
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Amorphous conjugated polymer networks as an emerging class of polymer nanostructures
Yuya
Oaki
 *a and
Kosuke
Sato
bc
*a and
Kosuke
Sato
bc
aDepartment of Applied Chemistry, Faculty of Science and Technology, Keio University, 3-14-1 Hiyoshi, Kohoku-ku, Yokohama 223-8522, Japan. E-mail: oakiyuya@applc.keio.ac.jp
bOrganic Materials Chemistry Group, Sagami Chemical Research Institute, 2743-1 Hayakawa, Ayase, Kanagawa 252-1193, Japan
cDepartment of Chemical Science and Engineering, School of Materials and Chemical Technology, Institute of Tokyo Science, 4259 Nagatsuta, Midori-ku, Yokohama 226-8502, Japan
First published on 19th November 2024
Abstract
The organizational states of functional monomer molecules have a significant impact on the properties of polymer materials. This Perspective summarizes amorphous conjugated polymer networks (CPNs) as a new family of polymerized structures. CPNs have been studied since the 1990s. The number of papers about CPNs increased in the 2010s after the earlier work was summarized in a review in 2005. However, the amorphous types had not attracted much attention in previous articles. Amorphous CPNs have potential structural advantages compared with conventional crystalline polymers and framework materials. Diverse combinations of monomers and linkers are used to synthesize amorphous CPNs. Conjugated monomers as functional units are not densely aggregated but are homogeneously dispersed in the network. The amorphous network contributes to structural flexibility for molecular motion related to dynamic properties. The low-crystalline nature affords control over the nanoscale morphology. This Perspective starts with a brief summary of polymerized structures. Our recent work is introduced to show the structures and properties of amorphous CPNs. Simultaneous and random copolymerization of multiple conjugated monomers provides amorphous CPNs. Enhanced electrochemical performance in energy-related applications was extracted from the resultant amorphous CPNs containing redox-active moieties, thanks to their structural characteristics. These results imply that a variety of advanced functional materials can be developed based on the concept of amorphous CPNs.
1. Introduction
Polymers have a history going back 100 years since macromolecule theory was proposed by Staudinger.1 The sequences of monomers have infinite patterns. This Perspective focuses on polymers based on covalently linked conjugated monomers (Fig. 1). Conjugated molecules exhibit unique photochemical, electrochemical, mechanical, electrical, and catalytic properties. For example, redox-active properties, such as doping–dedoping of a conjugated system and the reversible conversion of quinone–hydroquinone, mean they have been used as active materials in charge-storage devices.2 The heteroaromatic rings serve as active sites to interact with protons and oxygen for electrocatalytic water-splitting reactions.3 When these molecules are used as functional materials, their state of assembly is significant for their properties.4 Main-chain and side-chain polymers are the classical assembly states (Fig. 1a and b). Functional units are covalently introduced into the one-dimensional (1D) main and side chains. These macromolecules form crystalline assemblies via stacking. In recent years, covalently linked crystalline frameworks, such as ladder polymer networks, covalent organic frameworks (COFs), and porous organic polymers (POPs), have attracted a lot of interest as a new assembly state possessing both functional units and an interior pore space (Fig. 1c).5,6 The unit molecules are embedded in two-dimensional (2D) or three-dimensional (3D) covalent framework. The syntheses, structural characterization, and applications of these polymers have been extensively studied. Very recently, noncovalent and dynamic covalent approaches have been applied to generate and regenerate network polymers.7 However, the functional units are densely stacked and aggregated in rigid crystalline frameworks. In addition, their highly crystalline nature lowers the flexibility of organized states for dynamic motion. If a less crystalline structure containing dispersed functional monomers is designed, enhanced properties can be extracted from the structural flexibility: i.e. dynamic motion of the organized states and functional molecules.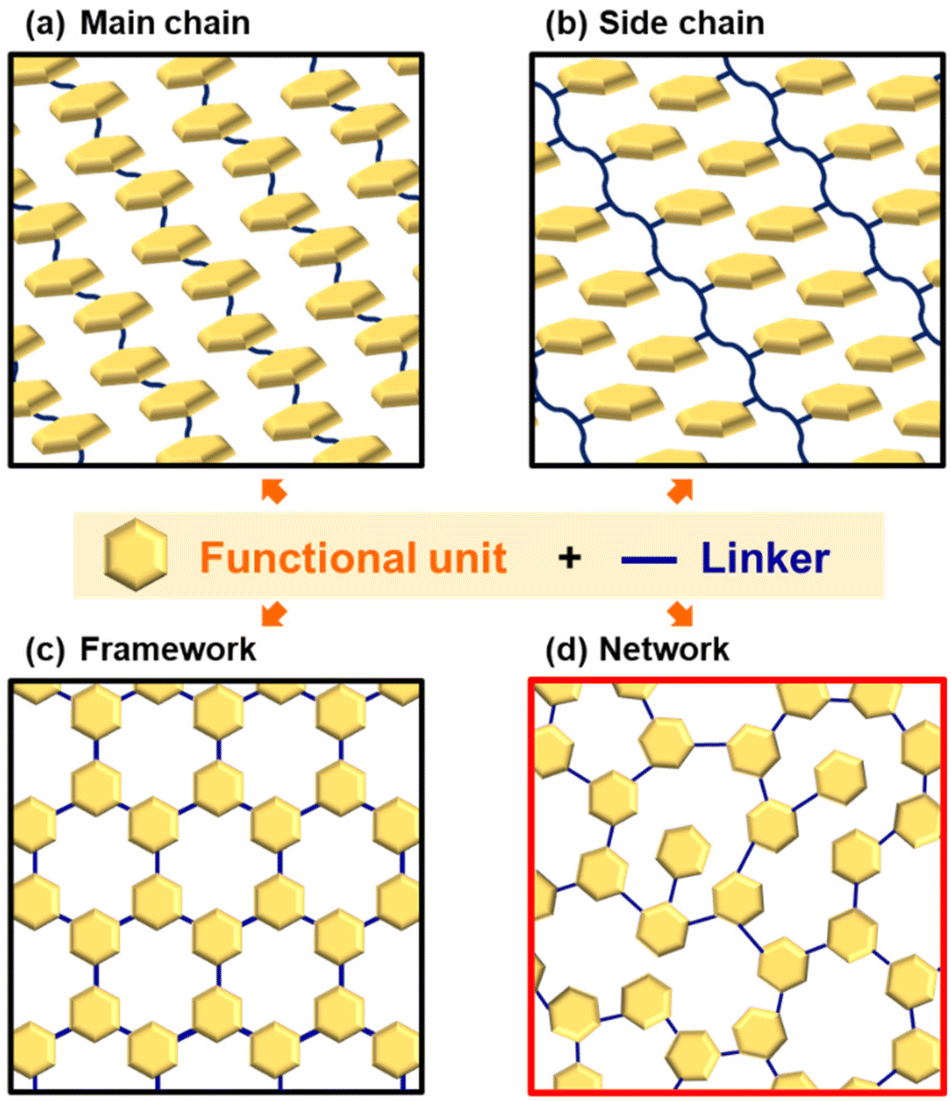 | ||
| Fig. 1 Organizational states of functional molecules. (a) Main-chain type. (b) Side-chain type. (c) Crystalline framework. (d) Amorphous CPN. | ||
Our group has proposed amorphous CPNs and their energy-related applications (Fig. 1d).8–12 The conjugated monomers as a functional unit are dispersed in the network structure. The networks form low-crystalline stackings. The synthetic strategy is simple. When multiple conjugated monomers are simultaneously reacted in multiple directions, the polymerization spontaneously provides random networking and low-crystalline stacking. The crystallinity is lower than that of typical carbon materials such as graphite, stacked graphene, or glassy carbon. The specific surface area is not available for typical gas-adsorption techniques because the pores are enclosed by the stacking. Although the resultant polymers are obtained as bulk particles, nanostructures, such as flakes and sheets, are easily obtained by disrupting the aggregation states by dispersion in organic solvents because of the low-crystalline weak stacking. In our previous work, amorphous CPNs containing redox-active moieties showed enhanced electrochemical performance, such as specific capacity for aqueous capacitors and electrocatalysts for the hydrogen evaluation reaction.8–12 In this Perspective, section 2 summarizes a brief history of CPNs. Section 3 introduces the synthetic processes and characterization of amorphous CPNs. Section 4 shows the structural characteristics and applications of reported CPNs. This Perspective shows the potential of amorphous CPNs as an emerging polymer material.
2. Conjugated polymer networks
2.1. Brief history of network polymers
Network polymers, a class of polymer in which the main chains are covalently interconnected, have been studied in the field of polymer science. Resins, a representative network polymer material, have been widely used in our everyday lives since the first development of phenolic resin in 1907.13 Improved properties, such as insolubility, thermal durability, and mechanical strength, are achieved by the cross-linking network of the polymer chains. Conventional resins, such as phenolic and epoxy resins, are composed of aromatic rings. However, networking was not easily controlled in the polymerization process, particularly in cross-linking reactions. As well-defined macromolecular structures were not obtained, the functions of these polymers were not fully extracted in the early stages.14 In particular, π-conjugated moieties were not introduced into network polymers to explore their electronic and electrochemical functions.Studies on conjugated polymers began in the 1950s and rapidly expanded to establish a field of organic optoelectronics.15 The main topic of the studies was a linear-chain-type structure, such as polyacetylene. A couple of earlier studies showed CPNs. The properties of CPNs were studied theoretically in 1979.16 For example, Manoharan et al. reported the synthesis of a cross-linked porphyrin network and its conductivity of 2.3 S cm−1 in 1988 (Fig. 2a).17 Vohlidal et al. demonstrated π-conjugated cross-linked polyacetylene in 1999 (Fig. 2b).18 However, their detailed structures and properties were not fully studied because characterization techniques for solid polymer materials had not been developed. Therefore, the numbers of papers related to CPNs did not increase much until the early 2000s (Fig. 3).
