
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A medicine-inspired hydroxyl-rich equimolar ZnSO4/D-mannitol electrolyte enables horizontally stacked Zn deposition for long-cycling aqueous batteries†
Jin
Xiao‡
,
Chenbo
Yuan‡
,
Cong
Liu
,
Ximei
Sun
,
Bing
Cheng
,
Zitong
Huang
,
Xiaowen
Zhan
 * and
Lingyun
Zhu
*
* and
Lingyun
Zhu
*
School of Materials Science and Engineering, Key Laboratory of Structure and Functional Regulation of Hybrid Materials of Ministry of Education, Anhui University, 230601 Hefei, Anhui, PR China. E-mail: xiaowen.zhan@ahu.edu.cn; 22149@ahu.edu.cn
First published on 15th November 2024
Abstract
Dendrite growth and adverse side reactions at the Zn anode often compromise the cycling stability and lifespan of aqueous Zn-metal batteries (AZMBs). Inspired by the high osmotic dehydration effect in medicine, this study introduces an equimolar ZnSO4/D-mannitol (MNT) electrolyte to stabilize the Zn anodes. Comprehensive spectrochemical characterization and theoretical calculations, coupled with rigorous electrochemical and electroanalytical tests, show that the hydroxyl-rich MNT strongly interacts with water, reducing hydrogen bonds and reshaping solvation structures of hydrated Zn2+, thereby effectively suppressing corrosion and side reactions. Furthermore, the preferential adsorption of MNT at the Zn anode surface regulates the interfacial reaction environment, enabling dendrite-free, orderly-stacked and corrosion-less Zn deposition. This is confirmed by backscattered-electron scanning electron microscopy observations on cycled electrodes prepared using a cross-section polisher. As a result, the use of a D-mannitol-enriched ZnSO4 electrolyte extends the cycle life of Zn||Zn symmetric cells to over 2980 hours and significantly enhances the long-term cycling performance of Zn||PANI (polypyrrole–polyaniline) full cells at a low N/P ratio of 22.48.
1. Introduction
Aqueous zinc-metal batteries (AZMBs) have emerged as strong contenders for the next generation of large-scale energy storage due to the low redox potential of zinc (Zn) anodes (−0.762 V vs. standard hydrogen electrode, SHE), their high theoretical capacity (820 mA h g−1, 5851 mA h cm−3) and their inherent safety in water-based environments.1 However, challenges arise due to the strong affinity of Zn2+ ions for forming [Zn(H2O)6]2+ solvation structures with water molecules in aqueous electrolytes, leading to uneven Zn deposition and dendrite growth.2–4 Moreover, the thermodynamically unstable Zn in aqueous solutions is prone to corrosion and side reactions, such as hydrogen evolution reactions (HERs), which cause battery volume expansion and potential failure.5 These issues significantly compromise the cycling stability and lifespan of AZMBs, posing obstacles to their practical use.Numerous strategies have been developed to address these challenges faced by AZMBs, including the construction of artificial interface layers,6–9 designing anode architectures,10,11 and electrolyte modifications.12–14 While creating artificial interface layers and designing anode architectures are often complex and time-intensive processes, electrolyte engineering such as adding electrolyte additives presents a simpler, direct, and cost-effective alternative.15,16 Currently, electrolyte modifications cover five main approaches: (1) forming hydrogen bonds with water to reduce its activity and thus mitigate corrosion and HERs;17,18 (2) altering the solvation shell of Zn2+ ions to regulate their solvation structures;19,20 (3) adsorbing onto the Zn anode surface to promote uniform Zn2+deposition;21,22 (4) creating in situ solid electrolyte interphase (SEI) layers on the Zn anode to protect against electrolyte attack;23–26 (5) accumulating at the tips of the Zn anode to establish an electrostatic shield.27,28 Although these strategies have notably improved the stability of Zn anodes, their effectiveness is generally focused on one aspect and falls short of the comprehensive requirements for high-performance AZMBs. Therefore, there is still a critical need to develop an environmentally sustainable, cost-effective, and versatile electrolyte modification strategy to advance AZMB technology.
In medicine, D-mannitol (MNT), a monosaccharide macromolecule, is widely used as a dehydrating agent due to its significant osmotic effects, which are also common in various plants.29 Its ability to promote water reflow from brain tissue into blood vessels results in brain tissue dehydration (Fig. 1a). Before this, Zhang et al. introduced trace amounts (0.02 M) of MNT into a 2 M ZnSO4 solution, demonstrating the ability of MNT to reconstruct the solvation structure of Zn2+ and inhibit dendritic growth on the Zn anode surface.30 However, the trace amount of MNT failed to disrupt the hydrogen bond network between active water molecules, and its role in modulating the anode/electrolyte interface was limited. Inspired by the effect of high concentrations of MNT molecules in increasing the attraction to water, we have incorporated MNT at an equimolar concentration into the conventional 1 M ZnSO4 aqueous electrolyte to explore its modification mechanism (Fig. 1b and c). Through spectral analysis, molecular dynamics (MD) simulations and density functional theory (DFT) calculations, we have found that MNT increases the electrolyte's affinity for water, forming hydrogen bonds with water molecules and disrupting the original electrolyte's hydrogen bond network. Furthermore, MNT can integrate into the solvation structure of Zn2+, replace an active water molecule, and reshape the solvation structure, effectively reducing corrosion and side reactions. Additionally, MNT's preferential adsorption on the Zn anode surface facilities uniform Zn2+ deposition along the (002) crystal plane. Leveraging an advanced backscattered-electron scanning electron microscope (BSE-SEM) coupled with a cross-section polisher (CP), we have observed a unique orderly stacked deposition structure free of dendrites and heavy corrosion enabled by MNT. As a result, both symmetric cells and full cells with MNT exhibit significantly improved cycling performances. The present work successfully applies the concept of MNT locking water for tissue dehydration to improving the performance of AZMBs, offering a promising avenue for future research.
