
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diesel soot oxidation over Mn–Pr–Ce oxide catalysts: structural changes and the impact of Mn doping
Sunaina S.
Patil
 a,
Hari Prasad
Dasari
*a,
Rahulkumar
Shirasangi
a and
Harshini
Dasari
*b
a,
Hari Prasad
Dasari
*a,
Rahulkumar
Shirasangi
a and
Harshini
Dasari
*b
aEnergy and Catalysis Materials Laboratory, Chemical Engineering Department, National Institute of Technology Karnataka, Surathkal, Mangalore-575025, Karnataka, India. E-mail: energyhari@nitk.edu.in; Tel: +91-8242473610
bChemical Engineering Department, Manipal Institute of Technology Manipal Academy of Higher Education, Manipal, 576104, Udupi, Karnataka, India. E-mail: harshini.dasari@manipal.edu; Tel: +91-8202924316
First published on 13th January 2025
Abstract
The soot oxidation activity of manganese-doped ceria-praseodymium catalysts, synthesized via solution combustion synthesis, was evaluated. The analyses performed with XRD and Raman spectroscopy indicated that the Mn-doped CP catalysts displayed the typical fluorite structure of CeO2. The addition of Mn to CP led to a reduction in crystallite size from 14 nm to below 10 nm. The F2g Raman active mode of fluorite-structured Ce and the oxygen vacancies resulting from the addition of Mn and Pr (bands ∼ 560 cm−1 to 580 cm−1) were consistently observed across all Mn-doped CP catalysts. 15 and 20 Mn-CP exhibited an additional secondary phase identified as Mn2O3. The analysis of BET surface area and BJH pore size revealed that the Mn-doped CP catalysts exhibited both micro and mesoporous characteristics. The H2-TPR and O2-TPD profiles indicated enhanced reducibility resulting from the incorporation of Mn and Pr into CeO2-doped catalysts. The improved T50 (365 ± 1 °C) for the 5 Mn-CP catalytic system is primarily due to its increased specific surface area of 45 m2 g−1 and the presence of active surface adsorbed oxygen species identified in the XPS and O2-TPD studies. 5 Mn-CP exhibited the lowest activation energy value compared to all other Mn-doped catalysts.
1. Introduction
Regenerating diesel particulate filters (DPFs) in the automotive industry is difficult and expensive since they use a lot of energy and operate at high temperatures (around 800 °C). A pressure drop develops when the diesel engine's exhaust flow is restricted due to a clogged or blocked DPF, which could lead to additional engine damage. Therefore, it is important to regenerate the filter carefully so that no harmful byproducts are released, and all particulates should be contained within the constraints of particle size restrictions and the engine's life.1Catalytic oxidation is the main technique used to reduce soot emissions. Adding catalysts lowers the oxidation temperature, whereas soot oxidizes at temperatures above 600 °C without catalysts.2–5 Ce-based mixed oxides are promising catalysts for redox processes, particularly in soot oxidation.6–14 The descriptors for Ce-based metal oxides that enhance the catalytic performance include the redox ability, the host structure, lattice oxygen species, site isolation, the nature of the metal–oxygen bond, multifunctionality, and phase cooperation.15 They are also known as the seven pillars, which regulate the activity based on the different reaction conditions. Additional criteria for better catalytic activity may include great oxygen storage capacity (OSC), porosity, and high surface area.16
Shuang et al.17 identified the parameters affecting activity and stability. The ceria-based catalysts oxidize soot via a Mars–van Krevelen mechanism, wherein gaseous O2 initially adsorbs onto Ce, dissociates into atomic O and then converts into Ox species. CeO2, whether utilized as a catalyst or as a support in the passive regeneration of particulate filters, is likely of minimal significance due to its inadequate textural stability under high-temperature reactions often present in exhaust gases. Upon exposure to elevated temperatures, the surface area of CeO2 significantly diminishes, concurrently resulting in the potential loss of its redox characteristics and oxygen storage capacity. The modification of CeO2 with diverse ions is recognized to enhance stability against sintering and augment the oxidation activity of the resultant catalysts. This alteration was ascribed to the variations in redox characteristics and the formation of oxygen vacancies, both enhancing oxygen exchange with the catalyst and increasing oxygen storage capacity.18 Some literature indicates that a positive correlation between catalytic performance and specific surface area is not assured.17,19 Instead, the catalytic soot oxidation depends on the interaction efficiency between active sites and soot on the surface.20 Rare earth and transition metals were doped with Ce to address sintering and thermal stability difficulties.7,21,22 In addition to Ce, the rare earth elements consist of praseodymium (Pr), terbium (Tb), dysprosium (Dy), lanthanum (La), gadolinium (Gd), samarium (Sm), neodymium (Nd), europium (Eu), scandium (Sc), yttrium (Y) and so on.6,7,23–27 Among them, La, Pr, Nd, Sm, Gd, and Tb were tested for soot oxidation activity, and most of them displayed significantly enhanced catalytic activity.6–9,28–35 It has been identified that ceria modified with La- and particularly Pr- remains highly effective as a thermally stable catalyst.6,18
Pr appears to exert a promotional effect on the catalytic behavior of ceria, potentially linked to the reducibility of PrOx.6,18,36 Praseodymium oxide exhibits the highest redox capability and promptly reduces adsorbed neutral dioxygen (O2) to the inert lattice oxide anion (O2−).36 Pr has larger ionic radii than Ce and is quickly reducible; Pr also exhibits a multi-oxidation state, which may improve its characteristics and reduce the temperature required for soot combustion.7
Additionally, transition metals such as chromium (Cr), iron (Ir), manganese (Mn), copper (Cu), cobalt (Co), hafnia (Hf), and zirconia (Zr) are chosen as dopants because most of them can exist in multiple oxidation states.21,37 Among them, Mn is one of the most accessible transition metals, with numerous applications.38 Liang et al.39 indicated that CuCe, followed by MnCe mixed oxides, are the most effective catalysts. In a loose contact mode, MnCe demonstrates the most remarkable activity, regardless of the presence of NOx.6,39 Mn's popularity in catalysis arises mostly from its multiple oxidation states.40 Research indicates that manganese oxide-based catalysts exhibit superior performance in soot oxidation activity.38,40–43 Mn has a substantially smaller ionic size than Ce and is easily reducible because it occurs in numerous valence states, passing on both extrinsic and intrinsic oxygen vacancies.44 Mn, as a dopant, results in increased oxygen storage capacity (OSC) and thermal stability.22,45 In the current study, the CeO2 is modified by doping with Pr and Mn, which possess redox ability.
Catalytic preparation techniques are crucial to fine-tuning the shape and physicochemical characteristics. Ce-based catalysts and complex oxides are synthesized using coprecipitation,46,47 hydrothermal,48 solution combustion synthesis,34,49,50 sol–gel,51 microemulsion,52 the EDTA-citrate method,53,54 and solvothermal methods.3,55 Among these, solution combustion synthesis (SCS) creates homogenous powders with high purity, which is a quick and straightforward procedure, saving time and energy.56,57 In this study, Mn-doped CP catalysts (Mnx(x=0–0.2)(Ce0.9Pr0.1)1−xO2−δ) with varying Mn concentrations were synthesized using the SCS method. The catalysts were characterized and tested for effectiveness towards soot oxidation.
2. Methodology
2.1. Synthesis methodology
The catalysts were synthesized via solution combustion synthesis (SCS). Metal nitrates [Ce(NO3)3·6H2O (Sigma-Aldrich ≥99%), Pr(NO3)3·6H2O (Sigma-Aldrich ≥99%), and Mn(NO3)3·6H2O (Sigma-Aldrich ≥98%)] and glycine were well dissolved in distilled water, according to the stoichiometric ratio maintaining a glycine (fuel) to nitrate (oxidizer) mole ratio (G/N) of 0.35. The solution was heated and agitated continuously at 70 °C until it became a viscous gel. The gel is then transferred to a preheated (350 °C) oven, where it is auto-ignited, and the obtained voluminous powder is calcined at 600 °C/5 h to eliminate impurities. The Mnx(x=0–0.2)(Ce0.9Pr0.1)1−xO2−δ catalysts with various Mn concentrations (5, 10, 15, 20 mol%, keeping ceria-praseodymium (CP) constant) were prepared and are herein denoted as 5 to 20 Mn-CP and Mn oxide.2.2. Characterization
The obtained catalysts are further characterized to determine the phase, structure, surface area, morphology, and occurrence of oxygen vacancies. The characterization tools such as X-ray diffraction (XRD) analysis (Malvern PAN analytical: Empyrean diffractometer, Kα radiation; λ = 1.54 Å), FT-Raman spectroscopy (LabRAM HR Horiba, France, Ex = 532 nm laser beam), BET surface area and BJH pore size analysis (Anton Paar- Autosorb iQ-XR-XR 195364 Quantachrome instruments), and X-ray photon spectroscopy (XPS) (Omicron ESCA+ – ultrahigh vacuum) were used in the present study. Before fitting the peaks for XPS, the baseline and carbon (C1s) peak correction (284.6 eV) were performed. The reducibility ratio and surface active oxygen species ratio were calculated by measuring the ratio of individual ions to total ions.H2-temperature programmed reduction (H2-TPR) and O2-temperature programmed desorption (O2-TPD) analyses of the catalysts were carried out with a chemisorption analyzer (BELCAT-II, M/s Microtrac, Japan). For the O2-TPD analysis, a 100 mg catalyst was pretreated at 300 °C/1 h in He and subsequently cooled to room temperature in He; the catalyst was then pretreated with a 5% O2/He mixture (30 mL min−1) at room temperature for 1 h. Following treatment in the oxidizing environment, the catalyst was heated from ambient temperature to 900 °C under He (30 mL min−1) at 10 °C min−1. For the H2-TPR analysis, a 100 mg catalyst was pretreated from room temperature to 500 °C in 5% O2/He at 10 °C min−1, held at 500 °C/1 h in the same environment and subsequently cooled to room temperature in He, and then the catalyst was heated to 900 °C at 10 °C min−1 in 10% H2/Ar gas mixture.
2.3. Soot oxidation activity
Soot oxidation reactions were performed in a Thermo Gravimetric Analyser (TGA, Exstar TGA/DTA 6300). Soot and catalyst (wt ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10) were mixed using an electric agate mortar and pestle for 45 min. The heating rate for the oxidation reaction is maintained at 10 °C min−1 with a temperature range from room temperature to 600 °C with an airflow rate of 100 mL min−1 (atm. pressure). To determine activation energy (Ea), the heating rate (β) for the soot oxidation reaction in TGA was varied at 5, 10, 15, and 20 °C min−1. The Flynn Wall Ozawa method58 was employed to determine average activation energy, calculated from the slope of log(β) vs. 1/T (K−1) plots where ‘T’ refers to the temperature in Kelvin.