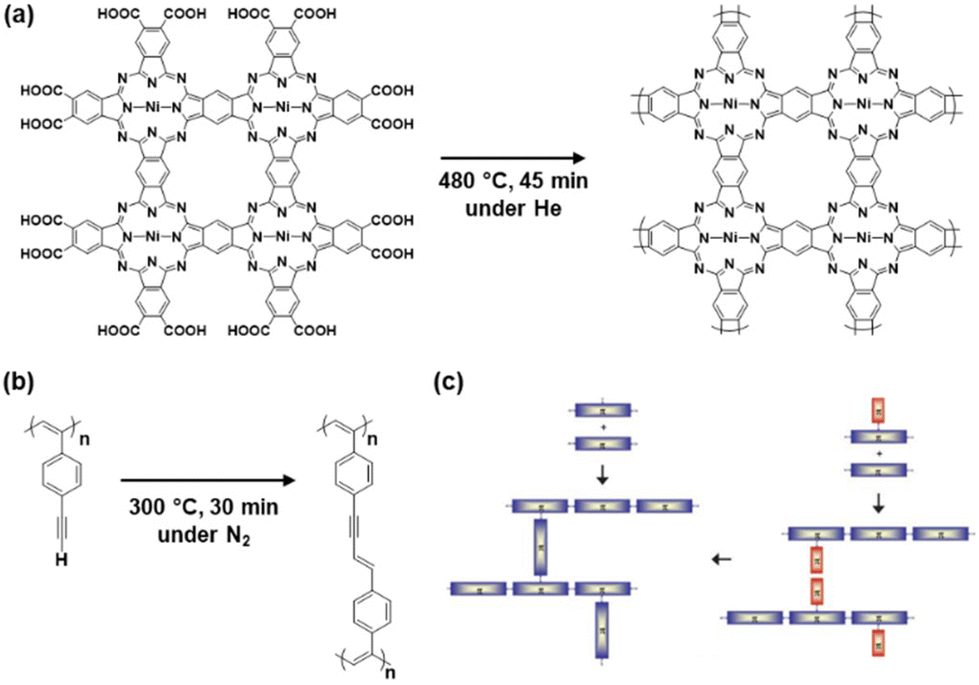 | ||
| Fig. 2 CPNs in pioneering work and reviews. (a) Conjugated porphyrin networks synthesized with heating of the pressed pellet. Reproduced from ref. 16 with permission from Elsevier. (b) Cross-linked conjugated polymer synthesized from poly(4-ethynyl)phenylacetylene. Reproduced from ref. 18 with permission (Copyright 1999, American Chemical Society). (c) Schematic illustrations for the synthesis of CPNs. Reproduced from ref. 19 with permission from the Royal Society of Chemistry. | ||
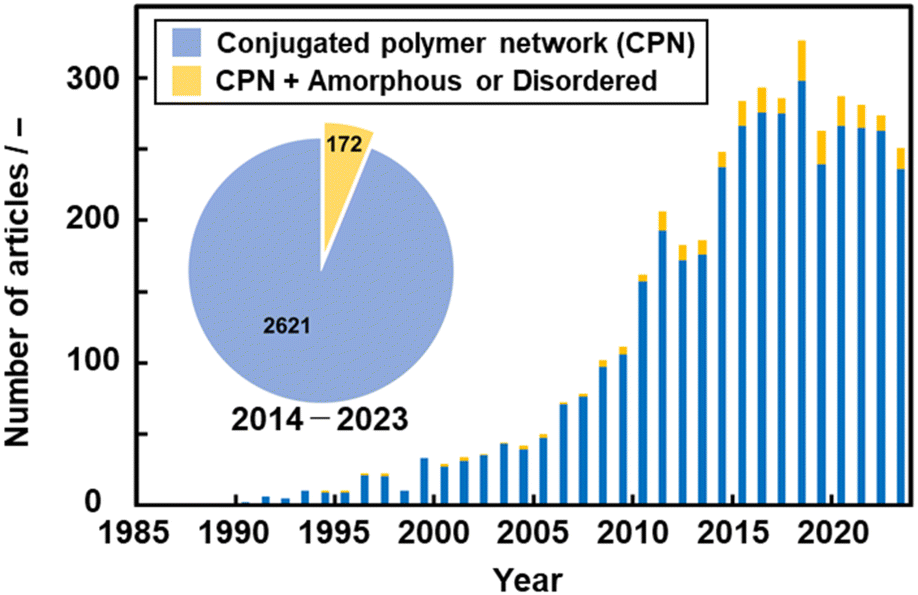 | ||
| Fig. 3 The number of published papers per year (bar chart) and in the last 10 years (pie chart, inset) about “conjugated polymer network (CPN, blue)” and “CPN with amorphous or disordered (yellow)” searched using Web of Science. | ||
In 2005, Weder summarized the significance of CPNs in a review paper.19 The incorporation of π-electron conjugation into the polymer network provides optical and electrical properties. In addition to these functions, durability originating from the nature of resins is ensured in the polymers. After that, the number of the papers related to CPNs increased in the 2010s in the fields not only of polymer chemistry but also of general materials science (Fig. 3). However, the number of publications has been almost constant in recent years because CPNs are sometimes called different names, such as conjugated covalent organic frameworks,20 pseudo-graphene,21 or conjugated microporous polymers.22 In recent years, our group has found new amorphous CPNs.8–12
2.2. Discovery of amorphous conjugated polymer network and its synthetic process
We found an amorphous CPN containing a redox-active moiety in 2019.8 This section focuses on synthetic processes at the molecular level. In earlier work, conductive polymers were synthesized on the solid crystal surface of organic oxidants by oxidative polymerization of heteroaromatic monomers, such as pyrrole (Py).23 A variety of quinone derivatives were used as oxidants for the polymerization of Py. At least one halogen substituent to benzoquinone (BQ) is required to initiate oxidative polymerization. This implies that polypyrrole (PPy) is not obtained using BQ without any substituents because of the lack of oxidizability. One of our group members carried out a reference experiment to confirm that PPy is not synthesized using BQ. However, interestingly, an amorphous CPN of BQ–Py was synthesized by the reactions of BQ and Py (Fig. 4). The reaction scheme is discussed in section 3.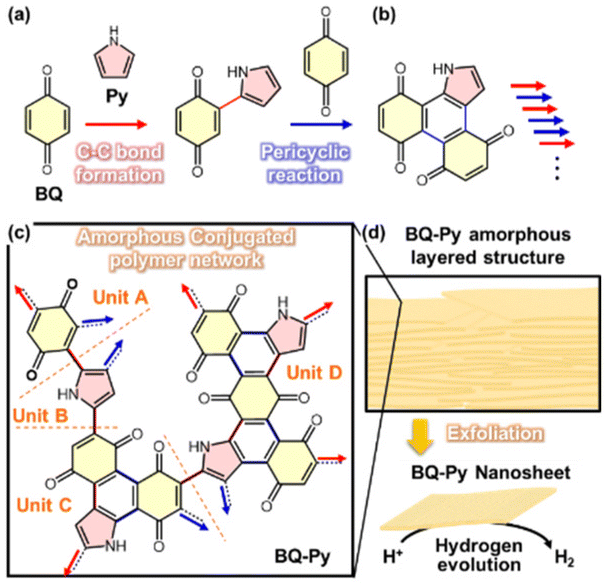 | ||
| Fig. 4 Schematic illustration of the BQ–Py amorphous CPN. (a and b) Successive C–C bond formation (red arrows) and pericyclic reaction between BQ and Py (blue arrows) for elongation and branching, respectively. (c) Four potential structural units of the BQ–Py polymer for networking. (d) Turbostratic layered structure and its exfoliation into nanosheets with enhanced catalytic properties for electrochemical HER. Reproduced from Ref. 8. | ||
2.3. Crystalline and amorphous CPNs
In the present article, CPNs based on conjugated polymers and network polymers are defined as cross-linked conjugated structures containing polymer chains or linkers (Fig. 5). If the linker molecule has a definite size and orientation, the ordered cross-linking provides crystalline 2D-conjugated COFs, such as graphene and graphdiyne (Fig. 5a).24 These structures are highly crystalline in both lateral and vertical stacking directions. All the other types containing non-crystalline (disordered) domains are classified as amorphous CPNs (Fig. 5b and c). In previous work, the introduction of π-electron conjugation into crystalline networks has attracted a lot of interest. Here we revisit the introduction of conjugation into amorphous networks. Flexible linkers provide amorphous CPNs with a low-crystalline nature in the lateral and/or vertical directions (Fig. 5b). Among amorphous CPNs, completely disordered types without any orientation or stacking in both lateral and vertical directions are prepared by highly flexible linkers (Fig. 5c). Therefore, most amorphous CPNs can be regarded as low-crystalline types in the present article (Fig. 5b). The classification is not discrete and quantitative but gradual and qualitative. Although not all reported CPNs can be classified into these types because of the lack of the data about structural characterization, we propose this classification and definition. A more detailed definition is required to classify these amorphous CPNs.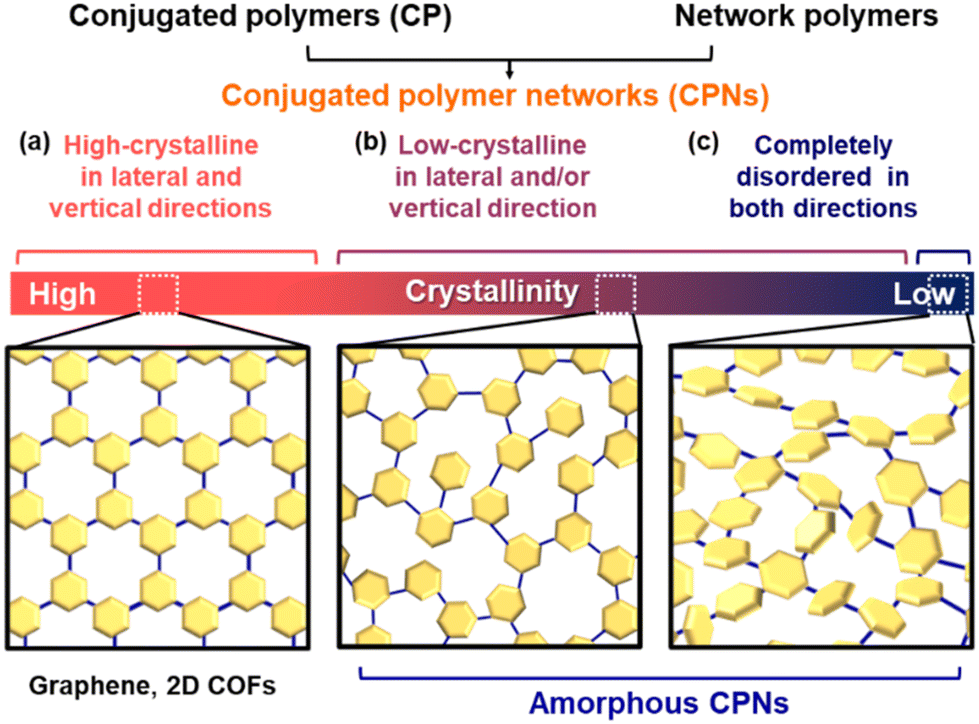 | ||
| Fig. 5 Schematic illustration of CPNs and their classifications in the present work. (a) High-crystalline CPNs in lateral and vertical directions. (b) Low-crystalline CPNs in lateral and/or vertical directions. (c) Disordered CPNs in lateral and vertical directions. | ||
3. Syntheses and characterization of amorphous CPNs
3.1. Reactions leading to amorphous CPNs
Here an amorphous CPN containing BQ and Py is introduced as a model case. Py vapor was supplied to the surface of a solid BQ crystal in a closed chamber at 60 °C under ambient pressure.8 The reactions of quinone moieties and Py derivatives were reported in previous work about the synthesis of dyes.25 However, repetitive reactions, i.e. polymerization, were not found in the previous reports. These studies imply that chain elongation and branching reactions can proceed simultaneously to achieve networking polymerization (Fig. 4a–c). The proposed polymerization process includes C–C bond formation, pericyclic reaction, and subsequent oxidation. In the first step, C–C bond formation between the active α-position of carbonyl and the 2-position of Py provides a BQ–Py diad. This C–C bond formation reaction is regarded as chain elongation for polymerization. Branching is formed by the pericyclic reaction between the BQ–Py diad (4π) and BQ (2π) with subsequent dehydrogenation by excess BQ recovering the π-conjugation. The branching corresponds to the cross-linking of the chain. In this manner, an amorphous CPN is synthesized by the random repetition of the chain elongation and branching reactions. Recent work indicates that the individual reactions for polymerization have negative Gibbs energy changes, based on density functional theory (DFT) calculation (Fig. 6).26 These reactions are facilitated by the electron-rich nature of the Py ring and the electron-deficient character of BQ.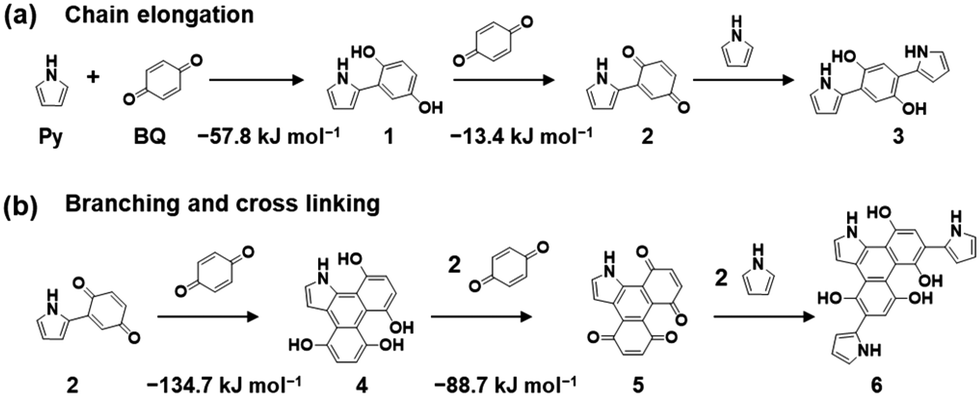 | ||
| Fig. 6 Reaction scheme for the synthesis of BQ–Py and changes in Gibbs energy in each step for chain elongation (a) and branching and cross-linking (b). Reproduced from ref. 26 with permission (Copyright 2022, American Chemical Society). | ||
