
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDensely atomic Co–Nx moiety-coupled hierarchical-carbon-microspheres for efficient oxygen electrodes in bioadaptable Mg–air batteries†
Yanru
Liu‡
a,
Taiqiang
Dai‡
a,
Jia
Wang
a,
Lirong
Zheng
 b and
Xiaogang
Fu
*c
b and
Xiaogang
Fu
*c
aState Key Laboratory of Oral & Maxillofacial Reconstruction and Regeneration, National Clinical Research Center for Oral Diseases, Shaanxi Key Laboratory of Stomatology, Department of Dental Materials, School of Stomatology, The Fourth Military Medical University, Xi'an, ShannXi 710032, P. R. China
bBeijing Synchrotron Radiation Facility (BSRF), Institute of High Energy Physics Chinese Academy of Sciences, Beijing 100049, P. R. China
cState Key Laboratory of Solidification Processing, Atomic Control & Catalysis Engineering Laboratory, School of Materials Science and Engineering, Northwestern Polytechnical University, Xi'an, Shaanxi 710072, P. R. China. E-mail: xiaogangfu@nwpu.edu.cn
First published on 5th February 2025
Abstract
The Mg–air battery is one of the promising candidates for the next generation of bioadaptable power sources. However, it suffers from low efficiency cathodes to facilitate the slow oxygen reduction reaction (ORR). Herein, this work reports a strategy to enhance the density of catalytic active sites and further facilitate their accessibility for advanced air electrodes. A prior spray-assisted stepwise pyrolysis strategy is employed to obtain the densely atomic Co–Nx active site-decorated hierarchically carbon microspheres (Co–Nx–HCM), which are further employed as advanced oxygen electrodes for primary Mg–air batteries. The resulting Co–Nx active sites impart electrodes with good bio-adaptability and decent ORR activity under neutral electrolytes. Furthermore, the hierarchical carbon microspheres endow the air electrode with smooth mass transport. In application of an assembled Mg–air flow battery, the SA Co–Nx–HCM electrode delivers a high open-circuit potential of 1.78 V, a high maximum power density of 34.2 mW cm−2, a decent gravimetric energy density of ca. 3441 Wh kgMg−1, and a long operating life over 100 h. This work paves a way to explore single atomic catalysts with hierarchically porous morphology as advanced air cathodes for bioadaptable electronic devices.
1. Introduction
The rapid development of bionic electronics has highlighted the requirements for exploring new energy storage systems.1–3 The magnesium (Mg)–air battery is a promising candidate due to its high theoretical capacity, low cost and high discharge voltage under neutral conditions.4–7 Also, Mg or Mg alloys have good bioresorbability, and Mg2+ ions generated during battery discharge are beneficial to the human body.8–10 However, one of the significant difficulties hindering the rapid development of Mg–air batteries is the lack of an efficient air cathode to facilitate the oxygen reduction reaction (ORR).11–14Single atomic transition metals and nitrogen co-dopped carbon matrix (SA M–Nx–C) have been reported as promising materials for catalyzing the sluggish ORR.15–21 Recently, metal–organic-frameworks, especially zeolite imidazole frameworks (ZIF-8), have been developed as ideal precursors to prepare SA M–Nx–C catalysts exhibiting remarkably ORR activity.22–26 Subsequently, various strategies based on ZIF-8, including the coordination effect,27–31 chemical bonding,32–34 and spatial confinement,35–37 have been explored to enhance the density of M–Nx active sites. Despite the great process, it remains challenging to further increase the active site density as the agglomeration/migration of single metal atoms into clusters or nanoparticles during high-temperature treatment leads to a relatively weak catalytic activity.38–40 Furthermore, the actual catalytic performance of these ZIF-8 developed SA M–Nx–C catalysts becomes limited due to the restricted diffusion of ions and air caused by the non-accessible and blocked structure of the cathodes.41,42 Therefore, three-dimensional (3D) porous structures in a catalyst for facilitating mass transport and building efficient three-phase interfaces for the ORR in air electrodes are highly desired.43–45 However, to achieve these 3D porous structures, the routine methods always used silicon-based templates, which inevitably introduce complicated removal treatments and are challenging for scalable production.46–48 Thus, developing a facile strategy for preparing dense atomically dispersed M–Nx–C catalysts with 3D hierarchical porosity is particularly appealing.
To address these challenges, we report a prior spray-assisted stepwise pyrolysis strategy to prepare densely single atomic Co–Nx active site-incorporated hierarchical-carbon-microspheres (SA Co–Nx–HCM) as competent oxygen electrodes for Mg–air batteries. The main preparation procedures include a prior spray drying of fresh synthesized zeolitic imidazolate frameworks (ZIF-8) to form a 3D hierarchical micro-spheric support followed by a stepwise pyrolysis treatment for stabilizing dense Co–Nx active sites. It is noteworthy that the SA Co–Nx–HCM shows good bio-adaptability and decent oxygen electrocatalytic performances in neutral electrolytes. The air electrode built with the SA Co–Nx–HCM catalyst possesses a 3D porous structure, facilitating the ion/air diffusion and imparting the assembled Mg–air battery with good activity and stability performance.