:10) were mixed using an electric agate mortar and pestle for 45 min. The heating rate for the oxidation reaction is maintained at 10 °C min−1 with a temperature range from room temperature to 600 °C with an airflow rate of 100 mL min−1 (atm. pressure). To determine activation energy (Ea), the heating rate (β) for the soot oxidation reaction in TGA was varied at 5, 10, 15, and 20 °C min−1. The Flynn Wall Ozawa method58 was employed to determine average activation energy, calculated from the slope of log(β) vs. 1/T (K−1) plots where ‘T’ refers to the temperature in Kelvin.
3. Results and discussion
3.1. Characterization of the developed catalysts
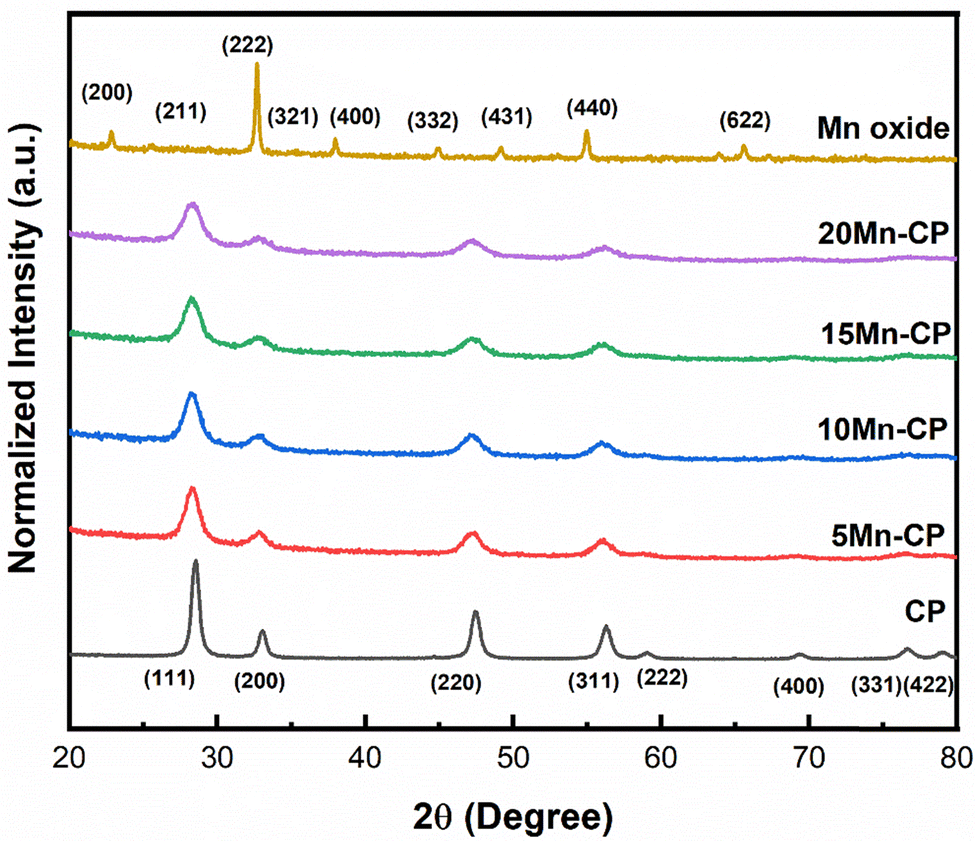 | ||
| Fig. 1 XRD spectra of Mnx(x=0–0.2)(Ce0.9Pr0.1)1−xO2−δ catalysts synthesised by SCS, calcined at 600 °C/5 h. | ||
Table 1 shows the crystallite size (obtained using the Scherrer equation65), lattice strain, and facet ratios ({100}/{111} and {110}/{111}) for the catalysts. It was discovered that 20 Mn-CP had the smallest crystallite size and the largest lattice strain, measuring 5.67 nm and 0.0262, respectively. Pure Mn oxide exhibited a greater crystallite size and a lower lattice strain (30.04 nm and 0.0043, respectively). As per the previous investigations,34 when Pr was added to CeO2, the crystallite size reduced from 21 nm to 14 nm. The further addition of Mn to the CP catalyst system reduced the crystallite size from 14 nm to less than ∼10 nm. Reactive facets are one of the most important characteristics in determining catalytic activity enhancement.66 It has been demonstrated that ceria exhibiting a higher number of exposed planes from the {100} and {110} facets shows enhanced activity in comparison to those with a predominance of {111}.67 Mn-loaded CP catalysts exhibit much higher lattice strain and facet ratios ({110/111}) than bare CP catalysts; hence, adding Mn to CP enhanced its physiochemical properties, which could contribute to improved catalytic efficiency. The addition of Mn to Ce indicated a reduction in the crystallinity of the Ce–Mn catalysts. The doped Ceria catalysts exhibited broad/wider diffraction peaks without any obvious shift, indicating MnOx in a well-dispersed phase.53,68
| Sample | Crystallite size (nm) | Lattice strain (ε) | Facet ratio | I Ov/IF2g | BET SA (m2 g−1) | Pore volume (cc g−1) | Avg. pore size (nm) | |
|---|---|---|---|---|---|---|---|---|
| {110}/{111} | {100}/{111} | |||||||
| CP | 14 | 0.0107 | 0.24 | 0.38 | 0.77 | 31 | 0.217 | 11.46 |
| 5 Mn-CP | 07 | 0.0218 | 0.35 | 0.37 | 0.75 | 45 | 0.110 | 4.92 |
| 10 Mn-CP | 07 | 0.0220 | 0.37 | 0.38 | 0.78 | 36 | 0.109 | 6.39 |
| 15 Mn-CP | 06 | 0.0234 | 0.39 | 0.36 | 0.57 | 33 | 0.333 | 5.92 |
| 20 Mn-CP | 06 | 0.0262 | 0.40 | 0.38 | 0.60 | 35 | 0.118 | 6.94 |
| Mn | 30 | 0.0043 | — | — | — | 20 | 0.039 | 4.08 |
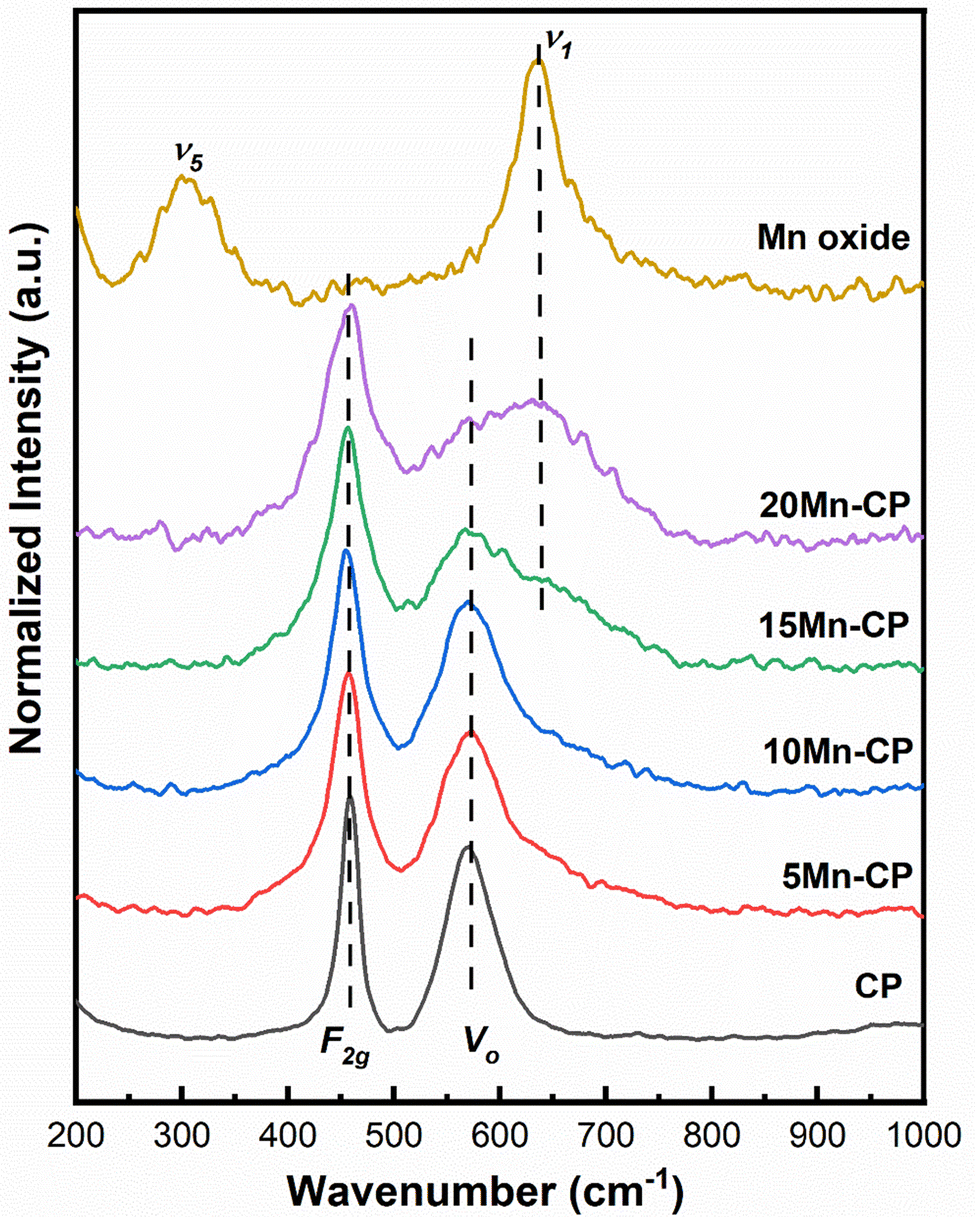 | ||
| Fig. 2 Raman spectra of Mnx(x=0–0.2)(Ce0.9Pr0.1)1−xO2−δ catalysts. | ||
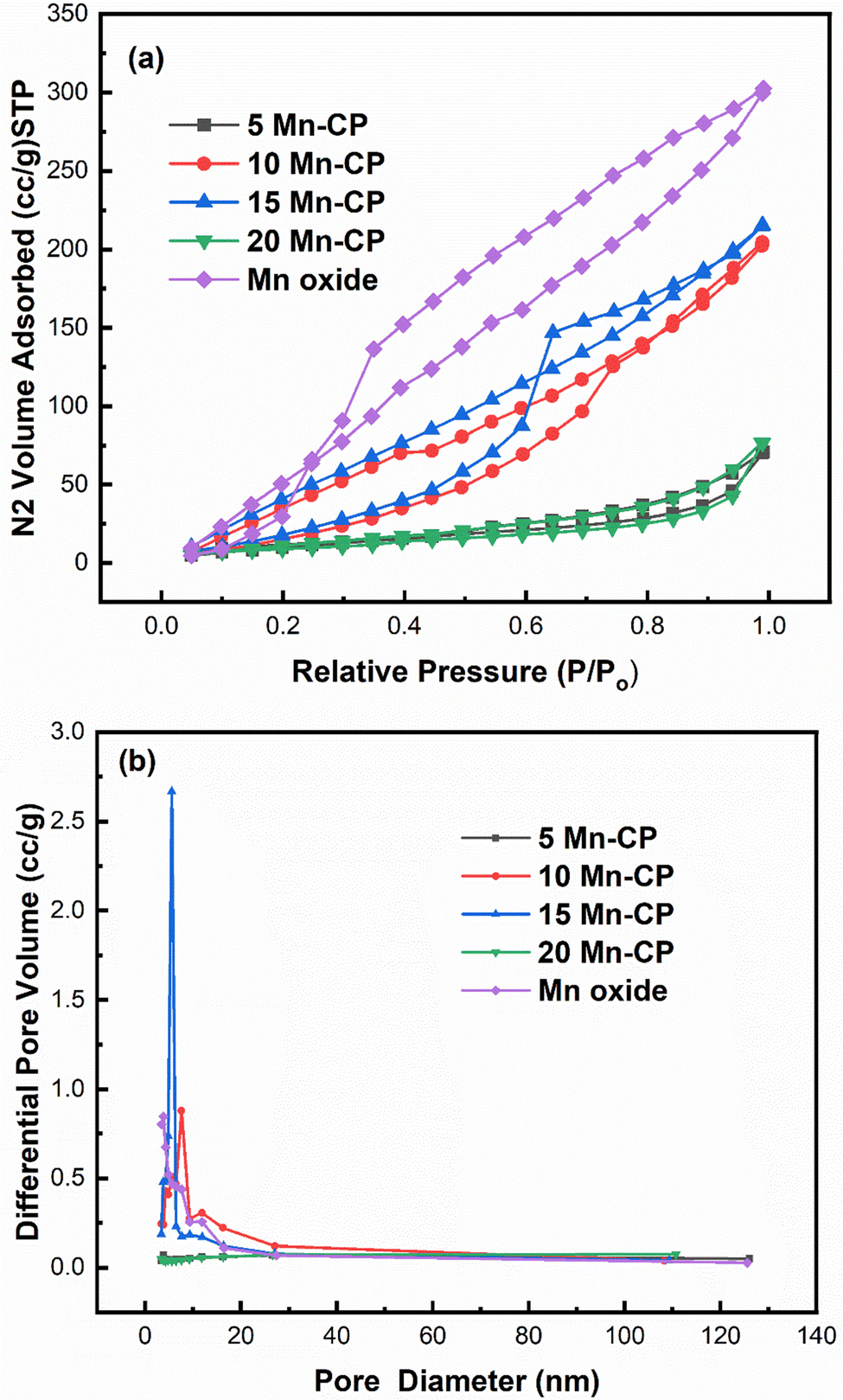 | ||
| Fig. 3 (a) Adsorption–desorption isotherm; (b) BJH pore size distribution of Mnx(x=0–0.2)(Ce0.9Pr0.1)1−xO2−δ catalysts. | ||
