
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient oxidative coupling of amines to imines under natural sunlight using a benzothiadiazole-based molecular photocatalyst†
Ajeet
Singh
 a,
Bidisa
Das
*b and
Saumi
Ray
*a
a,
Bidisa
Das
*b and
Saumi
Ray
*a
aDepartment of Chemistry Birla Institute of Technology and Science (BITS), Pilani, Pilani Campus, Rajasthan 333031, India. E-mail: p20200434@pilani.bits-pilani.ac.in; saumi@pilani.bits-pilani.ac.in
bResearch Institute for Sustainable Energy (RISE), TCG-CREST, Salt Lake, 700091, Kolkata, India. E-mail: bidisa.das@tcgcrest.org
First published on 29th January 2025
Abstract
Developing a ‘greener’ avenue for organic synthesis is a key challenge, which must focus on energy efficiency as well as sustainability. Harnessing solar energy to chemical energy is an efficient way to utilize renewable energy resources. Herein, we report a D–A-type (donor–acceptor-type) small organic molecular photocatalyst (SOMP) “Ph-BT-Ph” with benzothiadiazole as the primary photoactive unit for oxidative coupling of amines to synthesize imines. Photocatalyst Ph-BT-Ph is synthesized using a Suzuki–Miyaura coupling reaction and thoroughly characterized by 1H-NMR, HRMS, and cyclic voltammetry studies. Photoluminescence and lifetime studies of Ph-BT-Ph show a high excited state reduction potential (−1.37 V vs. Ag/AgCl) and longer lifetime (12.64 ns) which make it suitable for photocatalytic organic transformations. The photocatalytic activity of the catalyst has been evaluated on the direct oxidative coupling reaction of amines to synthesize imines in the presence of natural sunlight and O2 as a green oxidant. Catalyst Ph-BT-Ph exhibits excellent photocatalytic performance under optimal reaction conditions by converting >99% amine to imine with >98% selectivity within 2 hours. This high photocatalytic efficiency has been achieved by purging oxygen only for 2 minutes and without any mechanical energy input (no stirring). Quite a moderate amount of catalyst (0.13 mol%) has been employed which results in a high catalytic turnover frequency of 381 h−1. EPR spectroscopy and theoretical studies are performed to understand the reaction mechanism and to determine the active sites of the catalyst. The Ph-BT-Ph catalyst surpasses the photocatalytic efficiencies of many reported metal-free catalysts for oxidative coupling of amines. Such SOMPs, with easily tunable absorption range and well-defined energy-band positions, offer a new class of metal-free and photoactive catalysts for organic synthesis with outstanding performance under greener reaction conditions.
Introduction
Organic synthetic chemistry has made a substantial contribution to enhancing the quality of everyday life; however, the conventional methods of organic synthesis come with disadvantages like harsh reaction conditions for high energy requirements and usage of toxic chemicals which contribute to environmental pollution on one hand and cost more on the other. Therefore, developing a sustainable technique for organic synthesis is crucial,1,2 and photocatalysis plays an important role in averting these challenges.3 Photocatalysis is considered an important and efficient strategic approach for organic transformations as it promotes an atom economic reaction and is able to operate under moderate reaction conditions with the utilization of renewable energy sources, such as solar energy.4 Homogeneous photocatalysts, such as metal complexes5–8 (Ru, Ir, Cr, Cu, Mn, and Co) and organic dyes9–14 (xanthenes, cyanoarenes, and acridinium), have received a lot of attention in recent decades because of their high activity, well-defined active sites, and superior selectivity. Therefore, these homogeneous photocatalysts are attractive choices for various organic transformations6,15,16 such as oxidation, reduction, and coupling processes.17Benzothiadiazole-based18 donor–acceptor (D–A) type photoactive materials have achieved a lot of attention because of their efficient response to visible light, robust electron-withdrawing capacity, semiconductor-like electronic structure with tunable band-gap, outstanding stability and tailorable excited-state lifespan.19 These photoactive materials have been utilized in various applications, including sensors,20 OLEDs,21 photovoltaic cells,22 biological imaging,23 and optical and electrical devices.24 While conjugated microporous polymers (CMPs) based on benzothiadiazole have been used in various organic transformation processes, such as redox reactions, coupling reactions, etc., the semiconducting nature can be exploited for easy band engineering by synthetic approaches. According to the literature, the reduction potential of most of the benzothiazole-based photoactive compounds is higher than −0.6 eV which is sufficient enough to activate the molecular oxygen and these compounds are characterized by a longer excited-state lifetime.25–27 A combination of these characteristics makes them an excellent choice for molecular oxygen activation to reactive oxygen species. Superoxide radical anion, hydroxyl radical, hydroperoxyl radical, and singlet oxygen are the reactive oxygen species generated through either electron transfer or energy transfer processes catalysed by benzothiazole-based photoactive compounds.
Direct photocatalytic conversion of amines to synthesize imines is a promising technique for organic transformation as it utilizes oxygen as a green oxidant and photon energy, which eliminates the need for harsh reaction conditions.28–32 Imines have paramount significance as intermediates of other biologically and medicinally important chemicals, such as α-amino acids, α-amino alcohols, and heterocyclic molecules.33–37 In recent years, many photocatalytic systems have been developed which are based on benzothiadiazole as the core photoactive unit functioning under visible light, such as supramolecular coordination polymers (SCPs),38 covalent organic frameworks (COFs),39–41 and conjugated microporous polymers (CMPs),42–46 which convert amines to imines using a direct photocatalytic method. Herein, we report a small organic molecular photocatalyst (SOMP) “Ph-BT-Ph” synthesized using Suzuki–Miyaura coupling of 4,7-dibromobenzo[c]-1,2,5-thiadiazole and phenylboronic acid. This photocatalyst shows excellent catalytic efficacy for the transformation of amines under natural sunlight using a molecular oxygen oxidant and without any mechanical energy input (scheme shown in Fig. 1). A few studies in the existing literature reported on utilizing only natural sunlight to drive chemical reactions. The use of expensive light sources and a continuous supply of oxygen limit their practical use in large-scale organic synthesis. Our study maximizes the use of solar energy consisting of ∼52% infrared (IR, 700–2500 nm) ∼43% visible (400–700 nm), and ∼5% ultraviolet (UV, 300–400 nm) light. The utilization of natural sunlight as an energy source and oxygen as an oxidant in the present study shows promise for sustainable large-scale organic synthesis.
 | ||
| Fig. 1 Schematic representation of the photocatalytic oxidative coupling of amines to imines using Ph-BT-Ph under natural sunlight. | ||
Experimental section
Synthesis of the photocatalyst (Ph-BT-Ph)
The photocatalyst was synthesized following a reported method.47 To a 15 mL Schlenk tube, 1 mmol of 4,7-dibromobenzo[c]-1,2,5-thiadiazole, 3 mmol of phenylboronic acid, and 4 mol% (0.04 mmol) Pd(PPh3)4 in 5 mL of toluene were added (shown in Scheme 1). The solution was then sonicated for 2 minutes and purged with N2 gas to create an inert environment. Then, the reaction mixture was heated at 90 °C for an hour and 2 mL of 2 M K2CO3 solution was added to the reaction mixture and heated for an additional 18 hours at 90 °C. After that, the reaction mixture was poured into water, extracted with ethyl acetate and dried over BaSO4. After the removal of the solvent, the resulting powder was washed with hot methanol to obtain a greenish-yellow compound. (254 mg, 88% yield). | ||
| Scheme 1 Synthesis of Ph-BT-Ph. | ||
1H NMR (400 MHz, CDCl3) δH ppm: 7.98 (m, 4H), 7.81 (s, 2H), 7.57 (m, 4H), 7.49 (m, 2H). (Fig. S1, ESI†).
13C NMR (101 MHz, CDCl3) δC ppm: 154.12, 134.44, 133.39, 129.26, 128.65, 128.39, 128.15. (Fig. S2, ESI†).
ESI-HRMS: m/z = 289.0785 [M + H]+ and calculated [M + H]+ = 289.0794 (Fig. S3, ESI†).
Procedure for photocatalytic oxidative coupling of amines to imines
The photocatalytic activity of the prepared photocatalyst, Ph-BT-Ph, under natural sunlight was tested for the oxidative coupling of amines to imines and the reaction conditions were optimized. Under the optimized conditions, 0.13 mol% of Ph-BT-Ph and 4 mL of acetonitrile (ACN) solvent were mixed and then, 0.2 mmol of the amine derivative was added to a 15 mL dried Schlenk tube. Then, the solution was purged with O2 for 2 minutes using a purging needle. After that, the Schlenk tube was placed under natural sunlight for 2 hours without using any mechanical stirring. We then analyzed the percentage conversion and selectivity of the reaction using the gas chromatography (GC) technique, with bromobenzene as an internal standard (digital photograph of the reaction setup shown in Fig. S4, ESI†) (Scheme 2). | ||
| Scheme 2 Photocatalytic oxidative coupling of amine to imine. | ||
Results and discussion
Photophysical study
A UV-visible absorption study was performed using acetonitrile as the solvent to determine the ground-state photophysical properties of the Ph-BT-Ph compound. It absorbs in the range of 190 nm to 450 nm and exhibits an absorption maximum of 366 nm (shown in Fig. 2(a)). The peak at 366 nm corresponds to the intramolecular charge transfer (ICT) transition from the donor to the acceptor unit.48 A high molar extinction coefficient of 9.25 × 103 M−1 cm−1 indicates its excellent light harvesting properties. The calculated optical bandgap is 2.99 eV, obtained by Tauc plot transformation, as shown in Fig. 2(b). A comparatively low optical band gap makes the catalyst suitable for absorbing visible light to show photocatalytic activity. The excited state emission properties and its lifetime have been evaluated by photoluminescence (PL) spectroscopy and time-correlated single photon count (TCSPC) studies. The PL spectrum of the catalyst exhibits a highly emissive fluorescence peak at 508 nm in degassed acetonitrile solution. The catalyst shows a 12.64 ns average excited-state lifetime, which is indicative of a long-lived photo-generated electron–hole pair. A high exciton lifetime makes a photocatalyst efficient, because the higher the excited state lifetime, the higher the possibility of an excited state electron interacting with the quencher. Then, we carried out PL spectroscopic studies in oxygen and nitrogen purged solutions and the spectra are presented in Fig. 2(c), as in this case, molecular oxygen is used as an oxidant for the reaction. The oxygenated solution is characterized by a drastic decrease in fluorescence intensity in comparison with the nitrogen-purged solution, clearly indicating the charge separation. This result is also well-supported by the TCSPC study as the average lifetime of the exciton is decreased on going from a nitrogen purged solution to an oxygen purged solution (shown in Fig. 2(d)). Hence, it can be concluded that the excited state electrons get trapped by molecular oxygen. Importantly, a dramatic decrease in the fluorescence intensity on going from a nitrogen purged solution to an oxygen purged solution also indicates that the dominant pathway is the electron transfer pathway and not the energy transfer pathway.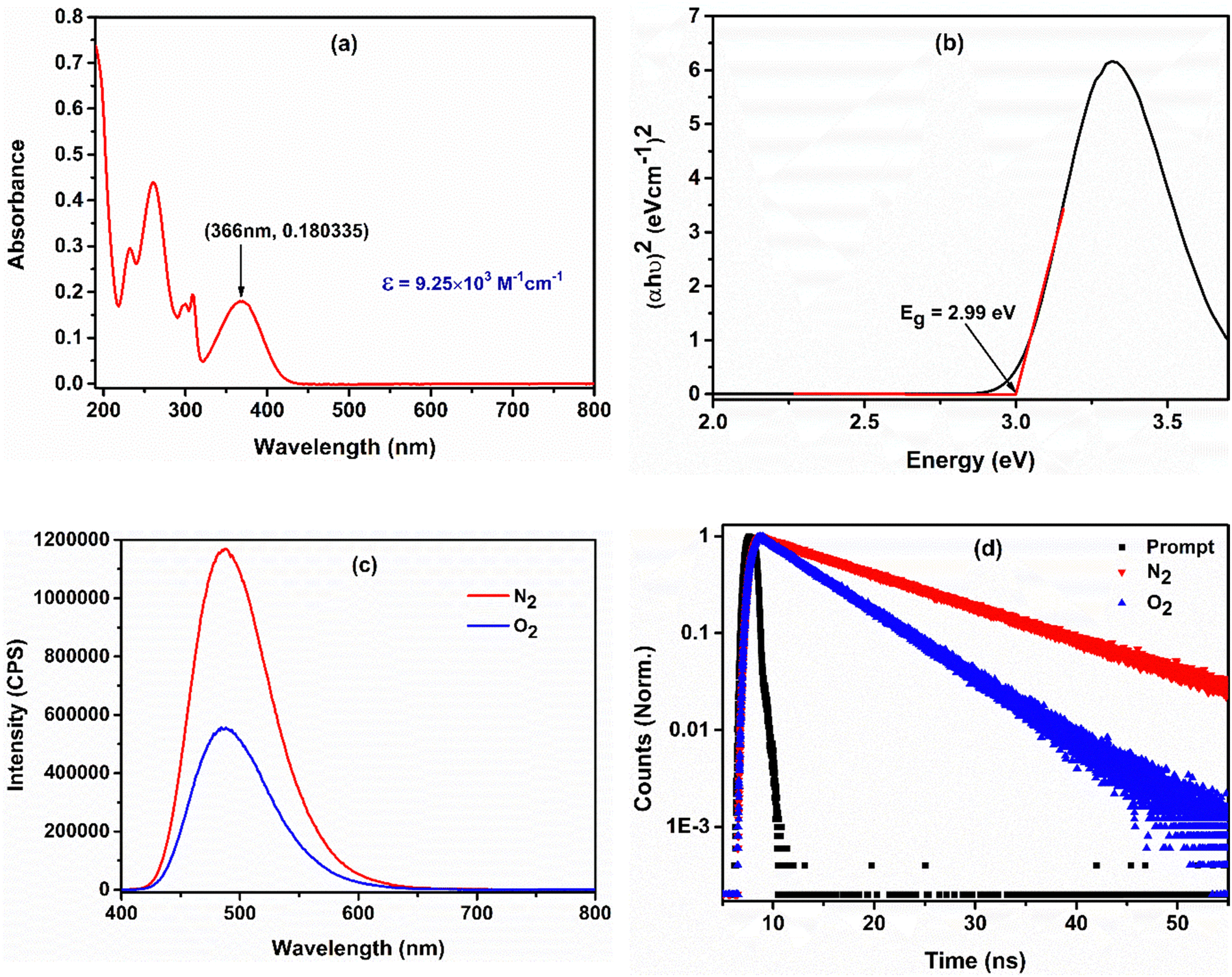 | ||
| Fig. 2 (a) UV-visible spectrum of Ph-BT-Ph in acetonitrile. (b) Bandgap estimation using Tauc plot transformation. (c) and (d) Fluorescence and lifetime spectra of Ph-BT-Ph in acetonitrile. | ||
Electrochemical properties
Cyclic voltammetric studies are performed in an acetonitrile solvent to determine the redox properties of the catalyst as shown in Fig. 3(a). The catalyst shows a high oxidation potential (HOMO) +1.56 V vs. Ag/AgCl and reduction potential (LUMO) −1.37 V vs. Ag/AgCl. The higher redox potential of the catalyst makes it an excellent alternative to reduce the molecular oxygen to the superoxide radical anion. The reduction potential of the O2/˙O2− system is −0.24 V vs. Ag/AgCl and the LUMO of the catalyst is more negative than the reduction potential of O2/˙O2−. Under these circumstances, molecular oxygen can be easily reduced by the catalyst to generate ROS. Moreover, the HOMO of the catalyst is more positive than the oxidation potential of benzylamine, and therefore the catalyst oxidizes benzylamine (BNA) to the benzylamine radical cation (BNA˙+), as the oxidation potential for BNA/BNA˙+ is +0.56 V vs. Ag/AgCl. Hence, appropriate energetic alignment of the HOMO and the LUMO makes the catalyst suitable for the photocatalytic oxidative coupling of benzylamine to benzylimine, as shown in Fig. 3(b).39,46,49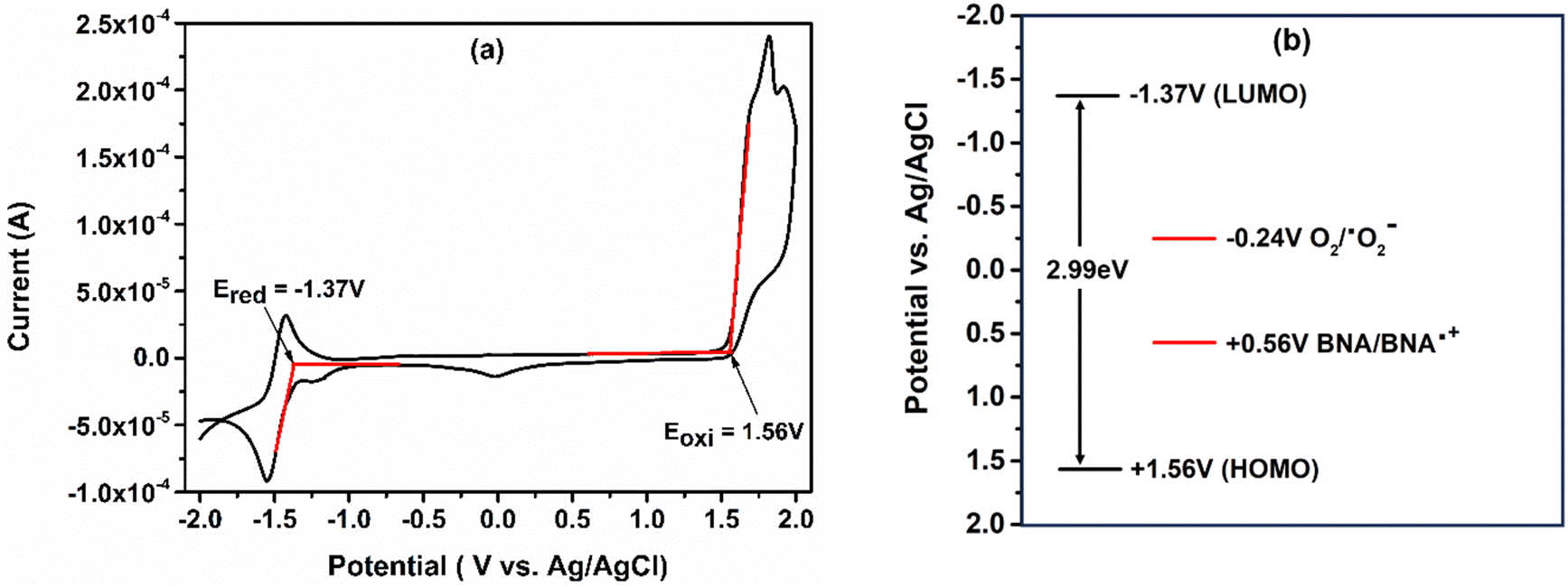 | ||
| Fig. 3 (a) Cyclic voltammogram of Ph-BT-Ph in an acetonitrile solvent. (b) Schematic representation of the HOMO–LUMO position of Ph-BT-Ph relative to the reduction potential O2/˙O2− system and oxidation potential of benzylamine. | ||
Photocatalytic oxidative coupling using “Ph-BT-Ph”
We have explored the photocatalytic efficiency of Ph-BT-Ph on the oxidative coupling of amine to imine. All the reactions were carried out in the presence of natural sunlight after purging oxygen into the reaction mixture just for 2 minutes and without any mechanical stirring. Conversion and selectivity of the photocatalytic reaction under various conditions are shown in Table 1, along with the reaction conditions mentioned. Optimization of the reaction time and amount of catalyst is presented in Fig. S5 (ESI†). The reaction does not proceed in the absence of either the catalyst or the sunlight, which ensures the importance of the catalyst and its photocatalytic activity. The reaction is attempted under an inert atmosphere as well as in air and there is a stark decrease observed in the conversion, especially when the reaction is attempted under an N2 atmosphere, which confirms that O2 is the sole oxidant to drive the reaction in the presence of a catalyst and light. In order to explore the reusability of the catalyst after a fresh catalytic cycle, we have performed the catalyst longevity experiment under a 250 W visible light lamp. Five consecutive cycles were performed after the fresh catalytic cycle by adding the same amount of reactant as used in the fresh catalytic cycle to the reaction mixture. The conversion of the reaction is decreased from 98% to 82% as shown in Fig. S38 (ESI†). Hence, the catalyst remains fairly active after a fresh catalytic cycle.|
|
||||
|---|---|---|---|---|
| Entry | Condition | Light source | Conversion/selectivity | TOF (h−1) |
| Standardized reaction condition: 0.2 mmol benzylamine, 0.13 mol% Ph-BT-Ph, 4 mL of acetonitrile, 2 min O2 purging, 2 h sunlight, atmospheric pressure, without stirring. Conversion determined by GC using Bromobenzene as an internal standard. TOF = mmol of amine converted per mmol of catalyst per hour. D = distance between the light source and the reaction vessel. | ||||
| 1 | Standard | Sunlight | >99%/>98% | 381 |
| 2 | Without catalyst | Sunlight | — | — |
| 3 | Dark | — | — | — |
| 4 | N2 | Sunlight | 4% | — |
| 5 | Air | Sunlight | 40% | 156 |
| 6 | Standard | 6 W UV-lamp (365 nm) | 11% | — |
| 7 | Standard | 12 W white LED | 4% | — |
| 8 | Standard | 250 W UV light lamp | 16% | — |
| 9 | Standard | 250 W visible light lamp | >99%/>98% | 381 |
| 10 | 1 h | 250 W visible light lamp | 75% | 577 |
| 11 | D = 5 cm | 150 W Hg lamp | 84% | 323 |
| 12 | D = 10 cm | 150 W Hg lamp | 48% | 185 |
| 13 | D = 20 cm | 150 W Hg lamp | 29% | 111 |
| 14 | D = 30 cm | 150 W Hg lamp | 16% | 61 |
Photocatalytic mechanism study
Scavenger study
To get a better insight into the reaction mechanism, we have performed scavenger studies to identify the active species involved in the reaction mechanism. When studying the reaction in the presence of radical scavenger hydroquinone (HQ), as shown in Fig. 4, there is a drastic decrease in the conversion from >99% to 27%, indicating that the reaction mechanism follows a radical pathway. We further used CuCl2 as an electron (e−) and KI as a hole (h+) scavenger for further studies. Drastic drops in conversion are again observed for both the cases which proves the involvement of the photo-generated electron and hole in the reaction mechanism. To explore the role of photo-generated electrons and holes to generate reactive oxygen species (ROS) from molecular oxygen, studies using benzoquinone (BQ) as the superoxide radical anion (˙O2−) scavenger and 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) as the singlet oxygen scavenger (1O2) were also performed. The reduction in the conversion conclusively shows the participation of ROS in the reaction mechanism. The experiment for hydroxyl radical scavenging using tBu-OH as a hydroxyl radical scavenger (HO˙) however shows no decrease in the conversion and it was concluded that HO˙ is not generated during the reaction.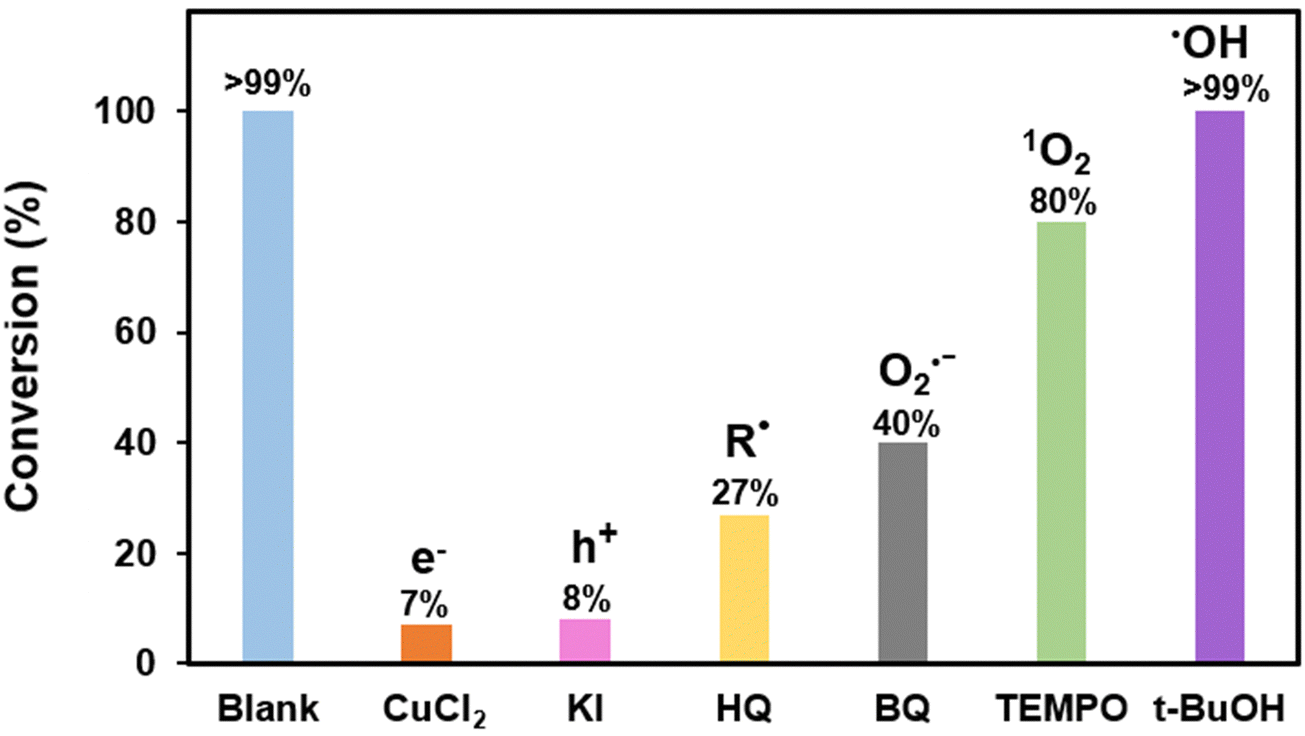 | ||
| Fig. 4 Scavenger study results for photocatalytic oxidative coupling of amines. | ||
EPR spectroscopy study
It is apparent from the scavenger studies that photo-generated electrons and holes participate in the reaction. To further confirm the ROS generation ability of the photo-catalyst, we have studied the coupling reaction by EPR spectroscopy, as shown in Fig. 5. For the study, ROS are trapped by using the trapping reagent TEMP (2,2,6,6-tetramethyl-4-piperidone) for singlet oxygen and DMPO (5,5-dimethyl-1-pyrroline N-oxide) for superoxide radical anion. We observe the characteristic EPR signals for both singlet oxygen and superoxide radical anion in an acetonitrile solution of Ph-BT-Ph when irradiated for 5 min by visible light in the presence of a trapping reagent. In addition to BNA, there is a lowering of signal intensity which shows that ROS is participating in the reaction. Furthermore, there is no EPR signal observed without light, which means ROS is generated only under illumination conditions by the reaction of oxygen molecules and photo-generated electrons.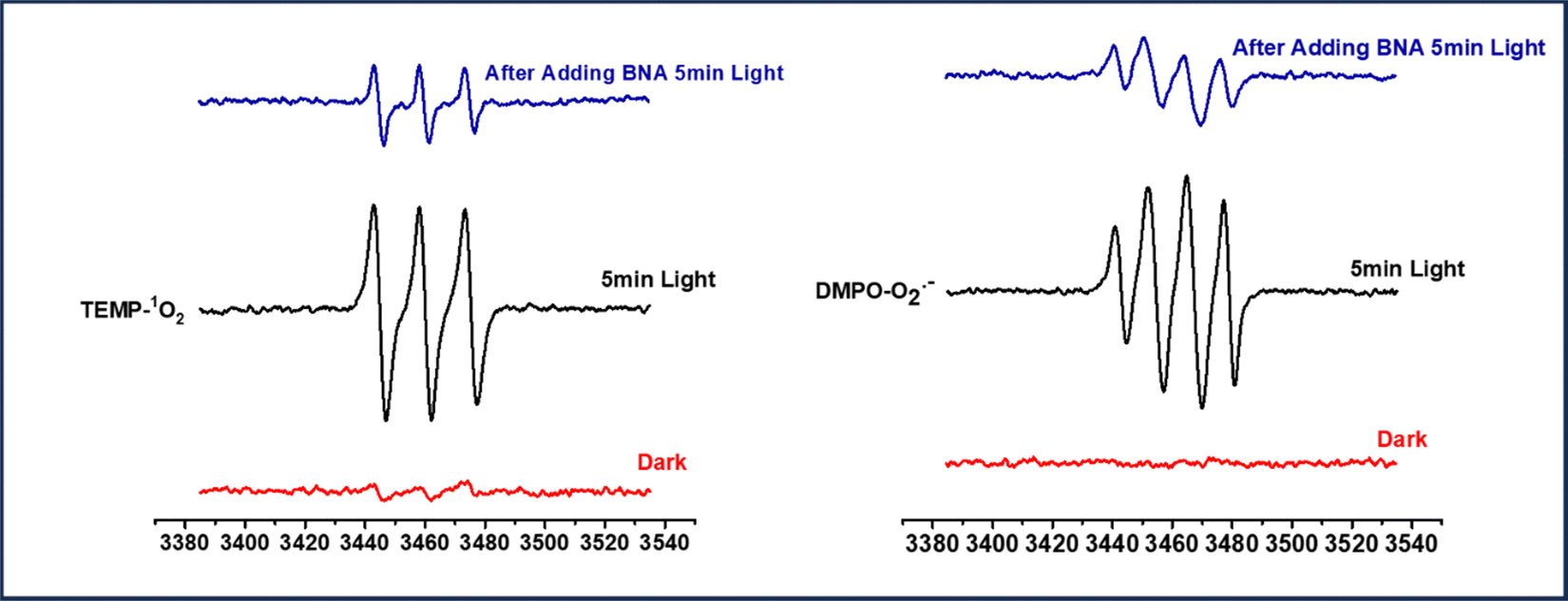 | ||
| Fig. 5 In situ ESR/EPR spectra of 1O2 & ˙O2− using TEMP and DMPO as trapping reagents. | ||
Substrate scope
We have conducted the substrate scope study to assess the versatility of the photocatalyst for various amine derivatives, and the results are shown in Table 2. The catalyst exhibited excellent selectivity and conversion within 2 hours of the reaction time for all the substrates. The catalyst exhibited turnover frequencies ranging from 508 h−1 to 215 h−1 for different amine substrates (shown in Table 2) indicating its high photocatalytic efficiency. Then, the shorter reaction time of 1 hour has also been attempted to investigate the substituent effects on different arylated amine derivatives and the activity for arylated and heterocyclic amine derivatives is also compared. No significant effect is notified especially with the electron-withdrawing substituent as the reaction with the electron-withdrawing substituent in the arylated amine takes place at a similar rate to that for the unsubstituted and methyl substituent aryl amine derivatives. However, in the case of aryl amines with –OMe substituent, the reaction takes place at a slower rate. In the case of heterocyclic amine derivates, the pyridine derivative reaction takes place much faster than the thiophene amine derivative.| Reaction condition: 0.2 mmol amine derivative, 0.13 mol% Ph-BT-Ph, 4 mL of acetonitrile, 2 min O2 purging, atmospheric pressure, sunlight, without stirring. Conversion and selectivity are determined by GC using bromobenzene as an internal standard (*, unable to record NMR spectra). TOF = mmol of amine converted per mmol of catalyst per hour. |
|---|
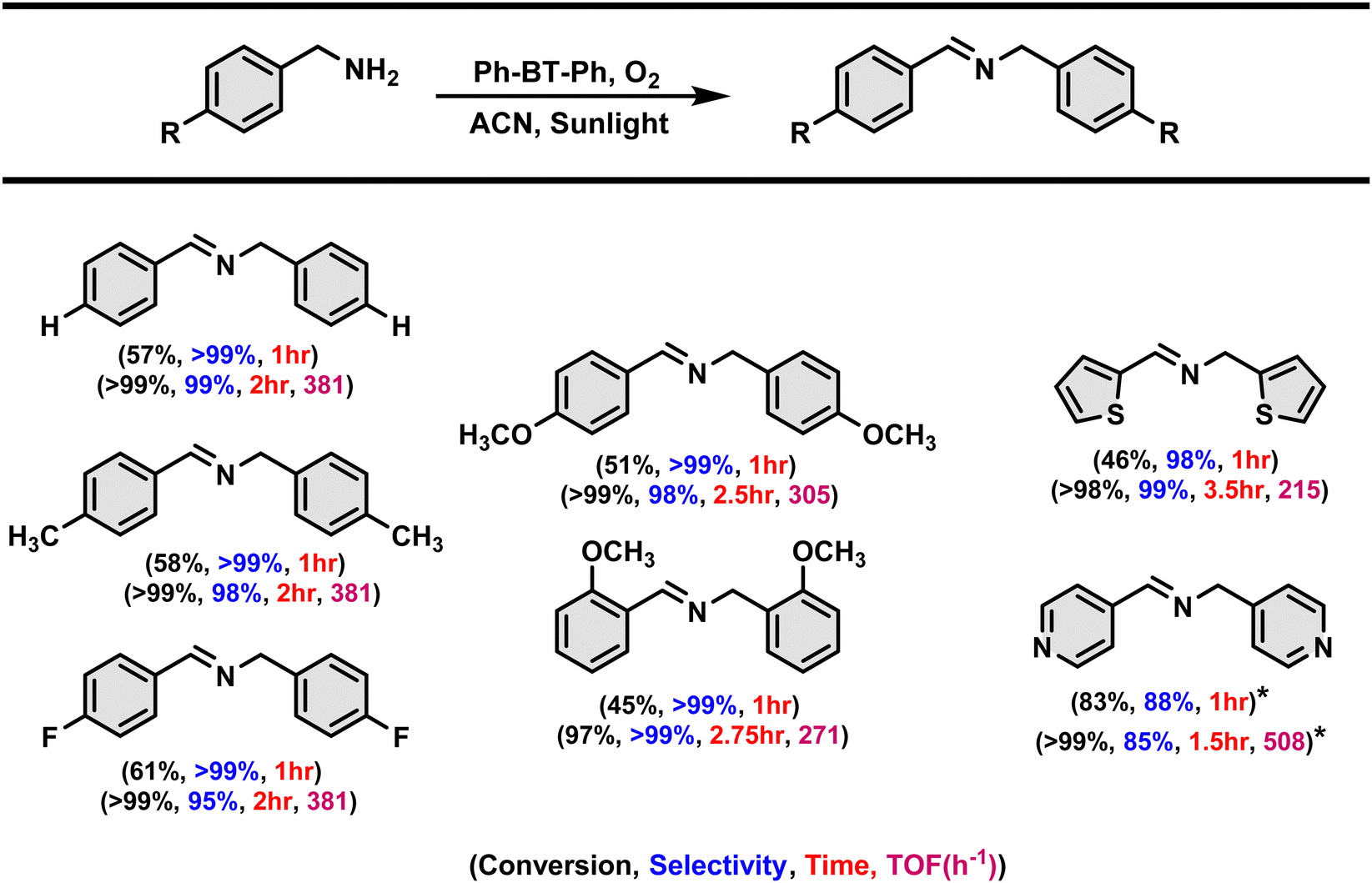
|
Proposed mechanism and insights from DFT
In the present study, the photocatalyst molecule, Ph-BT-Ph comprising the benzothiadiazole unit, is used to harvest the energy from sunlight to activate oxygen molecules and catalyse the oxidative coupling reaction. We propose the mechanism for photocatalytic oxidative coupling of amines to imines based on the outcomes of scavenger studies and EPR experiments described in the previous sections and considering reported studies.39 To further understand the role of the photocatalyst in the oxidative coupling reaction, a thorough theoretical study was carried out using DFT.As shown in the scheme (Fig. 6), upon photoexcitation, the catalyst generates the electron–hole pair. The appropriate alignment of the excited state energy level of the electron matches the required condition to reduce the molecular oxygen to generate ROS, i.e., ˙O2− by an electron transfer pathway and 1O2 by an energy transfer pathway, which is also obvious from the electrochemical properties of the catalyst (see Fig. 3(b) and 7(d)). The hole present in the ground state oxidizes benzylamine (BNA) to a benzylamine radical cation (BNA˙+) by a single electron transfer (SET) mechanism, as the catalyst has a higher oxidation potential than that of the amine. Benzylamine radical formation is confirmed by adding TEMPO50 during the photocatalytic reaction, and the BNA-TEMPO adduct was detected in the HRMS study (shown in Fig. S33, ESI†). Then, the BNA˙+ reacts with the reactive oxygen species, ˙O2− and 1O2, forming the imine and H2O2. Generation of H2O2 during the reaction is confirmed through the UV-visible KMnO4 spectroscopic titration51 and also by KI colorimetery52 after completion of the reaction (results shown in Fig. S34 and S35, ESI†). Additional BNA then reacts with the imine, liberating the NH3 gas and forming benzylimine (BNI) as the final product. Liberation of the ammonia gas is detected after the completion of the photocatalytic reaction by the Nessler reagent colorimetry test53 shown in Fig. S36 (ESI†).
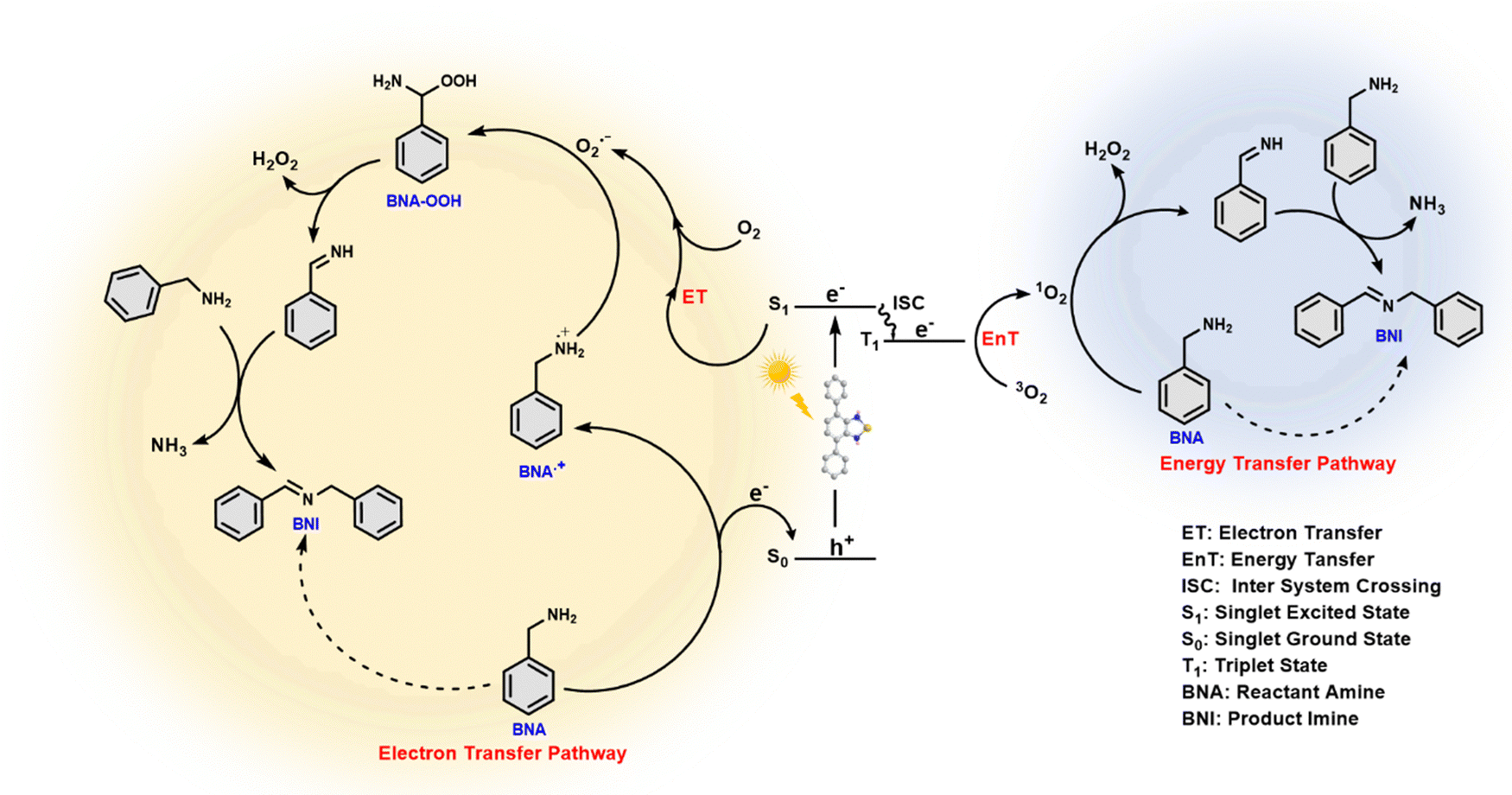 | ||
| Fig. 6 Plausible reaction mechanism for the oxidative coupling of amines to imines by Ph-BT-Ph. | ||
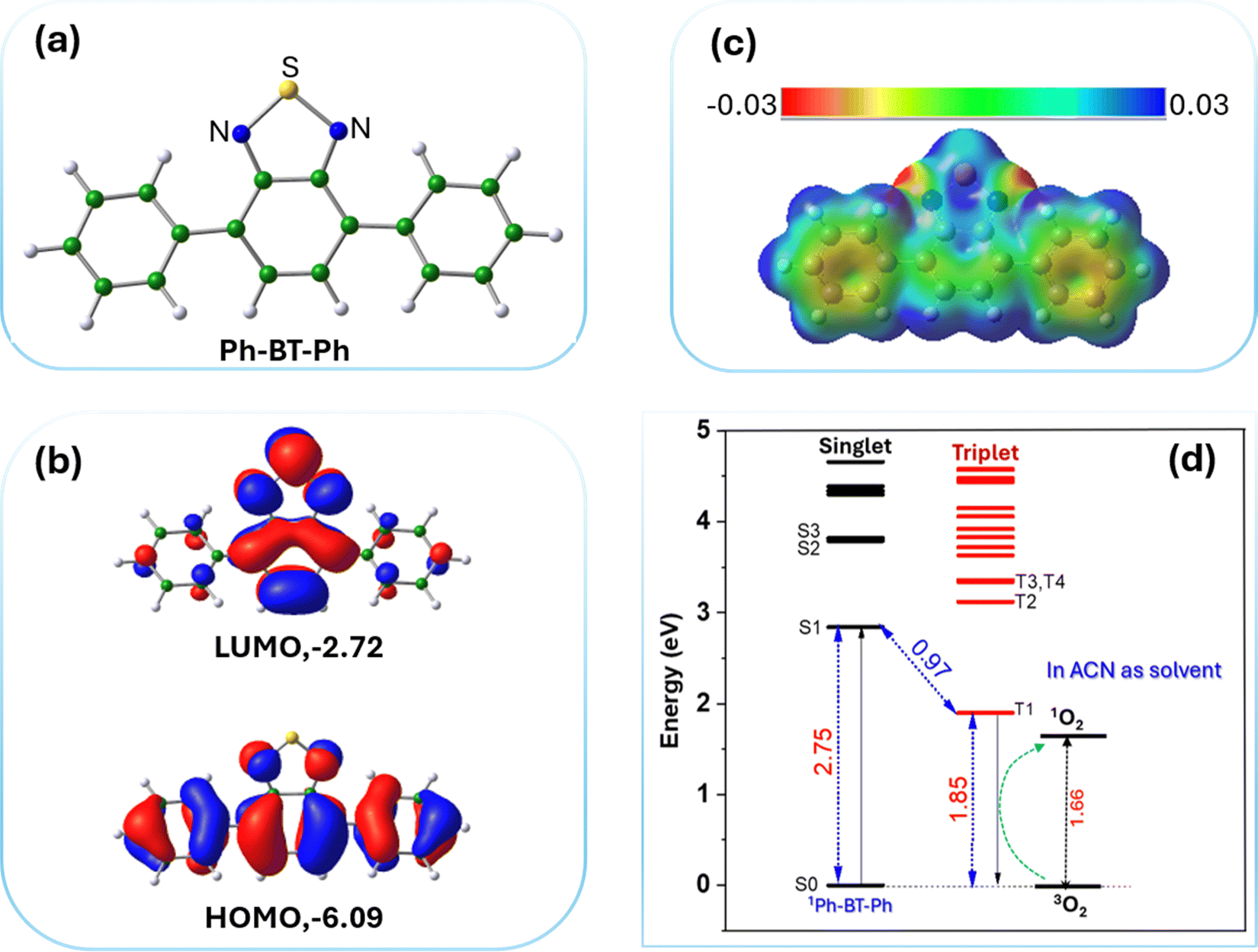 | ||
| Fig. 7 (a) Optimized structure of Ph-BT-Ph. (b) Plots of the HOMO and LUMO of Ph-BT-Ph, the color codes show different signs of wavefunctions. (c) Electrostatic potential fit (ESP) for Ph-BT-Ph, with the color bar shown. (d) The calculated ground state and excited states (singlet and triplet) of Ph-BT-Ph. Singlet states are denoted as S(n) and triplet states are denoted as T(n) with n being the n-th excited state. | ||
The optimized structure of the photocatalyst Ph-BT-Ph molecule is shown in Fig. 7(a). To understand the most electron-rich and most electron-deficient regions of Ph-BT-Ph, we have first calculated the electrostatic potential (ESP) map for Ph-BT-Ph (Fig. 7(c)) using DFT, which determines the active sites interacting with BNA or O2. The ESP map shows N atoms on the catalyst as electron rich, and the S site as slightly electron deficient. DFT studies also show that Ph-BT-Ph (Fig. 7(a)) has a suitable energetic alignment of the molecular orbitals (HOMO: −6.09 eV, LUMO: −2.72 eV) and an optimum HOMO–LUMO gap of 3.37 eV, (Fig. 7(b)) to absorb the sunlight. The energized Ph-BT-Ph undergoes electronic transitions which were calculated using the TD-DFT method and the spectra are shown in ESI† (Fig. S41). The calculated HOMO–LUMO gap is slightly higher than the experimentally obtained optical gap of 2.99 eV from the Tauc plot. The HOMO is extended throughout the molecule, showing good conjugation; however, for the LUMO the conjugation is affected as the central ring of Ph-BT-Ph is out-of-plane from the rest of the molecule (structural details are shown in ESI,† Table S2). Extended π-conjugation of the frontier molecular orbitals of Ph-BT-Ph is advantageous for electron transfer. The calculated energy difference between the ground-sate S0 and first excited-state S1 of Ph-BT-Ph is 2.75 eV calculated from TD-DFT studies, while the energy difference between the lowest triplet state and S0 state is 1.85 eV (Fig. 7d). The singlet and triplet excited states for Ph-BT-Ph along are shown in Fig. 7(d) while the obtained TD-DFT spectra are shown in ESI† (Fig. S41).
It is interesting to note that the triplet excited state of the photocatalyst 3Ph-BT-Ph can interact with an oxygen molecule in triplet state (3O2) resulting in high energy singlet oxygen (1O2) species as shown below which is utilized in the course of the reactions.
| 1Ph-BT-Ph + hυ → 1Ph-BT-Ph* → 3Ph-BT-Ph |
| 3Ph-BT-Ph + 3O2 → 1Ph-BT-Ph + 1O2 (singlet oxygen) |
Electron transfer pathway
To explore the underlying mechanism of the photocatalytic reaction as proposed in Fig. 6, we studied the interactions of BNA with Ph-BT-Ph using DFT and we found that BNA molecule interacts favorably with Ph-BT-Ph with the formation of a complex (BNA–[Ph-BT-Ph]) as shown in Fig. 8(a). Formation of the BNA–[Ph-BT-Ph] complex, with BNA and [Ph-BT-Ph] held in close proximity (closest distance N⋯H: 2.47 Å) is central to the electron transfer pathway generating ROS. The binding energy of BNA with ground state Ph-BT-Ph to form the BNA–[Ph-BT-Ph] complex is small, and it is expected to interact more with excited singlet Ph-BT-Ph in the presence of sunlight. Therefore, we have optimized the structure of Ph-BT-Ph in the singlet excited state S1 and calculated a binding energy of −0.014 eV (Ebind: −0.0137 eV in ACN) for BNA–[Ph-BT-Ph] complex formation (details are shown in ESI†). A close observation of the optimized structure of the complex shows that the H atoms from the –NH2 group of BNA are closer to the N atom of Ph-BT-Ph, indicating that the N atom of the photocatalyst acts as the active site in this case. The top view of the complex however shows the benzene ring of the BNA molecule on-top, and almost parallel to Ph-BT-Ph, indicative of modest π–π interactions as well. We then analyzed the frontier molecular orbitals of the BNA–[Ph-BT-Ph] complex (Fig. 8(b)) to probe the nature of the interactions between BNA and Ph-BT-Ph moieties. We note that while the three higher lying unoccupied MOs (LUMO, LUMO+1, LUMO+2) are localized on the Ph-BT-Ph portion of the complex, the HOMO−1 and HOMO−2 are both localized on the BNA portion of the BNA–[Ph-BT-Ph] complex (see Fig. 8(b) for the MOs).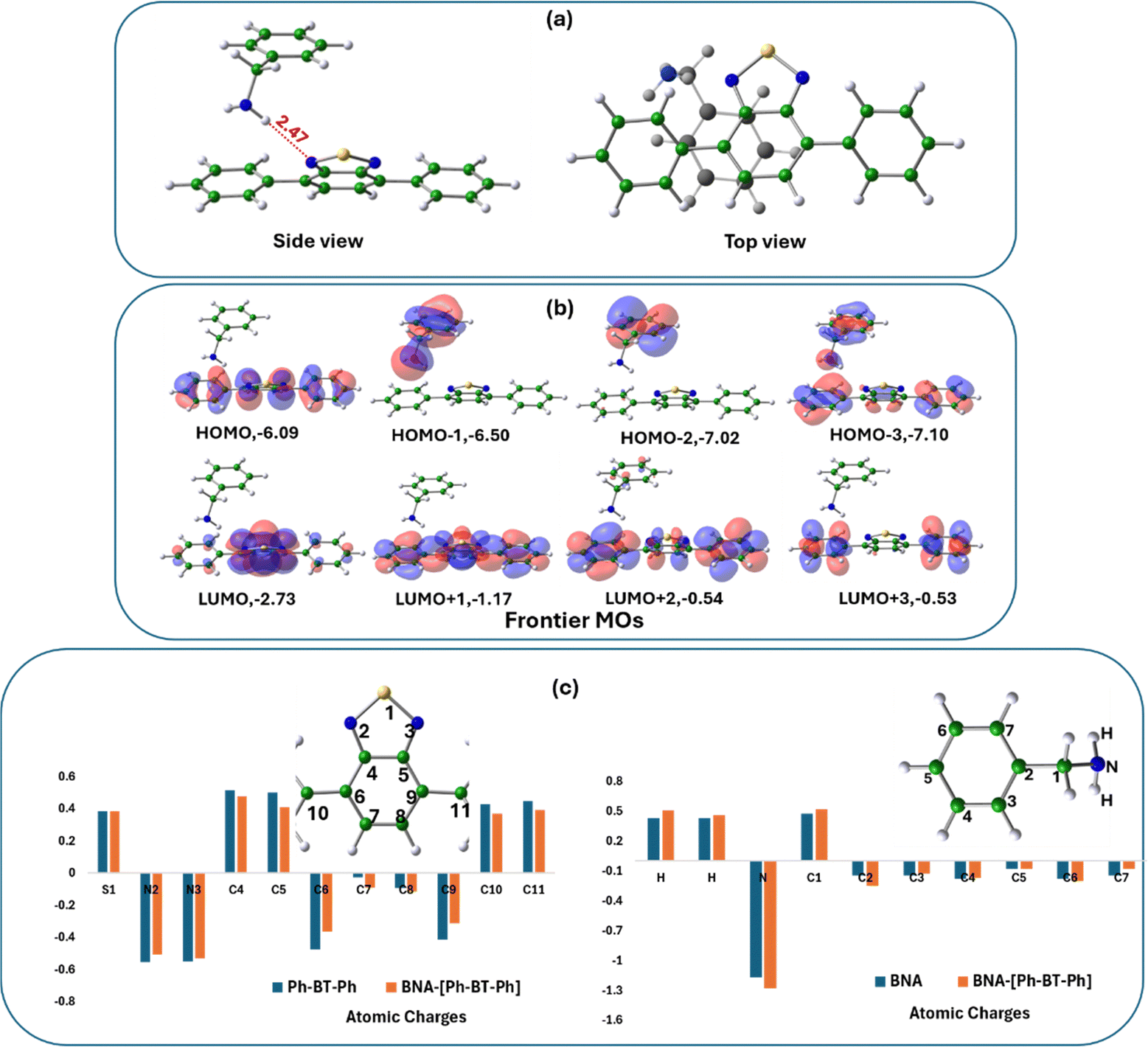 | ||
| Fig. 8 (a) The optimized structure of the BNA–[Ph-BT-Ph] complex; both side view and top views are shown. (b) Plots of frontier molecular orbitals including the HOMO and the LUMO of the BNA–[Ph-BT-Ph] complex; the color codes show different signs of wavefunctions. The energies of the respective MOs in eV are written alongside. (c) Bar chart showing the changes in atomic charges of BNA and Ph-BT-Ph before and after BNA–[Ph-BT-Ph] complex formation. The atom numbers and the respective molecular units are shown in the inset. | ||
The distribution of occupied and unoccupied MOs shows the partial donor characteristic of BNA, which can donate electrons to Ph-BT-Ph, which has a partial acceptor character. We further analyzed the changes in calculated atomic charges on the BNA–[Ph-BT-Ph] complex with respect to the free Ph-BT-Ph molecule (see Fig. 8c, left panel) and some changes in atomic charges on two N atoms and S atoms belonging to the Ph-BT-Ph molecule were observed along with changes on C atoms too. We also analyzed the changes in the atomic charges of BNA before and after BNA–[Ph-BT-Ph] complex formation (right panel, Fig. 8c), which indicates strong interactions of BNA and Ph-BT-Ph in the complex formed, which can indeed lead to electron transfer as shown in Fig. 6. The BNA–[Ph-BT-Ph] complex thus formed paves the way for electron transfer from BNA to Ph-BT-Ph in the excited state.
Energy transfer pathway
In light of the scheme shown in Fig. 6, we now use DFT to investigate the energy transfer pathway and consider the binding of the oxygen molecule (3O2) to Ph-BT-Ph. Because of spin constraints, this binding is unfavorable for Ph-BT-Ph in the ground singlet state. This can however become feasible upon electronic excitation, and the calculated excited energy states from TD-DFT studies are shown in Fig. 7(d). The electronic energy difference of Ph-BT-Ph between a singlet, ground state and an excited, triplet state is 1.85 eV, and such electronic excitation is possible after light irradiation: 1Ph-BT-Ph + hυ → 1Ph-BT-Ph* → 3Ph-BT-Ph, where ‘*’ represents the excited state.The 3Ph-BT-Ph molecule in the excited triplet state interacts with the triplet oxygen molecule without any spin-restrictions to give rise to a complex, [Ph-BT-Ph]–O2, which then breaks down to form reactive singlet oxygen species. The formation of this complex is crucial in the energy transfer pathway.
| 3Ph-BT-Ph + 3O2 → [Ph-BT-Ph]–O2 (Ph-BT-Ph oxygen complex) → Ph-BT-Ph + 1O2 |
The binding energy for O2 in the ‘bridge’ configuration in [Ph-BT-Ph]–O2 is negative (Ebind: −0.25 eV in ACN), showing favorable interactions between the two (structure shown in Fig. 9(a)). Interestingly, this is a complex with oxygen molecule held in the bridge position at a distance of 2.67 Å, but not bonded. Bridge bonding of molecular oxygen, however, is not uncommon in inorganic complexes.54 The close interaction between the two moieties in [Ph-BT-Ph]–O2 is also clear from the plots of the molecular orbitals (Fig. 9(b)) which shows that the HOMO and the LUMO of the [Ph-BT-Ph]–O2 complex reside on oxygen and the Ph-BT-Ph portion of the complex respectively, indicating an electronic charge redistribution as a result of mutual interactions. A quantitative understanding of the mutual interactions is again obtained from the change in atomic charges of Ph-BT-Ph before and after interaction as shown in Fig. 9(c). As oxygen binds in a bridge position on the benzene ring, there are significant changes in atomic charges on these C atoms along with some changes in atomic charges on oxygen atoms of O2. This interaction also has small effects on the atomic charges of the N and S atoms of the benzothiadiazole ring. We also studied another possible oxygen complex of Ph-BT-Ph where it interacts sideways (Fig. S42, ESI†) as reported in an earlier study,41 but this structure is slightly less probable energetically than the bridge configuration (Fig. 9(a)), but shows some changes in atomic charges on S and N atoms like the bridge bonded case discussed above. It is interesting to note that for the ‘bridge’ configuration of oxygen binding the active sites are C atoms on Ph-BT-Ph, while for the ‘side-on’ binding of oxygen, the active sites are the N atoms on Ph-BT-Ph.
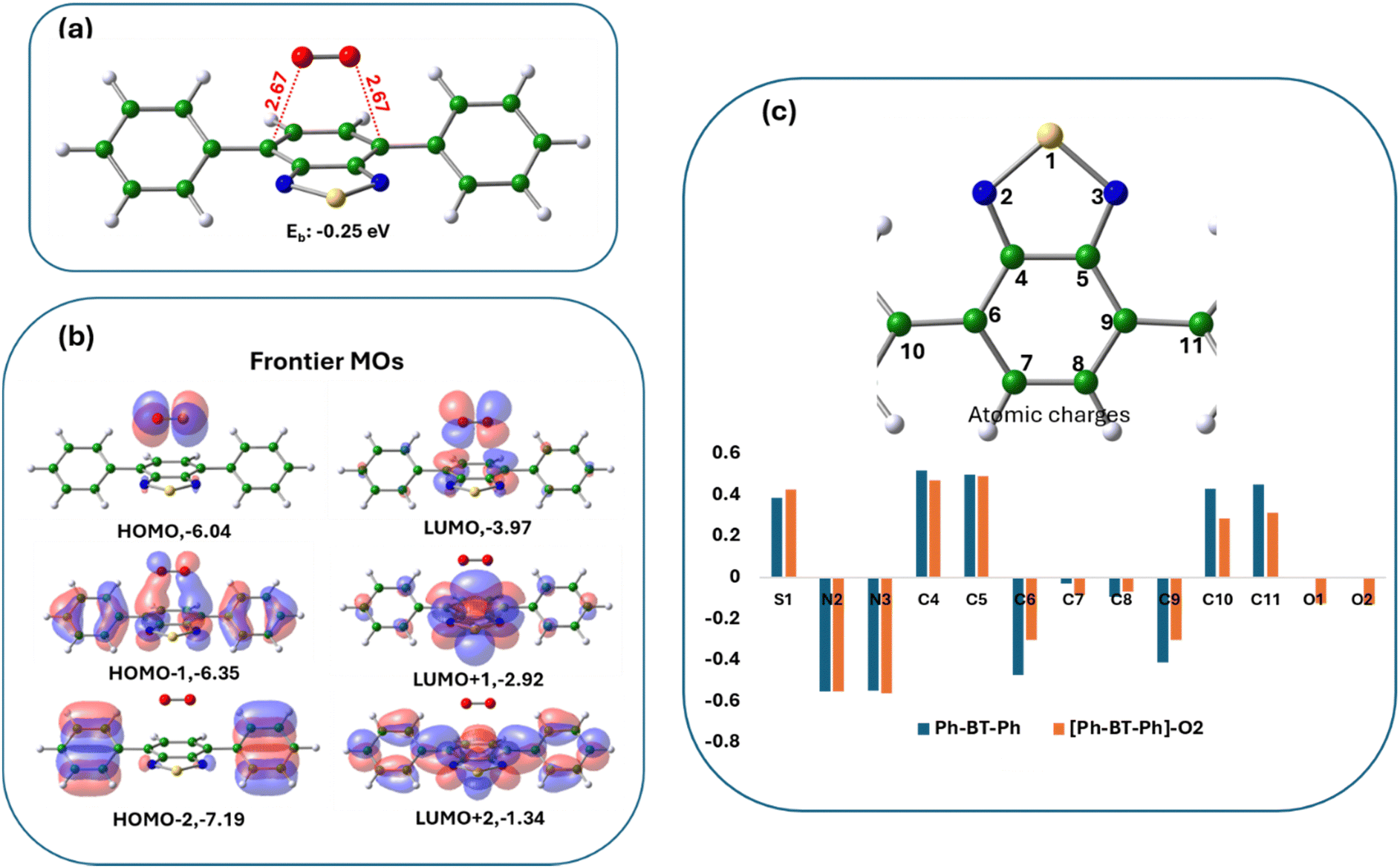 | ||
| Fig. 9 (a) The optimized structure of the [Ph-BT-Ph]–O2 complex with oxygen bound in bridge position interacting with C atoms of the benzene ring. (b) Plots of frontier molecular orbitals including the HOMO and LUMO of the [Ph-BT-Ph]–O2 complex; the color codes show different signs of wavefunctions. The energies of the respective MOs in the eV are written alongside. (c) Bar chart showing the changes in atomic charges of Ph-BT-Ph before and after interaction with oxygen. The atom numbers are shown upside. | ||
Therefore, we conclude that in the presence of light, oxygen interacts with Ph-BT-Ph and possibly forms a bridge complex, which can help in energy transfer from triplet Ph-BT-Ph to a triplet oxygen molecule giving rise to singlet oxygen. The singlet oxygen formed then reacts with BNA to form Ph–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH and H2O2 at first, which then further reacts with BNA to form NH3 and BNI and the calculated reaction free energies are negative (details are shown in ESI†) and the reactions are spontaneous as indicated earlier in Fig. 6.
NH and H2O2 at first, which then further reacts with BNA to form NH3 and BNI and the calculated reaction free energies are negative (details are shown in ESI†) and the reactions are spontaneous as indicated earlier in Fig. 6.
The overall reaction combining both the electron transfer and the energy transfer pathways and involving both BNA and oxygen complexes of Ph-BT-Ph can be written as a consolidated reaction as below:
| BNA–[Ph-BT-Ph] + [Ph-BT-Ph]–O2 + BNA → BNI + H2O2 + NH3 + 2[Ph-BT-Ph], ΔGACN = −0.11 eV |
The Gibb's free energy of the reaction above is −0.11 eV, which shows that the overall reaction is thermodynamically more feasible compared to the uncatalyzed reaction, which does not take place without a catalyst/sunlight. The reaction Gibb's free energies calculated from DFT for all the intermediate steps are listed in the ESI.†
Conclusion
Herein we report the synthesis of a small organic molecular photocatalyst (SOMP) “Ph-BT-Ph” using benzothiadiazole as the core photoactive unit targeting photocatalytic oxidative coupling of amines to synthesize imines. In the literature, there is no report on this promising photocatalytic oxidative coupling reaction using a benzothiadiazole-based small organic molecule; however, this organic catalyst has been reported for the intermolecular C–H functionalization of electron-rich heteroaromatic molecules.47 The catalyst was synthesized via the Suzuki–Miyaura coupling reaction protocol and we characterized the catalyst with various analytical techniques such as 1H NMR, HRMS, cyclic voltammetry, UV-visible spectroscopy, photoluminescence spectroscopy and TCSPC. The catalyst shows a high excited state redox potential (−1.37/+1.56 V vs. Ag/AgCl) and strong fluorescence. It is characterized by the generation of long-lived exciton which can be attributed to its donor–acceptor (D–A) type structure. All these characteristics make it a potential photocatalyst. Subsequently, we applied a significantly low amount (0.13 mol%) of catalyst for direct oxidative coupling of amines to imines in the presence of natural sunlight, by purging O2 only for 2 minutes. The catalyst exhibited excellent conversion and selectivity without any other external energy input, which makes the conversion not only green and sustainable, but also highly scalable. Control experiments, scavenger studies, and EPR measurement results proved that the oxidative coupling is mediated by the reactive oxygen species (ROS) and theoretical investigation revealed the active sites on the catalyst and the underlying mechanism for its unprecedented performance in photocatalytic oxidative coupling. This work paves the way for a cost-effective, scalable and eco-friendly route for organic transformation by using small organic molecular photocatalysts (SOMPs) that operate under natural sunlight.Author contributions
Ajeet Singh: conceptualization, overall research work, data acquisition, formal analysis, and manuscript writing and editing. Bidisa Das: computational work, supervision, and manuscript writing and editing. Saumi Ray: overall design of the research idea, data acquisition, formal analysis, supervision, funding acquisition, and manuscript writing and editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
A. S. is grateful to BITS-Pilani, Pilani campus for the institute research fellowship. S. R. is thankful to DST-SERB (CRG/2021/004126), Government of India, New Delhi and the BITS-Pilani CDRF-Saumi Ray-2023 (C1/23/165) project for financial support. We would like to thank DST-FIST for the HRMS facility at BITS Pilani, Pilani campus. We would like to acknowledge CRF IIT-Delhi for the EPR facility. We would like to thank Dr Mrinmoyee Basu for helping us with sunlight intensity measurement. We would also like to thank Dr Partha Sarathi Addy for providing different light sources for the photocatalytic study. We would like to acknowledge the Department of Chemistry, Birla Institute of Technology and Science (BITS), Pilani, for providing the instrumentation facilities and infrastructure. BD acknowledges SERB grant #CRG/2023/000093 for the computational resources used for DFT studies presented here.References
- P. T. Anastas and M. M. Kirchhoff, Acc. Chem. Res., 2002, 35, 686–694 CrossRef CAS PubMed.
- P. T. Anastas, M. M. Kirchhoff and T. C. Williamson, Appl. Catal., A, 2001, 221, 3–13 CrossRef CAS.
- G. Ciamician, Science, 1912, 36, 385–394 CrossRef CAS PubMed.
- J.-M. Herrmann, J. Photochem. Photobiol., A, 2010, 216, 85–93 CrossRef CAS.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- S. M. Stevenson, M. P. Shores and E. M. Ferreira, Angew. Chem., Int. Ed., 2015, 54, 6506–6510 CrossRef CAS PubMed.
- O. Reiser, Acc. Chem. Res., 2016, 49, 1990–1996 CrossRef CAS PubMed.
- D. P. Hari and B. König, Chem. Commun., 2014, 50, 6688–6699 RSC.
- S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600–1601 CrossRef CAS PubMed.
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS PubMed.
- I. Ghosh and B. König, Angew. Chem., Int. Ed., 2016, 55, 7676–7679 CrossRef CAS PubMed.
- I. Ghosh, L. Marzo, A. Das, R. Shaikh and B. König, Acc. Chem. Res., 2016, 49, 1566–1577 CrossRef CAS PubMed.
- K. Ohkubo, K. Mizushima, R. Iwata, K. Souma, N. Suzuki and S. Fukuzumi, Chem. Commun., 2010, 46, 601–603 RSC.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed.
- K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed.
- M. Fagnoni, D. Dondi, D. Ravelli and A. Albini, Chem. Rev., 2007, 107, 2725–2756 CrossRef CAS PubMed.
- B. A. D. Neto, A. A. M. Lapis, E. N. da Silva Júnior and J. Dupont, Eur. J. Org. Chem., 2013, 228–255 CrossRef CAS.
- R. Li, J. Byun, W. Huang, C. Ayed, L. Wang and K. A. I. Zhang, ACS Catal., 2018, 8, 4735–4750 CrossRef CAS.
- J. Wu, G. Lai, Z. Li, Y. Lu, T. Leng, Y. Shen and C. Wang, Dyes Pigm., 2016, 124, 268–276 CrossRef CAS.
- D. Zhang, T. Yang, H. Xu, Y. Miao, R. Chen, R. Shinar, J. Shinar, H. Wang, B. Xu and J. Yu, J. Mater. Chem. C, 2021, 9, 4921–4926 RSC.
- M. Jeffries-El, B. M. Kobilka and B. J. Hale, Macromolecules, 2014, 47, 7253–7271 CrossRef CAS.
- B. A. D. Neto, P. H. P. R. Carvalho and J. R. Correa, Acc. Chem. Res., 2015, 48, 1560–1569 CrossRef CAS PubMed.
- J. Zhang, W. Chen, A. J. Rojas, E. V. Jucov, T. V. Timofeeva, T. C. Parker, S. Barlow and S. R. Marder, J. Am. Chem. Soc., 2013, 135, 16376–16379 CrossRef CAS PubMed.
- K. Ohkubo, K. Mizushima, R. Iwata and S. Fukuzumi, Chem. Sci., 2011, 2, 715–722 RSC.
- A. U. Meyer, T. Slanina, C.-J. Yao and B. König, ACS Catal., 2016, 6, 369–375 CrossRef CAS.
- P. S. Rao and E. Hayon, J. Phys. Chem., 1975, 79, 397–402 CrossRef CAS.
- F. Su, S. C. Mathew, L. Möhlmann, M. Antonietti, X. Wang and S. Blechert, Angew. Chem., Int. Ed., 2011, 50, 657–660 CrossRef CAS PubMed.
- J. Yu, Q. Liu, W. Qiao, D. Lv, Y. Li, C. Liu, Y. Yu, Y. Li, H. Niemantsverdriet, B. Zhang and R. Su, ACS Catal., 2021, 11, 6656–6661 CrossRef CAS.
- L. Liu, S. Zhang, X. Fu and C.-H. Yan, Chem. Commun., 2011, 47, 10148–10150 RSC.
- Z. Wang and X. Lang, Appl. Catal., B, 2018, 224, 404–409 CrossRef CAS.
- Y. Xu, Y. Chen and W.-F. Fu, Appl. Catal., B, 2018, 236, 176–183 CrossRef CAS.
- S.-I. Murahashi, Angew. Chem., Int. Ed. Engl., 1995, 34, 2443–2465 CrossRef CAS.
- J. S. M. Samec, A. H. Éll and J.-E. Bäckvall, Chem. – Eur. J., 2005, 11, 2327–2334 CrossRef CAS PubMed.
- B. Chen, L. Wang and S. Gao, ACS Catal., 2015, 5, 5851–5876 CrossRef CAS.
- D. Dissanayake, L. A. Achola, P. Kerns, D. Rathnayake, J. He, J. Macharia and S. L. Suib, Appl. Catal., B, 2019, 249, 32–41 CrossRef CAS.
- P. Chen, L.-H. Meng, L. Chen, J.-K. Guo, S. Shen, C.-T. Au and S.-F. Yin, ACS Sustainable Chem. Eng., 2019, 7, 14203–14209 CrossRef CAS.
- S. Ahmed, A. Kumar and P. S. Mukherjee, Chem. Commun., 2023, 59, 3229–3232 RSC.
- Q. Li, J. Wang, Y. Zhang, L. Ricardez-Sandoval, G. Bai and X. Lan, ACS Appl. Mater. Interfaces, 2021, 13, 39291–39303 CrossRef CAS PubMed.
- S. Yang, X. Li, Y. Qin, Y. Cheng, W. Fan, X. Lang, L. Zheng and Q. Cao, ACS Appl. Mater. Interfaces, 2021, 13, 29471–29481 CrossRef CAS PubMed.
- M.-Y. Yang, S.-B. Zhang, M. Zhang, Z.-H. Li, Y.-F. Liu, X. Liao, M. Lu, S.-L. Li and Y.-Q. Lan, J. Am. Chem. Soc., 2024, 146, 3396–3404 CrossRef CAS PubMed.
- Z. J. Wang, K. Garth, S. Ghasimi, K. Landfester and K. A. I. Zhang, ChemSusChem, 2015, 8, 3459–3464 CrossRef CAS PubMed.
- C. Chu, Y. Qin, C. Ni and J. Zou, Chin. Chem. Lett., 2022, 33, 2736–2740 CrossRef CAS.
- J. J. Lee, W. Noh, T.-H. Huh, Y.-J. Kwark and T. S. Lee, Polymer, 2020, 211, 123060 CrossRef CAS.
- K. Wu, X.-Y. Liu, M. Xie, P.-W. Cheng, J. Zheng, W. Lu and D. Li, Appl. Catal., B, 2023, 334, 122847 CrossRef CAS.
- Y. Xiao, G. Tian, W. Li, Y. Xie, B. Jiang, C. Tian, D. Zhao and H. Fu, J. Am. Chem. Soc., 2019, 141, 2508–2515 CrossRef CAS PubMed.
- L. Wang, W. Huang, R. Li, D. Gehrig, P. W. M. Blom, K. Landfester and K. A. I. Zhang, Angew. Chem., Int. Ed., 2016, 55, 9783–9787 CrossRef CAS PubMed.
- R. Li, D. W. Gehrig, C. Ramanan, P. W. M. Blom, F. F. Kohl, M. Wagner, K. Landfester and K. A. I. Zhang, Adv. Synth. Catal., 2019, 361, 3852–3859 CrossRef CAS.
- H. Liu, C. Xu, D. Li and H.-L. Jiang, Angew. Chem., Int. Ed., 2018, 57, 5379–5383 CrossRef CAS PubMed.
- S. Mondal, L. Sahoo, C. P. Vinod and U. K. Gautam, Appl. Catal., B, 2021, 286, 119927 CrossRef CAS.
- R. Garg, S. Mondal, L. Sahoo, C. P. Vinod and U. K. Gautam, ACS Appl. Mater. Interfaces., 2020, 12, 29324–29334 CAS.
- G. He, Y. Lai, Y. Guo, H. Yin, B. Chang, M. Liu, S. Zhang, B. Yang and J. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 53724–53735 CrossRef CAS PubMed.
- X. Liu, R. Qi, S. Li, W. Liu, Y. Yu, J. Wang, S. Wu, K. Ding and Y. Yu, J. Am. Chem. Soc., 2022, 144, 23396–23404 CrossRef CAS PubMed.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00990h |
| This journal is © The Royal Society of Chemistry 2025 |