2. Experimental
2.1. Synthetic procedures
The ZIF-8 crystals were synthesized according to the previous literature.46 Then, 10.0 g of the pristine ZIF-8 nanoparticles were dispersed in deionized water and then spray dried at 200 °C to construct ZIF-8-based hierarchical porous microspheres.49 After heat-treating at 700 °C for 1 h in Ar, the hierarchical-carbon-microsphere (HCM) support was obtained. Then, 100.0 g of HCM was dispersed in 30.0 mL of n-hexane while sonicating for 30 min. Next, Co(NO3)2 aqueous solution (300 mg ml−1, 30 μL) was added to the mixed solution and sonicated for 3 h. Then, the obtained precipitate was washed with deionized water 3 times and dried. Finally, the resulting materials were heated at 900 °C in Ar for 2 h to yield SA Co–Nx–HCM. For comparison, pristine ZIF-8 nanocrystals were directly used without spray drying and were treated in the same way as SA Co–Nx–HCM to fabricate SA Co–Nx–C. The sample prepared by directly pyrolyzing ZIF-8 and Co(NO3)2 was denoted as Co–N–C.2.2. Material characterization
All the materials' elemental compositions were detected using X-ray photoelectron spectroscopy (XPS). The microstructures and morphology were obtained by using a high-resolution transmission electron microscopy instrument (JEOL 2010F microscope). Raman spectra were acquired using a Bruker Senterra Raman instrument. The crystalline phase was identified by powder X-ray diffraction (XRD) (Bruker D8, Cu Kα radiation). A Micromeritics ASAP 3020 instrument was used to analyze the Brunauer–Emmett–Teller (BET) surface area and pore size distribution data. The Co content was measured by inductively coupled plasma optical emission spectrometry (ICP-OES, Thermo Fisher Scientific). The Co K-edge XAS measurements were conducted at the beamline 1W1B of the Beijing Synchrotron Radiation Facility (BSRF, Beijing, China). The Athena program was used to process data.2.3. Evaluation of cytocompatibility
To investigate the biocompatibility of SA Co–Nx–HCM, the CCK8 test and live/dead staining were performed.50 For the cell counting kit-8 (CCK8) test, third passage bone mesenchymal stem cells (BMSCs) were seeded in 96 well plates (2000 cells per well, 100 μl) and cultured for 24 hours. The culture medium was changed with fresh α-MEM medium containing 1.5 μg ml−1 of SA Co–Nx–HCM. At 1, 2, 3, 5, and 7 days, 10 μL CCK8 (Plant Chem Med, China) was added to the well, and cells were incubated for another 2 hours. An ELISA reader measured the absorbance at 450 nm.To assess the viability of cells cultured with SA Co–Nx–HCM, live/dead assay was conducted. Briefly, BMSCs were cultured in confocal dishes for 24 hours, and the culture medium was switched to fresh medium with 1.5 μg ml−1 of SA Co–Nx–HCM. After 7 days, the cells were incubated with Calcein-AM/Propidium Iodide (AM/PI) (Beyotime Biotechnology, China) staining solution at 37 °C for 30 minutes in the dark according to the manufacturer's instructions. Finally, the cells were observed and photographed under a laser confocal microscope (Nikon A1+, Japan).
3. Results and discussion
As shown in Fig. 1, we developed a spray-assisted stepwise pyrolysis approach to obtain densely single atomic Co–Nx active site-decorated hierarchically carbon microspheres. First, pristine ZIF-8 nanocrystals were subjected to a spray-drying treatment to form porous microspheres with diameter sizes of ca. 0.5 μm (Fig. S1a†). Next, heat treatment at 700 °C was applied to maintain the integrity of this spheric morphology (Fig. S1b†) and prevent it from crushing during the post-treatment. Moreover, abundant nitrogen-doped microporous defects were generated after pyrolysis, as evidenced by BET and XPS data. The t-plot analysis indicates that the micropore surface area (390 m2 g−1) accounts for 81.3% of the overall surface area (480 m2 g−1, Table S1†). Also, the XPS results show the overall N content is ca. 5.41 at%. Such ample micropores with N dopants are ideal carbon matrices to fabricate atomic active sites effectively.51–53 Finally, the nitrogen-doped carbon spheres were subjected to absorbing Co2+ ions and undergoing a second pyrolysis and post-treatment, obtaining SA Co–Nx–HCM. Obviously, the SA Co–Nx–HCM catalyst maintains the spherical and porous morphology (Fig. 2a and b). The generated pore voids between neighbouring HCM and within HCM can be observed, which are supposed to promote mass transport in the air cathode. In addition, obvious disordered and amorphous structures were detected in the carbon matrix (Fig. S2†), which are supposed to benefit ORR catalysis.54 The reference sample SA Co–Nx–C (Fig. S1c†) was prepared