BQ plays multiple important roles: a monomer with an active α-position of carbonyl for C–C bond formation, a reactant with 2π for the pericyclic reaction, and a moderate oxidant to recover the conjugated structure with dehydrogenation. BQ is one of the simplest structures containing two electron-deficient C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds in a molecule. The four equivalent α-positions act as a polymerizable monomeric unit. The two 2π moieties afford the pericyclic reaction twice for polymerization. Moreover, the moderate reduction potential facilitates the formation of conjugated units in the polymer network via dehydrogenation. When BQ derivatives with a higher reduction potential, such as 2,6-dichloro-1,4-benzoquinone, are used, oxidative polymerization provides PPy.27 A reduction potential lower than that needed to oxidize Py is preferred to obtain the conjugated structure in the polymer network.
C bonds in a molecule. The four equivalent α-positions act as a polymerizable monomeric unit. The two 2π moieties afford the pericyclic reaction twice for polymerization. Moreover, the moderate reduction potential facilitates the formation of conjugated units in the polymer network via dehydrogenation. When BQ derivatives with a higher reduction potential, such as 2,6-dichloro-1,4-benzoquinone, are used, oxidative polymerization provides PPy.27 A reduction potential lower than that needed to oxidize Py is preferred to obtain the conjugated structure in the polymer network.
The amorphous network is obtained by the simultaneous multiple reactions of a number of monomers in different directions. In addition, the specific conjugated monomers and their reactions afford conjugated moieties in the polymer network. The network structures, including planarity and steric conformations, direct the low-crystalline stacking. Therefore, the crystallinity is lower than that of other macromolecular assemblies, such as crystalline main-chain and side-chain polymers, MOFs, and COFs. These structural characteristics enable molecular motion related to the dynamic functions of amorphous CPNs. In this manner, these consecutive reactions provide an amorphous CPN.
3.2. Structural analyses of amorphous CPNs
Amorphous CPNs are synthesized by the schemes discussed in section 3.1. However, structural analyses of the CPNs are not easily achieved by conventional techniques because of the insoluble and infusible macromolecules. Therefore, structural analysis requires a combination of several techniques.8–12 Here we summarize structural analyses of BQ–Py as a model case. An increase in the molecular weight, i.e. polymerization, is studied by the shift in the weight-loss temperature using thermogravimetry (TG) in an air atmosphere. The temperature of the weight loss with combustion is shifted to the higher temperature region compared with that of the monomers and linear PPy. The temperature is comparable to that of commercial graphene with a network structure. In addition to the TG curves, low solubility implies an increase in molecular weight. An estimated chemical structure can be drawn based on the potential reaction schemes, as discussed in section 3.1. The chemical bonding states are analyzed using infrared absorption (IR) spectroscopy, X-ray photoelectron spectroscopy (XPS), and solid-state 13C-nuclear magnetic resonance spectroscopy (NMR). These analyses are used to study the consistency between the estimated and actual structures, particularly for the substituents and main-chain framework. The estimated partial structure of the amorphous CPNs is modified based on these spectroscopic analyses. In addition, the monomer units and their proportions are calculated from the ratio of C, H, and N using elemental analysis. The proportion of hydrated water is estimated from the TG curve. The estimated proportion is modified to coincide with the calculated one within 0.5%.Raman and UV-Vis-NIR spectroscopy are used to study the extension of the conjugated framework. As the graphitic conjugated structures are randomly distributed in the low-crystalline network, broadened D and G bands are observed in the Raman spectra. The amorphous CPN shows a shift of the absorption edge to the NIR region originating from the extended conjugated main chain. In addition, X-ray diffraction (XRD) shows weak and broadened peaks corresponding to the low-crystalline stacking of the quasi-planar layers. The distance between the stackings is estimated from the peak position. The distance is larger than that of crystalline graphite because of the turbostratic stacking originating from the amorphous nature. If the simulated XRD profile based on the estimated structure is coincident with the experimental one, the structure can be identified more precisely.
In recent years, various nanocarbons with graphitic structures have been synthesized by thermal treatment of the precursors. As CPN is synthesized from defined precursor monomers, the structure and composition can be estimated more easily. More detailed analyses, including measurements and simulations, contribute to elucidating the real structures of amorphous CPNs.
4. Syntheses and applications of amorphous CPNs
Section 4 shows several model cases for amorphous CPNs and their applications. Section 4.1. introduces the characteristic layered structure of BQ–Py and its application as an electrocatalyst for the hydrogen evolution reaction (HER). Based on the finding of BQ–Py, Py is altered to the other conjugated linkers, such as benzoxazole (BO), to synthesize different types of the BQ-based amorphous CPNs (sections 4.2 and 4.3). Section 4.4 shows the design and synthesis of a ternary amorphous CPN containing pyridine (Pyd), thiophene (Th), and triazine (TAz). Our recent work indicates that these amorphous CPNs can be used for energy-related applications, thanks to their redox activity.4.1. Amorphous CPN of BQ and Py with layered structure
The molecular structures were discussed in the previous sections. The BQ–Py polymer network exhibits the characteristic structure originating from cross-conjugation and quasi-planarity.8 The XRD pattern indicates that BQ–Py has a turbostratic layered structure with an interlayer distance of 0.37 nm (Fig. 7a–c). The optimized structure of the BQ–Py unit shows a planar structure. The intramolecular hydrogen bonding between N–H in Py and CO in BQ stabilizes the planar structure (orange arrows in Fig. 7d). On the other hand, two adjacent CO groups in the fused ring protrude out of the plane because of steric hindrance (Fig. 7e). Therefore, the roughly stacked layered structure shows an interlayer distance larger than that of graphite (0.335 nm). The quasi-layered structure was exfoliated into nanosheets in dispersion media. Bulk particles of several-tens of micrometers in size were obtained after synthesis (Fig. 7f). Exfoliation induces the formation of sheet-like morphologies of different sizes, depending on the dispersion media (Fig. 7g–j).
