
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Methacrylate-based copolymers as tunable hosts for triplet–triplet annihilation upconversion†
Michael J.
Bennison
 a,
Abigail R.
Collins
a,
Larissa
Gomes Franca
a,
Georgina H.
Burgoyne Morris
a,
Niamh
Willis-Fox
b,
Ronan
Daly
b,
Joshua K. G.
Karlsson
a,
Bethan L.
Charles
a and
Rachel C.
Evans
*a
a,
Abigail R.
Collins
a,
Larissa
Gomes Franca
a,
Georgina H.
Burgoyne Morris
a,
Niamh
Willis-Fox
b,
Ronan
Daly
b,
Joshua K. G.
Karlsson
a,
Bethan L.
Charles
a and
Rachel C.
Evans
*a
aDepartment of Materials Science and Metallurgy, University of Cambridge, CB3 0FS, UK. E-mail: rce26@cam.ac.uk
bInstitute for Manufacturing, Department of Engineering, University of Cambridge, 17 Charles Babbage Rd, Cambridge, CB3 0FS, UK
First published on 10th January 2025
Abstract
The ability to convert light to higher energies through triplet–triplet annihilation upconversion (TTA-UC) is attractive for a range of applications including solar energy harvesting, bioimaging and anti-counterfeiting. Practical applications require integration of the TTA-UC chromophores within a suitable host, which leads to a compromise between the high upconversion efficiencies achievable in liquids and the durability of solids. Herein, we present a series of methacrylate copolymers as TTA-UC hosts, in which the glass transition temperature (Tg), and hence upconversion efficiency can be tuned by varying the co-monomer ratios (n-hexyl methacrylate (HMA) and 2,2,2-trifluoroethyl methacrylate (TFEMA)). Using the model sensitiser/emitter pair of palladium(II) octaethylporphyrin (PdOEP) and diphenylanthracene (DPA), the upconversion quantum yield was found to increase with decreasing glass transition temperature, reaching a maximum of 1.6 ± 0.2% in air at room temperature. Kinetic analysis of the upconversion and phosphorescence decays reveal that increased PdOEP aggregation in the glassy polymers leads to a competitive non-radiative relaxation pathway that quenches the triplet state. Notably, the threshold intensity is highly sensitive to the glass transition temperature, ranging from 1250 mW cm−2 for PHMA90TFEMA10 (Tg = −9.4 °C) to ∼200 mW cm−2 for more ‘glassy’ hosts, e.g. PHMA33TFEMA67 (Tg = 20.1 °C), suggesting the TTA-UC mechanism switches from diffusion-based collisions to triplet exciton migration at localised sensitiser–emitter pairs.
Introduction
Two low-energy photons can be converted to one high-energy photon using sensitised triplet–triplet annihilation upconversion (TTA-UC).1,2 This process has wide-reaching applications including solar energy harvesting,3–5 photo-catalysis,6 sensors,7–9 bioimaging,10–14 and anti-counterfeiting.15 However, the uptake for some of these applications has been limited by the practicality of incorporating suitable TTA-UC systems in mass-produced devices.16,17 The complexity of the TTA-UC mechanism places particular demands on both the luminophores and the host materials in which it is intended to occur.Fig. 1 illustrates the TTA-UC mechanism, which uses a pair of luminophores: a sensitiser and an emitter. For efficient TTA-UC, the sensitiser should strongly absorb the incident photons to populate the first singlet excited state (S1), before undergoing intersystem crossing (ISC) at a high rate to populate the first triplet excited state (T1). On collision between a triplet-excited sensitiser and a ground-state emitter, a good energy match between T1 states should facilitate effective triplet–triplet energy transfer (TTET). Two triplet-excited emitters may then collide and undergo triplet–triplet annihilation (TTA), such that one emitter populates the higher-energy S1 state, while the second relaxes to the ground-state.18 Finally, this singlet-excited emitter should fluoresce with a high photoluminescence quantum yield (ΦPL), emitting a photon at the desired upconverted energy. These multimolecular processes require a high degree of luminophore mobility for the necessary collisions to occur. High rates of chromophore diffusion have been found to be the main factor in enhancement of the TTET efficiency, providing the emitter concentration is not too large (>10−1 M).19
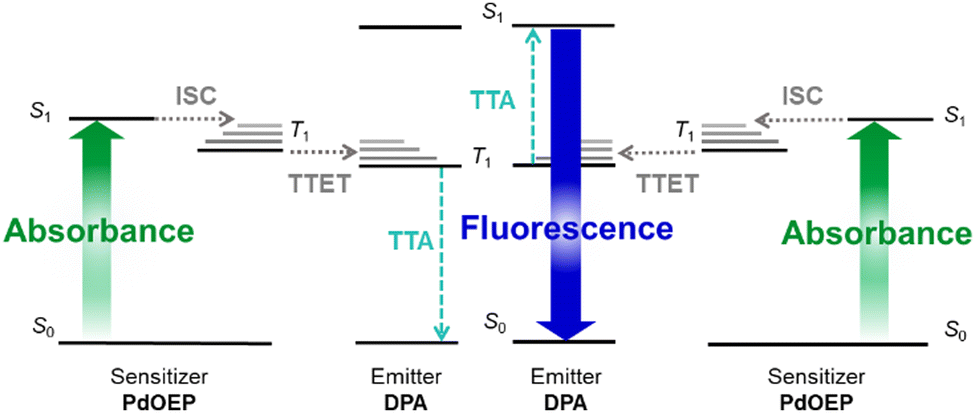 | ||
| Fig. 1 Energy level diagram illustrating the TTA-UC mechanism: absorption of an incident photon (green arrow) by a sensitiser, such as palladium(II) octaethylporphyrin (PdOEP), resulting in excitation from the ground state (S0) to the first singlet excited state (S1); intersystem crossing (ISC, grey dashed arrow), to the first triplet excited state (T1); triplet–triplet energy transfer (TTET, grey dashed arrow) from the sensitiser to an emitter such as 9,10-diphenylanthracene (DPA); triplet–triplet annihilation (TTA, turquoise dashed arrows) between two T1 excited emitters, such that one populates the S1 state and the other returns to S0; fluorescence (blue arrow) of the S1 emitter to return to the ground state. | ||
As a result, efficient TTA-UC has been demonstrated in liquid solutions, with upconversion quantum yields (ΦUC) exceeding 35% (as a two-photon process ΦUC has a maximum cap of 50%).20,21 However, potential issues with leakage and solvent evaporation make liquid systems impractical for device integration, so the development of solid-state hosts is desirable. These hosts should facilitate triplet exciton diffusion, allow homogeneous distribution of luminophores without aggregation, and possess high optical transparency, whilst retaining a robust yet flexible mechanical scaffold.22 Furthermore, for systems operating in air, ingress of molecular oxygen is detrimental due to quenching of triplet excited states.23 Therefore, unless the system is to be fabricated and encapsulated in air-free conditions,24,25 an ideal host should also act as an effective barrier to oxygen.26,27
Due to their tuneable properties, organic polymers have emerged as promising TTA-UC hosts.22 The host performance strongly depends on the glass transition temperature, Tg. Typically the TTA-UC efficiency is maximised using a polymer host whose Tg is below the required operating temperature.28 Chromophore mobility has been shown to have a crucial effect on UC activity, previously shown by lowering the operating temperature of a low Tg ethyleneoxide-epichlorohydrin polymer host.28 Above room temperature, the UC emission was clearly visible, whereas no UC was observed below 280 K which was comparable to the Tg of the material. It was concluded that for low chromophore concentrations where triplet exciton migration is not possible, fast mobility is essential for successful TTA-UC and the polymer host should be in its rubbery state. Cross-linked elastomers such as polyurethanes and alkyl acrylates are in their rubbery state at usual operating temperatures and provide excellent mechanical stability;28,29 however, they are difficult to recycle or reuse, which poses ethical questions around their sustainable use.30 Meanwhile, uncrosslinked polymers such as siloxanes operating well above their Tg behave increasingly as liquids, presenting the same issues of leakage and instability.31 To achieve a compromise between stability, efficiency, and processability, the ideal host would therefore be an uncrosslinked polymer whose Tg is close to, but still below, the intended working temperature.
Methacrylate polymers have been previously investigated as TTA-UC hosts.32–34 While upconversion was observed in poly(methyl methacrylate) (PMMA) glasses (a Tg = 92 °C), a high chromophore concentration (0.005% w/w PdOEP and 25% w/w DPA) was required.34 The strategy of increasing emitter concentration can lead to aggregation and subsequent quenching of emission.33 Polyacrylate elastomers of decreasing Tg were investigated by Monguzzi et al.29 at low chromophore concentrations (0.1 mM PdOEP, 10 mM DPA), where it was concluded that the diffusion length of excited chromophores is extended as the rigidity of host decreases. The host with the lowest Tg of −62 °C achieved the highest TTA-UC efficiency of 21%, highlighting the importance of the host state on overall performance.
Previous studies have focused on the development of a specific host system whose properties are tuned to suit specific operation conditions.19,35,36 Here, we take an alternative approach, in which we design a series of structurally-related polymer hosts, whose properties can be tuned to the device requirements. This is achieved through copolymerisation of a low Tg methacrylate monomer with a significantly higher Tg comonomer such that, by varying the ratio, a spectrum of intermediate Tg values may be targeted. The resultant copolymer hosts were doped with the benchmark TTA-UC sensitiser–emitter pair of palladium(II) octaethylporphyrin (PdOEP) and 9,10-diphenylanthracene (DPA) to assess the correlation between the thermal properties of the host, and the TTA-UC characteristics. Using a combination of steady-state and time-resolved spectroscopic analysis, we propose that the TTA-UC mechanism transitions from diffusion-based collisions to triplet exciton migration at localised sensitiser as the glass transition temperature of the host increases.
Results and discussion
Design of the copolymer host system
The choice of host system was determined by several requirements: (i) the polymer class should satisfy the outlined general host demands for TTA-UC; (ii) there should be a wide selection of functional monomers available; (iii) the identified monomers should be compatible with copolymerisation. The methacrylate homopolymer family has previously been demonstrated to be a suitable host for TTA-UC, with ΦUC from 0.08–3.5%,37–39 primarily using alkyl-functional monomers due to their low glass transition temperatures (Tg).29 Moreover, there is a wide variety of available methacrylate monomers suited to copolymerisation, which affords tunability in the Tg. We opted to use n-hexyl methacrylate (HMA), and 2,2,2-trifluoroethyl methacrylate (TFEMA) as monomers (Fig. 2), since their respective homopolymers show significant difference in their Tgs (−5 °C and 74 °C, respectively).40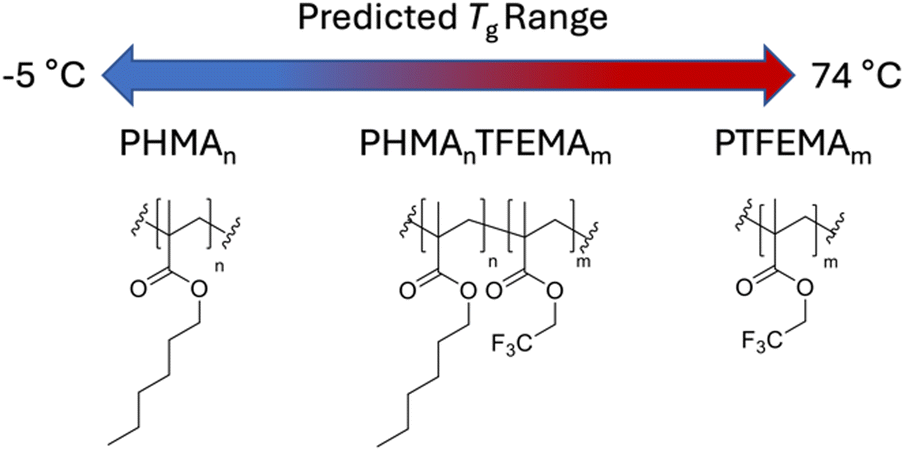 | ||
| Fig. 2 Chemical structures of the homopolymers, PHMA and PTFEMA, and the resultant copolymer poly(n-hexyl methacrylate-co-2,2,2-trifluoroethyl methacrylate) (PHMAnTFEMAm). The monomer ratios, n and m, were varied to access a range of glass transition temperatures, Tg. | ||
A series of poly(n-hexyl methacrylate-co-2,2,2-trifluoroethyl methacrylate) copolymers was synthesised using reversible-addition fragmentation transfer (RAFT) polymerisation, using 2-cyanopropan-2-yl dodecyl trithiocarbonate as a chain transfer agent (CTA)41 and varying ratios of TFEMA (m) and HMA (n) to tune the Tg, as outlined in Fig. 3 and Table 1. RAFT polymerisation was chosen to give control over the molecular weight (Mw) of the synthesised polymers, and as such minimise any contribution from Mw variation to the photophysical properties. All synthesised polymers were in the range 7400–16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 g mol−1 (Table 1). A key drawback of the RAFT method is that the CTA can give strong colouration to the final material, ranging from pink to yellow depending on the exact CTA used.42 In our case, the CTA resulted in a strong yellow colouration (see Fig. S4, ESI†), which would lead to parasitic absorption that would be detrimental to the TTA-UC efficiency. To overcome this, the CTA end group was removed post-polymerisation via reduction with azobisisobutyronitrile (AIBN) and tributyltin hydride to leave a single hydrogen as the chain end (Fig. 3, step 2). This mechanism has been reported to have a quantitative yield and to be particularly effective for methacrylic polymers.43 The resulting end-reduced polymers showed no coloration and retained good optical clarity (Fig. S4, ESI†). The final copolymers are denoted as PHMAnTFEMAm, where n and m are the molar percentages for HMA and TFEMA, respectively. All polymers, before and after end-reduction were fully characterised by 1H and 13C nuclear magnetic resonance (NMR) spectroscopy (Tables S1, S2 and Fig. S8–S39, ESI†) and size-exclusion chromatography (SEC, Fig. S40, ESI†). Full details of the synthetic methodology and characterisation data are available in the ESI† (Sections 1–3).