Fig. 3(a) indicates that for Mn-doped CP catalysts, 5 Mn-CP and 20 Mn-CP exhibited identical Type IV isotherms with H4 hysteresis and minimal adsorption. Mn Oxide and 10 Mn-CP also demonstrated Type IV isotherms with H3 and H2 hysteresis, respectively. They display strong N2 adsorption; in contrast, 10 Mn-CP displays delayed desorption. When the pore type exhibits a pore size smaller than the critical size of the adsorbent, cavitation occurs in the larger region, leading to increased challenges in fluid release from the pore during the desorption process, and such materials exhibit H2 hysteresis characteristics.75 In contrast, materials exhibiting nonrigid aggregates, slit-shaped or plate-like pores that show unlimited absorption (at elevated P/Po levels) are categorized under type H3 hysteresis. H4 hysteresis loops are commonly observed in intricate materials with micropores and mesopores.75 Multiple loops in the 15 Mn CP type IV isotherms resulted in a step-wise isotherm associated with non-existing pores filled in a sub-step to genuine pores.76 The pore size distributions of the Mn-doped catalysts were computed using the BJH method from the absorption isotherm curves, as shown in Fig. 3(b). All produced catalysts showed a narrow pore size distribution from 5 to 20 nm. Yang et al.77 found that the produced Mn2O3 had a spongy nature and was extremely porous, with a large surface area, resulting in improved catalytic activity. Atmuri et al.22,53 synthesized a series of Ce materials doped with varying mol% of Mn, ranging from 0 to 100. The surface area for Mn-doped Ce varied between 12 and 50 m2 g−1. Mn3O4 exhibited a minimal surface area of 12 m2 g−1, while the samples with 5 mol% Mn and 60 mol% Mn-doped Ce demonstrated a significantly higher surface area of 50 m2 g−1. A linear correlation can be established between surface area and activity for BET surface areas less than 25 m2 g−1, regarded as a threshold for surface area.6
According to Krishna et al.,18 the BET surface area of CeO2 primarily originates from the micropores, which is difficult for soot particles to penetrate. The incorporation of rare-earth metals enhanced the meso/macro pore volume and the external surface area of CeO2.18
 | ||
| Fig. 4 FE-SEM analysis of Mnx(x=0–0.2)(Ce0.9Pr0.1)1−xO2−δ catalysts (200 nm). | ||
3.2. Redox property of the catalysts
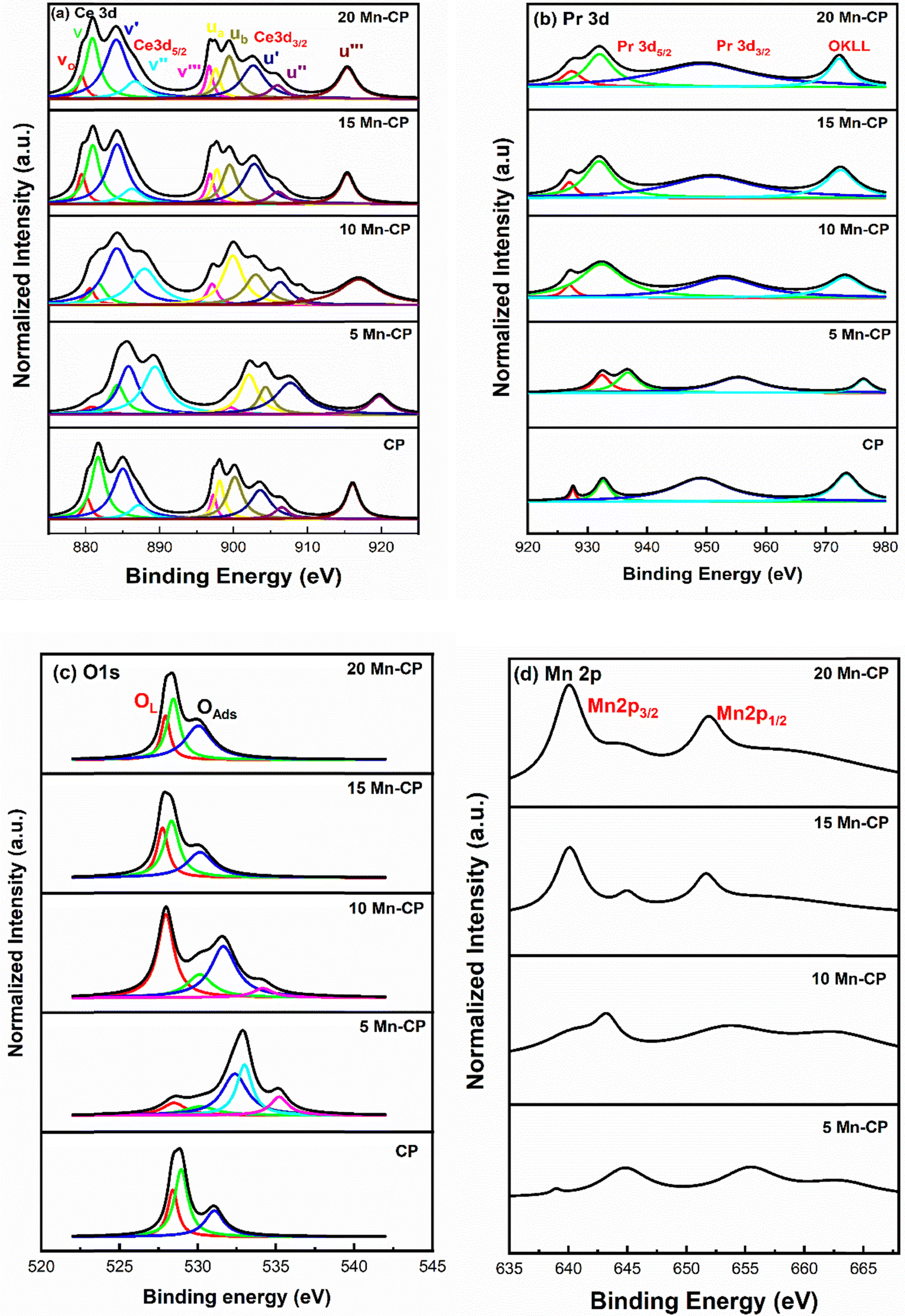 | ||
| Fig. 5 (a) Ce 3d, (b) Pr 3d, (c) O1s and (d) Mn 2p XPS spectra for the Mnx(x=0-0.2)(Ce0.9Pr0.1)1−xO2−δ catalysts. | ||
The presence of surface oxygen species is crucial for improving catalytic efficiency in soot oxidation reactions. The O1s spectra reveal the information corresponding to the lattice oxygen (OL) species and the surface-adsorbed oxygen species (OAds).90,91,93 From the O1s spectra of Mn-CP catalysts (Fig. 5c), the lattice oxygen peak was seen around 528.3 eV in CP and 5 and 10 Mn-CP; however, it shifted slightly to a lower binding energy of roughly 527.9 eV in the 15 and 20 Mn-CP.
The O1s spectra were analyzed for Co-doped transition metals such as Fe, Mn, and Cr, and the spectra were fitted into two distinct component peaks within the ranges of 529.7 to 531.5 eV (lattice oxygen) and 532.7 to 533.5 eV (surface adsorbed oxygen). O1s core level spectra of various ranges of Ce-based catalysts with dopants, including rare earth materials and transition metals, were studied by Mukherjee et al.7 The peak identified at approximately 530.2 eV corresponds to lattice oxygen (Oα). In contrast, the peaks detected from 531.9 eV to 533.5 eV include adsorbed oxygen species, which comprise surface adsorbed oxygen (Oβ), hydroxyls, chemisorbed water, and carbonates (Oλ).7 Surface oxygen encompasses O2− (superoxide) and O22− (peroxide) intermediates, which develop on the surface of CeO2 when gaseous oxygen is gradually integrated into lattice oxygen.7 Diez et al.83 reported that the peak at lower binding energy (528.7 eV) is attributable to lattice oxygen, whereas the peak at higher binding energy (above 530 eV) is due to oxygen species formed by water interaction with the ceria surface.
