
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDNA-guided interparticle energy transfer between rare earth doped nanoparticles†
Mingrui
Guo
b,
Vivienne
Tam
 b,
Fiorenzo
Vetrone
*a and
Marta
Cerruti
*b
b,
Fiorenzo
Vetrone
*a and
Marta
Cerruti
*b
aCentre Énergie, Matériaux et Télécommunications, Institut National de la Recherche Scientifique, Université du Québec, Varennes, Québec J3X 1P7, Canada. E-mail: fiorenzo.vetrone@inrs.ca
bDepartment of Mining and Materials Engineering, McGill University, Quebec H3A 0C5, Canada. E-mail: marta.cerruti@mcgill.ca
First published on 8th May 2025
Abstract
Rare earth doped nanoparticles (RENPs) emit upconverted radiation in the ultraviolet (UV), visible (vis), and higher energy near-infrared (NIR) range, and downshifted, lower NIR energy. Most RENP-based optical sensors depend on energy transfer between the RENP and another entity that emits UV/vis light upon detection of the target of interest. However, attenuation of the emitted UV/vis light by biological tissue components prevents in vivo sensing in deep tissue regions. To avoid this, we propose a sensor that works entirely in the NIR range, composed of two RENPs. The sensor is based on core@shell RENPs comprising Tm3+ (energy donor) and Nd3+ (energy absorber) ions doped in a LiYF4 host matrix, functionalized with complementary DNA strands. Hybridization of the DNA strands reduces interparticle distance allowing interparticle energy transfer (IPET) to occur, as shown by a decrease in intensity of the NIR emission from the Tm3+ doped RENPs observed only in the presence of DNA-functionalized Nd3+ doped RENPs. While previous works showed IPET occurring between RENPs held in close proximity by hydrophobic interactions between surface molecules, this work shows that responsive linkers (in our case, DNA) can be used to enable IPET. This result lays the foundation for an RENP-based NIR-excited and NIR-emitting optical sensor for deep tissue sensing.
1. Introduction
Optical sensors convert light, or a change in intensity of light, into a detectable signal in response to changes in the physical environment.1 Most optical sensors consist of a nanostructure (NS) that can be excited by ultraviolet (UV) or visible (vis) light, and a responsive linker that connects the NS with the target molecule to be detected.2 In the presence of the target, the responsive linker brings the NS and target close enough for energy transfer to occur, causing the target to emit in wavelengths different from the excitation source (Fig. 1A and B). While this design has been widely used and has proved effective in sensing various target molecules,2 absorption of UV/vis light by human tissue and water often leads to cell death and limited penetration depth.3 This limits in vivo application of these devices and requires sample purification to remove tissue components and ensure detection accuracy even in in vitro applications.4 Scattering of the emitted UV/vis by the target can also weaken the signal and lead to false negative results.5 | ||
| Fig. 1 Schematic representations of the mechanism of a typical optical sensor in the (A) absence and (B) presence of the target molecule, and of the proposed RENP-based NIRex–NIRem optical sensor in the (C) absence and (D) presence of Nd3+ core@shell RENP. DNA strands 1 and 2 are complementary. (E) Energy level diagram of Yb3+, and Tm3+ and Nd3+. Highlighted energy levels are where IPET is expected to occur. | ||
One solution to these problems is to move into the near-infrared (NIR) region, a range of wavelengths from 800 to 2500 nm.6 As water and human tissue have minimum absorption of light within this range, shifting both the excitation and emission wavelengths towards NIR light can effectively reduce the extent of scattering and biological autofluorescence,7 assuring larger intensity of the emitted signal.8
Rare earth doped nanoparticles (RENPs) are guest–host systems wherein the triply charged RE ions are dispersed as dopants in an appropriate dielectric host matrix. By carefully selecting the dopant ions, RENPs can emit higher energy photons in the UV/vis/NIR range upon excitation with two or more lower energy photons in the NIR range through a multiphoton process called upconversion (UC). Moreover, when excited with NIR light, they can also emit further in the NIR range, through a process referred to as downshifting (single photon luminescence).
RENPs have a wide range of applications in fields such as bioimaging,9,10 photodynamic therapy,11,12 nanothermometry,13–15 photocatalysis16 and sensing.17 RENP-based optical sensors have been used in both in vitro and in vivo detection.18 Among many analytes detected, some important examples include bacteria,19 ATP,20 and specific biomarkers expressed on cancer cells.21 Researchers have also expanded the functionality of RENP-based optical sensors to Raman by using RENPs as optical agents, gold nanourchins as enhancing substrates, and the organic dye TAMRA as a quencher and Raman reporter.20
RENP-based sensors usually consist of three components: the RENP as the luminescent donor, a luminescent acceptor that emits light upon receiving energy from the RENP and a responsive linker. Binding of the target to the responsive linker induces a conformational change in the linker that brings the RENP and the luminescent acceptor into proximity (preferably <10 nm) for energy transfer to occur via a process known as interparticle energy transfer (IPET).22 Examples of acceptors used so far in RENP-based sensors are Au NPs,23 dye molecules24 and carbon nanodots;23 all of them emit UV/vis radiation upon absorbing the RENP donor energy, with all the associated drawbacks previously discussed. To our knowledge, there are no reported RENP-based sensors that operate entirely in the NIR region. This constitutes a clear gap which our work aims to fill.
In this work, for the first time we build an NIR-excited and NIR-emitting (NIRex–NIRem) RENP-based optical sensor where RENPs are used both as donors and as acceptors. This paves the way towards an optical sensor that will allow deep tissue sensing in vivo and simpler detection in vitro without the need for purification steps.
Of the various energy transfer mechanisms reported in the literature for RENP-based optical sensors, only a few reported works have shown the occurrence of IPET between RENPs (used as a both donor and an acceptor): Sarkar et al. added Yb3+ and Tm3+ ions separately as dopants in BaLuF5 nanocrystals, and reported upconversion emission in the blue, red, and NIR regions from Tm3+ upon NIR irradiation only in colloidal mixtures of these two types of nanocrystals.22 Chen et al. observed IPET from NaNdF4 to NaYbF4 as long as the two RENPs were less than 8.5 nm apart.25 While these were very important steps towards the design of dual RENP-based sensors, in none of the studies were the RENPs linked together by responsive linkers that could act as recognition elements for target molecule binding; rather, the authors relied on hydrophobic interactions between molecules on the surface of RENPs to bring the RENPs close to each other and allow for IPET.22,25 This is a major limitation to using these RENPs for in vivo sensing, which would require successful dispersion of the RENPs in aqueous media.
Different from these works, here we functionalize the donor and acceptor RENPs with complementary single-stranded DNA (ss-DNA), aiming to prove the occurrence of what we call “DNA-guided IPET” between RENPs. We hypothesize that upon DNA hybridization, the RENPs will get close enough to result in energy transfer between the two RENPs (Fig. 1C and D).