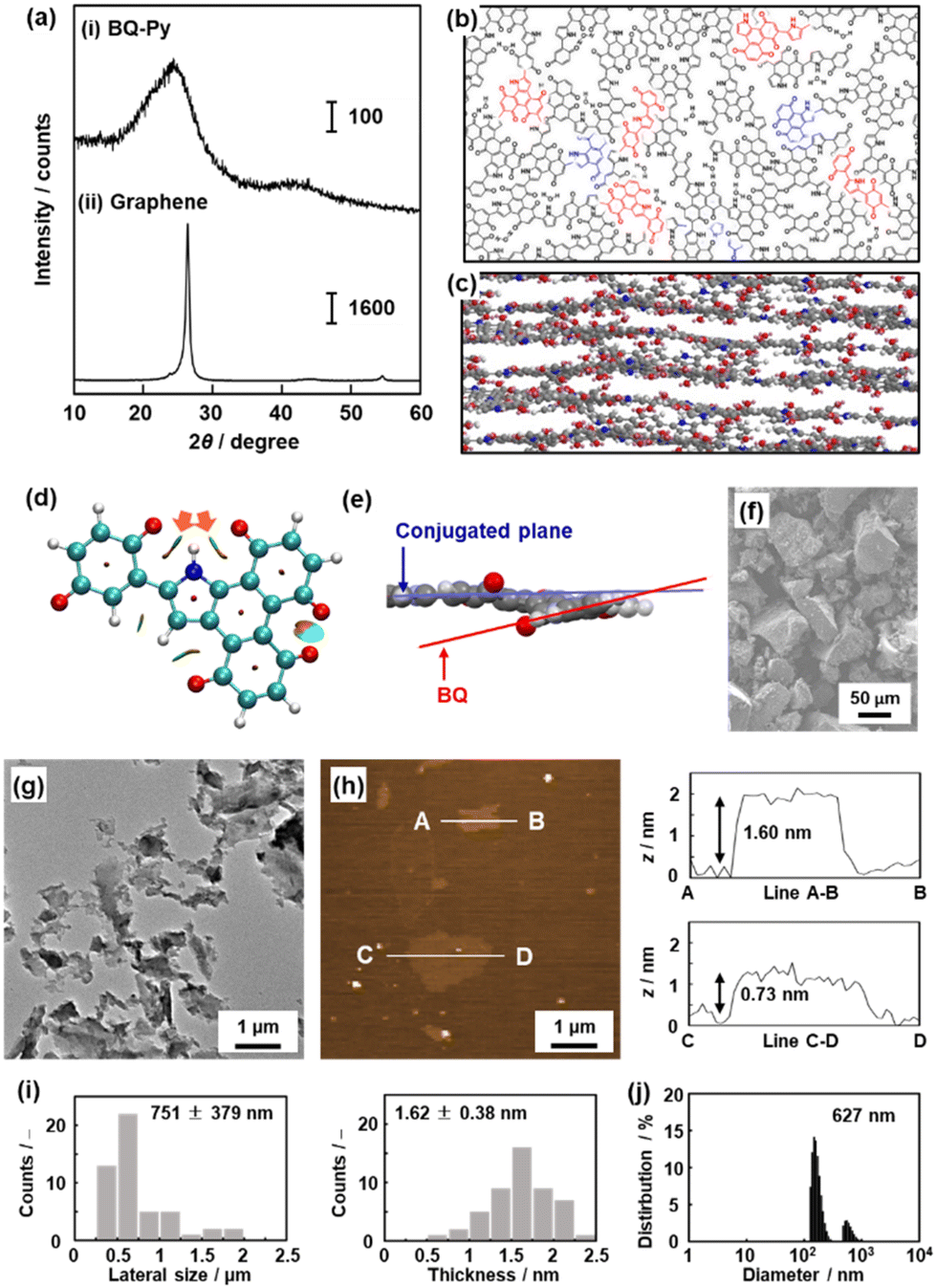 | ||
| Fig. 7 Layered structure of the BQ–Py polymer (a–f) and its exfoliation into nanosheets (g–j). (a) XRD patterns of layered BQ–Py (i) and commercial graphene powder (ii). (b and c) Schematic illustration of top (b) and side (c) views of the layered structure. The upper and lower layers are grown from the red and blue parts in panel (b), respectively. (d) Noncovalent interaction analysis of the BQ–Py oligomer. (e) Magnified side view of the conjugated plane and twisted BQ unit. (f) Scanning electron microscopy (SEM) image of the resultant layered BQ–Py. (g and h) Transmission electron microscopy (TEM) image (g) and atomic force microscopy (AFM) image (h) and its height profiles of the exfoliated nanosheets in water. (i) Histograms of the lateral size and thickness estimated from the TEM and AFM images, respectively. (j) Particle-size distribution measured using dynamic light scattering (DLS). Reproduced from ref. 8. | ||
The BQ–Py nanosheets are used as a metal-free electrocatalyst for HER. In recent years, metal-free HER catalysts have attracted a lot of interest.28 Nanocarbons and their heteroatom-doped types were studied as candidates for metal-free catalysts. However, the syntheses are performed at high temperature. Although HER catalysts are used for the production of hydrogen as a clean energy source, the high synthetic temperature of the catalyst with emission of carbon dioxide (CO2) defeats the objective. The catalytic performance of BQ–Py is measured on a glassy carbon (GC) electrode in 0.5 mol dm−3 sulfuric acid (H2SO4). Prior to measurement, electrochemical reduction of the BQ moiety to HQ is carried out by chronoamperometry at −0.30 V vs. RHE for 5 h to recover the conjugated system (Fig. 8). The overpotential (ΔE) at 10 mA cm−2 in the curve of linear sweep voltammetry (LSV), used as a metric for the catalytic activity, is 421 mV for bulk BQ–Py, 277 mV for BQ–Py nanosheets, and 58 mV for the reference platinum (Pt). In contrast, the current density of 20 mA cm−2 is not achieved using commercial graphene and a bare GC electrode in the potential range of the LSV measurement. The BQ–Py nanosheets show catalytic performance as a metal-free HER electrocatalyst. Moreover, the BQ–Py amorphous CPN is used for other energy-related applications, such as batteries23,29 and as a photocathode for water reduction,30 arising from its redox-active nature.
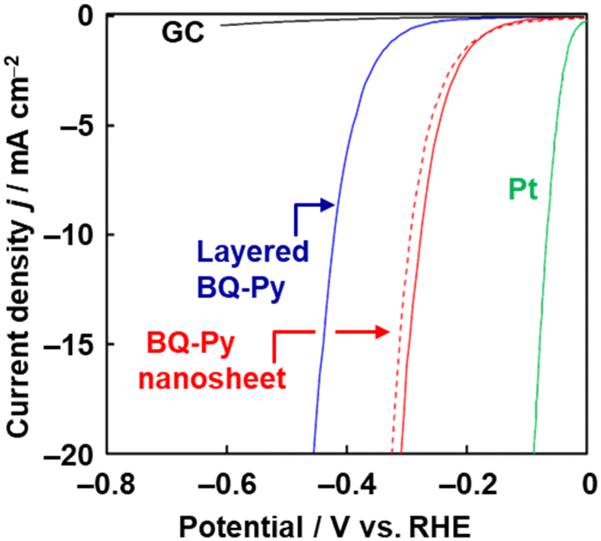 | ||
| Fig. 8 LSV curve of bare GC electrode (black line), layered BQ–Py (blue line), BQ–Py nanosheets (red line), and BQ–Py nanosheets after cyclic voltammetry in the range 0 to –0.3 V (vs. RHE) for 500 cycles (red dashed line). Reproduced from ref. 8. | ||
The morphology and size of the BQ–Py amorphous CPN are controlled by precipitation polymerization (Fig. 9).11 In our previous work, Py vapor was supplied to the surface of solid BQ.8 BQ–Py was formed in the inhomogeneous polymerization system using BQ solid and Py vapor. As an alternative method, precursor solutions containing BQ and Py were prepared and then mixed to synthesize the precipitates at 60 °C. As a proton initiates the first step of the reactions (Fig. 6a), changes in pH have effects on reactivity, morphology, and size.11 BQ–Py microspheres in the range of 70 nm and 1 μm are obtained with changes in the pH and precursor concentration (Fig. 9). Nanoparticles of around 100 nm in size show HER catalytic activity higher than that of the bulk particles. The results imply that precipitation polymerization as a homogeneous reaction system enables morphological and size control over amorphous CPNs.
 | ||
| Fig. 9 Morphological and size control over BQ–Py amorphous CPN. (a–c) SEM images of BQ–Py particles synthesized from precursor HCl solution containing the monomer with changing pH: 2.0 (a), 3.0 (b), and 4.0 (c). (d) Relationship between the particle diameter and synthetic conditions (pH and monomer concentrations). Reproduced from ref. 11 with permission from the Royal Society of Chemistry. | ||
4.2. Amorphous CPN of BQ and BO with improved catalytic performance
Higher catalytic performance was achieved using BQ–BO as another amorphous CPN (Fig. 10).10 Based on BQ–Py,8 a variety of heteroaromatic monomers as alternatives to Py are candidates for copolymerization with BQ. Prior to the experiments, a prediction model of HER catalytic activity is prepared to explore the monomers. Our group has developed sparse modeling for small data to construct prediction models combining machine learning and chemical insights in small data.31 A ΔE predictor is constructed using the small data for the ΔE of metal-free HER catalysts in the literature (Fig. 10a).10 The predicted results indicate that furane derivatives show potential for high catalytic activity. In addition, the diads of BQ and furane derivatives have specific hydrophilicity compared with those of BQ and other heteroaromatic compounds because calculation of the Hansen solubility (similarity) parameter (HSP) distance shows smaller values. Four furane derivatives were selected to study reactivity with BQ. BQ–BO was synthesized with the highest average yield of 13.3% among four furane derivatives. BQ and BO are reacted under microwave irradiation at 200 °C for 1 h. The structural analyses support the formation of an amorphous CPN containing BQ and BO (Fig. 10b). The resultant BQ–BO consists of bulk particles of around 10 μm in size. After dispersion in an organic solvent, a mixture of bulk and nanoparticles less than 500 nm forms through dissolution and reprecipitation. The BQ–BO bulk particles and bulk-nano mixture show ΔE = 279 and 230 mV at 10 mA cm−2, respectively (Fig. 11). The catalytic performance has one of the highest values among metal-free HER catalysts synthesized at a low temperature under 200 °C.10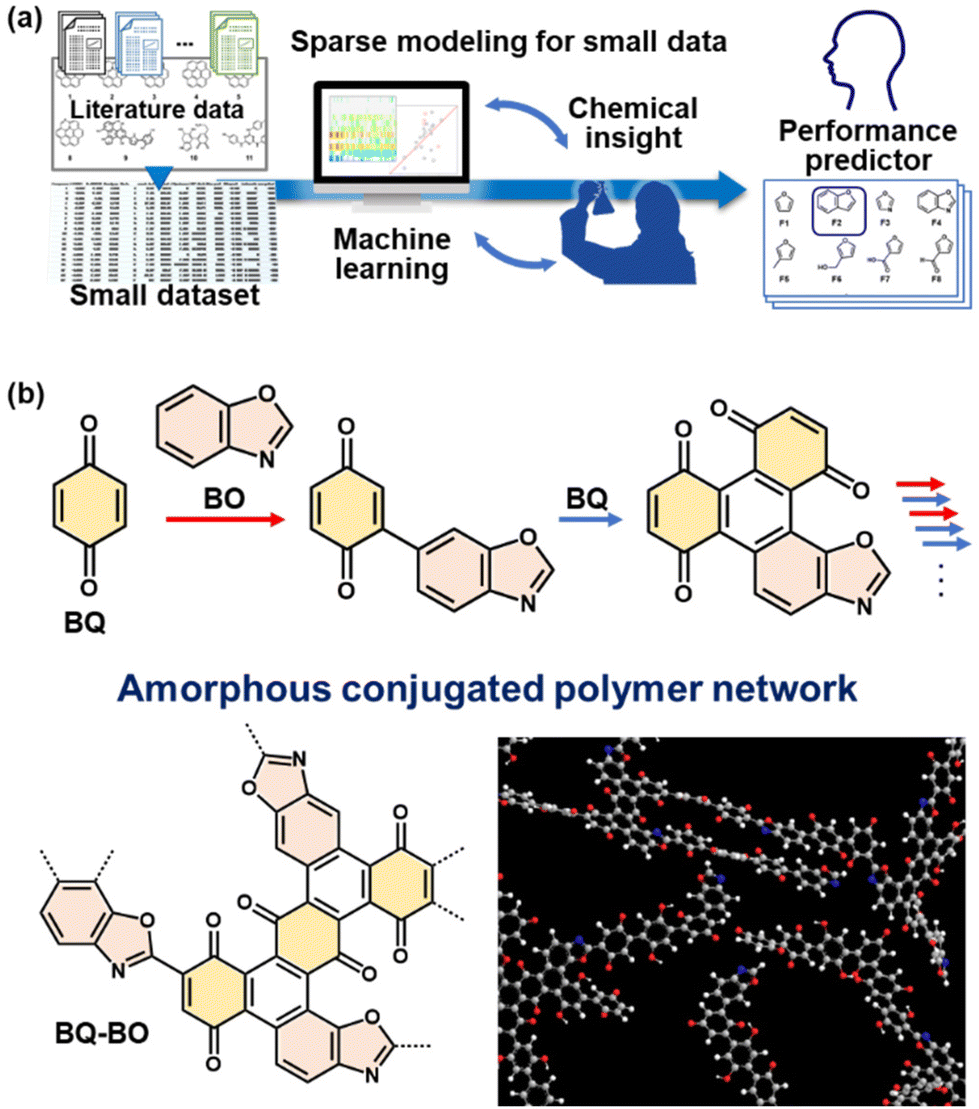 | ||
