
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of a mixed-linker Ce-UiO-67 metal–organic framework†
V.
Finelli
 ab,
G.
Mastronardi
a,
N. G.
Porcaro
a,
M.
Signorile
a,
F.
Bonino
a,
P. Á.
Szilágyi
c and
S.
Bordiga
*a
ab,
G.
Mastronardi
a,
N. G.
Porcaro
a,
M.
Signorile
a,
F.
Bonino
a,
P. Á.
Szilágyi
c and
S.
Bordiga
*a
aDepartment of Chemistry, NIS and INSTM Reference Centre, Università di Torino, Via G. Quarello 15/A, I-10135, and Via P. Giuria 7, I-10125, Turin, Italy. E-mail: silvia.bordiga@unito.it
bUniversity School for Advanced Studies, IUSS Pavia, Palazzo del Broletto, Piazza della Vittoria 15, I-27100, Pavia, Italy
cCentre for Materials Science and Nanotechnology, Department of Chemistry, University of Oslo, Sem Sælands vei 26, N-0315 Oslo, Norway
First published on 7th October 2024
Abstract
Ce-based metal–organic frameworks (MOFs) have recently gained scientific interest, since Ce is the most abundant rare-earth element in the Earth's crust and since their synthesis has some advantages, including first of all their redox activity, the high porosity of these crystalline materials, and Ce availability. In particular, Ce(IV)-based MOFs, such as Ce-UiO-66 and Ce-UiO-67, are synthesised under mild conditions. For most applications, the presence of functional groups in the frameworks is needed; in this context, linkers containing N-functionalities have been highlighted, as they allow for the incorporation of a large variety of metal cations. In order to insert N-functionalities for the sake of successive metal-functionalization of materials as Ce-UiO-67, we have successfully synthesised a mixed-linker version of this MOF, by incorporating 2,2′-bipyridine-5,5′-dicarboxylic acid together with the conventional biphenyl-4,4′-dicarboxylic acid linker; we have worked on a reproducible and upscalable procedure using benzoic acid as the modulator, without altering the original framework topology. Mixed-linker Ce-UiO-67 MOFs exhibit good thermal stability, high Brunauer–Emmett–Teller (BET) SSAs and microporosity. The pristine samples are highly stable if stored in a desiccator, as demonstrated by the preservation of their high crystallinity for at least 18 months.
1. Introduction
Metal–organic frameworks (MOFs) are hybrid organic–inorganic materials hypothesized in 1989 as an innovative way for constructing three-dimensional (3D) frameworks1 and then synthesised for the first time ten years later,2 opening the way for the widespread and prompt development of this research field. They are made up of Secondary Building Units (SBUs) of metal cations or oxide clusters connected together by organic molecules, i.e. linkers.3 MOFs are extremely versatile materials since their structure and properties can be finely engineered by, among others, introducing mixed-metal nodes, various ligands, and functional groups, making them suitable for several applications, ranging from gas sorption through drug delivery to catalysis.3Of all the metal candidates for SBUs, rare-earth elements have recently gained scientific interest, especially Ce that is their most abundant representative in the Earth's crust.4 Constructing MOFs with Ce nodes has some advantages, including the availability of its metal source,5 the redox activity of Ce species and the high porosity of these crystalline materials. As well reported in the review by J. Jacobsen et al.,3 the chemistry of Ce(IV)-MOFs exhibits many similarities to those of Zr-MOFs (i.e. UiO-66 and UiO-676 even if the synthesis protocols require a lower temperature and shorter time. In particular, Ce-UiO-66 and Ce-UiO-677,8 have been synthesised under mild conditions, including a short reaction time (15 min), low temperature (100 °C) and under autogenous pressure. Such a synthetic strategy opens up the possibility of easy scale-up of the procedure that often remains a challenge for MOFs. The so-obtained crystalline 3D structures (space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, fcu topology) are constructed of hexanuclear [Ce6(μ3-O)4(μ3-OH)4]12+ clusters interconnected by twelve terephthalate (BDC2−) or biphenyl dicarboxylate (BPDC2−) organic linkers for Ce-UiO-66 and for Ce-UiO-67, respectively. Even if their chemical and thermal stability is lower7,8 than their formerly discovered Zr-counterparts, Ce(IV)-based MOFs may be successfully used in catalysis, also exploiting the possibility of affecting the oxidation state of some Ce species, obtaining mixed Ce(III) and Ce(IV) species in a wide range.
m, fcu topology) are constructed of hexanuclear [Ce6(μ3-O)4(μ3-OH)4]12+ clusters interconnected by twelve terephthalate (BDC2−) or biphenyl dicarboxylate (BPDC2−) organic linkers for Ce-UiO-66 and for Ce-UiO-67, respectively. Even if their chemical and thermal stability is lower7,8 than their formerly discovered Zr-counterparts, Ce(IV)-based MOFs may be successfully used in catalysis, also exploiting the possibility of affecting the oxidation state of some Ce species, obtaining mixed Ce(III) and Ce(IV) species in a wide range.