He et al.94 indicated that the lattice oxygen species referred to as O2− exhibit a peak at approximately 528.5 to 529 eV.94,95 The other peaks are assigned as follows: surface adsorbed oxygen species O− and O2− which is generally observed at 529.8 to 530.2 eV, the carbonates (CO3−) and hydroxyl species (OH−) observed around 530.9–531.2 eV, and the latter peaks are dedicated as adsorbed molecular water (H–O–H). Fig. 5c shows that 5 Mn-CP and 10 Mn-CP showed the additional oxygen species at ∼534 eV, and the binding energy at 534 eV is assigned to the adsorbed H2O or adsorbed molecular water.96
Li et al.84 studied the O1s XPS spectra and demonstrated that the oxygen species are divided into six types. The Mn–Ce on carbon nanotube catalyst has binding energies of 529.6 and 530.3 eV, which correspond to the lattice oxygen of Ce–O and Mn–O, respectively. The peaks at 531.1 and 532.2 eV correspond to surface hydroxyls and adsorbed molecular water, respectively. The signal at 531.6 eV indicates a double bond between oxygen and carbon (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) from a carbonyl group, while the peak at 533.5 eV indicates a single bond. Paunović et al.88 also investigated O1s peaks for Pr-doped CeO2 samples and identified the peak corresponding to lower binding energy at 529.2 eV, while the peak associated with higher binding energy was found at 531.1 eV. With Pr doping, an increase in the higher binding energy peak was observed. Dimitrov et al.97 indicated that the O1s chemical shift corresponds to the varying degree of ionicity in the M–O bonds. The reduction in binding energy in the XPS spectra of simple oxides can be attributed to an enhancement in the electron charge density of the oxide ions, resulting from an increase in their electronic polarizability.97 Bonding an element to a different element causes a chemical change in its core levels compared to bonding with the same element. In metal oxides, metal atoms link with the more electronegative oxygen atom, resulting in greater binding energies at the core levels compared to the pure metallic state.98 It was also indicated by Mukherjee et al.7 that the lattice oxygen's surrounding environment in doped CeO2 differs from the undoped one, which is attributed to the variation in electronegativity between the dopant and CeO2. This leads to a variation in the peak position to varying degrees and in their study, the lattice oxygen peak of Ce–Mn had the lowest binding energy, demonstrating the existence of loosely bound lattice oxygen.7
O) from a carbonyl group, while the peak at 533.5 eV indicates a single bond. Paunović et al.88 also investigated O1s peaks for Pr-doped CeO2 samples and identified the peak corresponding to lower binding energy at 529.2 eV, while the peak associated with higher binding energy was found at 531.1 eV. With Pr doping, an increase in the higher binding energy peak was observed. Dimitrov et al.97 indicated that the O1s chemical shift corresponds to the varying degree of ionicity in the M–O bonds. The reduction in binding energy in the XPS spectra of simple oxides can be attributed to an enhancement in the electron charge density of the oxide ions, resulting from an increase in their electronic polarizability.97 Bonding an element to a different element causes a chemical change in its core levels compared to bonding with the same element. In metal oxides, metal atoms link with the more electronegative oxygen atom, resulting in greater binding energies at the core levels compared to the pure metallic state.98 It was also indicated by Mukherjee et al.7 that the lattice oxygen's surrounding environment in doped CeO2 differs from the undoped one, which is attributed to the variation in electronegativity between the dopant and CeO2. This leads to a variation in the peak position to varying degrees and in their study, the lattice oxygen peak of Ce–Mn had the lowest binding energy, demonstrating the existence of loosely bound lattice oxygen.7
The XPS spectra of Mn 2p (Fig. 5d) also consist of two groups, 2p1/2 and 2p3/2.48,71 According to the literature, the Mn 2p spectra may be deconvoluted into four peaks at ∼642.35 eV and 643.70 eV (belonging to Mn 2p3/2), 653.95 eV, and 655.30 eV (refers to Mn 2p1/2). The peak observed at ∼660 eV is assigned as a satellite peak.89,99 The binding energies of Mn2p3/2 at 640.9, 641.8, and 642.5 for MnO, Mn2O3, and MnO2, respectively, are too close to allow for a clear differentiation.99 Even in the present spectra, the peaks of Mn species overlap, it is difficult to effectively differentiate the valence states and measure the Mn species.
The area under the curve was utilized to compute the reducibility ratio [(Ce3+/(Ce3+ + Ce4+); Pr3+/(Pr3+ + Pr4+)], and the surface oxygen species ratio listed in Table 2. The table shows that CP showed a high reducibility ratio for Ce3+ (0.42), and 5, 10 Mn-CP revealed a high concentration of Pr3+ (0.43) compared to all the Mn-doped catalysts. Thus, the presence of Pr contributed to the enhancement of Ce3+ species on the surface. Furthermore, doping with modest levels of Mn increased surface Pr3+ ions.
| Catalyst | Reducibility ratio (Ce3+ and Pr3+) | Lattice oxygen (OL) | Adsorbed oxygen species (OAds) | ||
|---|---|---|---|---|---|
| Ce3+ | Pr3+ | OL | OSAds | H–O–H | |
| CP | 0.42 | 0.15 | 0.26 | 0.74 | — |
| 5 Mn-CP | 0.25 | 0.43 | 0.10 | 0.79 | 0.11 |
| 10 Mn-CP | 0.39 | 0.43 | 0.39 | 0.56 | 0.05 |
| 15 Mn-CP | 0.33 | 0.32 | 0.23 | 0.77 | — |
| 20 Mn-CP | 0.32 | 0.30 | 0.27 | 0.73 | — |
The ratios for lattice oxygen (OL, 528.3 to 527.9 eV) and the surface adsorbed oxygen (OSAds, 530.1 to 532.9 eV) along with adsorbed molecular water (H–O–H, 534.9 to 535.1 eV) are presented in Table 2. Adding Mn to Ce–Pr also significantly enhanced the concentration of surface oxygen species. It is observed that 10 Mn-CP exhibited the highest (0.39) lattice oxygen (OL) species, whereas 5 Mn-CP demonstrated a marginally greater quantity (0.79) of surface adsorbed oxygen species (OSAds).
Generally, Ce-based catalysts express more than a single form of oxygen ion species,100 and the active oxygen species obtained near binding energies of 530–532 eV play a critical role in oxidation reactions.7,93,95 The surface-active oxygen species (O22− and O−) are also critical in improving catalytic activity at high temperatures101 whereas the presence of lattice oxygen species (O2−) plays a significant role in catalytic soot oxidation if the surface area is low.16 In many instances, both oxygen species play a vital role in the soot oxidation reactions. However, the species that are adsorbed on the surface are actively involved in soot oxidation, and hence, a greater presence of surface-adsorbed oxygen suggests enhanced soot oxidation activity.7
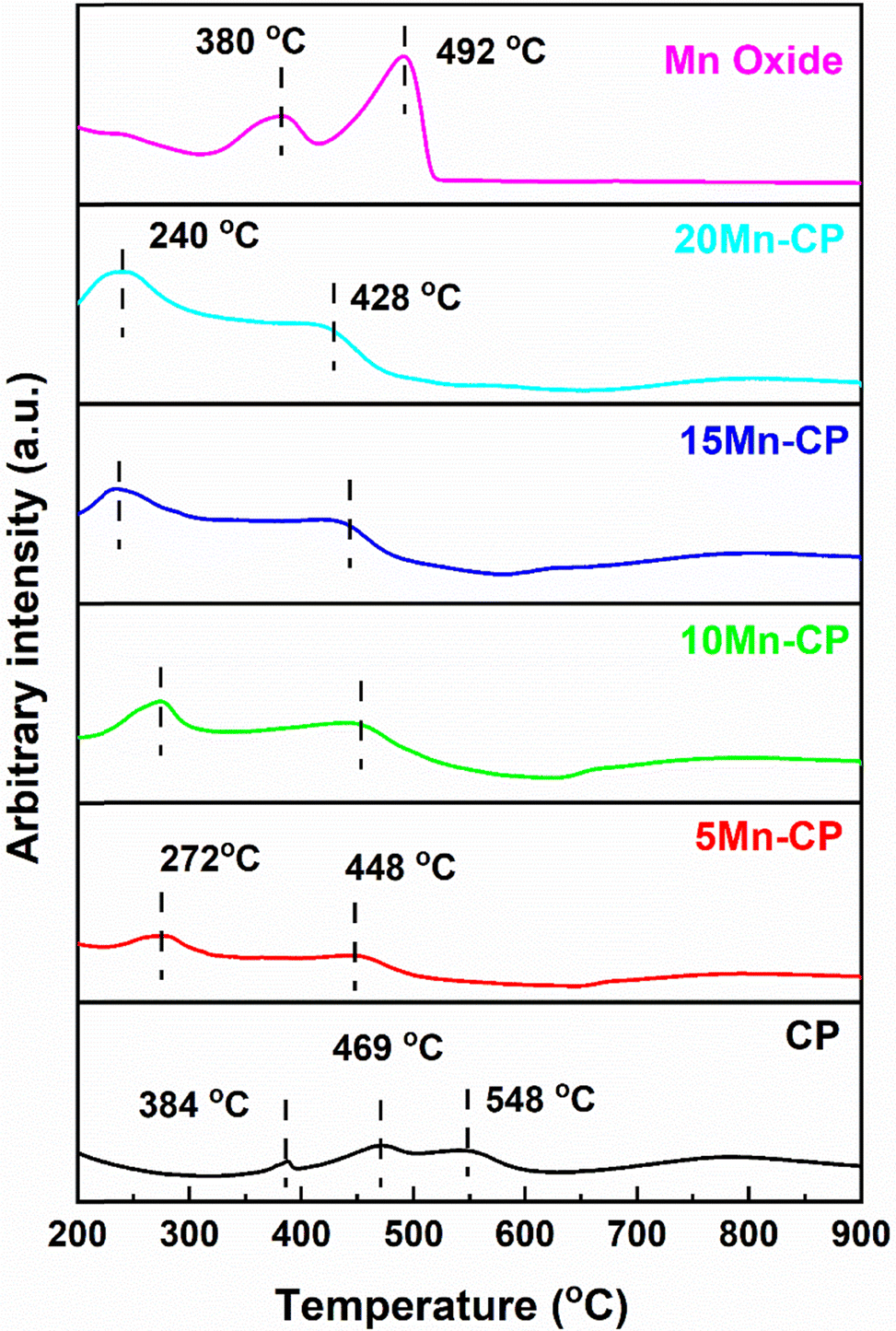 | ||
| Fig. 6 H2-TPR profiles of Mn(x=5–20)(Ce0.9Pr0.1)O2−δ catalysts. | ||
For CeO2, the low-temperature peak indicates the reduction of surface Ce4+ ions, and higher temperature contributes to the reduction of surface and lattice oxygen.105,106 According to Mukherjee et al.,7 CeO2 reduction is anticipated to occur gradually. At temperatures below 527 °C, the outermost layers of Ce4+ undergo surface reduction, followed by bulk reduction at higher temperatures ∼777 °C. They discovered a low-temperature reduction pattern for Ce–Mn due to solid solution formation and interactions between Mn–O and Ce–O. This increases oxygen mobility from the bulk to the surface, resulting in greater sites on the surface for hydrogen adsorption.7 Jan et al.104 studied H2-TPR analysis for pure CeO2 and platinum-doped CeO2 catalysts. Pure ceria demonstrates a two-step reduction process characterized by peaks at 540 °C and 820 °C, which are associated with the reduction of surface oxygen and bulk oxygen of ceria, respectively. Bulk oxygen exhibits a stronger bond with CeO2 than surface oxygen; thus, it reacts with H2 at very high temperatures. The reactant hydrogen gas is adsorbed onto platinum, which transfers the adsorbed hydrogen to the surface of cerium oxide, thereby promoting the reduction of oxygen.104
Mn2O3 has two reduction peaks, first changing into Mn3O4 and then MnO. Typically. Mn2O3 shows a reduction peak at 288 and 385 °C. Zhang et al.107 conducted H2-TPR studies on α-, β-, γ-, and δ-MnO2 catalysts. Gong et al.108 found that α-Mn2O3 exhibited reduction peaks at 378 °C and 481 °C, while γ-Mn2O3 showed reduction at 367 °C and 466 °C. These peaks were consistent with the current study's findings for pure Mn oxide. The reduction peaks for CP (at 384 and 469 °C) shifted to lower temperatures for all Mn-doped CP catalysts, showing increased reduction capacity and oxygen species mobility. At lower temperatures, the active oxygen species increase and are reduced by H2.109 Hence, including Mn and Pr eases the reducibility and doping of Mn and Pr with Ce, allowing oxygen to circulate more freely. Table 3 presents the total hydrogen consumption at reduction peaks, and it can be seen that Mn oxide displays high consumption. For catalytic activity, the H2 consumption at a reduction temperature below 450 °C is considered, and the order among doped catalysts is as follows: CP < 5 Mn-CP < 10 Mn-CP < 15 Mn-CP < 20 Mn-CP indicates that doping Mn and Pr facilitates the breakage of Ce–O.