| Fig. 10 Design of a new amorphous CPN with improved HER electrocatalytic performance. (a) A scheme of sparse modelling for small data to construct a performance predictor combining machine learning and our chemical insights based on small data. (b) Simultaneous multiple reactions of BQ and BO, including electrophilic substitution (red arrows) and pericyclic (blue arrows) reactions for the synthesis of a BQ–BO amorphous conjugated polymer network. Reproduced from ref. 10 with permission from the Royal Society of Chemistry. | ||
 | ||
| Fig. 11 Morphology and catalytic performance of BQ–BO. (a) SEM image of BQ–BO in a mixture of the bulk and nanoparticles after dispersion in acetone. (b) LSV curves of the bulk (green) and bulk-nano (blue) BQ–BO samples and reference Pt and GC (black). Reproduced from ref. 10 with permission from the Royal Society of Chemistry. | ||
The catalytic performance of the BQ–BO amorphous CPN is achieved by the appropriate active sites, conductivity, and hydrophilicity.10 Density function theory (DFT) calculation implies that the carbons neighboring the heteroatoms are the potential active sites. Electrochemical impedance analysis indicates conductivity originating from the conjugated structure. In addition, hydrophilicity based on BQ and BO is supported by terahertz time-domain spectroscopy. In this manner, the BQ-based amorphous CPNs show potential as metal-free HER electrocatalysts. The prediction model can help the design of new structures with enhanced catalytic performance.
4.3. Amorphous CPNs of BQ and conjugated linkers for energy-storage applications
The redox reactions of quinone moieties show potential as active materials for energy-related applications, such as batteries and capacitors.32 However, quinone derivatives with low molecular weight, such as BQ and hydroquinone (HQ), dissolve in electrolyte solution. In previous work, the quinone derivatives were loaded on conductive and/or porous materials, such as nanocarbons, to suppress dissolution.33 Another effective approach is polymerization of quinone derivatives to lower the solubility.34 When the quinone moieties are polymerized, the electrochemically inert linker lowers the specific capacity (mA h g−1). A linker with smaller molecular weight has potential to enhance the specific capacity. On the other hand, a shorter linker causes aggregation of the quinone moieties and the formation of rigid structures without flexibility.In our previous study,9 BQ was polymerized with three different conjugated linkers: 1,9-dihydroxyanthracene (DAT), 1,4-divinylbenzene (DVB), and HQ (Fig. 12). Amorphous CPNs of BQ–DAT, BQ–DVB, and BQ–HQ were synthesized. All the samples show irregular-shaped particles around 1 μm in size. The nanostructures were obtained by dispersion in organic media (Fig. 13a and b). These BQ-based amorphous CPNs show redox reactions with protons in H2SO4 as an electrolyte. After exfoliation, the specific capacity is improved with an increase in specific surface area (Fig. 13c and d). This work shows one of the highest specific capacities: i.e. the utilization rates for the redox reactions of the quinone moieties.
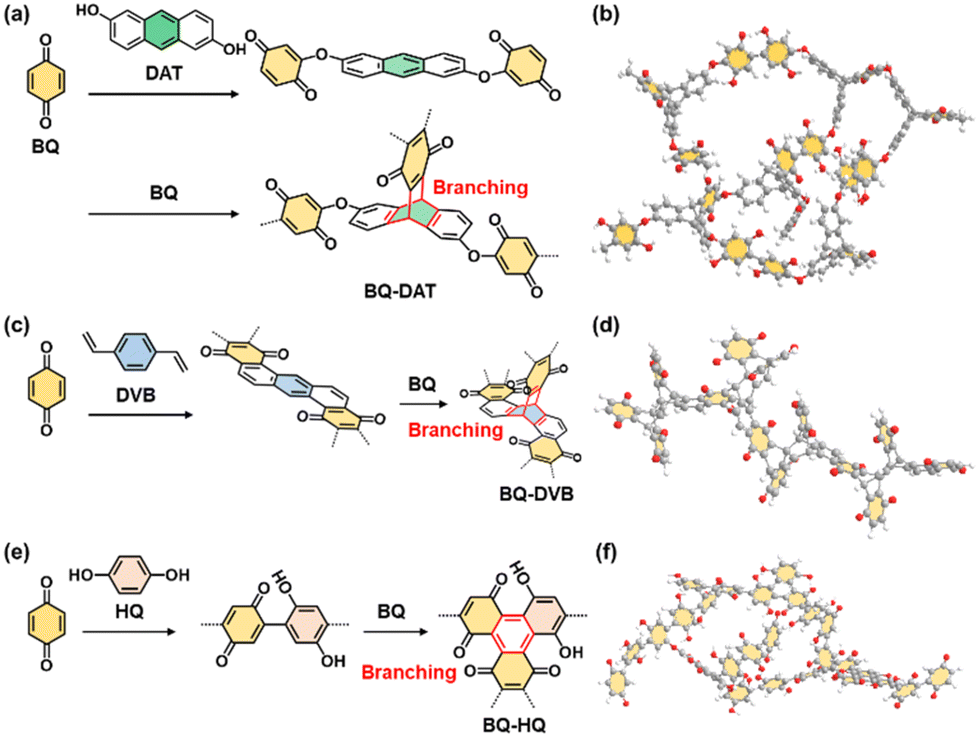 | ||
| Fig. 12 Molecular design of amorphous CPNs containing redox-active BQ moieties. (a and b) BQ–DAT. (c and d) BQ–DVB. (e and f) BQ–HQ. Reproduced from ref. 9 with permission from the Royal Society of Chemistry. | ||
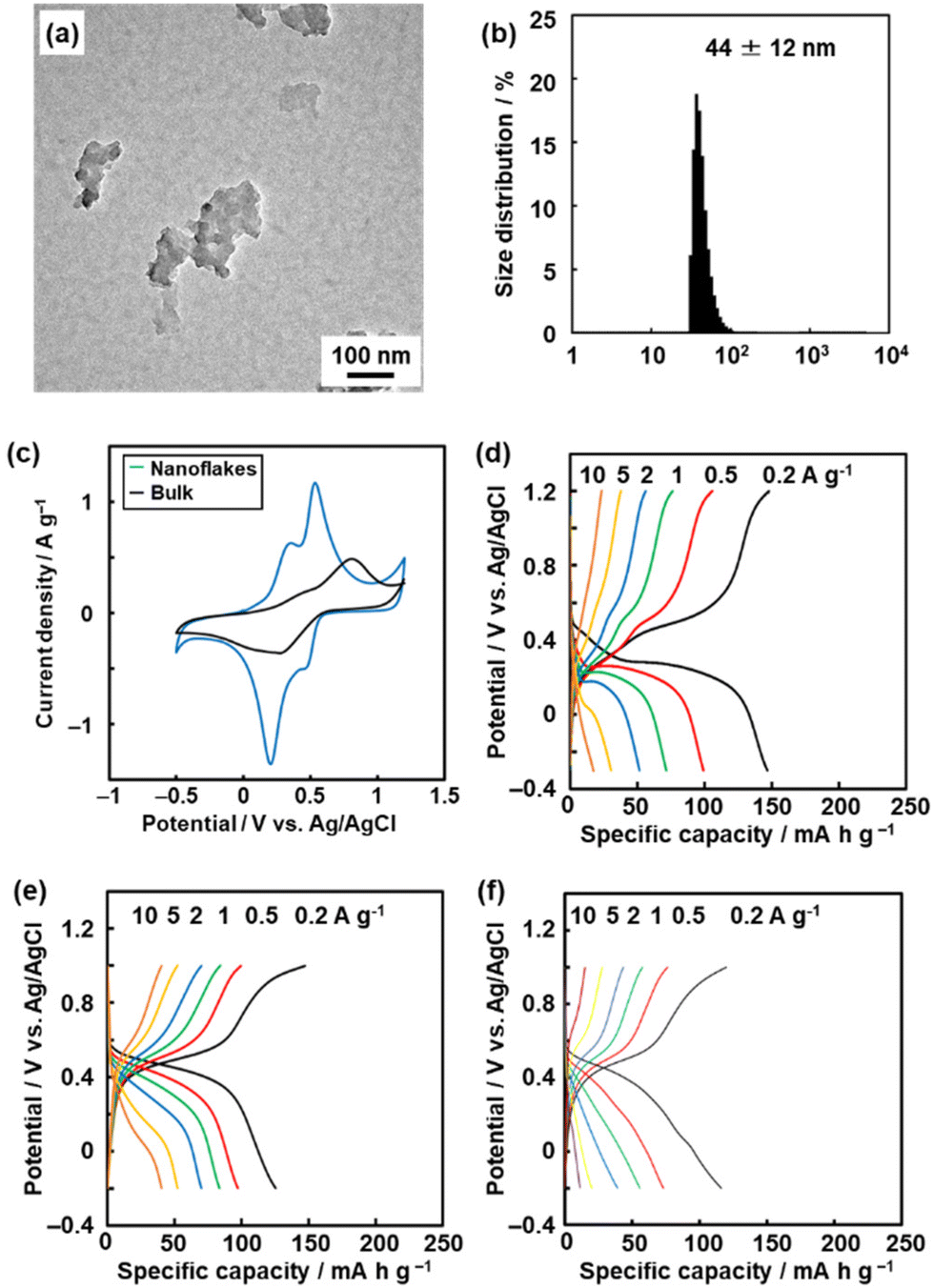 | ||
| Fig. 13 Amorphous CPNs containing BQ for application to aqueous supercapacitors. (a and b) TEM image (a) and DLS chart (b) of BQ–DVB after exfoliation. (c) Cyclic voltammograms of BQ–DVB before (bulk) and after (nanoflakes) exfoliation. (d) Charge–discharge curves of BQ–DVB nanoflakes. (e and f) Charge–discharge curves of BQ–HQ before exfoliation and the more branched polymer (f). Reproduced from ref. 9 with permission from the Royal Society of Chemistry. | ||
The amorphous CPN contributes to inhibiting dissolution. Moreover, the flexibility of the network structure plays an important role in achieving high specific capacity. BQ–HQ before exfoliation shows a specific capacity of 125 mA h g−1 at 0.2 A g−1 (Fig. 13e). A more frequently branched BQ–HQ is synthesized by changing the synthetic conditions. The specific capacity is lowered by about 20% (Fig. 13f). This implies that the flexibility of the network polymer has an effect on its performance. In addition, flexibility related to molecular motion in amorphous CPN can be controlled by the synthetic conditions.
