefore, we opted for the CV method to produce reduced GO electrodes for the subsequent experiments. | ||
| Fig. 2 Electrode modification optimisations. CV measurement of 1200 μM NADH solution on modified and bare gold electrodes as a result of ERGO with amperometry and CV (0–1 V, 100 mV s−1, single cycle) (a). Measurement of 5 mM Fe(CN)63−/4− on CV ERGO modified electrodes with 2 mg mL−1 GO at 25, 50, 75 cycles (CV measurement, −0.5–0.8 V, 100 mV s−1, single cycle) (b). Measurement of 1200 μM NADH on ERGO-modified electrodes with 1.5, 2, and 2.5 mg mL−1 GO (CV measurement, 0–1 V, 100 mV s−1, single cycle) (c). | ||
In the current study, we aimed to optimise the ERGO modification method by testing different GO concentrations (1.5, 2, 2.5 mg mL−1) and cycle numbers (25, 50, 75). Initially, a 2 mg mL−1 GO solution was electrochemically reduced using varying cycle numbers. A 5 mM Fe(CN)63−/4− solution was then measured using cyclic voltammetry (CV) at a single cycle and a scanning rate of 100 mV s−1 within the potential range of −0.5 to 0.8 V (Fig. S1†). The electrodes modified with 75 cycles exhibited the highest conductivity (Fig. 2b). Subsequently, a 1200 μM NADH solution was measured on three electrodes prepared using different GO amounts and reduced with 75 cycles. The measurements were performed using CV at a single cycle and a scanning speed of 100 mV s−1 within the potential range of 0 to 1 V. The results indicated that 2 mg mL−1 GO reduced with 75 cycles yielded the best performance (Fig. 2c).
The specified reduction process resulted in the formation of more intense, robust, and stable ERGO sheets. The modified electrode surfaces were examined using SEM (Fig. 3). The ERGO on the gold electrode created a rougher and darker surface compared to the bare gold electrode (Fig. 3a), and the ERGO sheets were evenly distributed (Fig. 3b) as similarly reported in previous studies.32,33
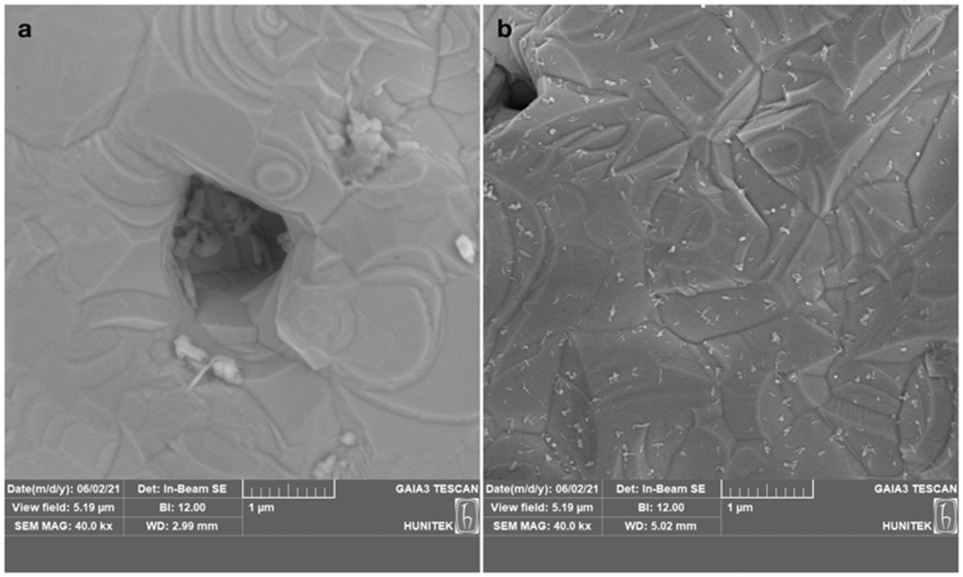 | ||
| Fig. 3 Representative SEM images of bare gold (a), and ERGO-modified gold electrodes (b). | ||
Design and operation of the CD cartridge
The cartridges were designed to contain three chambers for sample loading (sampling chamber), plasma separation (sedimentation chamber) and electrochemical measurement (analysis chamber). Centrifugal force, using the siphon effect, was utilised to move liquid between chambers without valves. When the rotational velocity of the CD was increased, the centrifugal force exerted on the fluid within the capillary channel opposed its progression. Conversely, as the rotational velocity decreased, the capillary action exerted a dominant influence, propelling the fluid beyond the apex of the capillary channel. Consequently, upon descending below the liquid level within the first channel, the fluid was entirely transferred to the second channel. This method is more tolerant to manufacturing variations than traditional valve-based methods, which require precise control of channel size and surface properties.23The pneumatic siphon dimensions for the CD (Fig. 1c) were calculated using eqn (1), resulting in the values listed in Table 1.