In our design, the donor is a Yb3+, Tm3+ co-doped RENP while the acceptor is a Nd3+-doped RENP. Excitation at 980 nm can be absorbed by Yb3+ ions and transferred to the Tm3+ ions, which in turn emit upconverted light in the UV/vis and NIR region. If IPET occurs, Tm3+ emission in the NIR region at 800 nm from the donor RENP would be absorbed by the Nd3+ ions in the acceptor RENP due to similarity in energy levels26 (Fig. 1E), resulting in most notably a decrease of emission intensity at 800 nm and potentially re-emission wavelengths from Nd3+ at 1060 and 1340 nm. Nd3+ ions are frequently used as dopants in NIR-emitting materials for laser applications and infrared bioimaging, as has been shown in previous studies.27,28
We synthesize both RENPs using a LiYF4 host matrix as it is widely used in RENP literature due to its low phonon energy.29,30 Additionally, we chose to coat the active RE-doped core with an inert shell to decrease quenching by surface defects, solvents and other molecules at the surface of the NPs. As previously shown, the inert shell increases the distance between RE ions and solvent and surface molecules, and effectively removes surface defects such as vacancies and interstitials.31,32 We design the shell to be as thin as possible, so that the RENP cores can get close enough upon DNA hybridization. The decrease in Tm3+ emission intensity observed in the presence of donor and acceptor RENPs functionalized with complementary DNA strands proves the occurrence of DNA-guided IPET in this system.
2. Materials and methods
Materials
Yttrium oxide (Y2O3, 99.99+%), ytterbium oxide (Yb2O3, 99.99+%), neodymium oxide (Nd2O3, 99.99+%), thulium oxide (Tm2O3, 99.99+%), trifluoroacetic acid (TFA, 99%), lithium trifluoroacetate (LiTFA, 97%), oleic acid (OA, 90%), 1-octadecene (ODE, 90%), and hexane (99%) were purchased from Alfa Aesar. Oleylamine (OM, 70%), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC, 97%), toluene (99%), magnesium chloride (MgCl2, 98+%), isopropanol (99.5+%), nitrosonium tetrafluoroborate (NOBF4, 95%) and acetone (99%) were purchased from Sigma Aldrich. Ethanol (99%) was obtained from Commercial Alcohols. N-Hydroxysulfosuccinimide (sulfo-NHS) and dichloromethane (DCM, 99.6%) were purchased from Thermo Fisher. All chemicals were used as received. Complementary DNA strands (sequence 5′-/5AmMC6/GGC TAT CCG A-3′ and sequence 5′-/5AmMC6/TCG GAT AGC C-3′) were purchased from Integrated DNA Technologies, Inc (Iowa, USA).Precursor preparation
2.5 mmol of lithium trifluoroacetate (LiTFA) precursor for the synthesis of Nd3+ doped RENPs was prepared by mixing 1.25 mmol Nd2O3 and 1.25 mmol Y2O3 with 6 mL water and 5 mL TFA in a 50 mL three-neck round bottom flask. The mixture was refluxed under vigorous stirring at 80 °C overnight; after this, the temperature was decreased to 60 °C to allow for residual TFA and water evaporation. Precursors for the synthesis of Tm3+ doped RENPs were prepared in a similar fashion by replacing Nd2O3 with Tm2O3 and Yb2O3. Yttrium trifluoroacetate precursor for the subsequent shelling experiment was prepared by replacing Nd2O3 with Y2O3.Synthesis of Nd3+ and Tm3+ doped RENP first nuclei via thermal decomposition
The ultra-small RENPs synthesized in this step are known as first nuclei (FN). To synthesize them, a mixture of 7 mL each of OA and OM, and 14 mL of ODE, was prepared in a 100 mL three-neck round bottom flask (solution A). Solution A was degassed under vacuum at 110 °C for 30 min, after which it was backfilled with argon (Ar) gas and the temperature was raised to 330 °C. Meanwhile, 2.5 mmol of LiTFA was added to the dried core precursor together with 3 mL of OA and 6 mL of ODE (solution B). Solution B was then degassed under vacuum and heated at 125 °C by placing the flask into an oil bath for 30 min. 3 mL of OM was added in solution B after the precursors were completely dissolved, and left to degas under stirring for an additional 10 min. Once solution A reached 330 °C, solution B was injected into solution A using a pump-syringe system (Harvard Apparatus Pump 11 Elite, USA) at an injection rate of 1.0 mL min−1. The looped wire used to connect the syringe and the flask containing solution A had to be submerged in the oil bath so that solution B remained hot during the hot injection process. After 1 h of vigorous stirring at 330 °C, the mixture was cooled down to room temperature (RT). The synthesized FN were stored in Falcon centrifuge tubes (50 mL) for subsequent stabilization. Due to evaporation of impurities in the starting materials (such as OA and ODE) and reaction of by-products, as well as minor losses accrued from intermediate steps of liquid handling, the final volume of FN mixtures was 36 mL.Stabilization of RENP FN
Core RENPs were formed by stabilizing FN with an excess of OA. The FN mixture, solidified upon storage at RT, was liquefied by submerging the Falcon tube in warm water prior to stabilization. 1.25 mmol of FN (18 mL of stock solution) was mixed with 16 mL each of OA and ODE in a 100 mL three-neck round bottom flask. The solution was degassed at 110 °C under vacuum and magnetic stirring for 30 min. The flask was then backfilled with Ar gas and the temperature was raised to 315 °C, at which point the reaction continued for 1 h. After cooling to room temperature, 1 mL of core NPs was sampled for characterization using TEM. Stabilized RENPs were called Nd3+ core and Tm3+ core RENPs respectively.Synthesis of core@shell RENPs
Core@shell RENPs were prepared by epitaxial growth of the shelling material (LiYF4) on the cores during hot injection. 0.5 mmol of core RENPs was mixed in a 100 mL three-neck round bottom flask together with equal parts of OA and ODE up to a total volume of 20 mL (solution A). Solution B was prepared by mixing 0.25 mmol of yttrium trifluoroacetate precursor together with 0.25 mmol of LiTFA, and 7.5 mL each of OA and ODE. Both solutions were degassed under vacuum and magnetic stirring at 110 °C for 30 min. After degassing, solution A was backfilled with Ar gas and the temperature was raised to 290 °C. Solution B was then injected into the reaction flask containing solution A using a syringe pump at an injection rate of 1.0 mL min−1. After cooling down to room temperature, RENPs were precipitated with ethanol and washed three times with equal parts of hexane and acetone up to 30 mL, followed by centrifugation (7500 relative centrifugal force, RCF) for 15 min. Finally, RENPs were redispersed in hexane for further structural and optical characterization. Nd3+ and Tm3+ core@shell RENPs dispersed in hexane had a concentration of 15 mg mL−1 and 18 mg mL−1 respectively. After shell addition, RENPs were called Nd3+ core@shell and Tm3+ core@shell RENPs respectively.Oleic acid removal