| Catalysts | Reduction peak temperature (°C) | Total H2 consumption (mmol g−1) | Desorption peak temperature (°C) | Total O2 desorption (mmol g−1) | ||||
|---|---|---|---|---|---|---|---|---|
| Peak | Peak | Peak | Peak | Peak | Peak | |||
| 1 | 2 | 3 | 1 | 2 | 3 | |||
| CP | 384 (0.053) | 469 (0.203) | 548 (0.192) | 0.448 | 402 (0.002) | 658 (0.032) | — | 0.034 |
| 5 Mn-CP | 272 (0.287) | 448 (0.127) | — | 0.414 | 247 (0.016) | 431 (0.021) | 759 (0.019) | 0.039 |
| 10 Mn-CP | 274 (0.478) | 450 (0.237) | — | 0.715 | 248 (0.009) | 441 (0.010) | 775 (0.015) | 0.034 |
| 15 Mn-CP | 236 (0.496) | 442 (0.355) | — | 0.815 | 252 (0.010) | 439 (0.015) | 758 (0.037) | 0.062 |
| 20 Mn-CP | 240 (0.792) | 428 (0.398) | — | 1.190 | 250 (0.008) | 434 (0.017) | 745 (0.088) | 0.113 |
| Mn oxide | 380 (3.831) | 492 (11.283) | — | 15.11 | — | — | 831 (0.836) | 0.836 |
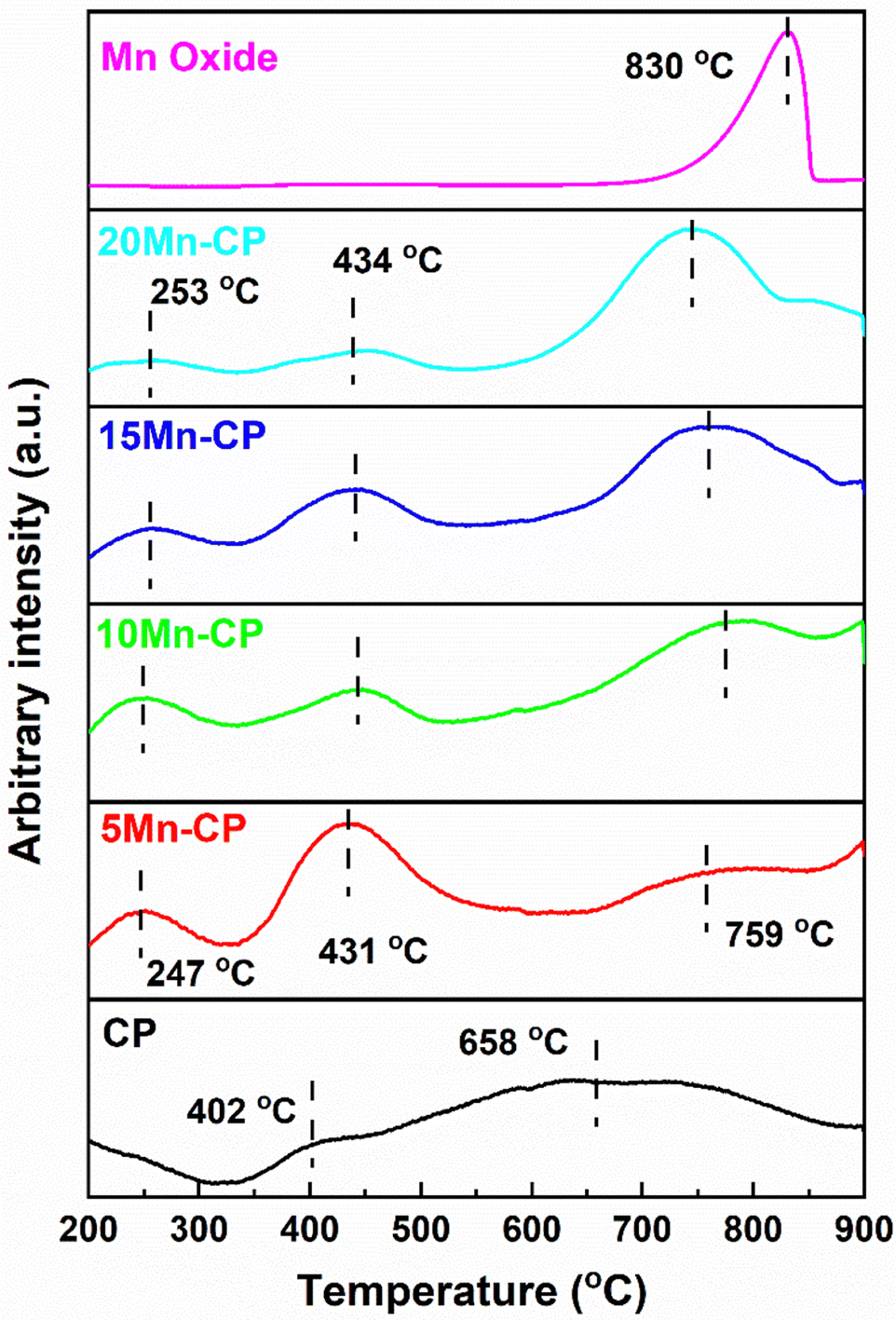 | ||
| Fig. 7 O2-TPD profiles of Mn(x=5–20)(Ce0.9Pr0.1)O2−δ catalysts. | ||
Pure Mn oxide displayed strong desorption peaks related to lattice oxygen species (O2−) at 830 °C. This peak is shifted to the lower temperature of Mn-doped-CP catalysts, indicating enhanced O2 mobility. Ma et al.111 reported the desorption peaks at 327, 594, and 654 °C for the Mn-doped Ce catalyst, indicating that it has better oxygen mobility than pure CeO2 and MnOx. According to Wei et al.,112 the peak identified between 650 and 850 °C results from the reduction of the internal Ce4+ layer and lattice oxygen. Cerium-rich catalysts exhibit enhanced efficacy in transferring oxygen from the lattice to soot surfaces over an extensive temperature range. This is attributable to the elevated mobility of lattice oxygen. Zhao et al.113 assert that incorporating Ni into Co3O4 induces structural distortion, enhances oxygen vacancy density, and augments lattice oxygen mobility. It generates a greater quantity of surface-active oxygen species. The reducibility of a catalyst might signify its capacity to absorb or eliminate oxygen, referred to as the mobility of lattice oxygen, which is frequently linked to catalytic efficacy in soot oxidation.114
Soot oxidation typically takes place within the temperature range of 200 to 600 °C, indicating that the medium temperature range associated with active oxygen is crucial. Table 3 provides O2 desorption at each peak, and it reveals that 5 Mn-CP displayed higher O2 desorption of 0.037 mmol g−1 and 10 Mn-CP displayed low O2 desorption of 0.019 mmol g−1 for surface adsorbed oxygen species, which is consistent with XPS analysis, however, the conditions were different for lattice and bulk oxygen species.
