
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering macroporous carbon film supports for freestanding Fe–N–C cathodes at high current densities†
Simon
Kellner
a,
Mengnan
Wang
 *ab,
Ying
Wang
a,
Jesús
Barrio
a,
Guangmeimei
Yang
c,
Jingyu
Feng
a,
Sandrine E. M.
Heutz
c,
Ifan E. L.
Stephens
c and
Maria-Magdalena
Titirici
*ad
*ab,
Ying
Wang
a,
Jesús
Barrio
a,
Guangmeimei
Yang
c,
Jingyu
Feng
a,
Sandrine E. M.
Heutz
c,
Ifan E. L.
Stephens
c and
Maria-Magdalena
Titirici
*ad
aDepartment of Chemical Engineering, Imperial College London, London SW7 2AZ, UK. E-mail: m.titirici@imperial.ac.uk
bDepartment of Chemical Engineering, Swansea University, Swansea, SA1 8EN, UK. E-mail: mengnan.wang@swansea.ac.uk
cDepartment of Materials, Royal School of Mines, Imperial College London, London SW7 2AZ, UK
dAdvanced Institute for Materials Research (AIMR), Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai, 980-8577, Japan
First published on 6th February 2025
Abstract
As the oxygen reduction reaction (ORR) kinetics account for the largest share of performance losses for fuel cells, most research in platinum group metal (PGM)-free catalysts prioritize on improving the activity of catalysts by maximizing the active site density and by engineering of the local coordination environment of the active sites to meet the activity targets. Thereby, the mass-transport capabilities of the catalyst are usually neglected at early stages of catalyst development. In this work, the reverse approach is taken: a freestanding carbon film support with an interconnected macropore network is prepared to improve the mass and charge transport of conventional PGM-free particle based catalyst layers. Carbon precursors mesophase pitch and polyvinylalcohol (PVA) are combined with the macropore template polystyrene (PS) spheres in a ball-milling process to form a slurry for casting the film which is subsequently carbonized in different atmospheres to tune the micropore volume. The macroporous films are thoroughly characterized by means of SEM, N2 sorption, XPS and Mercury Intrusion Porosimetry (MIP) and tested as ORR catalyst support for a model FeN4 active site in a gas-diffusion-electrode (GDE) half-cell, which can operate at high current density conditions. This study is the first to compare two support classes, a new in-house synthesized carbon film support and commercial Vulcan XC 72 particle support, in cathode catalyst layers by keeping the molecular catalyst (FePc) on both supports the same. Special focus is directed to the mass transport performances of both type of supports, which are compared at high current densities at 2 A cm−2 in the GDE cell.
1. Introduction
The widespread commercialization of low temperature fuel cell technologies depends on the stack production and materials costs, to which the cathode catalyst contributes up to 40%.1 Cheaper catalysts for the oxygen reduction reaction at the cathode are either obtained by a reduction of the platinum content with ultralow loading platinum group metal (PGM) catalysts,2 or by eliminating the PGM costs with platinum group metal free (PGM-free) catalysts.3 While the activity of Fe–N–C catalysts, the best performing PGM-free catalysts, has recently improved tremendously in both acidic4,5 and alkaline6,7 pH environments, the volumetric activity of less bulk dense Fe–N–C catalysts is still significantly inferior to that of commercial Pt/C catalysts due to their low active site density.8 Consequently, high loadings of Fe–N–C catalysts are applied compared to Pt/C cathodes. Typically, Fe–N–C catalysts show superior activity at high pH,9 due to a change of ORR mechanism from proton-coupled electron transfer to outer-sphere electron transfer going from low pH to high pH affecting the electrochemical activity.10 Therefore, when integrating Fe–N–C into the cathode of AEMFCs loadings of 1 mgFe–N–C cm−2 are sufficient,11,12 whereas in PEMFCs loadings of around 3–4 mgFe–N–C cm−2 are required to meet the DOE targets (44 mA cm−2 at 0.9 V (iR-free)) in the kinetics-controlled region.13–15 Consequently, the cathode catalyst layers (CL) for Fe–N–C catalysts are thicker than that of Pt-based catalysts, which manifests itself with 60–100 μm of CL for Fe–N–C in contrast to 10 μm for identical performance for Pt-based catalysts in PEMFCs.16 As a result, thick Fe–N–C CLs suffer from severe mass-transport losses at high current densities with reported overpotentials of about 100 mV at a current density of 1 A cm−2.17 Two main strategies are pursued to improve the mass-transport in Fe–N–C CL including the tailoring of mesopores18–20 content in the catalyst structure for efficient mass-transport pathways and the fine tuning of hydrophilic/hydrophobic properties of catalyst to achieve better water management.21,22 While these approaches focused on the material and structure design at catalyst level (<100 nm) might benefit the actual fuel cell performance, the effect of morphology design at catalyst layer level (>100 nm) on transport losses with limiting current measurements have rarely been investigated yet.23,24 Most spatial inhomogeneities are found on the macroscale with three-dimensional micro X-ray computed tomography.25 The observation of large voids and catalyst aggregates with sizes up to 10 μm raise questions whether such isolated aggregates are electrically conductively integrated into the CL and thus, are participating in the ORR. Conventional methods for cathode preparation such as slurry casting and spraying lead to random packing of catalyst particles, ionomer and pores, and thus, are restricting the control of the final catalyst layer morphology for efficient mass transport and accessibility of all available active sites. Therefore, it is suggested to overcome these issues with a freestanding carbon film cathode support with interconnected macropores penetrating the structure continuously. Such a structure is expected to benefit the reactant accessibility to the active sites in the catalyst layer, guaranteeing electrical connectivity, and allowing continuous mass transport.26 Inspired by the development of porous freestanding carbon film cathode based on mesophase pitch as a support for platinum,27 in this work the concept is extended to PGM-free electrode development. In previous work, mesophase pitch-templated carbon films were prepared using SiO2 templates, which required time-consuming and harsh chemical etching for removal. In this study, we substitute SiO2 with in-house synthesized polystyrene spheres, which not only save time but also eliminate the need for harsh etching conditions, as they can be easily washed off with organic solvent or decomposed during the carbonization process, offering a more sustainable and safer approach. Initially, the macroporous morphology of carbon film cathode support is characterized with SEM imaging complemented by detailed pore characterization with nitrogen adsorption and mercury intrusion porosimetry to prove the successful creation of a continuous macropore network. Adjusting the carbonization atmosphere to 1% oxygen in nitrogen affects the microporosity and changes the surface composition as revealed with X-ray photoelectron spectroscopy (XPS). The freestanding film support is compared with a conventional powder support of Vulcan XC 72. For sake of simplicity, both supports are equipped with active sites of a model FeN4 site, iron phthalocyanine (FePc), to guarantee that powder and film catalysts have the same active sites and the performance of the catalyst layer morphology is not distorted by heterogeneous active site composition. The catalysts are first screened in RDE, followed by characterization of the mass transport of performance at 2 A cm−2 in a gas-diffusion-electrode setup, which enables to study the catalyst layer morphology of powder and film cathode at realistic gas transport conditions as present in fuel cell catalyst layers.2. Results and discussion
2.1. Identification of the drawbacks of SiO2 template
The carbon sources for the carbon film include mesophase pitch, which can be obtained from pitch, a byproduct of crude oil and coal refining processes. This residue contains a heterogeneous mixture of polycyclic aromatic hydrocarbon structures with high C/H ratio, as shown in a representative molecular structure drawing (Scheme S1†).28 Mesophase pitches are produced by thermal polymerization of an aromatic rich fraction of pitch. Alternatively, aromatic resin mesophase pitch is produced by the catalytic treatment of aromatic molecules such as naphthalene.29 Due to the orientational order adopted by the stacking of planar aromatic molecular structures to domains, optical anisotropy is observed under an optical microscope with polarized light, defining the intermediate liquid crystal structure as mesophase.30Hard templates or activating agents31 are commonly used to endow structure on mesophase pitch-based carbons. Typical methods employed for the preparation of meso- and macroporous carbons from mesophase pitch such as colloid-imprinted carbon method32,33 or melt impregnation34,35 rely on silica templates. These meso- and macroporous carbons are relevant for applications in electrocatalysis such as support for Pt for the ORR.36 The commercial PGM-free catalyst by Pajarito Powder also uses a silica-based sacrificial support method.37 While silica templates can be effectively removed using hydrofluoric acid (HF), its high toxicity poses significant environmental and safety risks. Sodium hydroxide (NaOH) has been explored as a milder alternative, offering the added benefit of improving Fe–N–C catalyst activity through carbon activation.38 However, the etching process can take up to three days to remove SiO2 in its entirety, which hinders its scalability. Therefore, despite the progress made by introducing NaOH as a more sustainable alternative to HF, the etching step remains both time-intensive and resource-consuming. To highlight, the disadvantages of the SiO2 based templating method, a carbon film with SiO2 CL LUDOX (12 nm average particle size) according to the published protocol is prepared.27 A study on the timeline of the SiO2 etching process is carried out by tracking the residual silica in the carbon film by burning off the carbon in air and measuring the residual weight of silica leftover (Fig. 1a).
