
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reactivity of gaseous PtO2 and RhO2 with LaNiO3 thin films: a systematic XPS study†
Julie
Hessevik
 *,
Henrik H.
Sønsteby
,
Helmer
Fjellvåg
and
Anja O.
Sjåstad
*
*,
Henrik H.
Sønsteby
,
Helmer
Fjellvåg
and
Anja O.
Sjåstad
*
Department of Chemistry, Centre for Materials Science and Nanotechnology, University of Oslo, N-0318 Oslo, Norway. E-mail: juliehes@kjemi.uio.no; a.o.sjastad@kjemi.uio.no
First published on 13th January 2025
Abstract
The reaction between volatile platinum and rhodium gas species and bulk materials (alloys or oxides) is of high practical importance in the chemical process industry. Herein, an X-ray photoelectron spectroscopy (XPS) study has been conducted to understand how PtO2(g) and RhO2(g) react with LaNiO3 (LNO) thin films grown via atomic layer deposition (ALD) at elevated temperatures (900 °C). In this study, XPS data for reference powders of La2NiPtO6, LaNi0.95Pt0.05O3, LaNi0.88Rh0.12O3 and LaRhO3 provide a library for the interpretation of oxidation states of platinum and rhodium after reaction with LNO thin films. Upon short exposure to PtO2(g) and RhO2(g), platinum appear as Pt(III) in the LNO surface, but after prolonged exposure, Pt(IV) appears. Complementary diffraction analysis shows that LaNi1−xMxO3 solid solution forms initially where Ni, Pt and Rh are predominantly +III, however, quickly accompanied by the formation of a La2Ni2−2xM2xO6 (M = Pt, Rh) double perovskite phase with its characteristic (004) peak. Rhodium appears as Rh(III) and Rh(IV) in both the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c and the double perovskite like phases. Furthermore, platinum and rhodium depth profiles were generated by the combination of XPS and Ar+ sputtering. Platinum depth profiles reveal major differences between as-grown ALD films and post-annealed films when exposed to PtO2(g). Pt penetrates faster into the LNO material for films without post annealing, probably due to its smaller grain size and larger surface area and grain boundaries. When exposed to a mixture of PtO2(g) and RhO2(g), the Rh
c and the double perovskite like phases. Furthermore, platinum and rhodium depth profiles were generated by the combination of XPS and Ar+ sputtering. Platinum depth profiles reveal major differences between as-grown ALD films and post-annealed films when exposed to PtO2(g). Pt penetrates faster into the LNO material for films without post annealing, probably due to its smaller grain size and larger surface area and grain boundaries. When exposed to a mixture of PtO2(g) and RhO2(g), the Rh![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pt ratio in LNO increases upon prolonged reaction time. These results may have implications on the use of LNO as platinum and rhodium capture material in the Ostwald process.
:Pt ratio in LNO increases upon prolonged reaction time. These results may have implications on the use of LNO as platinum and rhodium capture material in the Ostwald process.
1. Introduction
The reaction between volatile PtO2 and RhO2 gas species and bulk materials (alloys or oxides) is of high practical importance to the chemical process industry, for instance in the Ostwald process for N-based fertilizer production.1,2 Herein, a X-ray photoelectron spectroscopy (XPS) study has been conducted to develop our understanding of the interplay between volatile platinum and rhodium oxide species and a redox active perovskite (ABO3) material, exemplified by LaNiO3 (LNO), at elevated temperatures. Of interest is to clarify the oxidation states of platinum and rhodium after the reaction with an ABO3 oxide surface and how the noble metal oxides enter as cations into the perovskite-type phase.Previous studies of the reaction between gaseous PtO2 and porous pellets of LNO at 800 and 900 °C have shown the formation of a monoclinic La2Ni2−2xPt2xO6 phase.3 This finding is fully in line with the described LaNi1−xPtxO3 system which includes a rhombohedral LaNi1−xPtxO3 solid solution (x ≤ 0.075, space group Rc), a two-phase region and a monoclinic phase La2Ni2−2xPt2xO6 (0.20 ≤ x ≤ 0.50; space group P21/n) with an ideal (ordered) double perovskite La2NiPtO6 as its end composition.4 Likewise, major solid solubility is reported between LaNiO3 and LaRhO3.5 For this LaNi1−xRhxO3 system, the crystal structure remains rhombohedral for x ≤ 0.15 (Rc) but becomes orthorhombic (Pnma) for higher Rh-contents, 0.25 ≤ x ≤ 1.00.5 This can be explained with a decrease in the Goldschmidt tolerance factor as a result of the larger size of the Rh(III)/Rh(IV) ions compared to Ni(II)/Ni(III). The short- and long-range ordering for platinum cations in La2Ni2−2xPt2xO6 probably occurs owing to the quite different oxidation states for the B-site elements. This is not the case for the LaNi1−xRhxO3 system, although mixed oxidation states with both Ni(III)/Rh(III) and Ni(II)/Rh(IV) have been reported with high rhodium contents.5
When exposed to reactive gases under Ostwald process conditions, pellets of LNO are found to incorporate both platinum and rhodium as verified by scanning electron microscopy–energy dispersive X-ray (SEM–EDX) and powder X-ray diffraction (PXRD) analyses.6 Although systematic studies of platinum incorporation in LNO pellets are already in hand,3 no such data exist for volatile rhodium species.
In this work, we use X-ray photoelectron spectroscopy (XPS) to investigate the chemical state of the surface as well as the incorporation and diffusion of platinum and rhodium into LNO(100) films, after reaction with gaseous PtO2 and RhO2. The LNO(100) thin films are epitaxially grown on SrTiO3(100) (STO) substrates via atomic layer deposition (ALD), following our recent LNO process for application in complex oxide electronics.7 SrTiO3 was selected as the substrate rather than the alternative of LaAlO3 since the latter is more likely to undergo interdiffusion owing to having isovalent cations with LNO.
To interpret the XPS spectra and assign the binding energies (BEs) of platinum and rhodium in LNO thin films, a library of reference XPS data of LaNi1−xMxO3 (M = Pt, Rh) is needed. Since data from the literature are limited, XPS reference data are collected for powders of well-defined LaNi0.95Pt0.05O3, La2NiPtO6, LaNi0.88Rh0.12O3 and LaRhO3. No XPS data have been reported previously for LaNi0.95Pt0.05O3 and La2NiPtO6 or any intermediate compositions. Likewise, no data exist for any other LaNi1−xRhxO3 compositions. To the best of our knowledge, only two former XPS studies on LaRhO3 are reported, with diverging results, stating BEs of Rh 3d5/2 = 308.6 eV (C 1s = 284.6 eV)8 and Rh 3d5/2 = 311.0 eV (C 1s = 285.0 eV).9
The depth profiles of platinum and rhodium in the LNO thin films were achieved by combining XPS measurements with Ar+ sputtering. The results were subsequently complemented with PXRD analyses to facilitate interpretations on how platinum and rhodium are incorporated into the LNO thin films. We thereby obtained insights into the platinum and/or rhodium containing solid solutions of LNO and the creation of new phases, along with information on the oxidation states of platinum and rhodium prior to the Ar+ sputtering. A key question is whether oxidation states may change in these systems containing redox active Ni, Pt and Rh cations. Since the LNO thin films are grown at 225 °C, we also explore how post deposition annealing at 900 °C affects LNO reactivity towards gas species at high temperatures, exemplified in this study by PtO2(g). The current work includes a comparison of the reactivity between situations with PtO2(g) exposure and simultaneous PtO2(g) and RhO2(g) exposure.