In addition to these properties, the possibility of synthesising mixed-linker MOFs could be advantageous in catalysis, in order to offer isolated anchoring points for post-synthetic metalation. Mixed-linker MOFs can be achieved by either post-synthetic linker exchange (PSLE) or via one-pot synthesis: this latter way was successfully applied for obtaining a mixed-linker version of Zr-UiO-67.9 Partially replacing (10% nominally) the original 4,4′-biphenyl dicarboxylic acid (H2BPDC) linker with 2,2′-bipyridine-5,5′-dicarboxylic acid (H2BPyDC), Kaur et al.9 introduced N-functionalities in the framework, without any detectable alteration in its topology. This one-pot synthesis allows avoiding successive addition steps as in the PSLE approach, granting an easier synthetic procedure, but a controlled and tuned insertion of the second linker is needed. To the best of our knowledge, a study concerning the one-pot synthesis of mixed-linker Ce-UiO-6X is instead still missing in the literature.
In this context, our contribution aims at synthesising mixed-linker Ce-UiO-67 frameworks replacing the original H2BPDC linker with 5% nominal H2BPyDC, targeting a specific % of insertion and offering a convenient possibility for functionalization and subsequent metalation. We selected a 5% loading based on the foreseen application of this material (i.e. insertion of metal sites at BPyDC coordination sites), aiming at increasing the dispersion of the second linker within the framework and, accordingly, at decreasing the probability of producing neighbouring metal sites in the linker-exchanged materials. These activities allowed us to familiarize with the insertion of a commercial linker in a Ce-UiO-67 framework. Our final aim is to incorporate linkers of synthetic origin and then obtain metalated MOFs for catalytic applications, investigating reactions happening both at solid–liquid and at solid–gas interfaces. Our scope is sharing that mixing-linker purposes are attainable even for the kinetically driven extremely rapid Ce-UiO-67 synthesis, and not proposing a generalized synthetic procedure. After the synthesis optimization, we managed to reproduce and upscale our procedure for obtaining a crystalline and thermally stable MOF.
2. Experimental
2.1. Materials
All the chemicals were employed without further treatment, and they are listed here: ammonium cerium(IV) nitrate (NH4)2Ce(NO3)6, 99.0%, Fluorochem; 4,4′-biphenyldicarboxylic acid (H2BPDC), 98%, Apollo Scientific; 2,2′-bipyridine-5,5′-dicarboxylic acid (H2BPyDC), >98%, TCI; benzoic acid (BA), ACS reagent ≥99.5%, Sigma Aldrich; N,N-dimethylformamide (DMF), ≥99.8% AnalaR NORMAPUR®, VWR Chemicals; dimethyl sulfoxide (DMSO), ≥99% GPR RECTAPUR®, VWR Chemicals.2.2. Basic characterization techniques
Powder X-Ray Diffraction (PXRD) patterns of MOFs were collected on a PANalytical X'Pert instrument with Cu Kα radiation corresponding to an incident wavelength λ = 1.5405 Å. The patterns were acquired in the 2θ range of 3–30° with a step size of 0.02°, using the Bragg–Brentano geometry.Thermo-gravimetric analyses (TGA) were performed on a TA Instruments SDT-Q600 under synthetic air (80% N2–20% O2) flow (100 ml min−1), equilibrating at 35 °C for 30 min and then increasing the temperature to 500 °C with a heating ramp of 10 °C min−1.
For NMR analysis, typically 20 mg of the samples was placed in 1 mL of a 0.1 M D2O solution of NaOD for overnight digestion. Once the digested suspension was centrifuged (12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 15 °C, 15 min), the supernatant was pipetted into the NMR tube. 1H-NMR spectra were recorded on a Jeol ECZ-R 600 MHz instrument, averaging 32 scans with a relaxation delay of 2 s (offset 6.5 ppm, sweep 15 ppm, x points 32 k, and 90° pulse 8.15 μs).
000 rpm, 15 °C, 15 min), the supernatant was pipetted into the NMR tube. 1H-NMR spectra were recorded on a Jeol ECZ-R 600 MHz instrument, averaging 32 scans with a relaxation delay of 2 s (offset 6.5 ppm, sweep 15 ppm, x points 32 k, and 90° pulse 8.15 μs).
The textural properties of the materials were evaluated on a Micromeritics 3FLEX instrument measuring the N2 adsorption–desorption isotherms at liquid N2 temperature (77 K), by using the software MicroActive provided by Micromeritics. Their Specific Surface Areas (SSAs) were calculated adopting the Rouquerol consistency criteria10 and applying the Brunauer–Emmet–Teller (BET) equation in the 0.001–0.03 p/p0 range for all the materials. The micropore volume of the MOFs (vide infra and the ESI† for the plots) was evaluated by using the t-plot method (with a density conversion factor on average equal to 0.0015492) applying the Harkins and Jura thickness curve in the 0.39–0.43 nm range, which is the one that allows the correlation coefficient being closer to 1 for all the materials, for the sake of consistency. The adsorption information format (.aif) files are available in the ESI† material.
2.3. Spectroscopic characterization techniques
Infrared (IR) spectra were collected in transmission mode, using a home-made quartz cell, combined with a Bruker INVENIO R FT-IR spectrometer, equipped with a mercury–cadmium–telluride (MCT) cryogenic detector. Prior to the transmission IR measurements, the samples were pelletized at a pressure of 0.5 ton; the use of higher pressures was avoided to prevent (partial) sample amorphization. The self-supported pellets were placed in a quartz cell, then connected to a vacuum line for the activation of the materials under dynamic vacuum. Once the activation temperature (110 °C) was reached with a heating rate of 3 °C min−1, the samples were outgassed for 4 h (residual pressure <10−4 mbar). The IR spectra were collected with a resolution of 2 cm−1 by averaging 32 scans. After IR measurements, PXRD patterns were collected to check the structural integrity of the pelletized samples re-exposed to air at the end of the spectroscopic experiments.Diffuse reflectance (DR) UV/vis-NIR ex situ spectra were recorded on a Varian Cary5000 spectrophotometer equipped with a reflectance sphere. Spectralon® powder was used as a standard for 100% reflectance. The pristine MOFs in powder form were directly placed inside the sample holder for ex situ measurements of the samples as such. The DR UV-vis spectra of the materials activated with the above-mentioned protocol were also acquired. All the spectra were collected in reflectance mode and then converted using the Kubelka–Munk F(R) function.
Attenuated Total Reflection Infrared (ATR-IR) spectra were collected with a resolution of 2 cm−1, accumulating 32 scans, by means of a Bruker Alpha instrument, equipped with a MCT cryogenic detector and single reflection diamond ATR accessory.
Raman spectra were measured with a Renishaw InVia Raman microscope spectrometer, equipped with a 1200 lines mm−1 grating monochromator and a Peltier-cooled CCD detector, working with a 785 nm excitation beam that was focused on the samples using a 50× objective, at 0.1% laser power (<1 mW at sample). Three spectra were collected for each material to check their stability under measurement conditions and, since identical spectra have been obtained, they have been averaged to improve spectral quality.
2.4. Computational details
Computational investigation has been carried on using the ORCA program11,12 (version 5.0.4) with the aim to underpin the interpretation of the experimental vibrational spectra. The presence of BPyDC linkers has been confirmed thanks to the molecular computation of its vibrational spectra, avoiding any type of periodic calculation considered unnecessary for our scope. The structures of BPDC2− and BPyDC2− ions have been fully optimized by the density functional theory (DFT) method using the B3LYP functional associated with the all electrons def2-TZVP basis set as proposed by Ahlrichs.13 Resolution of identity approximation (RIJCOSX scheme14) was employed to speed up the DFT calculations in combination with the def2/j auxiliary basis set. Default conditions for DFT grid generations and Self-Consistent Field (SCF) convergence criteria have been set (SETGRID 4 and TIGHTSCF). Furthermore, frequencies have been calculated numerically to simulate the Raman spectra of the linkers adopting the same conditions.3. Results and discussion
3.1. Optimization of the synthesis procedure
The synthesis of a mixed-linker Ce-UiO-67-5% BPyDC was optimized by combining the synthetic route reported by Lammert et al.,8 which reported the synthesis of Ce-UiO-67-100% BPyDC, with the addition of benzoic acid as the acid modulator, as reported by Kaur et al.9 who have optimized the synthesis for the traditional Zr-UiO-67 and the mixed-linker Zr-UiO-67 frameworks.First, 1.1781 g of H2BPDC, 0.0625 g of H2BPyDC, and 1.8756 g of BA were mixed with 28.8 mL of DMF under stirring at room temperature. After the preparation of a deionized water solution 0.5333 M of ammonium cerium(IV) nitrate (NH4)2Ce(NO3)6, 9.6 mL of it was added to the mixture at room temperature. The resulting stoichiometry for the synthetic mixture was 1 (NH4)2Ce(NO3)6:0.95 H2BPDC:0.05 H2BPyDC:3 BA:73 DMF. After 15 min under stirring at 100 °C in an oil bath, the mother liquor was removed by centrifugation of the product (10000 rpm, 15 °C, 5 min). The material underwent the following washing procedure: at first, it was suspended in DMSO (20 mL) at room temperature and sonicated for 5 min at 60 °C. After centrifugation and removal of the liquid phase, the procedure was repeated twice using DMF in place of DMSO. After recovering the solid phase, the obtained material was dried in a static oven overnight at 70 °C. This procedure corresponded to a range of yields (39–51%), depending on the synthetic batch. The desolvated MOF yields, corrected by solvent loss from TGA profiles (vide infra) at 250 °C, corresponded to the range 24–30%, depending on the synthetic batch.
In the synthetic field, one of the greatest challenges is evidently the reproducibility of the syntheses themselves, impinging on their upscalability. To address this, in this study three batches of upscaled Ce-UiO-67-5% BPyDC have been successfully synthesised, named #1, #2 and #3. The adopted washing procedure is effective in the removal of unreacted linkers, which can be checked by comparing the PXRD patterns of the synthesised MOFs and the linkers. Indeed, Fig. 1 shows that the reflections of the linkers are not present in the PXRD pattern of #1, taken as an example.
 | ||
| Fig. 1 A) PXRD pattern of #1 Ce-UiO-67-5% BPyDC (blue line) compared to those of the linkers H2BPDC (dashed pink line) and H2ByPDC (dashed violet line), showing no unreacted linkers residues. B) and C) show magnifications of the same PXRD patterns. For the MOF, intensities normalized to the 5.58° peak. | ||
The use of benzoic acid as the modulator was revealed to be necessary in order to obtain a mixed-linker material displaying good crystallinity; indeed, an attempt of synthesising mixed-linker Ce-UiO-67-5% BPyDC without any acid modulator is illustrated in Fig. S1,† showing a non-crystalline pattern.
Moreover, acetone wash of the resultant mixed-linker MOFs, as suggested by Lammert et al.8 for their Ce-UiO-67-100% BPyDC, rendered the material extremely electrostatic to the point of huge material losses that were unavoidable even on simple sample handling and preparation as for PXRD data collection. Therefore, DMF wash was prioritised for the sake of ease of handling.
After the washing procedure and before further handling and use, the materials were stored in a desiccator, since they are sensitive to moisture. The desiccator is filled with silica gels as the water sorbent and cobalt(II) chloride as the humidity indicator, both activated at 100 °C in an oven; these chemicals are promptly renewed as soon as the first amounts of cobalt(II) chloride turn pink. This precaution helped the preservation of the properties and structural integrity of the materials for at least 18 months after their synthesis (vide infra).
To explore the synthetic parameter space, a synthesis attempt was carried out decreasing the reaction temperature to 80 °C in a bid to slow down the nucleation rate and thereby increase the crystallite size. The PXRD pattern of the corresponding result shows a non-crystalline material in Fig. S2;† the non-reacted BPDC linker dominates the diffractogram.
Using longer reaction times, we have investigated their effect on the synthesis of mixed-linker Ce-UiO-67 frameworks. The PXRD patterns for crystalline #2 Ce-UiO-67-5% BPyDC synthesised for 15 min, 30 min and 1 h are depicted in Fig. S3a–c,† while even longer synthesis times (3 h, 6 h and 24 h) were proved to be detrimental to gaining insight, since we have observed changes in the resulting materials; additional diffraction peaks arose in their PXRD patterns (in Fig. S3d–f†), which could be attributed to the decomposition of the solvent, leading to the formation of Ce(III) formate [Ce(O2CH)3] with the evolution of one of its more intense corresponding peaks at 16.6°, as reported in the literature.15
For the sake of comparison, the upscaled synthesis of the pristine Ce-UiO-67 was also performed affording higher yields, especially for big batches of the material (vide the ESI,† section S1).
3.2. Basic multitechnique characterization
Fig. 2 illustrates a comparison of the PXRD patterns of the three synthetic attempts for Ce-UiO-67-5% BPyDC alongside that of the pristine Ce-UiO-67. Matching with the latter one, the mixed-linker material also crystallizes in the Fmm space group, where the [Ce6(μ3-O)4(μ3-OH)4]12+ clusters are ideally connected to twelve organic linkers, corresponding to the cubic close-packing fcu topology,8 resulting in the following ideal formula for the materials: [Ce6(μ3-O)4(μ3-OH)4(BPDC)6(1−x)(BPyDC)x], with x = 0.05 for nominal 5% linker exchange.
 | ||
| Fig. 2 PXRD patterns of the three batches of Ce-UiO-67-5% BPyDC, #1 (blue line), #2 (red line) and #3 (green line), compared to that of Ce-UiO-67 (black line). Intensities normalized to the 5.58° peak. | ||
To investigate the thermal stability of the materials, TGAs were measured in the temperature range 20–500 °C. Fig. 3 shows the TGA profiles characterized by two well-defined steps: the first weight loss is assigned to the dehydroxylation and desolvation (namely, loss of DMF) of the materials, which are stable in their dehydroxylated form in the temperature range of approx. 200–330 °C, on average. DMF constitutes approx. 40 wt% of the MOFs. The second one is related to the combustion of the organic materials, ultimately leaving ceria (CeO2) as solid inorganic residue, i.e. inorganic end-of-life natural form. Ce-UiO-67-5% BPyDC materials display good thermal stability, comparable to that of the pristine Ce-UiO-67 and to the material based on 100% BPyDC reported in the literature.8 The TGA curves for all the materials are also reported together with the corresponding differential thermal analysis (DTA) curves in Fig. S4.†
 | ||
| Fig. 3 TGA profiles of the three batches of Ce-UiO-67-5% BPyDC, #1 (blue line), #2 (red line) and #3 (green line), compared to that of Ce-UiO-67 (black line), reported as weight loss (%) related to CeO2, set as 100%. | ||
The nominal amount of BPyDC to be introduced was equal to 5% of the original BPDC linker. Table 1 summarises the actual percentages of BPyDC that match the nominally introduced amount in the mixed-linker materials; the gap between these results and the nominal value is not large, showing that with this acid modulated procedure we are able to control the mixing of the linkers in the structure. It should be noted, however, that the values are affected by a slight error, due to the manual integration of the 1H-NMR peaks of the BPyDC linker that are characterized by weak intensities.
| Material | % BPyDC inserted |
|---|---|
| #1 Ce-UiO-67-5% BPyDC | 5.1 |
| #2 Ce-UiO-67-5% BPyDC | 5.7 |
| #3 Ce-UiO-67-5% BPyDC | 6.8 |
To obtain mixed-linker MOFs, using longer synthesis times is not beneficial; in fact, the 1H-NMR spectra in Fig. S9–S10,† allowing the quantification of BPyDC inserted, evidence lower amounts than the nominal ones for longer synthesis times, as shown in terms of the percentage of BPyDC insertion in Table S1.† The defectivity of the materials has been computed by the combined approach of TGA and NMR reported by Shearer et al.,16 and it has been estimated in the range of 2.5–3.6% in terms of missing linkers17 for the mixed-linker materials, while it is around 1.5% for Ce-UiO-67. The calculation and the corresponding data are included in the ESI,† Table S2. At the present stage, we do not have any insights into the nature of the missing linker defects (BPDC or BPyDC).
Focusing on the assessment of the porosity and textural properties of all four materials synthesised for 15 min (also including Ce-UiO-67), their N2 adsorption–desorption isotherms were measured at liquid N2 temperature (77 K), as illustrated in Fig. 4, and for Ce-UiO-67 in Fig. S11.† According to IUPAC,18 the isotherms can be classified as type I, characteristic of microporous materials. The materials display a small amount of inter-particle voids, noticeable from hysteresis loops in the high relative pressure range, highlighted in Fig. S12.† This phenomenon is especially apparent for Ce-UiO-67 and #1 Ce-UiO-67-5% BPyDC, while less so for the other batches of the mixed-linker MOFs. They show comparable or even higher BET SSAs (BET fits reported in Fig. S13–S14†) and micropore volume, according to t-plot analysis (Fig. S15†), if compared to the literature8 and these values are summarised in Table 2. Applying the t-plot method to a type I isotherm, in order to obtain pieces of information on the micropore volume, the most linear part of the isotherm needs to be fitted; in particular, the y-axis intercept of the resulting fitting line offers hints on the quantity adsorbed by the micropores in cm3 g−1 STP. By means of multiplication to the density conversion factor provided by the software MicroActive by Micromeritics, it is possible to obtain the micropore volume of the materials. PXRD patterns were collected for crystallinity-check purposes after the isotherm measurements on Ce-UiO-67 and #3 Ce-UiO-67-5% BPyDC (as an example for the mixed-linker MOFs) and the crystallinity of the materials is retained after this characterization step as Fig. S16† displays.
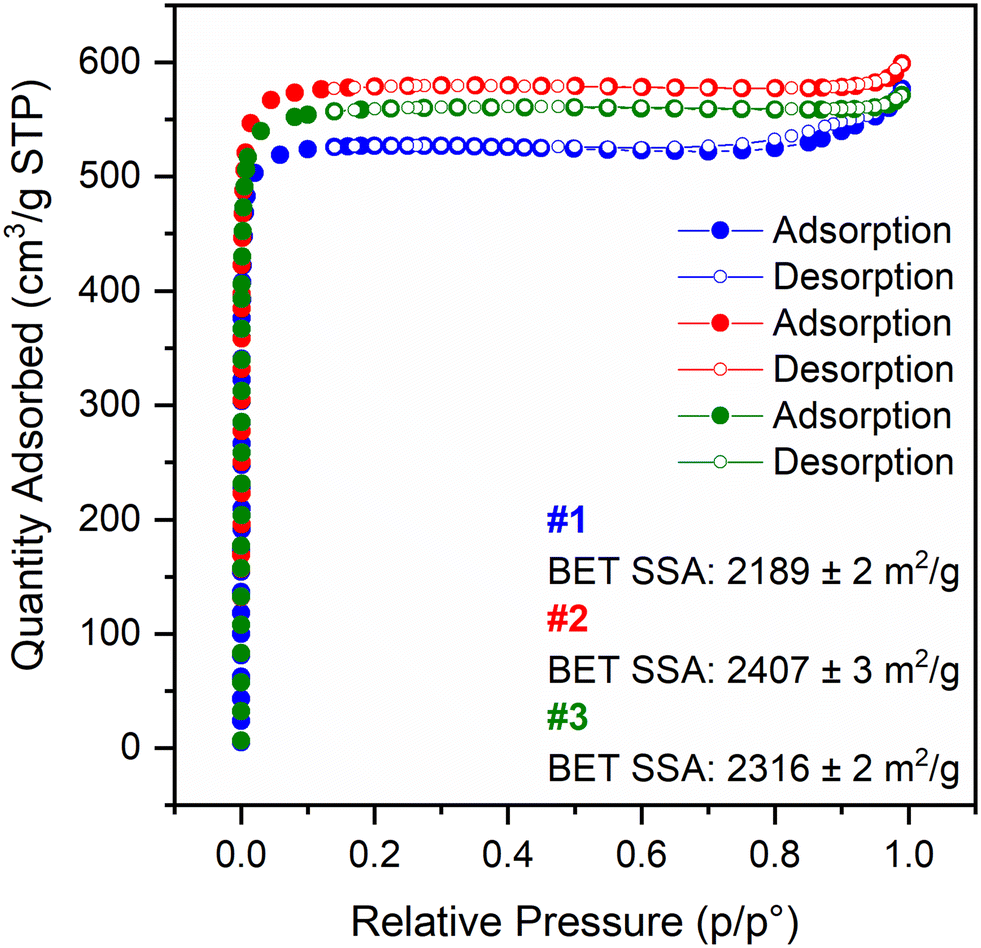 | ||
| Fig. 4 N2 adsorption–desorption isotherms at 77 K for the three batches of Ce-UiO-67-5% BPyDC: #1 in blue, #2 in red and #3 in green, respectively. For the sake of clarity, we have selected one of each three adsorption points up to 150 cm3 g−1 STP and then one every other. | ||
| Material | BET SSA (m2 g−1) | Micropore SSA (m2 g−1) | Micropore volume (cm3 g−1 STP) |
|---|---|---|---|
| Ce-UiO-67 | 2242 | 2159 | 0.80 |
| #1 Ce-UiO-67-5% BPyDC | 2189 | 2132 | 0.79 |
| #2 Ce-UiO-67-5% BPyDC | 2407 | 2334 | 0.86 |
| #3 Ce-UiO-67-5% BPyDC | 2316 | 2234 | 0.83 |
The stability of the materials, stored in a desiccator, was checked by means of PXRD patterns and N2 isotherm measurements after 18 months from the synthesis of the materials: #1 Ce-UiO-67-5% BPyDC was taken as an example. Its PXRD pattern (Fig. S17†) highlights that its crystallinity, despite diminished, is retained showing the same reflections as the as-synthesized material. The textural properties of the material after 18 months are depicted in Fig. S18,† with an inset in the inter-particle void region as seen in Fig. S19,† its BET linear fit is seen in Fig. S20† and its t-plot in Fig. S21.† Table S3† summarises the textural properties of #1 Ce-UiO-67-5% BPyDC compared to its aged version. There seems to be a slight decrease in the BET SSA of the material, together with its pore volumes, without altering the pore sizes. However, these values still remain highly comparable to the ones of the as-synthesized material, thereby demonstrating good stability under the said storage conditions.
3.3. Spectroscopic characterization
The insertion of BPyDC in the MOF network is also confirmed by the comparison of the Raman spectra of Ce-UiO-67 with those of Ce-UiO-67-5% BPyDC.
Fig. 5 compares the Raman spectra of Ce-UiO-67 (black curve) and Ce-UiO-67-5% BPyDC (green curve) with those of the H2BPDC (dashed pink curve) and H2BPyDC (dashed purple curve) linkers, all at room temperature in powder form. In the 1800–1100 cm−1 Raman shift range, the spectra of the MOFs show similarities: the most intense Raman active features include the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching of aromatic rings at 1619–1614 cm−1; the aromatic ring stretching at 1288 cm−1; the aromatic rings' breathing mode at 1154 cm−1.19–21 Moreover, there are two pairs of well-resolved features: at a higher Raman shift, the O–C–O in-phase symmetric stretching in carboxylates (not detectable in IR spectra because of its excessive intensity) in two ranges, namely, 1452–1445 cm−1 and 1426–1424 cm−1, and, at a lower Raman shift, the C–C symmetric breathing mode at 857–837 cm−1 (only Raman active19–21). Ce-UiO-67-5% BPyDC modes correspond well with three H2BPyDC features centred at 1601 cm−1, 1493 cm−1, and 1240 cm−1, marked with an asterisk in Fig. 5. Based on our simulations (Fig. S22†), these vibrational modes can be attributed to CC stretching modes coupled with C–H in-plane bending modes with different symmetries within the terephthalate rings. Similarities in some bands can also be observed in the ATR-IR spectra of the linkers and the transmission IR spectra of the materials, as depicted in Fig. S23.† In this case, merging pieces of information obtained from the spectral simulations (Fig. S24†), two H2BPyDC bands, centred at 1054 cm−1 and 811 cm−1 and marked with an asterisk, can be attributed to the asymmetric terephthalate ring stretching and out-of-plane terephthalate ring bending, respectively.
C stretching of aromatic rings at 1619–1614 cm−1; the aromatic ring stretching at 1288 cm−1; the aromatic rings' breathing mode at 1154 cm−1.19–21 Moreover, there are two pairs of well-resolved features: at a higher Raman shift, the O–C–O in-phase symmetric stretching in carboxylates (not detectable in IR spectra because of its excessive intensity) in two ranges, namely, 1452–1445 cm−1 and 1426–1424 cm−1, and, at a lower Raman shift, the C–C symmetric breathing mode at 857–837 cm−1 (only Raman active19–21). Ce-UiO-67-5% BPyDC modes correspond well with three H2BPyDC features centred at 1601 cm−1, 1493 cm−1, and 1240 cm−1, marked with an asterisk in Fig. 5. Based on our simulations (Fig. S22†), these vibrational modes can be attributed to CC stretching modes coupled with C–H in-plane bending modes with different symmetries within the terephthalate rings. Similarities in some bands can also be observed in the ATR-IR spectra of the linkers and the transmission IR spectra of the materials, as depicted in Fig. S23.† In this case, merging pieces of information obtained from the spectral simulations (Fig. S24†), two H2BPyDC bands, centred at 1054 cm−1 and 811 cm−1 and marked with an asterisk, can be attributed to the asymmetric terephthalate ring stretching and out-of-plane terephthalate ring bending, respectively.
 | ||
| Fig. 5 Raman spectra of the H2BPDC linker (dashed pink curve) and H2BPyDC linker (dashed purple curve), Ce-UiO-67 (black) and #3 Ce-UiO-67-5% BPyDC (green) in powder form. The spectra are reported in arbitrary units. Asterisks underline the band similarities at 1602 cm−1, 1494 cm−1 and 1240 cm−1 between #3 Ce-UiO-67-5% BPyDC (green) and H2BPyDC linkers (dashed purple curve). The right panel reports the data magnified by a factor of 2 in respect to the left panel. | ||
The IR spectra of pristine Ce-UiO-67 and mixed-linker Ce-UiO-67-5% BPyDC after a treatment of 4 h at 110 °C in dynamic vacuum, to remove the solvent, were collected (Fig. S25–S26,† respectively). The spectra of both materials are dominated by the carboxylate vibrations (bands in the range 1640–1215 cm−1 out of scale) and by an intense band at 3647 cm−1, associated with the stretching of the isolated μ3-OH species on the cornerstones of the Ce6-cluster of the MOFs.22 The bands at 3150–3000 cm−1 are characteristic of aromatic C–H stretching vibrations, while the ones in the range 2950–2830 cm−1 refer to the stretching of aliphatic C–H stretching vibrations peculiar of residual DMF,23 with another band slightly observable at around 1720 cm−1 related to its CO stretching mode.19 Closely, the band centred at 1645 cm−1 refers to the O–H bending of residual water.19 At lower wavenumbers, the band at around 1020 cm−1 and common to all the materials is due to the collective bending mode of the hydrogen atoms of the ring of the linkers.19
The same pretreatment was followed before the collection of the Diffuse Reflectance (DR) UV-vis spectra of the pristine Ce-UiO-67 (black curve) and Ce-UiO-67-5% BPyDC (green curve). Fig. S27† compares the DR-UV-vis spectra collected before (dashed ones) and after (solid ones) activation at 110 °C in dynamic vacuum for 4 h. For the pristine materials, the spectra are dominated by absorption edges at around 24000 cm−1, corresponding to a ligand-to-metal charge transfer (LMCT).24 After sample activation, the edges shift to lower wavenumbers, to 22800 cm−1 for Ce-UiO-67 and to 23400 cm−1 for Ce-UiO-67-5% BPyDC. According to the literature,25 this behaviour is associated with the formation of Ce(III) species derived from the partial reduction of the framework Ce(IV) species during the activation and dehydration process.
The PXRD patterns collected after the activation of the materials for IR experiments underline the stability and retention of the crystallinity of both the materials (Fig. S28†).
4. Conclusion
We have successfully synthesized three batches of mixed-linker Ce-UiO-67 MOFs and their synthesis was optimized resulting in a reproducible and upscalable procedure modulated using benzoic acid, without altering the original framework topology. These materials display good thermal stability, high BET SSAs and microporosity. We attribute their long-lifetime and stability to DMF molecules trapped in the MOF pores, effectively shielding them. These are significant results in terms of overcoming the practical challenges of storing such interesting functional materials.The insertion of N-functionalities, present in the BPyDC linker, in the Ce-UiO-67 framework could in principle be useful as anchoring points for the successive metalation of the MOFs that can be employed in heterogeneous catalysis for various applications. Moreover, the presence of Ce in the MOF nodes is also (photo-)catalytically interesting24 and paves the way for more complex and tandem reactions.
Data availability
The data concerning the present manuscript are available on request. The adsorption information format (.aif) files are available in the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement no. 856446 – CuBE). The authors acknowledge support from the Project CH4.0 under the MUR program “Dipartimenti di Eccellenza 2023-2027” (CUP: D13C22003520001). Dr Centrella is acknowledged for her support in understanding NMR spectroscopy at the first stages at the University of Turin. The authors deeply acknowledge their co-workers both at the University of Turin, especially Prof. Barolo, and at the University of Oslo, especially Prof. Olsbye, for their support during V.F.'s visiting period and further. Concerning V.F.'s activities, this paper and related research have been conducted during and with the support of the Italian inter-university PhD of national interest in Sustainable Development and Climate change (PhD-SDC).References
- B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1989, 111, 5962–5964 CrossRef CAS.
- H. Li, M. Eddaoudi and M. O'Keeffe, et al. , Nature, 1999, 402, 276–279 CrossRef CAS.
- J. Jacobsen, A. Ienco and R. D'Amato, et al. , Dalton Trans., 2020, 49, 16551–16586 RSC.
- https://www.usgs.gov/centers/national-minerals-information-center/rare-earths-statistics-and-information, (accessed 5 April 2024).
- https://www.euchems.eu/euchems-periodic-table/, (accessed 6 September 2024).
- J. H. Cavka, S. Jakobsen and U. Olsbye, et al. , J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef.
- M. Lammert, M. T. Wharmby and S. Smolders, et al. , Chem. Commun., 2015, 51, 12578–12581 RSC.
- M. Lammert, C. Glißmann and H. Reinsch, et al. , Cryst. Growth Des., 2017, 17, 1125–1131 CrossRef CAS.
- G. Kaur, S. Øien-Ødegaard and A. Lazzarini, et al. , Cryst. Growth Des., 2019, 19, 4246–4251 CrossRef CAS.
- J. Rouquerol, P. Llewellyn and F. Rouquerol, Stud. Surf. Sci. Catal., 2007, 160, 49–56 CrossRef CAS.
- F. Neese, F. Wennmohs and U. Becker, et al. , J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- F. Neese, WIREs Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- F. Neese, F. Wennmohs and A. Hansen, et al. , Chem. Phys., 2009, 356, 98–109 CrossRef CAS.
- N. Heidenreich, S. Waitschat and H. Reinsch, Z. Anorg. Allg. Chem., 2018, 644, 1826–1831 CrossRef CAS.
- G. C. Shearer, S. Chavan and S. Bordiga, et al. , Chem. Mater., 2016, 28, 3749–3761 CrossRef CAS.
- D. K. Sannes, S. Øien-Ødegaard and E. Aunan, et al. , Chem. Mater., 2023, 35, 3793–3800 CrossRef CAS.
- M. Thommes, K. Kaneko and A. V. Neimark, et al. , Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- S. Chavan, J. G. Vitillo and D. Gianolio, et al. , Phys. Chem. Chem. Phys., 2012, 14, 1614–1626 RSC.
- C. Atzori, G. C. Shearer and L. Maschio, et al. , J. Phys. Chem. C, 2017, 121, 9312–9324 CrossRef CAS.
- X. Zhang, X. Shi and Q. Zhao, et al. , Chem. Eng. J., 2022, 427, 131573 CrossRef CAS.
- C. Atzori, K. A. Lomachenko and J. Jacobsen, et al. , Dalton Trans., 2020, 49, 5794–5797 RSC.
- G. C. Shearer, S. Forselv and S. Chavan, et al. , Top. Catal., 2013, 56, 770–782 CrossRef CAS.
- X.-P. Wu, L. Gagliardi and D. G. Truhlar, J. Am. Chem. Soc., 2018, 140, 7904–7912 CrossRef CAS PubMed.
- S. Rojas-Buzo, D. Salusso and F. Bonino, et al. , Mater. Today Chem., 2023, 27, 101337 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lf00278d |
| This journal is © The Royal Society of Chemistry 2025 |