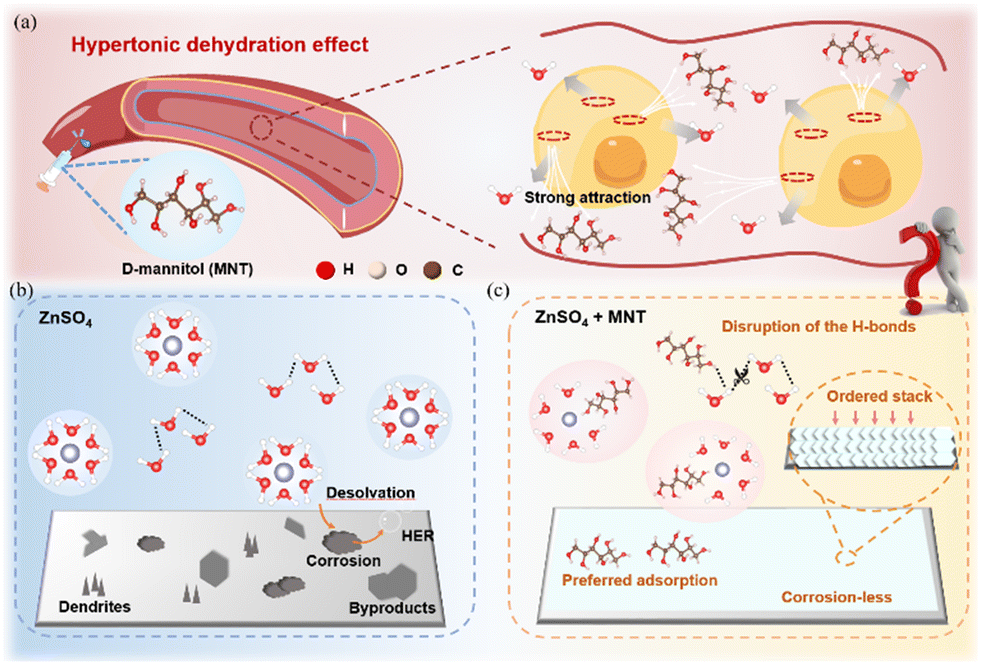 | ||
| Fig. 1 (a) Schematic diagram of the mechanism by which increased osmotic pressure in MNT causes tissue dehydration. Schematic illustration of Zn2+ solvation structures and interfacial chemistry at Zn anodes in (b) ZnSO4 and (c) ZnSO4 + MNT electrolytes. | ||
2. Results and discussion
2.1 Strong MNT/H2O interactions and reshaped solvation structures of Zn2+
MNT is a polyol, each molecule of which contains six hydroxyl groups. We prepared 1 M ZnSO4 aqueous electrolytes with varying concentrations of MNT (denoted as “x-MNT”, x = 0.01, 0.1, 0.5, 1, 2). As depicted in Fig. S1a,† the pure 1 M ZnSO4 solution is clear and transparent. However, as the concentration of added MNT increases to 2 M, the solution becomes turbid, indicating the oversaturation of MNT at this concentration. Additionally, we measured the pH and ion conductivity of the above solutions (Fig. S1b†). With the increase of MNT concentration, both the pH value and ion conductivity of the solutions slightly decrease, indicating that MNT primarily exists in the solutions in molecular form.Hydroxyl groups in MNT can form hydrogen bonds with active water molecules in aqueous solution, thereby suppressing water activity and enhancing thermodynamic stability.31 To explore the influence of the abundant hydroxyl groups in MNT molecules on the hydrogen bond network, we conducted nuclear magnetic resonance (NMR) tests on electrolyte solutions with different MNT concentrations. As shown in Fig. 2a, as the MNT concentration increases, the 2H peak shifts from 4.681 ppm to 4.683 ppm, indicating a reduction in the number of free water molecules.32 This suggests the formation of hydrogen bonds between MNT and H2O, disrupting the original hydrogen bond network among water molecules. Further definitive evidence is provided by FT-IR and Raman spectroscopy. The FT-IR spectra (Fig. 2b) show a blue shift in the stretching vibration peak of O–H with increasing MNT concentration, confirming a decrease in water activity.12 Peaks corresponding to hydrogen bond vibrations in the Raman spectra also shifts to higher wavenumbers (Fig. 2c), further corroborating the strong interaction between MNT and H2O.
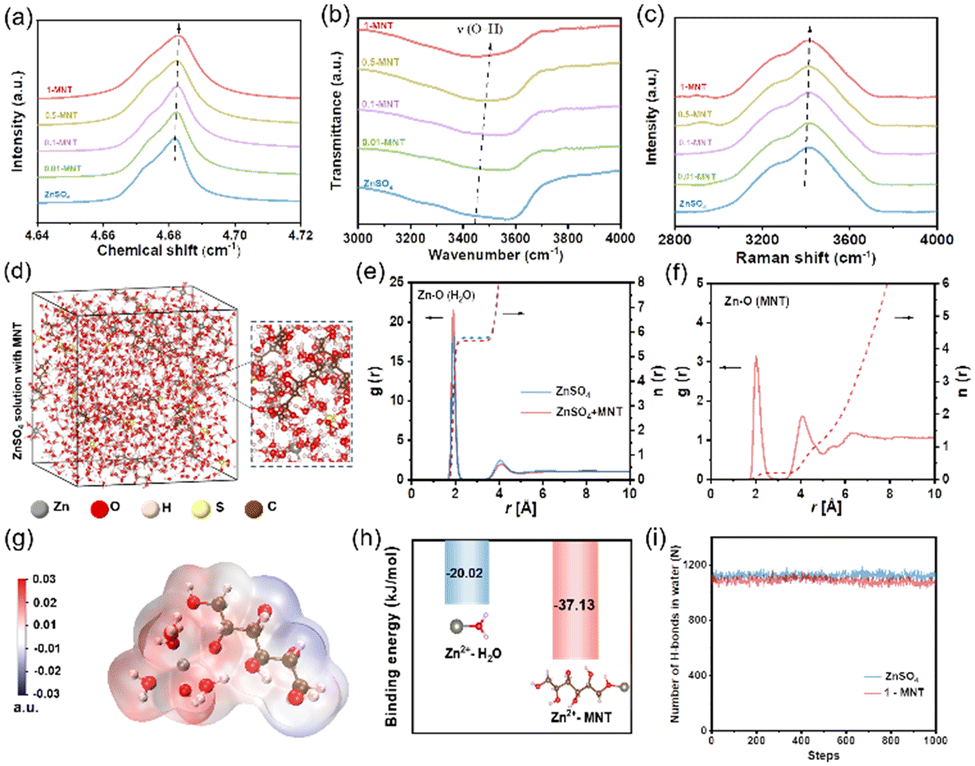 | ||
| Fig. 2 (a) 2H NMR spectra of D2O, (b) FT-IR spectra and (c) Raman spectra (v-OH band) of 1 M ZnSO4 electrolytes with various MNT concentrations. (d) Snapshot from MD simulation of the 1-MNT electrolyte and the local enlargement. RDFs of (e) Zn–O (H2O) and (f) Zn–O (MNT) in different systems. (g) ESP distribution of the [Zn(H2O)5MNT]2+ solvation structure. (h) Binding energy of Zn2+–H2O and Zn2+-MNT. (i) Number counts for hydrogen-bonds inside ZnSO4 and 1-MNT electrolytes. | ||
To explore the modulation effect of MNT on the solvation structure of Zn2+, we conducted molecular dynamics (MD) simulations and constructed models for pure ZnSO4 and 1-MNT solution systems. In the ZnSO4 system, the typical solvation structure for Zn2+ is represented as [Zn(H2O)6]2+ (Fig. S2a†). Upon the addition of MNT, one MNT molecule integrates into the solvation shell, displacing a water molecule (Fig. 2d). This change was further substantiated by the analysis of radial distribution functions (RDFs) and coordination number (CN) distribution functions. A Zn2+–O (H2O) peak appears at the first solvation shell with a radius of 2. With MNT present, the average CN of Zn2+ with H2O decreases from 5.75 to 5.62 (Fig. 2e), and a new peak with a CN of 0.2 emerges for Zn2+–O (MNT) (Fig. 2f), confirming the formation of [Zn(H2O)5MNT]2+. The electrostatic potential (ESP) of the solvation shell, as derived from MD simulations, was also calculated (Fig. 2g and S2b†). The substitution of one H2O molecule by MNT in the original [Zn(H2O)6]2+ structure results in the ESP of [Zn(H2O)5MNT]2+ being divided into distinct positive and negative regions, which enhances the transmission speed of solvated molecules under electrostatic interactions from the external environment. Further investigations using density functional theory (DFT) examined the interactions between Zn2+, H2O and MNT molecules. The binding energy between Zn2+ and MNT was found to be significantly higher than that between Zn2+ and H2O (−37.13 kJ mol−1vs. −20.02 kJ mol−1, Fig. 2h), indicating a more energetically favorable interaction between Zn2+ and MNT molecules. Additionally, the number of hydrogen bonds within each system was quantified (Fig. 2i). The addition of MNT resulted in a decrease in the number of hydrogen bonds among H2O molecules, which aligns with the spectral analysis previously discussed.