The BQ–Py polymer was applied as an active material for the organic cathode of a lithium-ion battery.26 In recent years, polymers containing a BQ moiety have shown reversible redox reactions with the insertion and deinsertion of Li+. The electrochemical properties of BQ–Py as a cathode material were summarized and compared with COF-related materials (Table 1).26,35 The BQ–Py electrode showed the highest specific capacity among the other related cathodes and cycle stability. The high theoretical capacity originates from the use of the low-molecular-weight Py linker. In contrast, the COF-related materials use a symmetrical linker moiety with higher molecular weight, resulting in low density of the redox-active unit. Moreover, BQ–Py shows potential for post lithium-ion batteries, such as magnesium (Mg2+) and aluminum (Al3+) ion batteries.36 The densely assembled redox-active sites can interact with multi-valent anions, whereas the typical COF only has isolated single-anion sites.
| Name | Unit structure | State | Capacity/mA h g−1 | Potential V/vs. Li/Li+ | Cycle stability | Ref. |
|---|---|---|---|---|---|---|
| BQ–Py |
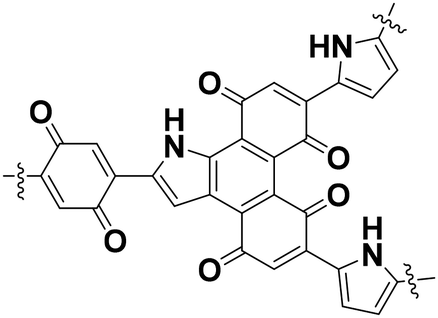
|
Low-crystalline | 255 (1.0 A g−1) | 2.3 | 81% (1000 cycles) | 26 |
| BQ–COF |
|
Amorphous | 207 (1.4 A g−1) | 2.2 | 86% (300 cycles) | 35a |
| PTO–COF |
|
Crystalline | 147 (1.0 A g−1) | 2.5 | 82% (3000 cycles) | 35b |
| Py–AQ–COF |
|
Crystalline | 142 (1.81 A g−1) | 2.3 | 75% (400 cycles) | 35c |
| BQbTPL |
|
Amorphous | 96 (1.0 A g−1) | 2.2 | 95% (1500 cycles) | 35d |
| DAAQ–ECOF |
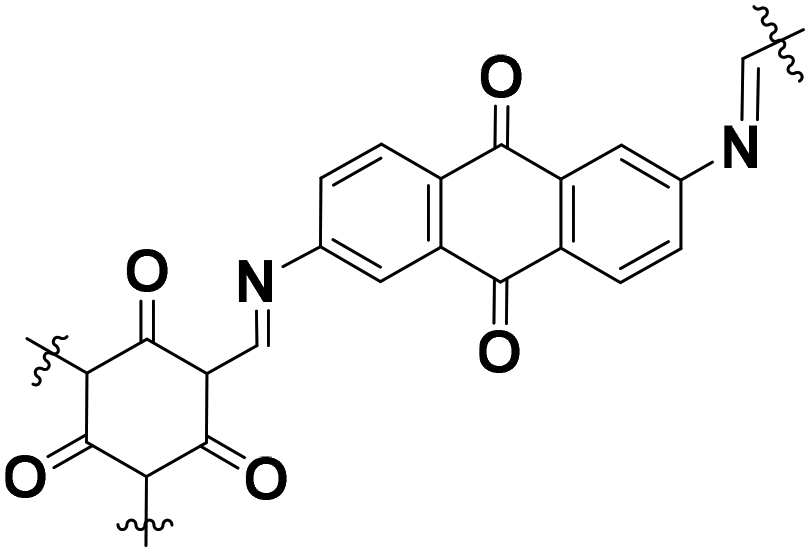
|
Low-crystalline | 99 (1.0 A g−1) | 2.2 | 98% (1800 cycles) | 35e |
| IEP-11-E |
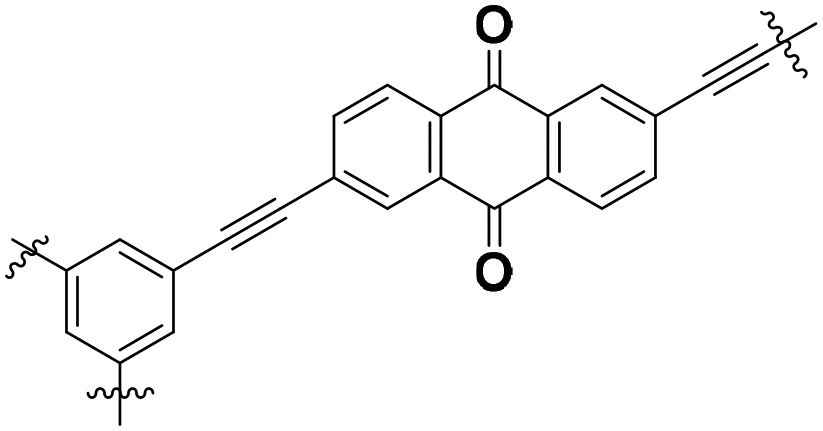
|
Crystalline | 104 (1.49 A g−1) | 2.2 | 90% (5000 cycles) | 35f |
4.4. Amorphous CPN of ternary heteroaromatic rings as an improved electrocatalyst for the oxygen reduction reaction
A variety of COFs with well-defined pore and framework structures have been developed. In addition, unique properties are reported for low-crystalline COFs.37 For example, layer slip is an interesting approach for preparing disordered vertical stacking in a layered 2D COF.38 Disordered stacking is formed with adsorption of gas and solvent.39,40 Perfect interlayer periodicity is not always needed for the adsorption properties of a COF. Amorphous COF layers form a thin film on a substrate via electrophoresis.41 A thin film based on amorphous COF enables efficient sieving of charged and neutral molecules with analogous structures. Although perfect and highly crystalline COFs have attracted a lot of interest, their low crystalline structure can exhibit unique properties.42The covalent triazine framework (CTF) is a notable material in CPNs.43 The periodic network structure is constructed with aromatic 1,3,5-triazine (TAz) rings in the π-conjugated plane. Porous and semiconductive CTFs promise applications in adsorption, separation, and catalysis.44 In general, reversibility of the polymerization reaction is a significant factor in forming crystalline frameworks because the steady bond formation and dissociation enable the repair of crystal dislocation and mismatch during the reaction.45 As formation of the TAz ring is mostly irreversible under mild conditions, highly crystalline layered structures are not obtained. CTF with a roughly-stacked layered structure is synthesized by the acid-catalysed cyclization of nitriles at moderate temperature.46 The roughly-stacked layered CTF is exfoliated into nanosheets and applied to the cathode material of a sodium-ion battery. Perfectly amorphous CTF, i.e. with disordering in both lateral and vertical directions without any anisotropy, is prepared by transition-metal-catalysed reactions, such as Suzuki–Miyaura and Stille cross-coupling reactions.44,47 The amorphous CTFs have a variety of applications, such as photocatalysts for water splitting and active materials for batteries. Unique properties can be extracted from both crystalline and non-crystalline CFT structures.