without spray-drying, and Co–N–C (Fig. S1d†) was synthesized by directly pyrolyzing ZIF-8 and cobalt salt. Apparently, all SA Co–Nx–HCM, SA Co–Nx–C and Co–N–C show similar adsorption hysteresis loops at high pressure (Fig. 2c), while the SA Co–Nx–HCM (894 m2 g−1) has a superior surface area compared to SA Co–Nx–C (712 m2 g−1) and Co–N–C (682 m2 g−1). The enhanced surface area of SA Co–Nx–HCM is ascribed to the voids between single ZIF-8-derived carbon crystals and the porosity within the assembled microspheres, as its external surface area (317 m2 g−1) is higher than that of SA Co–Nx–C (213 m2 g−1) and Co–N–C (197 m2 g−1, Table S1†). Also, these three samples show significant differences in pore size distribution (Fig. 2d). The SA Co–Nx–C and Co–N–C catalysts prepared without the spray-drying process show similar and narrow pore size distribution, and a large number of pore sizes centred at ca. 4 nm. As for the spray-drying processed SA Co–Nx–HCM catalyst, ample hierarchical pores ranging from 5–120 nm were detected. Such abundant and hierarchical meso/macropores could be employed as effective channels to facilitate reactants and products toward and away from Co–Nx active moieties and improve catalytic performance. However, some portions of the BET surface area cannot be effectively utilized, as electrolytes cannot reach them. Hence, the electrochemically active surface area (ECSA) was obtained to evaluate electrocatalysts' actual active surface areas during the ORR.55 As shown in Fig. 2e and f, the ECSA of SA Co–Nx–HCM (525 m2 g−1) is about 2.2 and 3.4 times higher than those of SA Co–Nx–HCM (238 m2 g−1) and Co–N–C (153 m2 g−1), respectively, which is in agreement to the trend observed with the BET surface areas. The superior ECSA is related to a high BET surface area and abundant porosity, suggesting that SA Co–Nx–HCM can provide good active site accessibility, smooth mass transport, and therefore better catalytic behaviours. To assess the tissue adaptability of the SA Co–Nx–HCM oxygen electrode, CCK8 tests were applied to evaluate the cytocompatibility in this study. As shown in Fig. 2g, SA Co–Nx–HCM exhibited low cytotoxicity, as the absorbance value at 450 nm significantly decreased when compared with the control sample. In addition, almost no or only a few dead cells were observed in control groups and groups with SA Co–Nx–HCM after 7 days (Fig. 2h and i), suggesting its good cell compatibility. These results guarantee its application potential in oxygen electrodes for bio-adaptable Mg–air batteries. | ||
| Fig. 1 Synthetic scheme for the preparation of SA Co–Nx–HCM catalyst. | ||
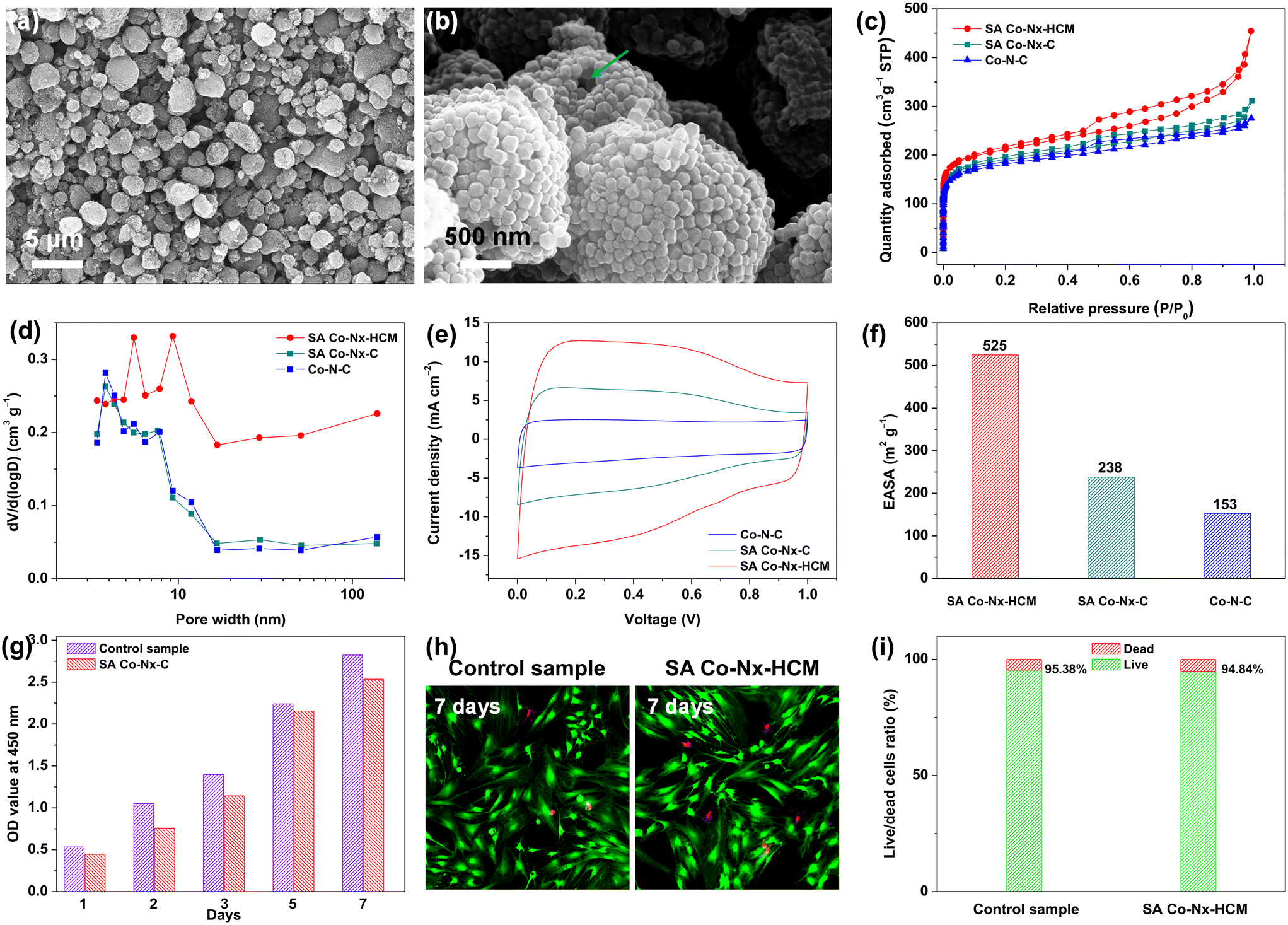 | ||
| Fig. 2 (a) and (b) SEM images of SA Co–Nx–MPCS. (c) N2 sorption isotherms, (d) the pore size distribution, (e) ECSAs determined in the PBH electrolyte, and (f) comparation of ECSAs for the prepared catalysts. In vitro cytocompatibility analysis of BMSCs cocultured with 1.5 μg ml−1 SA Co–Nx–HCM. (g) CCK8 test for BMSCs cocultured with SA Co–Nx–HCM. (h) Representative images of live/dead staining. (i) Live/dead cell ratio of BMSCs for 7 days. | ||
This spray-assisted stepwise pyrolysis approach is feasible for fabricating dense Co–Nx active sites. The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images indicate that no observable Co-based aggregates are presented in SA Co–Nx–HCM (Fig. 3a). Also, the elemental mapping reveals that the C, N and Co elements are uniformly distributed. Further, the atomic resolution HAADF-STEM image indicates that Co atoms are atomically dispersed on the carbon spheres (Fig. 3b). These results are in accord with the XRD results, as, except for the broad peak assigned to the (002) facet of graphitic carbon, there is no other diffraction peak of crystalline Co species shown in SA Co–Nx–HCM and SA Co–Nx–C (Fig. 3c), confirming the non-formation of cobalt crystals. By contrast, the Co–N–C catalyst prepared by directly pyrolyzing ZIF-8 and Co(NO3)2 precursor contains a small amount of CoO metallic particles, as demonstrated by the emerged peak at 45° (Fig. 3c) and the TEM analysis (Fig. S3†). The Raman spectra indicate that the ID/IG ratio values in SA Co–Nx–HCM (0.97) and SA Co–Nx–HCM (0.98) are higher than that in Co–N–C (0.92), suggesting that Co–N–C has a relatively higher graphitic structure (Fig. 3d). This is probably because trace Co metallic particles increase the degree of graphitization for the Co–N–C catalyst during heat treatment. All these results further indicate that the preheating of ZIF-8 at 700 °C is essential to avoid Co agglomeration and generate atomic Co–Nx sites.