To render the RENPs hydrophilic and allow for further modification with PAA, the oleic acid molecules were removed using a ligand exchange method as reported by Himmelstoß et al.33 0.01 M NOBF4 solution was prepared by dissolving 58.405 mg of NOBF4 in 50 mL of CH2Cl2. 5 mL of NOBF4 solution was added to 5 mL of hexane containing 25 mg washed RENPs. The mixture was shaken gently for 5 min, followed by centrifugation (7500 RCF) for 15 min. The resulting pellet was further dissolved in a mixture of 5 mL of toluene and 5 mL of hexane, followed by centrifugation (7500 RCF) for 15 min. Finally, RENPs free of OA but weakly bound by NOBF4 were dispersed in deionised (DI) water. After OA removal, RENPs were called Nd3+ core@shell@NOBF4 and Tm3+ core@shell@NOBF4 RENPs respectively. Nd3+ and Tm3+ core@shell@NOBF4 RENPs dispersed in DI water had a concentration of 10 mg mL−1 and 12 mg mL−1 respectively.Polyacrylic acid (PAA) coating
The BF4− stabilized RENPs were further modified with PAA using a method previously reported by Liu et al.34 20 μL of 200 mg mL−1 PAA (molecular weight: 1800 g mol−1) solution and 45 μL of 2 M NH4OH solution prepared in DI water were added to 50 mg of OA-free core@shell NPs dispersed in 10 mL DI water. The mixture was sonicated for 45 min before being transferred to a 100 mL round bottom flask. 40 mL of isopropanol was added to the round bottom flask in a dropwise manner under magnetic stirring at 420 revolutions per minute (RPM). The PAA-coated RENPs were then centrifuged at 8000 RCF for 20 min before being redispersed in DI water. These PAA-coated RENPs were called Nd3+ core@shell@PAA and Tm3+ core@shell@PAA RENPs respectively. They each had a final concentration of 3.6 mg mL−1 and 4.8 mg mL−1 respectively.DNA conjugation via EDC–NHS coupling
120 μL of 0.25 M EDC solution and 80 μL of 0.2 M sulfo-NHS solution prepared in 1 mL DI water were added to 1 mL of PAA coated core@shell RENPs dispersed in DI water. The mixture was gently shaken for 1 h before adding 5 μL of an aqueous solution containing 1000 nmol mL−1 of the desired DNA strands. After overnight incubation at RT, the mixture was centrifuged three times with water (7500 RCF). After this step, RENPs were redispersed in DI water. These DNA conjugated RENPs were called Nd3+ core@shell@DNA and Tm3+ core@shell@DNA RENPs and each had a final concentration of 3.0 mg mL−1 and 4.0 mg mL−1 respectively.RENP morphological structure characterization via transmission electron microscopy (TEM)
Samples of RENPs were first prepared by submerging TEM copper grids (Carbon Film on Square Mesh Grids, Electron Microscopy Sciences, Pennsylvania, USA) into solutions containing core RENPs, core@shell RENPs, and surface modified RENPs. Prior to TEM image capture, core@shell RENPs in hexane as well as core@shell@NOBF4 RENPs in DI water dispersions were diluted to 4 mg mL−1 to avoid stacking of RENPs on copper grids, thus ensuring the quality of TEM images. For core@shell@PAA RENPs and core@shell@DNA RENPs, copper grids were submerged into the original dispersions, at the concentrations previously mentioned. Images of samples were taken with a Tecnai™ Spirit transmission electron microscope (TEM) (FEI, Oregon, USA). RENP size was determined from TEM images using ImageJ image analysis software (Version 1.53) with a set size of at least 100 individual particles per sample. Size distribution graphs were plotted using Prism 9 graphing software (Version 9.5.0).X-ray diffraction (XRD)
XRD diffractograms were collected on a Bruker D8 Advance Diffractometer (Germany) equipped with a Cu Kα X-ray source (λ = 1.54 Å). Data were collected over a 2θ range from 4° to 60°.Dynamic light scattering (DLS)
Zeta potential and size of RENPs were measured using a Zetasizer Nano ZS (Version 7.13, Malvern Panalytical, UK). Parameters for DI water, such as dispersant refractive index of 1.333, dispersant dielectric constant of 80 and viscosity of 0.01 poise, were inputted into the Zetasizer Nano software prior to measurement. Three measurements were performed on each sample.Ultraviolet-visible (UV/vis) spectroscopy
UV/vis absorption spectra of DNA conjugated RENPs were obtained between 220 nm and 430 nm with a step size of 2 nm using a BioTek Cytation 5 Cell Imaging Multimode Reader (Agilent Technologies, California, USA). Spectra were plotted with the associated BioTek Gen5 software (Version 3.02).Near-infrared (NIR) luminescence
Spectra of Nd3+ core@shell RENPs and Tm3+ core@shell RENPs were obtained at RT under 793 nm or 980 nm continuous-wave laser diode excitation respectively (BTW, China). Laser power densities were 5 W cm−2 and 30 W cm−2, respectively. The downshifted emission spectra of Nd3+ core@shell RENPs in the NIR region was collected with a Shamrock 500i monochromator (Andor, Ireland) equipped with an iDus InGaAs 1.7 NIR detector (Andor, Ireland). Stray light was removed using a long-pass 980 nm filter (Newport Corp., USA).Upconversion luminescence
The upconversion emission of Tm3+ core@shell RENPs was collected using a lens at a 90° angle from the excitation beam and recorded with an Avaspec-ULS2048L spectrometer (Avantes, The Netherlands). Acquired data was spectrally corrected using black body emission approximation, and stray light from the excitation source was removed with a short-pass 825 nm filter (Newport Corp., USA).IPET experiment
Three sets of experiments, namely dilution, mixing and hybridization, were designed to study interparticle energy transfer (IPET) between Nd3+ core@shell and Tm3+ core@shell RENPs. In the dilution experiment, 1 mL of Tm3+ core@shell@PAA RENPs dispersed in DI water (4.8 mg mL−1) was placed in a cuvette. An increasing volume of DI water was added to the cuvette in increments of 100 μL. In the mixing experiment, Nd3+ core@shell@PAA RENPs dispersed in DI water (3.6 mg mL−1) were added in 100 μl increments to the cuvette containing the Tm3+ core@shell@PAA RENPs (4.8 mg mL−1). In the hybridization experiment, a cuvette was filled with 1 mL of Tm3+ core@shell@DNA RENPs dispersed in DI water (4.0 mg mL−1) and a solution containing Nd3+ core@shell@DNA NPs in DI water (3.0 mg mL−1) was added in 100 μL increments to the cuvette.Spectra measurements from 336 nm to 838 nm were recorded after the addition of each solution using an excitation wavelength of 980 nm and a power density of 30 W cm−2. Each set of experiments was repeated three times. The area under curve (AUC) for different spectral regions corresponding to Tm3+ emission peaks was calculated using the trapezoid rule using the AUC function in the Prism 9 graphing software (Version 9.5.0).
AUC% decrease was calculated based on this formula:

3. Results
3.1 Morphological and structural characterization of RENPs
TEM analysis showed both LiYF4:10%Nd3+ RENPs and LiYF4:2%Tm3+, 20%Yb3+ RENPs as monodispersed particles with bipyramidal shape (Fig. 2A and E), which is one of the common morphologies for this family of lithium-based RENPs.35 The growth of the inert LiYF4 shell did not change the shape and monodispersity of RENPs (Fig. 2D and H). The small debris found close to the RENPs in Fig. 2B and D are likely to be first nuclei (FN) that failed to coalesce into bigger RENPs.36 As detailed below, the presence of these FN did not negatively affect RENP optical properties. The success of shell addition on both types of RENPs was confirmed by comparing RENP dimensions before and after shelling. As shown in Fig. 2I–J, the major axes dimensions of Nd3+ core@shell and Tm3+ core@shell RENPs were 47.6 ± 2.4 and 25.0 ± 1.5 nm respectively (Fig. 2I–J). The increase in dimension observed for the two types of particles indicated the shell thickness was approximately 1.2 nm for the Nd3+-doped RENPs and 3.7 nm for the Tm3+-doped RENPs.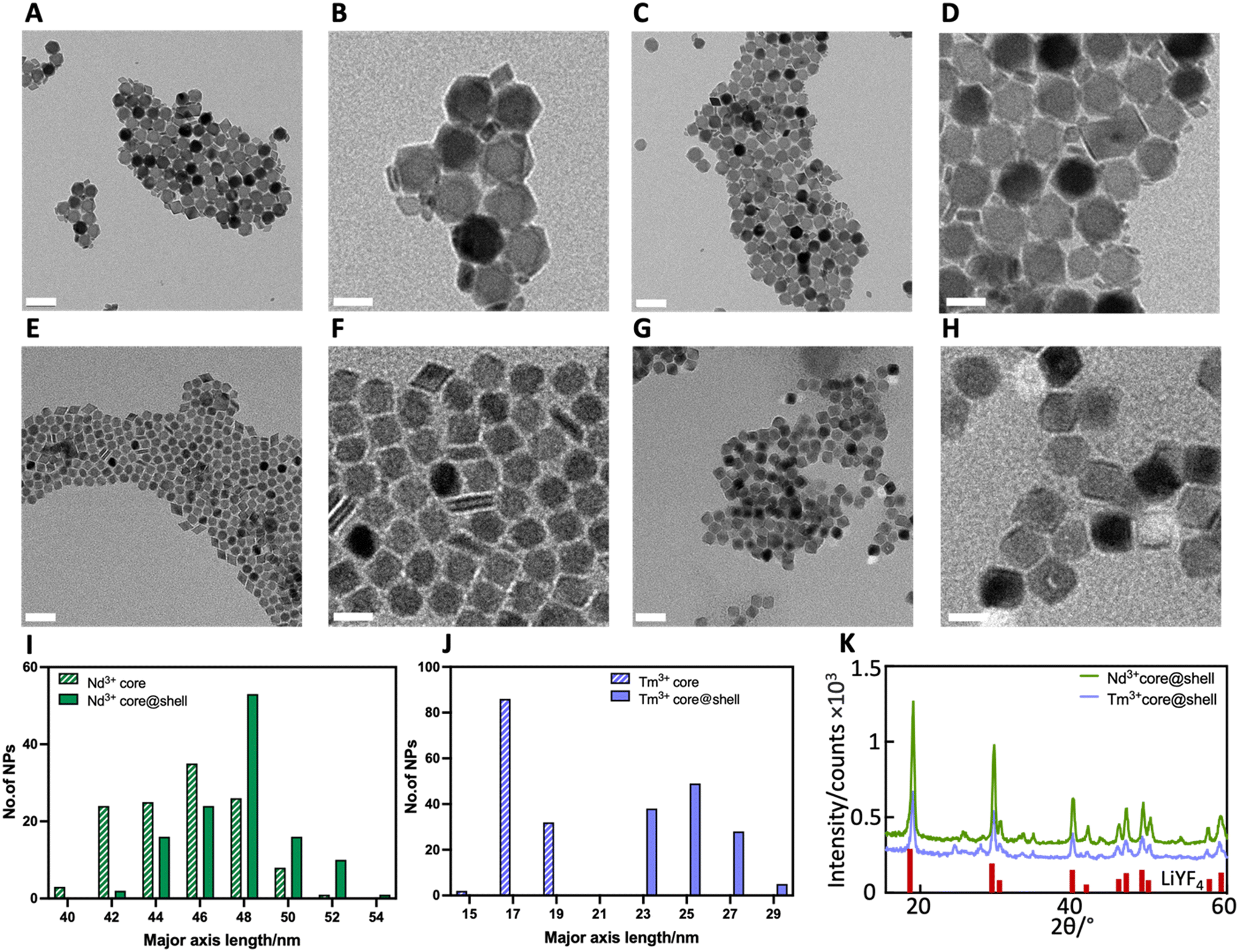 | ||
| Fig. 2 TEM images of (A and B) LiYF4:10%Nd3+ core (Nd3+ core), (C and D) LiYF4:10%Nd3+@LiYF4 (Nd3+ core@shell), (E and F) LiYF4:2%Tm3+, 20%Yb3+ core (Tm3+ core) and (G and H) LiYF4:2%Tm3+, 20%Yb3+@LiYF4 (Tm3+ core@shell) RENPs at different magnifications. Scale bars are 100 nm in A, C, E and G and 50 nm in B, D, F and H. Histograms showing size distribution for (I) Nd3+ core and Nd3+ core@shell RENPs and (J) Tm3+ core and Tm3+ core@shell RENPs. (K) XRD spectra of Nd3+ core@shell and Tm3+ core@shell RENPs. Diffraction peaks of pure LiYF4 are shown in red (PDF: 01-077-0816). | ||
Phase purity of the LiYF4 host matrix is desired since a single, crystalline phase offers high upconversion luminescence efficiency.37 The XRD diffractograms of Nd3+ core@shell and Tm3+ core@shell RENPs (Fig. 2K) showed sharp and narrow diffraction peaks corresponding to the tetragonal crystal structure of LiYF4.38 The absence of other peaks proved that a pure phase of LiYF4 was successfully synthesized, while the peak sharpness indicated the synthesized RENPs were highly crystalline, and that doping with Nd3+ ions, Yb3+ ions and Tm3+ ions did not cause changes in the host structure.
3.2 Spectral characterization of RENPs
We measured the emission intensity of Nd3+ core and Nd3+ core@shell RENPs upon excitation at 793 nm. Both samples exhibited NIR emission bands around 1060 nm and 1340 nm (Fig. 3A and B), which corresponded to the 4F3/2 → 4I11/2, and 4F3/2 → 4I13/2 radiative transitions of Nd3+ respectively (Fig. 3C). Similarly, under 980 nm excitation, Tm3+ core and Tm3+ core@shell RENPs exhibited the characteristic Tm3+ emission bands at 476 nm, 680 nm and 800 nm as shown in Fig. 3D and E, which corresponded to the radiative transitions 1G4 → 3H6, 1G4 → 3F4 and 3H4 → 3H6 respectively (Fig. 3F). These bands are not single peaks due to the so-called Stark effect: the energy levels shown for isolated ions in panels C and F are split into different sublevels due to the presence of an electrical field caused by the crystalline host matrix into which the Nd3+ and Tm3+ ions are doped;39 thus, multiple peaks close to each other are observed instead of only one. | ||
| Fig. 3 Photoluminescence spectra of Nd3+ core and Nd3+ core@shell RENPs in the (A) 1000–1400 nm and (B) the 1300 nm to 1400 nm region (λex = 793 nm). Photoluminescence spectra of Tm3+ core and Tm3+ core@shell RENPs in the (D) 350–850 nm and (E) the 400–750 nm region (λex = 980 nm). Energy level diagrams of free (C) Nd3+ and (F) Tm3+ ions. | ||
While the peak positions in the core and core@shell RENPs were the same for both types of RENPs, the intensity was much higher (approximately 6 and 4 times for Nd3+ and Tm3+ doped RENPs, respectively) in the core@shell structure compared to the core alone. Water is a notorious quencher of energy emitted from RE ions. Vibrations of the O–H bond at 3200–3600 cm−1 can result in an increased non-radiative relaxation of photons at the excited state and a subsequent decrease in emission intensity.40 Our results show the inert shell successfully helped in preventing luminescence quenching.