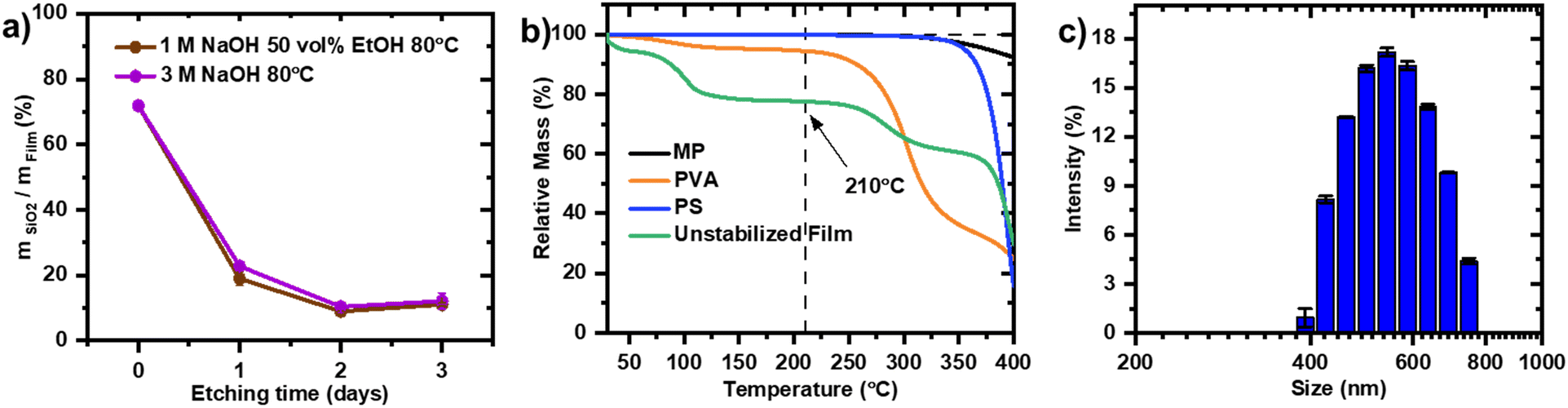 | ||
| Fig. 1 a) Tracking the etching of SiO2 from the carbon support in alkaline solutions by measuring the residual SiO2 after oxidative burn off of the carbon with TGA in compressed air (CA) atmosphere. b) TGA of MP, PVA, PS and unstabilized film in nitrogen (N2) atmosphere. c) Particle size distribution of synthesized PS spheres. | ||
The initial SiO2 weight proportion before starting the etching process of 72% decreases to 20–23% after 24 hours. After two days, the residual SiO2 in the templated carbon is around 10% and remains in that range with no further etching between 48 and 72 hours. The residual SiO2 is suspected to be isolated in the carbon matrix and not accessible for the etching solution. It is noteworthy that the low concentration 1 M NaOH (50 vol% EtOH) achieves a comparable etching as 3 M NaOH. The opening of the mesopores is confirmed with nitrogen adsorption measurement with sixfold increase of mesopores with 10 nm pore width (Fig. S1†).
2.2. Preparation of macroporous carbon films with polystyrene template
Polystyrene (PS) is an alternative spherical hard template for the preparation of ordered interconnected porous carbon matrix of Fe–N–C powder catalysts.39,40 PS nanospheres can be synthesized with diameter ranging from 15 nm to 1000 μm,41–45 allowing to tailor pore structures of inverse opal carbon. Pitch fibers cannot be directly carbonized without prior infusible stabilization treatments owing to their melting behaviour at higher temperature, which disrupt their morphology.46 Consequently, the carbon structures of the films in this work are solidified in an oxidative stabilization step before the removal of the template. During the oxidative stabilization step crosslinking of the functional groups of the various polycyclic aromatic hydrocarbons in the pitch takes place increasing its thermal stability and preventing it from melting at higher temperatures.47 The oxidative stabilization of mesophase pitch is carried out at a temperature higher than the softening point of the mesophase pitch,48 but at a temperature lower than the onset of thermal decomposition of the PS template. Thermomechanical analysis is used to determine the initial softening point of mesophase pitch as 175 °C (Fig. S2†) and is expected to increase as the oxidation treatment progresses.49 Thermogravimetric analysis (TGA) is used to identify the temperature of onsets of decomposition of the components of the envisioned carbon films. While MP and PS don't show any weight loss up to 300 °C, the partially hydrolyzed (86–89%) PVA shows a weight loss in the temperature region between 80 °C and 110 °C due to evaporation of water. A second, more pronounced weight loss begins at 230 °C, corresponding to the thermal decomposition of PVA (Fig. 1b). Therefore, 210 °C has been chosen for the stabilization process of the carbon film. The PS synthesis is carried out according to an additive-free emulsion polymerization method50 and the size distribution of the product is analysed with dynamic light scattering, where broad distribution of PS particles sizes ranging from 400 to 800 nm is obtained (Fig. 2c). Consequently, they were introduced as a macropore-directing hard template to substitute SiO2 nanoparticles. Additionally, the negatively charged surface (zeta potential −41.8 mV) measured for the PS particles facilitates the adsorption of positively charged Fex+ onto the template surface51 and thus, exposing the iron in pores after template removal. | ||
| Fig. 2 Upper row: Films with different proportions of PS a) 1.05 g, b) 1.25 g and c) 1.64 g after the oxidative stabilization step at 210 °C and subsequent dissolution of the PS template. Bottom row: Film with 1.64 g PS carbonized at 900 °C with d) top view, e) cross section view and f) edge view. | ||
A general outline of the preparation of the conductive macroporous film support (Scheme 1), is complemented by a detailed description in the ESI† (Scheme S1). The precursor reactants are mixed to produce a slurry for the casting of the film. The mesophase pitch is ball milled in 1-butanol, while an aqueous suspension of PS particles with PVA as a surfactant is probe sonicated. Then, the two liquids are unified and the plasticizer 1,3-propanediol is added for a second round of ball milling. The slurry is casted, dried and peeled off for the oxidative stabilization step. During the oxidative stabilization, oxygen-containing functional group are formed with simultaneous dehydrogenation, and polymerization reactions forming crosslinking bridges between polycyclic aromatic hydrocarbons.52 The introduction of oxygen into the pitch usually leads to a minimal weight gain, whereas other reactions such as dehydrogenation, cyclization and cross-linking might compensate the weight gain by emitting CO, CO2 and H2O and low molecular weight hydrocarbons. As a result of the stabilization process, MP in the film changes from a thermoplastic solid to a thermosetting material, which maintains the morphology of carbon film, as it can effectively inhibit the deformation and melting of the pitch at high temperature heat treatment.53
 | ||
| Scheme 1 Preparation steps for the conductive macroporous film. | ||
2.3. Adjusting the PS content for interconnected macropore network
The film's PS volume ratio is optimized to endow a well-connected macroporous framework on the solid carbon precursor mixture of MP and PVA. After the stabilization step, the PS template is dissolved in toluene within 1 hour, leaving the moulded carbon structure of MP and PVA intact (Fig. 2 upper row). The films containing 64% (1.05 g) and 68% (1.25 g) PS exhibit a lack of pore connectivity, particularly evident in the uppermost layer, despite having interconnected pores within the film bulk phase. Lower PS contents in the film are also studied (Fig. S3†). According to the theory of close-packing of equal spheres, 74% represents the maximum volume occupied by sphere of equal size. Remarkably, the film with 75% PS (1.64 g) confirms this theory, resulting in a macroporous network omnipresent in the film (Fig. 2c). The pore structure is preserved after carbonization in nitrogen atmosphere at 900 °C. Even though on the surface the macropores are coalescing to larger pores (Fig. 2d), the interpenetrating channel network remains unaltered as can be seen from the cross-sectional and edge imaging analysis (Fig. 2e and f). Through carbonization, the initially resistive film becomes an electron-conductive support, achieving a conductivity of 3.02 S m−1 (Fig. S4†). Flexibility is crucial for the film's effectiveness as a cathode in fuel cells. Notably, a stabilization temperature of 210 °C yields carbon fibers with the highest modulus,46 a key factor in selecting the stabilization temperature.2.4. Effect of heating protocol and carbonization atmosphere on the film properties
The heating rate has a decisive effect on the brittleness of the carbonized mesophase pitch carbon films.54 While 0.5 °C min−1 and 1 °C min−1 limited the bendability of the film, the heating rate of 0.1 °C min−1 enabled greater flexibility (Fig. S5†).The composition of the carbonization atmosphere can modify the pore structure and elemental surface composition of the macroporous carbon film.55Carbonization of the macroporous film under a nitrogen (N2) atmosphere results in a BET surface area of 112 m2 g−1 with a porosity of 79%, primarily originating from macropores, as shown by the nitrogen adsorption and mercury intrusion porosimetry (Fig. 3a and b). While a subtle peak around 2.5–3 nm is observed in the PSD (Fig. 3b), the corresponding mesopore volume is minimal (∼0.06 cm3 g−1, Table 1), and the film can still be classified as predominantly macroporous.