2. Experimental
2.1 Synthesis of powder reference samples
LaNi0.88Rh0.12O3 and La2NiPtO6 XPS reference samples were synthesized with the citric acid complexation method according to previously reported studies.4,5 Starting materials were La2O3 (99.99%, Molycorp inc.), Ni(CH3COO)2·4H2O (purity ≥ 99.0%, Sigma-Aldrich), Pt metal (99.9%, K. A. Rasmussen), Rh(NO3)3·xH2O (32% Rh, Sigma-Aldrich), C6H8O7·H2O (citric acid monohydrate, purity ≥ 99.5%, Sigma-Aldrich) and HNO3 (65 wt%, Merck KGaA). Prior to synthesis, La2O3 was annealed at 900 °C and cooled to ambient temperature in a desiccator. Exact formula masses of Ni acetate tetrahydrate and Rh nitrate hydrate were determined using thermogravimetry (see in-depth description in the ESI,† Section S1). La2O3 and the two Ni- and Rh salts were dissolved in 6 M HNO3, while the Pt metal was dissolved in aqua regia. Stoichiometric amounts of the precursors were mixed, and 50 g of citric acid was added per gram oxide product. The solutions were boiled until water and nitrous gases had evaporated, followed by an overnight heat treatment at 180 °C. Calcination was done at 400 °C in static air for 12 h in a muffle furnace. Calcined powders were pressed into cylindrical pellets at a static pressure of 100 bar. La2NiPtO6 was sintered twice at 1000 °C in 1 atm O2 (99.999%, AGA) for 2 + 3 days, with crushing and re-pelletizing in-between heat treatments. LaNi0.88Rh0.12O3 was sintered first at 850 °C and then at 1000 °C for 12 h in 1 atm O2 (99.999%, AGA), with crushing and re-pelletizing in-between heat treatments.2.2 ALD growth of LNO thin films on STO
STO(100) substrates were acquired from Crystal Gmbh with surface roughness (Ra < 0.5 nm) and dimensions (l × w × h = 10 mm × 3 mm × 1 mm). The ALD growth recipe of LNO on STO is previously reported.7 See Fig. S2 (ESI†) for a photography of LNO thin films on the STO substrate. As their dimensions were too small for characterization by home lab X-ray fluorescence (XRF) spectroscopy, spectroscopic ellipsometry and X-ray reflectometry (XRR), LNO was grown on an added Si substrate (30 mm × 30 mm) in the ALD chamber, next to the STO substrates. The LNO films were grown in batches during two campaigns.The films were grown in an F-120 Sat (ASM Microchemistry) reactor at a chamber temperature of 225 °C and a base pressure of 2.3 mbar with N2 (99.999%, Nippon gases) as inert carrier gas. La(thd)3 (thd = 2,2,6,6-tetramethyl-3,5-heptanedionate, Volatec Oy) and Ni(acac)2 (acac = acetylacetonate = C5H7O2−, Merck) were used as precursors supplied from open boats inside the reactor. O3 was used as the oxygen source and was generated using an In USA AC-2505 generator, supplied with 99.6% O2 (Praxair) giving 15 wt% O3 in O2. The pulse sequence was [X·(La(thd)3 + O3) + Y·(Ni(acac)2 + O3)]·Z. The X:Y ratio was optimized to achieve close to a 1:1 ratio of La:Ni in the deposited films. Z is the total number of cation pulse sequence cycles and was selected as to control the thickness of the LNO films.
Both batch 1 and batch 2 were grown using an X![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :Y ratio of 7:2, but batch 1 was grown with Z = 2000 while batch 2 was grown with Z = 1000. Batch 1 should ideally have approximately double thickness compared to batch 2. A handful of the LNO films from batch 1 were post-annealed at 900 °C prior to PtO2(g) exposure. The post deposition annealing was performed using a horizontal multi-zone furnace in a flow of O2 (444 mL min−1, 99.999%, AGA) for 24 h.
:Y ratio of 7:2, but batch 1 was grown with Z = 2000 while batch 2 was grown with Z = 1000. Batch 1 should ideally have approximately double thickness compared to batch 2. A handful of the LNO films from batch 1 were post-annealed at 900 °C prior to PtO2(g) exposure. The post deposition annealing was performed using a horizontal multi-zone furnace in a flow of O2 (444 mL min−1, 99.999%, AGA) for 24 h.
2.3 PtO2(g) and RhO2(g) reactivity studies
LNO thin films were exposed to PtO2(g) or to a mixture of PtO2(g) and RhO2(g). These experiments were performed in a horizontal multi-zone furnace under a flow of dry air (444 mL min−1, < 100 ppm H2O) at 900 °C equivalent to previously reported study.3 In the case of simultaneous PtO2(g) and RhO2(g) exposure, a rolled-up net of ∼10 wt% Rh and 90 wt% Pt (K. A. Rasmussen) was placed upstream to the LNO samples at 1000 °C. Experiments that included individual exposure to PtO2(g) were performed with LNO thin films from batch 2, both with and without post-annealing treatment. The duration of the exposure was 1, 4, 8 and 24 h for the as-grown films, while 1, 8, 24 and 48 h for the post-annealed samples. Two sets of experiments with simultaneous exposure to PtO2(g) and RhO2(g) were performed for LNO films of different thicknesses and without post-annealing treatment. The exposure times were 1, 8 and 24 h (batch 1) and 1, 4 and 24 h (batch 2).2.4 Characterization
Powder X-ray diffraction (PXRD) data for oxide reference powders and LNO thin films were collected using a Bruker D8 Discover diffractometer with Cu Kα1 radiation [Ge (111) Johansson monochromator and Lynxeye detector]. Data analyses were performed with the software packages EVA (V6.0) and TOPAS (V6). Rietveld refinements were based on input from cif-files of La2NiPtO610 and LaNi0.90Rh0.10O3.5The thickness of LNO films was measured in two ways: (1) ellipsometry with an Alpha SE spectrometric ellipsometer (J.A. Woollam) in the wavelength range of 390–900 nm at an incident angle of 70°, using the CompleteEASE software and a Cauchy fitting model to fit the data and (2) XRR with a Rigaku SmartLab 3 kW high-resolution X-ray diffractometer (Cu anode) and a HyPix 3000 high-energy resolution 2D detector. The measurements were carried out in continuous mode with a scan step of 0.005° and a scan speed of 1.11° min−1 and analysed using GenX 3.5 software.11 The incident parallel beam was obtained by means of a 5° Soller slit, a 10 mm in-plane parallel slit collimator and an incident slit (0.1°). The receiving optics included two receiver slits (0.3 and 0.4 mm) and a 5° Soller slit. The collected data was analyzed using GenX 3.5 software.11
XRF data was collected using an Axios Max minerals X-ray fluorescence system (Panalytical), equipped with a 4 kW Rh-tube, to determine the La:Ni atomic ratio of the as-grown LNO thin films. The system uses Omnian and Stratos options for standardless measurements, and quantification was carried out using the SuperQ software suite (Panalytical).
Field emission scanning electron microscopy (FE SEM) was carried out using a Hitachi SU-8230 microscope. Secondary electrons, created from an acceleration voltage of 3 keV, were used to image LNO thin film surfaces.
XPS data was collected on reference powder samples of La2NiPtO6, LaNi0.95Pt0.05O3, LaNi0.88Rh0.12O3 and LaRhO3, using an Axis Ultra DLD instrument from Kratos Analytical. The samples were pressed into indium foil fastened by carbon tape to a glass plate on a sample bar. The pressure was in the mid 10−9 Torr region and charge neutralization with low energy electrons was applied during acquisition. Survey spectra were acquired using a pass energy of 160 eV and a step length of 1 eV. Pt 4f and Pt 4d narrow scans were collected with pass energies of 40 eV and 0.1 eV in step length for LaNi0.95Pt0.05O3 and La2NiPtO6. Rh 3d narrow scans were collected with pass energies of 20 eV and 0.1 eV in step length for LaNi0.88Rh0.12O3 and LaRhO3.
XPS spectra were furthermore collected using a Theta Probe Angle-Resolved XPS system (Thermo Scientific; monochromatic Al Kα 1486.6 eV and an analysis chamber at ≈10−8 mbar pressure) to study the chemical state at the surface after exposure to PtO2/RhO2 gas species and to obtain depth profiles for the exposed LNO films after Ar+ sputtering (sequential XPS acquisition + sputtering cycles, Fig. 1).