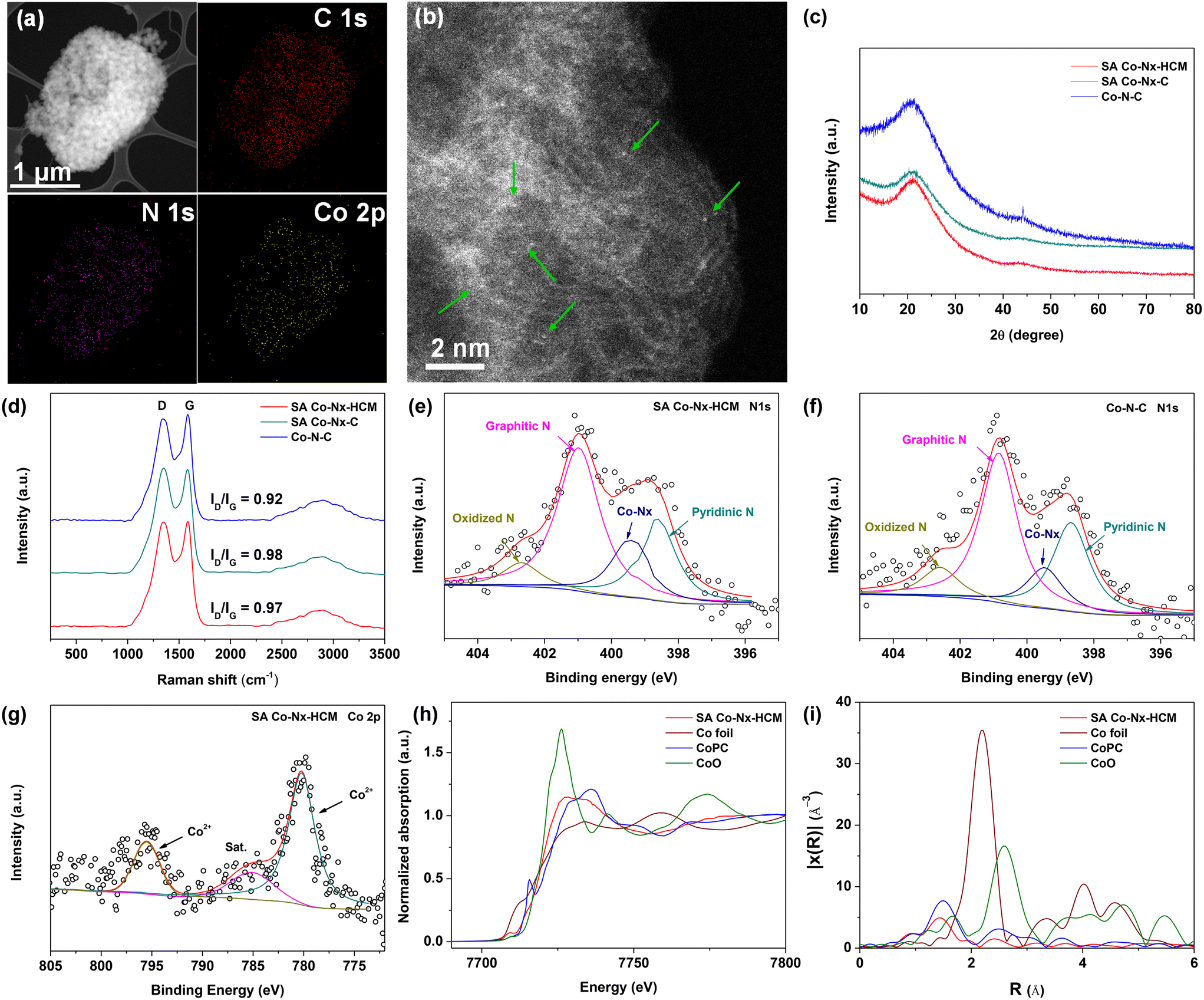 | ||
| Fig. 3 (a) STEM image and (b) the corresponding elemental mapping of SA Co–Nx–HCM. (c) XRD and (d) Raman patterns of the prepared catalysts. XPS N 1s of (e) SA Co–Nx–HCM and (f) Co–N–C. (g) XPS Co 2p of SA Co–Nx–HCM. (h) Fe K-edge XANES and (i) Fourier transforms of EXAFS spectra of prepared catalysts and references. | ||
XPS analyses were conducted to evaluate the prepared catalysts' surface chemical composition and elemental bonding configuration. The N and Co contents in SA Co–Nx–HCM are 4.41 and 0.22 at%, respectively, similar to those in the SA Co–Nx–C catalyst (4.34 at% and 0.24 at%, Table S2†). The N 1s fitting results indicate the coexistence of pyridinic N (398.5 eV), Co–Nx (399.3 eV), pyrrolic N (400.3 eV), graphitic N (401.2 eV), and oxidized N (402.5 eV), respectively (Fig. 3e and f, S4†). Furthermore, the Co–Nx contents in the stepwise pyrolyzed samples (SA Co–Nx–HCM, 0.78 at%; SA Co–Nx–C, 0.73 at%) are higher compared to that in the direct–carbonized sample (Co–N–C, 0.63 at%, Table S3†). The Co 2p spectrum for SA Co–Nx–HCM exhibit two prominent peaks at 780.3 and 795.7 eV corresponding to the Co2+–N coordinates with their satellite peaks centred at 785.2 eV (Fig. 3g). Then, X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) analyses were performed to further analyze the chemical environments of Co–Nx moieties in the catalysts. The Co K-edge spectra in Fig. 3h suggest that the features of SA Co–Nx–HCM are more similar to CoPC but different from Co foil and CoO, which further indicates the valence state of SA Co–Nx–HCM is situated between that of Co0 and CoII.28 Further, Fourier-transformed k2-weighted EXAFS (Fig. 3i) show that the SA Co–Nx–HCM exhibits one apparent peak at ca. 1.5 Å, which was ascribed to the Co–N first coordination shell, and there is almost no Co–Co peak at 2.2 Å identified. These results suggest that the Co atoms are atomically dispersed in the Co–Nx–HCM catalyst. Moreover, this strategy is able to generate a relatively high SA Co content (1.13 wt%) compared to previously reported catalysts (Table S4†), which is beneficial for boosting the ORR.
Considering the unique assembled-spheres with a hierarchical-porous structure and the highly dense active SA Co–Nx sites, the explored SA Co–Nx–HCM catalyst could be a good candidate for catalyzing the ORR in natural electrolytes. A commercial Pt/C catalyst, Co–N–C, and SA Co–Nx–C were evaluated for comparison. The SA Co–Nx–HCM samples prepared at 900 °C exhibit the highest half-wave potential (Fig. S5†). As shown in Fig. 4a, no obvious reduction peaks were observed in the N2-saturated PBS electrolyte, while characteristic cathode peaks appeared in the O2-saturated electrolyte, indicating the ORR electrocatalytic activity of the prepared Co-based samples. Further, SA Co–Nx–C exhibits a more positive half-wave potential (E1/2) of 0.69 V than Co–N–C (E1/2 = 0.67 V), suggesting the stepwise pyrolysis could significantly improve the ORR activity (Fig. 4b). The prior spray-assisted treatment could further enhance the catalytic performance for SA Co–Nx–HCM with the best E1/2 up to 0.71 V compared to the other catalysts. Meanwhile, SA Co–Nx–HCM also shows the largest diffusion-limiting current density (5.81 mA cm−2) compared with SA Co–Nx–C (5.41 mA cm−2) and Co–N–C (5.30 mA cm−2), indicating the smoothly accessible and high utilization of Co–Nx active sites. Further, the E1/2 of SA Co–Nx–HCM is only ca.10 mV less than that of Pt/C (E1/2 = 0.72 V), and the limiting current density is approaching the Pt/C catalyst (5.92 mA cm−2). At 0.85 V, the kinetic current density of the ORR on SA Co–Nx–HCM is 0.47 mA cm−2, which is much higher than that of Co–N–C (0.14 mA cm−2) and SA Co–Nx–C (0.31 mA cm−2), and lower than that of Pt/C (0.93 mA cm−2) (Fig. 4c). In addition, a rotating ring disk electrode (RRDE) study was conducted to evaluate the selectivity of these Co-based catalysts in a neutral electrolyte. The electron transfer number of the SA Co–Nx–HCM catalyst is above 3.97 (Fig. 4d), which is superior to those of the SA Co–Nx–C (3.95) and Co–N–C (3.93) catalysts and approaching that of the Pt/C catalyst (3.98), suggesting a high selectivity toward a four electron reaction. Also, the SA Co–Nx–HCM catalyst shows a H2O2 yield below 1.2% at the potential range from 0.4 to 0.9 V, which is superior to the SA Co–Nx–C (1.4%) and Co–N–C (2.0%) catalysts, suggesting the good selectivity of the oxygen reduction toward H2O (Fig. 4d). The decent ORR activity of SA Co–Nx–HCM was further confirmed by its smaller Tafel slope (82 mV dec−1) compared to SA Co–Nx–C (89 mV dec−1) and Co–N–C (97 mV dec−1), demonstrating that the SA Co–Nx–HCM electrocatalyst manifested improved ORR kinetics (Fig. 4e). Then, the electrochemical durability was evaluated to assess the catalyst's practical application. The SA Co–Nx–HCM and Pt/C benchmark were assessed and compared after 3000 CV cycles between 0.6 and 1.0 V under O2 atmosphere. It shows that the E1/2 difference is 26 mV for Pt/C after the accelerated degradation test, while there is almost no E1/2 change for the SA Co–Nx–HCM catalyst, unambiguously demonstrating its superior durability (Fig. 4f).
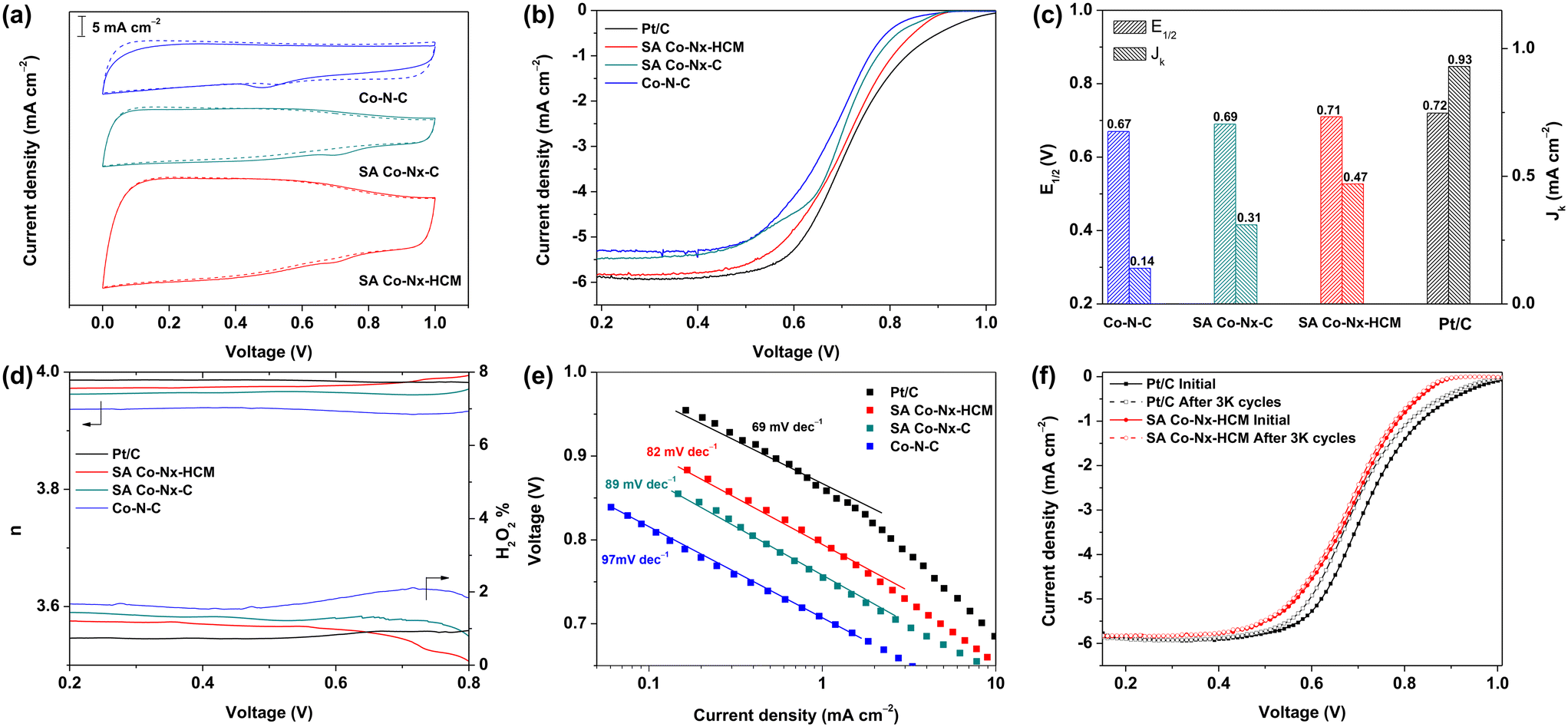 | ||
| Fig. 4 (a) CV curves of the prepared catalysts in O2- and N2-saturated PBS electrolytes. (b) ORR polarization plots, (c) Jk at 0.85 V and E1/2, (d) electron transfer number and H2O2 yield and (e) Tafel plots of the prepared catalysts. (f) ORR polarization curves of SA Co–Nx-HCM and Pt/C catalysts before and after 3000 cycles. | ||
To verify the application potential of the SA Co–Nx–HCM catalyst, homemade aqueous Mg–air batteries were assembled utilizing SA Co–Nx–HCM coated carbon paper as the air-cathode in a neutral PBS electrolyte (Fig. 5a). Fig. 5b shows the surface structures of air electrodes built with SA Co–Nx–HCM and SA Co–Nx–C. It is noticeable that the microspheres make the SA Co–Nx–HCM air cathode possess ample macro-sized pores and form a 3D open porous framework, which could facilitate the rapid diffusion of air to all accessible Co–Nx active sites and thus enhance the Mg–air battery performances. As for the SA Co–Nx–C cathode, the isolated ZIF-8-derived carbon particles form a dense and blocked catalyst layer with negligible large pores, leading to a decreased mass transport capability. As depicted in Fig. 5c, the open-circuit potential of the Mg–air battery equipped with SA Co–Nx–HCM is as large as ca. 1.78 V, approaching that of the Pt/C catalyst of 1.81 V. The maximum power density of SA Co–Nx–HCM is up to 34.2 mW cm−2, which is higher than that of SA Co–Nx–C (29.5 mW cm−2), Co–N–C (23.5 mW cm−2) and is approaching that of Pt/C (35.9 mW cm−2). The enhanced battery performance could be ascribed to the facilitated mass transfer abilities of the SA Co–Nx–HCM cathode, which was supported by electrochemical impedance spectra in Fig. 5d. The Nyquist plots indicate that the SA Co–Nx–HCM (0.67 Ω) cathode exhibits a much lower charge/mass transfer resistance than SA Co–Nx–C (1.52 Ω) and Co–N–C (1.55 Ω), suggesting that the 3D open porous electrode could sharply enhance mass transfer efficiency and thus construct robust triple phase interfaces for the ORR. Then the discharge performances for air electrodes at different current densities were evaluated. (Fig. 5f). Notably, SA Co–Nx–HCM shows a small voltage drop at the current density range of 0.05–8.0 mA cm−2. The SA Co–Nx–HCM air cathode delivers voltage plateaus of 1.76 and 1.24 V at the discharge current densities of 0.05 and 8 mA cm−2, which are superior to the SA Co–Nx–C (1.72 and 1.20 V) and Co–N–C (1.68 and 1.16 V) catalyst assembled batteries. Meanwhile, the SA Co–Nx–HCM based battery delivers an energy density of 3441 Wh kgMg−1 at 0.1 mA cm−2, which is close to the Pt/C based battery (3573 Wh kgMg−1) (Fig. S6†). In addition, the SA Co–Nx–HCM electrocatalyst works stably in a primary Mg-air battery at a current density of 0.1 mA cm−2. After 100 h of operation, the discharge voltage remains nearly unchanged, indicating the superior stability of SA Co–Nx–HCM in the Mg–air battery. (Fig. 5f).
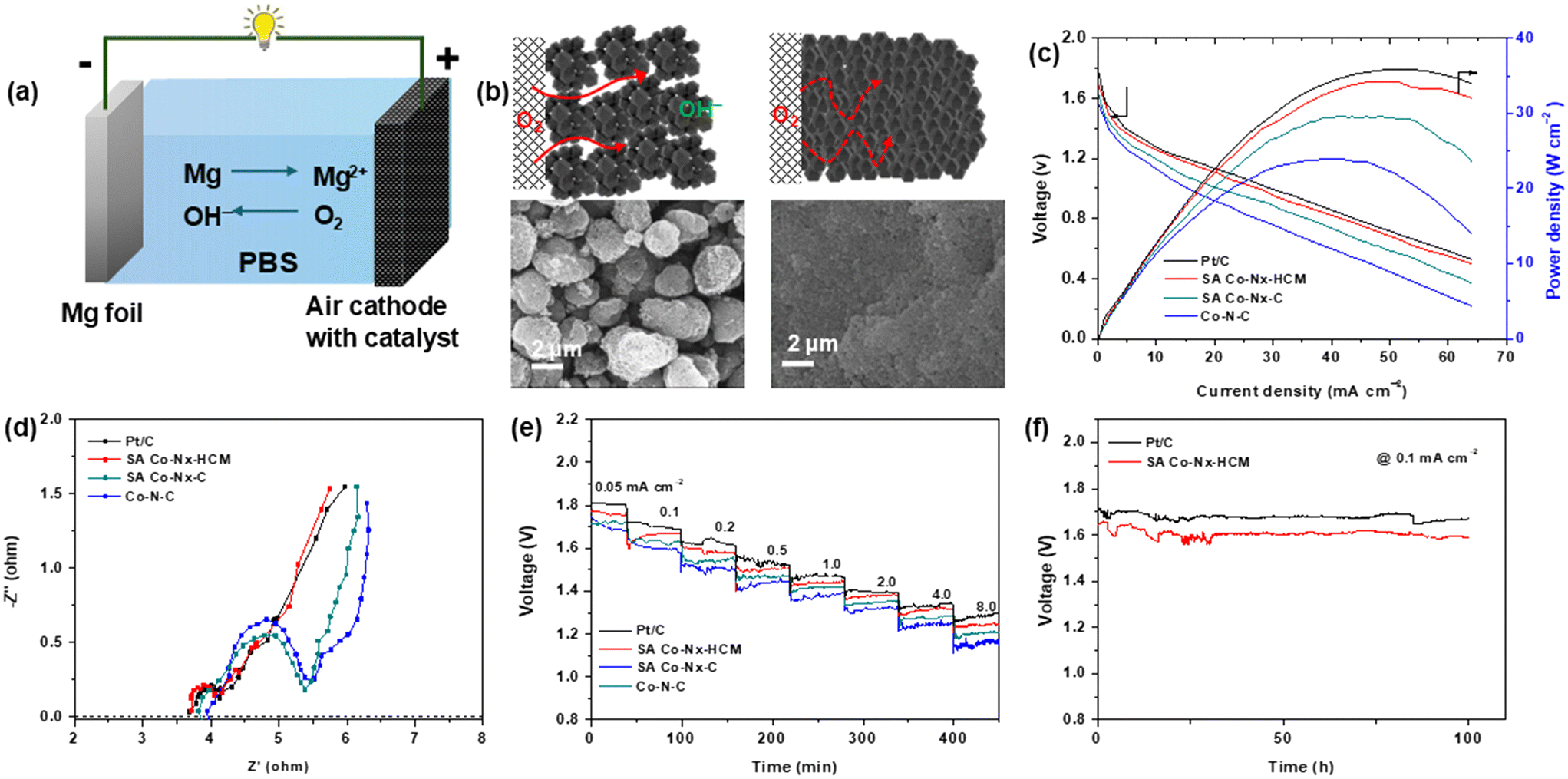 | ||
| Fig. 5 (a) Schematic illustration of the liquid Mg–air battery. (b) Schematic illustrations of the air electrode and their SEM images. (c) Polarization and power density curves of Mg–air batteries with the prepared catalysts and Pt/C. (d) Nyquist plots for batteries at 0.60 V. (e) The rate discharge performance of batteries at various current densities. (f) Discharge capacity of the Mg–air batteries with SA Co–Nx–HCM and Pt/C cathodes at a current density of 0.1 mA cm−2 in PBS. | ||
Conclusion
In summary, hierarchical carbon microspheres coupled with single atomic Co–Nx active sites were generated simultaneously by a spray-assisted stepwise pyrolysis strategy. The dense Co–Nx moieties endow the SA Co–Nx–HCM catalyst with good cytocompatibility, excellent ORR activity, and stability. In addition, the hierarchical carbon microspheres also manifested high mass transfer efficiency in the air cathode and boosted reaction kinetics. Moreover, using the SA Co–Nx–HCM catalyst assembly, a Mg–air battery delivers superior discharge ability and durability. This work provides a new way for rationally designing highly active electrocatalysts for the ORR and other energy-conversion-related catalytic reactions.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 22379123), the Natural Science Foundation of Shaanxi Province (Grant No. 2024JC-YBQN-0072, 2023-JC-YB-122), and the High-level Innovation and Entrepreneurship Talent Project from Qinchuangyuan of Shaanxi Province (Grant No. QCYRCXM-2022-226).References
- E. J. Bradbury, L. D. F. Moon, R. J. Popat, V. R. King, G. S. Bennett, P. N. Patel, J. W. Fawcett and S. B. McMahon, Nature, 2002, 416, 636–640 CrossRef CAS PubMed.
- T. Zhang, Z. Tao and J. Chen, Mater. Horiz., 2014, 1, 196–206 RSC.
- D. Yan, D. Tao, D. Xu, Y. Sun, B. Deng, G. Cao, J. Fang and W. Xu, Nano Energy, 2024, 129, 110000 CrossRef CAS.
- G. Song, Corros. Sci., 2007, 49, 1696–1701 CrossRef CAS.
- C. Yu, C. Wang, X. Liu, X. Jia, S. Naficy, K. Shu, M. Forsyth and G. G. Wallace, Adv. Mater., 2016, 28, 9349–9355 CrossRef CAS PubMed.
- J. Zhang, H. Zhang, Y. Zhang, X. Wang, H. Li, F. Feng, K. Wang, G. Zhang, S. Sun and Y. Zhang, J. Mater. Chem. A, 2023, 11, 7924–7948 RSC.
- Z. Xi, J. Han, Z. Jin, K. Hu, H.-J. Qiu and Y. Ito, Small, 2024, 20, 2308045 CrossRef CAS PubMed.
- M. P. Staiger, A. M. Pietak, J. Huadmai and G. Dias, Biomaterials, 2006, 27, 1728–1734 CrossRef CAS PubMed.
- Y. Yamasaki, Y. Yoshida, M. Okazaki, A. Shimazu, T. Kubo, Y. Akagawa and T. Uchida, Biomaterials, 2003, 24, 4913–4920 CrossRef CAS PubMed.
- X. Chen, X. Liu, Q. Le, M. Zhang, M. Liu and A. Atrens, J. Mater. Chem. A, 2021, 9, 12367–12399 RSC.
- J.-S. Lee, S. Tai Kim, R. Cao, N.-S. Choi, M. Liu, K. T. Lee and J. Cho, Adv. Energy Mater., 2011, 1, 34–50 CrossRef CAS.
- C.-S. Li, Y. Sun, F. Gebert and S.-L. Chou, Adv. Energy Mater., 2017, 7, 1700869 CrossRef.
- Y. Sun, X. Liu, Y. Jiang, J. Li, J. Ding, W. Hu and C. Zhong, J. Mater. Chem. A, 2019, 7, 18183–18208 RSC.
- Y. Dou, S. Xing, Z. Zhang and Z. Zhou, Electrochem. Energy Rev., 2024, 7, 6 CrossRef CAS.
- J. Li, S. Chen, N. Yang, M. Deng, S. Ibraheem, J. Deng, J. Li, L. Li and Z. Wei, Angew. Chem., Int. Ed., 2019, 58, 7035–7039 CrossRef CAS PubMed.
- Y. He, S. Liu, C. Priest, Q. Shi and G. Wu, Chem. Soc. Rev., 2020, 49, 3484–3524 RSC.
- X. Fu, G. Jiang, G. Wen, R. Gao, S. Li, M. Li, J. Zhu, Y. Zheng, Z. Li, Y. Hu, L. Yang, Z. Bai, A. Yu and Z. Chen, Appl. Catal., B, 2021, 293, 120176 CrossRef CAS.
- X. Li, X. Huang, S. Xi, S. Miao, J. Ding, W. Cai, S. Liu, X. Yang, H. Yang, J. Gao, J. Wang, Y. Huang, T. Zhang and B. Liu, J. Am. Chem. Soc., 2018, 140, 12469–12475 CrossRef CAS PubMed.
- J. Hu, W. Liu, C. Xin, J. Guo, X. Cheng, J. Wei, C. Hao, G. Zhang and Y. Shi, J. Mater. Chem. A, 2021, 9, 24803–24829 RSC.
- M. Wu, F. Dong, Y. Yang, X. Cui, X. Liu, Y. Zhu, D. Li, S. Omanovic, S. Sun and G. Zhang, Electrochem. Energy Rev., 2024, 7, 10 CrossRef CAS.
- W.-X. Hong, W.-H. Wang, Y.-H. Chang, H. Pourzolfaghar, I. H. Tseng and Y.-Y. Li, Nano Energy, 2024, 121, 109236 CrossRef CAS.
- L. Jiao, G. Wan, R. Zhang, H. Zhou, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2018, 57, 8525–8529 CrossRef CAS PubMed.
- H. Zhang, H. T. Chung, D. A. Cullen, S. Wagner, U. I. Kramm, K. L. More, P. Zelenay and G. Wu, Energy Environ. Sci., 2019, 12, 2548–2558 RSC.
- S. Shen, Y. Sun, H. Sun, Y. Pang, S. Xia, T. Chen, S. Zheng and T. Yuan, Catalysts, 2022, 12, 525 CrossRef CAS.
- Q. Zhang, P. Liu, X. Fu, Y. Yuan, L. Wang, R. Gao, L. Zheng, L. Yang and Z. Bai, Adv. Funct. Mater., 2022, 32, 2112805 CrossRef CAS.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- Z. Wang, H. Jin, T. Meng, K. Liao, W. Meng, J. Yang, D. He, Y. Xiong and S. Mu, Adv. Funct. Mater., 2018, 28, 1802596 CrossRef.
- Z. Lu, B. Wang, Y. Hu, W. Liu, Y. Zhao, R. Yang, Z. Li, J. Luo, B. Chi, Z. Jiang, M. Li, S. Mu, S. Liao, J. Zhang and X. Sun, Angew. Chem., Int. Ed., 2019, 58, 2622–2626 CrossRef CAS PubMed.
- L. Zhang, J. M. T. A. Fischer, Y. Jia, X. Yan, W. Xu, X. Wang, J. Chen, D. Yang, H. Liu, L. Zhuang, M. Hankel, D. J. Searles, K. Huang, S. Feng, C. L. Brown and X. Yao, J. Am. Chem. Soc., 2018, 140, 10757–10763 CrossRef CAS PubMed.