3.3 Surface modification and characterization of RENPs
We performed a series of surface modifications to not only render RENPs dispersible in water but also allow DNA conjugation to occur (Fig. 4A). The RENPs were synthesized in oleic acid (OA, the coordinating solvent) along with octadecene (OCD, the non-coordinating solvent) due to their high boiling point that allowed for thermal decomposition to occur at 360 °C. The OA also served to promote growth of monodispersed RENPs by preventing their coalescence during the growth step.41 However, its long non-polar, hydrophobic hydrocarbon chains prevented RENPs from further dispersal in polar solvents.42 To render the RENPs hydrophilic, we used NOBF4 to replace OA on RENP surfaces, followed by the exchange of the NOBF4 molecules with PAA via electrostatic interactions.43 In the last step, we conjugated the carboxylic acid groups on PAA with the amine groups on DNA strands through EDC–NHS coupling. We performed TEM after each stage of surface modification and found no changes in morphology throughout all the steps (Fig. 4B–G). | ||
| Fig. 4 (A) Schematic representation of surface modification of RENPs. TEM images of (B) Nd3+ core@shell@NOBF4, (C) Nd3+ core@shell@PAA, (D) Nd3+ core@shell@DNA, (E) Tm3+ core@shell@NOBF4, (F) Tm3+ core@shell@PAA and (G) Tm3+ core@shell@DNA RENPs. Scale bars are 100 nm in main panels and 20 nm in insets. (H) Zeta potential of Nd3+ core@shell RENPs and Tm3+ core@shell RENPs after OA removal with NOBF4, PAA coating and DNA conjugation. UV/vis spectra of (I) Nd3+ core@shell and (J) Tm3+ core@shell before and after DNA conjugation. | ||
Surface modification greatly affected the RENP zeta potential (Fig. 4H). Both Nd3+ core@shell@NOBF4 and Tm3+ core@shell@NOBF4 RENPs had a positive zeta potential (23.3 ± 0.4 and 31.3 ± 0.8 mV respectively). A similar result was also found by Dong et al., who reported a positive zeta potential for the BF4−-modified NaYF4 RENPs dispersed in N,N-dimethylformamide. These authors did not find any band associated with NO+ cations in the FTIR spectra of their RENPs, and thus hypothesized the positive zeta potential was caused by uncoordinated metal cations exposed on the RENP surface after the removal of OA.43 Secondary ligand exchange with PAA caused a significant shift in surface charge from positive to negative for both Nd3+ and Tm3+ core@shell@PAA RENPs (−8.6 ± 0.4 and −11.2 ± 0.5 mV respectively), due to the presence of many negatively charged carboxylate groups on PAA upon deprotonation.44 DNA-conjugated RENPs also had a negative surface charge, as expected due to the negatively charged phosphate rich DNA backbone.
To verify the successful conjugation of DNA strands onto the surface of RENPs, we performed UV-vis spectroscopy (Fig. 4I and J). The peak observed at around 260 nm for the core@shell@DNA RENPs is indicative of the presence of DNA, suggesting that EDC–NHS coupling successfully conjugated DNA strands to the PAA molecules on the surface of these RENPs.45
3.4 IPET experiment
At last, we measured the spectra of samples collected during three sets of experiments to test whether IPET between the Tm3+ and Nd3+ core@shell RENPs could occur when the distance between the RENPs decreased due to DNA hybridization. We named these experiments “dilution”, “mixing” and “hybridization” (Fig. 5A). In the dilution and the mixing experiment, which served as control experiments, we added DI water or aqueous dispersions of Nd3+ core@shell@PAA RENPs to cuvettes containing Tm3+ core@shell@PAA RENPs, respectively; in the hybridization experiment, we added water dispersions of Nd3+ core@shell@DNA RENPs to Tm3+ core@shell@DNA RENPs. We expected all experiments to show a decrease in intensity of the peaks related to Tm3+ emission, since the concentration of Tm3+ core@shell RENPs would have decreased in all of them. As well, random motions due to thermal fluctuations could bring the Nd3+ core@shell@PAA and Tm3+ core@shell@PAA close to each other and allow IPET to occur to some extent in the mixing experiment. However, we expected the greatest decrease in emission intensity in the hybridization experiment, since complementary DNA base pairing should bring the Nd3+ and Tm3+ RENPs close enough for IPET to occur consistently, for all RENPs. | ||
| Fig. 5 (A) Schematic representation of dilution, mixing and hybridization experiments. Emission spectra in the range of 700 nm to 850 nm collected during the (B) dilution, (C) mixing and (D) hybridization experiments (λex = 980 nm). (E) Cumulative% decrease of area under the curve (AUC) for the Tm3+ emission peaks corresponding to the transition 3H4 → 3H6 as a function of the volume of solution added in the dilution, mixing and hybridization experiments. * Indicates p < 0.05, ** indicates p < 0.01, *** indicates p < 0.001, **** indicates p < 0.0001, as measured using an unpaired t-test between the dilution and hybridization experiments. | ||
As expected, we observed a decrease in emission intensity of the 800 nm band, corresponding to the 3H4 → 3H6 transition, in the dilution and mixing experiments (Fig. 5B and C), but the greatest decrease was seen in the hybridization experiment (Fig. 5D), suggesting the occurrence of DNA-guided IPET. Similar decreases were observed for the bands corresponding to the 1I6 → 3F41G4 → 3H6 and 1G4 → 3F4, although to a lesser extent (Fig. S1†). To quantify whether the decrease in intensity observed in the hybridization experiment was significantly different from that observed in the other two, we evaluated the % decrease in the area under the curve (AUC) for peaks relative to Tm3+ emission in all experiments, by comparing the AUC measured after each addition to the AUC measured in the spectrum initially recorded for the Tm3+ RENPs alone at the beginning of each experiment (Fig. 5E).
A trend showing larger AUC% decreases in the hybridization experiments compared to the dilution or mixing experiments was found for all Tm3+ emission peaks as long as more than 100 μL solution was added (Fig. 5E and S2†). The AUC% decrease in the hybridization experiment was significantly greater for the 3H4 → 3H6 transition (Fig. 5E) starting from the 200 μL additions, while significant differences were found only when at least 600 μL were added for the 1I6 → 3F4 and 1G4 → 3H6 transitions (Fig. S2†). No differences between the dilution and mixing experiments were observed for any Tm3+ peak at any volume of solution added.
These results confirm that IPET occurred only between Tm3+ core@shell@DNA and Nd3+ core@shell@DNA RENPs, showing that (i) at the RENP concentrations used in this experiment, the probability that random motions brought the RENPs close enough for IPET to occur in aqueous solution were very low, and (ii) DNA-induced hybridization was effective in bringing the two types of RENPs close enough (<10 nm) to enable IPET.
