
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA diatom frustule-based Mn2SiO4@C@SiO2 multilayer-structure composite as a high-performance anode electrode material for lithium-ion batteries†
Shimao
Sun
a,
Hongchang
Liu
 *ab,
Yuxin
Chen
a,
Hongwei
Liu
ab,
Rongda
Yu
c,
Xingfu
Zheng
a,
Yunchao
Li
d,
Jian
Zhu
d,
Jinlan
Xia
ab and
Jun
Wang
ab
*ab,
Yuxin
Chen
a,
Hongwei
Liu
ab,
Rongda
Yu
c,
Xingfu
Zheng
a,
Yunchao
Li
d,
Jian
Zhu
d,
Jinlan
Xia
ab and
Jun
Wang
ab
aSchool of Minerals Processing and Bioengineering, Central South University, Changsha 410083, China. E-mail: hchliu2050@csu.edu.cn
bKey Lab of Biometallurgy of Ministry of Education of China, Central South University, Changsha 410083, China
cGuangzhou Institute of Geochemistry, Chinese Academy of Science, Guangzhou 510640, China
dDepartment for the History of Science and Scientific Archaeology, University of Science and Technology of China, Hefei 230026, China
First published on 18th November 2024
Abstract
Owing to its abundant reserves and high theoretical specific capacity, silica has been tested as an anode material for lithium-ion batteries. However, its utilization is limited by volume expansion during cycling and low electrical conductivity. Most studies have focused on designing nanostructures of SiO2 or combining them with conductive phases to solve this problem. In this work, diatom-based biological silica with a natural hollow porous structure was used as a template to prepare diatom-based silica anode materials coated with Mn2SiO4 nanoclusters via a hydrothermal method. A composite material with a structure of Mn2SiO4@C@SiO2 was obtained. The Mn/SiO2@C@SiO2 sandwich structure derived during electrochemical reduction has a high capacity and excellent rate performance and significantly inhibits the volume expansion of SiO2. The prepared anode material (AFD@C-Mn-40) with the Mn/SiO2@C@SiO2 structure retained a specific discharge capacity of approximately 1112 mA h g−1 after 100 cycles at 100 mA g−1, which provides new prospects for the large-scale application of silica.
Introduction
Lithium-ion batteries (LIBs) have been widely used in various fields because of their high energy density and long cycle life.1–4 However, the rapid advancement of the new energy industry has imposed greater demands on the capacity of lithium batteries. Traditional graphite anode materials (372 mA h g−1)5 can no longer meet market requirements, necessitating the development of novel anode materials that hold significant importance for the progress of lithium-ion batteries. Among them are alloy materials, silicon-based compounds, and carbon-based compounds.6 Si is the most promising alternative anode material among silicon-based materials because of its high theoretical capacity (4200 mA h g−1).7 Nevertheless, the extreme volume expansion (up to 400%) during the lithiation/delithiation process limits its application.8–10 As a substitute for Si, SiO2 has become a promising anode material because the volume expansion of SiO2 is the lowest (approximately 150%), and its relatively high theoretical specific capacity (1965 mA h g−1)11 is several times greater than that of graphite.Among the many silica composite materials, diatom-based biosilica has been reported to be a good source of silica material because of its natural hollow porous structure.12–15 Diatoms can absorb monosilicic acid from the environment during growth and convert it into their own SiO2 shell structure.16–19 The biogenic silica of diatoms possesses a sophisticated and complex hollow porous structure, which is much more regular and intricate than any artificially designed SiO2 nanostructure available today.20 This unique structure effectively mitigates the volume expansion of the silica anode material during cycling, and diatoms are widely distributed on Earth and are abundant in sources.21 Unfortunately, low conductivity is still the “Achilles' heel” of diatom-based biosilica (DBS) anode electrode materials. The low intrinsic conductivity of silica results in a low initial coulombic efficiency, which is not recognized in commercial large-scale production.22,23 Different strategies have been combined to enhance the overall electrochemical performance of DBS anode materials. The most common method is to combine DBS with conductive carbon. In accordance with Norberg et al.24 diatom frustules were coated with conventional carbon black, and a potentiation holding step was applied. After 50 cycles, the composite electrode maintained a theoretical specific capacity of approximately 723 mA h g−1. Wang et al.25 utilized three kinds of diatoms (Chaetoceros, Navicula, and Nitzschia) as sources of silica material, and the diatom biomass was transformed into a uniform conductive carbon layer coated on the surface of the diatom shell through high-temperature carbonization. The composite anode material has a high initial coulombic efficiency, high performance, and reversible specific capacity of up to 1200 mA h g−1. However, compared with other traditional silica-based anode materials,26–30 DBS coated with a conductive carbon layer still limits the electrochemical efficiency of DBS anode materials.
Researchers have begun to develop methods to combine DBS with more conductive metals to further increase the electrochemical efficiency of DBS anode materials. For example, Chen et al.31 obtained a DBS@C-Co composite material modified with Co nanoparticles by the precipitation of Co(II)–EDTA chelated on the surface of a diatom biosilica (DBS) visual cone and calcination in a nonreducing atmosphere. Co nanoparticles effectively improve the conductivity and electrochemical activity of SiO2. Through the synergistic action of the hollow porous structure, carbon layer, and Co nanoparticles, the DBS@C-Co-60 composite has a high reversible specific capacity of 620 mA h g−1 after 270 cycles at 100 mA g−1. Chen et al.32 also used organosilane as the silicon source required for diatom growth and adsorbed Ag+ with the active site (–SH) of S formed by organosilane on the diatom shell to achieve efficient loading of Ag+ on the shell. After 1000 cycles at a current density of 1 A g−1, the diatom shell had a specific discharge capacity of approximately 660 mA h g−1. Notably, the existing metal phase composite methods can often achieve only single-point loading of metal elements; that is, these metals are almost all in the form of metal nanoparticles embedded on the surface of SiO2 or are mesoporous or microporous. Although these modifications improve the conductivity of SiO2, there is still a certain loss in the transfer of electrons between these dispersed metal elements and a limited contribution to improving only the electrical conductivity of the electrode material, and the high cost of Co and Ag also limits their large-scale applications. Therefore, more efforts are needed to improve DBS anode materials.
In this work, we used diatom frustules as the active anode electrode material source for lithium-ion batteries and functionalized diatom frustules with amino groups through a simple hydrothermal method, forming a Mn2SiO4@C@SiO2 structure. The multifunctional Mn2SiO4 coating not only provides an anode electrode material for lithium-ion batteries but also significantly enhances the conductivity of SiO2. Additionally, it utilizes the Mn nanoparticles of its electrochemical reaction products to recycle Li+ ions, greatly reducing the irreversible loss of capacity and enabling the high capacity and rapid activation of SiO2 composite materials.