| ω (rpm) | V T (μL) | V D (μL) | r o (cm) | r i (cm) | Explanation |
|---|---|---|---|---|---|
| 4232.28 | 288.18 | 64.88 | 3.298 | 2.82 | Estimated rpm for blood separation |
| 327.33 | 288.18 | 0.5 | 3.298 | 2.82 | Conditions necessary for initiating the siphon process |
The functionality of the fabricated cartridge was evaluated as follows: initially, 200 μL of diluted blood sample was loaded into the sampling chamber, and the cartridge was then rotated at 5500 rpm. The sample was transferred from the sampling chamber to the sedimentation chamber (Fig. 4a). The trapped air pressure above the siphon in the sedimentation chamber remained lower than the centrifugal force, causing it to be compressed by the blood entering the said chamber. This resulted in the separation of blood into plasma within the sedimentation chamber while preventing leakage into the analysis chamber. The complete separation of blood into plasma occurred in approximately 20 s (Fig. 4b).
 | ||
| Fig. 4 The complete process of Phe detection in blood using the developed cartridges. Loading of blood into the sampling chamber (a); centrifugation at 5500 rpm to transfer blood to the sedimentation chamber and compress the air above it (b); initiation of siphoning by reducing the centrifuge speed to 300 rpm (c); addition of reagents to the analysis chamber (d); and electrochemical measurement of Phe in blood samples using CD cartridges (0.65 V, 2000 s), as illustrated in the amperometric measurement graph (e). The images were captured using a stroboscopic light source and an SLR camera (Canon, EOS450D) while the CD was rotating. | ||
Once the plasma separation was complete, the centrifuge speed was reduced to 300 rpm and kept constant for 40 s (Fig. 4c). During this phase, the air trapped by the blood exerted a pressure that exceeded the centrifugal force, causing it to expand. This expanding air pushed the plasma into the siphon channel, and the plasma was subsequently transferred into the analysis chamber. In conclusion, a CD cartridge with a functional siphon system and an appropriate micro-channel design was successfully fabricated.
Validation of the electrochemical method
Additionally, the effect of pH on the enzymatic response of our biosensor was examined at 25 °C within a pH range of 7.4 to 10.5. The maximum response was observed at pH 10.5. To optimise the measurement time, the electrochemical oxidation of enzymatically generated NADH was monitored by applying 0.65 V to the electrode for 2000 s. The current response exhibited a linear increase approximately 10 min after adding the reagents to the sample.
Reagents were added in different ways to increase the range of current values between concentrations and find the best method for cartridges (Fig. 5). The reaction was started by adding PheDH alone (Fig. 5a), both PheDH and NAD+ together (Fig. 5b), or NAD+ alone (Fig. 5c). In each test, diluted plasma samples with the same Phe concentrations were loaded on the electrode, and reagents were added after the measurement stabilized at 0.65 V.
 | ||
| Fig. 5 Amperometric measurements of different Phe concentrations (0, 2, 4, 10, 20 mg dL−1) in blood with ERGO-modified gold electrodes and various reaction initiation methods (0.65 V, 2000 s, 1.6 U mL−1 PheDH, 2.5 mM NAD+). The addition of PheDH alone (a), PheDH and NAD+ in the solution together (b), and NAD+ alone (c) were tested. Current increases in response to different reaction initiation methods were plotted against Phe concentrations (1:4 diluted) in a calibration graph (d). | ||
The relationship between the current increase and Phe concentration for all reagent addition methods is given in Fig. 5d. Adding PheDH and NAD+ together yielded the most suitable R2 and slope combination for cartridges produced in this study. Using the chosen method and within the Phe measurement range of 0–20 mg dL−1, 0.0524 mg dL−1 of limit of detection (LOD), 0.1587 mg dL−1 of limit of quantification (LOQ), and 0.3338 μA (mg dL−1)−1 sensitivity were obtained.