3.3. Soot oxidation activity and activation energy (Ea) determination by the FWO method
Fig. 8 displays the soot conversion curves with the temperature increase. The T50 temperature is recorded for each catalyst and is shown in Table 4. The table shows that adding Mn to the CP catalyst system resulted in a considerable drop in T50. Mn-loaded CP catalysts had a higher surface area and T50 values ranging from 365 to 395 °C. The T50 for the bare CP catalyst was noticed around 408 ± 4 °C,34 whereas the 5 Mn-CP catalyst had the lowest T50 at 365 ± 1 °C, with Mn displaying a higher T50 at 433 ± 1 °C. 5-Mn CP has the highest surface area (SA) of 45 m2 g−1. Replacing greater ionic radii “Ce” with a smaller ionic radius “Mn” in the fluorite structure would reduce the lattice constant while boosting catalytic activity.115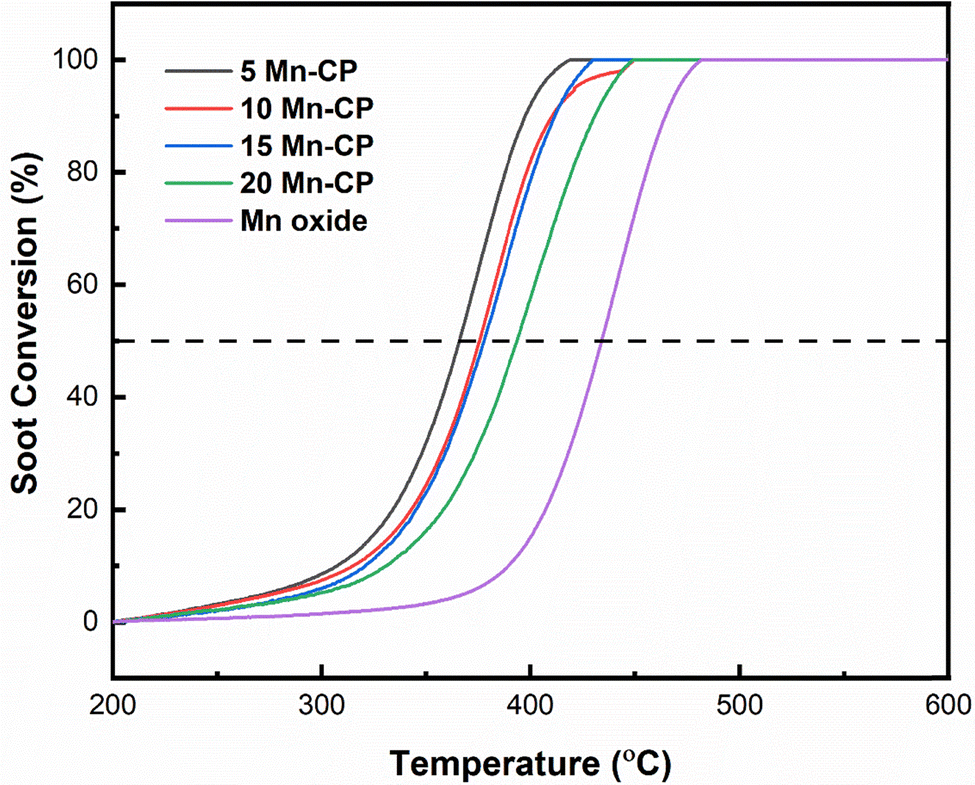 | ||
| Fig. 8 Soot oxidation activity of Mn(x=5–20)(Ce0.9Pr0.1)O2−δ catalysts. | ||
| Catalysts | Synthesis method | T 50 (°C) | E a (kJ mol−1) FWO method | Ref. |
|---|---|---|---|---|
| CP | Solution combustion synthesis | 408 ± 4 | 111 | This study |
| 5 Mn-CP | 365 ± 1 | 94 | ||
| 10 Mn-CP | 372 ± 2 | 138 | ||
| 15 Mn-CP | 377 ± 2 | 143 | ||
| 20 Mn-CP | 395 ± 3 | 139 | ||
| Mn oxide | 433 ± 1 | 151 | ||
| Ce0.95Mn0.05 | EDTA citrate method | 360 | 118 | 53 |
| Ce0.5Mn0.5O2 | Sol–gel method | 383 | 24.86 (Coats–Redfern integral method) | 116 |
| Mn doped Ce0.5Zr0.5O2 | Incipient wetness impregnation method | 360 | 117 | |
| Ce0.95Mn0.05 | Hydrothermal synthesis | 451 | 118 | |
| Ce0.95Mn0.025Cu0.025 | 516 | |||
| CeO2–MnO2 | Coprecipitation method | 396 | 119 | |
| CeO2–MnO2/TiO2 | 298 | |||
| Ceria-manganese | Coprecipitation method | 396 | 7 | |
| Ceria-praseodymium | 438 | |||
| Ce0.9Pr0.1 | Solid-phase grinding method | 398 | 110 | |
| Ce0.9Mn0.1 | 389 | |||
| Ce0.9Pr0.1 | Sol–gel method | 414 | ||
| Ce0.9Mn0.1 | 399 | |||
| Mn-doped ceria | Coprecipitation method | 484 | 120 | |
| Mn–Co doped ceria | 363 |
The activation energy (Ea) is determined by plotting Ozawa plots (Fig. S1) and is provided in Table 4. It reveals a modest variance in Ea values and activation energy for Mn-doped Ce catalysts, ranging from 94 to 151 kJ mol−1. Pure Mn oxide (151 ± 3 kJ mol−1) had a somewhat higher Ea value than Mn-doped catalysts. 5 Mn-CP had the lowest Ea value (94 ± 1 kJ mol−1) among all Mn-doped CP catalysts. The uncatalyzed soot oxidation activation energy values range from 150 to 160 kJ mol−1.4,18Table 4 also contains existing literature of various Mn-doped Ce-based catalysts synthesized by various techniques and their T50 temperatures for soot oxidation reactions. The lowest T50 of 298 °C was reported for CeO2–MnO2/TiO2 produced via co-precipitation.119 CeO2–MnO2/TiO2 exhibits much higher bulk and surface defects, likely because of the CeO2/TiO2 interface, which leads to defect formation.119 Shourya and Dasari53 reported soot oxidation activity for Mn-doped Ceria, and the improved catalytic activity of Ce0.95Mn0.05 was related to its increased surface area, which raises the number of reactive sites. Huang et al.116 reported that the Ce–Mn crystal structure is more stable, while Mn doping into Ce increases lattice defects, and with increasing Mn concentrations, the oxygen vacancy formation energy decreases. Mukherjee et al.7 investigated the effect of transition metals (Zr, Hf, Fe, Mn) and rare earth metals (La, Pr) as dopants in Ceria on soot and CO oxidation. Mn doped ceria exhibited better CO and soot oxidation activity, principally due to a significant reduction in lattice oxygen binding energy and a larger concentration of surface adsorbed oxygen species.7 Zhou et al.110 reported that the lattice distortion of CeO2 occurs when Pr is introduced; lattice defects create oxygen vacancies, which aid in the adsorption, movement, and desorption of oxygen. Rao et al.120 investigated the effect of manganese and cobalt co-doped ceria on soot oxidation activity. Their H2-TPR analysis revealed that the codoping of ceria remarkably increases the redox behaviour of ceria, as well as surface area and enhanced surface adsorbed oxygen species. Govardhan et al.86 indicated that Ag/Pr–Ce catalysts exhibit high porosity, which is associated with an increased surface area and pore diameter, enhancing their activity for the oxidation reaction. The elevated concentration of surface Ce3+ and the presence of surface chemisorbed oxygen in the 5 Ag/Pr–Ce and 15 Ag/Pr–Ce catalysts played a significant role in enhancing soot oxidation performance. Similarly, the lattice defects and the oxygen vacancies served as crucial descriptors in demonstrating the enhanced soot oxidation activity of the 10 Ni–Pr–Ce catalyst in the studies conducted by Rajvanshi et al.12 The role of Pr appears to contribute to the enhancement of the catalyst's properties. The inclusion of transition metals further enhances the improved surface parameters.
The descriptors for Ce-based metal oxide that enhance the catalytic performance include the redox ability, the host structure, lattice oxygen species, site isolation, the nature of the metal–oxygen bond, multifunctionality, and phase cooperation.15 In the case of ceria-based materials, it is not feasible to fixate on a single descriptor; rather, multiple descriptors govern the activity, influenced by the differing reaction conditions. The present investigation demonstrates that the addition of Mn to the Ce–Pr catalyst system improves the catalytic oxidation of soot.
In this study, the catalysts exhibited surface areas varying from 45 to 15 m2 g−1, with 5 Mn CP demonstrating the highest surface area of 45 m2 g−1. 5 Mn-CP showed a higher presence of active surface adsorbed species, as demonstrated by XPS and O2-TPD analysis which influenced catalytic performance displaying the least T50 of 365 ± 1 °C. Whereas 10 Mn-CP which had a very slight decrease in T50 of 372 ± 2 °C revealed the highest IOv/IF2g value of 0.78, along with lattice oxygen species as examined through Raman spectroscopy and XPS. CP exhibited a significant reducibility ratio for Ce3+, while the 5 and 10 Mn-CP samples demonstrated a notable concentration of Pr3+. Therefore, the presence of Pr played a significant role in enhancing the Ce3+ species on the surface and moderate amounts of Mn enhanced the presence of surface Pr3+ ions. Furthermore, the H2-reduction peaks of 15 Mn-CP and 20 Mn-CP exhibited a shift to lower temperatures, suggesting improved reducibility. Conversely, 20 Mn-CP exhibited the smallest crystallite size along with the largest lattice strain. However, with an increase in the Mn concentration within the Mn-CP catalytic system, a secondary phase emerges, leading to a reduction in activity.
4. Conclusions
Mnx(x=0–0.2)(Ce0.9Pr0.1)(1−x)O2−δ and pure Mn oxide catalysts were effectively produced using the SCS approach. The XRD and Raman analyses demonstrated the fluorite structure of ceria, with the crystallite size for Mn-CP catalysts measured between approximately 5 to 10 nm. 10 Mn-CP exhibited a slightly elevated oxygen vacancy ratio (IOv/IF2g) of 0.78. The catalysts exhibited significant porosity, and the particle size for pure Mn oxide was notably large, as evidenced by the SEM micrographs. The catalysts showed surface areas ranging from 45 to 15 m2 g−1, with 5 Mn CP displaying the maximum surface area of 45 m2 g−1. CP exhibited superior reducibility when compared to all Mn-doped CP catalysts that had a high concentration of Ce3+ surface ions. Conversely, 5 Mn-CP exhibited superior active surface adsorbed oxygen species, which are essential for the oxidation of soot. The 5 Mn-CP catalyst demonstrated enhanced catalytic activity, with a T50 of 365 ± 1 °C for the soot oxidation reaction. The factors influencing the soot oxidation activity in the current investigation include surface area, crystallite size, and the presence of active surface adsorbed oxygen species. As the Mn content in the Mn-CP catalytic system rises, a secondary phase emerges, leading to a reduction in soot oxidation activity. 5 Mn-CP showed the lowest activation energy value of 94 kJ mol−1 among all the Mn-doped catalysts, whereas Mn2O3 exhibited a slightly higher activation energy value in comparison to the Mn-doped catalysts.Author contributions
Sunaina S Patil: writing – original draft preparation, review, and editing, resources and methodology, formal analysis, and investigation; Hari Prasad Dasari: conceptualization, formal analysis and investigation, review, supervision, funding acquisition; Rahulkumar Shirasangi: H2-TPR and O2-TPD methodology and investigation. Harshini Dasari: formal analysis investigation, and review.Data availability
The data can be provided and made available upon request.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
The current study is funded by a Science and Engineering Research Board (SERB), India – Core Research Grant (CRG/2020/000425). The authors greatly acknowledge Central Research Facility (CRF), NITK, and Surathkal for facilitating XRD analysis, Raman spectroscopy, FE-SEM analysis, BET-BJH analysis, and H2-TPR/O2-TPD data. The authors sincerely thank Material Research Facility (MRC), MNIT, Jaipur for providing XPS data.References
- B. A. A. L. Van Setten, M. Makkee and J. A. Moulijn, Catal. Rev.: Sci. Eng., 2001, 43, 489–564 CrossRef CAS.