Results and discussion
Fig. 1 illustrates the preparation process of AFD@C-Mn. Surface modifications based on SiO2 templates have been widely reported.33 Using diatom-based biosilica as a template, a network nanostructure composed of Mn2SiO4, C, and SiO2 was prepared via a simple hydrothermal method. The hydrolysis product (3-aminopropyl)silanetriol (APST) of APTES is grafted onto the SiO2 surface by condensation of Si–OH groups on the surface of SiO2, and the transition metal Mn2+ then coordinates with amino groups of APTES to form Mn–APST complexes, which then form crystalline Mn2SiO4-like nanoclusters after calcination. The organic carbon skeleton in APST and the organic biomass in the diatom frustules are transformed into a uniform carbon layer coated on the surface of the hollow porous SiO2 structure after high-temperature carbonization in an Ar atmosphere, thus forming a silica-based electrode material with a Mn2SiO4@C@SiO2 structure.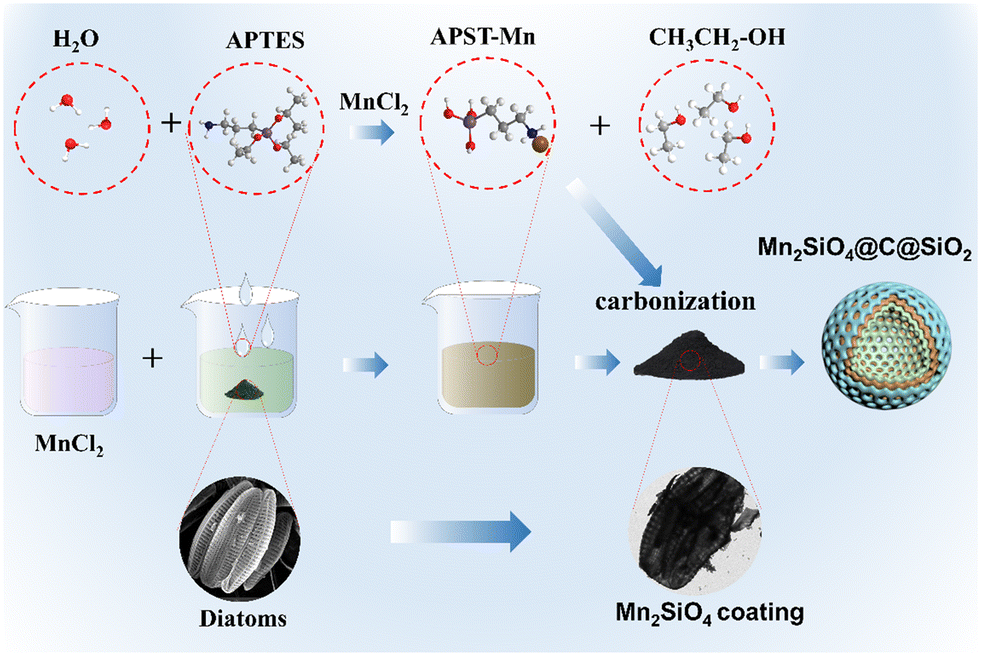 | ||
| Fig. 1 AFD@C-Mn material preparation diagram. | ||
The XRD patterns of the samples with different Mn contents are shown in Fig. 2(a). The broad peak at 22° corresponds to amorphous SiO2 in DBS@C. While the carbon diffraction peak appears at 24° in DBS@C, the appearance of the carbon peak cannot be observed in the sample with Mn, indicating that the addition of Mn promotes the transformation of the crystal structure of carbon toward disordered. As the Mn content gradually increases, diffraction peaks corresponding to Mn2SiO4 appear, indicating that amino silanol reacts with Mn2+ after carbonization. The Mn content of AFD@C-Mn-20 is low, and the supersaturation conditions required for crystal nucleation cannot be reached during the monomer accumulation stage, so crystal nuclei cannot be formed. The sharp diffraction peaks corresponding to AFD@C-Mn-40 and AFD@C-Mn-40–60 at 24.5°, 31.2°, 34.4°, 35° and 50.3° correspond to the (111), (031), (131), (211) and (222) crystal faces of Mn2SiO4, respectively. The amino silane molecular layer has abundant Mn affinity sites (–NH2).34 Therefore, when APTES hydrolyzes to form APST, the APST molecules grafted onto the surface of SiO2 can coordinate with Mn2+ to form chiral structures and generate Mn2SiO4 molecules after carbonization.
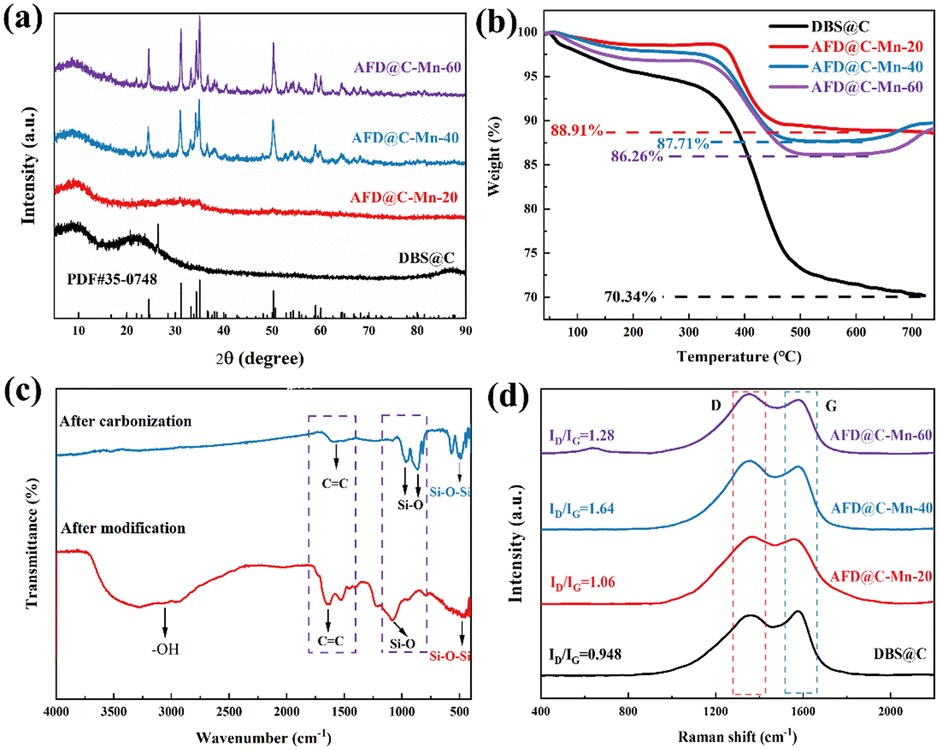 | ||
| Fig. 2 (a) XRD patterns of AFD@C-Mn composites prepared with different Mn contents, (b) TG curves of AFD@C-Mn composites, (c) FTIR spectrum of AFD@C-Mn-40, and (d) Raman spectra of the samples. | ||
Fig. 2(b) shows the TG curves of the composite materials. The TG curve can be divided into three stages over the entire temperature range. Under an O2 atmosphere, SiO2 has a stable structure and almost no mass loss. The mass loss in the first stage (40–200 °C) is attributed to the evaporation of H2O and then corresponds to the salvage of the carbon layer (200–600 °C, C + O2 = CO2). Notably, in the TG curves of AFD@C-Mn-40 and AFD@C-Mn-60, a small increase in mass can be observed above 650 °C, which may be caused by the oxidation of Mn2+. The Si content in the composite was analyzed via ICP-OES. The mass percentages of Si in DBS@C, AFD@C-Mn-20, AFD@C-Mn-40, and AFD@C-Mn-60 were 26.72 wt%, 16.31 wt%, 11.29 wt%, and 7.94 wt%, respectively. This result indicates that as the Mn content increases, the proportion of Si gradually decreases, indicating that the loading efficiency of Mn2+ gradually increases. However, the Mn content of AFD@C-Mn-60 did not increase substantially relative to that of AFD@C-Mn-40. As the coordination efficiency of APTES for Mn2+ reached its maximum, higher levels of Mn2+ could no longer be loaded. Fig. 2(c) shows the FTIR spectra of AFD@C-Mn-40 before and after carbonization. After carbonization, –OH disappears at 3750–2500 cm−1, which is attributed to the destruction of the –OH bond during high-temperature carbonization. The peak at 1600 cm−1 is attributed to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, and the peak decreases after carbonization, which shows that Mn catalyzes the pyrolysis of C in the process of high-temperature carbonization, which is also consistent with the XRD results. The peaks at 958 cm−1 and 866 cm−1 correspond to the symmetric stretching vibration of the Si–O bond in Mn2SiO4.35 The peak at 470 cm−1 corresponds to flexural vibrations from Si–O–Si in SiO2. Raman spectroscopy was used to characterize the carbon composition of the sample.36 Peak D corresponds to crystal defects, and peak G represents the ordered graphitic crystal structure in the carbon phase. As shown in Fig. 2(d), AFD@C-Mn-40 has the largest integral intensity ratio (ID/IG) between the D-band and the G-band. The D band at ≈1350 cm−1 and the G band at ≈1580 cm−1 are attributed to defects and long-range ordered structures in the carbon phase, respectively. The increase in the strength of the D-band indicates an increase in the disorder and defects of the carbon phase in the samples, which may be due to the catalytic cracking of the carbon phase by Mn during calcination.
C, and the peak decreases after carbonization, which shows that Mn catalyzes the pyrolysis of C in the process of high-temperature carbonization, which is also consistent with the XRD results. The peaks at 958 cm−1 and 866 cm−1 correspond to the symmetric stretching vibration of the Si–O bond in Mn2SiO4.35 The peak at 470 cm−1 corresponds to flexural vibrations from Si–O–Si in SiO2. Raman spectroscopy was used to characterize the carbon composition of the sample.36 Peak D corresponds to crystal defects, and peak G represents the ordered graphitic crystal structure in the carbon phase. As shown in Fig. 2(d), AFD@C-Mn-40 has the largest integral intensity ratio (ID/IG) between the D-band and the G-band. The D band at ≈1350 cm−1 and the G band at ≈1580 cm−1 are attributed to defects and long-range ordered structures in the carbon phase, respectively. The increase in the strength of the D-band indicates an increase in the disorder and defects of the carbon phase in the samples, which may be due to the catalytic cracking of the carbon phase by Mn during calcination.
XPS was used to examine samples of amino-functionalized diatom frustules before modification (DBS), after modification (AFD-Mn-40), and after calcination (AFD@C-Mn-40). Through wide scanning spectroscopy, it was confirmed that the modified and calcined composites contained Mn, indicating the successful loading of Mn and that all the samples contained C, N, O and Si. After carbonization, the spectrum of Mn 2p has two main peaks at 652.78 eV and 641.9 eV, corresponding to 2p1/2 and Mn 2p3/2 of Mn, respectively, and further deconvolution of Mn 2p3/2 gives Mn2+ (641.42 eV) and Mn3+ (640.17 eV). The appearance of Mn3+ indicates the inevitable oxidation of Mn2+ in the crystal structure of Mn2SiO4,37 or in the process of synthesizing Mn2SiO4 by high-temperature calcination, Mn2+ is transformed into Mn3+ by the Jahn–Teller effect.38 The characteristic peak of AFD@C-Mn-40 before and after carbonization at 645.78 eV and 643.81 eV should be the satellite peak of Mn. As shown in Fig. 3(c), a single band in the Si 2p spectrum of the sample before adsorption corresponds to the Si–O bond, which proves the existence of the original SiO2. After modification, the surface of SiO2 is covered by dense Mn2SiO4, resulting in the Si 2p peak of the sample after adsorption and calcination (101.87 eV, 101.26 eV), which corresponds to the characteristic Si4+ peak of the tetrahedron in Mn2SiO4, and the C 1s, N 1s, and O 1s XPS spectra are shown in Fig. S1 (ESI†). Moreover, we scraped the composite material of the AFD@C-Mn-40 electrode after 200 cycles from the electrode sheet for the XPS test. Combined with the Auger spectrum peak of Mn, we found that the characteristic peak corresponding to Mn3+ of Mn 2p3/2 disappeared after cycling, and the content of Mn2+ further increased. This shows that the reduction reaction of Mn3+ occurs during cycling (Mn3+ → Mn2+).
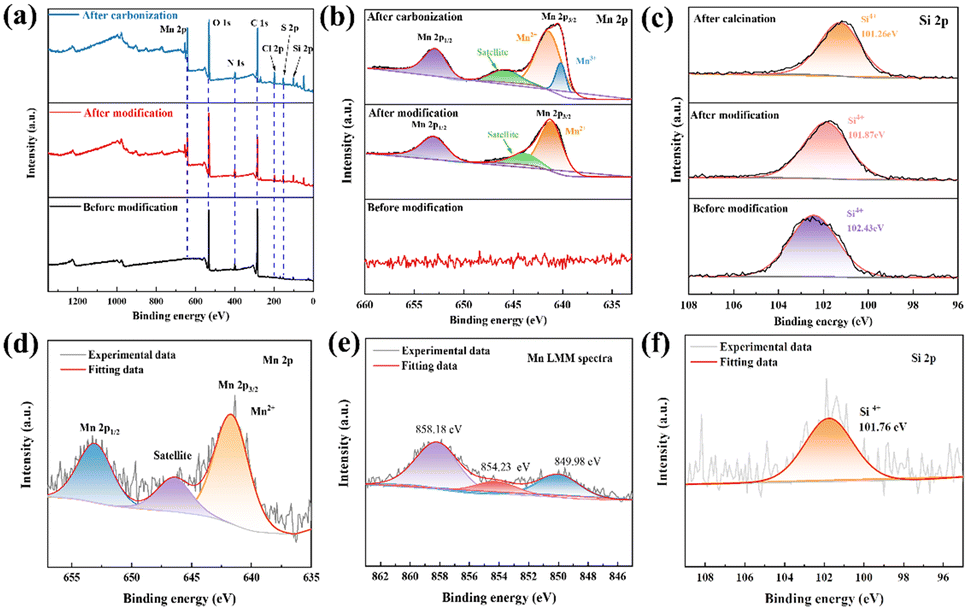 | ||
| Fig. 3 XPS spectra and fitted results of DBS, AFD-Mn-40, and AFD@C-Mn-40. (a) Survey, (b) Mn 2p, (c) Si 2p and (d) Mn 2p peaks of AFD@C-Mn-40 after 200 cycles. (e) Auger electron spectrum of the Mn LMM and (f) Si 2p peak of AFD@C-Mn-40 after 200 cycles. | ||
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images (Fig. 4) revealed that the average size of the diatom-based biosilica used in this study was approximately 5–10 μm, and SEM and TEM images clearly demonstrated the complex, regular, and ordered hollow and porous structure of the diatom-based biosilica. As shown in Fig. 4(b)–(d), after Mn2SiO4 formed, the surface of the diatom frustules became rough. Only a small amount of Mn2SiO4 can be observed on the AFD@C-Mn-20 surface (indicated by the oval in Fig. 4(b)), as shown by TEM-EDX (Fig. S3a, ESI†), and as the content of Mn2+ increases, the surface of the diatom frustules becomes rougher, and more Mn2SiO4 forms (indicated by the I-type areas in Fig. 4(c) and the circle in Fig. 4(d)). We observed a layer of Mn2SiO4 coating with a thickness of approximately 50–100 nm on the surface of the diatoms, High-resolution transmission electron microscopy (HRTEM) images of AFD@C-Mn-40 clearly revealed that a uniform Mn2SiO4 coating with C formed by carbonization of the organic matter on the diatom surface, forming a hollow porous structure composed of Mn2SiO4, C and SiO2, as shown in Fig. 4(c). In addition, the lattice fringes and diffraction rings of Mn2SiO4 can be observed in the HRTEM images of AFD@C-Mn-40 and the electron diffraction pattern, revealing the polycrystalline nature of Mn2SiO4 (Fig. S2, ESI†). Lattice spacings of 0.384 nm, 0.391 nm and 0.551 nm correspond to the (131), (211) and (222) crystal faces of the Mn2SiO4 crystal, respectively. TEM-EDX (Fig. S3b, ESI†) also clearly revealed the uniform distribution of Mn on the surface of the diatom frustules and the porous structure on the surface of the diatom frustules. This three-dimensional structure is conducive to improving the conductivity of SiO2 and alleviating the volumetric strain of silica during cycling. Mn2SiO4 can also act as an anode electrode material for lithium-ion batteries.35,39–41 A lithium-ion diffusion path forms from the outside to the inside, greatly decreasing the diffusion time of Li+ and improving the rate performance.