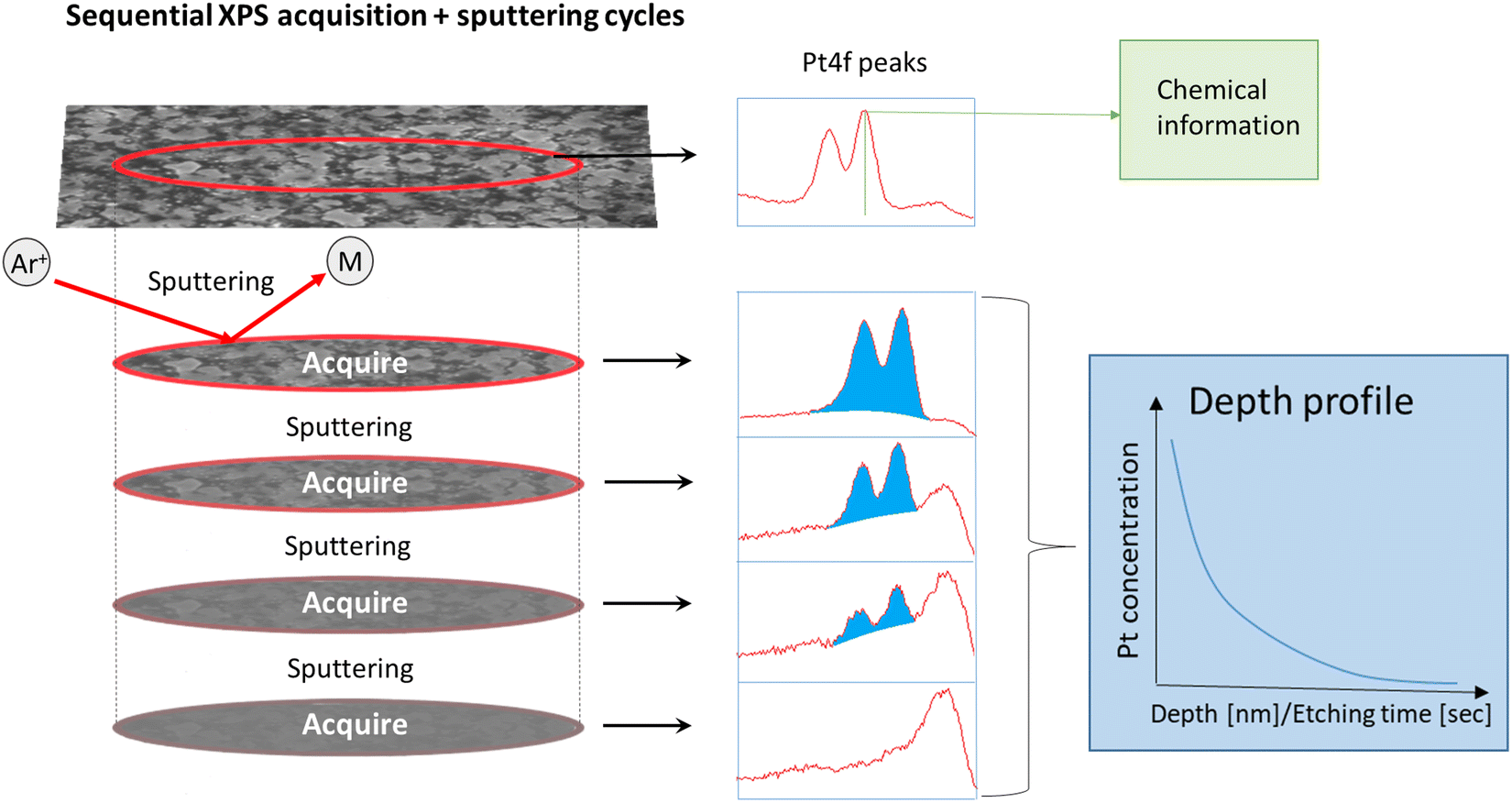 | ||
| Fig. 1 Illustration of sequential Ar+ sputtering and XPS collection of ALD thin films. | ||
The binding energy scales were calibrated to the main component of adventitious carbon set at 284.8 eV. All XPS spectra were analyzed using the Thermo Avantage software suite (V5.9931), and a smart background was employed (advanced Shirley model) for all fits. Survey spectra were collected with a pass energy of 200 eV and a step length of 1 eV. For LNO batch 1 samples exposed to PtO2(g), narrow scans of Pt 4f (64–87 eV) with a pass energy of 60 eV were collected and analyzed. For the samples exposed to both PtO2(g) and RhO2(g), narrow scans of Pt 4d and Rh 3d (300–350 eV) were collected and analyzed. A pass energy of 60 eV was used for narrow scans of LNO samples from batch 1, while a pass energy of 100 eV was used for LNO samples from batch 2. A step length of 0.1 eV was employed for all narrow scans.
The LNO films were sputtered with Ar+ in several sequences before the STO substrate was finally reached. Eight and nine sputtering sequences were employed for the films of batch 1 and batch 2, respectively. The films had different thicknesses and hence different Ar+ sputtering rates were employed, implying that the vertical resolution of the resulting depth profiles differs. Each sputtering sequence removed about 10 nm and 6 nm materials from the batch 1 and batch 2 films, respectively. It was assumed that the sputtering rates for platinum and rhodium are similar. Chemical information is lost once Ar+ sputtering has been employed, since this triggers reduction (e.g. the BE of Pt 4f7/2 after sputtering was 71.4 ± 0.1 eV). However, quantitative data were obtained to provide Pt and Rh depth profiles, given that the area (in counts per second (CPS)) under fitted peaks at different sputtering levels is proportional to the platinum and rhodium concentrations in the corresponding part of the film.
3. Results and discussion
3.1 XPS data of reference samples
Powders of LaNi0.95Pt0.05O3, La2NiPtO6, LaNi0.88Rh0.12O3 and LaRhO3 were synthesized and characterized as described above. LaNi0.88Rh0.12O3 (Fig. S3, ESI†) was phase pure, taking rhombohedral (Rc) symmetry. Powders of LaNi0.95Pt0.05O3, La2NiPtO6 and LaRhO3 were synthesized previously,4,5,12 and the materials crystallize in rhombohedral (Rc), monoclinic (P21/n) and orthorhombic (Pnma) symmetries, respectively. LaNi0.95Pt0.05O3 and LaRhO3 were phase pure, while La2NiPtO6 had ∼2 wt% Pt impurity.
The Pt 4f, Pt 4d and Rh 3d XPS peaks of the four reference samples were determined using peak fitting models. The Pt 4d and the Rh 3d peaks do not overlap with other peaks; however, the Pt 4f peaks for LaNi0.95Pt0.05O3 and La2NiPtO6 overlap with Ni 3p peaks. To help creating a proper peak fitting model, pristine LNO thin films were used as a reference for how to fit the Ni 3p peaks. An in-depth explanation of the peak fitting procedure and the fits are given in the ESI,† Section S2, and the obtained BEs are summarized along with literature data in Table 1. Some unresolved discussion of BEs of Rh(IV) in RhO2 exists in the literature.13–15 In this study, we rely on the literature values reported by Weng-Sieh et al.,15 which are in agreement with internal unpublished results of the same compounds. Interestingly, the BE of Pt 4f7/2 (73.3 eV) for LaNi0.95Pt0.05O3 has a value intermediate of what is reported for Pt(II) in PtO (72.2 eV) and Pt(IV) in PtO2 (74.0 eV). This suggests that platinum exists in the oxidation state of +III in the B-site solid solution LaNi0.95Pt0.05O3. Platinum rarely occurs in the oxidation state of +III in oxide crystal structures, as it usually appears in the oxidation state of +II in square planar geometry or +IV in octahedral geometry.16 However, Pt(III) has often been reported in binuclear complexes and in few rare cases as mononuclear species.17 Finally, it should be noted that the curve fit of the BE of Pt 4d5/2 for LaNi0.95Pt0.05O3 gives no conclusive result due to the low signal-to-noise ratio of the weak Pt 4d signal.
| Reference samples | BE (Pt 4d5/2) [eV] | BE (Pt 4f7/2) [eV] | BE (Rh 3d5/2) [eV] | |
|---|---|---|---|---|
| a Values are referenced using C 1s = 284.8 eV to facilitate comparison to results of the present study. Numbers in parentheses are original values in the paper before correction to C 1s = 284.8 eV. | ||||
| La2NiPtO6 | This study | 317.1 | 74.2 | — |
| LaNi0.95Pt0.05O3 | This study | — | 73.3 | — |
| Pt | Kaushik et al.18 | 314.0a (314.2) | 69.9a (70.1) | — |
| PtO | Kaushik et al.18 | 315.3a (315.5) | 72.2a (72.4) | — |
| PtO2 | Kaushik et al.18 | 317.1a (317.3) | 74.0a (74.2) | — |
| LaNi0.88Rh0.12O3 | This study | — | — | 308.0 and 309.1–309.5 |
| LaRhO3 | This study | — | — | 308.4 and 309.5 |
| Gysling et al.8 | — | — | 308.8a (308.6) | |
| Watson et al.9 | — | — | 310.8a (311.0) | |
| Rh2O3 | Weng-Sieh et al.15 | — | — | 308.1–308.4 |
| Kibis et al.13 and Abe et al.14 | — | — | 308.2 | |
| RhO2 | Weng-Sieh et al.15 | — | — | 309.4 |
| Kibis et al.13 and Abe et al.14 | — | — | 308.4–308.5 | |
In the case of La2NiPtO6, the XPS peak positions suggest that platinum exists in the oxidation state of +IV. We justify this with the fact that the Pt 4d5/2 and Pt 4f7/2 BEs show similar values as reported for PtO2, see Table 1 and references therein.