We found a new molecular design strategy for a low-periodic CTF with long functional linkers.12 In general, CTFs containing two or more ring linkers are formed in a perfectly amorphous state. Free rotation around the single bond can allow interpenetration of the large macrocyclic structure.48 Our intention is the introduction of both Pyd and Th rings enabling intramolecular interactions with each other (Fig. 14a). Furthermore, a base-catalysed liquid-phase polymerization is developed for the introduction of both Pyd and Th into the CTF network (Fig. 14b). The DFT calculation reveals that the combination of Pyd and Th rings realizes a stable planar conformation in the TAz–Pyd–Th network (Fig. 14c). The planar primal layers have the potential to stack into a layered structure (Fig. 14d). The Pyd–Th linker forms a roughly stacked layered structure, which is different from the perfectly amorphous structure using the Ph–Th linker (Fig. 14e). The intramolecular interaction can lead to the layered assembly of TAz–Pyd–Th, similar to TAz–2Pyd composed of single-ring linkers, even with simple changes in the structure from Ph to Pyd rings. Therefore, the resultant TAz–Pyd–Th as a low-crystalline CTF can be regarded as a family of amorphous CPNs.
 | ||
| Fig. 14 Amorphous CPNs containing TAz, Pyd, and Th. (a) Molecular design of CTF containing Pyd, Ph, and Th rings by base-catalysed TAz formation. (b) Structures of amorphous CTFs containing Pyd and Th. (c) Optimized conformations and their dihedral angles of Pyd, Ph–Th, Pyd–Th, and its metastable confirmation (Pyd–Th′). (d) Optimized unit cell of Pyd–Th and simulated layered structure of Pyd–Th with staggered stacking. (e) XRD profiles of the synthesized CTFs with different linkers. (f) SEM image of Pyd–Th exhibiting a laminated structure (red arrow). (g) SEM image of exfoliated nanosheets of Pyd–Th. (h) LSV curves of CTF-modified electrodes and reference samples as electrocatalysts for ORR in basic aqueous electrolyte. Reproduced from ref. 12 with permission from the Royal Society of Chemistry. | ||
The resultant TAz–Pyd–Th exhibited an anisotropic particle shape with both smooth and pleated surfaces (Fig. 14f). Among the heteroaromatic-ring-based organic ORR electrocatalysts, Pyd–Th shows comparable catalytic properties in terms of onset potential and electron transfer number (Table 2).12,49 The low-crystalline stacking of TAz–Pyd–Th affords exfoliation into nanosheets under mild conditions (Fig. 14g). The exfoliation yield of TAz–Pyd–Th is higher than that of TAz–2Pyd. In general, a nanosheet morphology can enhance the properties relevant to interface-related electrochemical applications. Moreover, the heteroaromatic rings can serve as active sites of metal-free electrocatalysts for the oxygen reduction reaction (ORR). The nanosheets of TAz–Pyd–Th with conductive carbon show catalytic performance as a metal-free electrocatalyst (Fig. 14h). The low-crystalline nature of the TAz–Pyd–Th linker has effects on superior electrocatalytic properties, ensuring the tuneability of the morphologies. As some effective molecular structures for ORR have been reported,50 the proposed strategy, embedding the active moiety into low-crystalline networks, has potential to enhance their catalytic activity.
| Name | Unit structure | State | Onset potential/V vs. RHE | N ET | Ref. |
|---|---|---|---|---|---|
| Prd–Th–TAz |
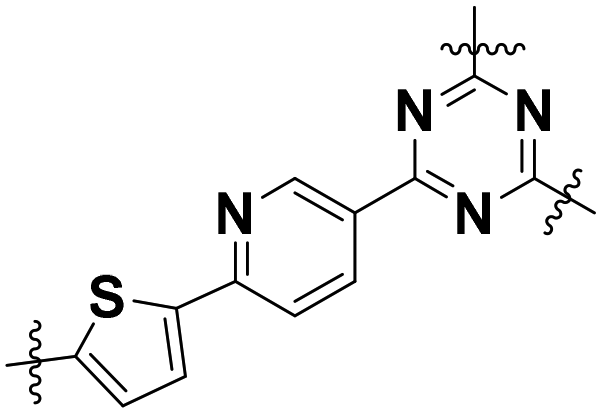
|
Low-crystalline/nanosheet | 0.77 | 3.77 | 12 |
| Cz–TAz |
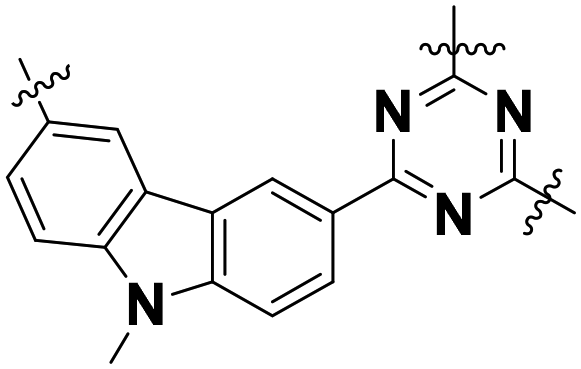
|
Amorphous/bulk particle | 0.74 | 2.60 | 49a |
| TAz–COP |

|
Crystalline/nanoparticle | 0.77 | 3.52 | 49b |
| BTT–TAPB COF |
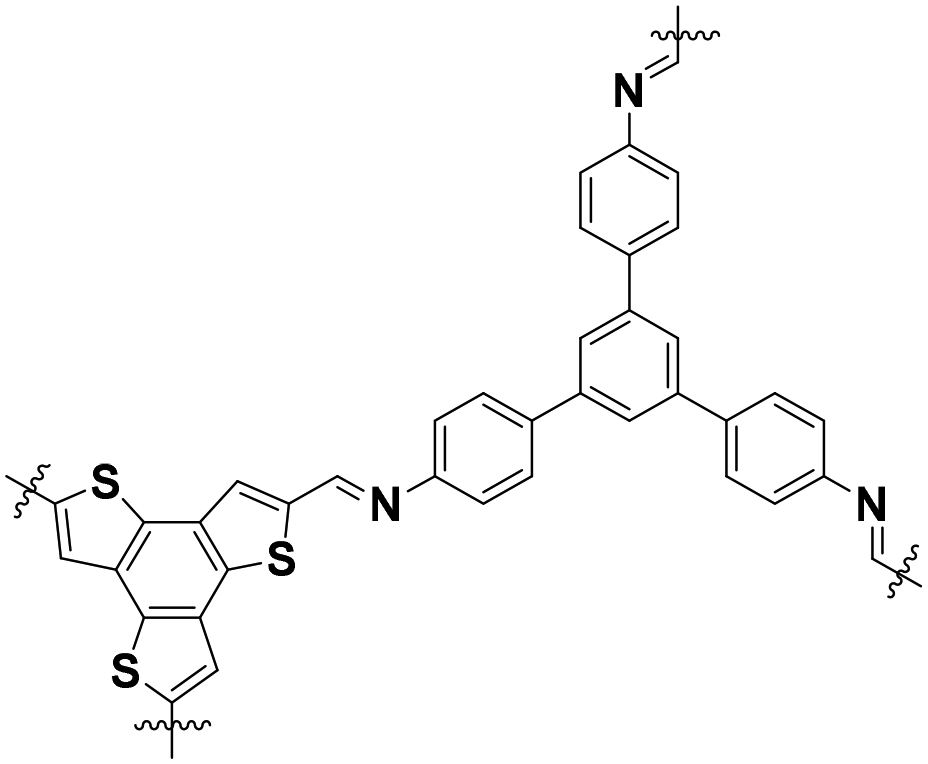
|
Crystalline/nanoparticle | 0.79 | 3.5 | 49c |
| TDC–TAPB COF |
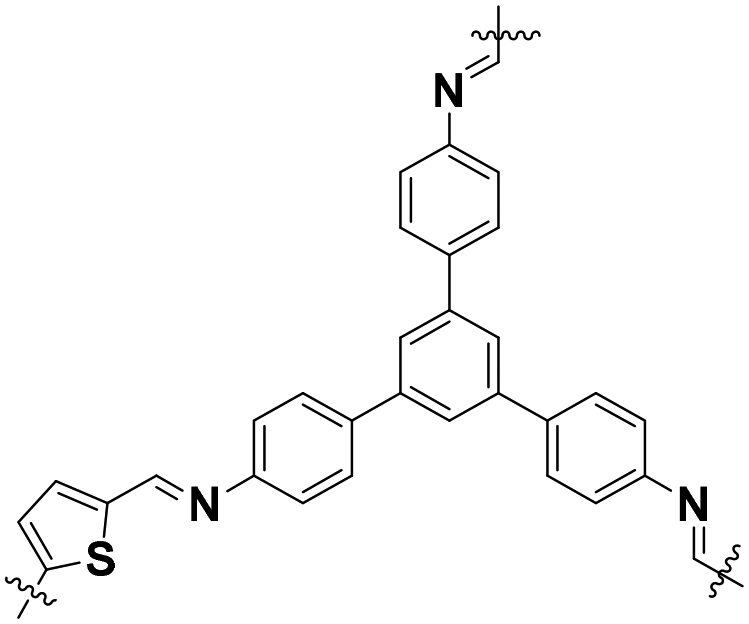
|
Crystalline/sub-μm particle | 0.76 | 3.46 | 49d |
| Azo–TPPy COF |

|
Low-crystalline/nanoparticle | 0.79 | 3.30 | 49e |
5. Summary and outlook
This Perspective focuses on amorphous CPN, as a new family of CPNs. In recent years, highly crystalline frameworks with conjugated structures, such as conjugated COFs, are promising nanostructures. However, amorphous types were not fully studied in previous work. Our group found that the simultaneous multiple reactions of the conjugated monomers provide random network polymers. The disordered structures are expanded in both the lateral networking and vertical stacking directions. In this article, amorphous CPNs, namely BQ–Py, BQ–BO, BQ–DVB, BQ–DAT, BQ–HQ, and TAz–Pyd–Th, were introduced to show their reaction processes, structural characteristics, and potential applications. In particular, these polymers can be effectively used for energy-related applications, such as electrocatalysts and redox capacitors. Whereas small functional molecules are easily dissolved in the solution phase, amorphous CPNs can contain and preserve the functional units in the polymer networks. The low-crystalline structures afford efficient utilization of the functional units because of the molecular motion originating from structural flexibility.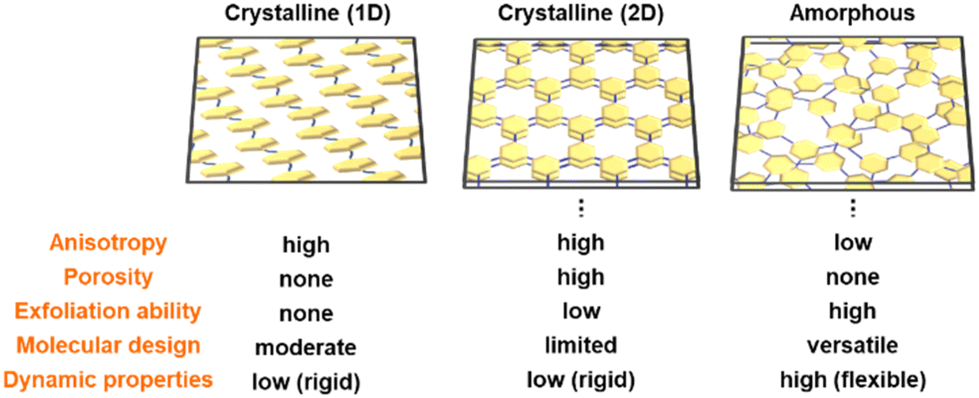 | ||
| Fig. 15 Structural advantages of amorphous CPNs compared with crystalline 1D conjugated polymers and 2D frameworks. | ||
Fig. 15 briefly summarizes the structural advantages of amorphous CPNs compared with crystalline 2D CPNs, such as COFs, and conventional conjugated polymers with crystalline 1D conjugated polymers and 2D the anisotropic arrangement of the functional moieties. The functional units are used as the assembled states. In amorphous CPNs, the isolated functional units that are homogenously dispersed without aggregation in the network can be used efficiently. Only the crystalline CPNs have definite pores for adsorption of gas molecules. On the other hand, amorphous CPNs with no pores for gas adsorption showed the inclusion of solution, such as electrolyte, in our previous work, even though the mechanisms are still unclear. The low-crystalline nature of the amorphous CPNs affords structural flexibility related to dynamic properties and ability for exfoliation. As shown in section 4, the amorphous CPNs have potentials for molecular design with various combinations of multiple functional monomers. New functions can be derived from unknown combinations of the monomers in amorphous CPNs. Compared with this versatility, conventional 1D conjugated polymers and crystalline 2D CPNs have limitations in forming an ordered state.