- D. Yu, Y. Ma, F. Hu, C.-C. Lin, L. Li, H.-Y. Chen, X. Han and S. Peng, Adv. Energy Mater., 2021, 11, 2101242 CrossRef CAS.
- C. P. K. Prabhu, K. R. Naveen and J. Hur, RSC Appl. Interfaces, 2024, 1, 301–312 RSC.
- Y. Zhou, R. Lu, X. Tao, Z. Qiu, G. Chen, J. Yang, Y. Zhao, X. Feng and K. Müllen, J. Am. Chem. Soc., 2023, 145, 3647–3655 CrossRef CAS PubMed.
- P. Yao, J. Cao, X. Gong, C. Han, M. Ruan, P. Song and W. Xu, J. Phys. Chem. C, 2021, 125, 9839–9846 CrossRef CAS.
- S. Son, D. Lim, D. Nam, J. Kim, S. E. Shim and S.-H. Baeck, J. Solid State Chem., 2019, 274, 237–242 CrossRef CAS.
- C. Gao, S. Mu, R. Yan, F. Chen, T. Ma, S. Cao, S. Li, L. Ma, Y. Wang and C. Cheng, Small, 2022, 18, 2105409 CrossRef CAS PubMed.
- M. Liu, Q. Xu, Q. Miao, S. Yang, P. Wu, G. Liu, J. He, C. Yu and G. Zeng, J. Mater. Chem. A, 2022, 10, 228–233 RSC.
- L. Zheng, Y. Zhao, H. Zhang, W. Xia and J. Tang, ChemSusChem, 2022, 15, e202102642 CrossRef CAS PubMed.
- Y. J. Sa, D.-J. Seo, J. Woo, J. T. Lim, J. Y. Cheon, S. Y. Yang, J. M. Lee, D. Kang, T. J. Shin, H. S. Shin, H. Y. Jeong, C. S. Kim, M. G. Kim, T.-Y. Kim and S. H. Joo, J. Am. Chem. Soc., 2016, 138, 15046–15056 CrossRef CAS PubMed.
- X. Fu, N. Li, B. Ren, G. Jiang, Y. Liu, F. M. Hassan, D. Su, J. Zhu, L. Yang, Z. Bai, Z. P. Cano, A. Yu and Z. Chen, Adv. Energy Mater., 2019, 9, 1803737 CrossRef.
- Y. He, Q. Shi, W. Shan, X. Li, A. J. Kropf, E. C. Wegener, J. Wright, S. Karakalos, D. Su, D. A. Cullen, G. Wang, D. J. Myers and G. Wu, Angew. Chem., Int. Ed., 2021, 60, 9516–9526 CrossRef CAS PubMed.
- S. Li, C. Cheng, X. Zhao, J. Schmidt and A. Thomas, Angew. Chem., Int. Ed., 2018, 57, 1856–1862 CrossRef CAS PubMed.
- X. Fu, P. Zamani, J.-Y. Choi, F. M. Hassan, G. Jiang, D. C. Higgins, Y. Zhang, M. A. Hoque and Z. Chen, Adv. Mater., 2017, 29, 1604456 CrossRef PubMed.
- Y. Han, Y.-G. Wang, W. Chen, R. Xu, L. Zheng, J. Zhang, J. Luo, R.-A. Shen, Y. Zhu, W.-C. Cheong, C. Chen, Q. Peng, D. Wang and Y. Li, J. Am. Chem. Soc., 2017, 139, 17269–17272 CrossRef CAS PubMed.
- G. Chen, P. Liu, Z. Liao, F. Sun, Y. He, H. Zhong, T. Zhang, E. Zschech, M. Chen, G. Wu, J. Zhang and X. Feng, Adv. Mater., 2020, 32, 1907399 CrossRef CAS PubMed.
- J. Yu, B.-Q. Li, C.-X. Zhao, J.-N. Liu and Q. Zhang, Adv. Mater., 2020, 32, 1908488 CrossRef CAS PubMed.
- X. Wan, X. Liu, Y. Li, R. Yu, L. Zheng, W. Yan, H. Wang, M. Xu and J. Shui, Nat. Catal., 2019, 2, 259–268 CrossRef CAS.
- A. H. A. Monteverde Videla, L. Osmieri, M. Armandi and S. Specchia, Electrochim. Acta, 2015, 177, 43–50 CrossRef CAS.
- X. Yang, Y. Wang, G. Zhang, L. Du, L. Yang, M. Markiewicz, J.-y. Choi, R. Chenitz and S. Sun, Appl. Catal., B, 2020, 264, 118523 CrossRef.
- X. Fu, R. Gao, G. Jiang, M. Li, S. Li, D. Luo, Y. Hu, Q. Yuan, W. Huang, N. Zhu, L. Yang, Z. Mao, J. Xiong, A. Yu, Z. Chen and Z. Bai, Nano Energy, 2021, 83, 105734 CrossRef CAS.
- X. Zhang, Y. Li, D. He, Z. Ma, K. Liu, K. Xue and H. Li, Chem. Eng. J., 2021, 425, 130677 CrossRef CAS.
- J. Li, L. Jiao, E. Wegener, L. L. Richard, E. Liu, A. Zitolo, M. T. Sougrati, S. Mukerjee, Z. Zhao, Y. Huang, F. Yang, S. Zhong, H. Xu, A. J. Kropf, F. Jaouen, D. J. Myers and Q. Jia, J. Am. Chem. Soc., 2020, 142, 1417–1423 CrossRef CAS PubMed.
- F. Jaouen, M. Lefèvre, J.-P. Dodelet and M. Cai, J. Phys. Chem. B, 2006, 110, 5553–5558 CrossRef CAS PubMed.
- J. Li, H. Zhang, W. Samarakoon, W. Shan, D. A. Cullen, S. Karakalos, M. Chen, D. Gu, K. L. More, G. Wang, Z. Feng, Z. Wang and G. Wu, Angew. Chem., Int. Ed., 2019, 58, 18971–18980 CrossRef CAS PubMed.
- H. Zhang, S. Hwang, M. Wang, Z. Feng, S. Karakalos, L. Luo, Z. Qiao, X. Xie, C. Wang, D. Su, Y. Shao and G. Wu, J. Am. Chem. Soc., 2017, 139, 14143–14149 CrossRef CAS PubMed.
- S. Gupta, S. Zhao, O. Ogoke, Y. Lin, H. Xu and G. Wu, ChemSusChem, 2017, 10, 774–785 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lf00408f |
| ‡ Yanru Liu and Taiqiang Dai contributed equally to this study. |
| This journal is © The Royal Society of Chemistry 2025 |