The BE of Rh 3d5/2 for LaRhO3 (308.4 eV) is in consensus with values reported for Rh2O3 (ref. 13–15 in Table 1), i.e., supporting the expected oxidation state, +III. The result is in line with previously reported values by Gysling et al.8 In addition, some Rh seem to be in the oxidation state of +IV as indicated by a small additional shoulder at a BE of 309.5 eV. This may possibly reflect higher valent rhodium at the surface being different from the bulk situation. Similar BE values were found for Rh 3d5/2 in LaNi0.88Rh0.12O3, but the Rh(IV)/Rh(III) ratio is higher for the latter material compared to LaRhO3. This is evidenced by a more pronounced shoulder at higher BEs (309.1–309.5 eV). We suggest that this is due to some possible Ni(II)/Rh(IV) redox activity at the top atomic surface terminating layers.
3.2 LNO thin films grown via ALD
Two batches of epitaxial LNO films with a targeted La:Ni atomic ratio close to 50:50 were grown on STO(100) substrates. The complexity of the process led to slight variations in the achieved La:Ni atomic ratio (Table 2). The quality of the films is documented by the sharp pseudo cubic (h00) LNO peaks in the powder X-ray diffractograms (Fig. S11, ESI†). The morphologies of the achieved films are however different, as seen from top view SEM images reported in Table 2. The grain size is larger for batch 2 (approx. 0.3–2 μm in diameter) than that for batch 1 (approx. 10–40 nm in diameter) specimens. This may reflect aspects like variations in the La:Ni ratio, degree of crystallinity, film thickness and more. The LNO film thicknesses (grown on Si substrates) were evaluated by means of ellipsometry and XRR. Since the LNO film absorbs in the wavelength range of the ellipsometer, we considered the most accurate result to be achieved by XRR, see Table 2. We note that the thickness of batch 1 films is approximately twice that of batch 2 films. This is in line with the doubled number of deposition cycles in the ALD recipe. The fitted raw XRR data are given in Fig. S12 and S13 (ESI†). The thickness should be accurately described by the well-fitted periodicity, despite difficulties in fitting the full set of data with respect to profile and intensity.
:Ni atomic ratio (XRF) and film thickness (ellipsometry and XRR) of LNO thin films grown by ALD on STO(100) substrates, and exposure to PtO2(g) or PtO2(g) and RhO2(g). Yes and no stand for experimentally executed or not
| Batch 1 | Batch 1 post annealed | Batch 2 | |
|---|---|---|---|
| SEM image |

|

|

|
| La:Ni atomic ratio |
46.3:53.7 |
No | 49.9:50.1 |
| Thickness ellipsometry [nm] | 75.1 | No | 43.7 |
| Thickness XRR [nm] | 88.2 | No | 45.3 |
| PtO2 exposure | Yes | Yes | No |
| PtO2 and RhO2 exposure | Yes | No | Yes |
The surface morphology of batch 1 samples changed upon post deposition annealing. NiO particles precipitated and the grain size of LNO increased to 1–2 μm. PXRD shows the same pseudo cubic (h00) Bragg reflections as initially, but the peak height relative to STO peaks is enhanced after annealing (Fig. S14, ESI†). This implies an improved crystallinity in the LNO film or relaxation leading to less variation in cell parameters.
3.3 LNO films exposed to PtO2(g) and effects of post-annealing
The as-grown and post-annealed LNO thin films were exposed to PtO2(g), see Table 2. In these cases, the PXRD of LNO on STO after exposure shows new Bragg reflections at lower 2θ values than what is expected for LNO and STO (see e.g. Fig. S17, ESI†). Their intensities increased upon prolonged exposure time, and the Bragg reflections appear to fit well with (002) and (004) of pseudo tetragonal La2NiPtO6 (see Fig. S18, ESI† for the crystal structure). Fig. 2 shows the development of (004) at 2θ ≈ 45.8° after different exposure times (a) without and (b) with post-annealing.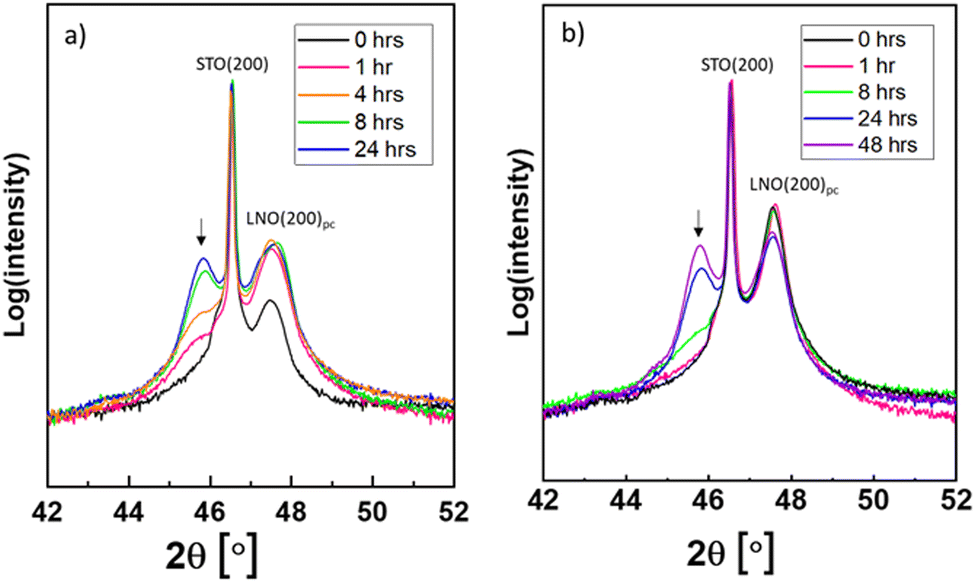 | ||
| Fig. 2 Powder X-ray diffractograms of LNO films on STO substrates after PtO2(g) exposure for various durations, see (a) without post-annealing and (b) with post-annealing. The emerging peaks ascribed to La2NiPtO6 are marked by arrows. The Bragg reflections (200) for STO and pseudocubic (200) for LNO are ascribed to the other peaks in the diffractograms. | ||
The measured BE of Pt 4f on the surface of the LNO thin film sample (without post-annealing) exposed to PtO2(g) for 24 h is representative for all LNO films individually exposed to PtO2(g). The XPS results are reported in Fig. 3, showing the Pt 4f doublet with overlapping peaks at lower BEs. The latter is believed to be Ni 3p peaks for nickel in +II and +III oxidation states. XPS of the pristine LNO film of batch 1 (Fig. S19a, ESI†) fits well with two doublets of nickel and an additional set of peaks at higher BEs that are probably nickel satellite/shake-up peaks. The BEs of the two Ni 3p doublets (67.2 and 69.2 eV, 71.1 and 73.5 eV) included in the peak fit for the sample exposed to PtO2(g) (Fig. 3) are in good correspondence with the literature values of Ni(II) and Ni(III).19 Details of the peak fitting are described in the ESI,† Section S4, together with an example survey spectrum (Fig. S20, ESI†). The combined peak fitting of Pt 4f and Ni 3p shows that platinum is most probably in the oxidation state of +IV since the Pt 4f7/2 peak at a BE of 74.0 eV is close to values reported for PtO2 (Table 1).