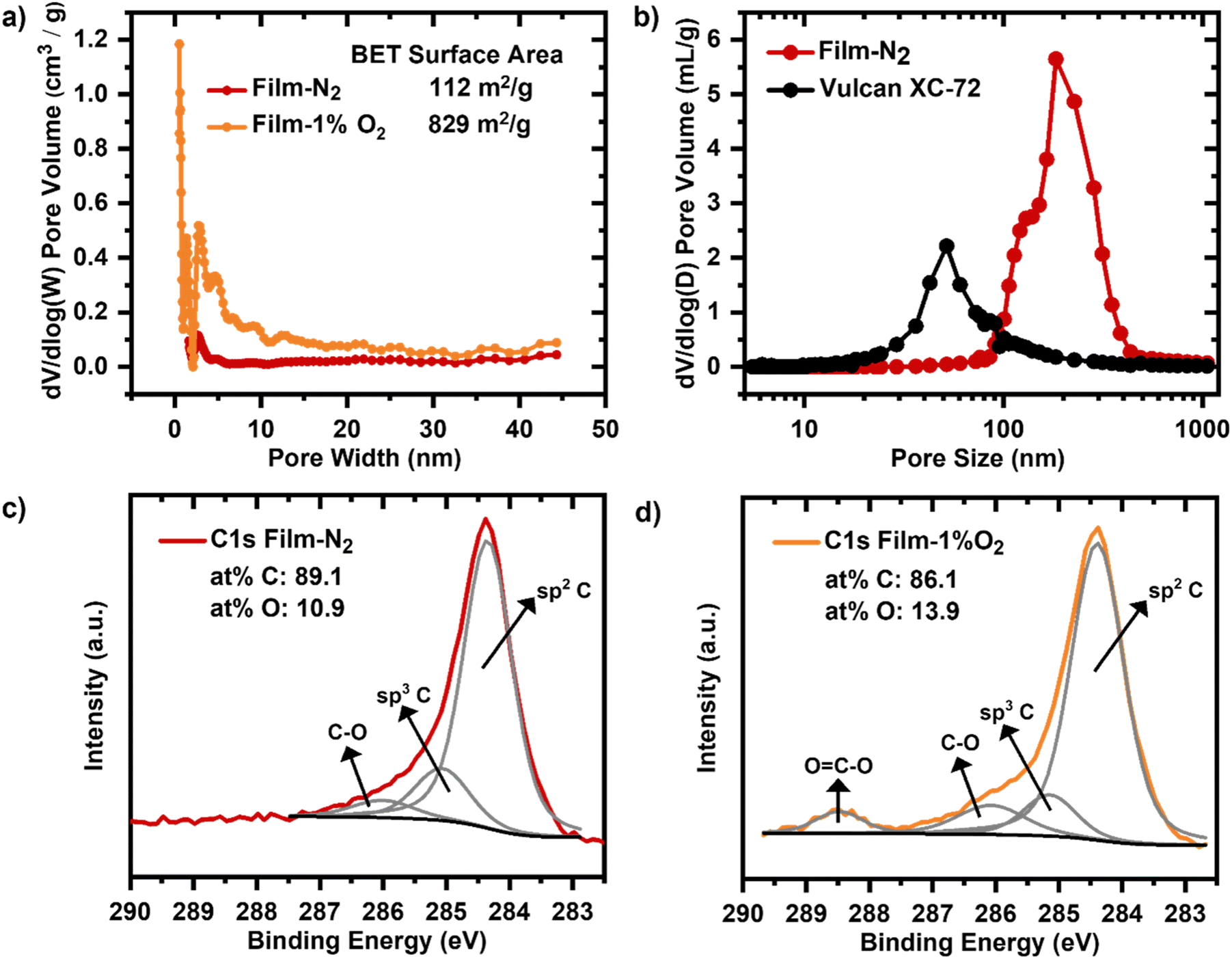 | ||
| Fig. 3 a) Pore size distribution in for micro- and mesopore range obtained by nitrogen adsorption before electrochemical characterization. b) Pore size distribution for cathodes based on Film-N2 and Vulcan XC-72/Sustainion obtained by MIP before electrochemical characterization. XPS spectra of the C1s for the film carbonized in c) N2 and in d) 1% O2/99% N2 before electrochemical characterization. | ||
| Film-N2 | Film-1%O2 | Vulcan | |
|---|---|---|---|
| Micropore volume via N2 sorption (cm3 g−1) | <0.01 | 0.31 | 0.06 |
| Mesopore volume via N2 sorption (cm3 g−1) | 0.06 | 0.24 | 0.29 |
| Macropore volume via MIP (cm3 g−1) | 2.56 | NA | NA |
When the film is carbonized under a 1% O2 atmosphere, the BET surface area increases significantly to 829 m2 g−1, primarily due to the introduction of micropores, as indicated by the enhanced nitrogen adsorption at low relative pressures (Fig. S15†). Although peaks in the mesopore range (2.5–5 nm) appear in the PSD of the O2-treated film (Fig. 3b), the increase in mesopore volume is relatively minor compared to the rise in micropore volume (Table 1). Therefore, the primary effect of the O2 treatment is attributed to the formation of micropores.
To complement the nitrogen sorption analysis, the macropore structure of the N2-carbonized film was further investigated using MIP. The MIP results confirm that the majority of the pore volume is in the macropore range, with pore sizes above 100 nm (Fig. 3c), consistent with the intended design of the film. A comparison with the PSD of Vulcan shows that the macroporous film exhibits larger pore sizes, which can facilitate enhanced mass transport under high current density conditions.
The high resolution XPS C1s spectra for the film carbonized in nitrogen (Film-N2) and the film carbonized with a 1% dosage of O2 in N2 atmosphere (Film-1%O2) exhibit both three common peaks (Fig. 3c) and d)). The peak centred at 284.5 eV corresponds to aromatic (sp2) carbon bonds. The peak at 285 eV can be assigned to sp3 carbon bonds, which is slightly decreased for the Film-1%O2.56,57 The peak at a binding energy of 286 eV is indicative of oxygen functional groups (C–O) on the carbon surface and the additional peak at 288.5 eV for carboxy fragments (O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) is detected for the Film-1%O2 sample (Fig. 3d)), which is absent in C1s spectrum of the Film-N2.58,59 Quantitative analysis reveals that the Film-N2 sample contains 89.1 at% carbon and 10.9 at% oxygen. As a consequence of the exposure to 1% oxygen during the carbonization, the degree of carbon oxidation for the Film-1%O2 sample is higher, as reflected also by an increased proportion of oxygen of 13.9 at% besides the abovementioned additional peak at 288.5 eV.
O) is detected for the Film-1%O2 sample (Fig. 3d)), which is absent in C1s spectrum of the Film-N2.58,59 Quantitative analysis reveals that the Film-N2 sample contains 89.1 at% carbon and 10.9 at% oxygen. As a consequence of the exposure to 1% oxygen during the carbonization, the degree of carbon oxidation for the Film-1%O2 sample is higher, as reflected also by an increased proportion of oxygen of 13.9 at% besides the abovementioned additional peak at 288.5 eV.
2.5. RDE study of the ORR performance of the ball-milled film
The carbon film supports were ball-milled into a powder for rotating disk electrode (RDE) testing to evaluate the intrinsic catalytic activity of the material under well-defined and controlled mass transport conditions. The bulk carbon films are inherently incompatible with the RDE setup, as they cannot form the uniform, thin catalyst layer required for precise electrochemical evaluation. By ball-milling the carbon films, we ensured the material could be uniformly dispersed in an ink with a Nafion binder, allowing for the preparation of a homogeneous thin layer on a glassy carbon electrode tip. While this process disrupted the macroporous structure of the original films, as shown by SEM imaging (Fig. S6†), it enabled reliable and reproducible evaluation of the material's catalytic performance using RDE.The ball milled (BM) supports BM-Film-N2 and BM-Film-1%O2 are compared to a commercial reference support Vulcan XC 72 (BET surface area: 250 m2 g−1). Metal macrocyclic compounds such as porphyrins and phthalocyanines are frequently used as molecular catalysts for the oxygen reduction reaction (ORR), as they can be readily solution-adsorbed onto supports. Iron centres in particular outperformed the other transition metals in terms of catalytic activity.60 The disadvantage of molecular iron phthalocyanine (FePc) is its susceptibility to degrade in acidic environment.61 Three demetallization processes of FePc were identified including the electrochemical oxidation of the ligand ring at high potentials (>0.65 V vs. RHE), Fenton reagents (between 0.6 and 0.2 V vs. RHE) and hydrogen peroxide at lower potentials.62 By heat treatment at temperatures (≥900 °C) of FePc/C, a more stable catalyst can be obtained.63 However, at high temperature FePc is converted to a heterogeneous mixture of active site structures including iron single sites coordinated to nitrogen (Fe–N4), metallic iron, and iron oxide (Fe3O4) nanoparticles.64,65 As a consequence of FePc's instability in acidic electrolytes, the majority of studies with FePc are conducted at alkaline pH.66 The effect of the support on the ORR performance can be easily elaborated with macrocyclic molecular catalyst, because the parameter of type of active sites can be kept constant due to their homogeneous nature.67 FePc weight loading was quantified and optimized on reference support Vulcan XC 72 (Fig. S7a†). Besides electron transport to the active sites, the main role of the carbon support is to disperse the molecular catalyst for increased availability of the active sites by preventing aggregation, a tendency that macrocyclic molecular catalysts without support show.68 The 1 wt% Fe sample showed the highest mass activity (at 0.85 V vs. RHE), whereas the other iron loadings had mass activities below 30 A g−1 (Fig. S7b†). The iron content of the sample with experimentally calculated FePc for 1 wt% Fe was verified as 0.45 ± 0.07 wt% Fe with ICP-MS (Fig. S7c†). It is suspected that for lower FePc loadings, the support is sparsely covered with FePc active sites. In contrast, at higher FePc loading (2 wt% Fe), active sites might be buried in molecular aggregates leading to reduced active site exposure. To understand the chemistry of the FePc-loaded carbon film before subjecting it to electrochemical tests, detailed characterizations were performed. X-ray diffraction (XRD) analysis (Fig. S13†) was conducted to investigate the crystalline structure of the catalyst. The absence of sharp peaks corresponding to Fe (111) at 43.8° or Fe (020) at 51.2° confirms the lack of metallic Fe nanoparticles.59 Additionally, no diffraction peak at 45° was observed, ruling out the presence of Fe3C (110), further supporting the conclusion that FePc active sites remain isolated without forming Fe nanoparticles or iron carbide. Furthermore, XPS analysis (Fig. S14†) provided additional insights into the chemical composition of the catalyst. The C1s spectra of the FePc-loaded carbon film exhibited binding energies similar to those of the pristine carbon film, indicating that the low loading of FePc had a minimal impact. This observation aligns with the absence of Fe peaks on XPS and confirms that Fe sites are present as isolated Fe–N configurations.69,70 Furthermore, the N1s spectra reveal two distinct nitrogen species consistent with the phthalocyanine structure,71 providing further evidence of the successful introduction of FePc as the model molecular catalyst.
The cyclic voltammograms (CVs) of FePc/BM-Film-N2 and FePc/BM-Film-1%O2 in oxygen-saturated 0.1 KOH are compared with FePc 1 wt% Vulcan XC 72 (Fig. 4a). In the kinetic region, FePc 1 wt% Vulcan XC 72 exhibits the lowest overpotential, followed by FePc/BM-Film-1%O2, and slightly higher values for BM-Film-N2. The minor improvement observed in FePc/BM-Film-1%O2 compared to BM-Film-N2 is likely due to its slightly higher oxygen surface content, which facilitates axial coordination of FePc to oxygen groups on the support, enabling better oxygen adsorption and activation.72,73 Besides the modification of the surface chemical composition, the presence of additional micropores in FePc/BM-Film-1%O2 may also contribute to the enhanced activity. However, the diffusion-limiting current density is significantly lower for FePc/BM-Film-1%O2 compared to Vulcan XC 72, likely due to the incompatibility of the ball-milled films with the thin-film RDE testing setup. The primary particles obtained after ball milling are significantly larger (a few hundred nm to micrometers, Fig. S6†) than those of Vulcan XC 72 (20–50 nm, Fig. S11†), which hinders the formation of a uniform thin film required for RDE testing. As a result, the full utilization of active sites in BM-Films is restricted under RDE conditions, impacting the limiting current density. Furthermore, the mass-activity of the three supports at 0.85 V (vs. RHE) is evaluated. The mass activity at 0.85 V (vs. RHE) for FePc 1 wt% Vulcan XC 72 with 51.15 A g−1 is approximately ten times greater than FePc/BM-Film-N2 and FePc/BM-Film-1%O2 with both having values below 5 A g−1 (Fig. 4b). Despite the BM-Films having twofold higher FePc loadings as determined by ICP-MS (Fig. 4c), the ball milling process disrupts the macroporous structure and prevents the films from matching the performance of the reference Vulcan XC 72 support under RDE conditions.
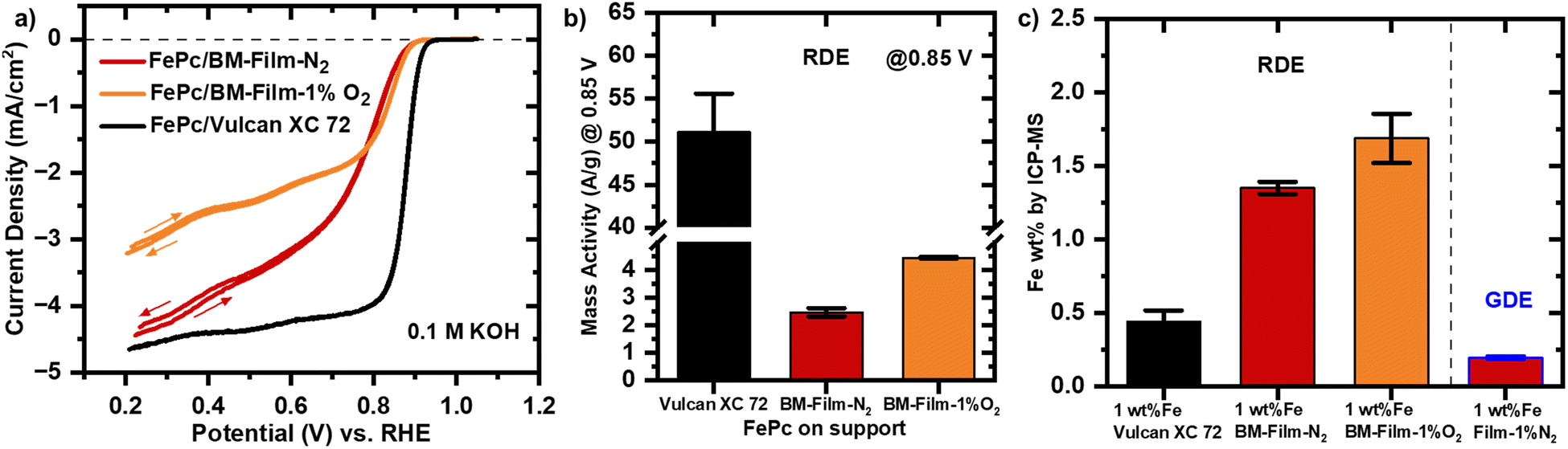 | ||
| Fig. 4 a) Cyclic voltammetry of ball milled film supports (BM-Film-N2, BM-Film-1%O2) with model active site FePc compared to FePc 1 wt% Vulcan XC 72 in oxygen-saturated 0.1 M KOH starts at a potential of 0.20 VRHE with a 10 mV s−1 scan rate in anodic scan direction at 1600 rpm. The catalyst loading 200 μg cm−2 is measured on a glassy carbon electrode (Ø = 5 mm) (WE) in a cell containing a glassy carbon rod (CE) and Hg/HgO (RE). The applied voltage is corrected for iR drop determined by EIS post measurement. b) Overview of the mass activities at a potential of 0.85 V in the RDE configuration. c) Iron content measured with ICP-MS for the catalyst prepared by physisorption of FePc on the respective powders for RDE and the Film-1%N2 for GDE. | ||
It can be concluded that the selection of the support for molecular catalysts is crucial for improving the ORR activity, which is in agreement with literature.74 The surface chemical properties and electronic structure of the carbon support interact with the electronic structure of the molecular catalyst adsorbed, and thus modifying the adsorption energy of the ORR intermediates. While a detailed DFT study is beyond the scope of this work, the adsorption energies reaction participants in the ORR on FePc on Vulcan XC 72 have been calculated elsewhere.75
2.6. ORR performance study of macroporous carbon film cathode in GDE
The section focuses on the comparison of two support materials as component of cathode catalyst layers. The effect of the support type in the assessment of catalytic activity of both support structures can be extracted by loading the same molecular catalyst on both supports. Even though non-heat-treated molecular catalysts with exceptional performance have been reported in the past, they were rarely rested in a fuel cell. In 2012, the performance of catalyst coated Tokuyama A901 membranes with phthalocyanines with different metal centres supported on Vulcan XC 72 were compared in an AEMFCs. While keeping the cathode loading constant at 2 mg cm−2, it was found that FePc/Vulcan XC 72 showed the poorest performance with a peak power density (PPD) of 88 mW cm−2.60 An optimization of the catalyst layer mass loading in the cathode, utilizing FePc supported on Ketjenblack, demonstrated that the lowest loading of 1.0 mg cm−2 led to improved performance. In numbers, 1 mg cm−2 loading with a PPD of 108 mW cm−2 surpassed the loadings of 2.0 mg cm−2 and 3.0 mg cm−2.67 Fluorinated iron phthalocyanine (FeFPc) supported on high surface area carbon BP2000 (FePPc@BP2000, 2 mg cm−2) exhibited the highest PPD with 166 mW cm−2 of molecular catalysts implemented on the cathode in AEMFCs.76 The ORR activity study is extended from the ball-milled powder RDE study to ORR performance evaluation of the carbon film cathode equipped with FePc active sites in the small GDE setup.77 The advantage of the GDE setup is that much larger current densities (up to 2 A cm−2) can be measured due to effective oxygen diffusion through the GDL. We note that established protocol might fail for the specific materials due to a different break in time compared with conventional powder catalyst.78 As Pt/C catalyst layers tested in GDE setup are usually thinner by several factors than Fe–N–C catalyst layers, the community established a pre-conditioning protocol, which might be too short for the thicker catalyst layer investigated in this work. At the end of preconditioning period, the thicker film samples might not be in their most ORR active state. The reported ORR data is not purely driven by potential but also by time as evidenced in ESI† (Fig. S10). Consequently, some of the data points at high current densities deviate from the slope and show slightly lower overpotentials at a current density value higher than the previous data point. The galvanostatic steps coupled with electrochemical impedance spectroscopy (GEIS) curve of the FePc/Film-N2 and FePc/Film-1%O2 with the same thickness of 70 μm superimpose at low current densities. At high current densities (1.5 A cm−2, 2.0 A cm−2), FePc/Film-1%O2 has a slightly lower overpotential than the FePc/Film-N2 (Fig. 5a). This is consistent with the enhanced activity observed in the RDE study, which is attributed to the increased accessibility of active sites resulting from the additional presence of micropores and mesopores. According to a modelling study, the thickness of the catalyst layer, proportional to the catalyst loading, is expected to be a main determinant of mass transport losses in PGM-free catalyst layers.79 The macroporous film supports have been designed with the intention to have the capability to increase the thickness of catalyst layers without suffering mass transport losses compared to conventional powder catalyst layers. The goal was to tailor the pore size distribution of the macroporous structure to enhance through-plane mass transport within the catalyst layer by increasing the macropore sizes, enabling improved mass transport capabilities. The macropore size distribution for the Vulcan XC 72 catalyst layer prepared with alkaline Sustainion ionomer is compared with the Film-N2 with MIP (Fig. 3b). The majority of macropores in the Vulcan XC 72 catalyst layer are between 40 nm and 60 nm, whereas macropores in the carbon film cathode fall into a range between 100 and 400 nm. The effect of adjusting the thickness of the macroporous carbon film cathode (FePc/Film-N2) for 30 μm and 70 μm is explored in GDE configuration. Notably, the 30 μm film outperforms the 70 μm slightly in all galvanostatic step by approximately 10 mV lower overpotential (Fig. 5b). To be able to determine the mass activities of the carbon film cathodes, catalyst layer mass loading is determined by weighing laser cut samples (Ø = 3 mm) and subsequent measuring of their geometrical electrode area in SEM (Fig. S8†). Reference catalyst layers of conventional FePc/Vulcan XC 72 with loadings of 2 mg cm−2 and 4 mg cm−2, corresponding to thicknesses of 50 μm and 100 μm, as determined with SEM cross section imaging, are prepared (Fig. S9†). At low current densities, the 2 mg cm−2 loading has lower overpotentials than the 4 mg cm−2. At high current densities, the error in the overpotential of the 2 mg cm−2 becomes large (±24 mV), which can be potentially explained by inhomogeneous catalyst layer loadings due to hand spraying. In general, it is observed that the 4 mg cm−2 catalyst with more active sites performs better at higher current densities above 1 A cm−2. In the entire current range, the powder-based catalysts layer of 4 mg cm−2 outperform the carbon film cathodes except for the data recorded for the 2 mg cm−2 at 2 A cm−2. At high current density of 2 A cm−2, where mass transport losses are expected to be most significant due to highest oxygen consumption, the thickness of the catalyst layer seems not to influence the mass transport for either support type, Vulcan XC 72 and Film-N2. The average overpotentials at 2 A cm−2 for the four types of samples fall into a range of 50 mV difference, which is within the error range (Fig. 5c). It has to be noted again that the established preconditioning protocol for powder catalysts might not be applicable to carbon films,75 as a repetition of the steps at high current densities in a second-round lead to an improvement of 50 mV except for the 2 A cm−2 value, which improved by 10 mV (Fig. S10†). The performance of the freestanding catalyst layer developed in this study was compared with previously reported Fe–N–C catalysts tested under similar GDE configurations, as summarized in Table S1,† highlighting its competitive potential under practical conditions despite its higher loading.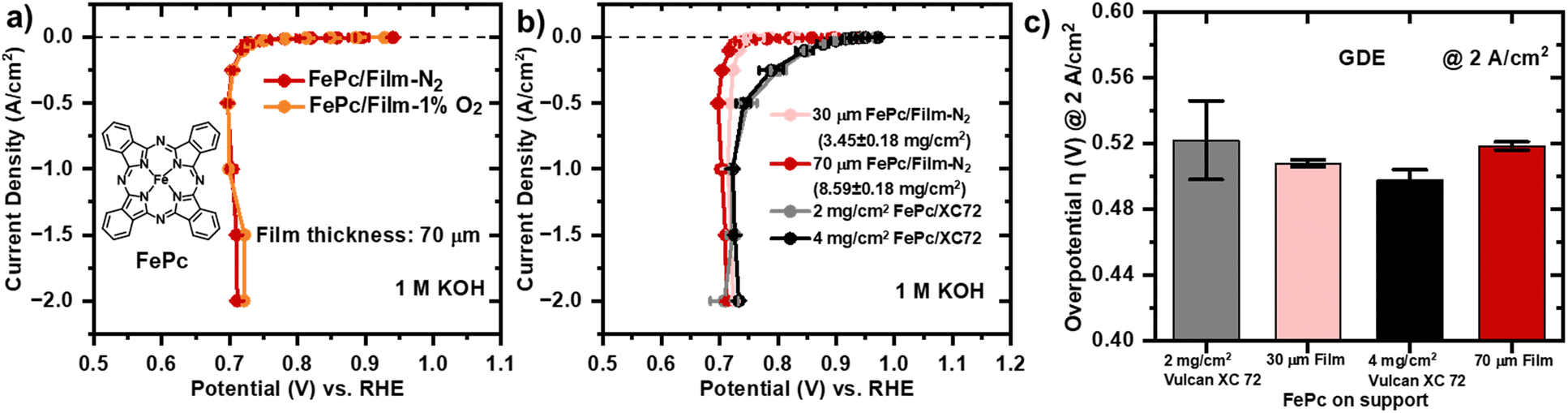 | ||
| Fig. 5 a) Oxygen reduction reaction polarization curves measured with the GEIS protocol for FePc supported on macroporous carbon films, Film-N2 and Film-1%O2, with the same thickness of 70 μm in 1 M KOH. b) Oxygen reduction reaction polarization curves measured with the GEIS protocol for different powder and film catalyst layer loadings. The data is recorded in a cell, in which the RHE (RE) is in a separate compartment and a the membrane separates the tested sample (WE) from platinum rod (CE). The GEIS protocol starts with galvanostatic steps and holding time from small current densities to high current densities in the order of −0.1 mA cm−2 (90 s), −1/−2.5/−5/−10 mA cm−2 (30 s), −25/−50/−10/−250 mA cm−2 (5 s), −0.5/−1.0/−1.5/−2.0 A cm−2 (5 s) at an oxygen flow rate of 300 mL min−1. The last recorded potential value at each galvanostatic step is taken. Each single potential value is iR-compensated with the value of the uncompensated resistance corresponding to the magnitude of the impedance measured after the corresponding galvanostatic step. Thereby, the single frequency for which the phase angle is closest to 0 in the high frequency region >1000 Hz is taken. At least two sets of independent GEIS measurements for each type of sample are carried out. It is noted that some of curves show slightly lower overpotentials falling out of the slope for some samples at high current densities (1, 1.5 and 2.0 A cm−2), which are time-dependent artefacts. The recorded data is a mixture of time and potential-dependent processes, which is elaborated in ESI† Fig. S10. c) Overview of the overpotentials at 2 A cm−2 for the catalyst layers tested in GDE configuration. | ||
3. Conclusions
This study represents the first exploration of macropore engineering in freestanding PGM-free carbon film cathodes using a sustainable polystyrene templating technique that eliminates the need for SiO2 template etching. By refining the heat treatment protocol, we optimized the templating process, achieving a conductive carbonized scaffold capable of efficient electron transport to all active sites. Oxygen activation during carbonization (1% O2) further enhanced the microporous structure and surface composition, contributing to improved ORR kinetics.The macroporous film supports demonstrated comparable mass transport performance to a conventional powder catalyst layer (Vulcan XC 72), with overpotentials at a high current density of 2 A cm−2 showing minimal variation between the two designs. This work establishes a platform for the development of a new class of freestanding film electrodes for Fe–N–C catalysts and alkaline fuel cells, offering promising opportunities for future advancements.
Future efforts will focus on further optimizing the pore structure by increasing mesopore abundance to enhance active site accessibility. Additionally, integrating Fe–N4 sites into the carbon surface could extend the functionality of these cathodes to acidic environments, broadening their applicability. Advanced electrochemical characterization will be employed to further investigate mass transport properties. Recognizing the importance of stability for practical applications, future work will also include long-term stability testing under relevant operating conditions.
Data availability
Additional data supporting this article is included in the ESI.† An excel sheet “Electrochemistry Data” containing the raw data from the electrochemical studies is included.Author contributions
S. K. contributed to the conceptualization, investigation, methodology, data curation, formal analysis and writing the original draft as well as reviewing the draft. M. W. contributed in the synthesis of PS, the data analysis and revision of the manuscript. Y. W. contributed to the preparation and the optimization of the film. J. B. contributed the XPS measurements and writing – review & editing. G. Y. contributed the conductivity measurements. J. F. contributed to writing – review & editing. I. E. L. S., S. H., and M.-M. T. contributed to writing – review & editing and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The thermomechanical analysis data were thankfully recorded by Koji Nakabayashi. S. K. acknowledges the financial support from Imperial College London through the Chemical Engineering departmental PhD Scholarship. M. W. acknowledges support from EPSRC Centre for Doctoral Training in the Advanced Characterisation of Materials (grant number EP/L015277/1). G. Y. would like to thank Imperial College London for the President's Scholarship. J. B. acknowledges financial support from Imperial College London through the Imperial College Research Fellowship. M.-M. T. acknowledges the Royal Academy of Engineering grant (grant number CiET1819\2\60).Notes and references
- L. Fan, H. Deng, Y. Zhang, Q. Du, D. Y. C. Leung, Y. Wang and K. Jiao, Energy Environ. Sci., 2023, 16, 1466–1479 RSC.
- K. Kodama, T. Nagai, A. Kuwaki and Y. Morimoto, Nat. Nanotechnol., 2021, 16, 140–147 CrossRef CAS PubMed.
- X. X. Wang, M. T. Swihart and G. Wu, Nat. Catal., 2019, 2, 578–589 CrossRef CAS.
- Y. Deng, J. Luo, B. Chi, H. Tang, J. Li, X. Qiao, Y. Shen, Y. Yang, C. Jia, P. Rao, S. Liao and X. Tian, Adv. Energy Mater., 2021, 11, 2101222 CrossRef CAS.
- L. Jiao, J. Li, L. L. Richard, Q. Sun, T. Stracensky, E. Liu, M.-T. Sougrati, F. Yang, S. Zhong, H. Xu, S. Mukerjee, Y. Huang, D. A. Cullen, J. H. Park, M. Ferrandon, D. J. Myers, F. Jaouen and Q. Jia, Nat. Mater., 2021, 20, 1385–1391 CrossRef CAS PubMed.