To further prove that hybridization of complementary DNA strands on RENP surfaces was crucial for IPET to occur, we measured the hydrodynamic diameter of the samples collected from the dilution, mixing and hybridization experiments after adding 1000 μL solution, i.e. at the end of the experiments (Fig. 6). The RENPs collected at the end of the mixing experiment were somewhat larger than those collected at the end of the dilution experiment (hydrodynamic diameters of 160 ± 3 nm vs. 155 ± 4 nm, p < 0.01); this can be explained by the fact that in the dilution experiments only Tm3+ core@shell@PAA RENPs were present, whereas in the mixing experiments the Nd3+ core@shell@PAA RENPs were also present, which are larger in size (see Fig. 2I–J). The much larger hydrodynamic diameter of the RENPs collected at the end of the hybridization experiment (3235 ± 588 nm) indicates the success of complementary base pairing between DNA strands conjugated onto the surface of Nd3+ and Tm3+ core@shell@DNA RENPs, leading to the formation of aggregates.
 | ||
| Fig. 6 Hydrodynamic diameter of samples collected from dilution, mixing and hybridization experiment after the addition of 1000 μL. ** Indicates p < 0.01, and **** indicates p < 0.0001. | ||
4. Discussion
In this work, we chose single-stranded complementary DNA strands as responsive linkers to bind two types of RENPs and aimed to prove DNA-guided IPET when complementary base pairing occurred between the DNA functionalized RENPs.To do so, we first synthesized monodispersed LiYF4:2%Tm3+, 20%Yb3+ (energy donor) and LiYF4:10%Nd3+ (energy acceptor) core RENPs with major axes 17.6 ± 0.7 nm and 45.3 ± 2.4 nm respectively. The size of RENPs is crucial to their performance as optical agents. To date, RENPs with size ranging from 5 nm to 200 nm have been synthesized.46 Even though their potential as optical probes has been demonstrated, sub-10 nm RENPs have low emission intensity due to high surface-to volume ratio which can cause quenching due to surface defects.47 On the other hand, RENPs larger than 100 nm in size have limited cellular uptake and internalization;48 hence, in the frame of biomedicine, RENPs smaller than 50 nm are preferred. The RENPs synthesized in this work project fall well within the range that ensures both high luminescence and possibility of cellular internalization.
Since water molecules are notorious quenchers of RENP emission intensity,37 we added an inert shell to the RENPs using the same host material as their core, to restore luminescence. The LiYF4:2%Tm3+, 20%Yb3+@LiYF4 and LiYF4:10%Nd3+@LiYF4 RENPs with a core@shell structure had major axes dimensions of 25.0 ± 1.5 nm and 47.6 ± 2.4 nm respectively, verifying the successful growth of their respective shell thicknesses of 3.7 nm and 1.2 nm. We designed this shell to be as thin as possible, so that it would help preserve RENP luminescence but still allow RENPs to get close enough to each other to enable IPET.
IPET is a distance-sensitive process, and the maximum distance over which IPET can occur was shown to be 10 nm.22 With this in mind, we selected the complementary ssDNA strands used for surface conjugation in the following step to be 10 bases long, which corresponds to a length of approximately 3 nm.49 This implies that, considering both shells and the connecting hybridized DNA strands, the distance between Tm3+ and Nd3+ ions in the two RENPs is on average approximately 7.9 nm, which is similar to the distance reported for effective IPET between RENPs by Chen et al.25
After confirming the presence of DNA on RENPs via UV/vis spectroscopy, we performed IPET experiments to verify whether DNA guided IPET could occur. Our IPET experiments showed the greatest decrease in the 800 nm emission intensity of Tm3+ ions doped in DNA conjugated RENPs, proving energy was transferred from Tm3+ ions to Nd3+ ions (Fig. 5B–D). The significant differences in AUC observed between DNA conjugated RENPs in the hybridization experiment compared to simply mixing or diluting DNA-free RENPs confirmed the role of DNA strands in bringing RENPs in close proximity and allowing energy transfer to occur via IPET (Fig. 5E). As expected, this decrease was seen to a greater degree for the 3H4 → 3H6 transition (Fig. 5E) compared to the 1I6 → 3F4 and 1G4 → 3H6 transitions (Fig. S2†), as the most efficient energy transfer between Tm3+ and Nd3+ is expected to occur between the 3H4 level of Tm3+ and the 4F3/2 level of Nd3+ (Fig. 1).50 However, as IPET occurs, the 3H4 level of Tm3+ can be depleted, disrupting the energy cascade and leading to higher energy emissions being quenched, though to a lesser degree.50
Further verification that the decrease in emission was due to IPET and not only to aggregate formation in the DNA-conjugated RENPs (shown by hydrodynamic measurements, see Fig. 6) which may have led to a decrease in intensity due to RENP settling, lays in the fact that the decrease in intensity was more obvious for the 3H4 → 3H6 transition. Only for this peak did we start seeing a significantly lower emission intensity for the DNA-conjugated RENPs compared to the DNA-free RENPs after adding as little as 200 mL of RENPs. Should the lower emission intensity have been due to RENP settling, one would have expected all transitions to decrease in intensity uniformly.51
We propose the following energy transfer route to explain the DNA-guided IPET in our work: upon excitation at 980 nm, Yb3+ ions in the LiYF4:2%Tm3+, 20%Yb3+@LiYF4 RENPs transfer their energy to a Tm3+ ion in close proximity in the same RENP which promotes the Tm3+ ion from its 3H6 ground state to the 3H4 excited state. Some of the Tm3+ ions in the 3H4 excited state then transfer energy to the 4F3/2 level of the Nd3+ ions in the LiYF4:10%Nd3+@LiYF4 RENPs via DNA-guided IPET. This Yb3+–Tm3+–Nd3+ energy transfer route is different from the Yb3+–Tm3+ route22 and Nd3+–Yb3+ route25 shown in the 2 previous published works on IPET and offers a new choice of donor–acceptor combinations for NIRex–NIRem optical sensors. Additionally, while Sarkar's work showed IPET between RENPs in toluene22 and Chen's in a pressed tablet of well-mixed solid powder,25 in our work we showed the occurrence of IPET between RENPs dispersed in water, facilitated by a biological interaction (in our case, DNA hybridization). This represents the first step towards building a NIRex–NIRem optical sensor for biological applications.
A limitation of the current work is that we did not observe emission from the Nd3+ ions upon IPET (Fig. S3†). This was most likely because the intensity of the re-emitted bands was too low to be detected given the low quantum yield of the energy transfer in water, as has been previously reported.22 Furthermore, if energy transfer occurred from lower energy levels of the Tm3+ ion (e.g.3F4 instead of 3H6) as shown by Xue et al.,52 there would be no re-emission from Nd3+ as its 4F3/2 level would not have been excited. These reasons, along with the typical pathways favoring non-radiative relaxation as compared to radiative emission, could all explain the lack of Nd3+ re-emission bands despite successful IPET. This may be overcome in the future by finetuning the shell thickness, adjusting the dopant concentration and sensitizing RENP surfaces with dye molecules to enhance energy absorption and increase the chance of seeing emission from the Nd3+ ions.
5. Conclusion
We successfully synthesized monodispersed LiYF4:10%Nd3+@LiYF4 and LiYF4:2%Tm3+, 20%Yb3+@LiYF4 RENPs with a core@shell structure using the thermal decomposition method. We coated these RENPs with PAA to render them hydrophilic, followed by conjugation with complementary single stranded DNA. We demonstrated the occurrence of IPET between Tm3+ core@shell and Nd3+ core@shell RENPs enabled by hybridization of the complementary DNA strands on the two RENPs, as evidenced by a decrease in the intensity of Tm3+ 800 nm emission band due to absorption by Nd3+ ions. We also proved the essential role of DNA in bringing RENPs close together for IPET to occur. Our work is the first report on all-NIR DNA-guided IPET between RENPs and lays the foundation for the construction of NIRex–NIRem sensors. Beyond complementary DNAs, RENPs in these sensors could be linked with an aptamer that could change conformation upon binding to the target; if designed correctly, a conformational change of the aptamer could bring the RENPs less than 10 nm apart and allow IPET only in the presence of the target, thus enabling sensing of a large variety of molecules and ions. Besides in vivo testing, the possibility of using these sensors to test complex biological samples such as blood without need for purification makes them promising tools for point of care diagnosis. This would enable analytical information to be provided remotely and ensure more timely, affordable and accessible diagnosis.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Author contributions
The responsibilities of Mingrui Guo included designing and carrying out experiments, collecting and analyzing data, as well as writing the manuscript draft. Vivienne Tam helped to write and revise the manuscript draft, as well as revise certain figures. Prof. Cerruti provided guidance on experimental design, results analysis, and draft revision. Prof. Fiorenzo Vetrone provided guidance on experimental design and helpful discussions on result analysis.Conflicts of interest
The authors declared no conflict of interests.Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery grants from both FV and MC. MG also acknowledges support from the McGill Engineering Undergraduate Student Masters Award (MEUSMA). Additionally, we thank Dr. Andrea Greschner and Kevin Coutu for their help in UV/vis spectra measurement.References
- M. Andrejevic and M. Burdon, Defining the sensor society, Telev. New Media, 2014, 16(1), 19–36, DOI:10.1177/1527476414541552.
- R. Ulber, J.-G. Frerichs and S. Beutel, Optical sensor systems for bioprocess monitoring, Anal. Bioanal. Chem., 2003, 376(3), 342–348, DOI:10.1007/s00216-003-1930-1.
- L. Schober and H.-G. Löhmannsröben, Determination of optical parameters for light penetration in particulate materials and soils with diffuse reflectance (DR) spectroscopy, J. Environ. Monit., 2000, 2(6), 651–655, 10.1039/b004127k.
- R. D. McDowall, Sample preparation for biomedical analysis, J. Chromatogr. B: Biomed. Sci. Appl., 1989, 492, 3–58, DOI:10.1016/s0378-4347(00)84463-1.
- Y. T. Lim, S. Kim, A. Nakayama, N. E. Stott, M. G. Bawendi and J. V. Frangioni, Selection of Quantum Dot Wavelengths for Biomedical Assays and Imaging, Mol. Imaging, 2003, 2(1), 153535002003021, DOI:10.1162/15353500200302163.
- S. Shabahang, S. Kim and S. Yun, Light-Guiding Biomaterials for Biomedical Applications, Adv. Funct. Mater., 2018, 28(24), 1706635, DOI:10.1002/adfm.201706635.
- Z. Feng, et al., Perfecting and extending the near-infrared imaging window, Light: Sci. Appl., 2021, 10(1), 197, DOI:10.1038/s41377-021-00628-0.
- R. J. Clarke and A. Oprysa, Fluorescence and Light Scattering, J. Chem. Educ., 2004, 81(5), 705, DOI:10.1021/ed081p705.
- G. Liang, et al., Recent progress in the development of upconversion nanomaterials in bioimaging and disease treatment, J. Nanobiotechnol., 2020, 18(1), 154, DOI:10.1186/s12951-020-00713-3.
- V. Patsula, et al., Polymer-coated hexagonal upconverting nanoparticles: chemical stability and cytotoxicity, Front. Chem., 2023, 11, 1207984, DOI:10.3389/fchem.2023.1207984.
- D. E. J. G. J. Dolmans, et al., Photodynamic therapy for cancer, Nat. Rev. Cancer, 2003, 3(5), 380–387, DOI:10.1038/nrc1071.
- J. G. Jesu Raj, M. Quintanilla and F. Vetrone, Near-infrared triggered generation of reactive oxygen species from upconverting nanoparticles decorated with an organoiridium complex, J. Mater. Chem. B, 2016, 4(18), 3113–3120, 10.1039/c5tb02555a.
- A. R. N. Bastos, et al., Thermal Properties of Lipid Bilayers Determined Using Upconversion Nanothermometry, Adv. Funct. Mater., 2019, 29, 1905474, DOI:10.1002/adfm.201905474.
- S. A. Ferdinandus, S. Takeoka, S. Ishiwata, M. Suzuki and H. Sato, Facilely Fabricated Luminescent Nanoparticle Thermosensor for Real-Time Microthermography in Living Animals, ACS Sens., 2016, 1(10), 1222–1227, DOI:10.1021/acssensors.6b00320.
- X. Zhu, et al., Upconversion nanocomposite for programming combination cancer therapy by precise control of microscopic temperature, Nat. Commun., 2018, 9, 2176, DOI:10.1038/s41467-018-04571-4.
- M. Mishra and D.-M. Chun, α-Fe 2 O 3 as a photocatalytic material: A review, Appl. Catal., A, 2015, 498, 126–141, DOI:10.1016/j.apcata.2015.03.023.
- W. Mok and Y. Li, Recent Progress in Nucleic Acid Aptamer-Based Biosensors and Bioassays, Sensors, 2008, 8(11), 7050–7084, DOI:10.3390/s8117050.
- N. Duan, et al., Dual-color upconversion fluorescence and aptamer-functionalized magnetic nanoparticles-based bioassay for the simultaneous detection of Salmonella Typhimurium and Staphylococcus aureus, Anal. Chim. Acta, 2012, 723, 1–6, DOI:10.1016/j.aca.2012.02.011.
- X. Liu, et al., Hybrid material for enrofloxacin sensing based on aptamer-functionalized magnetic nanoparticle conjugated with upconversion nanoprobes, Sens. Actuators, B, 2016, 233, 394–401, DOI:10.1016/j.snb.2016.04.096.
- J. Zhao, et al., Upconversion Luminescence-Activated DNA Nanodevice for ATP Sensing in Living Cells, J. Am. Chem. Soc., 2018, 140(2), 578–581, DOI:10.1021/jacs.7b11161.
- X. Jin, Q. Zeng, J. Zheng, D. Xing and T. Zhang, Aptamer-Functionalized Upconverting Nanoformulations for Light-Switching Cancer-Specific Recognition and In Situ Photodynamic–Chemo Sequential Theranostics, ACS Appl. Mater. Interfaces, 2020, 13(8), 9316–9328, DOI:10.1021/acsami.0c14730.
- S. Sarkar, B. Meesaragandla, C. Hazra and V. Mahalingam, Sub-5 nm Ln3+-doped BaLuF5 Nanocrystals: A Platform to Realize Upconversion via Interparticle Energy Transfer (IPET), Adv. Mater., 2012, 25(6), 856–860, DOI:10.1002/adma.201203641.
- Q. Ouyang, Y. Yang, S. Ali, L. Wang, H. Li and Q. Chen, Upconversion nanoparticles-based FRET system for sensitive detection of Staphylococcus aureus, Spectrochim. Acta, Part A, 2021, 255, 119734, DOI:10.1016/j.saa.2021.119734.
- L. Matlock-Colangelo and A. J. Baeumner, Recent progress in the design of nanofiber-based biosensing devices, Lab Chip, 2012, 12(15), 2612, 10.1039/c2lc21240d.
- X. Chen, et al., Interparticle energy transfer between NaNdF4 and NaYbF4 in self-assembled nanostructures, J. Rare Earths, 2024, 42(6), 1029–1035, DOI:10.1016/j.jre.2023.03.011.
- N. Dubey, et al., Upconversion nanoparticles: Recent strategies and mechanism based applications, J. Rare Earths, 2022, 40(9), 1343–1359, DOI:10.1016/j.jre.2022.04.015.
- S. P. Dhale, et al., Synthesis and spectroscopic analysis of NaCaYF6:Nd3+, Yb3+NIR emitting phosphor, J. Mol. Struct., 2025, 1323, 140588 CrossRef CAS.
- S. Kshetrapal, et al., Electronic structure analysis of Bi2WO6 and observation of near infrared emission on Nd3+doping, J. Alloys Compd., 2024, 985, 173966 CrossRef CAS.
- V. Mahalingam, F. Vetrone, R. Naccache, A. Speghini and J. A. Capobianco, Colloidal Tm3+/Yb3+-Doped LiYF4 nanocrystals: Multiple luminescence spanning the UV to NIR regions via low-energy excitation, Adv. Mater., 2009, 21, 4025–4028 CrossRef CAS.
- V. Tam, et al., Upconverting Nanoparticles Coated with Light-Breakable Mesoporous Silica for NIR-Triggered Release of Hydrophobic Molecules, ACS Appl. Mater. Interfaces, 2024, 16, 29029–29041 CrossRef CAS PubMed.
- N. J. J. Johnson and F. C. J. M. van Veggel, Sodium lanthanide fluoride core-shell nanocrystals: A general perspective on epitaxial shell growth, Nano Res., 2013, 6(8), 547–561, DOI:10.1007/s12274-013-0333-7.
- A. Skripka, et al., Inert Shell Effect on the Quantum Yield of Neodymium-Doped Near-Infrared Nanoparticles: The Necessary Shield in an Aqueous Dispersion, Nano Lett., 2020, 20(10), 7648–7654, DOI:10.1021/acs.nanolett.0c03187.
- S. F. Himmelstoß and T. Hirsch, Long-Term Colloidal and Chemical Stability in Aqueous Media of NaYF4-Type Upconversion Nanoparticles Modified by Ligand-Exchange, Part. Part. Syst. Charact., 2019, 36, 1900235 CrossRef.
- B. Liu, et al., Poly (Acrylic Acid) Modification of Nd3+-Sensitized Upconversion Nanophosphors for Highly Efficient UCL Imaging and pH-Responsive Drug Delivery, Adv. Funct. Mater., 2015, 25, 4717–4729 CrossRef CAS.
- Y. Qiao, et al., Plant tissue imaging with bipyramidal upconversion nanocrystals by introducing Tm 3+ ions as energy trapping centers, Nanoscale, 2021, 13(17), 8181–8187, 10.1039/D0NR07399G.
- T. Cheng, R. Marin, A. Skripka and F. Vetrone, Small and Bright Lithium-Based Upconverting Nanoparticles, J. Am. Chem. Soc., 2018, 140(40), 12890–12899, DOI:10.1021/jacs.8b07086.
- S. Park, et al., Liquid Crystalline Order and Electric Switching of Upconversion Luminescence in Colloidal Nanorod Suspensions, Adv. Opt. Mater., 2019, 7(9), 1900041, DOI:10.1002/adom.201900041.
- H.-W. Chien, C.-H. Wu, C.-H. Yang and T.-L. Wang, Multiple doping effect of LiYF4:Yb3+/Er3+/Ho3+/Tm3+@LiYF4:Yb3+ core/shell nanoparticles and its application in Hg2+ sensing detection, J. Alloys Compd., 2019, 806, 272–282, DOI:10.1016/j.jallcom.2019.07.258.
- D. A. Harmin, Theory of the Stark effect, Phys. Rev. A: At., Mol., Opt. Phys., 1982, 26(5), 2656–2681, DOI:10.1103/physreva.26.2656.
- J.-C. Boyer, M.-P. Manseau, J. I. Murray and F. C. J. M. van Veggel, Surface Modification of Upconverting NaYF4 Nanoparticles with PEG−Phosphate Ligands for NIR (800 nm) Biolabeling within the Biological Window, Langmuir, 2009, 26(2), 1157–1164, DOI:10.1021/la902260j.
- N. Jurga, D. Przybylska, P. Kamiński, A. Tymiński, B. F. Grześkowiak and T. Grzyb, Influence of the synthesis route on the spectroscopic, cytotoxic, and temperature-sensing properties of oleate-capped and ligand-free core/shell nanoparticles, J. Colloid Interface Sci., 2022, 606, 1421–1434, DOI:10.1016/j.jcis.2021.08.093.
- S. Gyergyek, D. Makovec and M. Drofenik, Colloidal stability of oleic- and ricinoleic-acid-coated magnetic nanoparticles in organic solvents, J. Colloid Interface Sci., 2011, 354(2), 498–505, DOI:10.1016/j.jcis.2010.11.043.
- A. Dong, et al., A Generalized Ligand-Exchange Strategy Enabling Sequential Surface Functionalization of Colloidal Nanocrystals, J. Am. Chem. Soc., 2010, 133(4), 998–1006, DOI:10.1021/ja108948z.
- T. N. Pashirova, et al., Nanogels for Drug Delivery: Physiocochemical Properties, Biological Behavior, and In Vivo Applications, in Nanotechnology and Regenerative Medicine: History, Techniques, Frontiers and Applications, ed. M. H. Santana, et al., Academic Press, 2023, pp. 111–127 Search PubMed.
- H. Su, et al., Near Infrared Fluorescence Imaging Probes for Cancer Diagnosis and Treatment, in Cancer Theranostics, ed. Chenet al., Academic Press, 2014, pp. 55–67 Search PubMed.
- L. Kolokolova, L. Nagdimunov and D. Mackowski, Light scattering by hierarchical aggregates, J. Quant. Spectrosc. Radiat. Transfer, 2018, 204, 138–143, DOI:10.1016/j.jqsrt.2017.09.019.
- D. He, et al., A fluorometric method for aptamer-based simultaneous determination of two kinds of the fusarium mycotoxins zearalenone and fumonisin B1 making use of gold nanorods and upconversion nanoparticles, Microchim. Acta, 2020, 187(4), 254, DOI:10.1007/s00604-020-04236-4.
- X. Jin, Q. Zeng, J. Zheng, D. Xing and T. Zhang, Aptamer-Functionalized Upconverting Nanoformulations for Light-Switching Cancer-Specific Recognition and In Situ Photodynamic–Chemo Sequential Theranostics, ACS Appl. Mater. Interfaces, 2020, 13(8), 9316–9328, DOI:10.1021/acsami.0c14730.
- C.-H. Shen, Diagnostic molecular biology, Elsevier/Academic Press, London, 2019 Search PubMed.
- M. Majak and A. Bednarkiewicz, The mechanisms behind the extreme susceptibility of photon avalanche emission to quenching, Mater. Horiz., 2024, 11, 4791–4801 RSC.
- A. Xiang, et al., Poly(acrylic acid) coating induced 2-line ferrihydrite nanoparticle transport in saturated porous media, J. Nanopart. Res., 2013, 15, 1705, DOI:10.1007/s11051-013-1705-3.
- X. Xue, M. Thitsa, T. Cheng, W. Gao and D. Deng, Laser power density dependent energy transfer between Tm3+ and Tb3+: tunable upconversion emissions in NaYF4:Tm3+, Tb3+, Yb3+ microcrystals, Opt. Express, 2016, 24, 270–278 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5lf00083a |
| This journal is © The Royal Society of Chemistry 2025 |