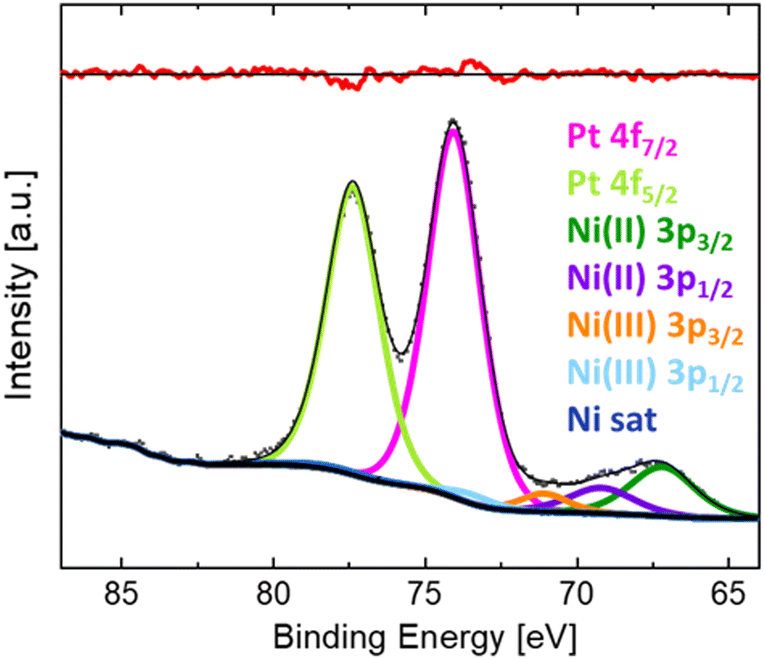 | ||
| Fig. 3 XPS narrow scan of Pt 4f and Ni 3p and fitted peaks for LNO thin films (without post-annealing) after exposure to PtO2(g) for 24 h. | ||
Platinum depth profiles were obtained according to the procedure in Fig. 1. All spectra for different sputtering levels are presented in Fig. S21 and S22 (ESI†). The data clearly show that platinum is incorporated into the bulk of the film. Quantification of platinum contents was based on fitting the entire Pt 4f narrow scan including a doublet of Pt 4f peaks and the Ni 3p peaks. Since platinum is reduced due to the Ar+-sputtering, asymmetrical functions were needed to fit the spectra. In depth description is given in the ESI,† Section S4, exemplified with fits for LNO exposed to gaseous PtO2 for 24 h after 78 and 182 s of Ar+-sputtering (Fig. S23, ESI†). Quantifications of platinum to lanthanum ratios have not been performed due to challenges of multiplet splitting of lanthanum and a strong overlap between Ni 2p and La 3d peaks in LaNiO3 (see Fig. S24, ESI†).20
The resulting platinum depth profiles are plotted as area CPS versus sputtering time, as well as normalized curves (Fig. 4). When comparing samples with and without post-annealing, we note that less platinum has been incorporated into the films that have been post-annealed prior to the PtO2(g) exposure. This is probably an effect of the grain size of the LNO material. The smaller grain size and more numerous grain boundaries for the pristine LNO films (SEM images in Table 2) can probably explain a faster reaction and transformation to La2NiPtO6, since grain boundary- and surface diffusion are expectedly much faster than bulk diffusion.21 However, it seems that less platinum is present at the surface after prolonged exposure times while more platinum is found deeper into the film. Possibly, this reflects an equilibrium between the rate of incoming PtO2(g) in the gas phase and platinum grain boundary diffusion into the bulk of the thin film.
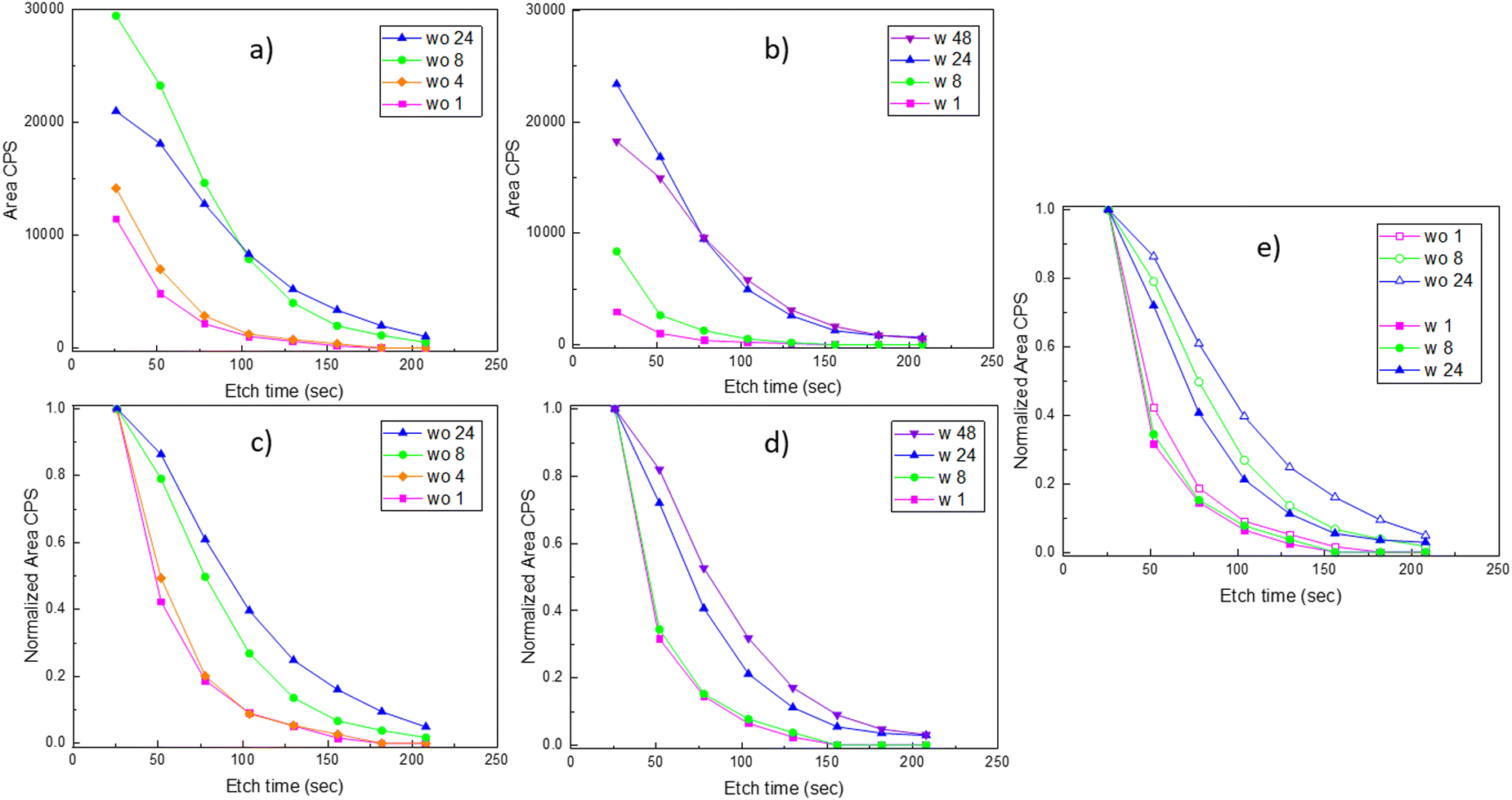 | ||
| Fig. 4 Platinum depth profiles in LNO samples after exposure to PtO2(g). (a) and (c) from a set of experiments on LNO samples without (wo) post-annealing after 1, 4, 8 and 24 h exposure to PtO2(g), where (a) shows the area CPS of Pt 4f peaks measured at certain etch time intervals, while (b) shows normalized curves of the same data. (b) and (d) From another set of experiments on LNO samples with (w) post-annealing after 1, 8, 24 and 48 h exposure to PtO2(g), where (b) shows the area CPS of Pt 4f peaks measured at certain etch time intervals, while (d) shows the normalized curves of the same data. (e) The normalized depth profiles shown in (c) and (d) with common experimental exposure times (1, 8 and 24 h) are repeated here for easier comparison of LNO samples w and wo post-annealing. | ||
3.4 LNO films exposed to a mixture of PtO2(g) and RhO2(g)
The two batches of as-grown LNO thin films with different thicknesses were exposed to a mixture of PtO2(g) and RhO2(g). The measured PXRD patterns are similar to those described with PtO2(g) exposure. A selected part is reported in Fig. S25 (ESI†), which can be compared to the zoomed-in view of the diffractograms of samples exposed to only PtO2(g) (Fig. 2). All diffractograms (Fig. 2 and Fig. S25, ESI†) show the characteristic (004) Bragg reflection of a La2NiPtO6 like phase, possibly also containing rhodium. Interestingly, an additional Bragg reflection is observed after the combined Pt–Rh exposure for batch 2, at a slightly lower diffraction angle than the (200)pc (pc = pseudo cubic) LNO peak. This Bragg reflection is more pronounced for short exposure times, indicating the existence of a second rhombohedral/pseudo cubic LaNi1−xMxO3 phase that is different from LNO. This second phase contains some platinum and/or rhodium, presumably M = Pt; x ≤ 0.075 and/or M = Rh, x ≤ 0.15, based on published data for such solid solution systems,4,5 and has a (200) Bragg reflection at a lower diffraction angle than that for LNO. The (200) Bragg reflection disappears when more platinum and/or rhodium has been incorporated into the thin film, possibly due to a conversion into a double perovskite-like mixed oxide.XPS narrow scans (BE: 300–345 eV) of the exposed thin films show peaks of Pt 4d and Rh 3d (Fig. 5). During the following analysis, we address the chemical state of platinum and rhodium; furthermore, depth profiles are evaluated based on repeated sputtering and XPS data collection.
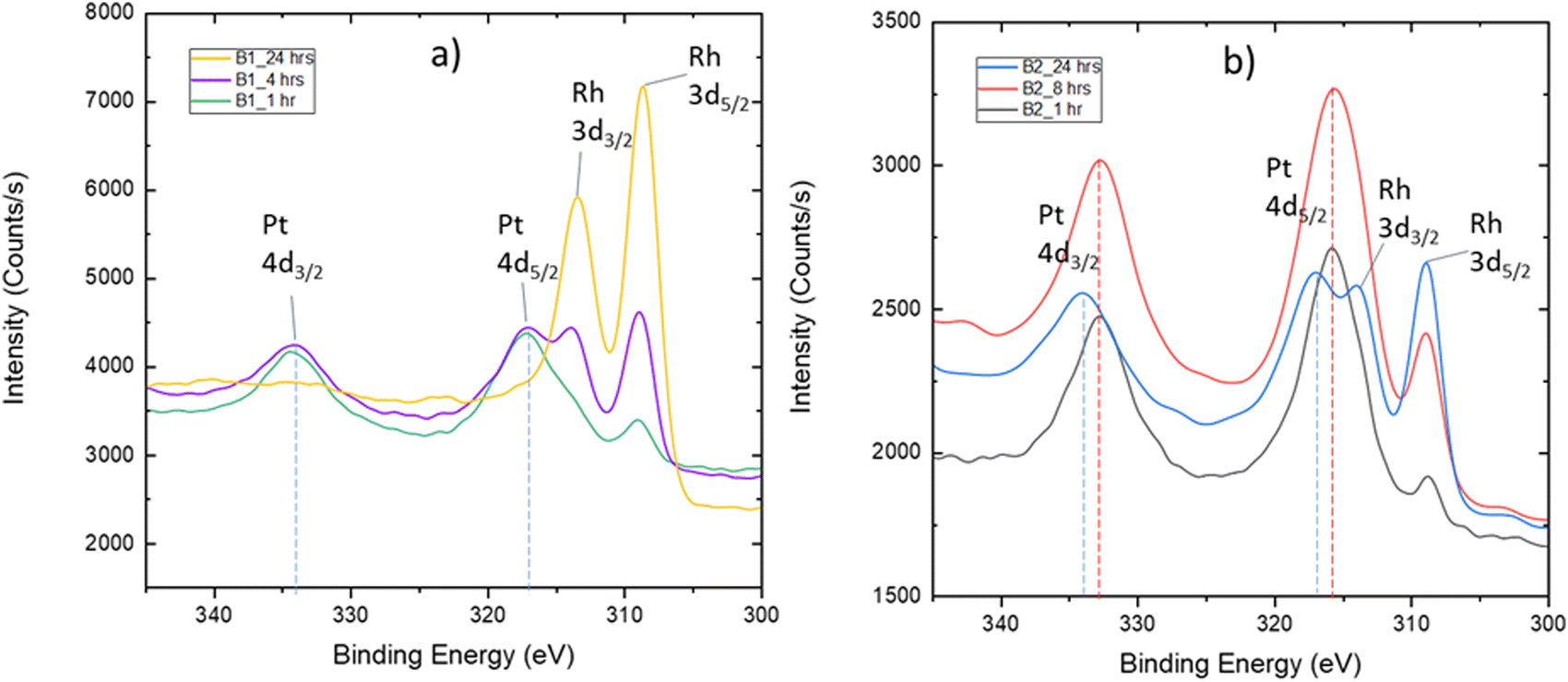 | ||
| Fig. 5 XPS narrow scans of LNO surfaces after simultaneous exposure to PtO2(g) and RhO2(g) for (a) 1, 4 and 24 h in the case of batch 1 (B1) and for (b) 1, 8 and 24 h in the case of batch 2 (B2). | ||
The quantification and identification of peak positions for the spectra reported in Fig. 5 required fitting of the entire spectrum since Rh 3d3/2 and Pt 4d5/2 peaks overlap. Details are given in the ESI,† Section S5. This procedure was applied to quantify changes in the Pt 4d peak position as a function of exposure time and was found to be up to 1 eV (see Fig. 5b). The fits are shown in Fig. S27 and S28 (ESI†). These changes are considered in the perspective of the Pt 4f BE for the powder reference samples LaNi0.95Pt0.05O3 and La2NiPtO6 (Table 1). On this basis, we suggest that platinum initially is incorporated into the rhombohedral perovskite phase as +III. Thereafter, platinum becomes oxidized to +IV and Ni(III) is reduced to Ni(II) as a result of the formation of a La2NiPtO6 like double perovskite, either as well-defined crystallites or as small domains with a B-site cation ordering.
The sample from batch 1 exposed to PtO2(g) and RhO2(g) for 24 h shows a high rhodium amount at the surface (Fig. 5a). In this case, it is not possible to observe the Pt 4d peaks. However, analysis of platinum can easily be done based on narrow scans for the stronger Pt 4f peaks (Fig. S29, ESI†), see the ESI,† Section S5, giving a BE of 72.6 eV for Pt 4f7/2. This value is in-between the literature value of Pt(II) in PtO (72.2 eV) and the proposed Pt(III) species in the LaNi0.95Pt0.05O3 (73.3 eV) reference sample, see Table 1. An oxidation state between +II and +III for platinum is not fully in line with our expectations regarding the solid solution LaNi1−xMxO3 (M = Pt, Rh), with trivalent platinum.
For the batch 2 samples, exposed for 1 and 24 h, it is possible to fit one set of Rh 3d peaks in addition to one set of Pt 4d peaks (Fig. S27 and S28, ESI†). The obtained BEs for Rh 3d5/2 are 308.6 and 308.8 eV, respectively. These BEs are higher than the Rh(III) value for the LaNi0.88Rh0.12O3 (308.0 eV) and LaRhO3 reference samples (308.4 eV). One can speculate whether rhodium exists as both Rh(III) and Rh(IV) in the LNO films, as part of a regular solid solution in the rhombohedral phase, or as part of the double perovskite phase.
Interestingly, the quantification shows that the molar Rh:Pt ratio in the surface region increases significantly for prolonged exposure time (Fig. 5). In order to shed light on this observation, a procedure with sequential Ar+ sputtering and XPS acquisition was adopted. All spectra acquired for the different sputtering levels are presented in Fig. S30 (ESI†). A simplified fit, using the Pt 4d3/2 and Rh 3d5/2 peaks, was performed for the quantification of platinum and rhodium as the basis for depth profiles, see Fig. 6. Despite that these depth profiles must be viewed as approximate owing to the peak overlap and a simplified fit, it seems clear that the observations for the surfaces (Fig. 5) are also reflected in the depth of the films. Assuming that the relative composition of the Pt–Rh source of 10 wt% Rh and 90 wt% Pt (corresponding to a Rh:Pt molar ratio of 20:80) is maintained for the gas phase and hence the incoming RhO2(g) and PtO2(g), there is an enhanced amount of Rh in the surface relative to the Pt/Rh content of the incoming gas species. It is well known that rhodium is more attracted to oxygen than platinum and that could explain why more rhodium accumulates in the top surface.22,23 This in turn could also explain why the LNO films become enriched on the Rh sub-surface, but other factors as thermal stability of Rh- versus Pt containing mixed oxides and mobility of Rh- and Pt species in oxide lattices could be important factors to fully explain the observations in Fig. 6.
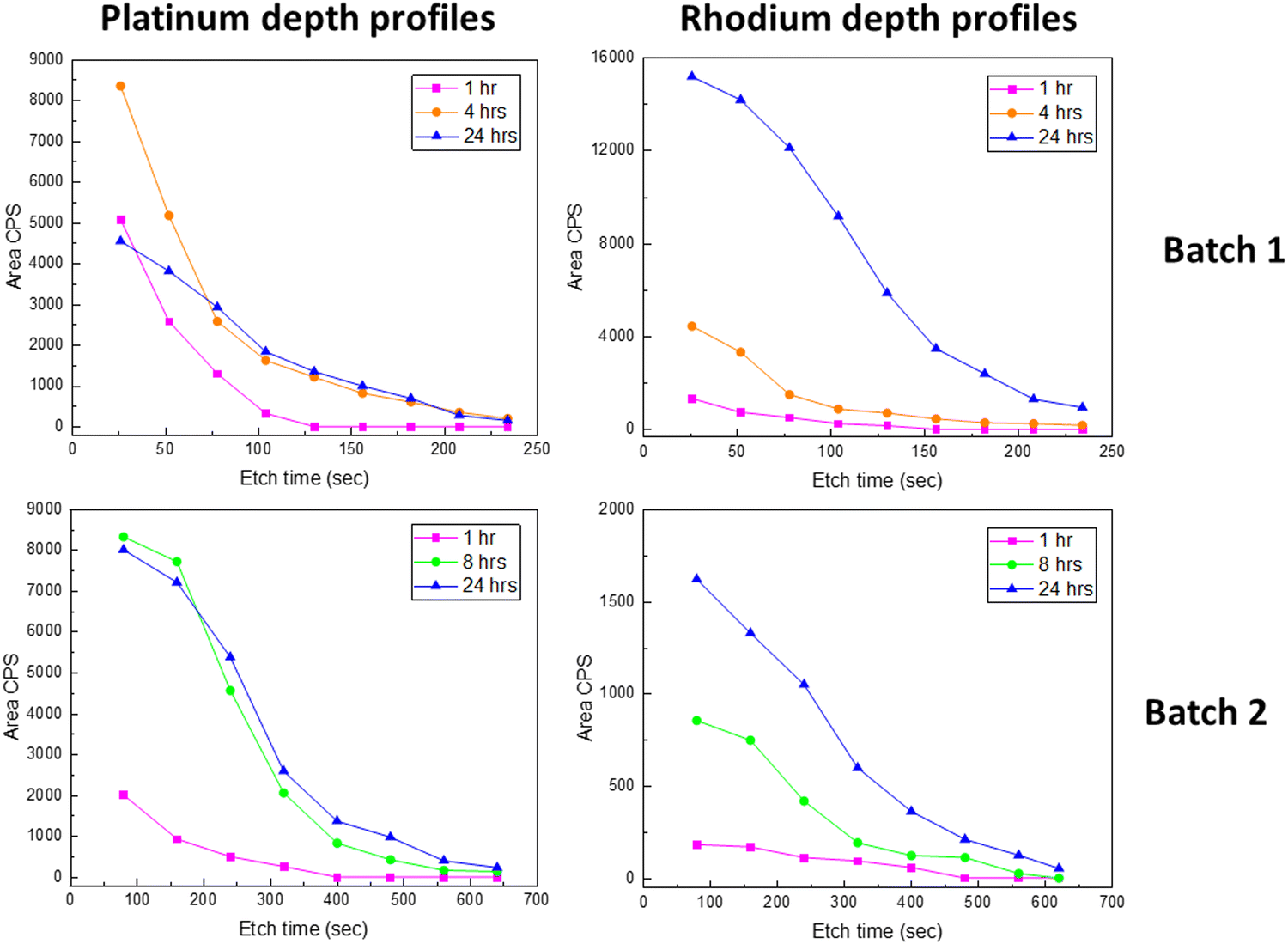 | ||
| Fig. 6 Platinum depth profiles and rhodium depth profiles in LNO films (without post-annealing) after 1, 4 (batch 1)/8 (batch 2) and 24 h PtO2(g) and RhO2(g) exposure, shown with the area CPS of Pt 4d3/2 and Rh 3d5/2 for batch 1 and batch 2. | ||
An additional observation from Fig. 5 and 6 could also be an effect of the grain size. The grain size of batch 1 (10–40 nm) was significantly smaller than the grain size of batch 2 (0.3–2 μm), as shown in Section 3.2. The effect on the amount of platinum and rhodium incorporation due to the experimental duration time and the increased grain size seems to be comparable. The fact that rhodium accumulation is occurring to a larger extent in batch 1 compared to batch 2 might be due to batch 1 having a morphology of a smaller grain size compared to batch 2.
The platinum depth profiles (Fig. 4) indicate a restriction to PtO2(g) uptake as more platinum enters the LNO thin film, and faster reactivity seems to be the case in the presence of more surfaces and grain boundaries. For the application of LNO as the platinum catchment material in e.g. the Ostwald process, it seems important with a high surface area, also for better contact between the gas phase and LNO. The restriction to RhO2(g) uptake is not as evident (Fig. 6); however, there seem to be limitations to how fast platinum and rhodium can diffuse into the LNO material. As such, the ALD technique is also interesting because it can facilitate growth of LNO on high-aspect ratio substrates and could be an improved design for higher platinum and rhodium capture efficiencies. The observation of an increased Rh:Pt atomic ratio in the surface after prolonged exposure to PtO2(g) and RhO2(g) (Fig. 5 and 6) is also of interest when considering the possibility to use LNO for simultaneous platinum and rhodium capture in industries such as the Ostwald process.
4. Conclusions
XPS data of powders of La2NiPtO6, LaNi0.95Pt0.05O3, LaNi0.88Rh0.12O3 and LaRhO3 provide insight into platinum and rhodium electronic states as well as serve as a library for later interpretation of XPS spectra of these compounds. It is found that the platinum oxidation state is +IV in La2NiPtO6 (P21/n) due to the BE of 74.2 eV being similar to that of PtO2(s) (74.0 eV). For LaNi0.95Pt0.05O3 (Rc), the platinum oxidation state is considered +III due to the BE of 73.3 eV, which is between values for PtO (72.2 eV) and PtO2 (74.0 eV). For LaRhO3 (Pnma), a strong peak with a BE of 308.4 eV corresponding to an oxidation state +III is observed, in agreement with data for Rh2O3. Small shoulders indicate minor amounts of rhodium in the oxidation state +IV (BE = 309.5 eV; cf. RhO2 with 309.4 eV). Finally, LaNi0.88Rh0.12O3 shows a larger shoulder indicating the presence of Rh(IV), thus containing both +III and +IV species in the analyzed surface region.
XPS measurements complimented with PXRD data suggest PtO2(g) and RhO2(g) to react with LNO forming solid solutions where platinum and/or rhodium substitutes for nickel, providing a rhombohedral perovskite (Rc) or a B-site ordered double perovskite (P21/n). For short exposure times, platinum is believed to occur as Pt(III) in the LNO surface but after prolonged exposure time Pt(IV) becomes dominant. The diffraction analysis shows that LaNi1−xMxO3 solid solution forms initially where Ni, Pt and Rh are predominantly +III, however, soon being accompanied by the formation of a La2Ni2−2xM2xO6 (M = Pt, Rh) double perovskite phase. Rhodium appears to exist as Rh(III) and Rh(IV) in both the Rc and the double perovskite like phases. Some Rh(IV) could probably be assigned to surface termination effects; however, PXRD data indicate that rhodium probably enters the double perovskite and then Rh(IV) could be a possible oxidation state also in the bulk.
The platinum and rhodium depth profiles show major differences between the as-grown and post-annealed ALD films. Platinum penetrates faster into the LNO material for films without post-annealing, probably due to its smaller grain size and larger surface area and grain boundaries. For simultaneous PtO2(g) and RhO2(g) exposure, the Rh:Pt ratio in LNO increases upon prolonged reaction times. This may reflect PtO2 and RhO2 gas equilibria with the solid as well as thermal stability of the platinum- and rhodium-containing phases.