400 g mol−1 (Table 1). A key drawback of the RAFT method is that the CTA can give strong colouration to the final material, ranging from pink to yellow depending on the exact CTA used.42 In our case, the CTA resulted in a strong yellow colouration (see Fig. S4, ESI†), which would lead to parasitic absorption that would be detrimental to the TTA-UC efficiency. To overcome this, the CTA end group was removed post-polymerisation via reduction with azobisisobutyronitrile (AIBN) and tributyltin hydride to leave a single hydrogen as the chain end (Fig. 3, step 2). This mechanism has been reported to have a quantitative yield and to be particularly effective for methacrylic polymers.43 The resulting end-reduced polymers showed no coloration and retained good optical clarity (Fig. S4, ESI†). The final copolymers are denoted as PHMAnTFEMAm, where n and m are the molar percentages for HMA and TFEMA, respectively. All polymers, before and after end-reduction were fully characterised by 1H and 13C nuclear magnetic resonance (NMR) spectroscopy (Tables S1, S2 and Fig. S8–S39, ESI†) and size-exclusion chromatography (SEC, Fig. S40, ESI†). Full details of the synthetic methodology and characterisation data are available in the ESI† (Sections 1–3).
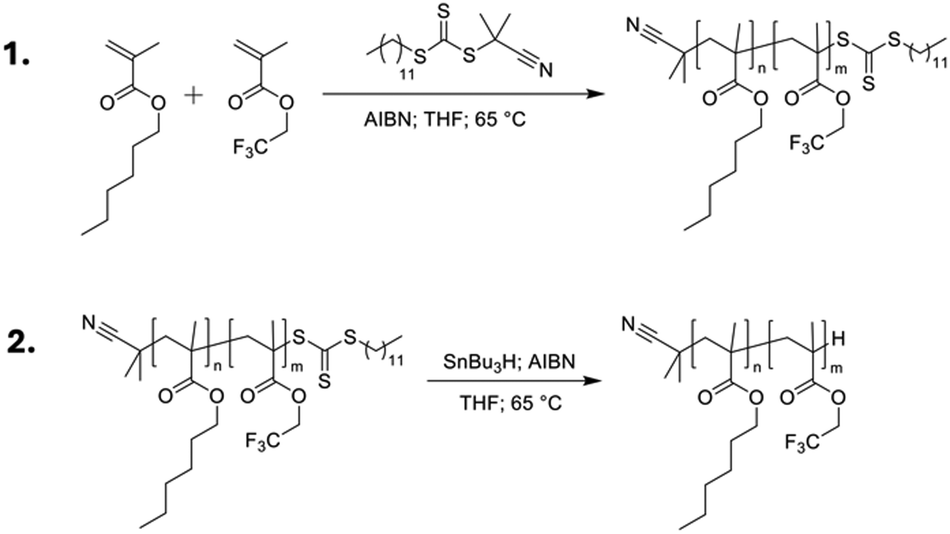 | ||
| Fig. 3 Synthesis of methacrylate copolymers. Step 1: polymerisation of n-hexyl methacrylate and 2,2,2-trifluoroethyl methacrylate via RAFT polymerisation. Step 2: Radical reduction of trithiocarbonate using AIBN and tributyl tin hydride to remove coloration from the final polymers. | ||
| Polymer | Form | T g(Calc) (°C) | T g (°C) | M n (g mol−1) | M w (g mol−1) | Đ |
|---|---|---|---|---|---|---|
| a Calculated glass transition temperature (from eqn (1)). b Measured glass transition temperature (from DSC). c Number-average molecular weight (from SEC). d Weight-average molecular weight (from SEC). e Dispersity, calculated as Mw/Mn. f Form varied with laboratory temperature due to overlap with Tg. | ||||||
| PTFEMA100 | Glassy solid | 74 | 43.3 | 7400 | 10300 |
1.40 |
| PHMA33TFEMA67 | Glassy solidf | 43 | 20.1 | 10200 |
13600 |
1.33 |
| PHMA50TFEMA50 | Rubbery solid | 29 | 16.6 | 10700 |
14300 |
1.34 |
| PHMA60TFEMA40 | Highly viscous liquid | 22 | 10.4 | 11500 |
15000 |
1.31 |
| PHMA67TFEMA33 | Highly viscous liquid | 17 | 1.0 | 12100 |
15600 |
1.29 |
| PHMA80TFEMA20 | Viscous liquid | 8 | −8.49 | 14300 |
18200 |
1.27 |
| PHMA90TFEMA10 | Viscous liquid | 1 | −9.42 | 15700 |
19500 |
1.24 |
| PHMA100 | Viscous liquid | −5 | −10.26 | 16400 |
20500 |
1.25 |
Thermal and flow properties
The ratios of HMA and TFEMA used to fabricate the copolymers were identified using the using the Fox equation (eqn (1)),44 which predicts the expected Tg of the copolymer based on the weight fractions Wi of each comonomer and the glass transition temperatures of their respective homopolymers (Tg,i):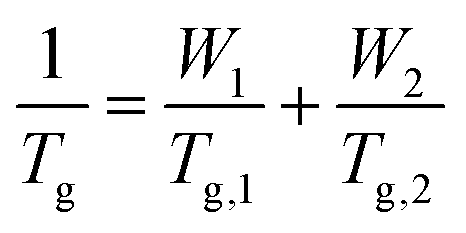 | (1) |
Differential scanning calorimetry (DSC) was performed to assess the validity of the predicted to experiment Tg values. As shown in Table 1, the measured Tg values are all notably lower than those predicted, which can be rationalised based on the relatively low Mw of the polymers, which results in a higher free volume due to the chain ends and hence lower Tg than those calculated for infinite chains. While these lower Tg values will result in a shift towards more liquid-like behaviour for all copolymers, the compositional region in which the behaviour of the synthesised polymers shifts from glassy to viscous liquid remains around 40–50% HMA. Crucially, the trend is as predicted: the Tg decreases as HMA content is increased, such that the PHMAnTFEMAm series spans a range of properties from glassy solids to viscous liquids at room temperature.
Steady-state optical properties
Polymers were doped with PdOEP (0.3 mM) and DPA (30 mM) as the TTA-UC sensitiser–emitter pair and cast as thick films (∼200 μm) on glass substrates. This chromophore pair has been extensively studied for green-blue upconversion, providing a useful benchmark to assess the host performance.35,45,46 The sensitiser-to-emitter ratio was selected based on previous studies using samples of similar thickness to facilitate comparison.26,29Fig. 4a presents the transmittance spectra for both undoped and doped PHMA50TFEMA50 copolymer films (see Fig. S42–S44, ESI† for corresponding data for all other samples). The undoped copolymers exhibit good optical transmittance in the visible range (85–95%). When doped with the TTA-UC luminophore pair, the polymer films retain high transmittance above 600 nm, but absorb strongly from 490–560 nm (PdOEP, Q band) and 320–420 nm (DPA S0 → S1 and PdOEP Soret band) as expected. The individual absorption and emission spectra of DPA and PdOEP in tetrahydrofuran solutions (5 μM) are provided in Fig. S45, ESI† for reference.Photoluminescence quantum yields (ΦPL) were measured for polymer films doped with 30 mM of DPA (DPA-only, Table S4, ESI†) and for the TTA-UC doped films (PdOEP:DPA, Table 2) to understand the impact of the host. In DPA-only samples, upon direct excitation at 375 nm, the ΦPL values exceeded 85% for all hosts (Fig. 5a), with the PHMA67TFEMA33 sample reaching 96%, comparable to that of DPA in solvents.47 In contrast, the PdOEP:DPA-doped polymer films exhibited a significant decrease in ΦPL, ranging from 33 to 50% due to parasitic absorption from the Soret band of PdOEP. As shown in Fig. 5a, the ΦPL increases with increasing HMA content, from 49.7% in PHMA67TFEMA33 to 52.8% in PHMA100 - the lowest Tg matrix.
:DPA (0.3 mM:30 mM) doped PHMAnTFEMAm hosts
| Polymer | Φ PL (%) | Φ UC (%) | UC lifetimes (collection at 440 nm)c | Phosphorescence lifetimes (collection at 660 nm)d | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| τ 1 (ms)/f1 (%) | τ 2 (ms)/f2 (%) | 〈τUC〉 (ms) | χ 2 | τ 1 (ms)/f1 (%) | τ 2 (ms)/f2 (%) | 〈τphos〉a (ms) | χ 2 | |||
| a Photoluminescence quantum yield of the emitter DPA (λex = 375 nm, λem = 380–530 nm). b Upconversion quantum yield at excitation power intensity of 1 W cm−2 (532 nm). c Lifetime fitting data for UC decay curves (average lifetime (〈τUC〉)), individual lifetimes (τi) and fractional contributions (fi) and goodness-of-fit (χ2). λex = 532 nm, λem = 440 nm. d Lifetime fitting data for phosphorescence decay curves (average lifetime (τPhos)) individual lifetimes (τi) and fractional contributions (fi) and goodness-of-fit (χ2). λex = 532 nm, λem = 660 nm. *There is no detectable signal. | ||||||||||
| PHMA100 | 53 ± 2 | 1.6 ± 0.2 | 1.1/29 | 3.1/71 | 2.52 | 1.085 | 0.36/22 | 2.3/78 | 1.88 | 1.453 |
| PHMA90TFEMA10 | 42 ± 3 | 1.4 ± 0.1 | 0.2/38 | 0.59/62 | 0.44 | 1.381 | 0.51/17 | 1.4/83 | 1.24 | 1.362 |
| PHMA80TFEMA20 | 46 ± 4 | 1.2 ± 0.2 | 0.18/42 | 0.48/58 | 0.35 | 1.365 | 0.29/11 | 1.4/89 | 1.27 | 1.413 |
| PHMA67TFEMA33 | 50 ± 4 | 1.3 ± 0.2 | 0.18/35 | 0.61/65 | 0.46 | 1.522 | 0.15/22 | 1.5/78 | 1.21 | 1.451 |
| PHMA60TFEMA40 | 43 ± 4 | 1.2 ± 0.2 | 0.15/41 | 0.43/59 | 0.32 | 1.388 | * | * | * | * |
| PHMA50TFEMA50 | 42 ± 3 | 0.7 ± 0.2 | 0.16/39 | 0.45/61 | 0.34 | 1.247 | * | * | * | * |
| PHMA33TFEMA67 | 34 ± 6 | 0.5 ± 0.1 | 0.11/57 | 0.27/43 | 0.18 | 1.358 | 0.09/29 | 0.71/71 | 0.53 | 1.400 |
| PTFEMA100 | 34 ± 4 | — | 0.027/72 | 0.074/28 | 0.04 | 1.134 | * | * | * | * |
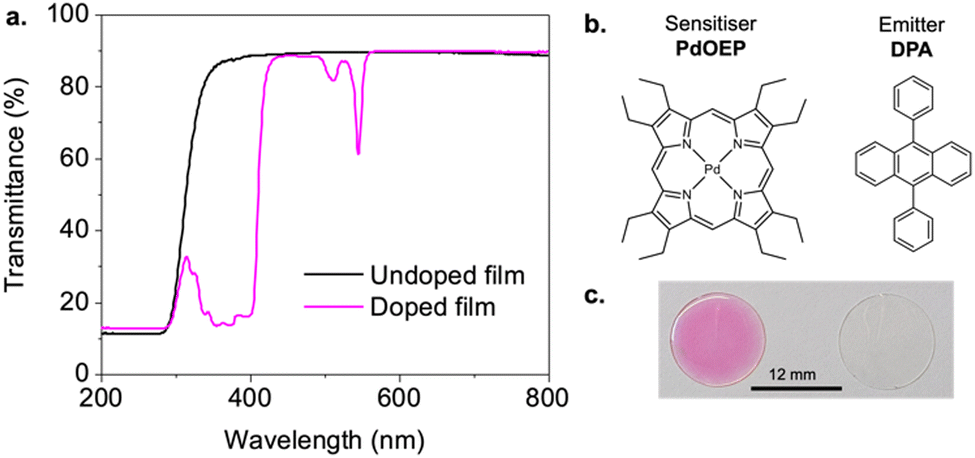 | ||
| Fig. 4 Optical properties of methacrylate copolymer hosts. (a) Transmittance spectra of undoped (black) and doped (pink) PHMA50TFEMA50 copolymer film. (b) Chemical structures of sensitiser PdOEP and emitter DPA. (c) Photographs of a lumophore-doped film (left), and undoped film (right). | ||
Upconversion characterisation
To quantify the upconversion activity in the different polymer hosts, the upconversion quantum yield was measured (see Section 2.6.1, ESI†). The ΦUC can be expressed as:35| ΦUC = ½fΦISCΦTTETΦTTAΦPL | (2) |
Fig. 5b illustrates how ΦUC varies as a function of HMA content in the polymer. The increase in ΦUC with HMA content can be attributed to the increase in chromophore mobility with decreasing Tg, with the highest ΦUC (1.6 ± 0.2%) obtained for PHMA100, the most liquid-like host. A transition can also be seen from a steep increase at lower HMA content to a shallower slope above ∼67% HMA. This corresponds to the transition in host properties, from glassy solids with Tgs above room temperature, to viscous liquids with sub-ambient Tg values. For the more liquid-like hosts, the diffusivity will be higher, and hence less limiting to the upconversion efficiency. Notably, PHMA67TFEMA33 presents promising results, as it combines a ΦUC of 1.3 ± 0.3%, comparable to those in lower-Tg hosts, with solid-like behaviour. This is preferable for applications at and above room temperature, compared to more liquid films like those of PHMA100. While these efficiencies are below that of the highest performing acrylate hosts of 21%,29 the purpose of this study is to investigate the tunability of host properties via copolymerisation to suit a variety of applications at varying operating temperatures. Such studies in methacrylate polymers are usually conducted in melt-processed glassy matrixes with high chromophore concentrations via triplet exciton migration,32–34 which rely on close chromophore proximity, and are avoided in the low Tg host and mechanism presented here.
Threshold intensity
Fig. 5c shows the threshold intensity (Ith) for three PdOEP:DPA-doped copolymers: PHMA90TFEMA10, PHMA67TFEMA33 and PHMA33TFEMA67. These hosts were selected to compare systems with low, ambient and high Tg values. Ith, also known as the power density threshold, is the excitation at which the ΦTTA is 50% of the maximum value.48,49 Experimentally, Ith is determined by locating the point at which the TTA-UC emission intensity shifts from a quadratic (where the slope of the emission intensity relative to power is 2) to a first-order dependence on incident light intensity (where the slope is 1).50 Systems with lower thresholds are generally favoured and will convert more efficiently under low-power sources such as natural sunlight (100 mW cm−2).48,51 It should be noted that, in our systems, at laser power densities >500 mW cm−2, photobleaching occurred before the full emission spectrum could be measured which led to a plateau at high intensities, which is not an uncommon issue with this measurement.52 Photodegradation at high power was quantified for each film (Fig. S46, ESI†), where copolymers show a higher resistance to degradation than the mono-polymers. All samples showed fast photodegradation at very high laser power (4 W cm−2) as demonstrated by the decreased UC emission intensity and the appearance of clear spot on the film at the point of laser incidence.For the more liquid-like PHMA90TFEMA10 (Tg = −9.4 °C) the transition from quadratic to first-order dependence transition is clearly discernible and corresponds to Ith = 995 mW cm−2. In contrast, for the more ‘glassy’ hosts, PHMA33TFEMA67 (Tg = 20.1 °C) and PHMA67TFEMA33 (Tg = 1.0 °C), this transition is more subtle and leads to significantly lower Ith values, around 220–250 mW cm−2. This large decrease in the Ith value indicates that changing the Tg of the host can completely alter the TTA-UC mechanism. Low threshold intensities have been reported for crystalline TTA-UC materials, which often exhibit large triplet exciton diffusion lengths of the acceptor molecules due to localised chromophore aggregation.53–56 It seems plausible that increased chromophore aggregation will occur in the higher Tg hosts (see following section), which would support a localised exciton diffusion mechanism and the observed decrease in Ith. Note that although the steady-state photoluminescence spectra indicate a reduction in the first vibronic peak due to reabsorption (Fig. S47, ESI†), lifetime measurements suggest DPA aggregation also makes a contribution (Fig. S48 and Table S5, ESI†). Compared to the monoexponential decay of DPA in dilute THF solution, DPA-only doped methacrylate copolymer films show a biexponential decay, where the short lifetime, τ1 (∼5 ns) corresponds to quenched species due to aggregation, and τ2 (∼10 ns) exhibits a longer lifetime associated with the reabsorption effect. Importantly, the contribution of τ1 is significantly higher for DPA-only PTFEMA100, supporting the argument that stronger aggregation is observed in the high Tg hosts. In contrast, in “soft” hosts, such as PHMA90TFEMA10, Brownian diffusion of the acceptor molecules is expected to determine and limit the maximum achievable triplet exciton diffusion length of the acceptor,57 leading to the large Ith. This change in mechanism would be expected to affect multiple parameters, such as the rates of ISC, TTET, and triplet lifetimes.49
Time-resolved emission measurements
Lifetime measurements were performed to gain better understanding of the variation in upconversion behaviour across the series of polymers. The polymers vary significantly in Tg and, consequently, in viscosity of the host matrix, which is expected to affect the kinetics of excited state decay of both luminophores (DPA and PdOEP). Fig. 6a shows the upconversion decay traces for the series of PdOEP:DPA-doped polymer films (collected at 440 nm). The UC lifetimes (τUC) values were estimated by tail-fitting a biexponential decay function. Fig. 6b and Table 2 summarise the average UC lifetimes (〈τUC〉), which ranged from 0.04 ms in PTFEMA100 to 2.51 ms in PHMA100. In agreement with the ΦUC values, a significant increase in the 〈τUC〉 is observed as the HMA content of the polymer films increased. This suggests that for the viscous liquid film (PHMA100), the higher ΦUC value is accompanied by a longer 〈τUC〉 indicating reduced competitive quenching mechanisms in this matrix.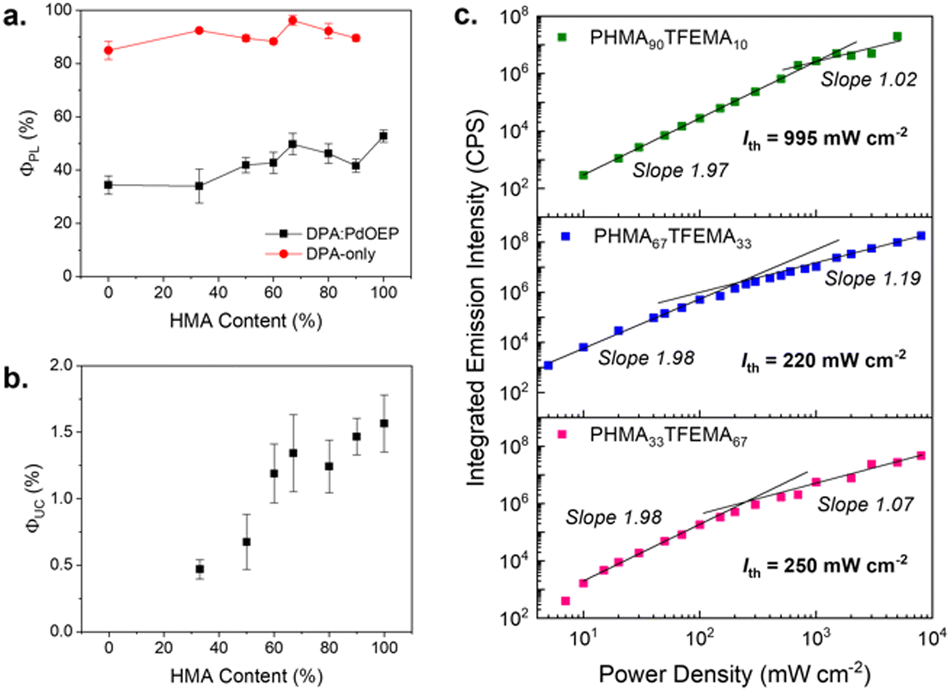 | ||
| Fig. 5 Upconversion and photoluminescence of methacrylate copolymers. (a) Photoluminescence quantum yield of the emitter DPA with sensitiser present (red circles) and without sensitiser (black squares) with increasing hexyl content of the copolymer, λex = 375 nm, λem = 380–530 nm. (b) Upconversion quantum yield at excitation intensity of 1 W cm−2, λex = 532 nm, λem = 380–500 nm. (c) Threshold intensity was determined from a logarithmic plot of integrated UC emission intensity at varying laser power (532 nm) for PHMA90TFEMA10 (green), PHMA67TFEMA33 (blue), and PHMA33TFEMA67 (pink). | ||
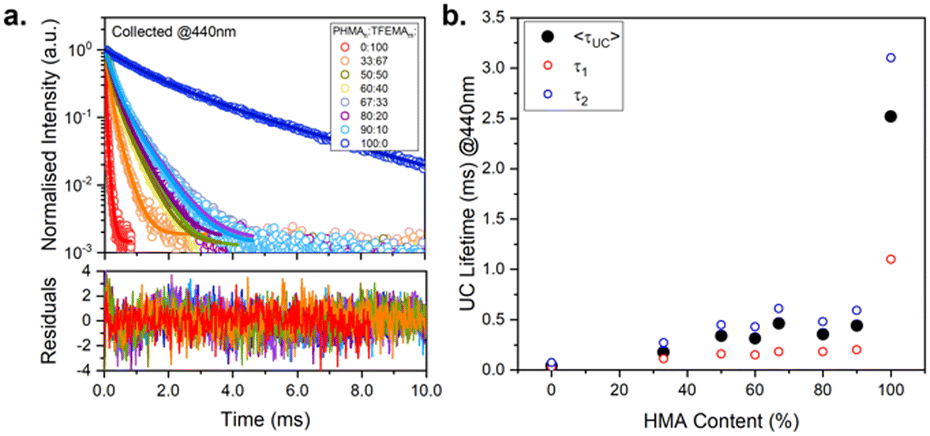 | ||
| Fig. 6 Upconversion kinetic studies on PdOEP:DPA (0.3 mM:30 mM) doped in different PHMAnTFEMAm host matrices. (a) Upconversion decay traces (open symbols) and biexponential decay fits (solid lines) and corresponding residuals. λex = 532 nm, λem = 440 nm. (b) Corresponding upconversion average lifetimes (filled black circles) and individual lifetime components (τ1 and τ2) obtained from decay fits. | ||
The average phosphorescence lifetimes in air (〈τphos〉, collection at 660 nm, Fig. S48, ESI†) were obtained for the same samples from biexponential tail fits, comprising a short lifetime component, τ1 ∼0.1–0.5 ms, and a longer-lived component, τ2 ∼1.4 ms, as summarised in Table 2. We assign τ2 to the natural phosphorescence lifetime, since in previous studies of PdOEP-only in polymer hosts, a characteristic τphos of 1.4 ms was observed in the absence of oxygen.58 The shorter lifetime observed in these PdOEP:DPA-doped polymer samples, τ1, is assigned to the quenched triplet state, which arises from competing relation pathways such as aggregation-caused quenching, oxygen quenching and TTET. We observed that polymer hosts with particularly low Tg, e.g. PHMA90TFEMA10, exhibit a high fractional contribution of τ2, even in air. Meanwhile, those with higher Tg – e.g. PHMA33TFEMA67-show greater fractional contribution from τ1, leading to a significantly shorter 〈τphos〉. This same trend is also observed in the analogous PdOEP-only doped films (Fig. S49 and Table S5, ESI†).
To further understand the triplet relaxation mechanisms, phosphorescence decay curves (Fig. 7 and Table S6, ESI†) were measured for representative low (PHMA100) and high (PTFEMA100) Tg homopolymer hosts doped with PdOEP-only, in air and N2 atmospheres. In both cases, τ2 became more prevalent with deaeration, further supporting the assignment of this component to the natural phosphorescence lifetime of the unquenched species (around 1.2–1.5 ms). Its presence even under ambient conditions suggests that the second coverslip used to aid sample handling prevented further oxygen ingress during the measurement, with any local oxygen already present inside the matrix being rapidly photoconsumed, especially in PHMA100 where oxygen is expected to diffuse more readily. On the other hand, for PTFEMA100, even in the absence of oxygen, the decay trace still exhibited a significant contribution from τ1, suggesting that aggregation-caused quenching is a significant relaxation pathway. We note that as DPA is not present in these samples, the contribution from TTET is also excluded. The phosphorescence lifetime analysis thus supports the presence of increased PdOEP aggregation in the high Tg polymer hosts. Aggregation is also evident in the DPA fluorescence lifetimes (Fig. S48 and Table S5, ESI†), where PdOEP:DPA-doped films exhibit shorter average lifetimes than DPA-only doped films, suggesting additional non-radiative pathways for these samples. This can result in longer triplet exciton diffusion lengths for the acceptors, consistent with the lower threshold intensity observed in these samples.
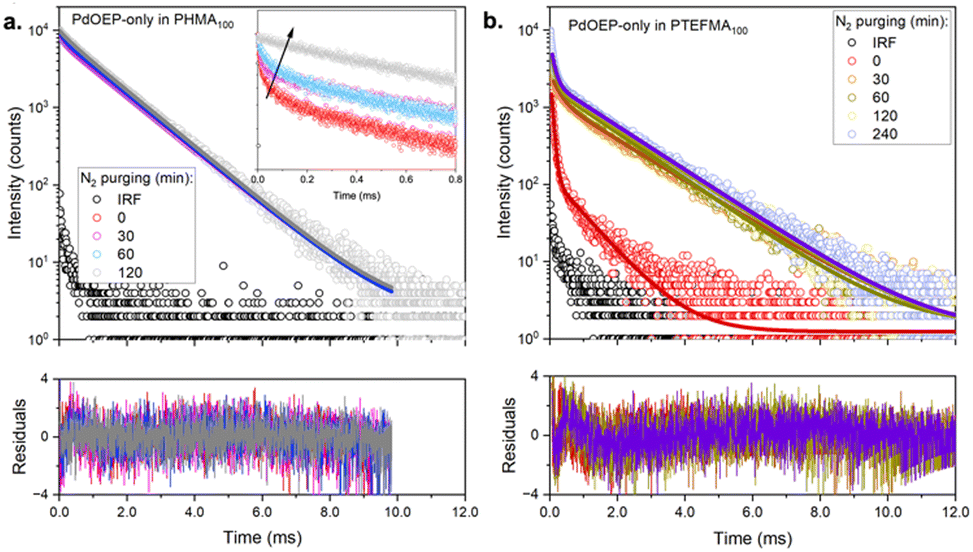 | ||
| Fig. 7 Phosphorescence decay traces of PdOEP-only (0.3 mM) doped in: (a) PHMA100 and (b) PTFEMA100 under varying durations of nitrogen (N2) purging. Inset: Zoom in on the first millisecond regimes to highlight the increase in the shorter lifetime after increased N2 purging. Measurements were performed using a 532 nm excitation source and collected at 660 nm. | ||
Conclusions
We have demonstrated the formation of tunable TTA-UC host materials through RAFT copolymerisation of two structurally-similar methacrylate monomers, HMA and TFEMA. Through variation of the co-monomer ratios, a series of PHMAnTFEMAm copolymers were synthesised, whose glass transition temperatures spanned a range from below to above room temperature. For the model TTA-UC pair (PdOEP:DPA), the upconversion quantum yields increased with decreasing glass transition temperature, across the range 0.5–1.6% in air at room temperature. This is accompanied by a five-fold decrease in the threshold intensity, which we propose is due to increased luminophore aggregation in the higher Tg hosts, leading to a switch in mechanism from diffusion-based collisions to triplet exciton migration at localised sensitiser–emitter pairs. Notably, this work demonstrates the versatility of copolymerisation to optimise the physical form to meet a required operating temperature or application, without dramatically changing the upconversion efficiency. Further tuning of the chromophore concentrations and limiting of oxygen diffusion and subsequent quenching could be employed in future work to improve efficiency of these hosts. The insight gained through this study will inform the design of next generation TTA-UC hosts, with the aim of increasing the upconversion efficiency in real-world conditions.Data availability
Data for this article are available at the University of Cambridge Apollo repository at https://www.repository.cam.ac.uk/home.Author contributions
M. J. B.: conceptualisation, methodology, investigation, data curation, formal analysis, writing – original draft, writing – reviewing & editing. A. R. C.: investigation, data curation, formal analysis, writing – original draft, reviewing & editing. L. G. F.: investigation, data curation, formal analysis, writing – original draft, writing – reviewing & editing. G. H. B. M.: data curation, formal analysis, writing – reviewing & editing. N. W. F.: investigation, data curation. R. D.: resources. J. J. G. K.: investigation and data curation. B. L. C.: investigation and data curation. R. C. E.: conceptualisation, project administration, resources, funding acquisition, supervision, writing – reviewing & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (Grant Agreement No. 818762 - SPECTRACON). LGF acknowledges the Research Fellowship awarded by the Royal Commission for the Exhibition of 1851 (RF2023/3). GBM thanks the EPSRC (EP/W524633/1) for a PhD studentship.Notes and references
- C. A. Parker, C. G. Hatchard and T. A. Joyce, Nature, 1965, 205, 1282–1284 CrossRef CAS.
- C. A. Parker and C. G. Hatchard, Proc. R. Soc. London, Ser. A, 1962, 269, 574–584 Search PubMed.
- M. J. Y. Tayebjee, D. R. McCamey and T. W. Schmidt, J. Phys. Chem. Lett., 2015, 6, 2367–2378 CrossRef CAS PubMed.
- J. Pedrini and A. Monguzzi, J. Photonics Energy, 2017, 8, 1 Search PubMed.
- D. Beery, T. W. Schmidt and K. Hanson, ACS Appl. Mater. Interfaces, 2021, 13, 32601–32605 CrossRef CAS PubMed.
- T. Yu, Y. Liu, Y. Zeng, J. Chen, G. Yang and Y. Li, Chem. – Eur. J., 2019, 25, 16270–16276 CrossRef CAS PubMed.
- Y. Sun, L. Wei, S. Zhu, P. Jin, C. He, Q. He, C. Yang and W. Wu, Sens. Actuators, B, 2023, 387, 133764 CrossRef CAS.
- M. Xu, X. Zou, Q. Su, W. Yuan, C. Cao, Q. Wang, X. Zhu, W. Feng and F. Li, Nat. Commun., 2018, 9, 2698 CrossRef PubMed.
- A. A. Mendonsa, C. C. Soeldner, N. E. Mudd, S. C. Saccomano and K. J. Cash, ACS Sens., 2023, 8, 3043–3050 CrossRef CAS PubMed.
- Q. Dou, L. Jiang, D. Kai, C. Owh and X. J. Loh, Drug Discovery Today, 2017, 22, 1400–1411 CrossRef CAS PubMed.
- W. Lin, J. Li, H. Feng, F. Qi and L. Huang, J. Anal. Test., 2023, 7, 327–344 CrossRef.
- S. H. C. Askes, W. Pomp, S. L. Hopkins, A. Kros, S. Wu, T. Schmidt and S. Bonnet, Small, 2016, 12, 5579–5590 CrossRef CAS PubMed.
- O. S. Kwon, H. S. Song, J. Conde, H. Kim, N. Artzi and J.-H. Kim, ACS Nano, 2016, 10, 1512–1521 CrossRef CAS PubMed.
- B. Zhang, K. D. Richards, B. E. Jones, A. R. Collins, R. Sanders, S. R. Needham, P. Qian, A. Mahadevegowda, C. Ducati, S. W. Botchway and R. C. Evans, Angew. Chem., Int. Ed., 2023, 62, e202308602 CrossRef CAS PubMed.
- W. Yin, T. Yu, J. Chen, R. Hu, G. Yang, Y. Zeng and Y. Li, ACS Appl. Mater. Interfaces, 2021, 13, 57481–57488 CrossRef CAS PubMed.
- B. Joarder, N. Yanai and N. Kimizuka, J. Phys. Chem. Lett., 2018, 9, 4613–4624 CrossRef CAS PubMed.
- A. K. Williams, B. J. Davis, E. R. Crater, J. R. Lott, Y. C. Simon and J. D. Azoulay, J. Mater. Chem. C, 2018, 6, 3876–3881 RSC.
- L. G. Franca, D. G. Bossanyi, J. Clark and P. L. dos Santos, ACS Appl. Opt. Mater., 2024 DOI:10.1021/acsaom.4c00041.
- A. Monguzzi, R. Tubino and F. Meinardi, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 155122 CrossRef.
- Y. Zhou, F. N. Castellano, T. W. Schmidt and K. Hanson, ACS Energy Lett., 2020, 5, 2322–2326 CrossRef CAS.
- N. Yanai, K. Suzuki, T. Ogawa, Y. Sasaki, N. Harada and N. Kimizuka, J. Phys. Chem. A, 2019, 123, 10197–10203 CrossRef CAS PubMed.
- M. J. Bennison, A. R. Collins, B. Zhang and R. C. Evans, Macromolecules, 2021, 54, 5287–5303 CrossRef CAS PubMed.
- V. Gray, K. Moth-Poulsen, B. Albinsson and M. Abrahamsson, Coord. Chem. Rev., 2018, 362, 54–71 CrossRef CAS.
- S. Baluschev, K. Katta, Y. Avlasevich and K. Landfester, Mater. Horiz., 2016, 3, 478–486 RSC.
- D. Dzebo, K. Moth-Poulsen and B. Albinsson, Photochem. Photobiol. Sci., 2017, 16, 1327–1334 CrossRef CAS PubMed.
- A. R. Collins, B. Zhang, M. J. Bennison and R. C. Evans, J. Mater. Chem. C, 2024, 12, 6310–6318 RSC.
- A. J. Svagan, D. Busko, Y. Avlasevich, G. Glasser, S. Baluschev and K. Landfester, ACS Nano, 2014, 8, 8198–8207 CrossRef CAS PubMed.
- T. N. Singh-Rachford, J. Lott, C. Weder and F. N. Castellano, J. Am. Chem. Soc., 2009, 131, 12007–12014 CrossRef CAS PubMed.
- A. Monguzzi, M. Mauri, A. Bianchi, M. K. Dibbanti, R. Simonutti and F. Meinardi, J. Phys. Chem. C, 2016, 120, 2609–2614 CrossRef CAS.
- D. J. Fortman, J. P. Brutman, G. X. De Hoe, R. L. Snyder, W. R. Dichtel and M. A. Hillmyer, ACS Sustainable Chem. Eng., 2018, 6, 11145–11159 CrossRef CAS.
- J. Fang, C. Zhou, Y. Chen, L. Fang, W. Wang, C. Zhu, Y. Ni and C. Lu, ACS Appl. Mater. Interfaces, 2020, 12, 717–726 CrossRef CAS PubMed.
- S. H. Lee, M. A. Ayer, R. Vadrucci, C. Weder and Y. C. Simon, Polym. Chem., 2014, 5, 6898–6904 RSC.
- S. Raišys, S. Juršėnas and K. Kazlauskas, Sol. RRL, 2022, 6, 2100873 CrossRef.
- S. H. Lee, J. R. Lott, Y. C. Simon and C. Weder, J. Mater. Chem. C, 2013, 1, 5142 RSC.
- R. R. Islangulov, J. Lott, C. Weder and F. N. Castellano, J. Am. Chem. Soc., 2007, 129, 12652–12653 CrossRef CAS PubMed.
- S. H. Lee, D. C. Thévenaz, C. Weder and Y. C. Simon, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1629–1639 CrossRef CAS.
- A. Monguzzi, A. Oertel, D. Braga, A. Riedinger, D. K. Kim, P. N. Knüsel, A. Bianchi, M. Mauri, R. Simonutti, D. J. Norris and F. Meinardi, ACS Appl. Mater. Interfaces, 2017, 9, 40180–40186 CrossRef CAS PubMed.
- L. Jiang, L. Zeng, M. Zhang, L. Huang and D. Pang, Adv. Opt. Mater., 2024, 12, 2301879 CrossRef CAS.
- D. Yildiz, C. Baumann, A. Mikosch, A. J. C. Kuehne, A. Herrmann and R. Göstl, Angew. Chem., Int. Ed., 2019, 58, 12919–12923 CrossRef CAS PubMed.
- J. Brandrup, E. H. Immergut, E. A. Grulke, A. Abe and D. R. Bloch, Polymer Handbook, John Wiley & Sons, 4th edn, 2005 Search PubMed.
- B. A. Abel and C. L. McCormick, Macromolecules, 2016, 49, 465–474 CrossRef CAS.
- S. Perrier, P. Takolpuckdee and C. A. Mars, Macromolecules, 2005, 38, 2033–2036 CrossRef CAS.
- Y. K. Chong, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2007, 40, 4446–4455 CrossRef CAS.
- T. G. Fox, Bull. Am. Phys. Soc., 1956, 1, 123 CAS.
- T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560–2573 CrossRef CAS.
- V. Gray, D. Dzebo, M. Abrahamsson, B. Albinsson and K. Moth-Poulsen, Phys. Chem. Chem. Phys., 2014, 16, 10345–10352 RSC.
- L. Hou, A. Olesund, S. Thurakkal, X. Zhang and B. Albinsson, Adv. Funct. Mater., 2021, 31, 2106198 CrossRef CAS.
- A. Monguzzi, R. Tubino, S. Hoseinkhani, M. Campione and F. Meinardi, Phys. Chem. Chem. Phys., 2012, 14, 4322 RSC.
- N. A. Durandin, J. Isokuortti, A. Efimov, E. Vuorimaa-Laukkanen, N. V. Tkachenko and T. Laaksonen, J. Phys. Chem. C, 2019, 123, 22865–22872 CrossRef CAS.
- P. Duan, D. Asthana, T. Nakashima, T. Kawai, N. Yanai and N. Kimizuka, Faraday Discuss., 2017, 196, 305–316 RSC.
- T. Ogawa, N. Yanai, A. Monguzzi and N. Kimizuka, Sci. Rep., 2015, 5, 10882 CrossRef CAS PubMed.
- F. Edhborg, A. Olesund and B. Albinsson, Photochem. Photobiol. Sci., 2022, 21, 1143–1158 CrossRef CAS PubMed.
- P. B. Merkel and J. P. Dinnocenzo, J. Lumin., 2009, 129, 303–306 CrossRef CAS.
- P. Duan, N. Yanai, H. Nagatomi and N. Kimizuka, J. Am. Chem. Soc., 2015, 137, 1887–1894 CrossRef CAS PubMed.
- P. Duan, N. Yanai and N. Kimizuka, J. Am. Chem. Soc., 2013, 135, 19056–19059 CrossRef CAS PubMed.
- K. Narushima, S. Hirata and M. Vacha, Nanoscale, 2017, 9, 10653–10661 RSC.
- A. Monguzzi, J. Mezyk, F. Scotognella, R. Tubino and F. Meinardi, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 195112 CrossRef.
- P. Douglas and K. Eaton, Sens. Actuators, B, 2002, 82, 200–208 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic methodology and structural characterisation data of homo- and copolymers, pre- and post-chain end reduction; materials, instrumentation and experimental methods, polymer synthesis and characterisation, supporting experimental data: NMR spectra, SEC traces, DSC curves, steady-state optical properties, photoluminescence quantum yields, phosphorescence lifetimes in air and N2. See DOI: https://doi.org/10.1039/d4ma01221f |
| This journal is © The Royal Society of Chemistry 2025 |