2.2 Anode/electrolyte interface regulation induced by MNT adsorption
In addition to altering the solvation structure, MNT plays a crucial role in regulating the anode/electrolyte interface. Tafel curves were used to evaluate the corrosion resistance of Zn electrodes in the two electrolytes, and the corrosion current density was obtained by extrapolating polarization curves in the strong polarization region (Fig. 3a). The Zn electrode in the 1-MNT electrolyte exhibits a lower corrosion current density (1.826 mA cm−2) than that in pure ZnSO4 (3.121 mA cm−2). To further explore the anti-corrosion ability of MNT, polished Zn foils were immersed in ZnSO4 and x-MNT (x = 0.01, 0.1, 0.5, 1 M) solutions for 5 days. Scanning electron microscopy (SEM) images reveal the presence of corrosion products on the surfaces of Zn foils immersed in the abovementioned solutions (Fig. 3b and S3†). Intriguingly, with increasing MNT concentration, these corrosion products shift from standard hexagonal plate-like structures to irregular, fragmented shapes. At a concentration of 1 M MNT, the corrosion products on the Zn foil surfaces transform into relatively flat, leaf-like shapes (Fig. S4†). We employed X-ray Diffraction (XRD) to analyze the surface material composition of Zn plates immersed in ZnSO4 (denoted as “ZnSO4-Zn”) and 1-MNT (denoted as “MNT-Zn”) solutions for 5 days. The XRD results reveal that the corrosion products on ZnSO4-Zn primarily consists of Zn4SO4(OH)6·5H2O, while those on MNT-Zn are highly crystalline Zn4SO4(OH)6·H2O (Fig. 3c). This difference likely stems from the reduced amount of free water in the solution, due to the strong interaction between MNT and H2O.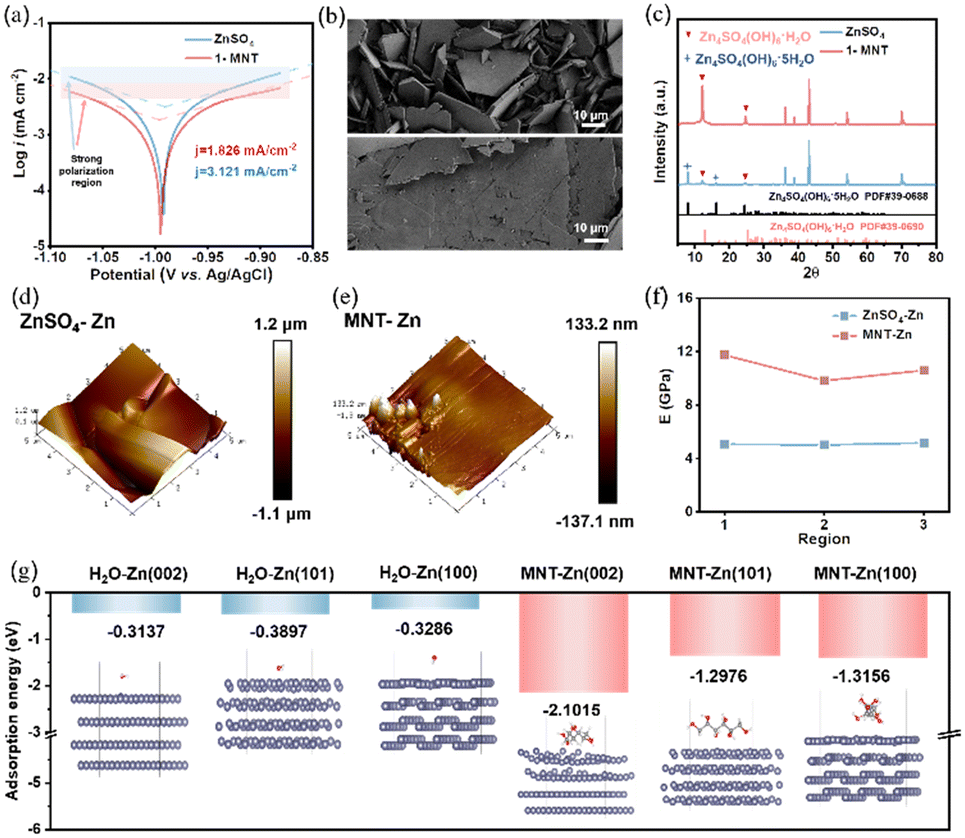 | ||
| Fig. 3 (a) Comparison of Tafel plots of different electrolyte systems. (b) SEM images and (c) XRD spectra of Zn electrodes after soaking in ZnSO4 and 1-MNT electrolytes for 5 days. AFM maps of the Zn surfaces after soaking in (d) ZnSO4 and (e) 1-MNT electrolytes for 5 days. (f) Young's modulus of three regions selected on Zn plates after soaking in ZnSO4 and 1-MNT electrolytes for 5 days. (g) Adsorption energy comparison of H2O and MNT molecules on various Zn crystal planes. | ||
To further examine the physical properties of the corrosion products, we utilized atomic force microscopy (AFM) to analyze the surface structures of both Zn foils. The ZnSO4-Zn surface is notably irregular and uneven, while the MNT-Zn surface appear relatively flat and smooth (Fig. 3d and e). We also measured the Young's modulus in three distinct regions of each sample (Fig. 3f and S5†). The findings indicate that the Young's modulus of MNT-Zn is more than twice that of ZnSO4-Zn, suggesting that the corrosion products formed in the 1-MNT solution are dense and compact, potentially enhancing the corrosion resistance of the Zn anode. To assess whether the corrosion products in the 1-MNT electrolyte impact the transport of Zn2+, we conducted electrochemical impedance spectroscopy (EIS) on symmetric cells left for different durations. Results illustrated in Fig. S6† indicate that the charge-transfer resistance of symmetric cells using the ZnSO4 electrolyte increased significantly after 5 days. In contrast, symmetric cells with the 1-MNT electrolyte display relatively stable charge-transfer resistance, suggesting that the corrosion products in the 1-MNT electrolyte enhance the stability of the Zn electrode. To further examine the protective effects of these corrosion products, Zn foils immersed in the ZnSO4 and 1-MNT electrolytes for five days were assembled into symmetric cells with the ZnSO4 electrolyte and tested (Fig. S7†). The finding demonstrates that the MNT-Zn||MNT-Zn symmetric cell maintained stable operation for over 2300 hours, confirming that the corrosion products in the MNT-modified electrolyte act as barriers for the Zn anode, mitigating adverse reactions between the electrolyte and the Zn anode and thus improving interfacial stability.