The gained insight is of relevance for understanding how oxides can be used for catchment of volatile noble metal species in industrial processes. The results indicate that LNO is capturing relatively more rhodium than platinum in the long run; however, more studies are needed to document this finding. A noble metal capture design could potentially involve ALD to grow LNO coatings with high aspect ratios to reduce transport distances for platinum and rhodium and thus enhance capture performance relative to compact LNO beads.
Author contributions
Julie Hessevik: conceptualization, methodology, formal analysis, investigation, data curation, visualization, writing – original draft, and writing – review and editing; Henrik H. Sønsteby: methodology, investigation, data curation, resources, and writing – review and editing; Helmer Fjellvåg: conceptualization, methodology, writing – review and editing, supervision, and funding acquisition; Anja O. Sjåstad: conceptualization, methodology, writing – review and editing, supervision, funding acquisition, and project administration.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge Cathinka S. Carlsen for synthesis of LaNi0.88Rh0.12O3 and the thin film group for growing LNO ALD thin films and performing the XRF and ellipsometry measurements, with a special thanks to Veronica Killi, Linn Rykkje and Anjali Choubey. The XPS measurements were performed on instrumentation at NICE (National Surface and Interface Characterization Laboratory, RCN grant no. 195565), XRR experiments were performed at NorFab (Norwegian Micro- and Nano-Fabrication Facility, RCN grant no. 295864) and XRD data collection was performed at RECX (Norwegian center for X-ray diffraction and scattering, RCN grant no. 208896). The authors acknowledge the iCSI center (industrial Catalysis, Science and Innovation center, RCN grant no. 237299) for financial support.Notes and references
- M. Warner, The Kinetics of Industrial Ammonia Combustion, PhD thesis, University of Sydney, 2013. https://hdl.handle.net/2123/9426.
- B. F. Sperner and W. Hohmann, Rhodium-Platinum Gauzes for Ammonia Oxidation: A Study by Scanning Electron Microscopy, Platinum Met. Rev., 1976, 20, 12–20, DOI:10.1595/003214076X2011220.
- J. Hessevik, A. S. Fjellvåg, O. Iveland, T. By, J. Skjelstad, D. Waller, H. Fjellvåg and A. O. Sjåstad, LaNiO3 as a Pt catchment material in the ammonia oxidation process, Mater. Today Commun., 2022, 33, 104084, DOI:10.1016/j.mtcomm.2022.104084.
- A. S. Fjellvåg, Ø. S. Fjellvåg, Y. Breard and A. O. Sjåstad, Structural disorder and antiferromagnetism in LaNi1−xPtxO3, J. Solid State Chem., 2021, 299, 122181, DOI:10.1016/j.jssc.2021.122181.
- A. S. Fjellvåg, Ø. S. Fjellvåg, S. Kumar, A. Ruud and A. O. Sjåstad, Interplay of valence states and magnetic interactions in the perovskite system LaNi1−xRhxO3, J. Solid State Chem., 2021, 298, 122124, DOI:10.1016/j.jssc.2021.122124.
- A. S. Fjellvåg, Platinum catchment by noble metal alloys and structural studies of Pt- and Rh-containing perovskites, PhD thesis, University of Oslo, 2022, ISSN 1501-7710.
- H. H. Sønsteby, E. Skaar, Ø. S. Fjellvåg, J. E. Bratvold, H. Fjellvåg and O. Nilsen, A foundation for complex oxide electronics -low temperature perovskite epitaxy, Nat. Commun., 2020, 11, 2872, DOI:10.1038/s41467-020-16654-2.
- H. J. Gysling, J. R. Monnier and G. Apai, Synthesis, characterization, and catalytic activity of LaRhO3, J. Catal., 1987, 103, 407–418, DOI:10.1016/0021-9517(87)90132-1.
- P. Watson, The formation of oxygen-containing organic molecules by the hydrogenation of carbon monoxide using a lanthanum rhodate catalyst, J. Catal., 1982, 74, 282–295, DOI:10.1016/0021-9517(82)90034-3.
- K. Ouchetto, F. Archaimbault, J. Choisnet and M. Et-Tabirou, New ordered and distorted perovskites: the mixed platinates Ln2MPtO6 (Ln = La, Pr, Nd, Sm, Eu, Gd; M = Mg, Co, Ni, Zn), Mater. Chem. Phys., 1997, 51, 117–124 CrossRef.
- A. Glavic and M. Björck, GenX 3: the latest generation of an established tool, J. Appl. Crystallogr., 2022, 55, 1063–1071, DOI:10.1107/S1600576722006653.
- J. Hessevik, C. S. Carlsen, O. Bestul, H. Fjellvåg and A. O. Sjåstad, Oxides for platinum capture in the ammonia oxidation process – a screening study, 2025.
- L. S. Kibis, A. I. Stadnichenko, S. V. Koscheev, V. I. Zaikovskii and A. I. Boronin, XPS Study of Nanostructured Rhodium Oxide Film Comprising Rh4+ Species, J. Phys. Chem. C, 2016, 120, 19142–19150, DOI:10.1021/acs.jpcc.6b05219.
- Y. A. Y. Abe, K. K. K. Kato, M. K. M. Kawamura and K. S. K. Sasaki, Electrical Properties of Amorphous Rh Oxide Thin Films Prepared by Reactive Sputtering, Jpn. J. Appl. Phys., 2000, 39, 245, DOI:10.1143/JJAP.39.245.
- Z. Weng-Sieh, R. Gronsky and A. T. Bell, Microstructural Evolution of γ-Alumina-Supported Rh upon Aging in Air, J. Catal., 1997, 170, 62–74, DOI:10.1006/jcat.1997.1738.
- K. B. Schwartz and C. T. Prewitt, Structural and electronic properties of binary and ternary platinum oxides, J. Phys. Chem. Solids, 1984, 45, 1–21, DOI:10.1016/0022-3697(84)90096-9.
- T. V. O’Halloran and S. J. Lippard, The Chemistry of Platinum in the +3 Oxidation State, Isr. J. Chem., 1985, 25, 130–137, DOI:10.1002/ijch.198500021.
- V. K. Kaushik, Identification of Mixed Platinum States and Electronic Effects of Support on Platinum in Supported Catalysts, Z. Phys. Chem., 1991, 173, 105–113, DOI:10.1524/zpch.1991.173.Part_1.105.
- L. Qiao and X. Bi, Direct observation of Ni3+ and Ni2+ in correlated LaNiO3−δ films, Europhys. Lett., 2011, 93, 57002, DOI:10.1209/0295-5075/93/57002.
- S. Mickevičius, S. Grebinskij, V. Bondarenka, B. Vengalis, K. Šliužienė, B. A. Orlowski, V. Osinniy and W. Drube, Investigation of epitaxial LaNiO3−x thin films by high-energy XPS, J. Alloys Compd., 2006, 423, 107–111, DOI:10.1016/j.jallcom.2005.12.038.
- A. Atkinson and C. Monty, Grain Boundary Diffusion in Ceramics, in Surf. Interfaces Ceram. Mater., ed. L.-C. Dufour, C. Monty and G. Petot-Ervas, Springer, Netherlands, Dordrecht, 1989, pp. 273–284 DOI:10.1007/978-94-009-1035-5_15.
- R. Westerström, J. G. Wang, M. D. Ackermann, J. Gustafson, A. Resta, A. Mikkelsen, J. N. Andersen, E. Lundgren, O. Balmes, X. Torrelles, J. W. M. Frenken and B. Hammer, Structure and reactivity of a model catalyst alloy under realistic conditions, J. Phys.:Condens. Matter, 2008, 20, 184018, DOI:10.1088/0953-8984/20/18/184018.
- O. Ivashenko, N. Johansson, C. Pettersen, M. Jensen, J. Zheng, J. Schnadt and A. O. Sjåstad, How Surface Species Drive Product Distribution during Ammonia Oxidation: An STM and Operando APXPS Study, ACS Catal., 2021, 11, 8261–8273, DOI:10.1021/acscatal.1c00956.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00974f |
| This journal is © The Royal Society of Chemistry 2025 |