- M. M. Hossen, M. S. Hasan, M. R. I. Sardar, J. b. Haider, Mottakin, K. Tammeveski and P. Attanassov, Appl. Catal., B, 2023, 325, 121733 CrossRef CAS.
- D. Menga, J. L. Low, A. G. Buzanich, B. Paulus and T.-P. Fellinger, Adv. Energy Mater., 2024, 2400482 CrossRef CAS.
- L. Wang, X. Wan, S. Liu, L. Xu and J. Shui, J. Energy Chem., 2019, 39, 77–87 CrossRef.
- M. Gong, A. Mehmood, B. Ali, K.-W. Nam and A. Kucernak, ACS Catal., 2023, 13, 6661–6674 CrossRef CAS PubMed.
- S. Rojas-Carbonell, K. Artyushkova, A. Serov, C. Santoro, I. Matanovic and P. Atanassov, ACS Catal., 2018, 8, 3041–3053 CrossRef CAS.
- H. Adabi, A. Shakouri, N. Ul Hassan, J. R. Varcoe, B. Zulevi, A. Serov, J. R. Regalbuto and W. E. Mustain, Nat. Energy, 2021, 6, 834–843 CrossRef CAS.
- H. Adabi, P. G. Santori, A. Shakouri, X. Peng, K. Yassin, I. G. Rasin, S. Brandon, D. R. Dekel, N. Ul Hassan, M.-T. Sougrati, A. Zitolo, J. R. Varcoe, K. R. Regalbuto, F. Jaouen and W. E. Mustain, Mater. Today Adv., 2021, 12, 100179 CrossRef CAS.
- A. Mehmood, M. Gong, F. Jaouen, A. Roy, A. Zitolo, A. Khan, M.-T. Sougrati, M. Primbs, A. Martinez Bonastre, D. Fongalland, G. Drazic, P. Strasser and A. Kucernak, Nat. Catal., 2022, 5, 311–323 CrossRef CAS.
- L. Jiao, J. Li, L. LaRochelle Richard, Q. Sun, T. Stracensky, E. Liu, M. T. Sougrati, Z. Zhao, F. Yang, S. Zhong, H. Xu, S. Mukerjee, Y. Huang, D. A. Cullen, J. H. Park, M. Ferrandon, D. J. Myers, F. Jaouen and Q. Jia, Nat. Mater., 2021, 20, 1385–1391 CrossRef CAS PubMed.
- S. Liu, C. Li, M. J. Zachman, Y. Zeng, H. Yu, B. Li, M. Wang, J. Braaten, J. Liu, H. M. Meyer III, M. Lucero, A. J. Kropf, E. E. Alp, Q. Gong, Q. Shi, Z. Feng, H. Xu, G. Wang, D. J. Myers, J. Xie, D. A. Cullen, S. Litster and G. Wu, Nat. Energy, 2022, 7, 652–663 CrossRef CAS.
- X. Zhang, L. Truong-Phuoc, T. Asset, S. Pronkin and C. Pham-Huu, ACS Catal., 2022, 12, 13853–13975 CrossRef CAS.
- T. Lopes, A. Kucernak, D. Malko and E. A. Ticianelli, ChemElectroChem, 2016, 3, 1580–1590 CrossRef CAS.
- X. Yang, Y. Wang, G. Zhang, L. Du, L. Yang, M. Markiewicz, J. Choi, R. Chenitz and S. Sun, Appl. Catal., B, 2020, 264, 118523 CrossRef.
- J. Barrio, A. Pedersen, S. C. Sarma, A. Bagger, M. Gong, S. Favero, C. Zhao, R. Garcia-Serres, A. Y. Li, Q. Zhang, F. Jaouen, F. Maillard, A. Kucernak, I. E. L. Stephens and M.-M. Titirici, Adv. Mater., 2023, 35, 2211022 CrossRef CAS PubMed.
- X. Wan, X. Liu, Y. Li, R. Yu, L. Zheng, W. Yan, H. Wang, M. Xu and J. Shui, Nat. Catal., 2019, 2, 259–268 CrossRef CAS.
- C. Shao, S. Zhuang, H. Zhang, Q. Jiang, X. Xu, J. Ye, B. Li, X. Wang and H. Zhang, Small, 2021, 17, 2006178 CrossRef CAS PubMed.
- S.-H. Park, D.-H. Park, J.-H. Byeon, M.-H. Kim, Y. Gu, D.-M. Lim, J.-H. Kim, J.-S. Jang, C.-E. Hong, D.-G. Seo, J.-I. Han and K.-W. Park, Carbon, 2024, 218, 118666 CrossRef CAS.
- S. Chen, S. Xiang, Z. Tan, H. Li, X. Yan, J. Yin, S. Shen and J. Zhang, Front. Energy, 2023, 17, 123–133 CrossRef.
- A. G. Star, G. Wang, S. Medina, S. Pylypenko and K. C. Neyerlin, J. Power Sources, 2020, 450, 227655 CrossRef CAS.
- A. Serov, A. D. Shum, X. Xiao, V. De Andrade, K. Artyushkova, I. V. Zenyuk and P. Atanassov, Appl. Catal., B, 2018, 237, 1139–1147 CrossRef CAS.
- M. Wang, J. Zhang, S. Kellner, I. E. L. Stephens and M.-M. Titirici, J. Mater. Chem. A, 2024, 12, 24878–24885 RSC.
- M. Atwa, X. Li, Z. Wang, S. Dull, S. Xu, X. Tong, R. Tang, H. Nishihara, F. Prinz and V. Birss, Mater. Horiz., 2021, 8, 2451–2462 RSC.
- S. Kumar and M. Srivastava, Carbon Lett., 2015, 16, 171–182 CrossRef.
- S. Lee, Y. Eom, B.-J. Kim, I. Mochida, S.-H. Yoon and B. C. Kim, Carbon, 2015, 81, 694–701 CrossRef CAS.
- J. D. Brooks and G. H. Taylor, Carbon, 1965, 3, 185–186 CrossRef CAS.
- S. Jerez, A. Pedersen, M. Ventura, L. Mazzoli, M. I. Pariente, M. Titirici, J. A. Melero and J. Barrio, Electrochim. Acta, 2024, 483, 144045 CrossRef CAS.
- Z. Li and M. Jaroniec, J. Am. Chem. Soc., 2001, 123, 9208–9209 CrossRef CAS PubMed.
- X. Li, D. Banham, F. Feng, F. Forouzandeh, S. Ye, D. Y. Kwok and V. Birss, Carbon, 2015, 87, 44–60 CrossRef CAS.
- H. Yang, Y. Yan, Y. Liu, F. Zhang, R. Zhang, Y. Meng, M. Li, S. Xie, B. Tu and D. Zhao, J. Phys. Chem. B, 2004, 108, 17320–17328 CrossRef CAS.
- P. Adelhelm, K. Cabrera and B. M. Smarsly, Sci. Technol. Adv. Mater., 2012, 13, 015010 CrossRef.
- G. Gupta, D. A. Slanac, P. Kumar, J. D. Wiggings-Camacho, J. Kim, R. Ryoo, K. J. Stevenson and K. P. Johnston, J. Phys. Chem. C, 2010, 114, 10796–10805 CrossRef CAS.
- A. Serov, K. Artyushkova and P. Atanassov, Adv. Energy Mater., 2014, 4, 1301735 CrossRef.
- C. Janson and A. E. C. Palmqvist, Catal. Lett., 2019, 149, 1297–1304 CrossRef CAS.
- J. Zou, C. Chen, Y. Chen, Y. Zhu, Q. Cheng, L. Zou, Z. Zou and H. Yang, ACS Catal., 2022, 12, 4517–4525 CrossRef CAS.
- M. Qiao, Y. Wang, Q. Wang, G. Hu, X. Mamat, S. Zhang and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 2688–2694 CrossRef CAS PubMed.
- G. He and Q. Pan, Macromol. Rapid Commun., 2004, 17, 1545–1548 CrossRef.