To verify the preferential adsorption of MNT on the Zn anode surface, the double-layer capacitance (EDLC) of the Zn anode was initially tested (Fig. S8†). The EDLC value significantly decreases from 0.21 to 0.14 mF cm−2 following the addition of MNT, indicating an increase in the thickness of the electric double layer (EDL) due to MNT adsorption.33 DFT calculations were performed to analyze the adsorption energies of H2O and MNT on three major crystal planes of Zn, further corroborating these observations. As depicted in Fig. 3g, the adsorption energies of MNT on all crystal planes significantly exceed those of H2O, highlighting the strong preferential surface adsorption of MNT on the Zn surface. Notably, the adsorption energy of MNT on Zn (002) is the highest at −2.1015 eV. Drawing on previous research, we deduce that the crystal growth direction of Zn metal is closely associated with the orientation of its crystal faces.34–36 This insight suggests that there are favorable conditions for plane deposition of Zn along the (002) direction. To provide macroscopic evidence of MNT's affinity to Zn, contact angle measurements were performed as displayed in Fig. S9.† The introduction of MNT reduces the contact angle from 90.1° to 60.2°. This significant decrease suggests that the 1-MNT electrolyte improves the wetting properties on the Zn foil, possibly due to MNT adsorption reducing the surface energy.37 These findings demonstrate that MNT effectively modifies the electrode/electrolyte interface environment by preferentially adsorbing onto the Zn foil surface, thereby mitigating side reactions and promoting uniform Zn deposition during cycling.
2.3 Effect of MNT on the kinetics and morphology of Zn2+ deposition
Building on the previous adjustments related to solvation and corrosion inhibition effects, as well as through a comprehensive comparison of the electrochemical performance of symmetrical cells, we have determined that the optimal concentration of MNT is 1 M, hereafter denoted as “ZnSO4 + MNT”. We tracked changes at the Zn electrode interface during the constant-current plating in both ZnSO4 and ZnSO4 + MNT electrolytes using in situ optical microscopy (Fig. 4a). In the ZnSO4 electrolyte, noticeable protrusions appeared on the surface after 30 minutes of electroplating, which worsened over time. Additionally, it was observed that dendritic growth was most vigorous at the initial sites of these protrusions, confirming that Zn2+ ions are preferentially plated at the tips due to the uneven distribution of the electric field. After 120 minutes, visible bubbles indicated the occurrence of the HER during plating. Conversely, uniform and dense Zn deposition was observed on the Zn electrode in the ZnSO4 + MNT electrolyte within two hours of electroplating. This confirms that the adsorption of MNT on the Zn electrode surface helps distribute the electric field, effectively regulating the electroplating behavior of Zn2+ and suppressing dendritic growth and the HER. Linear sweep voltammetry (LSV) was used to assess HER kinetics before and after the addition of MNT (Fig. S10†). Notably, the HER onset potential shifted from −1.164 V to −1.226 V (vs. Ag/AgCl) at 20 mA cm−2 after introducing MNT, indicating a significant inhibition of HER kinetics. | ||
| Fig. 4 (a) In situ optical observations of the Zn deposition process at a current density of 1 mA cm−2 in the ZnSO4 and ZnSO4 + MNT electrolytes. Scale bar: 200 μm (b) SEM images of Zn electrodes after different cycles at 1 mA cm−2 and 1 mA h cm−2 in ZnSO4 and ZnSO4 + MNT electrolytes. (c) Line chart for the ratio of I(002)/I(101) planes. Cross-sectional BSE-SEM images and corresponding EDX maps of the cycled Zn electrode after 100 cycles in (d) ZnSO4 and (e) ZnSO4 + MNT electrolytes at 1 mA cm−2 and 1 mA h cm−2. (f) Schematic of the preferential growth of the (002) plane induced by MNT during cycling of AZMBs. | ||
The influence of MNT on the nucleation and growth of Zn was examined using chronoamperometry (CA) as depicted in Fig. S11.† The current response in symmetrical cells with the ZnSO4 electrolyte was noted to decline rapidly, indicating uncontrolled 2D diffusion of Zn2+ on the Zn surface. Conversely, with the ZnSO4 + MNT electrolyte, the current stabilized after 20 seconds, suggesting a swift transition from 2D disorderly diffusion to 3D stable diffusion.38 To quantitatively assess the kinetics of Zn2+ deposition, we calculated the dissolution activation energy (Ea) for Zn2+ as shown in Fig. S12.†39 Additionally, impedance measurements of symmetrical cells at different temperatures showed an increase in charge transfer resistance when MNT was added to the electrolyte. This suggests that MNT decelerates the charge transfer kinetics on the Zn anode surface. Furthermore, using the Arrhenius equation, we determined the dissolution activation energy (Ea) of Zn2+ on the anode surface. The results indicated that the energy barrier required for Zn2+ transfer in the ZnSO4 + MNT electrolyte is higher (29.90 vs. 24.13 kJ mol−1), thereby corroborating the adsorption of MNT on the Zn anode surface and effectively inhibiting the 2D disorderly diffusion of Zn2+ across the electrode surface.
To further elucidate MNT's role in regulating Zn2+ deposition, we examined the micro-morphological changes on the surface of Zn anodes in symmetric cells after various numbers of cycles using SEM. As depicted in Fig. 4b, after just one cycle in the ZnSO4 electrolyte, the Zn anode surface featured numerous irregularly arranged dendritic flakes. Moreover, with an increase in the number of cycles, the dendritic flakes' thickness continued to grow, accompanied by a significant presence of by-products scattered across the Zn anode surface. In contrast, the Zn anode cycled in the ZnSO4 + MNT electrolyte maintained a smooth and dense surface. The elemental composition of the Zn anode surface after 100 cycles was analyzed using energy dispersive X-ray spectroscopy (EDX). Notably, a sulfur (S) mass fraction as high as 4.3% was detected on the surface of the Zn anode cycled in the ZnSO4 electrolyte, indicating a substantial accumulation of Zn4SO4(OH)6·xH2O by-products (Fig. S13†). Meanwhile, the S content measured on the Zn anode cycled in the ZnSO4 + MNT electrolyte was significantly lower (Fig. S14†), which confirms MNT's effective role in suppressing by-product formation.
Interestingly, after cycling in the ZnSO4 + MNT electrolyte, the Zn deposition on the Zn anode surface showed a distinct preferred orientation, forming a layered morphology, as confirmed by corresponding XRD spectra (Fig. S15†). The intensity ratio of I(002)/I(101) on the Zn anode surface after cycling in the pure ZnSO4 electrolyte displayed no consistent trend. However, with the addition of MNT, this ratio gradually increased with each cycle (from 1.3 to 1.9) (Fig. 4c). The (002) crystal plane is known to promote the deposition of Zn2+ parallel to the Zn anode surface. To visually examine the deposition morphology of Zn2+, we conducted argon ion beam polishing on the cross-sections of the Zn anodes after 100 cycles and captured backscattered electron (BSE-SEM) images. The cross-sectional images from the Zn anode after 100 cycles in the ZnSO4 electrolyte (Fig. 4d) revealed the growth of large dendrites along with moss-like dead Zn and hexagonal by-products on the dendrite surfaces. The significant accumulation of sulfur (S) detected in the EDX spectra of the Zn anode surface corroborated this observation. In contrast, after the introduction of MNT, the Zn deposited on the Zn anode surface after 100 cycles was orderly stacked, with only trace amounts of by-products evident (Fig. 4e). Even at lower magnifications, dendrites were scarcely visible (Fig. S16†). Thus, the preferred (002) crystal plane in the ZnSO4 + MNT electrolyte highlights MNT's role in regulating the Zn2+ deposition morphology, which is crucial in suppressing dendritic growth that arises from uneven electric field distribution during Zn2+ plating/stripping processes.
2.4 Enhanced electrochemical performances of batteries with MNT
To validate the beneficial impact of MNT on enhancing the reversibility and stability of AZMBs, we assembled Zn||Zn symmetric cells with various concentrations of MNT to evaluate their long-term cycling performance (Fig. S17†). We can observe that the cycle life gradually increases with the rise in MNT concentration, which is consistent with the trend shown in Fig. 2a–c, indicating that the hydroxyl groups in MNT form hydrogen bonds with water, effectively suppressing side reactions and significantly enhancing the stability of the Zn anode. Notably, the symmetric cell with the 1 M MNT electrolyte demonstrated the longest cycle life, achieving over 2980 hours of cumulative plating/stripping time, which is five times longer than that of the pure ZnSO4 electrolyte (Fig. 5a). The voltage curve of the symmetric cell using the ZnSO4 electrolyte exhibited drastic fluctuations after 640 hours, which we suspect were due to dendrite formation and severe corrosion of the Zn anode, leading to short circuits. Examination of the cross-section of the Zn anode from the Zn||Zn symmetric cell with the ZnSO4 electrolyte post-short circuit revealed large clusters of dendrites and dead Zn that had grown on the surface, with dendrites tall enough to puncture the separator and cause battery short circuits (Fig. S18†). Additionally, the Zn plate experienced severe corrosion, resulting in significant holes and structural collapse, supporting our initial conjecture.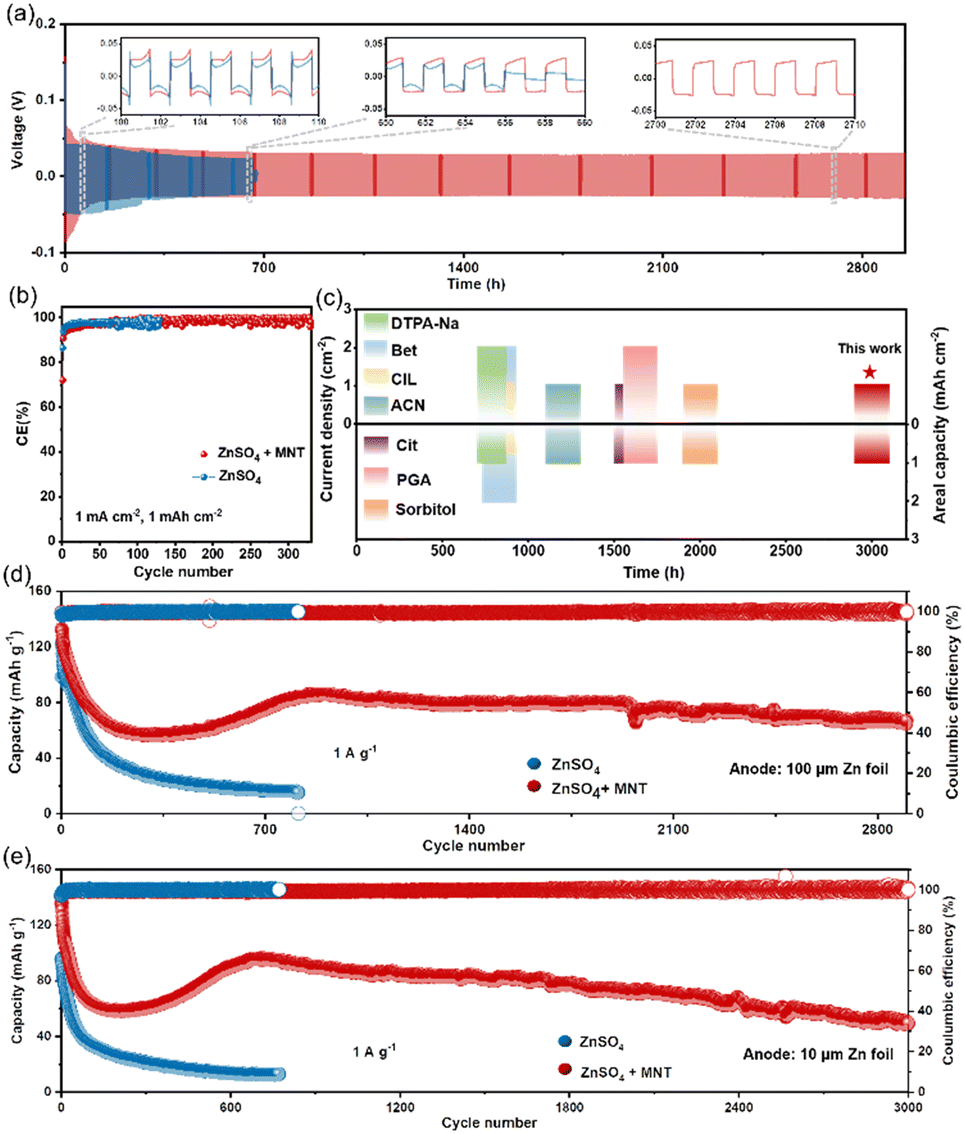 | ||
| Fig. 5 (a) Long-term cycling stability of symmetrical cells at 1 mA h cm−2 and 1 mA cm−2. The utilization of Zn: 1.81%. (b) Coulombic efficiencies of Zn plating/stripping in various electrolytes at 1 mA h cm−2 and 1 mA cm−2. (c) Long-term symmetric-cell cyclability enabled by the ZnSO4 + MNT electrolyte compared to those achieved via some representative electrolyte modifications under different cycling conditions. (d) Long-span cycling performances of Zn||PANI@CC full cells at 1 A g−1. (e) Cycling performances of Zn||PANI@CC cells at 1 A g−1 at a low N/P ratio of 22.48. | ||
It is noteworthy that the introduction of MNT slightly increased the initial overpotential from 56.7 mV to 64 mV, primarily due to the strong adsorption of MNT on the Zn anode surface (Fig. S19†). A higher nucleation overpotential provides a greater driving force for nucleation, which results in a reduced Zn nucleation radius and increased nucleation density, facilitating the formation of dense and flat deposition layers.40 Even when the current density was increased to 5 mA cm−2, the symmetric cell with the ZnSO4 + MNT electrolyte maintained good cycling stability up to 430 hours (Fig. S20†). Fig. S21† illustrates the rate performance of the Zn||Zn symmetric cell with a fixed capacity of 1 mA h cm−2 at current densities ranging from 0.2 to 10 mA cm−2. At lower current densities between 0.2 and 1 mA cm−2, the overpotentials of both cells remained similar. However, as the current density exceeded 2 mA cm−2, the overpotential of the symmetric cell with the ZnSO4 electrolyte increased significantly.
Zn||Ti asymmetric cells were assembled to evaluate the coulombic efficiency (CE) of the Zn anode during cycling (Fig. 5b). The Zn||Ti cell with the ZnSO4 + MNT electrolyte demonstrated an impressive average CE of 97.9% over 300 cycles at 1 mA cm−2 and 1 mA h cm−2, indicating excellent reversibility of Zn plating/stripping processes. In contrast, the CE of the cell with the ZnSO4 electrolyte plummeted to zero after just 100 cycles due to side reactions and dendritic growth. The detailed capacity–voltage curves are shown in Fig. S22.† The cell with the ZnSO4 + MNT electrolyte exhibits higher reversibility and lower voltage polarization. To highlight the effectiveness of the MNT addition, we compared the cycling life of symmetric cells using MNT with other published electrolyte modifications for AZMBs under identical conditions of 1 mA cm−2 and 1 mA h cm−2 (Fig. 5c).13,34,41–45 The results show that our ZnSO4 + MNT electrolyte significantly outperforms these other modified electrolytes.
To further validate the practical effectiveness of the ZnSO4 + MNT electrolyte, we employed PANI@CC (polypyrrole–polyaniline on carbon cloth) as the cathode material to assemble full cells for testing. SEM images and corresponding EDX mapping showed that PANI was uniformly dispersed on the fibers of the carbon cloth (Fig. S23†). In the cyclic voltammetry (CV) curves, the positions of the oxidation and reduction peaks overlapped, indicating a consistent charge storage mechanism of the PANI@CC cathode in both electrolyte systems (Fig. S24†). Long-term cycle performance tests of the full cells were conducted at a current load of 1 A g−1 (Fig. 5d), showcasing the robustness and reliability of the MNT-modified electrolyte in practical applications. The Zn||PANI@CC full cell with the ZnSO4 electrolyte experienced continuous capacity decay from the onset of cycling and abruptly failed after the 814th cycle. Meanwhile, the full cell using the ZnSO4 + MNT electrolyte achieved stable cycling for over 2900 cycles, with a capacity retention rate of 74.42% between cycles 830 and 2900. The voltage–capacity curves of the Zn||PANI@CC full cells with different electrolytes, as shown in Fig. S25,† indicate that the full cell with the ZnSO4 + MNT electrolyte maintained closer charge–discharge curve intervals across various cycle numbers, suggesting that a higher capacity retention rate was achieved. The rate performance at different current densities ranging from 0.05 to 1 A g−1 further demonstrates the enhanced stability of the battery with the MNT-containing electrolyte (Fig. S26†). Additionally, while previously reported high-performance AZMBs often used excess Zn (over 100 μm) to achieve long cycle life, we conducted a more realistic evaluation using thinner Zn foils (10 μm). Testing at a low N/P ratio of 22.48, the Zn||PANI@CC full cell with the ZnSO4 + MNT electrolyte still maintained stable cycling for over 3000 cycles at 1 A g−1, whereas the cell with the ZnSO4 electrolyte failed after just 772 cycles (Fig. 5e). These results underscore that the introduction of MNT significantly enhances the stability of the Zn anode, thereby improving the electrochemical performance of the full cell.