- M. P. A. Vijay, S. S. Patil, D. R. Madhura, A. P. Anantharaman, P. Gouramma, H. P. Dasari, S. B. Arya and H. Dasari, Mater. Today: Proc., 2022, 57, 1865–1870 CAS.
- A. P. Anantharaman, H. J. Gadiyar, M. Surendran, A. S. Rao, H. P. Dasari, H. Dasari and G. U. Babu, Chem. Pap., 2018, 72, 3179–3188 CrossRef CAS.
- C. S. Shenoy, S. S. Patil, P. Govardhan, A. Shourya and H. P. Dasari, Emiss. Control Sci. Technol., 2019, 5, 342 CrossRef CAS.
- S. Ganiger, S. S. Patil, H. P. Dasari, R. Priyanka and S. Kollimarla, Chem. Eng. Sci., 2022, 247, 117016 CrossRef CAS.
- L. I. U. Shuang, W. U. Xiaodong, W. Duan and R. A. N. Rui, J. Rare Earths, 2015, 33, 567–590 CrossRef.
- D. Mukherjee, B. G. Rao and B. M. Reddy, Appl. Catal., B, 2016, 197, 105–115 CrossRef CAS.
- B. M. Reddy, P. Bharali and G. Thrimurthulu, Catal. Lett., 2008, 123, 327–333 CrossRef CAS.
- S. S. Patil, R. Kumar and H. P. Dasari, J. Taiwan Inst. Chem. Eng., 2024, 105459 CrossRef CAS.
- G. Thrimurthulu, K. N. Rao and D. Devaiah, et al. , Res. Chem. Intermed., 2012, 38, 1847–1855 CrossRef CAS.
- S. S. Patil, H. Prasad and H. Dasari, Nano-Struct. Nano-Objects, 2019, 20, 100388 CrossRef CAS.
- K. Rajvanshi, S. S. Patil, Lakhanlal, H. P. Dasari, M. B. Saidutta and H. Dasari, Chem. Pap., 2020, 74, 4581–4592 CrossRef CAS.
- S. S. Patil, S. Naik, M. D. Ramesh, H. Dasari and H. P. Dasari, Chem. Eng. Technol., 2022, 45(3), 517–525 CrossRef CAS.
- A. P. Anantharaman, H. P. Dasari, H. Dasari and G. U. B. Babu, Appl. Catal., A, 2018, 566, 181–189 CrossRef CAS.
- R. K. Grasselli, Top. Catal., 2002, 21, 79–88 CrossRef CAS.
- Q. Liang, X. Wu, X. Wu and D. Weng, Catal. Lett., 2007, 119, 265–270 CrossRef CAS.
- S. Liu, X. Wu, W. Liu, W. Chen, R. Ran, M. Li and D. Weng, J. Catal., 2016, 337, 188–198 CrossRef CAS.
- K. Krishna, A. Bueno-López, M. Makkee and J. A. Moulijn, Appl. Catal., B, 2007, 75, 189–200 CrossRef CAS.
- M. Piumetti, S. Bensaid, N. Russo and D. Fino, Appl. Catal., B, 2015, 165, 742–751 CrossRef CAS.
- J. Gao, Y. Wang, S. Wang, X. Li, X. Chang, X. Wang, C. Yang and R. Xuan, Chem. Eng. J., 2022, 443, 136392 CrossRef CAS.
- E. Aneggi, C. De Leitenburg, G. Dolcetti and A. Trovarelli, Catal. Today, 2006, 114, 40–47 CrossRef CAS.
- A. Shourya and H. P. Dasari, Nano-Struct. Nano-Objects, 2023, 34, 100970 CrossRef CAS.
- K. Kim, J. Do Yoo, S. Lee, M. Bae, J. Bae, W. C. Jung and J. W. Han, ACS Appl. Mater. Interfaces, 2017, 9, 15449–15458 CrossRef CAS PubMed.
- Z. Hou, W. Pei, X. Zhang, K. Zhang, Y. Liu, J. Deng and L. Jing, J. Rare Earths, 2020, 38, 819–839 CrossRef CAS.
- P. Dulgheru and J. A. Sullivan, Top. Catal., 2013, 56, 504–510 CrossRef CAS.
- H. P. Dasari, J. S. Ahn, K. Ahn, S. Y. Park, J. Hong, H. Kim, K. J. Yoon, J. W. Son, H. W. Lee and J. H. Lee, Solid State Ionics, 2014, 263, 103–109 CrossRef CAS.
- H. P. Dasari, S. Y. Park, H.-I. Ji, H.-R. Kim, J.-W. Son, B.-K. Kim, H.-W. Lee and J.-H. Lee, J. Phys. Chem. C, 2012, 116, 3467–3476 Search PubMed.
- T. Vinod Kumar, D. Mukherjee, C. Subrahmanyam and B. M. Reddy, New J. Chem., 2018, 42, 5276–5283 RSC.
- A. Hernández-Giménez A, L. Xavier and A. Bueno-López, Appl. Catal., A, 2013, 462–463, 100–106 CrossRef.
- D. N. Durgasri, T. Vinodkumar, F. Lin, I. Alxneit and B. M. Reddy, Appl. Surf. Sci., 2014, 314, 592–598 CrossRef CAS.
- P. Sudarsanam, K. Kuntaiah and B. M. Reddy, New J. Chem., 2014, 38, 5991–6001 RSC.
- A. P. Anantharaman, J. Geethu, M. R. Rahul, H. P. Dasari and H. Dasari, Mol. Catal., 2018, 451, 247–254 CrossRef CAS.
- E. Aneggi, C. De Leitenburg, G. Dolcetti and A. Trovarelli, Top. Catal., 2007, 42–43, 319–322 CrossRef.
- S. S. Patil and H. P. Dasari, Braz. J. Chem. Eng., 2023, 41, 269–285 CrossRef.
- S. S. Patil and H. P. Dasari, Environ. Sci. Pollut. Res., 2024 DOI:10.1007/s11356-024-35652-1.
- M. Machida, Y. Murata, K. Kishikawa, D. Zhang and K. Ikeue, Chem. Mater., 2008, 20, 4489–4494 CrossRef CAS.
- T. Vinodkumar, D. Durgasr Naga and B. M. Reddy, Int. J. Adv. Eng. Sci., 2013, 5, 224–231 Search PubMed.
- A. A. Khaskheli, L. Xu and D. Liu, Energy Fuels, 2022, 36(4), 7219–7920 Search PubMed.
- Q. Liang, X. Wu, D. Weng and H. Xu, Catal. Today, 2008, 139, 113–118 CrossRef CAS.
- S. K. Ghosh, ACS Omega, 2020, 5, 25493–25504 CrossRef CAS PubMed.
- M. Fu, J. Lin, W. Zhu, J. Wu, L. Chen, B. Huang and D. Ye, J. Rare Earths, 2014, 32, 153–158 CrossRef CAS.
- S. D. Neelapala, A. Shetty, G. Gaggar, R. Mall and H. Dasari, Int. J. Appl. Eng. Res., 2018, 13, 245–251 Search PubMed.
- Y. Kuwahara, G. Kato, A. Fujibayashi, K. Mori and H. Yamashita, Chem. – Asian J., 2020, 15, 2005–2014 CrossRef CAS.
- P. Venkataswamy, D. Jampaiah, D. Mukherjee, C. U. Aniz and B. M. Reddy, Catal. Lett., 2016, 146, 2105–2118 CrossRef CAS.
- H. Zhang, J. Wang, Y. Cao, Y. Wang, M. Gong, Y. Chen and C. Xuebao, Chin. J. Catal., 2015, 36, 1333–1341 CrossRef CAS.
- X. Yue, D. Ye, M. Fu, J. Wu, J. Ouyang, B. Huang and H. Liang, Catal. Today, 2010, 153, 125–132 CrossRef.
- A. B. Aberkane, M. P. Yeste, D. Fayçal, D. Goma and M. Á. Cauqui, Materials, 2019, 12(20), 3436 CrossRef CAS PubMed.
- X. Niu, M. Li, B. Hao and H. Li, J. Mater. Sci.:Mater. Electron., 2016, 27, 6845–6848 CrossRef CAS.
- P. Palmisano, N. Russo, P. Fino, D. Fino and C. Badini, Appl. Catal., B, 2006, 69(1–2), 85–92 CrossRef CAS.
- S. Patil and H. P. Dasari, Mater. Sci. Energy Technol., 2019, 2, 485–489 Search PubMed.
- I. Shajahan, J. Ahn, P. Nair, S. Medisetti, S. Patil, V. Niveditha, G. Uday Bhaskar Babu, H. P. Dasari and J. H. Lee, Mater. Chem. Phys., 2018, 216, 136–142 CrossRef CAS.
- A. J. Zarur and J. Y. Ying, Nat. Lett., 2000, 403, 65–67 CrossRef CAS PubMed.
- A. Shourya and H. P. Dasari, Chem. Pap., 2022, 76(11), 7095–7110 CrossRef CAS.
- A. P. Anantharaman, H. P. Dasari, J. H. Lee, H. Dasari and G. U. B. Babu, Catal. Lett., 2017, 147, 3004–3016 CrossRef CAS.
- E. Kumar, P. Selvarajan and D. Muthuraj, Mater. Res., 2013, 16, 269–276 CrossRef CAS.
- A. Varma, A. S. Mukasyan, A. S. Rogachev and K. V. Manukyan, Chem. Rev., 2016, 116, 14493–14586 CrossRef CAS.
- A. Varma and A. S. Mukasyan, Korean J. Chem. Eng., 2004, 21, 527–536 CrossRef CAS.
- T. Ozawa, Thermochim. Acta, 1992, 203, 159–165 CrossRef CAS.
- B. M. Reddy, G. Thrimurthulu and L. Katta, Catal. Lett., 2011, 141, 572–581 CrossRef CAS.
- E. Aneggi, D. Wiater, C. De Leitenburg, J. Llorca and A. Trovarelli, ACS Catal., 2014, 4, 172–181 CrossRef CAS.
- A. S. Ryabova, S. Y. Istomin, K. A. Dosaev, A. Bonnefont, J. Hadermann, N. A. Arkharova, A. S. Orekhov, R. P. Sena, V. A. Saveleva, G. Kéranguéven, E. V. Antipov, E. R. Savinova and G. A. Tsirlina, Electrochim. Acta, 2020, 137378 Search PubMed.
