
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembly of phosphole-lipids in 2D films: the influence of π-interactions and steric constraints†
Zahra
Alinia
a,
Dandan
Miao
b,
Thomas
Baumgartner
 b and
Christine E.
DeWolf
*a
b and
Christine E.
DeWolf
*a
aDepartment of Chemistry and Biochemistry and Centre for NanoScience Research, Concordia University, 7141 Sherbrooke St. W., Montréal, Québec H4B 1R6, Canada. E-mail: christine.dewolf@concordia.ca
bDepartment of Chemistry, York University, 4700 Keele Street, Toronto, Ontario M3J 1P3, Canada
First published on 10th December 2024
Abstract
Promising photophysical properties of π-conjugated phosphole-based materials make them appealing building blocks for electronic and optoelectronic devices. In practical terms, a well-ordered 2D film organization is re4quired that can be obtained by deposition and/or self-assembly of thin films on a solid substrate. Manipulation of the existing noncovalent interactions within the films, via altering the chemical structure or environmental conditions to modify the molecular arrangements, is one approach to control the electronic properties of these thin films. The inter- and intramolecular π–π interactions influencing the 2D film structure in Langmuir and Langmuir–Blodgett films of a series of lipids with phosphole-based, π-conjugated headgroups is explored in the presence and absence of aromatic additives. Brewster angle and atomic force microscopy demonstrated the simultaneous formation of 3D aggregates and a condensed phase. GIXD measurements confirmed that the 3D material formation should be hindered to promote the formation of an ordered 2D film. Different approaches were considered to manipulate the π–π interactions in the film: addition of small-molecule aromatics, mixtures of phosphole-lipids with phenolic surfactants, as well as conjugation extension of phosphole-lipid headgroup. Such π–π interactions can modify the directional growth of domains within 2D film, however, it is not strong enough to completely eliminate the 3D aggregate formation.
1. Introduction
Polymeric materials with π-conjugated backbones have been used for electronic and optoelectronic applications for many years, where the photophysical performance depends on the molecular packing and crystallinity.1–5 Modification of the chemical structure through functional group selection, altering the conjugation length and incorporation of heteroatoms, provides the means to tune the photophysical properties for the final product fabrications.6–9 With respect to heteroatoms in these materials, systems with backbones of aniline, pyrrole, or thiophene have been studied in depth.10–13 Phosphole, as the main phosphorus-based building block, has emerged in recent years as a unique member of this heteroatom-based group, as it displays partial aromaticity and high electron delocalization.14 The chemical properties of phosphole-based π-conjugated materials are a function of both the conformation of the phosphorus centre and the conjugation of its substituents; these lead to unique multifunctional opportunities for optoelectronic device optimization including thermal stability, luminescence efficiency and charge-transport features.10,15–18 Rigid, fused-ring systems can be incorporated, extending the π-electron networks of the phospholes amplifying their light-emitting capacity.19–21 These building blocks can be accessed by flexible synthetic protocols and provide opportunities for functionalization of the phosphorus centre, including metal coordination and alkylation.19,20 Furthermore, the ensuing π-interactions can decrease the electronic energy levels (HOMO–LUMO),22–24 and enhance the charge- and energy-transfer properties through intermolecular electronic coupling.25–27The molecular orientation can directly influence properties like charge-carrier transport or light-absorption efficiency. For example, phosphorus-based materials employing phosphinine building blocks show directional charge transfer derived from their organized solid-state packing.16 Other properties like orientation-dependent ionization energies may be caused by packing-induced electronic energy levels.1 While the π–π interactions may strengthen the existing electrostatic and van der Waals interactions, they can also induce aggregation in solid-state and liquid-crystalline materials,28,29 which hinders the application of these building blocks. Aggregation becomes even more challenging as only a thin layer of these materials (on the nm scale) is needed for the majority of optoelectronic applications. For devices such as organic semiconductors and field-effect transistors, a major challenge remains the reproducible and controlled deposition of structurally well-defined thin films, as their charge-transfer properties are influenced by structure, orientation, and crystallinity.3 Thus, the deposition technique and the external conditions play an important role for proper application properties.
The Langmuir–Blodgett deposition technique (LB) provides high control over the thin-film orientation and characterization. For proper LB deposition (from air–water to air–solid interfaces), the phosphole-based materials should be surface-active, usually achieved via the addition of alkyl chains. For surfactants with π-interactions in the headgroup, the balance between the hydrophobic and hydrophilic properties impacts the film behaviour at the air–water interface, as it defines the packing configuration in the plane of the π system (edge-on or face-on).6,30 The chains also serve to promote liquid-crystalline over solid-phase formation. A series of phosphole-based surfactants (phosphole-lipid) with promising photophysical properties have been synthesized (Scheme 1).24 Within this series, the smallest headgroup promotes well-ordered lamellar architectures (mainly impacted by ionic interactions), while the extension of π-conjugation (additional aromatic rings in the larger headgroup) leads to stronger emission and quantum yield at both solution and solid state.24 We have recently reported the LB film behaviour of the smallest headgroup of the series (PL1) in presence of halides.31 Herein, we characterize the monolayer behaviour of these phosphole-lipids focusing on the intermolecular π-interaction (both from the extension of the headgroup conjugation and the introduction of additional aromatic moieties) and structure of the LB films formed.
 | ||
| Scheme 1 Chemical structure of previously reported phosphole-lipid with dithienophosphole headgroup (PL1), phosphole-lipid with benzo-fused dithienophosphole headgroup (PL2),24 dodecyl 4-hydroxybenzoate surfactant (C12HB), and the aromatic molecules phenol, naphthalene, and 2-naphthol. | ||
2. Materials and methods
Solution and subphases
Phosphole-lipids with dithienophosphole headgroup (PL1) and benzo-fused dithienophosphole headgroup (PL2) (Scheme 1) were synthesized as previously reported24 and their spreading solutions (0.6–1.2 mM) were prepared in chloroform (Certified ACS, approximately 0.75% ethanol as preservative) purchased from Fisher Scientific. For all subphases, ultrapure water with resistivity of 18.2 MΩ cm−1 (pH 5.6 at 25 °C) was obtained from a Milli-Q HX 7080 water purification system (HC). For solutions as the subphase, NaBr (ACS reagent, ≥99.0%), NaCl (ACS reagent, ≥99.0%), NaI (ACS reagent, ≥99.5%), and phenol (ACS reagent, 99.0–100.5%) were purchased from Sigma-Aldrich and concentrations from ∼1 μM (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of counterion to lipid molecules) to 100 mM were prepared in ultrapure water as needed. For lipid mixture spreading solutions, dodecyl 4-hydroxybenzoate 98.0+% (C12HB, Scheme 1) was purchased from Fisher Scientific and spreading solutions of PL1:C12HB and PL2:C12HB with different molar ratios were prepared.
:1 molar ratio of counterion to lipid molecules) to 100 mM were prepared in ultrapure water as needed. For lipid mixture spreading solutions, dodecyl 4-hydroxybenzoate 98.0+% (C12HB, Scheme 1) was purchased from Fisher Scientific and spreading solutions of PL1:C12HB and PL2:C12HB with different molar ratios were prepared.
Surface pressure-molecular area isotherm
Phosphole-lipids PL1 and PL2 were spread on ultrapure water and the other subphases in a Langmuir film balance (Nima Technology Ltd., Coventry, U.K.), with a maximum surface area of 300 cm2. All measurements were made at room temperature (21.5–22.5 °C) and ten minutes were allowed for complete solvent evaporation and relaxation of the surfactant for all the experiments. A symmetrical compression rate of 5–10 cm2 min−1 (2.5–6.0 Å2 (molecule min)−1 – depending on the initial molecular area) was used. A filter-paper (1 cm × 2 cm, Whatman No. 1 chromatography paper) based Wilhelmy plate was used to measure the surface pressure. To ensure the reproducibility, at least three separate isotherms were obtained for all experiments.Brewster angle microscopy imaging (BAM)
An I-Elli2000 imaging ellipsometer (Nanofilm Technologies GmbH, Göttingen, Germany) equipped with a 50 mW Nd:YAG laser (λ = 532 nm) was used to perform Brewster angle microscopy. The magnification was set at 20× with lateral resolution of 1 μm. All images were obtained at an incident angle of 53.15° (Brewster angle of water) with the laser output of 50% (analyzer, compensator and polarizer set to 0°). Three separate BAM imaging experiments were performed for all systems.Ellipsometry measurements
The I-Elli2000 was also used for measurement of the ellipsometric angles ψ and Δ with an incident angle of 50°, a laser output of 100%, and a compensator angle of 20°. Each value reported in the ellipsometric isotherm represents the average of measurements made on three independently formed films. For each independent film, the δΔ represents the average of 10 measurements of the same monolayer film at constant surface pressure.The initial angles of analyzer and polarizer were set to 14 and 20°, respectively, and later adjusted to have the reflection of the polarized beam eliminated. δΔ values (δΔ = Δfilm − Δsubphase) are reported in ellipsometric isotherms, representing how the ellipsometric angle Δ of the film is changing through the isotherm, relative to the ellipsometric angle Δ of the subphase alone. Both film thickness and refractive index impact δΔ, however, considering the relatively constant refractive index of the film at the air-water interface, δΔ is taken to represent the change in the monolayer thickness upon compression.
Atomic force microscopy imaging
For the AFM sample deposition, Mica sheets V1 quality were purchased from Electron Microscopy Science and freshly cleaved before usage. Monolayers were transferred onto the mica on the upstroke at constant pressures by Langmuir–Blodgett technique and dipping rate of 1 mm min−1 and the transfer ratios (ratio of the monolayer surface area loss over the substrate surface area during deposition) calculated. The AFM samples were allowed to dry for 30 minutes in ambient conditions followed by imaging within a 24-hour window. A Bruker multimode 8HR scanning probe microscope (Nanoscope 9.7, Digital Instruments, Santa Barbara, CA) was used to capture AFM images at the air-solid interface at room temperature. Peak-force tapping mode was used at a scan rate of 0.3–1 Hz with SCANASYST-AIR probes (silicon nitride cantilever, frequency 70 kHz, nominal spring constant 0.4 N m−1, and tip radius 2 nm). Nanoscope software version 2.0 was used to perform all AFM image processing and analysis.Grazing incidence X-ray diffraction (GIXD) and X-ray reflectivity (XRR)
All GIXD and XRR measurements at the air-water interface were conducted at 15-ID-C ChemMatCARS at the Advanced Photon Source (APS) in Argonne National Laboratory, with monochromatic X-rays and energy of 10 keV. A Nima Langmuir trough with 340 cm2 area with compression rate of the mobile barriers set at 2–5 cm2 min−1 (1–4 Å2 (molecule min)−1) was placed in the beam path in an air-tight enclosure with Kapton windows which allowed for purging the atmosphere with helium. The raw data were extracted and processed using software developed by Wei Bu, beamline scientist at ChemMatCARS.The following parameters were used for GIXD measurements:
X-ray beam wavelength: 1.239 Å, incidence angle: 0.0906°, horizontal size: 20 mm, vertical size: 120 mm, beam footprint: 20 mm by 7.6 cm. The two-dimensional Swiss Light source PILATUS 100 K set to single-photon counting mode was used as detector. To minimize intense low-angle scattering, two sets of slits were used, one was placed in front of the detector (to control the beam footprint), and the other was located 280.0 mm from the sample. The GIXD data measured were plotted as contour plots of the intensity as a function of the horizontal (Qxy) and the vertical (Qz) scattering vector components. The lattice spacing dhk was obtained from the in-plane diffraction data as dhk = 2π/qhkxy, where the Miller indices h and k were used to index the Bragg peaks needed to calculate the unit cell parameters for the in-plane lattice.32,33 The Bragg rods and peaks were fitted with Gaussian and Lorentzian function, respectively, using Origin lab graphing and analysis software. The full-width-at-half-maximum values obtained from the corrected Bragg peaks and rods were used to obtain the in-plane correlation length and vertical scattering length through Scherrer formula:32,34
 | (1) |
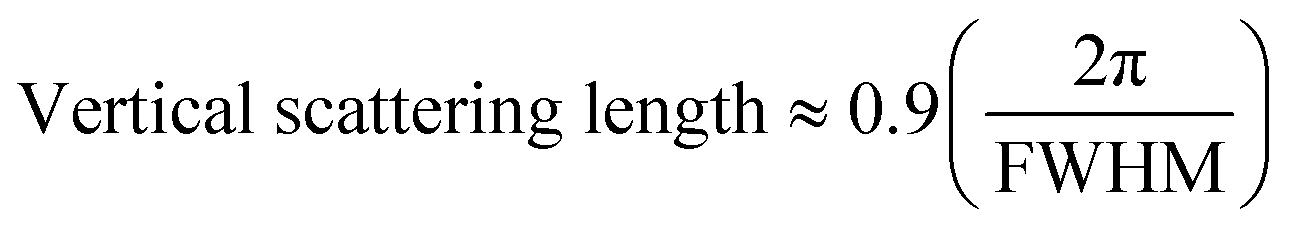 | (2) |
3. Results and discussion
While ionic interactions were proposed to be the main driving force of lamellar structure formation in the liquid crystalline formed in bulk for phosphole-lipid PL1,24 other intermolecular interactions such as π-interactions have been proposed to impact the formation and stability of these structures. With a headgroup comprising a conjugated π-system, an alkoxy benzyl connector between the alkyl chains and the dithienophospholium unit, the intermolecular interactions of these phosphole-lipids can be influenced by the presence of other aromatic rings to the environment or extension of the conjugation length.Our first approach to tailoring the π-interactions of the thin film at the air–water interface involves introduction of aromatics via the subphase. The surface pressure-molecular area isotherms of the PL1 on an ultrapure water and different concentrations of phenol in the subphase, at room temperature are shown in Fig. 1. As previously reported, the isotherm of PL1 on ultrapure water exhibits a pressure onset at 135 Å2 molecule−1, and a main phase transition plateau around 30 mN m−1, corresponding to a transition from the liquid-expanded phase (LE) to the simultaneous formation of 3D aggregates and condensed-phase domains, followed by a second transition at ∼40 mN m−1 at which a second population of 3D aggregates formed.31
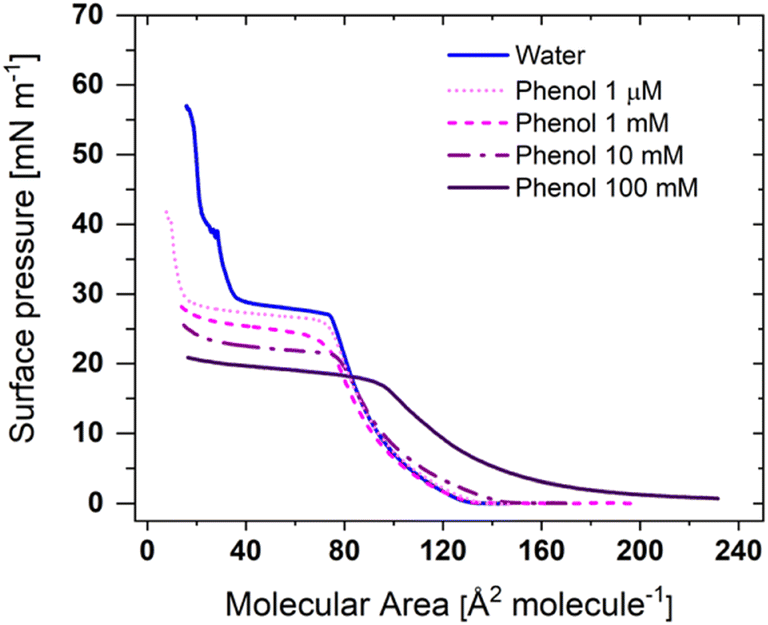 | ||
| Fig. 1 Surface pressure-molecular area isotherm of PL1 as a function of phenol concentration in the subphase. The isotherm for PL1 on water was previously reported,31 and is repeated here for easier comparison. | ||
The presence of phenol in the subphase at its lowest concentration (∼1 μM, corresponding to 10:1 molar ratio of phenol to phosphole-lipid) leads to the same pressure onset area and phase transition plateau surface pressure (136 Å2 molecule−1 and ∼30 mN m−1, respectively). However, the area reduction through the plateau is larger in presence of 1 μM phenol, leading to the observation of the second transition at smaller molecular areas and at ∼40 mN m−1. Increasing the subphase phenol concentration to 1 mM does not alter pressure onset area, however, the phase transition plateau is shifted to a lower surface pressure of ∼25 mN m−1. The phase-transition onset is noticeably less sharp and more rounded. Further increasing the phenol concentration (10 mM) leads to a slight increase of the pressure onset area of the isotherm (∼145 Å2 molecule−1) and a further decrease in the phase transition plateau pressure (∼21 mN m−1). At concentrations of 1 and 10 mM, phenol is theoretically in excess in the subphase (i.e., all bound to the film), but it is only the presence of 100 mM phenol that leads to the elongation of LE phase and the shift of the pressure onset to much larger areas (∼240 Å2 molecule−1). The 100 mM phenol subphase also exhibits the lowest phase transition plateau surface pressure at ∼18 mN m−1.
This noticeable shift in the pressure onset area is likely the result of the higher concentration driving a greater partitioning of phenol to the headgroup region such that it can interdigitate between the phosphole headgroups and shift the lipids further apart. The consequence of the interdigitation is to reduce the chain–chain interactions that are dominated by short-range van der Waals interactions (and hence film organization) and to promote the formation of 3D aggregates, which correlates with the decrease in the phase transition plateau pressure. The formation of 3D aggregates through the phase transition is expected for all concentrations of phenols, as the plateau reaches molecular areas smaller than a minimum cross-sectional area required for three alkyl chain.31,32 In contrast to the subphase counterions,31 the presence of the single aromatic ring of phenol with the same concentration in the subphase does not have a significant impact on the PL1 isotherm.
Phenol was initially chosen as a small aromatic ring, with practical water solubility35 to be used as a subphase solution. However, it may be that the phenol–phosphole-lipid interactions are weak compared to the existing headgroup interactions, including electrostatics and intra- or interlipid interactions, and consequently at low concentrations, it may only remain adsorbed at the headgroup surface rather than penetrating into the film.
Fig. 2 presents the BAM images of a PL1 monolayer on water and different concentrations of phenol in the subphase. For better comparison of the film changes relative to the isotherm, the BAM images corresponding the phase transition plateau surface pressures are aligned vertically. The BAM images are relatively similar for all subphases (in the absence or presence of phenol at different concentrations); bright and small 3D aggregates appear, forming as the film begins to reach the plateau. With further compression, these aggregates grow in number and brightness. The increase in the contrast (brighter aggregates) corresponds to an increase in height difference (film thickness).
 | ||
| Fig. 2 Representative BAM images of PL1 on water and different subphase concentrations of phenol. The images corresponding to the surface pressures of the main phase transition plateau are aligned vertically and framed (pink). | ||
The AFM images obtained for films formed by LB deposition from water and different concentrations of phenol are shown in Fig. 3. For both water and the lowest concentration of phenol (Fig. 3, first and second rows), the formation of relatively circular domains accompanied by the 3D aggregates begin at the end of the phase transition plateau (30 mN m−1). The number of domains and aggregates increase at higher surface pressures, and beyond the second phase transition, a second population of 3D aggregates is observed that are much smaller in height and width, and only form on the domain/background boundary (45 mN m−1). As the number of 3D aggregates increases, this leads to higher transfer ratios (Table S1†).
 | ||
| Fig. 3 Representative AFM images of PL1 deposited onto mica by LB at different surface pressures from water and phenol subphases (concentrations as indicated). All images are 50 × 50 μm2, with height scale of 10 nm. The height scale was selected to enable visualization of the monolayer domains which are otherwise obscured by the large height of the aggregates (average aggregate heights are provided in Fig. S1†). | ||
With 1 mM phenol (Fig. 3, third row), the domains initially adopt an elongated, ribbon-like shape (30 mN m−1) that are narrower than the domains formed at the same surface pressure with water and the lowest phenol concentration. These ribbons vary in length and degree of branching and cover approximately 10–15% of the image area. However, as the surface pressure increases and the domains are pushed to higher area coverage (∼50% at 35 mN m−1 and ∼65% at 40 mN m−1), their shape becomes more rounded (with both quasi-circular domains and residual ribbons observable), and they begin to coalesce.
With 10 mM phenol (fourth row), due to the shape of the isotherm, we could only obtain AFM images for one surface pressure beyond the phase transition plateau. The domains observed at this surface pressure (30 mN m−1) are even narrower than the ones for films deposited from a 1 mM phenol subphase. With 100 mM phenol, we could no longer pass the plateau to record the formation of condensed-phase domains. The only observation is the formation of 3D aggregates at the surface pressure of 20 mN m−1, in the phase transition plateau.
For all concentrations of phenol, the impact on the formation of domains and 3D aggregates is not as prominent as was observed with halide counterions with the same concentration (altered electrostatic interactions).31 Specifically, the presence of phenol does not help to form a uniform film, nor does it limit 3D aggregation, as was observed for the addition of bromide.31 In other aromatic compounds such as para-azoxyanisole with two aromatic rings, electrostatic interactions have been reported as the main driving force of the liquid-crystalline packing.36 Additionally, wherein high concentrations of halides significantly decreased domain heights,31 no specific trend in the change of height for domains or 3D aggregates as a function of phenol concentration could be discerned (Fig. S1†). Note that the aggregate heights range from <50 nm to almost 400 nm, with a large aggregate height variation for given conditions, despite their consistency in shape and lateral dimensions (attributed to line tension, given the three chains and a large hydrophobic volume). However, unlike the addition of halides, increasing concentrations of phenol leads to domain elongation (see the AFM images for surface pressure of 30 mN m−1 in Fig. 3). This elongation appears as ribbon-like domains that are first observed at concentrations >1 mM phenol, which is in agreement with the first observation of a shift in the pressure onset area in the isotherms, suggesting that at these concentrations the penetration of the phenol begins to impact the headgroup organization.
Any interaction that alters the balance of intermolecular forces in the monolayer can disturb the competition between line tension and electrostatic repulsion and result in the domain shape change.37–39 The narrowing of the domains as the phenol concentration increases, may be the result of an increase of the inter-headgroup (intermolecular) distance as the phenol moieties localize between the headgroups, leading to the formation of smaller domains (similar to how electrostatic repulsion would alter the shape of domains). The concurrent formation of extended and elongated domains may derive from the π–π interactions of phenol and aromatic rings of PL1; as the surfactant molecules align in the presence of phenol, oriented domain growth is observed. Nittmann described the formation of dendritic domains as an early stage of domain growth due to lattice anisotropy, but competitive interactions eventually lead to round domains.40 However, persistent directional growth has been reported for other surfactant headgroups exhibiting π-stacking interactions, albeit the directional growth in such systems was more prominent with the formation of dendric domains.41,42 π–π Stacking in surfactants has been reported to provoke long-range orientational order though edge-to-face arrangements of aryl rings in the headgroup region and further packing of the chains.43 In many cases, where nondirectional interactions dominate, this stage is not observed, and domains appear rounded from the onset. In this case, the strong, directional interactions, the elongated domains become more circular only at higher surface pressures. Such directional and oriented domain growth may be intentionally invoked through crystallization with the aim of better controlling the charge transfer properties and electrical conductivity in electronic devices.44
Fig. 4 shows the ellipsometric measurements performed on PL1 films at the air-subphase interface for the 1 mM and 100 mM phenol concentrations. The change in magnitude of δΔ normally correlates to changes in film thickness (assuming the refractive index is relatively constant). At all surface pressures, the lower concentration of phenol leads to larger δΔ magnitudes, with the greatest difference at a surface pressure of 0 mN m−1. However, as the films are compressed to the phase transition plateau, the δΔ magnitudes at the air-subphase interface, and consequently the film thickness, for both phenol concentrations converge. It should be mentioned that the lower δΔ values for 100 mM phenol may only be the result of the isotherm shape: as the isotherm is shifted to larger molecular areas and with the larger lateral space, the phosphole-lipids should form thinner films at the same surface pressure. Both phenol concentrations follow the same trend δΔ upon compression, as previously reported for a water subphase.31
 | ||
| Fig. 4 Ellipsometric measurements of PL1 at the air–subphase interface as a function of phenol concentration in the subphase. Each value represents the average of measurements made on three independently formed films. For each independent film, the δΔ is the average of 10 measurements of the same monolayer film at constant surface pressure. | ||
For Fig. 4, the values of δΔ are only reported up to the phase transition (the full δΔ-molecular area ellipsometric isotherms for all four phenol concentrations are shown in Fig. S2†). The ellipsometric isotherm for 1 μM phenol is similar, including larger errors, to that on a water subphase.31 The errors decrease substantially with higher phenol concentrations, suggesting that phenol, while not impacting the absolute heights of domains or aggregates, does create a more uniform distribution of phases within the region of interest that is measured. Although the 3D aggregates begin to form upon reaching the phase transition plateau, the ellipsometric measurements do not show an increase in the δΔ magnitude, however the formation of 3D aggregates leads to nonuniformity (multiple phases with different thicknesses) and increased errors in the measurement. The δΔ value represents the average measurement of the film at the region of study, and a larger area covered by the thinner background may balance the thickness increase due to the other higher phases present. For the 10 mM phenol subphase, the film could be compressed beyond the plateau and a significant increase in δΔ could then be observed.
GIXD measurements at 18 mN m−1 (below the plateau) and 26 mN m−1 (in the plateau) were carried out for PL1 with the 10 mM phenol subphase since this is the first concentration at which the surface pressure-area isotherm shifts to higher molecular areas. It was not possible to compress the film beyond the plateau for a higher surface pressure measurement. No diffraction signal was observed at either surface pressure (Fig. S3†) confirming that the condensed phase forms only after the end of the plateau.
The vertical structure of the film at the same surface pressures was also probed using X-ray reflectivity. The reflectivity was fit with a simple two-box model where the upper box corresponds to the chain region and the lower box to the headgroups. The fits to the X-ray reflectivity are shown in Fig. S4† and the fitted parameters are provided in Table S2.† The presence of phenol in the subphase does not alter the thickness or electron density of either region. This is in good agreement with ellipsometry that shows similar values of δΔ for all phenol concentrations indicative of similar total film thicknesses.
To evaluate, if the size of the aromatic ring present has an impact on the ability of the molecule to intercalate between headgroups and therefore the headgroup packing, naphthalene and 2-naphthol were introduced. Strong interactions have been reported when sodium 2-naphthalenesulfonate was mixed with aromatic surfactants, where the π–π interaction and edge-to-face stackings formed enhanced the naphthalene insertion and led to ordered conformations with less space requirements.45,46 As both naphthalene and 2-naphthol have lower solubility than phenol in water, they were cospread with the phosphole-lipid. The chloroform-based lipid mixtures of 1:10 molar ratio of phosphole-lipid:aromatic hydrocarbon were separately prepared (to match the ∼1 μM concentration phenol in subphase) and the surface pressure-molecular area isotherms of two lipid mixtures were obtained (Fig. 5, solid lines). Both lipid mixtures exhibit a pressure onset area of around 13 Å2 molecule−1, followed by a phase transition at slightly lower surface pressures than the film on a water subphase (∼24 mN m−1). These pressure onset areas are too small for even a single chain surfactant with a small headgroup. When the same isotherms are replotted as a function of molecular area of a pure phosphole-lipid (dashed line, Fig. 5, inset), they show the same pressure onset area as the water subphase. Although the conjugation of naphthalene or 2-naphthol may be expected to generate stronger interactions and better bridging between the headgroups, their size may not be optimal for penetration into the intermolecular space and disruption of the existing strong intra- and intermolecular interactions. Consequently, despite being cospread, naphthalene and 2-naphthol do not appear to remain intercalated within the film. Considering that all of the naphthalene or 2-naphthol must then dissolve into the subphase, the total concentration of naphthalene or 2-naphthol would be approximately 1 μM, which is still below their respective solubility limits of 0.241 mM and 5.24 mM.47,48 That said, similar to phenol at high concentrations, the isotherms exhibit a rounding of the plateau and a shift of the plateau to lower surface pressures, indicating that some of the naphthol or naphthalene may be surface bound (adsorbed).
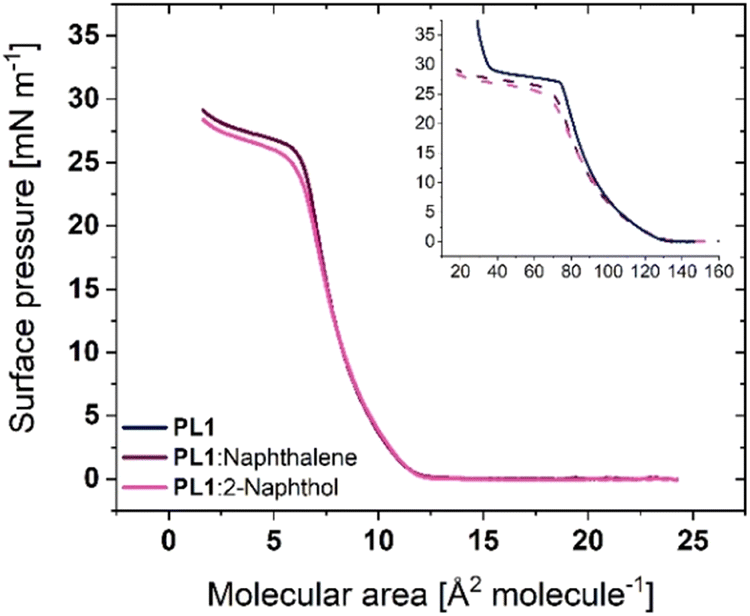 | ||
| Fig. 5 Surface pressure-molecular area isotherms of PL1 and lipid mixtures with 1:10 molar ratio of PL1:naphthalene and PL1:2-naphthol (solid lines, pressure onset area ∼13 Å2 molecule−1). Inset: the same isotherms are replotted as a function of pure phosphole-lipid molecular area (dashed lines). | ||
As the phenol and naphthalene solutions used in this work were within their solubility range in water, and it cannot be expected that all of the aromatic hydrocarbon molecules would remain at the subphase surface. Another approach to retain the aromatic species at the air-water interface, in the plane of phosphole-lipids, is to cospread another surfactant with an aromatic headgroup and similar alkyl chain length. For this approach, a lipid mixture of PL1 with the phenolic surfactant dodecyl 4-hydroxybenzoate (C12HB in Scheme 1) was studied. As shown in Fig. 6, the surface pressure-molecular area isotherm of a 1:1 molar ratio lipid mixture of PL1:C12HB is found in between the isotherms of each individual surfactant, with a phase transition plateau shifted to lower surface pressures than PL1. Although the 1:1 isotherm exhibits a negative excess area of −4.6 ± 0.1 Å2 molecule−1 (indicative of attractive interactions), the BAM images (Fig. S5a†) and ellipsometric measurements (Fig. S5b†) show no significant differences to those of the phosphole-lipid (i.e., no formation of domains and bright 3D aggregates that begin to form upon reaching the phase transition plateau and similar δΔ values).
 | ||
| Fig. 6 Surface pressure-molecular area isotherms of PL1, C12HB, and their mixture with 1:1 molar ratio (structures shown in Scheme 1). | ||
The AFM images (Fig. S5c†) show that the condensed-phase domains still begin to form at the end of plateau (25 mN m−1) accompanied by the formation of the 3D aggregates. The main difference observed is that the domains are slightly bigger in width and lower in height for the lipid mixture compared to PL1 and upon reaching higher surface pressures the condensed-phase domains of the lipid mixture coalesce to form larger domains (that was not observed for PL1). Additionally, even at these high surface pressures, significantly fewer aggregates are observed. Thus, the addition of a cosurfactant reduces the extent of aggregation but does not eliminate it and unlike high concentrations of bromide, the condensed phase formation and aggregate formation still occur simultaneously (i.e., the plateaus are not separated).
Fig. S6† shows isotherms and AFM images for both 10:1 and 1:10 ratios of PL1:C12HB. The 10:1 mixture is dominated by the PL1 properties as would be expected, with similar isotherms and morphologies. The 1:10 mixture exhibits a plateau at higher surface pressures than PL1 (surface pressures commensurate with the collapse of C12HB) and a homogeneous film even at these high surface pressure (up to 35 mN m−1). Thus, increased proportions of C12HB prevent aggregation. Further study would be required to determine an optimal amount of C12HB to promote retention in the plane of the film while still generating the desired film optoelectronic properties.
GIXD measurements were carried out for 1:1 lipid mixture of PL1:C12HB on water subphase at 17 mN m−1 (below the plateau) and 23 mN m−1 (in the plateau). Again, it was not possible to compress the film beyond the plateau precluding measurements at higher surface pressures. No diffraction signal was observed at either surface pressure (data not shown). Thus, the condensed phase forms only after the end of the plateau. The vertical structure of the lipid mixture film at the same surface pressures was also probed using X-ray reflectivity. The presence of phenolic surfactant of C12HB in the in the plane of phosphole-lipid at the air-water interface does not impact the length or electron density of chain and headgroup regions (Table S2†).
The final approach employed to modify π-interactions in the phosphole-lipids Langmuir films is the extension the conjugation length in their headgroup (PL2 in Scheme 1). This extension of the conjugation has been shown to alter the photophysical properties in both the solid and solution phases. The intermolecular π–π interactions of this larger scaffold have been reported to cause a large red-shift in emission wavelength for PL2 in the solid state compared to PL1. Although PL2 shows an organized orientation in the liquid crystalline phase, the alkyl chain interdigitation and ionic interactions of the headgroups are not as strong as those reported for PL1, due to the larger space requirements for the headgroup that increases the intermolecular distance and the layer thickness in liquid crystalline phase.24
The surface pressure-molecular area isotherm of PL2 is shown in Fig. 7. Although the headgroup is larger for PL2, its isotherm exhibits only a slight decrease in pressure onset area (∼6 Å2 molecule−1). This small change in onset area may be due to the dominance of three alkyl chains in the area requirements at low surface pressures. The impact of the headgroup becomes more significant at higher surface pressures. With the larger headgroup, the phase transition plateau begins at slightly larger molecular areas and lower surface pressures. Also, the isotherm does not reach surface pressures beyond the plateau, indicative of the stronger influence of aggregate formation as opposed to condensed phase formation in the isotherm. This lack of condensed phase is confirmed by BAM and AFM imaging and ellipsometry shows a similar trend along the isotherm as observed for PL1 (Fig. S7). Both the solution and liquid-crystalline phases for PL2 show evidence of inter- and intra headgroup π-stacking which might be expected to generate a condensed phase.24 Studies of lipids with nonaromatic headgroups have shown that an increase in the size of the headgroup relative to the alkyl chain cross-sectional area weakens the packing of the alkyl chains, increases the tilt angle, and decreases the monolayer thickness.49
 | ||
| Fig. 7 Surface pressure-molecular area isotherms of PL1, PL2, and their mixture with 1:1 molar ratio (structures shown in Scheme 1). | ||
A 1:1 mixture of PL1 and PL2 exhibits a pressure onset area that is slightly larger than either PL1 or PL2 and positive excess areas throughout the isotherm (+5.3 ± 0.2 Å2 molecule−1) indicative of repulsive interactions, which may be the result a mismatch of the headgroup space requirements. The isotherm is most similar to that of PL2 suggesting that the larger phosphole-lipid headgroup can dominate the film properties.
PL2 has been probed in the same range of subphases, including halide counterions and phenolic structures (Fig. S8†). The notable outcomes of these studies are that the films of PL2 lose their selectivity towards bromide counterions (i.e., there is no differentiation based on the halide selected), have reduced shifts upon incorporation of phenol, and exhibit small positive excess areas with naphthalene and 2-naphthol.
The presence of both aromatic rings and charged species can promote the possibility of charge-π interactions, and further impact the molecular orientation and structure.50–52 Factors like the aromatics' electron richness, or the components' polarity and electron affinity, and environmental factors such as pH or ionic strength have been reported as the defining elements for such charge-π interactions.53–55 Despite the wealth of potential interactions in the phosphole-lipid films studied here, it appears that the headgroup size may be the principal factor of PL2 behaviour with halide subphases. The headgroup size can also explain the shortening of the LE phase for PL2 compared to PL1 with 100 mM phenol subphase.
4. Conclusions
The impact of intra- and intermolecular interactions and arrangement of conjugated phosphole-based materials on the performance of the electronic devices provide opportunities for tailoring their self-assembly and organization and thus improving the final product functionality. Herein, interfacial characterization of the phosphole-lipids showed that an ordered 2D film (condensed phase) only begins to form after a nonclassical phase transition in the isotherms. However, obtaining a well-ordered film remains a challenge as the plateau is also associated with the formation of 3D aggregates.For better control over the formation of the desired ordered, 2D thin films, various aromatic additives were introduced to modify the π-interactions for which phosphole-lipid headgroup size, intermolecular space requirements, and the balance between all the noncovalent interactions play a key role. Introducing the single aromatic ring of phenol in the subphase (particularly, at higher concentrations) alters the growth pattern of the domains. However, the resultant interactions are not sufficient to retain the phosphole-lipid in the monolayer plane or prevent the formation of aggregation. Although the phenol-phosphole π-interactions showed the capacity to control the film formation through directional domain growth.
The polycyclic, fused rings of naphthalene and 2-naphthol did not remain intercalated between the headgroups when cospread with phosphole-lipids. However, anchoring the aromatic ring to the interface, via cospreading a mixture of the phosphole-lipid and a phenolic surfactant, reduced the formation of 3D aggregates while also generating larger domains. The extension of the conjugation in the headgroup, on the other hand, resulted in the loss of selectivity towards halide counterions, as the larger headgroup space requirements may lead to larger pores available. The larger area required for headgroups relative to the chains hinders the optimal packing and no domain formation was recorded. Although the conjugation extension generates promising photophysical properties (increased quantum yield and intensity), the larger headgroup induces a loss of distance-dependent (chain-chain) interactions and insufficient π-interactions to yield an organized film. It may be possible to achieve a well-ordered, aggregate-free thin films required for organic electronics through a combined approach of tuning headgroup interactions for both strong electrostatic interactions (via counterions) and weaker π-interactions (via phenolic surfactants).
LB films offer a unique path to investigate and control molecular organization due to the precise control over monolayer assembly, critical in the design of 2D thin films for applications in optoelectronics. However, achieving a balance between headgroup interactions, chain packing, and noncovalent interactions (such as π-interactions) remains a significant challenge to forming uniform, well-ordered, aggregate-free thin films. By introducing additives and mixtures, such as aromatic compounds and phenolic surfactants, we explored approaches to intentionally tune and direct molecular orientation and enhance interfacial order. This strategy supports the broader efforts in the field to engineer self-assembled systems with tailored structures and functions from the bottom-up. These understandings highlight the potential of combining molecular design and control over environmental conditions to optimize film organization, offering a pathway to eliminate aggregate formation and promote ordered domain growth, critical for functional self-assembled architectures.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. Author contributions are as follows: Zahra Alinia: investigation, formal analysis, writing – original draft and review & editing; Dandan Miao: resources; Thomas Baumgartner: resources, writing – review & editing; Christine DeWolf: conceptualization, supervision, resources, project administration, writing – original draft, reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. D. acknowledges funding from the Natural Sciences and Engineering Research Council (NSERC, RGPIN-2019-07043) of Canada and the Canada Foundation for Innovation (CFI). C. D. is a member of the multi-institutional Quebec Center for Advanced Materials (QCAM). T. B. is grateful to the NSERC, CFI, and the Canada Research Chairs program for funding this work. The authors acknowledge the Centre for NanoScience Research funded by Concordia University, Montreal, Canada. GIXD was carried out at NSF's ChemMatCARS, Sector 15 at the Advanced Photon Source (APS), Argonne National Laboratory (ANL) which is supported by the Divisions of Chemistry (CHE) and Materials Research (DMR), National Science Foundation, under grant number NSF/CHE- 1834750. This research used resources of the Advanced Photon Source; a U.S. Department of Energy (DOE) Office of Science user facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.References
- J. Zhang, M. Wang, Z. Yang and X. Zhang, Highly flexible and stretchable strain sensors based on conductive whisker carbon nanotube films, Carbon, 2021, 176, 139–147 CrossRef CAS.
- L. S. Price and S. L. Price, Packing Preferences of Chalcones: A Model Conjugated Pharmaceutical Scaffold, Cryst. Growth Des., 2022, 22, 1801–1816 CrossRef CAS.
- P. Gómez, J. Cerdá, M. Más-Montoya, S. Georgakopoulos, I. Da Silva, A. García, E. Ortí, J. Aragó and D. Curiel, Effect of molecular geometry and extended conjugation on the performance of hydrogen-bonded semiconductors in organic thin-film field-effect transistors, J. Mater. Chem. C, 2021, 9, 10819–10829 RSC.
- Z.-F. Yao, J.-Y. Wang and J. Pei, Control of π–π Stacking via Crystal Engineering in Organic Conjugated Small Molecule Crystals, Cryst. Growth Des., 2018, 18, 7–15 CrossRef CAS.
- R. Thakuria, N. K. Nath and B. K. Saha, The Nature and Applications of π–π Interactions: A Perspective, Cryst. Growth Des., 2019, 19, 523–528 CrossRef CAS.
- J. Y. Park and R. C. Advincula, in Nanocoatings and Ultra-Thin Films: 2 - Nanostructured thin films from amphiphilic molecules, ed. A. S. Hamdy Makhlouf and I. Tiginyanu, Woodhead Publishing, 2011, pp. 24–56 Search PubMed.
- J. Crassous and R. Réau, π-Conjugated phosphole derivatives: Synthesis, optoelectronic functions and coordination chemistry, Dalton Trans., 2008, 6865–6875 RSC.
- K. Bagchi and M. D. Ediger, Controlling Structure and Properties of Vapor-Deposited Glasses of Organic Semiconductors: Recent Advances and Challenges, J. Phys. Chem. Lett., 2020, 11, 6935–6945 CrossRef CAS PubMed.
- X. Feng, Y. Bai, M. Liu, Y. Li, H. Yang, X. Wang and C. Wu, Untangling the respective effects of heteroatom-doped carbon materials in batteries, supercapacitors and the ORR to design high performance materials, Energy Environ. Sci., 2021, 14, 2036–2089 RSC.
- H. C. Su, O. Fadhel, C. J. Yang, T. Y. Cho, C. Fave, M. Hissler, C. C. Wu and R. Réau, Toward functional π-conjugated organophosphorus materials: Design of phosphole-based oligomers for electroluminescent devices, J. Am. Chem. Soc., 2006, 128, 983–995 CrossRef CAS.
- A. Kraft, A. C. Grimsdale and A. B. Holmes, Angew. Chem., Int. Ed., 1998, 37, 402–428 CrossRef.
- A. P. Kulkarni, C. J. Tonzola, A. Babel and S. A. Jenekhe, Electron Transport Materials for Organic Light-Emitting Diodes, Chem. Mater., 2004, 16, 4556–4573 CrossRef CAS.
- H. Yan, P. Lee, N. R. Armstrong, A. Graham, G. A. Evmenenko, P. Dutta and T. J. Marks, High-Performance Hole-Transport Layers for Polymer Light-Emitting Diodes. Implementation of Organosiloxane Cross-Linking Chemistry in Polymeric Electroluminescent Devices, J. Am. Chem. Soc., 2005, 127, 3172–3183 CrossRef CAS.
- T. Baumgartner, Insights on the Design and Electron-Acceptor Properties of Conjugated Organophosphorus Materials, Acc. Chem. Res., 2014, 47, 1613–1622 CrossRef CAS.
- M. Stolar and T. Baumgartner, Phosphorus-Containing Materials for Organic Electronics, Chem. – Asian J., 2014, 9, 1212–1225 CrossRef CAS PubMed.
- X. He, L. Chen and T. Baumgartner, Modified Viologen- and Carbonylpyridinium-Based Electrodes for Organic Batteries, ACS Appl. Mater. Interfaces, 2024, 16, 48689–48705 CrossRef CAS PubMed.
- S. Yamaguchi, A. Fukazawa and M. Taki, Phosphole P-Oxide-Containing π-Electron Materials: Synthesis and Applications in Fluorescence Imaging, Yuki Gosei Kagaku Kyokaishi, 2017, 75, 1179–1187 CrossRef CAS.
- Z. Wang and T. Baumgartner, Organophosphorus Avenues toward Self-Assembled Conjugated Soft Materials, Chem. Rec., 2015, 15, 199–217 CrossRef CAS.
- Y. Matano, T. Miyajima, T. Fukushima, H. Kaji, Y. Kimura and H. Imahori, Comparative study on the structural, optical, and electrochemical properties of bithiophene-fused benzo[c]phospholes, Chem. – Eur. J., 2008, 14, 8102–8115 CrossRef CAS PubMed.
- C. Romero-Nieto and T. Baumgartner, Dithieno[3,2-b:2′,3′-d]phospholes: A Look Back at the First Decade, Synlett, 2013, 24, 920–937 CrossRef CAS.
- Y. Makioka, T. Hayashi and M. Tanaka, Poly[2,7-(9-oxo-9-phosphafluorenylene)-alt-co-(1,4-arylene)]s: Phosphorus-containing π-Conjugated Polymers, Chem. Lett., 2004, 33, 44–45 CrossRef CAS.
- T. Hatano and T. Kato, A columnar liquid crystal based on triphenylphosphine oxide-its structural changes upon interaction with alkaline metal cations, Chem. Commun., 2006, 1277–1279 RSC.
- M. Kimura, T. Hatano, T. Yasuda, J. Morita, Y. Akama, K. Minoura, T. Shimomura and T. Kato, Photoluminescent Liquid Crystals Based on Trithienylphosphine Oxides, Chem. Lett., 2009, 38, 800–801 CrossRef CAS.
- Y. Ren, W. H. Kan, M. A. Henderson, P. G. Bomben, C. P. Berlinguette, V. Thangadurai and T. Baumgartner, External-stimuli responsive photophysics and liquid crystal properties of self-assembled ‘phosphole-lipids’, J. Am. Chem. Soc., 2011, 133, 17014–17026 CrossRef CAS.
- Y. Ren and T. Baumgartner, Dually switchable heterotetracenes: Addressing the photophysical properties and self-organization of the P-S system, J. Am. Chem. Soc., 2011, 133, 1328–1340 CrossRef CAS.
- M. D. Curtis, J. Cao and J. W. Kampf, Solid-State Packing of Conjugated Oligomers: From π-Stacks to the Herringbone Structure, J. Am. Chem. Soc., 2004, 126, 4318–4328 CrossRef CAS PubMed.
- E. G. Kim, V. Coropceanu, N. E. Gruhn, R. S. Sánchez-Carrera, R. Snoeberger, A. J. Matzger and J. L. Brédas, Charge Transport Parameters of the pentathienoacene Crystal, J. Am. Chem. Soc., 2007, 129, 13072–13081 CrossRef CAS.
- A. West, in Interface Science and Technology, Elsevier B.V., 2017, vol. 21, pp. 49–130 Search PubMed.
- R. J. Abraham, F. Eivazi, H. Pearson and K. M. Smith, Mechanisms of aggregation in metalloporphyrins: demonstration of a mechanistic dichotomy, J. Chem. Soc., Chem. Commun., 1976, 698–699 RSC.
- D. T. McQuade, J. Kim and T. M. Swager, Two-dimensional conjugated polymer assemblies interchain spacing for control of photophysics, J. Am. Chem. Soc., 2000, 122, 5885–5886 CrossRef CAS.
- Z. Alinia, D. Miao, T. Baumgartner and C. E. DeWolf, Tuning Electrostatics to Promote Ordered Monolayers of Phosphole-Lipids, Langmuir, 2024, 40, 22301–22313 CrossRef CAS PubMed.
- V. M. Kaganer, H. Möhwald and P. Dutta, Structure and phase transitions in Langmuir monolayers, Rev. Mod. Phys., 1999, 71, 779–819 CrossRef CAS.
- D. Jacquemain, F. Leveiller, S. P. Weinbach, M. Lahav, L. Leiserowitz, K. Kjaer and J. Als-Nielsen, Crystal structure of self-aggregates of insoluble aliphatic amphiphilic molecules at the air-water interface. An x-ray synchrotron study, J. Am. Chem. Soc., 1991, 113, 7684–7691 CrossRef CAS.
- E. Alami, G. Beinert, P. Marie and R. Zana, Alkanediyl-α,ω-bis (dimethylalkylammonium bromide) Surfactants. 3. Behavior at the Air-Water Interface, Langmuir, 1993, 9, 1465–1467 CrossRef CAS.
- G. R. Southworth and J. L. Keller, Hydrophobic Sorption of Polar Organics by Low Organic Carbon Soils, Water, Air, Soil Pollut., 1986, 28, 239–248 CrossRef CAS.
- L. H. Mendoza Huizar, C. H. Rios-Reyes and E. García Sanchez, A theoretical study of the intermolecular interactions in the p-azoxyanisole liquid crystal, J. Mol. Liq., 2014, 199, 530–537 CrossRef CAS.
- G. Brezesinski, A. Dietrich, B. Dobner and H. Möhwald, Morphology and structures in double-, triple-and quadruple-chain phospholipid monolayers at the air/water interface, Prog. Colloid Polym. Sci., 1995, 98, 255–262 CAS.
- C. A. Helm and H. Möhwald, Equilibrium and nonequilibrium features determining superlattices in phospholipid monolayers, J. Phys. Chem., 1988, 92, 1262–1266 CrossRef CAS.
- H. M. McConnell, Structures and Transitions in Lipid Monolayers at the Air-Water Interface, Annu. Rev. Phys. Chem., 1991, 42, 171–195 CrossRef CAS.
- J. Nittmann and H. Eugene Stanley, Tip splitting without interfacial tension and dendritic growth patterns arising from molecular anisotropy, Nature, 1986, 321, 663–668 CrossRef.
- S. Behyan, D. Gritzalis, R. Schmidt, E. Kebede, L. A. Cuccia and C. Dewolf, Structural organization and phase behaviour of: Meta -substituted dioctadecylaminobenzoquinones at the air/water interface, Phys. Chem. Chem. Phys., 2019, 21, 2345–2350 RSC.
- R. Miclette Lamarche and C. Dewolf, Strong Headgroup Interactions Drive Highly Directional Growth and Unusual Phase Co-Existence in Self-Assembled Phenolic Films, ACS Appl. Mater. Interfaces, 2019, 11, 45354–45363 CrossRef CAS.
- M. Jurak, K. Szafran, P. Cea and S. Martín, Analysis of Molecular Interactions between Components in Phospholipid-Immunosuppressant-Antioxidant Mixed Langmuir Films, Langmuir, 2021, 37, 5601–5616 CrossRef CAS.
- L. H. Jimison, M. F. Toney, I. McCulloch, M. Heeney and A. Salleo, Charge-Transport Anisotropy Due to Grain Boundaries in Directionally Crystallized Thin Films of Regioregular Poly(3-hexylthiophene), Adv. Mater., 2009, 21, 1568–1572 CrossRef CAS.
- L. Zhao, H. Zhang and W. Wang, π-π stacking interaction in mixed surfactant solutions assembled by cationic surfactant and organic salt with a naphthalene nucleus, J. Mol. Liq., 2017, 240, 14–20 CrossRef CAS.
- C. F. Neumann and H. Sigel, Ternary Complexes in Solution. XVIII. The Stability Enhancement of Nucleotide-Containing Charge-Transfer Adducts through the Formation of a Metal Ion Bridge, J. Am. Chem. Soc., 1974, 96, 2750–2756 CrossRef.
- R. S. Pearlman, S. H. Yalkowsky and S. Banerjee, Water Solubilities of Polynuclear Aromatic and Heteroaromatic Compounds, J. Phys. Chem. Ref. Data, 1984, 13, 555–562 CrossRef CAS.
- S. H. Yalkowsky, Y. He and P. Jain, Handbook of Aqueous Solubility Data, CRC Press, Boca Raton, 2010 Search PubMed.
- E. Madrid and S. L. Horswell, Effect of headgroup on the physicochemical properties of phospholipid bilayers in electric fields: Size matters, Langmuir, 2013, 29, 1695–1708 CrossRef CAS PubMed.
- D. A. Dougherty, The Cation−π Interaction, Acc. Chem. Res., 2013, 46, 885–893 CrossRef CAS.
- H. T. Chifotides and K. R. Dunbar, Anion−π Interactions in Supramolecular Architectures, Acc. Chem. Res., 2013, 46, 894–906 CrossRef CAS PubMed.
- M. Giese, M. Albrecht and K. Rissanen, Experimental investigation of anion–π interactions – applications and biochemical relevance, Chem. Commun., 2016, 52, 1778–1795 RSC.
- M. Akamatsu, A. Kimura, K. Yamanaga, K. Sakai and H. Sakai, Anion-π interaction at the solid/water interfaces, Chem. Commun., 2021, 57, 4650–4653 RSC.
- M. Akamatsu, K. Yamanaga, K. Tanaka, Y. Kanehara, M. Sumita, K. Sakai and H. Sakai, Anion−π Interactions in Monolayers Formed by Amphiphilic Electron-Deficient Aromatic Compounds at Air/Water Interfaces, Langmuir, 2023, 39, 5833–5839 CrossRef CAS PubMed.
- J. Chen, Q. Peng, J. Liu and H. Zeng, Mussel-Inspired Cation−π Interactions: Wet Adhesion and Biomimetic Materials, Langmuir, 2023, 39, 17600–17610 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tabulated transfer ratios for AFM images, domain and 3D aggregate height measurements, ellipsometric measurements, additional GIXD contour plots and X-ray reflectivity plots, best fit X-ray reflectivity data (box thickness and electron densities), additional Langmuir film characterization of lipid mixtures and phosphole-lipid with extended conjugation (isotherm, BAM images, ellipsometric measurements and AFM images), surface pressure-molecular area isotherms of the PL2 as a function of different subphase components. See DOI: https://doi.org/10.1039/d4lf00361f |
| This journal is © The Royal Society of Chemistry 2025 |