- I. Milošević, V. Mauroy, H. Dabboue, S. Serieye, F. Warmount, J.-P. Salvetat, M.-L. Saboungi and S. Guillot, Microporous Mesoporous Mater., 2009, 120, 7–11 CrossRef.
- A. B. D. Nandiyanto, N. Hagura, F. Iskandar and K. Okuyama, Acta Mater., 2010, 58, 282–289 CrossRef CAS.
- K. Abitaev, Y. Qawasmi, P. Atanasova, C. Dargel, J. Bill, T. Hellweg and T. Sottmann, Colloid Polym. Sci., 2021, 299, 243–258 CrossRef CAS.
- S. Homaeigohar, R. Kabir and M. Elhbahri, Sci. Rep., 2020, 10, 5191 CrossRef CAS PubMed.
- P. Sieira, C. Guimarães, A. Braga, C. E. L. dos Santos, M. H. Pereira and L. E. Pizarro Borges, J. Ind. Eng. Chem., 2024, 246, 465–474 CrossRef.
- T. Senda, Y. Yamada, M. Morimoto, N. Nono, T. Sogabe, S. Kubo and S. Sato, Carbon, 2019, 142, 311–326 CrossRef CAS.
- J. Drbohlav and W. T. K. Stevenson, Carbon, 1995, 33, 693–711 CrossRef CAS.
- G. Yuan, X. Li, X. Xiong, Z. Dong, A. Westwood, B. Li, C. Ye, G. Ma, Z. Cui, Y. Cong, J. Zhang and Y. Li, Carbon, 2017, 115, 59–76 CrossRef CAS.
- A. Sandi, A. Sawitri, A. Rajak, A. Zulfi, D. Edikresnha, M. M. Munir and K. Khairurrijal, Mater. Sci. Eng., 2018, 367, 012015 Search PubMed.
- O. Larue, E. Vorobiev, C. Vu and B. Durand, Sep. Purif. Technol., 2003, 31, 177–192 CrossRef CAS.
- T. Qin, J. Zhao, R. Shi, C. Ge and Q. Li, Chem. Eng. J., 2020, 399, 125656 CrossRef CAS.
- X. Wang, G. Sun, F. Wang, X. Li, Z. Zhang, Y. Zhen, D. Wang, X. Ga, F. Fu and R. Chi, J. Colloid Interface Sci., 2023, 646, 228–237 CrossRef PubMed.
- V. Birss, X. Li, D. Banham and D. Y. Kwok, US Pat., 20170014780A1, 2019 Search PubMed.
- Z.-M. Wang, N. Yamashita, Z.-X. Wang, K. Hoshinoo and H. Kanoh, J. Colloid Interface Sci., 2004, 276, 143–150 CrossRef CAS PubMed.
- E. Desimoni, G. I. Casella, A. M. Salvi, T. R. I. Cataldi and A. Morone, Carbon, 1992, 30, 527–531 CrossRef CAS.
- X. Chen, X. Wang and D. Fang, Fullerenes, Nanotubes Carbon Nanostruct., 2020, 28, 1048–1058 CrossRef CAS.
- G.-F. Han, F. Li, W. Zou, M. Karamad, J.-P. Jeon, S.-W. Kim, S.-J. Kim, Y. Bu, Z. Fu, Y. Lu, S. Siahrostami and J.-B. Baek, Nat. Commun., 2020, 11, 2209 CrossRef CAS PubMed.
- L. Mazzoli, A. Pedersen, S. Kellner, R. D. Hunter, R. Cai, M. Wang, K. Sivula, S. J. Haigh and J. Barrio, Green Chem., 2024, 26, 3271–3280 RSC.
- J. Guo, H. He, D. Chu and R. Chen, Electrocatalysis, 2012, 3, 252–264 CrossRef CAS.
- S. Baranton, C. Coutanceau, C. Roux, F. Hahn and J.-M. Léger, J. Electroanal. Chem., 2005, 577, 223–234 CrossRef CAS.
- H. C. Honig and L. Elbaz, ChemElectroChem, 2023, 10, e202300042 CrossRef CAS.
- G. Lalande, G. Faubert, R. Côté, D. Guay, J. P. Dodelet, L. T. Weng and P. Bertrand, J. Power Sources, 1996, 61, 227–237 CrossRef CAS.
- H. A. Miller, M. Bellini, W. Oberhauser, X. Deng, H. Chen, Q. He, M. Passaponti, M. Innocenti, R. Yang, F. Sun, Z. Jiang and F. Vizza, Phys. Chem. Chem. Phys., 2016, 18, 33142–33151 RSC.
- M. Mubyuddin, E. Berretti, S. A. Mirshokraee, J. Orsili, R. Lorenzi, L. Capozzoli, F. D'Acapito, E. Murphy, S. Guo, P. Atanassov, A. Lavacchi and C. Santoro, Appl. Catal., B, 2024, 343, 123515 CrossRef.
- M. A. C. de Oliveira, V. C. A. Ficca, R. Gokhale, C. Santoro, B. Mecheri, A. D'Epifanio, S. Licoccia and P. Atanassov, J. Solid State Electrochem., 2021, 25, 93–104 CrossRef.
- J. Yang, J. Tao, T. Isomura, H. Yanagi, I. Moriguchi and N. Nakashima, Carbon, 2019, 145, 565–571 CrossRef CAS.
- Y. Jiang, Y. Lu, X. Lv, D. Han, Q. Zhang, L. Niu and W. Chen, ACS Catal., 2013, 3, 1263–1271 CrossRef CAS.
- M. Han, K. Zheng, J. Liu, Z. Zou, Y. Mu, H. Hu, F. Yu, W. Li, L. Wei, L. Zeng and T. Zhao, Adv. Energy Mater., 2024, 2403851 CrossRef.
- Z. Li, M. Han, J. Wang, L. Zhang, P. Yu, Q. Li, X. Bai and J. Yu, Adv. Funct. Mater., 2024, 34, 2404263 CrossRef CAS.
- J. H. Kim, Y. J. Sa, H. Y. Jeong and S. H. Joo, ACS Appl. Mater. Interfaces, 2017, 9(11), 9567–9575 CrossRef CAS PubMed.
- K. Chen, K. Liu, P. An, H. Li, Y. Lin, J. Hu, C. Jia, J. Fu, H. Li, H. Liu, Z. Lin, W. Li, J. Li, Y.-R. Lu, T.-S. Chan, N. Zhang and M. Liu, Nat. Commun., 2020, 11, 4173 CrossRef CAS PubMed.
- C. Z. Loyola and F. Tasca, Curr. Opin. Electrochem., 2023, 40, 101316 CrossRef.
- Y. Hong, L. Li, B. Huang, X. Tang, W. Zhai, T. Hu, K. Yuan and Y. Chen, Adv. Energy Mater., 2021, 11, 2100866 CrossRef CAS.
- R. Chen, H. Li, D. Chu and G. Wang, J. Phys. Chem. C, 2009, 113, 20689–20697 CrossRef CAS.
- A. Friedman, M. Mizrahi, N. Levy, N. Zion, M. Zachman and L. Elbaz, ACS Appl. Mater. Interfaces, 2021, 13, 58532–58538 CrossRef CAS PubMed.
- G. H. K. Wiberg, S. Nösberger and M. Arenz, Curr. Opin. Electrochem., 2022, 36, 101129 CrossRef.
- K. Ehelebe, N. Schmitt, G. Sievers, A. W. Jensen, A. Hrnjić, M. Geuß, Y.-P. Ku, P. Jovanovič, K. J. J. Mayrhofer, B. Etzold, N. Hodnik, M. Escudero-Escribano, M. Arenz and S. Cherevko, ACS Energy Lett., 2022, 7, 816–826 CrossRef CAS.
- S. Komini Babu, H. T. Chung, P. Zelenay and S. Litster, J. Electrochem. Soc., 2017, 164, F1037–F1049 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lf00380b |
| This journal is © The Royal Society of Chemistry 2025 |