 | ||
| Fig. 4 SEM, TEM, and HRTEM images of (a) DBS@C, (b) AFD@C-Mn-20, (c) AFD@C-Mn-40, and (d) AFD@C-Mn-60; the red circles indicate Mn2SiO4 crystals. | ||
We also used synchrotron radiation soft X-ray nano-CT to scan the 3D network structure of AFD@C-Mn-40 and obtain the three-dimensional distribution of Mn. As shown in Fig. 5, Mn is uniformly distributed and coated on the surface of the diatom shell. The density of Mn grains in the convex rib of the shell surface is relatively high. The structure of the convex rib of a diatom is conducive to offsetting the mechanical stress caused by the alloying reaction.42 A higher particle density further enhances the mechanical strength of the convex rib.
 | ||
| Fig. 5 Distribution of three-dimensional CT images and Mn in AFD@C-Mn-40 collected from a synchrotron radiation source. | ||
N2 adsorption/desorption isotherms were used to analyze the specific surface area of AFD@C-Mn-40. Fig. S4 (ESI†) shows that the curve of the AFD@C-Mn-40 sample is a type IV adsorption isotherm with an H4-type hysteresis loop, indicating that there are micropores, mesopores, and slits in the AFD@C-Mn-40 sample. As shown in Fig. S5,† the pore size distribution of AFD@C-Mn-40 is mainly between 2 and 20 nm, indicating that the pore types are mainly micropores and mesopores, and mesopores can be directly observed through the SEM and TEM images in Fig. 4. The gap between Mn2SiO4 and the carbon layer or between the carbon layer and SiO2 corresponds to the existence of a slit. By choosing a reasonable P/P0 range, the BET-specific surface area of AFD@C-Mn-40 was 66.8275 m2 g−1 (Fig. S6, ESI†). Detailed data on the BET surface area and micropore diameter of the other composites are shown in Table S1 (ESI†). The large specific surface area of AFD@C-Mn-40 is significantly greater than that of DBS@C (34.6288 m2 g−1), which means that AFD@C-Mn-40 has a greater contact area with Li+ and a greater activation area, which is beneficial for increasing the capacity of the SiO2 anode material.
Electrochemical characterization results
To investigate the electrochemical performance of AFD@C-Mn, button batteries were assembled with AFD@C-Mn and DBS@C as the working electrode and a lithium sheet as the counter electrode. In a previous report, the lithiation mechanism of SiO2 was described in detail:43,44| SiO2 + 4Li+ + 4e− → 2Li2O + Si | (1) |
| 2SiO2 + 4Li+ + 4e− → Li4SiO4 + Si | (2) |
| 5SiO2 + 4Li+ + 4e− ↔ Li2Si2O5 + Si | (3) |
| Si + xLi+ + xe− ↔ LixSi | (4) |
CV tests were conducted on AFD@C-Mn at a scanning speed of 0.5 mV s−1. Fig. 6(a)–(d) shows the results obtained for DBS@C, AFD@C-Mn-20, AFD@C-Mn-40, and AFD@C-Mn-60. The CV curve shows that during cathodic scanning, the reduction peak at 1.2 V for DBS@C and AFD@C-Mn-20 and at 1.5 V for AFD@C-Mn-40 and AFD@C-Mn-60 corresponds to the formation of an SEI. Owing to the incomplete coating of Mn2SiO4 in AFD@C-Mn-20, direct contact between the carbon layer of the SiO2 surface and Li+ may have occurred. As a result, similar reduction peaks appear at 0.75 V and 0.2 V in DBS@C and AFD@C-Mn-20, corresponding to irreversible reactions (1) and (2). The Mn2SiO4 coating is completely coated in AFD@C-Mn-40 and AFD@C-Mn-60; thus, the reduction peak at 0.5 V correspond to reactions (5) (Mn2+ → Mn0). Unlike Li et al.'s study,45 in our experimental system, the potential of the Mn2+ → Mn0 reaction is located at 0.5 V instead of 0.2 V. The reduction peak at 0.01 V is probably due to the alloying reaction between Li and Si (Si + xLi+ + xe− ↔ LixSi).46 During anodic scanning, the oxidation peak at 1.2 V in the first cycle corresponds to the reaction (Mn + Li2O ↔ MnO + 2Li+ + 2e−). In addition, additional oxidation peaks (approximately 1.75 V and 2.25 V) can be observed for the AFD@C-Mn composites, which may be caused by the oxidation of Mn. Notably, additional redox peaks different from those of DBS@C can be observed in the CV curves of the second cycle of AFD@C-Mn, which may be due to the lithiation of SiO2 obtained from the conversion of Mn2SiO4 (reaction (5)) after the first cycle. The detailed reaction is as follows:
| Mn2SiO4 + 4Li+ + 4e− → 2Mn + SiO2 + 2Li2O | (5) |
| Mn2SiO4 + 4Li+ + 4e− → 2MnO + Si + Li2O | (6) |
| Mn + Li2O ↔ MnO + 2Li+ + 2e− | (7) |
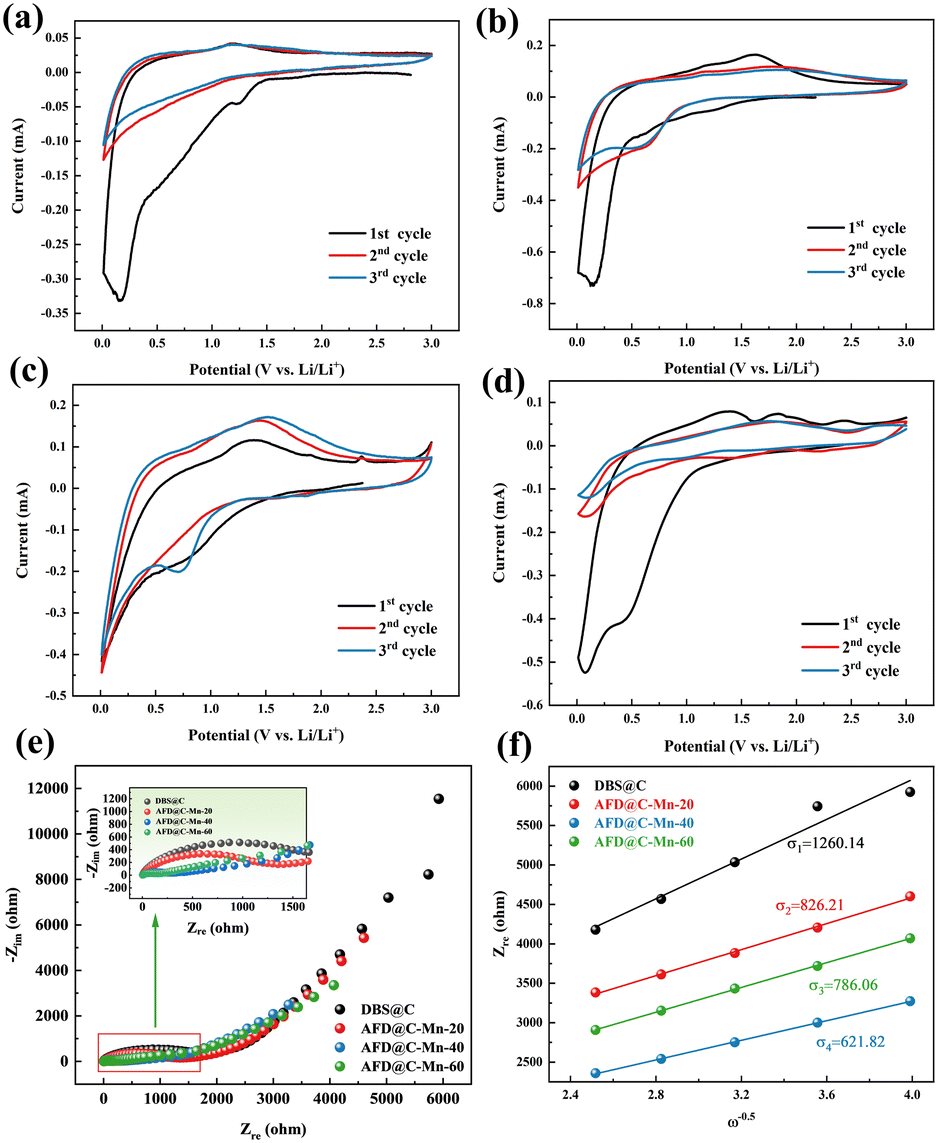 | ||
| Fig. 6 CV curves of (a) DBS@C, (b) AFD@C-Mn-20, (c) AFD@C-Mn-40, and (d) AFD@C-Mn-60; (e) EIS plot and (f) Warburg factors of the AFD@C-Mn anodes. | ||
The above reactions indicate that after a current is applied, a sandwich structure composed of Mn/SiO2@C@SiO2 forms on the surface of the electrode material after Mn2SiO4 is reduced. Mn is also embedded in the outermost SiO2 structure. This structure not only greatly promotes the electrochemical reduction rate of SiO2 but also improves its capacity and rate performance. For SiO2 anode materials, the large loss of Li+ caused by irreversible reactions and low initial coulombic efficiency in the first cycle are often discussed. However, in the AFD@C-Mn material designed in this study, the Mn produced by the reduction of Mn2SiO4 can react with the irreversible reaction product Li2O of SiO2 to recycle Li+ (Mn + Li2O ↔ MnO + 2Li+ + 2e−). The capacity loss caused by the irreversible reaction is further alleviated, which is consistent with the long cycling of the electrode material. After lithiation, the AFD@C-Mn material showed a greater capacity retention rate than DBS@C. The capacity retention rates of DBS@C, AFD@C-Mn-20, AFD@C-Mn-40, and AFD@C-Mn-60 were 29.61%, 49.03%, 49.14%, and 47.6% (after 50 cycles), respectively. Studies on the use of silicate as an anode electrode material for lithium-ion batteries have already been conducted;47 for example, Zhang et al.48 synthesized manganese silicate/C composites for SC electrodes with satisfactory performance. Owing to its thermal stability as a negative electrode material for lithium-ion batteries, this single-metal silicate material, which is based on the conversion reaction, suffers a large volumetric strain. Fortunately, the Mn2SiO4 coating does not alter the hollow porous structure of the original diatom frustules; instead, it forms a nanostructure that preserves the initial hollow porous architecture. This process plays a crucial role in effectively restraining the volume expansion of SiO2.
Fig. 6(e) and (f) shows the EIS plot and Warburg factor of the AFD@C-Mn composite material. The EIS spectra of the composite materials have similar characteristics, with the high-frequency region showing a typical semicircular shape and the low-frequency region showing a sloping line. The semicircle of the high-frequency region corresponds to the charge transfer impedance (Rct) between the electrode and the electrolyte, and the oblique line is the Warburg impedance related to the lithium-ion diffusion coefficient. According to the EIS results, AFD@C-Mn-40 has the lowest resistance, indicating that the Mn2SiO4 coating structure provides a faster charge transfer channel for Li+. The lithium-ion diffusion coefficient is calculated via the following formula:
| Zre ∝ σω−1/2 |
| DLi+ = R2T2/2A2n4F4C2σ2 |
The linear fit between Zre and ω−0.5 is shown in Fig. 6(f). The values of σ and DLi+ for the samples are summarized in Table S2 (ESI†). AFD@C-Mn-40 has the largest DLi+ (1.21 × 10−15), which indicates that the Mn2SiO4 layer significantly enhances the conductivity of the electrode materials and has the highest discharge specific capacity. To verify this result, we subjected the assembled half battery to GITT tests (Fig. S7, ESI†), and the test results were basically consistent with the EIS results.
Fig. 7(a) shows the long cycle performance curves of the AFD@C-Mn composite materials at a current density of 100 mA g−1, DBS@C, AFD@C-Mn-20, AFD@C-Mn-40, and AFD@C-Mn-60; the initial coulombic efficiencies were 28.82%, 39.05%, 46.21%, and 47.77%, respectively. This is consistent with the ICP-MS results; the higher the Mn2+ content is, the greater the initial coulombic efficiency. The Mn2SiO4 coating significantly improved the conductivity and initial coulombic efficiency of the electrode material. After 100 cycles, AFD@C-Mn-40 showed the highest discharge specific capacity, reaching 1112 mA h g−1. Compared with DBS@C, AFD@C-Mn-20, and AFD@C-Mn-60, the battery capacities are 481.2 mA h g−1, 736.8 mA h g−1, and 972.1 mA h g−1, respectively. This occurred because the crystallinity of the Mn2SiO4 coating on the middle surface of AFD@C-Mn-40 is greater, resulting in a denser coating layer. Irregular and uneven coatings lead to more Li+ consumption, and the Si content in these coatings contributes mainly to the electrode. The results in Table S3 (ESI†) show that the Mn content in AFD@C-Mn-40 is almost the same as that in AFD@C-Mn-60, but the Si content in AFD@C-Mn-40 is significantly greater than that in AFD@C-Mn-60 because the theoretical specific capacity of Si is significantly greater than that of Mn. Therefore, the decisive factor for the capacity of the composite material is the Si content, which directly leads to the highest specific discharge capacity of AFD@C-Mn-40. Although DBS@C and AFD@C-Mn-20 have relatively high Si contents, they are limited by the low conductivity of the original SiO2, resulting in a large irreversible capacity loss. Compared with Mn2SiO4 or other metallic silicates reported in other references (Table S4, ESI†), the diatomaceous composite with Mn2SiO4@C@SiO2 multi-layer hollow structure in this work has excellent electrochemical properties.
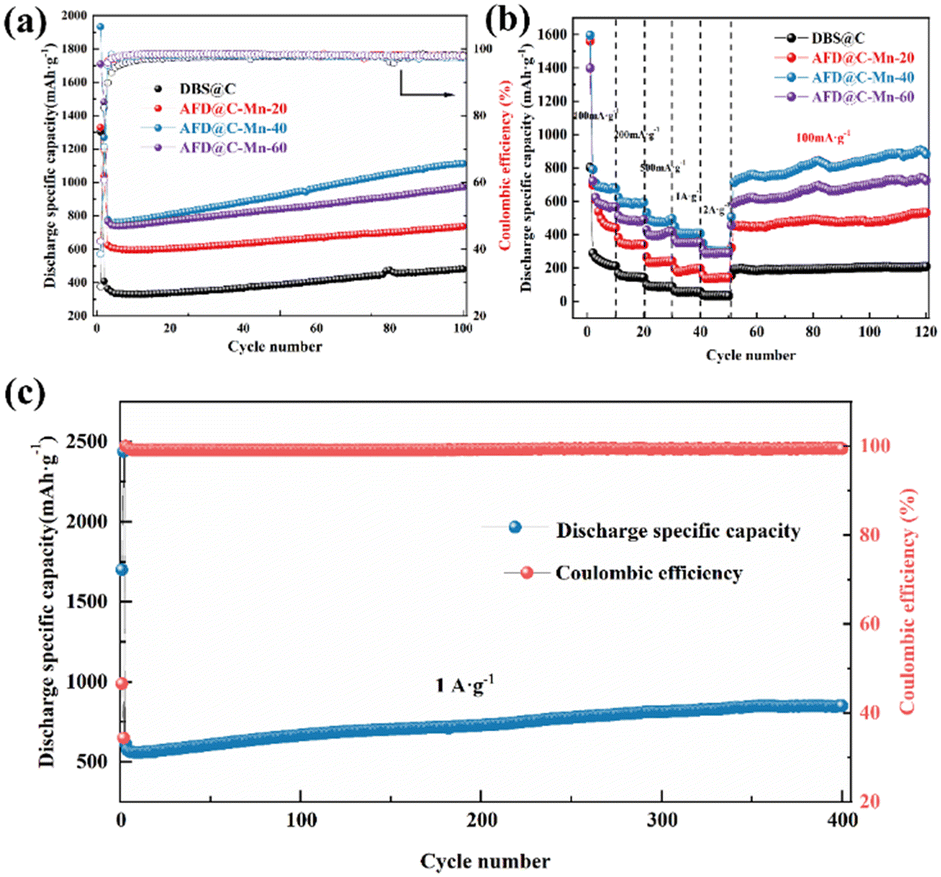 | ||
| Fig. 7 (a) Long cycle performance and (b) gravimetric capacity versus cycle number at different current densities of DBS@C, AFD@C-Mn-20, AFD@C-Mn-40, and AFD@C-Mn-60. (c) Long-term cycling performance of AFD@C-Mn-40 at 1 A g−1. | ||
Fig. 7(b) shows the rate performance of the AFD@C-Mn composite materials at different current densities. After 10 cycles at current densities of 100 mA g−1, 200 mA g−1, 500 mA g−1, 1 A g−1, and 2 A g−1, the optimal rate performances of AFD@C-Mn-40 were 676.81 mA h g−1, 593.06 mA h g−1, 494.84 mA h g−1, 406.86 mA h g−1, and 302.08 mA h g−1, respectively. The detailed rate performance data of the other electrode materials can be found in Table S5 (ESI†). After 70 cycles, the capacity of AFD@C-Mn-40 after the current density returned to 100 mA g−1 was 900.53 mA h g−1, and it continued to increase. The coulombic efficiency remains stable at approximately 98%, indicating excellent electrochemical properties. Fig. 7(c) shows that AFD@C-Mn-40 retains a specific discharge capacity of approximately 849 mA h g−1 after 400 cycles at a current density of 1 A g−1, and a potentiostatic step is performed in the second cycle (holding at 2 mV for 48 h). Overall, the capacity of diatom-based biosilica first decreases but then increases, which was explained in our previous study.49 This is because in SiO2-based anode materials, owing to the insulating nature of SiO2, the current cannot penetrate into the interior of the SiO2 material, so in the initial cycle, not all the active substances undergo a lithium reaction; instead, the core contraction mechanism is controlled by surface diffusion to activate and then participate in the lithium reaction, resulting in a gradual increase in capacity during the charge and discharge cycles. The activation process of SiO2 is proposed to be a crystallization-like process.
Fig. 8 shows that a similar voltage platform (0.5 V and 1.0 V)40 and capacity platform appear in the electrode material AFD@C-Mn, which corresponds to the oxidation–reduction peak in the CV curve, which comes from the oxidation–reduction reaction between Mn2SiO4 and Li+. Interestingly, the lithium potential of MnO is only approximately 0.5 V (MnO + 2Li+ + 2e− ↔ Mn + Li2O), which is the lowest among transition metal oxides. This also indirectly proves that the voltage plateau at 0.5 V in the charge and discharge curve of the AFD@C-Mn electrode may be caused by the reduction reaction of Mn2+, corresponding to reaction (7). We observe a similar trend in the charge and discharge curves of AFD@C-Mn except for the first cycle of DBS@C, where the presence of Mn leads to a decrease in the voltage platform, which further proves that the presence of SiO2 (the product of electrochemical reduction at the AFD@C-Mn electrode.) The derived Mn/SiO2@C@SiO2 sandwich structure enables the efficient activation and utilization of the SiO2 anode electrode.
 | ||
| Fig. 8 Charge and discharge curves of (a) DBS@C, (b) AFD@C-Mn-20, (c) AFD@C-Mn-40, and (d) AFD@C-Mn-60. | ||
Activation mechanism analysis
The activation process can be explained in Fig. 8, which is based on the conversion of Mn2SiO4 to SiO2 to form the Mn/SiO2@C@SiO2 sandwich structure shown in Fig. 4(c), as shown in reactions (5)–(7). The activation mechanism of AFD@C-Mn is divided into two stages: (i) Mn2SiO4 conversion and (ii) double-layer activation of Mn/SiO2@C@SiO2 (Fig. 9).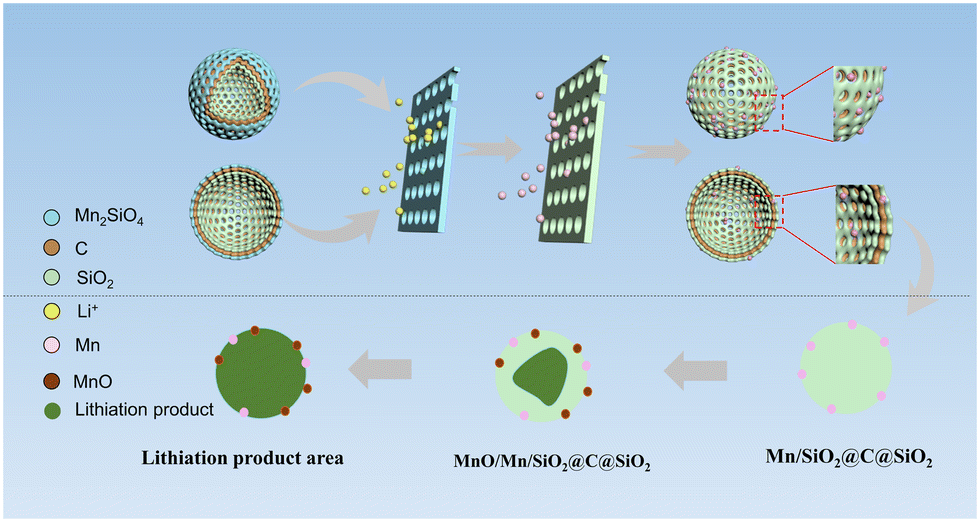 | ||
| Fig. 9 Schematic diagram of the rapid activation mechanism of the Mn2SiO4@C@SiO2 multilayer-structure composite. | ||
(i) After the application of current, the conversion of Mn2SiO4 can be divided into two parts: one is that the lithium reaction between Mn2SiO4 and Li+ generates SiO2, Mn, and Li2O (reaction (5)), and the other is the reaction of Mn2SiO4 with Li+ to produce Li2O and MnO. Li2O has been shown to exist in a crystalline form in lithium products. The XPS spectra (Fig. 3d–f) and XRD results (Fig. S8, ESI†) of AFD@C-Mn-40 after cycling show that the characteristic peaks of MnO appear in the cycled composites. Notably, the products of both conversion reactions have been proven to have stronger conductivity than SiO2. The resulting electron conduction zone composed of lithium products can accelerate the transfer of electrons, and the current can further penetrate the carbon layer inside the composite and the SiO2 on the surface of the diatom shell, which is favorable for the accelerated activation of silica. Therefore, the composite material transforms into a sandwich structure of Mn/SiO2@C@SiO2.
(ii) With increasing current depth, the newly generated SiO2 on the surface of the diatom shell also begins to undergo a lithium reaction. The mechanism of the lithium reaction of SiO2 and the activation process of SiO2 on the surface of the diatom shell have been described by researchers.50 Unlike in the previous studies, the Mn2SiO4 in this study was also derived from the metal Mn. Mn can further react with the lithium reaction products of SiO2 to produce Li2O and MnO. Li2O is one of the irreversible lithium reaction products of SiO2; although it can improve the conductivity of SiO2, it also leads to irreversible loss of Li+. In this work, Mn can react reversibly with Li2O to achieve the recycling of Li+. The results show that the specific discharge capacity of AFD@C-Mn is significantly greater than that of DBS@C. MnO can achieve the smallest volume expansion (∼186%) in various metal oxide anode materials.
Notably, the Si and LixSi produced by the activation of SiO2, which has been widely reported. According to reactions (1)–(4), the generation of lithium products is accompanied by their generation. Therefore, it is speculated that they may be dispersed in the lithium region. The porous SiO2 and the conductive carbon layer form a fast Li+ diffusion channel. The activation region gradually shrinks to SiO2 in the core to form a new activation region, so the increase in the capacity of the composite material begins to accelerate.
Conclusions
An electrode material with a network structure composed of Mn2SiO4@C@SiO2 prepared by using biobased SiO2 as a template was proposed. The good electrochemical activity, dense crystal layer, and thin and uniform structure of the outermost Mn2SiO4 coating not only significantly improved the conductivity of SiO2 and the initial coulombic efficiency but also effectively inhibited the volume expansion of SiO2 during cycling, and Mn2SiO4 transformed into SiO2 after electrochemical reduction, forming a sandwich structure of Mn/SiO2@C@SiO2 and further improving the capacity of the SiO2 anode electrode. The unique Mn2SiO4@C@SiO2 material was constructed in a one-pot, single-step synthesis. This process is facile and effective and can be easily scaled up for mass production. Notably, the metal Mn2+ used in this method has the best capacity performance in all the studies on metal ion-modified diatom-based biological silica. After only 100 cycles, the specific discharge capacity of AFD@C-Mn-40 was greater than 1112 mA h g−1 and continued to increase.Experimental section
Diatom cultivation and pretreatment
Diatom seeds (Navicula sp.) were purchased from the freshwater algae culture library of the Wuhan Institute of Aquatic Biology in China and cultured in the laboratory. The initial culture density was 120 cells per mL. The diatom cultivation device used was a conical flask with a capacity of 3 L. F/2 medium containing artificial seawater was used to cultivate diatoms under conditions of a light-to-dark ratio of 12 h![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12 h, a light intensity of 2300 lx, and a constant temperature of 25 °C. Cultured diatoms were collected, soaked in 5% HCl for 6 h, washed with anhydrous ethanol for 1–2 times, and finally freeze-dried at −70 °C for 24 h to obtain diatom powder.
:12 h, a light intensity of 2300 lx, and a constant temperature of 25 °C. Cultured diatoms were collected, soaked in 5% HCl for 6 h, washed with anhydrous ethanol for 1–2 times, and finally freeze-dried at −70 °C for 24 h to obtain diatom powder.
AFD@C-Mn material preparation
First, a 0.1 M MnCl2 solution was prepared, 0.2 g of diatom powder was evenly dispersed into 100 mL of anhydrous ethanol solute, the mixture was sonicated for 20 minutes, 2 mL of APTES was added to the above-mixed solution, and then 20, 40, and 60 mL of MnCl2 solution were added. The mixture was stirred and heated at 80 °C for 1 h, centrifuged to collect the diatom frustules, washed with anhydrous ethanol, freeze-dried at −70 °C for 24 h, and finally heated to 600 °C for 3 h under an Ar atmosphere at a rate of 5 °C min−1. The final products were referred to as AFD@C-Mn-20, AFD@C-Mn-40, and AFD@C-Mn-60. The unmodified diatom frustules were referred to as DBS@C.Materials characterization
X-ray powder diffraction (XRD, Rigaku Smart Lab SE) was used to detect the crystal structure of the materials at a scan rate of 5° min−1 with Cu Kα radiation. Scanning electron microscopy (SEM, MIRA3 LMH) and transmission electron microscopy (TEM, TF20) were used to observe the microscopic appearance of the composites, and TEM-EDX was used to characterize the elemental composition and distribution of the samples. Selective area electron diffraction (SAED) was used to observe the diffraction ring structure of Mn2SiO4, and the BET surface areas were calculated from nitrogen adsorption–desorption isotherms measured via a Micromeritics APSP 2460. TG curves were measured via a thermogravimetric analyzer (PerkinElmer TL8000). XPS analysis was performed with Al Kα radiation (Thermo Scientific K-Alpha). The element content was measured and calculated by ICP-OES (Agilent 5110). The chemical bonds of the samples were analyzed via Fourier transform infrared spectroscopy (FTIR, Nicolet Nexus 670) in the range of 400~4000 cm−1. Raman spectroscopy of the composite was performed with a LabRAM HR Evolution instrument (Horiba Japan). The soft X-ray nano-CT technique, which is based on synchrotron radiation, was used to scan and construct the three-dimensional distribution of Mn on the surface of the diatom shell. The dual-energy subtraction energies of Mn are 645 eV and 635 eV.Half-cell assembly
The electrochemical performance of AFD@C-Mn was tested via a CR2016 button battery. The working electrode was prepared with 70% AFD@C-Mn material, 20% conductive carbon black (Super black), 10% PVDF and N-methyl-2-pyrrolidine (NMP) solvents on a planetary ball mill at a rate of 500 r min−1 for 3 h, and the ball mill slurry was coated on a 10 μm copper foil. The coating thickness was 50 μm, and the coated copper foil was vacuum-dried at 80 °C for 24 h and cut into a circular pole sheet with a diameter of 12 mm. The electrolyte is a commercial lithium electrolyte purchased from the market and consists of 1 M LiPF6 in EC/DEC/EMC with a volume ratio of 1:1:1. Lithium foil was used as the counter and reference electrode, and a Celgard 2400 was used as the separator. Finally, CR2016-type coin cells were assembled in a glove box filled with Ar.
Electrochemical performance
Galvanostatic charge–discharge tests were carried out in the potential range of 0.001–3.0 V (vs. Li/Li+) on a multichannel battery testing system (LAND CT3002A) with different currents. CV was performed at a scan rate of 0.1 mV s−1 between 0.01 and 3 V (vs. Li+/Li) with a Gamry Interface 1010E electrochemical workstation and electrochemical impedance spectroscopy (EIS) with a frequency range from 0.01 to 10 MHz was tested by an electrochemical workstation (Gamry Interface 1010E).Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the National Natural Science Foundation of China (No. 41802038 & No. 41830318) and Key Project of Yunnan Precious Metals Laboratory Co., Ltd (No. YPML-2023050213).References
- S. Wu, C. Han, J. Iocozzia, M. Lu, R. Ge, R. Xu and Z. Lin, Angew. Chem., Int. Ed., 2016, 55, 7898–7922 CrossRef CAS.
- Z. Wen, G. Lu, S. Cui, H. Kim, S. Ci, J. Jiang, P. T. Hurley and J. Chen, Nanoscale, 2014, 6, 342–351 RSC.
- X. Sun and L. Qiao, J. Mater. Sci.: Mater. Electron., 2021, 32, 10277–10288 CrossRef CAS.
- R. V. Salvatierra, W. Chen and J. M. Tour, Adv. Energy Sustainability Res., 2021, 2, 2000110 CrossRef CAS.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef.
- P. U. Nzereogu, A. D. Omah, F. I. Ezema, E. I. Iwuoha and A. C. Nwanya, Appl. Surf. Sci., 2022, 9, 100233 CrossRef.
- Y. Ma, H. Qu, W. Wang, Y. Yu, X. Zhang, B. Li and L. Wang, Adv. Funct. Mater., 2023, 33, 2211648 CrossRef CAS.
- M. S. Al Ja'farawy, D. N. Hikmah, U. Riyadi, A. Purwanto and H. Widiyandari, J. Electron. Mater., 2021, 50, 6667–6687 CrossRef.
- X. Zhang, D. Kong, X. Li and L. Zhi, Adv. Funct. Mater., 2019, 29, 1806061 CrossRef.
- J. Yang, Y. Wang, W. Li, L. Wang, Y. Fan, W. Jiang, W. Luo, Y. Wang, B. Kong, C. Selomulya, H. K. Liu, S. X. Dou and D. Zhao, Adv. Mater., 2017, 29, 1700523 CrossRef PubMed.
- W. Wu, M. Wang, J. Wang, C. Wang and Y. Deng, ACS Appl. Energy Mater., 2020, 3, 3884–3892 CrossRef CAS.
- A. Lisowska-Oleksiak, A. P. Nowak and B. Wicikowska, RSC Adv., 2014, 4, 40439–40443 RSC.
- A. P. Nowak, A. Lisowska-Oleksiak, B. Wicikowska and M. Gazda, J. Solid State Electrochem., 2017, 21, 2251–2258 CrossRef CAS.
- A. P. Nowak, M. Sprynskyy, W. Brzozowska and A. Lisowska-Oleksiak, Algal Res., 2019, 41, 101538 CrossRef.
- M. S. Garapati, A. P. V. K. Saroja and R. Sundara, Mater. Sci. Eng., B, 2020, 261, 114695 CrossRef CAS.
- M. De Stefano, L. De Stefano and R. Congestri, Superlattices Microstruct., 2009, 46, 64–68 CrossRef CAS.
- D. Losic, R. J. Pillar, T. Dilger, J. G. Mitchell and N. H. Voelcker, J. Porous Mater., 2007, 14, 61–69 CrossRef CAS.
- C. Jeffryes, J. Campbell, H. Li, J. Jiao and G. Rorrer, Energy Environ. Sci., 2011, 4, 3930–3941 RSC.
- D. Liu, P. Yuan, Q. Tian, H. Liu, L. Deng, Y. Song, J. Zhou, D. Losic, J. Zhou, H. Song, H. Guo and W. Fan, Nat. Commun., 2019, 10, 4829 CrossRef PubMed.
- X. Liu, Y. Gao, R. Jin, H. Luo, P. Peng and Y. Liu, Nano Energy, 2014, 4, 31–38 CrossRef CAS.
- V. Renman, M. V. Blanco, A. N. Norberg, F. Vullum-Bruer and A. M. Svensson, Solid State Ionics, 2021, 371, 115766 CrossRef CAS.
- W. Hua, I.-E. Nylund, F. Cova, A. M. Svensson and M. V. Blanco, Sci. Rep., 2023, 13, 20447 CrossRef CAS PubMed.
- J. Luo, P. Liu, G. Peng and J. T. Zhang, ChemistrySelect, 2023, 8, e202204313 CrossRef CAS.
- A. N. Norberg, N. P. Wagner, H. Kaland, F. Vullum-Bruer and A. M. Svensson, RSC Adv., 2019, 9, 41228–41239 RSC.
- Z. Wang, J. Zhao, S. Liu, F. Cui, J. Luo, Y. Wang, S. Zhang, C. Zhang and X. Yang, ACS Sustainable Chem. Eng., 2021, 9, 844–852 CrossRef CAS.
- J. Kim, N. Dan Thien, J. Kang and S. Song, J. Alloys Compd., 2015, 633, 92–96 CrossRef CAS.
- D. Xu, Y. Yu, M. Zheng, G. Guo and Y. Tang, Electrochem. Commun., 2003, 5, 673–676 CrossRef CAS.
- F. Wan, W. Wang, Z. Zou, H. Xie, H. Ping and Z. Fu, J. Mater. Sci. Technol., 2021, 72, 61–68 CrossRef CAS.
- G. Hu, H. Liu, Y. Luo, K. Zhang, D. Guo, X. Liu and A. Qin, Mater. Lett., 2022, 308, 131288 CrossRef CAS.
- Z. Favors, W. Wang, H. Bay, A. George, M. Ozkan and C. S. Ozkan, Sci. Rep., 2014, 4, 4605 CrossRef.
- Y. Chen, H. Liu, W. Xie, Z. Shen, J. Xia, Z. Nie and J. Xie, Small, 2023, 19, 2300707 CrossRef CAS PubMed.
- Y. Chen, H. Liu, Z. Shen, K. Yang, J. Xia, P. Yuan, Z. Nie, H. Liu and J. Xie, iScience, 2024, 27, 108850 CrossRef CAS PubMed.
- B. M. Estevao, I. Miletto, N. Hioka, L. Marchese and E. Gianotti, ChemistryOpen, 2021, 10, 1251–1259 CrossRef CAS PubMed.
- Y. Wang, J. Xu, X. Xu, D. Yang, X. Zheng, J. Pan, T. Zhang, F. Qiu and C. Li, Appl. Organomet. Chem., 2018, 32, 4182 CrossRef.
- Y. Wang, T. Li, Y. Qi, R. Bai, L. Yin, H. Li, N. Lun and Y.-J. Bai, Electrochim. Acta, 2015, 186, 572–578 CrossRef CAS.
- A. P. Nowak, B. Wicikowska and A. Lisowska-Oleksiak, Solid State Ionics, 2014, 263, 131–139 CrossRef CAS.
- X. Dong, Y. Mu, L. Shen, H. Wang, C. Huang, C. Meng and Y. Zhang, Chem. Eng. J., 2023, 456, 141031 CrossRef CAS.
- R. Freitag, T. J. Muller and J. Conradie, J. Chem. Crystallogr., 2014, 44, 352–359 CrossRef CAS.
- C. Zhu, Y. Zhang, Y. Ye, G. Li and F. Cheng, ACS Appl. Mater. Interfaces, 2023, 39363–39373 CrossRef CAS PubMed.
- J. Zhu, C. Tang, Z. Zhuang, C. Shi, N. Li, L. Zhou and L. Mai, ACS Appl. Mater. Interfaces, 2017, 9, 24584–24590 CrossRef CAS PubMed.
- H. Wei, Z. Xia and D. Xia, ACS Appl. Mater. Interfaces, 2017, 9, 43657–43664 CrossRef CAS.
- A. P. Nowak, M. Sprynskyy, I. Wojtczak, K. Trzcinski, J. Wysocka, M. Szkoda, B. Buszewski and A. Lisowska-Oleksiak, Materials, 2020, 13, 101538 Search PubMed.
- Q. Sun, B. Zhang and Z.-W. Fu, Appl. Surf. Sci., 2008, 254, 3774–3779 CrossRef CAS.
- Z. Liu, Q. Yu, Y. Zhao, R. He, M. Xu, S. Feng, S. Li, L. Zhou and L. Mai, Chem. Soc. Rev., 2019, 48, 285–309 RSC.
- S.-R. Li, Y. Sun, S.-Y. Ge, Y. Qiao, Y.-M. Chen, I. Lieberwirth, Y. Yu and C.-H. Chen, Chem. Eng. J., 2012, 192, 226–231 CrossRef CAS.
- M. N. Obrovac and V. L. Chevrier, Chem. Rev., 2014, 114, 11444–11502 CrossRef CAS PubMed.
- S.-H. Yu, B. Quan, A. Jin, K.-S. Lee, S. H. Kang, K. Kang, Y. Piao and Y.-E. Sung, ACS Appl. Mater. Interfaces, 2015, 7, 25725–25732 CrossRef CAS PubMed.
- H. Jiang, Y. Zhang, C. Wang, Q. Wang, C. Meng and J. Wang, Inorg. Chem. Front., 2019, 6, 2788–2800 RSC.
- Y. Chen, H. Liu, K. Yang, Z. Nie, J. Xia, Z. Shen, J. Xie and H. Liu, J. Mater. Chem. A, 2023, 11, 16704–16713 RSC.
- J. E. Entwistle, S. G. Booth, D. S. Keeble, F. Ayub, M. Yan, S. A. Corr, D. J. Cumming and S. V. Patwardhan, Adv. Energy Mater., 2020, 10, 2001826 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lf00324a |
| This journal is © The Royal Society of Chemistry 2025 |