The presented Phe detection system using ERGO-modified gold electrodes has several advantages over other sensor systems in the literature, including a low detection limit using patient samples.3,34 While other studies have achieved lower detection limits in buffer solutions, the results may vary when using real samples.35,36 For instance, Moreira et al. reported a lower LOD of 0.0033 mg dL−1.25 However, a meaningful comparison is not possible due to the excessive enzyme use, complex sample preparation, and lack of specified current values in the aforementioned study. Other studies have also achieved lower LODs in real samples, but these methods are often complex and time-consuming.37,38 Here, we developed a sensor system combining microfluidics and electrochemical sensing and achieved a comparable LOD using a much more simplified and low-cost sensor architecture.
Upon the successful fabrication of the cartridge and the optimisation of the electrochemical method, Phe measurement in blood was performed. The blood sample was diluted 1:4 with buffer solution and loaded into the sampling chamber on the CD (Fig. 4a). The separation of plasma was performed as described previously. Once the sample was in the analysis chamber, the measurement began, and the system reached equilibrium in about 800 s. The reagent inlet in the analysis chamber was then punctured, and PheDH and NAD+ were added (Fig. 4d). After about 10 minutes, current increases that were directly proportional to the concentration were observed (Fig. 4e).
The performance of the developed test system was evaluated using both fabricated cartridges and the gold standard HPLC method with blood samples containing varying Phe concentrations (1.26, 3.38, 5.15, 10.65, 13.71, 19.63 mg dL−1). The accuracy of the test system was calculated to be 84.1%, while the HPLC method's accuracy was 83.1%. Although HPLC results have high accuracy at low concentrations, it could not maintain this level at higher concentrations (Fig. 6).
 | ||
| Fig. 6 A comparative study of HPLC and CD cartridges in measuring Phe, visualised through a bar graph. Blue dashed line is the parity line y = x. | ||
Overall, the results indicate that we successfully measured Phe in the range of 0.6–20 mg dL−1 with an LOD of 0.0524. This range is consistent with established diagnostic criteria for PKU, whereby blood phenylalanine levels below 2 mg dL−1 are considered normal, while levels exceeding 4 mg dL−1 are indicative of elevated phenylalanine and potential PKU, necessitating a preventative dietary intervention.39,40
Conclusions
In this study, a LoCD-based diagnostic platform was designed for the early detection and monitoring of PKU, which goes beyond the current state-of-the-art. This system incorporates disposable CD-shaped cartridges equipped with electrodes enhanced through ERGO modifications. The system offers user-friendly, cost-effective, and sample-to-answer solutions (under 20 min) as well as a sensitive and accurate sensing method. The resulting diagnostic tool is both portable and capable of delivering quantitative results. Moreover, the system can be customised according to different needs by simply changing the enzyme used to detect the target molecule. A crucial factor to consider in such applications is that the product of the enzymatic reaction (in this study, NADH) must be electroactive and capable of undergoing redox reactions within the working potential range of the electrode. Ensuring this compatibility is essential for accurate and reliable molecular detection. Therefore, this biomedical device, designed for molecular detection in patient body fluids, holds potential for broader applications in diagnosing various diseases.Data availability
Data are available upon request from the authors.Author contributions
Ipek Akyilmaz: investigation, methodology, validation, data curation, formal analysis, writing – original draft. Dilan Celebi-Birand: writing – original draft, visualisation. Naim Yagiz Demir: investigation, validation, formal analysis, writing – original draft. Deniz Bas: writing – review & editing, conceptualization. Caglar Elbuken: writing – review & editing, conceptualization. Memed Duman: conceptualization, funding acquisition, writing – review & editing, resources, supervision, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by The Scientific and Technological Research Council of Turkey (TUBITAK, Ankara, Turkey), ARDEB 1003, Grant no.: 118S047.References
- T. Arakawa, T. Koshida, T. Gessei, K. Miyajima, D. Takahashi, H. Kudo, K. Yano and K. Mitsubayashi, Microchim. Acta, 2011, 173, 199–205, DOI:10.1007/s00604-010-0536-5.
- R. Robinson, L. Wong, R. J. Monnat and E. Fu, Micromachines, 2016, 7, 2–11, DOI:10.3390/mi7020028.
- D. J. Weiss, M. Dorris, A. Loh and L. Peterson, Biosens. Bioelectron., 2007, 22, 2436–2441, DOI:10.1016/j.bios.2006.09.001.
- H. R. Shoraka, A. A. Haghdoost, M. R. Baneshi, Z. Bagherinezhad and F. Zolala, Clin. Exp. Pediatr., 2020, 63(2), 34–43, DOI:10.3345/kjp.2019.00465.
- C. M. Mak, H. C. H. Lee, A. Y. W. Chan and C. W. Lam, Crit. Rev. Clin. Lab. Sci., 2013, 50, 142–162, DOI:10.3109/10408363.2013.847896.
- H. Naruse, Y. Y. Ohashi, A. Tsuji, M. Maeda, K. Nakamura, T. Fujii, A. Yamaguchi, M. Matsumoto and M. Shibata, Screening, 1992, 1, 63–66, DOI:10.1016/0925-6164(92)90030-9.
- H. Shahbaz Mohammadi and E. Omidinia, J. Sci., Islamic Repub. Iran, 2011, 22, 15–20 Search PubMed.
- J. Wang, Biosens. Bioelectron., 2006, 21, 1887–1892, DOI:10.1016/j.bios.2005.10.027.
- C. D. Chin, V. Linder and S. K. Sia, Lab Chip, 2006, 7(1), 41–57, 10.1039/B611455E.
- W.-C. Lee, H.-Y. Ng, C.-Y. Hou, C.-T. Lee and L.-M. Fu, Lab Chip, 2021, 21(8), 1433–1453, 10.1039/D0LC01304H.
- E. O. Adekanmbi and S. K. Srivastava, Lab Chip, 2016, 16(12), 2148–2167, 10.1039/C6LC00355A.
- M. Madou, J. Zoval, G. Jia, H. Kido, J. Kim and N. Kim, Annu. Rev. Biomed. Eng., 2006, 8, 601–628, DOI:10.1146/annurev.bioeng.8.061505.095758.
- R. Gorkin, J. Park, J. Siegrist, M. Amasia, B. S. Lee, J. M. Park, J. Kim, H. Kim, M. Madou and Y. K. Cho, Lab Chip, 2010, 10, 1758–1773, 10.1039/B924109D.
- B. S. Lee, Y. U. Lee, H.-S. Kim, T.-H. Kim, J. Park, J.-G. Lee, J. Kim, H. Kim, W. G. Lee and Y.-K. Cho, Lab Chip, 2011, 11, 70–78, 10.1039/C0LC00205D.
- J. Ducrée, S. Haeberle, S. Lutz, S. Pausch, F. Von Stetten and R. Zengerle, J. Micromech. Microeng., 2007, 17, S103, DOI:10.1088/0960-1317/17/7/S07.
- T. H. Kim, K. Abi-Samra, V. Sunkara, D. K. Park, M. Amasia, N. Kim, J. Kim, H. Kim, M. Madou and Y. K. Cho, Lab Chip, 2013, 13, 3747–3754, 10.1039/c3lc50374g.
- T. Li, Electronic Thesis and Dissertation Repository, PhD thesis, 2012, vol. 696, p. 176 Search PubMed.
- C. Yi, Q. Zhang, C. W. Li, J. Yang, J. Zhao and M. Yang, Anal. Bioanal. Chem., 2006, 384, 1259–1268, DOI:10.1007/s00216-005-0252-x.
- A. Gencoglu and A. R. Minerick, Microfluid. Nanofluid., 2014, 17, 781–807, DOI:10.1007/s10404-014-1385-z.
- C. M. Miyazaki, E. Carthy and D. J. Kinahan, Processes, 2020, 8, 1–44, DOI:10.3390/pr8111360.
- T. Li, Y. Fan, Y. Cheng and J. Yang, Lab Chip, 2013, 13, 2634–2640, 10.1039/C3LC00020F.
- I. Akyilmaz, N. Y. Demir, D. Bas and M. Duman, RSC Adv., 2024, 14, 29874–29882, 10.1039/D4RA05045B.
- Y. Deng, J. Fan, S. Zhou, T. Zhou, J. Wu, Y. Li, Z. Liu, M. Xuan and Y. Wua, Biomicrofluidics, 2014, 8(2), 024101, DOI:10.1063/1.4867241.
- D. J. Kinahan, S. M. Kearney, N. A. Kilcawley, P. L. Early, M. T. Glynn and J. Ducrée, PLoS One, 2016, 11, 1–13, DOI:10.1371/journal.pone.0155545.
- C. M. Moreira, S. V. Pereira, J. Raba, F. A. Bertolino and G. A. Messina, Clin. Chim. Acta, 2018, 486, 59–65, DOI:10.1016/j.cca.2018.07.016.
- S. Jampasa, W. Siangproh, K. Duangmal and O. Chailapakul, Talanta, 2016, 160, 113–124, DOI:10.1016/j.talanta.2016.07.011.
- O. M. Istrate, L. Rotariu, V. E. Marinescu and C. Bala, Sens. Actuators, B, 2016, 223, 697–704, DOI:10.1016/j.snb.2015.09.142.
- E. Omidinia, N. Shadjou and M. Hasanzadeh, IEEE Sens. J., 2014, 14, 1081–1088, DOI:10.1109/JSEN.2013.2292875.
- B. R. Adhikari, M. Govindhan and A. Chen, Sensors, 2015, 15, 22490–22508, DOI:10.3390/s150922490.
- R. Kour, S. Arya, S.-J. Young, V. Gupta, P. Bandhoria and A. Khosla, J. Electrochem. Soc., 2020, 167, 037555, DOI:10.1149/1945-7111/ab6bc4.
- J. Ping, Y. Wang, K. Fan, J. Wu and Y. Ying, Biosens. Bioelectron., 2011, 28, 204–209, DOI:10.1016/j.bios.2011.07.018.
- M. Eryiğit, E. Çepni, B. Kurt Urhan, H. Öztürk Doğan and T. Öznülüer Özer, Synth. Met., 2020, 268, 116488, DOI:10.1016/j.synthmet.2020.116488.
- Y. Li, I. Martens, K. C. Cheung and D. Bizzotto, Electrochim. Acta, 2019, 319, 649–656, DOI:10.1016/j.electacta.2019.07.004.
- R. Villalonga, S. Tachibana, R. Cao, H. L. Ramirez and Y. Asano, Biochem. Eng. J., 2006, 30, 26–32, DOI:10.1016/j.bej.2006.01.013.
- X. Xu, D. Ji, Y. Zhang, X. Gao, P. Xu, X. Li, C. C. Liu and W. Wen, ACS Appl. Mater. Interfaces, 2019, 11, 20734–20742, DOI:10.1021/acsami.9b05431.
- Y. Yoshimi and N. Ishii, Anal. Chim. Acta, 2015, 862, 77–85, DOI:10.1016/j.aca.2015.01.001.
- Y.-F. Hu, Z.-H. Zhang, H.-B. Zhang, L.-J. Luo and S.-Z. Yao, Talanta, 2011, 84, 305–313, DOI:10.1016/j.talanta.2011.01.010.
- S. M. Naghib, M. Rabiee and E. Omidinia, Int. J. Electrochem. Sci., 2014, 9, 2341–2353 CrossRef.
- N. Blau, J. B. Hennermann, U. Langenbeck and U. Lichter-Konecki, Mol. Genet. Metab., 2011, 104(Suppl), S2–S9, DOI:10.1016/j.ymgme.2011.08.017.
- H. O. de Baulny, V. Abadie, F. Feillet and L. de Parscau, J. Nutr., 2007, 137(6 Suppl 1), 1561S–1575S, DOI:10.1093/jn/137.6.1561S.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lc00912f |
| This journal is © The Royal Society of Chemistry 2025 |