Now, amorphous CPNs are still in the development stage with challenges remaining. More detailed studies are required at the molecular and secondary-structure level. For example, why is superior redox performance achieved with penetration of the electrolytes in the solution phase, even though the gas molecules have not penetrated into the polymer network? Microscopy and simulation studies are helpful for understanding the structure. Another challenge is to understand the processes of simultaneous multiple reactions from the monomers for the growth of the polymer networks. These understandings will accelerate the syntheses and applications of amorphous CPNs in the future. In particular, amorphous CPNs can be used as metal-free high-performance active materials in energy-related applications. Moreover, new functional amorphous CPNs can be synthesized and found in the infinite combinations of monomers.
Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this Feature Article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank all the collaborators contributing to the related our previous works cited in this article. This work was supported by JSPS-KAKENHI (JP22H04559 to Y. O. and JP21K14734 to K. S.), Toray Science Foundation (Y. O.), and Mitsubishi Foundation (Y. O.).References
- H. Staudinger, Ber. Dtsch. Chem. Ges., 1920, 53, 1073 CrossRef
.
-
(a) D. Larcher and J.-M. Tarascon, Nat. Chem., 2015, 7, 19 CrossRef CAS PubMed
-
(a) P. Peng, Z. Zhou, J. Guo and Z. Xiang, ACS Energy Lett., 2017, 2, 1308 CrossRef CAS
-
(a) A. D. Shlüter, Adv. Mater., 1991, 3, 282 CrossRef
-
(a) S. L. James, Chem. Soc. Rev., 2003, 32, 276 RSC
-
(a) D. Wu, F. Xu, B. Sun, R. Fu, H. He and K. Matyjaszewski, Chem. Rev., 2012, 112, 3959 CrossRef CAS PubMed
-
(a) W. Zhang, L. Chen, S. Dai, C. Zhao, C. Ma, L. Wei, M. Zhu, S. Y. Chong, H. Yang, L. Liu, Y. Bai, M. Yu, Y. Xu, X. W. Zhu, Q. Zhu, S. An, R. S. Sprick, M. A. Little, X. Wu, S. Jiang, Y. Wu, Y. B. Zhang, H. Tian, W. H. Zhu and A. I. Cooper, Nature, 2022, 604, 72 CrossRef CAS PubMed
- S. Yano, K. Sato, J. Suzuki, H. Imai and Y. Oaki, Commun. Chem., 2019, 2, 97 CrossRef
- J. Suzuki, A. Ishizone, K. Sato, H. Imai, Y. J. Tseng, C. H. Peng and Y. Oaki, Chem. Sci., 2020, 11, 7003 RSC
- W. Hamada, M. Hishida, R. Sugiura, H. Tobita, H. Imai, Y. Igarashi and Y. Oaki, J. Mater. Chem. A, 2024, 12, 3294 RSC
- R. Sugiura, H. Imai and Y. Oaki, Nanoscale Adv., 2024, 6, 1084 RSC
- K. Sato, N. Osada and H. Aihara, RSC Adv., 2023, 13, 11794 RSC
-
L. Pilato, Phenolic Resins: A Century of Progress, Springer Nature, Berlin, 2010 Search PubMed
-
(a) K. C. Radford, J. Mater. Sci., 1971, 6, 1286 CrossRef CAS
-
(a) S. C. Rasmussen, ChemPlusChem, 2020, 85, 141 CrossRef
-
(a) M. Whangbo, R. Hoffmann and R. B. Woodward, Proc. R. Soc. London, Ser. A, 1979, 366, 23 CrossRef CAS
- S. Venkatachalam, K. V. C. Rao and P. T. Manoharan, Synth. Met., 1988, 26, 237 CrossRef CAS
- O. Lavastre, S. Cabioch, P. H. Dixneuf, J. Sedlacek and J. Vohlidal, Macromolecules, 1999, 32, 4477 CrossRef CAS
- C. Weder, Chem. Commun., 2005, 5378 RSC
-
(a) E. Jin, M. Asada, Q. Xu, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine, Q. Chen and D. Jiang, Science, 2017, 357, 673 CrossRef CAS PubMed
-
(a) X. Li, B. Li, Y. He and F. Kang, New Carbon Mater., 2020, 35, 619 CrossRef CAS
-
(a) J. S. M. Lee and A. I. Cooper, Chem. Rev., 2020, 120, 2171 CrossRef CAS PubMed
-
(a) Y. Oaki and K. Sato, Nanoscale Adv., 2022, 4, 2773 RSC
-
(a) R. Matsuoka, R. Sakamoto, K. Hoshiko, S. Sasaki, H. Masunaga, K. Nagashio and H. Nishihara, J. Am. Chem. Soc., 2017, 139, 3145 CrossRef CAS PubMed
-
(a) E. Bullock, Can. J. Chem., 1958, 36, 1744 CrossRef CAS
- J. Chu, G. Li, Y. Wang, X. Zhang, Z. Yang, Y. Han, T. Cai and Z. Song, ACS Appl. Mater. Interfaces, 2022, 14, 25566 CrossRef CAS PubMed
- K. Sato, H. Imai and Y. Oaki, ACS Appl. Nano Mater., 2018, 1, 4218 CrossRef CAS
-
(a) J. Duan, S. Chen, M. Jaroniec and S. Z. Qiao, ACS Catal., 2015, 5, 5207 CrossRef CAS
- D. Tripathy, H. M. Viswanatha, M. N. K. Hanish and S. Sampath, Chem. Commun., 2022, 58, 7821 RSC
- T. Shimamura, A. Kobayashi, Y. Oaki, M. Yoshida and M. Kato, Energy Fuels, 2022, 36, 11559 CrossRef CAS
-
(a) K. Noda, Y. Igarashi, H. Imai and Y. Oaki, Adv. Theory Simul., 2020, 3, 2000084 CrossRef CAS
-
(a) Z. P. Song and H. S. Zhou, Energy Environ. Sci., 2013, 6, 8 Search PubMed
-
(a) G. Pognon, C. Cougnon, D. Mayilukila and D. Belanger, ACS Appl. Mater. Interfaces, 2012, 4, 3788 CrossRef CAS PubMed
-
(a) K. Naoi, S. Suematsu and A. Manago, J. Electrochem. Soc., 2000, 147, 420 CrossRef CAS
-
(a) Z. Luo, L. Liu, J. Ning, K. Lei, Y. Lu, F. Li and J. Chen, Angew. Chem., Int. Ed., 2018, 57, 9443 CrossRef CAS PubMed
-
(a) D. Tripathy, H. M. Viswanatha, H. M. N. Kotresh, P. V. Babu and S. Sampath, ACS Appl. Mater. Interfaces, 2022, 14, 26671 CrossRef CAS PubMed
-
(a) G. Zhou, T. Yang and Z. Huang, Commun. Chem., 2023, 6, 116 CrossRef CAS PubMed
-
(a) C. Kessler, R. Schuldt, S. Emmerling, B. V. Lotsch, J. Kästner, J. Gross and N. Hansen, Microporous Mesoporous Mater., 2022, 336, 111796 CrossRef CAS
- C. Kang, Z. Zhang, A. K. Usadi, D. C. Calabro, L. S. Baugh, K. Chai, Y. Wang and D. Zhao, J. Am. Chem. Soc., 2022, 144, 20363 CrossRef CAS PubMed
- T. Sick, J. M. Rotter, S. Reuter, S. Kandambeth, N. N. Bach, M. Döblinger, J. Merz, T. Clark, T. B. Marder, T. Bein and D. D. Medina, J. Am. Chem. Soc., 2019, 141, 12570 CrossRef CAS PubMed
-
(a) X. Wang, J. Yang, X. Shi, Z. Zhang, C. Yin and Y. Wang, Small, 2022, 18, 2107108 CrossRef CAS PubMed
-
(a) R. Iqbal, M. K. Majeed, A. Hussain, A. Ahmad, M. Ahmad, B. Jabar, A. R. Akbar, S. Ali, S. Rauf and A. Saleem, Mater. Chem. Front., 2023, 7, 2464 RSC
-
(a) P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450 CrossRef CAS PubMed
-
(a) X. Hu, Z. Zhan, J. Zhang, I. Hussain and B. Tan, Nat. Commun., 2021, 12, 6596 CrossRef CAS PubMed
-
(a) V. S. Vyas, F. Haase, L. Stegbauer, G. Savasci, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Nat. Commun., 2015, 6, 8508 CrossRef CAS PubMed
- J. Liu, P. Lyu, Y. Zhang, P. Nachtigall and Y. Xu, Adv. Mater., 2018, 30, 1705401 CrossRef
-
(a) C. B. Meier, R. S. Sprick, A. Monti, P. Guiglion, J.-S. M. Lee, M. A. Zwijnenburg and A. I. Cooper, Polymer, 2017, 126, 283 CrossRef CAS
-
(a) M. S. Kim, C. S. Phang, Y. K. Jeong and J. K. Park, Polym. Chem., 2017, 8, 5655 RSC
-
(a) W. Yu, S. Gu, Y. Fu, S. Xiong, C. Pan, Y. Liu and G. Yu, J. Catal., 2018, 362, 1 CrossRef CAS
-
(a) Z. Yang, Y. Gao, L. Zuo, C. Long, C. Yang and X. Zhang, ACS Catal., 2023, 13, 4790 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2025 |