- X. Niu, H. Wei, K. Tang, W. Liu, G. Zhao and Y. Yang, RSC Adv., 2015, 5, 66271–66277 RSC.
- B. C. Yadav, M. Singh and C. D. Dwivedi, Sens. Transducers J., 2011, 125(2), 68–75 CAS.
- R. Lo Nigro, R. G. Toro, G. Malandrino, V. Raineri and I. L. Fragalà, Adv. Mater., 2003, 15(3), 1071–1075 CrossRef CAS.
- M. Shiraishi and M. Inagaki, Carbon Alloys, 2003, 10, 161–173 Search PubMed.
- H. Chi, P. Zhang, J. Xiong, Y. Wei, Y. Li and Z. Zhao, Appl. Surf. Sci., 2023, 608, 155116 CrossRef CAS.
- E. Aneggi, C. de Leitenburg, J. Llorca and A. Trovarelli, Catal. Today, 2012, 197, 119–126 CrossRef CAS.
- H. Zhao, X. Zhou, M. Wang, Z. Xie, H. Chen and J. Shi, RSC Adv., 2017, 7, 3233–3239 RSC.
- J. G. Kang, B. K. Min and Y. Sohn, J. Alloys Compd., 2015, 619, 165–171 CrossRef CAS.
- T. S. Soliman, S. I. Elkalashy and M. M. Hessien, J. Mater. Sci.: Mater. Electron., 2024, 35, 2083 CrossRef CAS.
- R. Naeem, M. Ali Ehsan, R. Yahya, M. Sohail, H. Khaledi and M. Mazhar, Dalton Trans., 2016, 45, 14928–14939 RSC.
- Y. Xin, H. Cao, C. Liu, J. Chen, P. Liu, Y. Lu and Z. Ling, J. Raman Spectrosc., 2022, 53, 340–355 CrossRef CAS.
- I. El Arrouji, C. Chen, J. Toyir, C. Larabi, K. C. Szeto, A. de Mallmann, M. Taoufik and A. Oulmekki, Catalysts, 2021, 11(8), 950 CrossRef CAS.
- M. AlKetbi, K. Polychronopoulou, M. Abi Jaoude, M. A. Vasiliades, V. Sebastian, S. J. Hinder, M. A. Baker, A. F. Zedan and A. M. Efstathiou, Appl. Surf. Sci., 2020, 505, 144474 CrossRef CAS.
- R. Kumar, Surface Characterization Techniques, 2022, ISBN 978-3-11-065599-5 Search PubMed.
- J. C. Groen, L. A. A. Peffer and J. Pérez-Ramírez, Microporous Mesoporous Mater., 2003, 60, 1–17 CrossRef CAS.
- S. Yang, J. Wang, W. Chai, J. Zhu and Y. Men, Catal. Sci. Technol., 2019, 9, 1699–1709 RSC.
- S. T. Aruna and A. S. Mukasyan, Curr. Opin. Solid State Mater. Sci., 2008, 12, 44–50 CrossRef CAS.
- T. Drake, P. Ji and W. Lin, Acc. Chem. Res., 2018, 51, 2129–2138 CrossRef CAS.
- M. Mittal, A. Gupta and O. P. Pandey, Sol. Energy, 2018, 165, 206–216 CrossRef CAS.
- S. Hassan, R. Kumar, A. Tiwari, W. Song, L. van Haandel, J. K. Pandey, E. Hensen and B. Chowdhury, Mol. Catal., 2018, 451, 238–246 CrossRef CAS.
- J. P. Holgado, R. Alvarez and G. Munuera, Appl. Surf. Sci., 2000, 161, 301–315 CrossRef CAS.
- A. S. Diez, M. Graziano-Mayer, G. Radivoy and M. A. Volpe, Appl. Catal., A, 2014, 482, 24–30 CrossRef CAS.
- C. Li, Y. Sun and A. Zhang, RSC Adv., 2015, 5, 36394–36403 RSC.
- E. Poggio-Fraccari, G. Baronetti and F. Mariño, J. Electron Spectrosc. Relat. Phenom., 2018, 222, 1–4 CrossRef CAS.
- P. Govardhan, A. P. Anantharaman, S. S. Patil, H. P. Dasari, H. Dasari and A. Shourya, Korean J. Chem. Eng., 2022, 39, 328–342 CrossRef CAS.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy, 1992, ISBN: 0-9627026-2-5 Search PubMed.
- N. Paunović, Z. Dohcevic-Mitrovic, R. Scurtu, S. Aškrabić, M. Prekajski, B. Matović and Z. V. Popović, Nanoscale, 2012, 4, 5469–5476 RSC.
- M. Wang, K. Chen, J. Liu, Q. He, G. Li and F. Li, Catalysts, 2018, 8(4), 138 CrossRef.
- R. Patel, A. H. Fakeeha, S. O. Kasim, M. L. Sofiu, A. A. Ibrahim, A. E. Abasaeed, R. Kumar and A. S. Al-Fatesh, Mol. Catal., 2021, 510, 111676 CrossRef CAS.
- A. S. Al-Fatesh, R. Kumar, S. O. Kasim, A. A. Ibrahim, A. H. Fakeeha, A. E. Abasaeed, R. Alrasheed, A. Bagabas, M. L. Chaudhary, F. Frusteri and B. Chowdhury, Catal. Today, 2020, 348, 236–242 CrossRef CAS.
- N. Paunović, Z. Dohčević-Mitrović, R. Scurtu, S. Aškrabić, M. Prekajski, B. Matović and Z. V. Popović, Nanoscale, 2012, 4(17), 5469–5476 RSC.
- T. E. Jones, T. C. R. Rocha, A. Knop-Gericke, C. Stampfl, R. Schlögl and S. Piccinin, ACS Catal., 2015, 5, 5846–5850 CrossRef CAS.
- J. He, T. Wang, X. Bi, Y. Tian, C. Huang, W. Xu, Y. Hu, Z. Wang, B. Jiang, Y. Gao, Y. Zhu and X. Wang, Nat. Commun., 2024, 15, 5422 CrossRef CAS.
- T. E. Jones, T. C. R. Rocha, A. Knop-Gericke, C. Stampfl, R. Schlögl and S. Piccinin, ACS Catal., 2015, 5, 5846–5850 CrossRef CAS.
- L. V. Yafarova, G. V. Mamontov, I. V. Chislova, O. I. Silyukov and I. A. Zvereva, Catalysts, 2021, 11(10), 1256 CrossRef CAS.
- V. Dimitrov, T. Komatsu and R. Sato, J. Ceram. Soc. Jpn., 1999, 107(1241), 21–26 CrossRef CAS.
- D. N. G. Krishna and J. Philip, Appl Surf. Sci. Adv., 2022, 12, 100332 CrossRef.
- M. Kantcheva, M. U. Kucukkal and S. Suzer, J. Mol. Struct., 1999, 482–483, 19–22 CrossRef CAS.
- P. Sudarsanam, K. Kuntaiah and B. M. Reddy, New J. Chem., 2014, 38, 5991–6001 RSC.
- Y. Wei, J. Liu, Z. Zhao, A. Duan, G. Jiang, C. Xu, J. Gao, H. He and X. Wang, Energy Environ. Sci., 2011, 4, 2959–2970 RSC.
- J. Chen, X. Chen, X. Chen, W. Xu, Z. Xu, H. Jia and J. Chen, Appl. Catal., B, 2018, 224, 825–835 CrossRef CAS.
- N. Guillén-Hurtado, J. Giménez-Mañogil, J. C. Martínez-Munuera, A. Bueno-López and A. García-García, Appl. Catal., A, 2020, 590, 117339 CrossRef.
- A. Jan, J. Shin, J. Ahn, S. Yang, K. J. Yoon, J. W. Son, H. Kim, J. H. Lee and H. Il Ji, RSC Adv., 2019, 9, 27002–27012 RSC.
- J. He, H. Zhang, W. Wang, P. Yao, Y. Jiao, J. Wang and Y. Chen, Environ. Sci. Pollut. Res., 2021, 28, 26018–26029 CrossRef CAS PubMed.
- A. S. Nayak, S. S. Patil, H. P. Dasari, D. Telaginatot, M. Rynjah and S. Cheruku, Chem. Eng. Res. Des., 2024, 208, 910–920 CrossRef CAS.
- J. Zhang, Y. Li, L. Wang, C. Zhang and H. He, Catal. Sci. Technol., 2015, 5, 2305–2313 RSC.
- P. Gong, J. Xie, D. Fang, F. He, F. Li and K. Qi, Mater. Res. Express, 2017, 4, 115036 CrossRef.
- L. Fan, K. Xi, Y. Zhou, Q. Zhu, Y. Chen and H. Lu, RSC Adv., 2017, 7, 20309–20319 RSC.
- B. Zhou, K. Xi, L. J. Fan, Y. Zhou, Y. Wang, Q. L. Zhu and H. F. Lu, Appl. Catal., A, 2018, 562, 1–10 CrossRef CAS.
- J. Ma, X. Li, C. Zhang, Q. Ma and H. He, Appl. Catal., B, 2020, 264, 118498 CrossRef CAS.
- Y. Wei, J. Liu, Z. Zhao, A. Duan and G. Jiang, J. Catal., 2012, 287, 13–29 CrossRef CAS.
- M. Zhao, J. Deng, J. Liu, Y. Li and J. Liu, et al. , Catalysis, 2019, 9(8), 7548–7567 CAS.
- P. Yao, J. He, X. Jiang, Y. Jiao, J. Wang and Y. Chen, J. Energy Inst., 2020, 93, 774–783 CrossRef CAS.
- X. Wu, S. Liu, D. Weng, F. Lin and R. Ran, J. Hazard. Mater., 2011, 187, 283–290 CrossRef CAS.
- H. Huang, J. Liu, P. Sun, S. Ye and B. Liu, RSC Adv., 2017, 7, 7406–7412 RSC.
- J. He, P. Yao, J. Qiu, H. Zhang, Y. Jiao, J. Wang and Y. Chen, Fuel, 2021, 286, 119359 CrossRef CAS.
- M. Dosa, M. Piumetti, S. Bensaid, T. Andana, C. Novara, F. Giorgis, D. Fino and N. Russo, Catal. Lett., 2018, 148, 298–311 CrossRef CAS.
- D. Mukherjee, P. Venkataswamy, D. Devaiah, A. Rangaswamy and B. M. Reddy, Catal. Sci. Technol., 2017, 7, 3045–3055 RSC.
- B. Govinda Rao, D. Jampaiah, P. Venkataswamy and B. M. Reddy, ChemistrySelect, 2016, 1, 6681–6691 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |