
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrofluidic digital focus assays for the quantification of infectious influenza virus†
Siddharth Raghu
Srimathi
ab,
Maxinne A.
Ignacio
c,
Maria
Rife
c,
Sheldon
Tai
d,
Donald K.
Milton
bd,
Margaret A.
Scull
*bc and
Don L.
DeVoe
 *abe
*abe
aDepartment of Chemical and Biomolecular Engineering, University of Maryland, College Park, USA
bFischell Institute for Biomedical Devices, University of Maryland, College Park, USA
cDepartment of Cell Biology and Molecular Genetics, Maryland Pathogen Research Institute (MPRI), University of Maryland, College Park, USA
dDepartment of Global, Environmental, and Occupational Health, University of Maryland, College Park, USA
eDepartment of Mechanical Engineering, University of Maryland, College Park, USA
First published on 31st January 2025
Abstract
Quantifying infectious virus is essential for vaccine development, clinical diagnostics, and infectious disease research, but current assays are constrained by long turnaround times, high costs, and laborious procedures. To address these limitations, we present a digital focus assay employing an array of independent nanoliter cell cultures. The microfluidic platform allows cells in each nanowell to be inoculated with virus, followed by oil discretization to prevent cross-contamination. After incubation, infected cells are visualized through immunofluorescence staining, and a binary map of wells positive for viral antigen is generated by automated image analysis, allowing infectious viral titer to be calculated by statistical analysis. The platform requires significantly smaller sample and reagent volumes than conventional focus assays while enhancing assay automation and endpoint time flexibility. The technology is applied to the quantification of infectious influenza A using both model virus and clinical specimens, demonstrating the digital platform as an accurate, rapid, cost-effective, and convenient tool for viral load quantification with broad utility in clinical, pharmaceutical, and research applications.
Introduction
Quantification of viral titer is important for a wide range of applications in infectious disease research. For example, characterization of viral load over time is necessary for establishing infection kinetics,1–7 improving the fundamental understanding of disease progression by informing dynamic models of virus replication within host cells,8 and identifying host cellular factors required for viral replication.9,10 Viral titer quantification is also critical for epidemiology and viral transmission studies,11–17 characterizing the relative pathogenicity of viral strains,16 and evaluating the mechanism and efficacy of antiviral treatments.18–26 In addition, quantifying infectious virus is important in the development of novel vaccine candidates,27–30 testing newer methods of vaccine production,31–34 and evaluating vaccines for safety and efficacy.35–38 It also finds application in newer areas of research such as in the production of recombinant adeno-associated virus (rAAV)-based gene therapy, where the infectious titer of the AAV particles is important to determine the efficacy of the product and thereby the potency of AAV-based gene therapy.39–41 Measurements of viral titer can also play a role in clinical settings as an approach to assessing the severity of infection.42–44The most common methods used for virus quantification are nucleic acid amplification tests (NAATs) including polymerase chain reaction (PCR) and loop-mediated isothermal amplification (LAMP). These assays can confirm the presence of virus-specific nucleic acids,11,14,15,45–50 while serological assays like the enzyme-linked immunosorbent assay (ELISA) can be designed to test for viral antigens such as the influenza A nucleoprotein51–54 or measure the level of virus specific antibodies in that are produced in response to an infection or vaccination.55–57 However, immunoassays can yield false-negative results due to the limited window of detection58 and are less sensitive than NAATs.59 Furthermore, like NAATs, they only provide a correlation to the infectious viral load and cannot be used to determine if functional viral particles are present. This is notable, since in the case of influenza virus, less than 1% of the total virus particle count has been reported to be infectious46 and the infection generation rate might be 1/300th of the total number of viral copies detected by PCR.14,47,48
Unlike NAATs and immunoassays, cell-culture based assays can provide direct measurements of viable virus by quantifying individual cell inoculation events leading to virus expansion. The plaque assay is considered a gold standard60 that provides a simple and inexpensive approach to quantifying viable virus through the counting of dead cell plaques that form in a monolayer cell culture after viral inoculation.61,62 However, plaque assays suffer from several drawbacks including long assay times of at least 2–3 days, labor intensive operation, subjectivity in counting plaques, and poor performance when evaluating viruses that generate small and/or unclear plaques, or simply no plaques at all.63–65 Furthermore, plaque assays can be unreliable when other sources of cytotoxicity are present in the sample, such as in clinical settings where an individual may carry multiple agents capable of causing cytopathic effects (CPE) in host cells.
One alternative to the plaque assay is the median tissue culture infectious dose (TCID50) assay. This assay is also cell-culture based, but quantifies viable virus by determining the dose required to infect half of the inoculated wells.65–67 Still, as with plaque assays, TCID50 assays are time consuming, with a 3–5 day turnaround for influenza A.65,66 Thus, another option is the focus-forming assay (FFA), which is a variation of the plaque assay in which viable virus is quantified through the counting of foci of infected cells that are visualized after staining with a virus-specific antibody. In addition to providing higher specificity though the use of antibody labeling, the FFA offers higher sensitivity than plaque assays together with the ability to quantify non-lytic viruses.68 It is also considerably faster than a plaque assay (e.g. 8–24 hours69vs. 3 days70 in the case of influenza virus). However, the FFA assay cost is relatively high due to the fluorescent dyes and antibodies required for staining,68 and assay endpoint times must be optimized for a given virus strain and cell line, since shorter incubation times suffer from undercounting of foci71,72 while longer incubation times can lead to undercounting due to the merging of foci.72 In addition, while the FFA readout analysis can be automated through the use of specialized instrumentation, manual counting is typically employed, introducing subjectivity in distinguishing between small foci and visual artifacts.73
Here we report a focus-forming assay that is performed in a digital format that overcomes the key limitations of conventional FFAs to enable faster, less expensive, more convenient, and more reliable quantification of infectious virus. Digital assays employ large numbers of discrete sample volumes where dilute assay targets are distributed across these volumes. The target concentration within the initial sample can then be determined by evaluating the fraction of volumes containing at least one target and assuming a Poisson distribution across the volumes. Digital assays employing droplets or microwell arrays for sample discretization have been broadly successful for nucleic acid quantification, with microfluidic digital PCR74–76 and digital LAMP77,78 being widely used for absolute quantification of nucleic acids with high sensitivity. Taking inspiration from digital NAATs, the digital focus assay (dFA) described here discretizes a virus-containing sample into a microwell array culture platform, with viral titer determined by evaluating the fraction of wells where viral replication occurs. To prevent cross-talk between adjacent microwells, which would compromise assay readout,79 an immiscible oil phase is used to discretize the microwells.80–82 While a conventional FFA requires foci counting to be performed within an optimal time window to ensure sufficient virus expansion for detection while avoiding excessive growth and subsequent overlapping of individual foci, the discretized dFA chips allow viral titer to be calculated through binary readout from individual wells after extended replication, with indirect quantification based on statistical analysis. As with a conventional FFA, the dFA was found to be significantly faster than a plaque assay while providing a high level of specificity to the virus through immunofluorescence staining and removing the requirement for the virus to cause CPE in the host cells. The microfluidic architecture of the dFA serves to reduce reagent and sample consumption by approximately 10-fold, reducing reagent costs compared with conventional FFAs while also making the platform compatible with highly limited sample volumes. Significantly, the digital nature of the platform isolates discrete infection events, allowing flexible incubation times without concern over excessive growth of foci that would otherwise add uncertainty to assay readout. Furthermore, the assay process including sample introduction and immunostaining is convenient and straightforward, and image analysis for digital readout is readily automated without the need for specialized instrumentation. Performance of the dFA platform is investigated for the quantification of influenza A virus, with viral titer measurements found to be in good agreement with a paired plaque assay making it a feasible, rapid, convenient, and cost-effective platform for the quantification of viable virus.
Experimental
Device design and fabrication
Devices were fabricated through PDMS soft lithography using multi-layered photoresist molds prepared by adapting a reported technique.83 Briefly, a 110 μm thick layer of SU-8 2075 photoresist (Kayaku Advanced Materials) was spin coated on a 4′′ silicon wafer (University Wafer). The coated wafer was soft-baked and the design corresponding to the channels and the pillar arrays for mixing were exposed using a maskless aligner (MLA 150, Heidelberg Instruments). Following a short post-exposure bake at 65 °C for 60 s, a second layer of 110 μm SU-8 was spin coated over the initial SU-8 layer. The spin speed employed for the second SU-8 film was 1.2 times higher than the spin speed employed on the bare silicon surface.83 The wafer was then soft-baked at 65 °C for 5 min followed by 95 °C for 20 min before mask alignment and UV exposure to pattern the microwell array. Following a post-exposure bake, development, and a hard-bake the final multi-layer SU-8 mold was ready for PDMS soft lithography. A separate single-layer SU-8 mold containing alignment marks for fluidic access ports was prepared using the above steps for fabrication of the sealing layer.PDMS (Sylgard 184, Dow) was prepared by mixing the base prepolymer and the crosslinker (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 weight ratio). Next, 1 μL of saturated Pluronic F-108 (Sigma Aldrich) solution in ethanol (200 mg mL−1) was added to every 1 g of the PDMS prepolymer and mixed together to support hydrophilic modification of the final PDMS surfaces.84 The mixture was degassed and then poured onto the multi-layered SU-8 microwell mold. It was cured at 80 °C for 3 h and peeled from the mold. Blocks corresponding to a single device were cut and bonded to a 1′′ × 3′′ glass slide (AmScope) through oxygen plasma activation of the surfaces at 200 W for 30 s (PE-25, Plasma Etch). Similarly, cured PDMS blocks for the enclosing layer were cut and holes punched for fluidic access using a 0.5 mm ID biopsy punch tool (World Precision Instruments), and bonded to the base PDMS layer through oxygen plasma activation. Sterile 24 gauge blunt-tip needle segments (SAI Infusion Technologies) were inserted into the fluidic access holes to enable the connection of flexible Tygon tubing (0.020′′ ID × 0.060′′ OD, Masterflex) for interfacing between the chip and a syringe pump (Fusion 100, Chemyx) for injection of fluids into the chip using 1 mL syringes (BD Luer-Lok).
:1 weight ratio). Next, 1 μL of saturated Pluronic F-108 (Sigma Aldrich) solution in ethanol (200 mg mL−1) was added to every 1 g of the PDMS prepolymer and mixed together to support hydrophilic modification of the final PDMS surfaces.84 The mixture was degassed and then poured onto the multi-layered SU-8 microwell mold. It was cured at 80 °C for 3 h and peeled from the mold. Blocks corresponding to a single device were cut and bonded to a 1′′ × 3′′ glass slide (AmScope) through oxygen plasma activation of the surfaces at 200 W for 30 s (PE-25, Plasma Etch). Similarly, cured PDMS blocks for the enclosing layer were cut and holes punched for fluidic access using a 0.5 mm ID biopsy punch tool (World Precision Instruments), and bonded to the base PDMS layer through oxygen plasma activation. Sterile 24 gauge blunt-tip needle segments (SAI Infusion Technologies) were inserted into the fluidic access holes to enable the connection of flexible Tygon tubing (0.020′′ ID × 0.060′′ OD, Masterflex) for interfacing between the chip and a syringe pump (Fusion 100, Chemyx) for injection of fluids into the chip using 1 mL syringes (BD Luer-Lok).
Following fabrication, deionized water was perfused through the chip, and the water-filled device was placed under a vacuum for 2 min to displace air from the microwells to ensure complete filling of the microwells with water. The water-filled chip was then incubated in a refrigerator at 4 °C for at least 24 h to facilitate the migration and reorientation of Pluronic F-108 molecules within the PDMS matrix to present hydrophilic domains at the exposed surfaces. Because the PDMS was observed to gradually recover its hydrophobicity over time, all chips used in this work were stored under water at 4 °C to slow the surface recovery process, and cell culture was initiated within a week of device fabrication.
Cell seeding
The first step for performing the dFA was seeding of Madin–Darby canine kidney (MDCK) cells into the microwells. The MDCK cells were kindly provided as a gift from Dr. Wendy Barclay (Imperial College London). A 50 μL suspension (106 cells per mL) of MDCK cells in culture medium (DMEM, high glucose, pyruvate, Gibco) supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific) and 1× penicillin–streptomycin (Thermo Fisher Scientific) was injected into the device. The cells were allowed to sediment into the microwells over a period of 2 min with the flow stopped. This seeding process was repeated until the desired cell density was reached in the microwells. At the end of the cell seeding step, 50 μL of cell-free medium was flowed into the device to nudge any cells that had sedimented in the plateau region between the microwells into the nearest microwell. This step also ensured the removal of cells from other parts of the channel, leaving the cells contained only within the microwell array. The chips were subsequently placed overnight in an incubator at 37 °C with 5% CO2 to facilitate the adhesion and growth of the MDCK cells on the base and walls of the microwells.Virus inoculation and well discretization
A laboratory stock of the influenza A virus (A/Perth/16/2009(H3N2)), produced by plasmid transfection in 293T cells and subsequent coculture with MDCK cells, was serially diluted in serum-free DMEM giving 500 μL of the virus inoculum each at 10−2, 10−3, and 10−4 dilution factors. Prior to introducing the influenza virus inoculum, the chip was flushed with serum-free DMEM to remove all FBS from the microwells. A 60 μL suspension of virus inoculum at each dilution factor was injected into each chip and incubated at 37 °C with 5% CO2 to facilitate the adsorption and attachment of the influenza virus to the MDCK cells. After 2 h incubation, an overlay media consisting of either maintenance media (MM) (DMEM/Nutrient Mixture F-12 Ham with L-glutamine, 15 mM HEPES, and sodium bicarbonate, Sigma Aldrich) containing 1.5 μg mL−1 TPCK-treated trypsin (Thermo Fisher Scientific) or 0.6% microcrystalline cellulose (MCC) (Sigma Aldrich) in the aforementioned MM was perfused through the chip at a flow rate of 100 μL min−1. In addition to providing an appropriate environment for cell culture, the overlay flow served to facilitate removal of any unbound virus from the upper plateau region of the microwell chip. Mineral oil (Light mineral oil, Sigma Aldrich) was next introduced at a flow rate of 50 μL min−1 into the chip to discretize the microwells, while retaining the overlay medium in each microwell owing to the hydrophilic modification through the embedded Pluronic. The oil-discretized chip was then placed in an incubator at 37 °C with 5% CO2 for an additional 22 h to support replication and spread of the progeny influenza virus within individual microwells.Fixing and staining
At the end of the infection incubation, the chips were flushed with phosphate-buffered saline (PBS) to remove all oil and overlay medium. To visualize infected cells, 60 μL of 4% paraformaldehyde (PFA) (Electron Microscopy Sciences) in PBS was flowed into each chip and incubated for 10 min to fix the cells. The PFA was flushed out by flowing PBS + 0.1% Tween 20 (PBS-T) into the chip. The PBS-T also helped to remove any residual mineral oil from the channels. The cells were then permeabilized by flowing 2.5% Triton-X in PBS to facilitate detection of intracellular targets. Cells were incubated for 15 min and then washed by flowing PBS into the chip. Blocking was performed by flowing 3% bovine serum albumin (BSA) (Sigma Aldrich) in PBS, preventing non-specific binding and thereby reducing background noise in the subsequent staining steps. After incubating for 1 h at room temperature, the blocking solution was flushed out by flowing PBS into the chip. A 60 μL volume of primary antibody (mouse anti-influenza A antibody, nucleoprotein, clones A1, A3 Blend, Sigma Aldrich) diluted 1:100 in PBS with 1% BSA was introduced into the chip and incubated at 4 °C overnight. Following a thorough wash with PBS, secondary antibody (donkey anti-mouse IgG, Alexa Fluor 488, Thermo Fisher) diluted 1:500 in PBS was flowed through the chip and incubated at room temperature for at least 1 hour. Following a thorough wash with PBS, 1 μg mL−1 of nuclear stain (Hoechst 33258, pentahydrate (bis-benzimide), Thermo Fisher) was flowed into the chip and incubated for 10 min before thoroughly washing away unbound dye with PBS. The chip was then visualized using epifluorescence microscopy (Nikon ECLIPSE Ti2-E) and images of the full microwell array were captured (pco.edge 4.2 bi USB sCMOS Camera).
Image analysis to determine the distribution of cells across the microwells through the Hoechst nuclear stain, as well as the distribution of viral antigen-positive cells through immunofluorescent staining, was automated using custom Python code. For final assay readout, raw fluorescence microscopy images were converted to an 8 bit grayscale format and divided into regions of interest (ROIs) aligned with the 13 × 13 array of microwells. A pixel intensity threshold was applied to each ROI to remove background fluorescence, and regions with at least 10 contiguous pixels above the threshold value were interpreted as a positive infection event.
Clinical specimen analysis
Nasal swab samples were collected from a volunteer donor, participating in a clinical research study approved by the University of Maryland Baltimore Institutional Review Board (HP-00103865), with an acute influenza A infection, confirmed by Cepheid Xpert Xpress CoV-2/Flu/RSV plus tests. Two samples were collected on subsequent days and separated into two fractions, with one fraction used for dFA assays and the second fraction reserved for digital PCR (dPCR) analysis. The first fraction of each sample was run independently on 3 replicate dFA chips. Cell seeding in the dFA chips followed the same procedure used for the model virus experiments, but with MDCK cells replaced with humanized MDCK (hCK) cells that are genetically engineered to express higher levels of human influenza virus receptors, making them more susceptible to infection by currently circulating wild-type influenza virus.85 Influenza A viral loads in clinical specimens were measured using the QIAcuity One dPCR system (Qiagen). The reactions were set up using the QIAcuity OneStep Advanced Probe Kit (Qiagen), influenza A virus-specific primers and probes,86 and nucleic acids freshly extracted using the MagMAX™ Pathogen RNA/DNA Kit (Applied Biosystems), following manufacturers' instructions. After reactions were distributed into QIAcuity 26k Nanoplates (Qiagen), the plates were incubated at 50 °C for 40 min, followed by 95 °C for 2 min and 45 cycles of 95 °C for 5 s and 55 °C for 30 s. The QIAcuity One instrument imaged the nanoplates after thermocycling completed and calculated RNA copy numbers in the reaction based on the number of positive partitions, which were back calculated to RNA copies per mL in the original sample. Multiple dilutions of the extracted nucleic acid were included to ensure at least one falls within the dynamic range of the dPCR assay.Results
Efficient discretization and uniform flow are enabled by the dFA chip design
The dFA chips were fabricated using two layers of polydimethylsiloxane (PDMS) patterned by soft lithography,87 and bonded together to form an enclosed microwell array and fluidic channel for cell, sample, and reagent delivery. Each chip contained a 13 × 13 array of square wells with 200 μm width, 120 μm depth, and 50 μm spacing between adjacent wells (Fig. S1†). The enclosing channel, centered around the array, had a height of 120 μm spanning 38 mm in length and 11 mm in width. The selection of PDMS as the chip substrate material was motivated by the high gas permeability,87 excellent biocompatibility,88–90 and low toxicity88,90 of PDMS, making it a suitable material to support on-chip cell culture. The high optical transparency87,90 and low auto-fluorescence91 of PDMS also ensures efficient assay readout from the fabricated devices. However, because PDMS is a hydrophobic material, the chips required hydrophilic surface modification to enable effective well filling, discretization, and cell attachment. While PDMS can be temporarily rendered hydrophilic by oxygen plasma treatment,92,93 the modified surfaces quickly revert to their native hydrophobic state. Thus, to provide a more robust hydrophilic modification, an embedded Pluronic block copolymer strategy was utilized.84,94 By mixing Pluronic surfactant with PDMS prepolymer during dFA chip fabrication, surfactant molecules at the PDMS surface tend to expose their hydrophilic chain segments while embedding their hydrophobic segments within the PDMS bulk, as summarized in Fig. S2.† When attempting to discretize culture media in dFA chips fabricated from native PDMS without the addition of Pluronic, the substrate surface energy was insufficient to prevent the oil phase from lubricating the PDMS surface, resulting in droplet formation and removal of media from the wells. In contrast, when combined with oxygen plasma activation to promote bonding of the PDMS layers during chip fabrication, the Pluronic-modified chips yielded consistent and complete retention of culture media within the microwells when flowing oil in the enclosing channel to discretize the wells.The dFA chips were designed with square arrays of 169 identical microwells residing within an open chamber aligned to the wells through the use of the multilayer mold fabrication process. While early chips were fabricated by manually aligning and bonding two patterned layers of PDMS, one containing the microwells and the other containing the channel structure, the final self-alignment process was found to prevent asymmetry within the flow field and avoid maldistribution of cells across the microwell array (data not shown). To further improve the seeding uniformity across the array, COMSOL Multiphysics simulations of the flow of cell suspensions through various channel geometries and mixing structures were performed. These simulations (Fig. S3†) revealed that the cylindrical pillar array serves to disrupt the initial streamlines in the upstream cell delivery channel, thereby improving the lateral dispersion of cells within the chamber. Similarly, chambers with symmetric pillar arrays at the inlet and outlet, together with designs employing a constant wall curvature, were also found to enhance seeding uniformity within the array. Each of these modifications was validated experimentally before selecting the chip design featuring a pair of micropillar arrays located at the inlet and outlet of the chamber (Fig. S3D†), for all further experiments. In the absence of these pillars, cells were observed to preferentially flow along the centerline of the channel, leading to lower cell seeding density within microwells located farther from the centerline (data not shown).
MDCK cells form confluent, isolated monolayers within the dFA platform
To next prepare the chip for virus introduction, a suspension of MDCK cells was perfused into the chip (Fig. 1A) to fill the microwell array chamber, after which the flow was halted and cells were allowed to sediment into the microwells and adhere to the well surfaces. Notably, the combined Pluronic and oxygen-plasma based hydrophilic modification of the PDMS surface was found to be effective in promoting cell adhesion, eliminating the need for complicated patterning and coating of extracellular matrix protein like collagen and fibronectin to support microwell culture.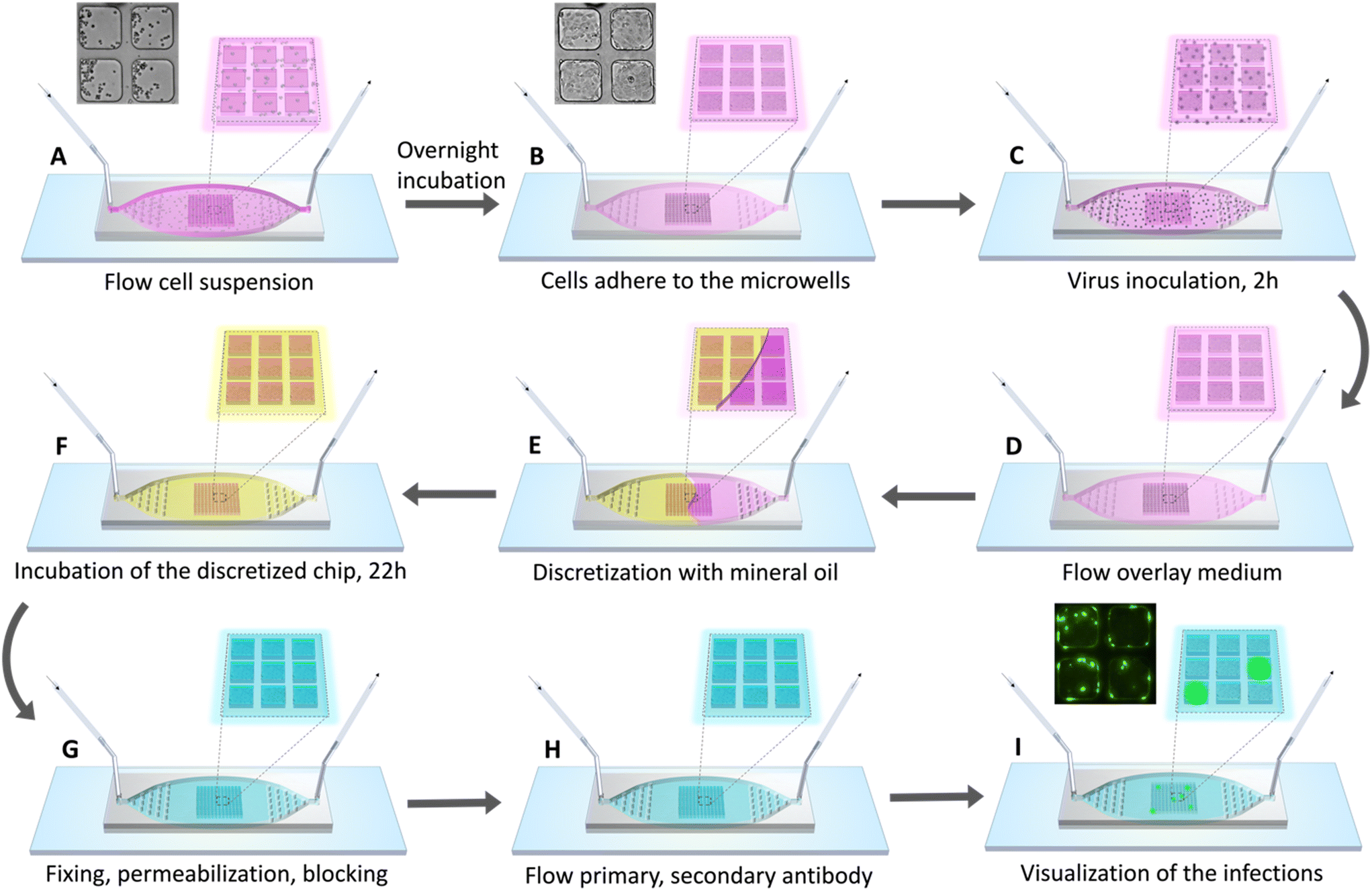 | ||
| Fig. 1 Digital focus assay process. (A) As MDCK cell suspension flows into the chip, the cells sediment into the microwells and adhere to the walls after incubation at 37 °C with 5% CO2. (B) Overnight incubation resulted in nearly confluent MDCK cell growth within individual microwells. (C) Influenza A virus inoculum is introduced into the chip and incubated for 2 h. (D) Overlay medium is introduced into the chip that flushed out any unadsorbed virus from the chip. (E) Mineral oil is introduced into the chip to discretize the microwells and prevent cross-contamination between their volumes. (F) Discretized microwell arrays containing the cultured MDCK cells inoculated with influenza A virus and filled with an overlay medium are incubated for 22 h. (G–I) The oil and overlay media are flushed out with PBS at the end of the assay to permit fixing, permeabilization, and immunofluorescent staining of the cells. | ||
The lateral well dimensions were selected to support approximately 30–50 MDCK cells within each well at confluence, and the well height was designed to ensure robust discretization while also trapping a sufficient volume of culture medium (4.8 nL) to maintain high cell viability during assay operation. Indeed, following overnight incubation, nearly confluent cell monolayers were achieved within the individual microwells (Fig. 1B and 2A). The distributions of cells for all 9 chips used for evaluation of the digital focus assay are displayed in Fig. 2B. As expected, most microwells contained between 30 and 50 cells 24 h after seeding. Specifically, 67.5% of the total microwells across the 9 chips contained between 30 and 50 cells while 84% of the microwells contained between 25 and 55 cells each. To better visualize the spatial distribution of the cells across the microwells, a heatmap of the distribution of cells per microwell in the 13 × 13 array averaged across all 9 chips along with the corresponding standard deviation of the distribution are presented in Fig. 2C and D.
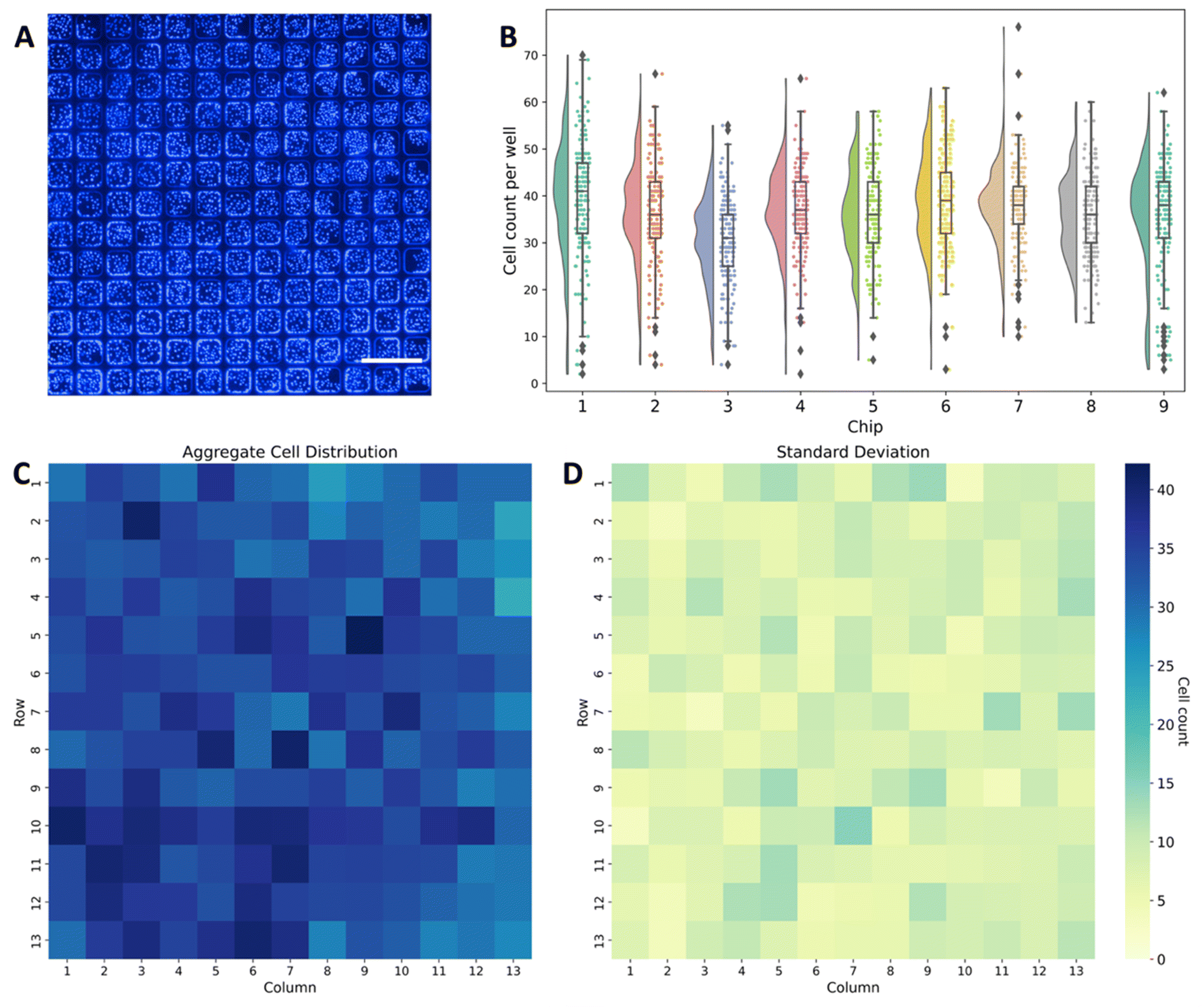 | ||
| Fig. 2 Distribution of MDCK cells. (A) Sample image displaying typical MDCK cell distribution across the 13 × 13 microwell array following cell seeding, expansion, and nuclear staining for visualization. Scale bar: 500 μm. (B) Distributions of cells within 9 individual chips. (C) Mean and (D) standard deviation for aggregate cell distributions within each array position across all 9 chips used for the digital focus assay with MM overlay. | ||
While any cells initially seeded onto the upper plateau of the microwell chip were found to be efficiently removed by shear forces during the flow of overlay media into the chip (data not shown), the hydrophilic surface modifications employed during device fabrication to promote effective microwell filling and cell attachment can potentially support migration of cells from the wells and onto the plateau following well discretization, leading to unwanted bridging of cells between adjacent wells during longer term culture. Bridging can potentially allow the contents of discretized microwells to enter neighboring wells, resulting in virus cross-contamination that may comprise assay accuracy. To quantify cell bridging experimentally, the bridging probability was defined as the total number of bridging events observed scaled by the total number of well gaps across which bridging may occur within a single array, with results calculated for assays performed with either MCC or MM overlays. As seen in Fig. 3, the average bridging probability is below 1% for chips prepared with MCC overlay regardless of the culture time, while chips prepared with MM overlay exhibited higher bridging at 8 h compared with 24 h. The lower initial bridging probability associated with the MCC overlay may be due to the media limiting the mobility of cells during the early stages of culture, while the decrease in bridging probability at 24 h for the MM overlay is presumably due to extended exposure of the cells in the plateau region to mineral oil, thereby reducing survival of these cells. While these results indicate that an MCC overlay may reduce cell migration during the early stages of culture, the minimal degree of bridging observed for each overlay at 24 h reveals that both MCC and MM are effective at preventing the migration of infected cells between adjacent microwells.
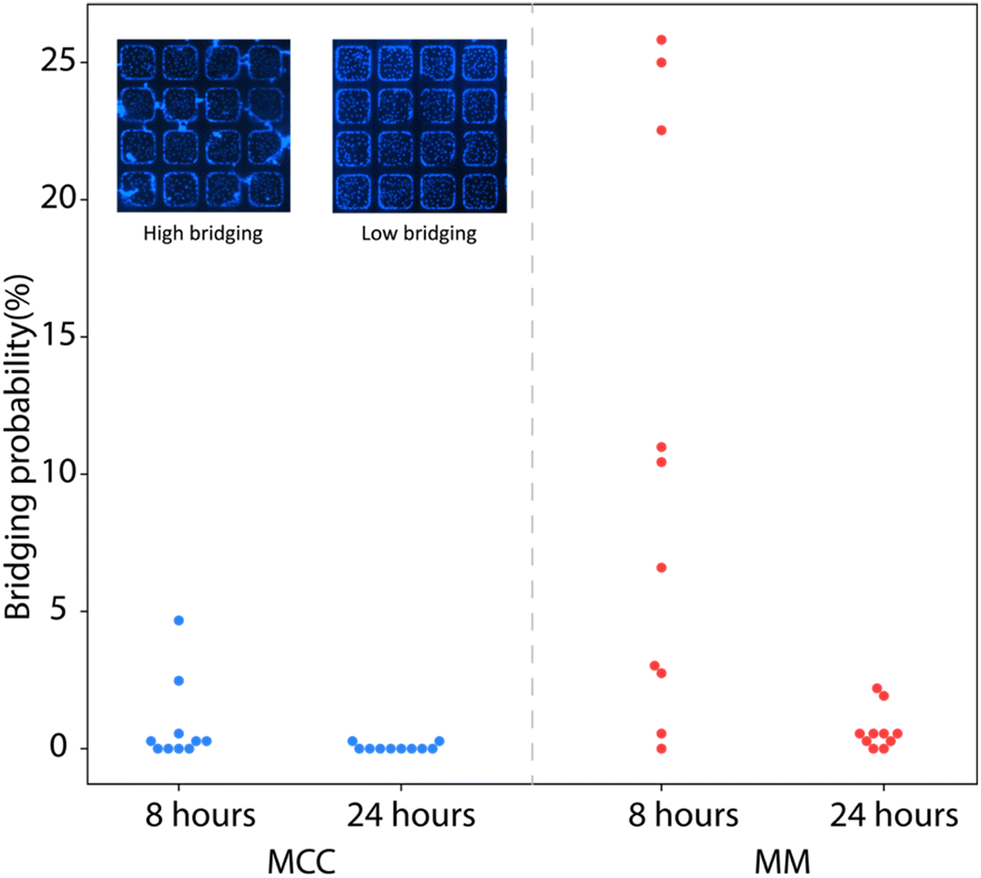 | ||
| Fig. 3 Impact of overlay medium on cell bridging across microwells. Quantification of cell bridging events between adjacent microwells for MCC and MM overlays at 8 h and 24 h time points. Bridging probability is defined as number of bridging events observed scaled by the total number of adjacent well gaps across which bridging may occur within a single array (n = 10 chips for each overlay). Insets of high and low bridging example cases are included. | ||
Infectious influenza A virus titers can be determined using the dFA platform
Following cell seeding, serial dilutions of an influenza A virus inoculum was then introduced into chips in parallel and incubated for 2 h to allow virus within the upper chamber to diffuse into the microwells for uptake by the MDCK cells (Fig. 1C). Residual virus was next removed from the system by flowing overlay media through the chip (Fig. 1D), followed by injection of mineral oil to discretize the wells (Fig. 1E). The chip was then incubated to achieve a total inoculation time of up to 24 h (Fig. 1F), including the initial incubation step.To image the resulting distribution of microwells containing infected cells, the oil and overlay media were flushed out with PBS to allow the introduction of reagents for fixing and permeabilizing the cells (Fig. 1G), and for immunofluorescence-mediated detection of influenza nucleoprotein within the wells (Fig. 1H and I). The flow-through architecture of the dFA chips allowed all fluid handling steps involved in fixing and staining the cells to be efficiently performed by manually swapping syringes containing different reagent solutions between each step, using a single syringe pump to control all injections with predictable and repeatable performance.
Fluorescence microscopy images of the 13 × 13 array with the MDCK cells stained for influenza A virus nucleoprotein were captured for all dilution factors. Image analysis was automated through a Python script designed to generate a binary map of infections across the array. The raw fluorescence images together with the mapping of positive wells at each dilution are presented in Fig. 4A.
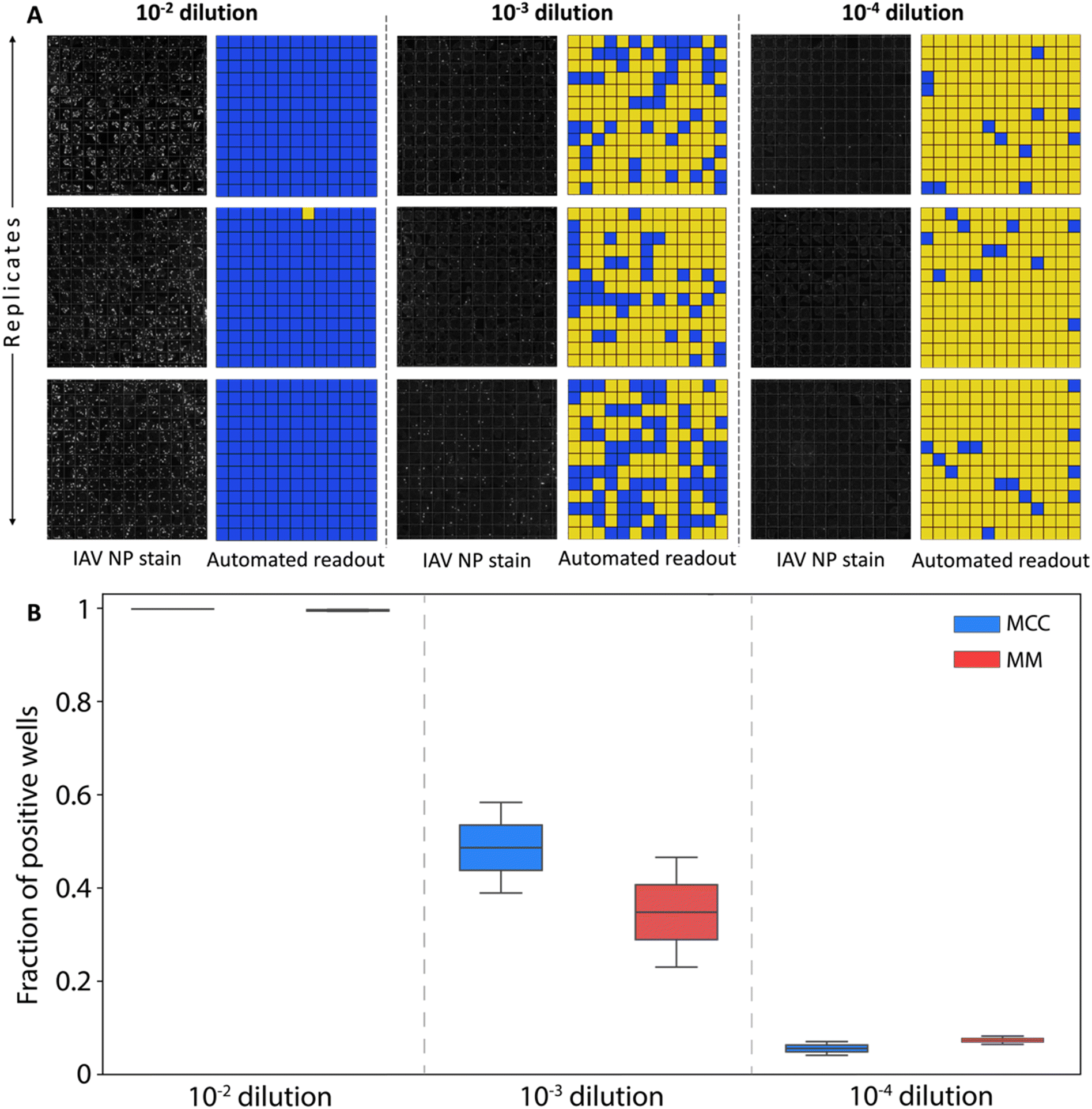 | ||
| Fig. 4 Digital assay readout. (A) Paired images of influenza A virus nucleoprotein stain and resulting binary readouts (yellow = negative, blue = positive) generated by automated image processing at increasing virus stock dilution factors of 10−2, 10−3, and 10−4 (MM overlay, 3 replicates for each case shown). (B) Microwell fractions exhibiting positive infection using MCC and MM overlays for each viral stock dilution. The error bars in the boxplots represent the variation in data between the replicate chips at each condition, with the box bounding the data between the first and third quartiles, while the whiskers on either end span 1.5 times the interquartile range. | ||
The distribution of the finite number of infectious virions in the sample among the microwells and the resulting infections can be approximated as a Poisson distribution, with the probability of n virions residing within a particular microwell given by,95
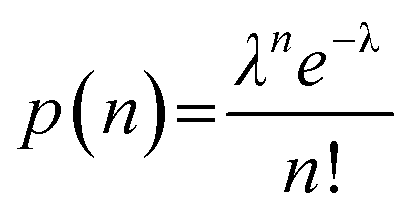 | (1) |
| p(0) = e−λ | (2) |
| λ = −ln(p(0)) | (3) |
λ = −ln(1 − ![[p with combining circumflex]](https://www.rsc.org/images/entities/i_char_0070_0302.gif) ) ) | (4) |
is the probability of a well containing at least one virus particle. Since the mean count of virions per microwell is simply the product of the virus concentration (c) and microwell volume (V), the viral titer of an unknown sample can be determined from eqn (4) as,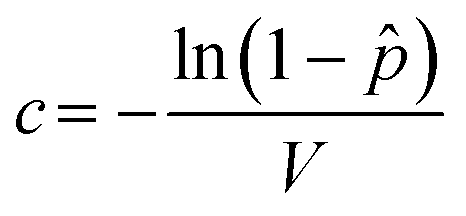 | (5) |
under the assumption that a single virion is capable of infecting a target cell and leading to replication within the well. Note that the effective well volume is the volume of sample from which virions are free to infect the MDCK cells during the initial inoculation event. To account for virus that can diffuse into the wells from the sample delivery channel, the effective well volume was calculated as the sum of the geometric volume of the microwell and the volume of the column in the enclosing channel directly over the microwell, assuming that the initial volume of culture media within each well is fully replaced by virus suspension during sample perfusion. Since the channel height is the same as the microwell depth, the effective volume of virus sample delivered to each well is twice the geometric well volume, and is calculated to be V = 9.6 nL.
Unlike a conventional focus assay, in which individual foci are counted, the digital assay introduces uncertainty in the quantification process due to statistical variability in virus distribution. The confidence interval for the predicted value of can be approximated for large values of n by:96
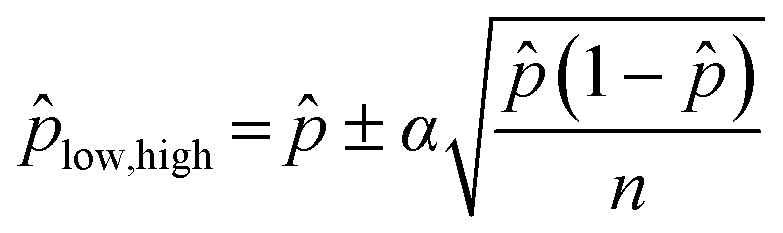 | (6) |
low and high in eqn (5).
For each experiment, the value of was determined from the ratio of positive to negative wells in the binary readout. Identical experiments were performed using both MCC and MM overlay media at each dilution factor to evaluate the impact of the overlay on assay performance. The resulting viral titers and confidence limits were calculated using eqn (5) and (6), with results presented in Table 1 and Fig. 4B.
| Assay format: | Digital focus assay | Plaque assay | |||||
|---|---|---|---|---|---|---|---|
| Dilution factor: | 10−2 | 10−3 | 10−4 | 10−6 | |||
| Overlay: | MCC | MM | MCC | MM | MCC | MM | Agar |
| Mean positive well fraction (): |
1.00 | 0.998 | 0.414 | 0.331 | 0.051 | 0.073 | — |
| Mean viral titer (PFU mL−1): | — | — | 5.9 × 107 | 4.4 × 107 | 5.5 × 107 | 7.9 × 107 | 5.6 × 107 |
| 95% confidence interval: | — | — | 4.8–6.4 × 107 | 3.6–4.8 × 107 | 3.4–7.6 × 107 | 5.4–10.5 × 107 | — |
At the lowest dilution factor of 10−2, the high virus concentration was sufficient to yield infected cells in all microwells for the majority of replicates, preventing the viral titer from being determined using Poisson statistics. At dilution factors of both 10−3 and 10−4, positive well fractions were within a range that enabled virus concentration to be estimated (Fig. 5). Good agreement was found between the resulting values and the reference titer determined through a plaque assay, with average titer predicted from all assays falling within 10% of the reference value. Further, no clear difference was observed between assays utilizing either MCC or MM overlays.
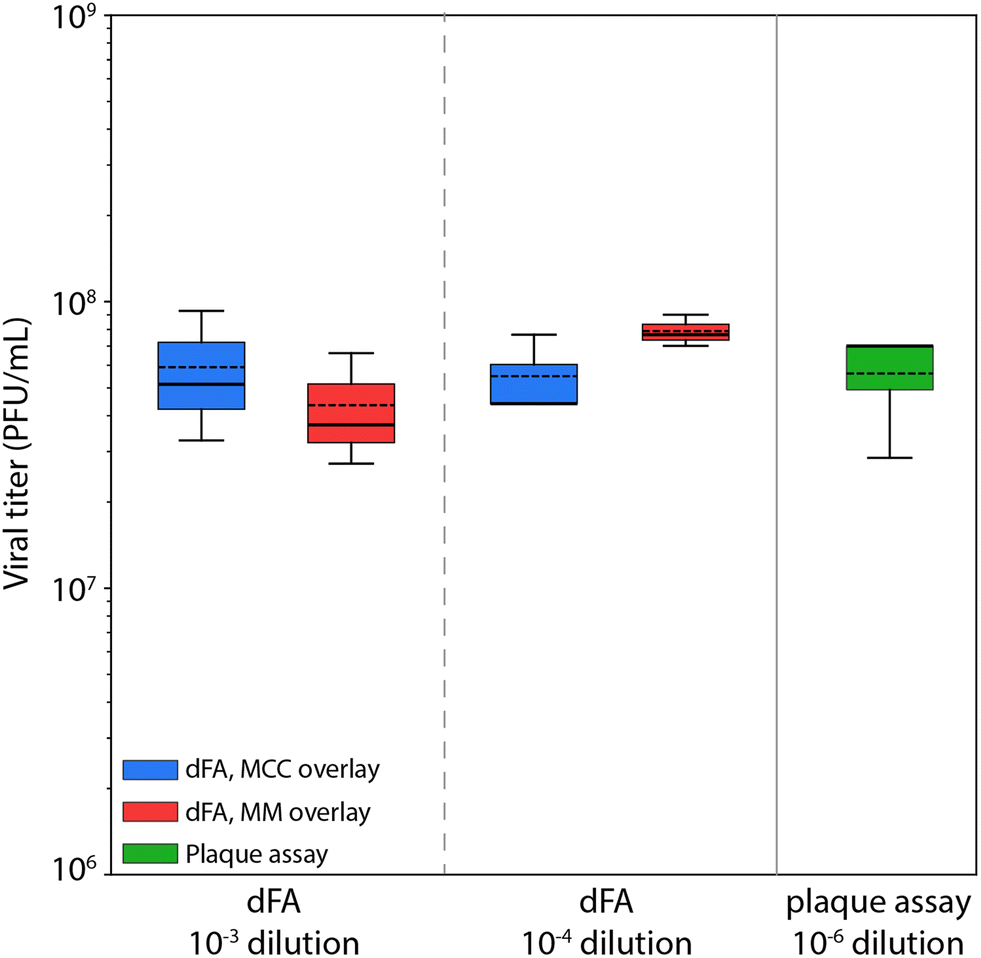 | ||
| Fig. 5 Viral titers determined from dFA and plaque assay. Predicted viral titers determined by the digital focus assay when utilizing different initial dilution factors. Assay results are presented for both MCC and MM overlays, and compared with the readout from a plaque assay for the same viral stock. The error bars in the boxplots represent the variation in data between the replicate assays at each condition, with the box bounding the data between the first and third quartiles, while the whiskers on either end span 1.5 times the interquartile range. Solid and dashed lines represent median and mean viral titers, respectively. A Z-test was applied to compare each dFA distributions against the reference plaque assay, yielding Z-scores between 0.07–1.5. | ||
The dFA platform is compatible with clinical specimens
Following validation with a model H3N2 virus, the dFA platform was evaluated using clinical specimens collected as part of an ongoing influenza transmission study. Samples were collected from a single donor on two subsequent days for characterization by dFA. Aliquots of each sample were also reserved for matching dPCR analysis. The first specimen collected on the day of admission into the study exhibited 100% average positive well counts from all three dFA replicates. This result corresponds to a minimum titer of 5.9 × 105 PFU mL−1, but does not allow an upper bound for the titer to be specified. The second specimen collected on the subsequent day yielded an average positive well count of 67.7%, resulting in a predicted viral titer of 1.2 × 105 PFU mL−1. Quantification by dPCR indicated that these specimens contained 3.9 × 108 and 1.3 × 107 influenza A virus RNA copies per mL, respectively.Discussion
Various cell culture-based methods currently exist to quantify infectious virus. Compared with these conventional assays, the dFA offers several advantages. The microfluidic architecture of the device, for example, serves to simplify assay implementation, while the small volume of the combined microwell array and fluid delivery channel (approximately 30 μL) minimizes the quantities of both sample and reagents required for assay operation. Multiple chips can be easily run in parallel to increase the number of microwells if desired, or alternatively, the microwell design can be modified to increase the array density to improve readout confidence with only a minor increase in reagent consumption.An important advantage of the dFA compared to a traditional focus assay is its flexible assay endpoint time. With a traditional FFA, short endpoint times (≤8 h for IAV with MDCK cells) yield foci that are composed of single infected cells, making it hard to distinguish virus expansion from other visual artifacts. Moreover, not all infectious foci are visible at short endpoint times,71,72 making the readout unreliable. On the other hand, at long endpoint times (>24 h for IAV with MDCK cells), excessive proliferation of progeny virus risks readout inaccuracy due to the merging of neighboring foci. As a result, focus assay times must be optimized for specific viruses, cells, and culture conditions. In contrast, in the absence of well bridging or cell death, the dFA has no strict upper bound on assay endpoint time, since the volume of each microwell is physically isolated by the overlay media, preventing readout integrity from being compromised through crosstalk between the microwells.
The dFA platform also simplifies the process of assay readout. In a traditional FFA, large areas within each well of a 24 or 96 well plate must be manually scanned under a microscope, a laborious process that can introduce subjectivity in quantification. Automated foci counting is possible with specialized instruments, but these tools are cost prohibitive for many end users. In contrast, the dFA only requires imaging across the small microwell array area, with a binary readout that supports robust automation through simple image processing code without the need to determine the precise number of infection events within each well.
While the chip design was optimized to reduce variability in cell seeding density, differences in the number of cells initially delivered to each microwell should not significantly affect assay readout. In our experiments, between 30 and 50 cells were typically observed in each microwell following the overnight cell expansion step, providing sufficient cell density to allow infectious virus to diffuse to at least one cell during the initial 2 hour incubation window, noting that the diffusion time scale for influenza virus in a 200 μm microwell is calculated from the Stokes–Einstein equation to be approximately 75 min. For microwells exhibiting either limited cell expansion or cell loss during reagent perfusion, the selected wells may simply be omitted from analysis to prevent bias in the digital quantification process.
Assay results for the nasal swab samples demonstrate that the dFA can successfully be applied to clinical specimens containing contemporary influenza strains. Unfortunately, the low sample volumes available for these clinical specimens prevented us from running the assay at multiple dilution levels to establish suitable dilutions for optimal quantification of infectious virus within each sample. As a result, the exceptionally high titer for the first sample prohibited a quantitative comparison between the assays. Regardless, consistent quantitative results were seen across all replicates for the lower titer sample, and a positive correlation between measured titers and RNA copy numbers between both clinical specimens was observed.
Opportunities remain for further optimization of the dFA platform. For our current chip design with 169 wells, the confidence interval range increases dramatically as the positive well fraction approaches the lower ( → 0) and upper ( → 1) limits and the confidence interval limits remain within ±50% of the mean concentration value when 0.10 < < 0.96. Depending on the required assay range and precision, it may be desirable to increase the number of microwells in the array in order to reduce measurement uncertainty and extend the limits of detection. For the current microwell array design, the theoretical range of detection for the dFA chip that maintains a ±50% confidence interval with 0.10 < < 0.96 is between 1.1 × 104 and 3.4 × 105 PFU mL−1. In contrast, by combining data from 3 chips yielding a total of 507 wells, the same confidence interval limits are achieved for a significantly wider range of positive well fractions given by 0.03 < < 0.99. In this case, the detection range is extended to between 3.2 × 103 and 4.8 × 105 PFU mL−1. Enhanced detection limits can alternately be achieved by increasing the well volume through modification of the microwell geometry.
Further, in its current implementation, the tubing connecting the syringe pump to the chip introduces significant dead volume, resulting in unnecessary reagent waste. In the present study, the dead volume associated with the inlet tubing was approximately 3× higher than the full on-chip volume. Modifying the apparatus to employ pneumatic-based flow control rather than displacement-based control using a syringe pump may further reduce reagent waste, allowing reagents to be directly pipetted into the chip inlet port prior to pneumatic injection into the microwell array, thereby eliminating the need for added tubing. Modifications to the integrated format of the microfluidic device can also deliver benefits the characterization of higher biosafety-level pathogens where the elimination of sharps is desirable to reduce operator risk. For example, the use of sharps can be avoided by replacing the glass substrate used as a rigid chip support with a thermoplastic layer, while the blunt needle interface used for the delivery of fluids into the chip may be replaced with an elastomeric pipette interface to support manual or automated media, sample, and reagent perfusion. Similarly, the compact chip format can allow assays to be operated in an enclosed chamber or holder to reduce the risk of bioaerosol formation.
With its low sample and reagent volume requirements, simple assay implementation, and flexible endpoint timing, together with its small form factor, scalable format, and compatibility with any suitable fluorescence microscope for assay readout, the dFA technology is a cost-effective and accessible alternative to traditional focus assays that can also be adapted for use with other adherent cell lines and pathogens, making it a promising platform for a range of infectious disease research and diagnostic applications.
Data availability
The data supporting this article have been included as part of the main text and figures. Additional details including quantitative measurements and imaging results will be made available upon reasonable request.Author contributions
Conceptualization: DLD; methodology: SRS, DLD, MAS; investigation: SRS, MAI, MR; resources: ST, DKM; supervision: DLD, MAS; funding acquisition: DKM, MAS, DLD; writing – original draft: SRS, DLD, MAS; writing – review & editing: all authors.Conflicts of interest
The authors declare no competing interests. DKM is a scientific advisor and reports stock options in Lumen Bioscience, Inc.Acknowledgements
The reverse genetics system for A/Perth/16/2009(H3N2) was a generous gift from Jesse Bloom with the Fred Hutchinson Cancer Research Center. The hCK cells used for viral culture from clinical specimens were kindly provided by Yoshihiro Kawaoka with the University of Wisconsin-Madison. This research was supported by the National Institutes of Health cooperative agreement 1U19AI162130 (DKM, MAS, DLD).References
- A. M. Smith and A. S. Perelson, Wiley Interdiscip. Rev.:Syst. Biol. Med., 2011, 3, 429–445 CAS.
- H. Miao, J. A. Hollenbaugh, M. S. Zand, J. Holden-Wiltse, T. R. Mosmann, A. S. Perelson, H. Wu and D. J. Topham, J. Virol., 2010, 84, 6687–6698 CrossRef CAS PubMed.
- L. Canini and F. Carrat, J. Virol., 2011, 85, 2764–2770 CrossRef CAS PubMed.
- R. A. Saenz, M. Quinlivan, D. Elton, S. MacRae, A. S. Blunden, J. A. Mumford, J. M. Daly, P. Digard, A. Cullinane, B. T. Grenfell, J. W. McCauley, J. L. N. Wood and J. R. Gog, J. Virol., 2010, 84, 3974–3983 CrossRef CAS PubMed.
- K. A. Pawelek, G. T. Huynh, M. Quinlivan, A. Cullinane, L. Rong and A. S. Perelson, PLoS Comput. Biol., 2012, 8, e1002588 CrossRef CAS PubMed.
- C. Hadjichrysanthou, E. Cauët, E. Lawrence, C. Vegvari, F. De Wolf and R. M. Anderson, J. R. Soc., Interface, 2016, 13(119), 20160289 CrossRef PubMed.
- P. Baccam, C. Beauchemin, C. A. Macken, F. G. Hayden and A. S. Perelson, J. Virol., 2006, 80, 7590–7599 CrossRef CAS PubMed.
- Y. Sidorenko and U. Reichl, Biotechnol. Bioeng., 2004, 88, 1–14 CrossRef CAS PubMed.
- R. König, S. Stertz, Y. Zhou, A. Inoue, H. H. Hoffmann, S. Bhattacharyya, J. G. Alamares, D. M. Tscherne, M. B. Ortigoza, Y. Liang, Q. Gao, S. E. Andrews, S. Bandyopadhyay, P. De Jesus, B. P. Tu, L. Pache, C. Shih, A. Orth, G. Bonamy, L. Miraglia, T. Ideker, A. García-Sastre, J. A. T. Young, P. Palese, M. L. Shaw and S. K. Chanda, Nature, 2009, 463, 813–817 CrossRef PubMed.
- A. Karlas, N. MacHuy, Y. Shin, K. P. Pleissner, A. Artarini, D. Heuer, D. Becker, H. Khalil, L. A. Ogilvie, S. Hess, A. P. Mäurer, E. Müller, T. Wolff, T. Rudel and T. F. Meyer, Nature, 2010, 463, 818–822 CrossRef CAS PubMed.
- R. Tellier, J. R. Soc., Interface, 2009, 6(suppl_6), S783–S790 CrossRef PubMed.
- J. Yan, M. Grantham, J. Pantelic, P. J. B. De Mesquita, B. Albert, F. Liu, S. Ehrman and D. K. Milton, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 1081–1086 CrossRef CAS PubMed.
- B. J. Cowling, D. K. M. Ip, V. J. Fang, P. Suntarattiwong, S. J. Olsen, J. Levy, T. M. Uyeki, G. M. Leung, J. S. Malik Peiris, T. Chotpitayasunondh, H. Nishiura and J. Mark Simmerman, Nat. Commun., 2013, 4, 1–6 Search PubMed.
- P. Fabian, J. J. McDevitt, W. H. DeHaan, R. O. P. Fung, B. J. Cowling, K. H. Chan, G. M. Leung and D. K. Milton, PLoS One, 2008, 3, e2691 CrossRef PubMed.
- N. Nikitin, E. Petrova, E. Trifonova and O. Karpova, Adv. Virol., 2014, 2014(1), 859090 Search PubMed.
- A. M. Smith, F. R. Adler, J. L. McAuley, R. N. Gutenkunst, R. M. Ribeiro, J. A. McCullers and A. S. Perelson, PLoS Comput. Biol., 2011, 7, e1001081 CrossRef CAS PubMed.
- A. W. C. Yan, J. Zhou, C. A. A. Beauchemin, C. A. Russell, W. S. Barclay and S. Riley, Epidemics, 2020, 33, 100406 CrossRef PubMed.
- C. A. A. Beauchemin, J. J. McSharry, G. L. Drusano, J. T. Nguyen, G. T. Went, R. M. Ribeiro and A. S. Perelson, J. Theor. Biol., 2008, 254, 439–451 CrossRef CAS PubMed.
- A. Handel, I. M. Longini and R. Antia, PLoS Comput. Biol., 2007, 3, e240 CrossRef PubMed.
- M. Matrosovich, T. Matrosovich, J. Carr, N. A. Roberts and H.-D. Klenk, J. Virol., 2003, 77, 8418–8425 CrossRef CAS PubMed.
- R. B. Moss, C. Hansen, R. L. Sanders, S. Hawley, T. Li and R. T. Steigbigel, J. Infect. Dis., 2012, 206, 1844–1851 CrossRef CAS PubMed.
- Y. Furuta, K. Takahashi, Y. Fukuda, M. Kuno, T. Kamiyama, K. Kozaki, N. Nomura, H. Egawa, S. Minami, Y. Watanabe, H. Narita and K. Shiraki, Antimicrob. Agents Chemother., 2002, 46, 977–981 CrossRef CAS PubMed.
- F. G. Hayden, K. M. Cote and R. G. Douglas, Antimicrob. Agents Chemother., 1980, 17, 865–870 CrossRef CAS PubMed.
- E. J. Mifsud, F. G. Hayden and A. C. Hurt, Antiviral Res., 2019, 169, 104545 CrossRef CAS PubMed.
- C. J. Dunn and K. L. Gon, Drugs, 1999, 58, 761–784 CrossRef CAS PubMed.
- N. Lee, P. K. S. Chan, D. S. C. Hui, T. H. Rainer, E. Wong, K. W. Choi, G. C. Y. Lui, B. C. K. Wong, R. Y. K. Wong, W. Y. Lam, I. M. T. Chu, R. W. M. Lai, C. S. Cockram and J. J. Y. Sung, J. Infect. Dis., 2009, 200, 492–500 CrossRef PubMed.
- J. Herrera-Rodriguez, T. Meijerhof, H. G. Niesters, G. Stjernholm, A. O. Hovden, B. Sørensen, M. Ökvist, M. A. Sommerfelt and A. Huckriede, Virology, 2018, 515, 21–28 CrossRef CAS PubMed.
- Y. Hatta, D. Boltz, S. Sarawar, Y. Kawaoka, G. Neumann and P. Bilsel, Vaccine, 2018, 36, 5097–5103 CrossRef CAS PubMed.
- M. C. Eichelberger, D. M. Morens and J. K. Taubenberger, Curr. Opin. Immunol., 2018, 53, 38–44 CrossRef CAS PubMed.
- V. Wacheck, A. Egorov, F. Groiss, A. Pfeiffer, T. Fuereder, D. Hoeflmayer, M. Kundi, T. Popow-Kraupp, M. Redlberger-Fritz, C. A. Mueller, J. Cinatl, M. Michaelis, J. Geiler, M. Bergmann, J. Romanova, E. Roethl, A. Morokutti, M. Wolschek, B. Ferko, J. Seipelt, R. Dick-Gudenus and T. Muster, J. Infect. Dis., 2010, 201, 354–362 CrossRef CAS PubMed.
- J. Liu, X. Shi, R. Schwartz and G. Kemble, Vaccine, 2009, 27, 6460–6463 CrossRef CAS PubMed.
- A. Le Ru, D. Jacob, J. Transfiguracion, S. Ansorge, O. Henry and A. A. Kamen, Vaccine, 2010, 28, 3661–3671 CrossRef CAS PubMed.
- Y. Genzel, C. Dietzsch, E. Rapp, J. Schwarzer and U. Reichl, Appl. Microbiol. Biotechnol., 2010, 88, 461–475 CrossRef CAS PubMed.
- M. G. Pau, C. Ophorst, M. H. Koldijk, G. Schouten, M. Mehtali and F. Uytdehaag, Vaccine, 2001, 19, 2716–2721 CrossRef CAS PubMed.
- P. M. Mendelman, J. Cordova and I. Cho, Vaccine, 2001, 19, 2221–2226 CrossRef CAS PubMed.
- R. A. Karron, K. Talaat, C. Luke, K. Callahan, B. Thumar, S. DiLorenzo, J. McAuliffe, E. Schappell, A. Suguitan, K. Mills, G. Chen, E. Lamirande, K. Coelingh, H. Jin, B. R. Murphy, G. Kemble and K. Subbarao, Vaccine, 2009, 27, 4953–4960 CrossRef CAS PubMed.
- A. Hawksworth, R. Lockhart, J. Crowe, R. Maeso, L. Ritter, O. Dibben and H. Bright, Vaccine, 2020, 38, 4209–4218 CrossRef CAS PubMed.
- K. Debbink, J. T. McCrone, J. G. Petrie, R. Truscon, E. Johnson, E. K. Mantlo, A. S. Monto and A. S. Lauring, PLoS Pathog., 2017, 13, e1006194 CrossRef PubMed.
- A. François, M. Bouzelha, E. Lecomte, F. Broucque, M. Penaud-Budloo, O. Adjali, P. Moullier, V. Blouin and E. Ayuso, Mol. Ther.--Methods Clin. Dev., 2018, 10, 223–236 CrossRef PubMed.
- A.-L. Shiau, P.-S. Liu and C.-L. Wu, J. Virol., 2005, 79, 193–201 CrossRef CAS PubMed.
- N. Mittereder, K. L. March and B. C. Trapnell, J. Virol., 1996, 70, 7498–7509 CrossRef CAS PubMed.
- N. Lee, P. K. S. Chan, D. S. C. Hui, T. H. Rainer, E. Wong, K. W. Choi, G. C. Y. Lui, B. C. K. Wong, R. Y. K. Wong, W. Y. Lam, I. M. T. Chu, R. W. M. Lai, C. S. Cockram and J. J. Y. Sung, J. Infect. Dis., 2009, 200, 492–500 CrossRef PubMed.
- L. L. H. Lau, B. J. Cowling, V. J. Fang, K. H. Chan, E. H. Y. Lau, M. Lipsitch, C. K. Y. Cheng, P. M. Houck, T. M. Uyeki, J. S. Malik Peiris and G. M. Leung, J. Infect. Dis., 2010, 201, 1509–1516 CrossRef PubMed.
- D. R. Hijano, J. B. De Cardenas, G. Maron, C. D. Garner, J. A. Ferrolino, R. H. Dallas, Z. Gu and R. T. Hayden, PLoS One, 2019, 14, e0220908 CrossRef CAS PubMed.
- L. García-Arroyo, N. Prim, N. Martí, M. C. Roig, F. Navarro and N. Rabella, J. Med. Virol., 2016, 88, 45–50 CrossRef PubMed.
- Z. Wei, M. Mcevoy, V. Razinkov, A. Polozova, E. Li, J. Casas-Finet, G. I. Tous, P. Balu, A. A. Pan, H. Mehta and M. A. Schenerman, J. Virol. Methods, 2007, 144, 122–132 CrossRef CAS PubMed.
- L. J. R. Van Elden, M. Nijhuis, P. Schipper, R. Schuurman and A. M. Van Loon, J. Clin. Microbiol., 2001, 39, 196–200 CrossRef CAS PubMed.
- B. Schweiger, I. Zadow, R. Heckler, H. Timm and G. Pauli, J. Clin. Microbiol., 2000, 38, 1552–1558 CrossRef CAS PubMed.
- M. Pan, J. A. Lednicky and C. Y. Wu, J. Appl. Microbiol., 2019, 127, 1596–1611 CrossRef CAS PubMed.
- O. Puhach, B. Meyer and I. Eckerle, Nat. Rev. Microbiol., 2022, 21, 147–161 Search PubMed.
- K. Leirs, P. Tewari Kumar, D. Decrop, E. Pérez-Ruiz, P. Leblebici, B. Van Kelst, G. Compernolle, H. Meeuws, L. Van Wesenbeeck, O. Lagatie, L. Stuyver, A. Gils, J. Lammertyn and D. Spasic, Anal. Chem., 2016, 88, 8450–8458 CrossRef CAS PubMed.
- D. S. Song, Y. J. Lee, O. M. Jeong, Y. J. Kim, C. H. Park, J. E. Yoo, W. J. Jeon, J. H. Kwon, G. W. Ha, B. K. Kang, C. S. Lee, H. K. Kim, B. Y. Jung, J. H. Kim and J. S. Oh, J. Vet. Sci., 2009, 10, 323–329 CrossRef PubMed.
- E. M. Zhou, M. Chan, R. A. Heckert, J. Riva and M. F. Cantin, Avian Dis., 1998, 42, 517–522 CrossRef CAS PubMed.
- G. F. de Boer, W. Back and A. D. M. E. Osterhaus, Arch. Virol., 1990, 115, 47–61 CrossRef CAS PubMed.
- A. Meijer, A. Bosman, E. E. H. M. Van De Kamp, B. Wilbrink, M. D. R. Van Beest Holle and M. Koopmans, J. Virol. Methods, 2006, 132, 113–120 CrossRef CAS PubMed.
- S. E. Ohmit, J. G. Petrie, R. T. Cross, E. Johnson and A. S. Monto, J. Infect. Dis., 2011, 204, 1879–1885 CrossRef CAS PubMed.
- J. C. Pedersen, Methods Mol. Biol., 2014, 1161, 11–25 CrossRef PubMed.
- S. Lu, S. Lin, H. Zhang, L. Liang and S. Shen, Micromachines, 2021, 12, 697 CrossRef PubMed.
- S. V. Vemula, J. Zhao, J. Liu, X. W. Xue, S. Biswas and I. Hewlett, Viruses, 2016, 8, 96 CrossRef PubMed.
- D. Juarez, K. C. Long, P. Aguilar, T. J. Kochel and E. S. Halsey, J. Virol. Methods, 2013, 187, 185–189 CrossRef CAS PubMed.
- K. Tobita, A. Sugiura, C. Enomoto and M. Furuyama, Med. Microbiol. Immunol., 1975, 162, 9–14 CrossRef CAS PubMed.
- C. R. Gaush and T. F. Smith, Appl. Microbiol., 1968, 16, 588–594 CrossRef CAS PubMed.
- Y. Lin, Y. Gu, S. A. Wharton, L. Whittaker, V. Gregory, X. Li, S. Metin, N. Cattle, R. S. Daniels, A. J. Hay and J. W. Mccauley, Influenza Other Respir. Viruses, 2015, 9, 331–340 CrossRef CAS PubMed.
- S. Hatakeyama, Y. Sakai-Tagawa, M. Kiso, H. Goto, C. Kawakami, K. Mitamura, N. Sugaya, Y. Suzuki and Y. Kawaoka, J. Clin. Microbiol., 2005, 43, 4139–4146 CrossRef CAS PubMed.
- J. Hollý, M. Fogelová, L. Jakubcová, K. Tomčíková, M. Vozárová, E. Varečková and F. Kostolanský, J. Virol. Methods, 2017, 247, 107–113 CrossRef PubMed.
- U. Karakus, M. Crameri, C. Lanz and E. Yángüez, Methods Mol. Biol., 2018, 1836, 59–88 CrossRef CAS PubMed.
- A. Klimov, A. Balish, V. Veguilla, H. Sun, J. Schiffer, X. Lu, J. M. Katz and K. Hancock, Methods Mol. Biol., 2012, 865, 25–51 CrossRef CAS PubMed.
- A. Baer and K. Kehn-Hall, J. Visualized Exp., 2014, e52065 Search PubMed.
- C. E. Stokes, J. M. Bernstein, S. A. Kyger and F. G. Hayden, J. Clin. Microbiol., 1988, 26, 1263–1266 CrossRef CAS PubMed.
- K. Tobita, A. Sugiura, C. Enomoto and M. Furuyama, Med. Microbiol. Immunol., 1975, 162, 9–14 CrossRef CAS PubMed.
- D. Tilmanis, C. van Baalen, D. Y. Oh, J. F. Rossignol and A. C. Hurt, Antiviral Res., 2017, 147, 142–148 CrossRef CAS PubMed.
- E. T. Stone, A. J. Hirsch, J. L. Smith, J. D. Brien and A. K. Pinto, STAR Protoc., 2022, 3, 101473 CrossRef CAS PubMed.
- D. Cresta, D. C. Warren, C. Quirouette, A. P. Smith, L. C. Lane, A. M. Smith and C. A. A. Beauchemin, PLoS Comput. Biol., 2021, 17, e1009480 CrossRef CAS PubMed.
- M. M. Kiss, L. Ortoleva-Donnelly, N. R. Beer, J. Warner, C. G. Bailey, B. W. Colston, J. M. Rothberg, D. R. Link and J. H. Leamon, Anal. Chem., 2008, 80, 8975–8981 CrossRef CAS PubMed.
- C. D. Ahrberg, J. M. Lee and B. G. Chung, BioChip J., 2019, 13, 269–276 CrossRef CAS.
- J. E. Kreutz, T. Munson, T. Huynh, F. Shen, W. Du and R. F. Ismagilov, Anal. Chem., 2011, 83, 8158–8168 CrossRef CAS PubMed.
- Q. Zhu, Y. Gao, B. Yu, H. Ren, L. Qiu, S. Han, W. Jin, Q. Jin and Y. Mu, Lab Chip, 2012, 12, 4755–4763 RSC.
- Y. Hu, P. Xu, J. Luo, H. He and W. Du, Anal. Chem., 2017, 89, 745–750 CrossRef CAS PubMed.
- S. Lindström, K. Mori, T. Ohashi and H. Andersson-Svahn, Electrophoresis, 2009, 30, 4166–4171 CrossRef PubMed.
- G. Amselem, C. Guermonprez, B. Drogue, S. Michelin and C. N. Baroud, Lab Chip, 2016, 16, 4200–4211 RSC.
- S. Padmanabhan, J. Y. Han, I. Nanayankkara, K. Tran, P. Ho, N. Mesfin, I. White and D. L. DeVoe, Biomicrofluidics, 2020, 14, 014113 CrossRef CAS PubMed.
- S. Padmanabhan, A. Sposito, M. Yeh, M. Everitt, I. White and D. L. DeVoe, Biomicrofluidics, 2021, 15, 014103 CrossRef CAS PubMed.
- B. Bohl, R. Steger, R. Zengerle and P. Koltay, J. Micromech. Microeng., 2005, 15, 1125 CrossRef.
- Z. Wu and K. Hjort, Lab Chip, 2009, 9, 1500–1503 RSC.
- K. Takada, C. Kawakami, S. Fan, S. Chiba, G. Zhong, C. Gu, K. Shimizu, S. Takasaki, Y. Sakai-Tagawa, T. J. S. Lopes, J. Dutta, Z. Khan, D. Kriti, H. van Bakel, S. Yamada, T. Watanabe, M. Imai and Y. Kawaoka, Nat. Microbiol., 2019, 4, 1268–1273 CrossRef CAS PubMed.
- B. Shu, M. K. Kirby, W. G. Davis, C. Warnes, J. Liddell, J. Liu, K. H. Wu, N. Hassell, A. J. Benitez, M. M. Wilson, M. W. Keller, B. L. Rambo-Martin, Y. Camara, J. Winter, R. J. Kondor, B. Zhou, S. Spies, L. E. Rose, J. M. Winchell, B. M. Limbago, D. E. Wentworth and J. R. Barnes, Emerging Infect. Dis., 2021, 27, 1821–1830 CrossRef CAS PubMed.
- Y. Xia and G. M. Whitesides, Annu. Rev. Mater. Sci., 1998, 28, 153–184 CrossRef CAS.
- L. Bordenave, R. Bareille, F. Lefebvre, J. Caix and C. H. Baquey, J. Biomater. Sci., Polym. Ed., 1992, 3, 509–516 CrossRef CAS PubMed.
- J. H. Park, K. D. Park and Y. H. Bae, Biomaterials, 1999, 20, 943–953 CrossRef CAS PubMed.
- J. N. Lee, X. Jiang, D. Ryan and G. M. Whitesides, Langmuir, 2004, 20(26), 11684–11691 CrossRef CAS PubMed.
- A. Piruska, I. Nikcevic, S. H. Lee, C. Ahn, W. R. Heineman, P. A. Limbach and C. J. Seliskar, Lab Chip, 2005, 5, 1348–1354 RSC.
- I. Wong and C. M. Ho, Microfluid. Nanofluid., 2009, 7, 291–306 CrossRef CAS PubMed.
- S. Halldorsson, E. Lucumi, R. Gómez-Sjöberg and R. M. T. Fleming, Biosens. Bioelectron., 2015, 63, 218–231 CrossRef CAS PubMed.
- S. H. Au, P. Kumar and A. R. Wheeler, Langmuir, 2011, 27, 8586–8594 CrossRef CAS PubMed.
- A. S. Basu, SLAS Technol., 2017, 22, 369–386 CrossRef PubMed.
- S. Dube, J. Qin and R. Ramakrishnan, PLoS One, 2008, 3, e2876 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lc00940a |
| This journal is © The Royal Society of Chemistry 2025 |