3. Conclusion
In this work, we proposed a novel equimolar aqueous ZnSO4/MNT electrolyte, inspired by the hypertonic dehydration effect observed in medicine, to address the critical issues of dendrite growth and side reactions troubling the Zn anodes in aqueous batteries. In the ZnSO4 + MNT electrolyte, the hypertonic dehydration effect significantly decreases the active water content, effectively inhibiting the water-induced corrosion and side reactions. Moreover, MNT plays a vital role in promoting the preferred (002) deposition orientation of Zn2+, resulting in a unique and orderly stacked morphology free of dendritic structures. Zn||Zn symmetric cells utilizing the ZnSO4 + MNT electrolyte demonstrate an ultra-long cycling life exceeding 2980 hours at 1 mA cm−2 and 1 mA h cm−2. Furthermore, Zn||PANI@CC full cells show markedly improved cycling performances in terms of capacity output, retention rate and cycle life at both high and low N/P ratios. This study offers a fresh perspective on developing environmentally friendly and cost-effective electrolyte modification strategies, merging biomedical and chemical sciences to support sustainable energy utilization and green environmental protection.Data availability
All the data related to the current submission entitled “A medicine-inspired hydroxyl-rich equimolar ZnSO4/D-mannitol electrolyte enables horizontally stacked Zn deposition for long-cycling aqueous batteries” are available upon reasonable request directly from the authors via email to the corresponding authors.Author contributions
J. Xiao: conceptualization, investigation, methodology, formal analysis, writing – original draft. C. Yuan: software, methodology, formal analysis. C. Liu: software, investigation, formal analysis. X. Sun: methodology, software. B. Cheng: investigation. Z. Huang: methodology. X. Zhan: conceptualization, methodology, data curation, writing – review & editing, supervision. L. Zhu: conceptualization, methodology, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for financial support from the National Natural Science Foundation of China (52202199), Anhui Provincial Natural Science Foundation (2108085QE202), and start-up grants from Anhui University (S020318008/007).References
- J. F. Parker, C. N. Chervin, I. R. Pala, M. Machler, M. F. Burz, J. W. Long and D. R. Rolison, Science, 2017, 356, 415–418 CrossRef CAS PubMed.
- L. Ding, L. Wang, J. Gao, T. Yan, H. Li, J. Mao, F. Song, S. Fedotov, L.-Y. Chang, N. Li, Y. Su, T. Liu and L. Zhang, Adv. Funct. Mater., 2023, 33, 2301648 CrossRef CAS.
- J. Xiao, C. Yuan, L. Xiang, X. Li, L. Zhu and X. Zhan, Chem. – Eur. J., 2024, 30, e202304149 CrossRef CAS PubMed.
- V. Yufit, F. Tariq, D. S. Eastwood, M. Biton, B. Wu, P. D. Lee and N. P. Brandon, Joule, 2019, 3, 485–502 CrossRef.
- M. Li, Z. Li, X. Wang, J. Meng, X. Liu, B. Wu, C. Han and L. Mai, Energy Environ. Sci., 2021, 14, 3796–3839 RSC.
- A. Chen, C. Zhao, J. Gao, Z. Guo, X. Lu, J. Zhang, Z. Liu, M. Wang, N. Liu, L. Fan, Y. Zhang and N. Zhang, Energy Environ. Sci., 2022, 16, 275–284 RSC.
- J. Duan, J. Dong, R. Cao, H. Yang, K. Fang, Y. Liu, Z. Shen, F. Li, R. Liu, H. Li and C. Chen, Adv. Sci., 2023, 10, 2303343 CrossRef CAS PubMed.
- M. Zhao, Y. Lv, S. Zhao, Y. Xiao, J. Niu, Q. Yang, J. Qiu, F. Wang and S. Chen, Adv. Mater., 2022, 34, 2206239 CrossRef CAS PubMed.
- C. Yuan, C. Liu, X. Li, Y. Zhi, X. Zhan and S. Gao, ACS Appl. Energy Mater., 2023, 6, 1897–1905 CrossRef CAS.
- C. Cao, K. Zhou, W. Du, C. C. Li, M. Ye, Y. Zhang, Y. Tang and X. Liu, Adv. Energy Mater., 2023, 13, 2301835 CrossRef CAS.
- J. Yang, X. Xu, Y. Gao, Y. Wang, Q. Cao, J. Pu, F. Bu, T. Meng and C. Guan, Adv. Energy Mater., 2023, 13, 2301997 CrossRef CAS.
- G. Duan, Y. Wang, B. Luo, L. Sun, S. Zheng, J. Huang and Z. Ye, Energy Storage Mater., 2023, 61, 102882 CrossRef.
- H. Ren, S. Li, B. Wang, Y. Zhang, T. Wang, Q. Lv, X. Zhang, L. Wang, X. Han, F. Jin, C. Bao, P. Yan, N. Zhang, D. Wang, T. Cheng, H. Liu and S. Dou, Adv. Mater., 2023, 35, 2208237 CrossRef CAS.
- Z. Cheng, K. Wang, J. Fu, F. Mo, P. Lu, J. Gao, D. Ho, B. Li and H. Hu, Adv. Energy Mater., 2024, 14, 2304003 CrossRef CAS.
- Z. Li and A. W. Robertson, Battery Energy, 2023, 2, 20220029 CrossRef CAS.
- Z. Ye, Z. Cao, M. O. Lam Chee, P. Dong, P. M. Ajayan, J. Shen and M. Ye, Energy Storage Mater., 2020, 32, 290–305 CrossRef.
- J. Xie, Z. Liang and Y.-C. Lu, Nat. Mater., 2020, 19, 1006–1011 CrossRef CAS PubMed.
- J. Cui, X. Liu, Y. Xie, K. Wu, Y. Wang, Y. Liu, J. Zhang, J. Yi and Y. Xia, Mater. Today Energy, 2020, 18, 100563 CrossRef CAS.
- W. Deng, Z. Xu and X. Wang, Energy Storage Mater., 2022, 52, 52–60 CrossRef.
- J. Hao, L. Yuan, C. Ye, D. Chao, K. Davey, Z. Guo and S.-Z. Qiao, Angew. Chem., 2021, 60, 7366–7375 CrossRef CAS PubMed.
- Y. Jin, K. S. Han, Y. Shao, M. L. Sushko, J. Xiao, H. Pan and J. Liu, Adv. Funct. Mater., 2020, 30, 2003932 CrossRef CAS.
- C. Sun, C. Wu, X. Gu, C. Wang and Q. Wang, Nano-Micro Lett., 2021, 13, 89 CrossRef CAS PubMed.
- C. Huang, X. Zhao, S. Liu, Y. Hao, Q. Tang, A. Hu, Z. Liu and X. Chen, Adv. Mater., 2021, 33, 2100445 CrossRef CAS PubMed.
- W. Yuan, G. Ma, X. Nie, Y. Wang, S. Di, L. Wang, J. Wang, S. Shen and N. Zhang, Chem. Eng. J., 2022, 431, 134076 CrossRef CAS.
- H. Dai, T. Sun, J. Zhou, J. Wang, Z. Chen, G. Zhang and S. Sun, Nat. Commun., 2024, 15, 8577 CrossRef CAS PubMed.
- D. Li, L. Cao, T. Deng, S. Liu and C. Wang, Angew. Chem., Int. Ed., 2021, 60, 13035–13041 CrossRef CAS PubMed.
- Z. Hu, F. Zhang, Y. Zhao, H. Wang, Y. Huang, F. Wu, R. Chen and L. Li, Adv. Mater., 2022, 34, 2203104 CrossRef CAS PubMed.
- W. Xu, K. Zhao, W. Huo, Y. Wang, G. Yao, X. Gu, H. Cheng, L. Mai, C. Hu and X. Wang, Nano Energy, 2019, 62, 275–281 CrossRef CAS.
- W. Su, N. Jia, H. Li, H. Hao and C. Li, Chin. J. Chem. Eng., 2017, 25, 358–362 CrossRef CAS.
- D. Zhang, J. Cao, Z. Dai, R. Chanajaree, C. Yang, X. Wu, X. Zhang and J. Qin, J. Mater. Chem. A, 2023, 11, 23779–23786 RSC.
- S. Bai, Z. Huang, G. Liang, R. Yang, D. Liu, W. Wen, X. Jin, C. Zhi and X. Wang, Adv. Sci., 2024, 11, 2304549 CrossRef CAS PubMed.
- Y. Liu, Y. An, L. Wu, J. Sun, F. Xiong, H. Tang, S. Chen, Y. Guo, L. Zhang, Q. An and L. Mai, ACS Nano, 2023, 17, 552–560 CrossRef CAS PubMed.
- L. M. Da Silva, L. A. De Faria and J. F. C. Boodts, Electrochim. Acta, 2001, 47, 395–403 CrossRef CAS.
- C. Huang, J. Mao, S. Li, W. Zhang, X. Wang, Z. Shen, S. Zhang, J. Guo, Y. Xu, Y. Lu and J. Lu, Adv. Funct. Mater., 2021, 33, 2315855 Search PubMed.
- Z. Cai, J. Wang, Z. Lu, R. Zhan, Y. Ou, L. Wang, M. Dahbi, J. Alami, J. Lu, K. Amine and Y. Sun, Angew. Chem., Int. Ed., 2022, 61, e202116560 CrossRef CAS PubMed.
- W. Feng, Z. Liang, W. Zhou, X. Li, W. Wang, Y. Chi, W. Liu, D. Gengzang, G. Zhang, Q. Chen, P. Wang, W. Chen and S. Zhang, Dalton Trans., 2023, 52, 7457–7463 RSC.
- J. Hao, J. Long, B. Li, X. Li, S. Zhang, F. Yang, X. Zeng, Z. Yang, W. K. Pang and Z. Guo, Adv. Funct. Mater., 2019, 29, 1903605 CrossRef.
- M. Zhou, S. Guo, J. Li, X. Luo, Z. Liu, T. Zhang, X. Cao, M. Long, B. Lu, A. Pan, G. Fang, J. Zhou and S. Liang, Adv. Mater., 2021, 33, 2100187 CrossRef CAS PubMed.
- Y. Yang, C. Liu, Z. Lv, H. Yang, Y. Zhang, M. Ye, L. Chen, J. Zhao and C. C. Li, Adv. Mater., 2021, 33, 2007388 CrossRef CAS PubMed.
- H. Wang, H. Li, Y. Tang, Z. Xu, K. Wang, Q. Li, B. He, Y. Liu, M. Ge, S. Chen, T. Hao, G. Xing and Y. Zhang, Adv. Funct. Mater., 2022, 32, 2207898 CrossRef CAS.
- Y. Xia, R. Tong, J. Zhang, M. Xu, G. Shao, H. Wang, Y. Dong and C.-A. Wang, Nano-Micro Lett., 2024, 16, 82 CrossRef CAS PubMed.
- Z. Zheng, D. Ren, Y. Li, F. Kang, X. Li, X. Peng and L. Dong, Adv. Funct. Mater., 2024, 34, 2312855 CrossRef CAS.
- J. Zheng, B. Zhang, X. Chen, W. Hao, J. Yao, J. Li, Y. Gan, X. Wang, X. Liu, Z. Wu, Y. Liu, L. Lv, L. Tao, P. Liang, X. Ji, H. Wang and H. Wan, Nano-Micro Lett., 2024, 16, 145 CrossRef PubMed.
- J. Chen, N. Liu, W. Dong, Y. Xu, Y. Cao, S. Zhang, J. Hou, H. Bi, T. Lin and F.-Q. Huang, Adv. Funct. Mater., 2024, 2313925 CrossRef CAS.
- P. Sun, L. Ma, W. Zhou, M. Qiu, Z. Wang, D. Chao and W. Mai, Angew. Chem., Int. Ed., 2021, 60, 18247–18255 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lf00328d |
| ‡ J. Xiao and C. Yuan contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |