
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic imidazolium phosphate@titanium dioxide ionogels as environmental pollutant removers†
Mohamed
Boundor
a,
Nadia
Katir
 *a and
Abdelkrim
El Kadib
*ab
*a and
Abdelkrim
El Kadib
*ab
aEuromed University of Fes, UEMF, Morocco. E-mail: n.katir@ueuromed.org
bHassan II Academy of Science and Technology, Rabat, Morocco. E-mail: a.elkadib@ueuromed.org
First published on 27th January 2025
Abstract
Phosphate-containing ionic liquids display interesting inherent properties including versatile chemical reactivity, thermal and hydrolytic stability and biological response. This contribution focuses on synthesizing diverse imidazolium dialkylphosphates through a simple method involving N-quaternization of 1-alkylimidazole with trialkylphosphate. The use of these building-blocks during sol–gel polymerization of titanium alkoxide affords novel imidazolium phosphate@TiO2 hybrid ionogels. The highest affinity of phosphate towards titanium dioxide results in covalent functionalization of the two phases through stable P–O–Ti bridges, without compromising the molecular dynamic of the embedded ionic liquids. The cationic moiety can be fully exchanged as illustrated using Na+ and NH4+ providing access to ionomaterials with adjustable properties. The beneficial effect of the as-prepared dynamic hybrid ionogels was clearly demonstrated by their superior efficiency in removing representative cationic dyes, pharmaceutical pollutants, and metal ions compared to unmodified, native TiO2.
Introduction
Phosphate-containing ionic liquids stand as a new class of hydrophilic ionic liquids with notable attributes including outstanding reactivity, good thermal stability, enhanced hydrolysis resistance, and low toxicity.1–5 They find applications in diverse fields such as biomass processing,6–11 oil desulfurization,12–14 medical biotechnology,15,16 azeotropic system separation,17–20 and organic synthesis as solvents and catalysts.21–26 Remarkably, phosphate ionic liquids are credited for dissolving recalcitrant microcrystalline cellulose, opening exciting avenues in green and sustainable chemistry processing.2,27–30Akin to conventional ionic liquids, their liquid state often impedes their use as advanced solvents and electrolytes, especially in applications requiring solid forms.31,32 Challenges include high viscosity, low diffusion coefficients, purification and recycling difficulties, and high costs.33
One strategy to address these issues, particularly fluidity, is to immobilize ionic liquids on porous matrices.34–36 This can be achieved through physical confinement inside of the porous network or covalent grafting on the surface.34,35 The final properties can be adjusted for specific applications by modifying either the cationic or the anionic group of the starting ionic liquid as well as the selected porous carrier.34,35,37,38 Covalent grafting requires ionic liquids bearing specific groups like trimethoxysilyl, dimethylphosphonate, or thiol, along with supports having reactive surfaces, e.g. hydroxyl groups in the case of silica.34,35,39,40 For inert porous matrices like porous carbon and metal, additional surface pretreatment is required to introduce functional groups like hydroxyls.41 Covalent attachment prevents ionic liquid migration, and mitigates drawbacks like high viscosity and slow gas diffusion while lowering costs and improving thermal and chemical stability. The fruitful combination of high surface area supports with unique ionic liquid properties lends utility to various domains including catalysis, gas separation, energy storage, lubrication enhancement, and carbon material production.34,35,37,39,40
While the interaction of ionic liquids and silica hosts has been deeply studied, including either the entrapped or covalently linked forms,42–50 this was not the case for non-siliceous metal oxide carriers, where few examples were comparatively reported.51–53 With this aim, we herein report a straightforward synthesis of phosphate-containing ionic liquids (PILn) featuring 1,3-dialkylimidazolium paired with various dialkylphosphates. These ionic building blocks were used to develop novel nanostructured hybrid ionogels (PILn@TiO2) through sol–gel processing, involving the co-condensation of the phosphate anion with TiO2. Owing to the highest affinity of phosphate to titanium dioxide, the embedded ionic liquids form stable Ti–O–P bonds with the ceramic phase. The dynamic character of these hybrid ionogels was substantiated through spectroscopic analysis and further corroborated through cationic exchange. The imidazolium cation was indeed replaced by Na+ and NH4+ cations, without disrupting the way that anionic phosphate binds to titanium dioxide. We further demonstrate the efficiency of PILn@TiO2 for the removal of methylene blue as a representative cationic dye, tetracycline as a representative pharmaceutical antibiotic, and Fe(III) and Au(III) as metal ions from aqueous solutions.
Results and discussion
Synthesis of phosphate ionic liquids (PILn)
Phosphate ionic liquids were prepared via direct alkylation, involving the N-quaternization of 1-alkylimidazole with a slight excess of trialkylphosphate (1.2 eq.) under a nitrogen atmosphere (Scheme 1a). 1H NMR monitoring showed that the quaternization kinetics slowed as the phosphate's alkyl chain length increased. In a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2 ratio of alkylimidazole to trialkylphosphate, complete conversion was achieved after two days at 80 °C with trimethylphosphate versus three days at 120 °C with tributylphosphate. All ionic liquids were isolated in a quantitative yield of ∼99% and are liquid at room temperature.
:1.2 ratio of alkylimidazole to trialkylphosphate, complete conversion was achieved after two days at 80 °C with trimethylphosphate versus three days at 120 °C with tributylphosphate. All ionic liquids were isolated in a quantitative yield of ∼99% and are liquid at room temperature.
 | ||
| Scheme 1 (a) Synthesis of the PILn used in this study, (b) sol–gel method for preparing PILn@TiO2 ionogels and (c) cation exchange carried out on PILn@TiO2. | ||
The FTIR spectra of the synthesized phosphate ionic liquids show bands between 2800 and 3000 cm−1 corresponding to the stretching vibrations of alkyl groups (–CH2– and –CH3), and bands between 3100 and 3200 cm−1 for the imidazolium ring (–C–H). C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C vibrations appear at 1575 cm−1, while the PO vibration is observed between 1177 and 1239 cm−1.54 Additionally, a band at ∼3400 cm−1 indicates the presence of absorbed water, showing that the hydrophilicity of phosphate ionic liquids decreases with increasing carbon chain length (Fig. S1, ESI†).
C vibrations appear at 1575 cm−1, while the PO vibration is observed between 1177 and 1239 cm−1.54 Additionally, a band at ∼3400 cm−1 indicates the presence of absorbed water, showing that the hydrophilicity of phosphate ionic liquids decreases with increasing carbon chain length (Fig. S1, ESI†).
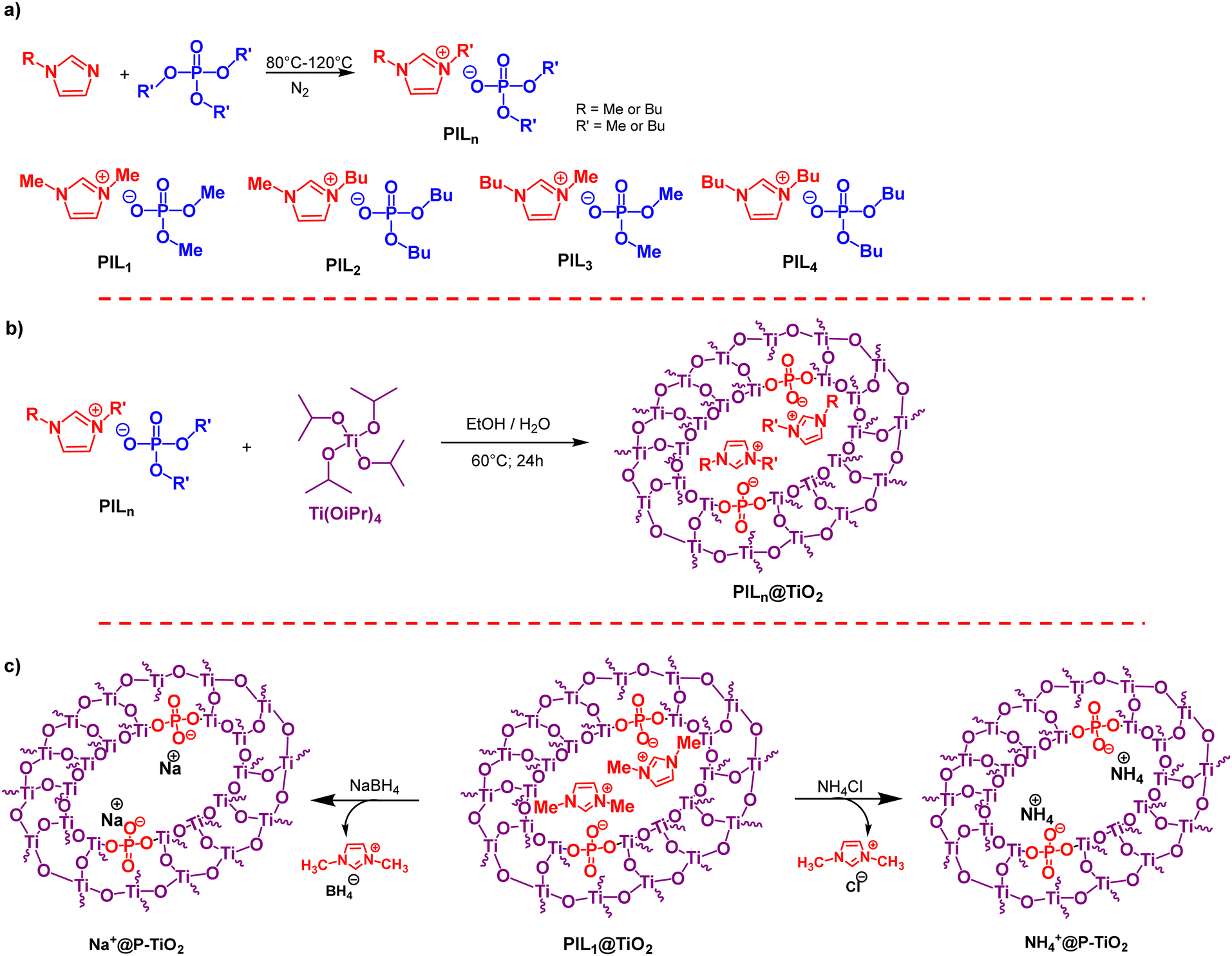
Multinuclear NMR spectroscopy confirmed the structure of the synthesized phosphate ionic liquids. The 31P NMR spectrum of PIL1 reveals a complete disappearance of the signal of the starting trimethylphosphate at 3.8 ppm and the appearance in turn of a new septuplet signal at 2.87 ppm (Fig. 1a), due to the coupling of phosphorus with six equivalent protons from the methoxy groups, with a coupling constant of 3JP–H = 10.7 MHz.54 This multiplicity indicates that each phosphate anion is counterbalanced by one imidazolium cation, which consequently excludes the formation of ionic liquids in dimeric or trimeric states during alkylation. In the 1H NMR spectrum of PIL1, the imidazolium proton appears at 8.55 ppm, with symmetric protons at positions 4 and 5 showing a doublet at 7.32 ppm. A doublet at 3.97 ppm corresponds to the six symmetric hydrogens on the two methyl groups at positions 1 and 3, with a weak coupling constant of 0.7 Hz. The dimethylphosphate anion signal was shown at 3.46 ppm, with a coupling constant of 3JP–H = 10.7 Hz that is fully consistent with the 31P NMR spectrum as discussed above. The 13C NMR spectrum of PIL1 displays peaks at 136.49, 123.35, and 35.55 ppm, corresponding to the carbons C2, C3, and N–CH3, respectively. The signal from the dimethylphosphate carbon appears as a doublet at 52.67 ppm, indicating a coupling constant of 2JC–P = 5.7 Hz between carbon and phosphorus (Fig. 1a). The chemical structure of the other phosphate ionic liquids was also elucidated by multinuclear (1H, 13C, 31P) NMR spectroscopy and provided in the experimental section (Fig. S2–S4, ESI†).
 | ||
| Fig. 1 (a) Liquid-state (31P, 1H and 13C) NMR of PIL1 and (b) solid-state (31P, 1H and 13C) MAS NMR of PIL1@TiO2. | ||
Preparation of PLIn@TiO2 hybrid ionogels
The synthesis of PILn@TiO2 was achieved through a two-step process involving hydrolysis and condensation of titanium tetraisopropoxide (Ti(OiPr)4) in the presence of PILn (Scheme 1b). Initially, phosphate-containing ionic liquid was dissolved in ethanol, and then Ti(OiPr)4 was added at a 1:20 molar ratio. After stirring for 15 minutes, water (2/5 the volume of ethanol) was introduced, resulting in a cloudy solution because of the extent of titanium alkoxide hydrolysis and condensation. The gel was subsequently heated at 60 °C for 24 hours. The resulting solid was filtered, washed with ethanol, and dried at 60 °C to yield a white solid powder of PILn@TiO2 ionogel.
The structural composition of the resulting materials has been elucidated through infrared spectroscopy (FTIR) and multinuclear solid-state MAS NMR spectroscopy, including 31P, 1H and 13C nuclei. The FTIR spectrum of PIL1@TiO2 reveals key bands, including Ti–O–Ti bending vibration at 445 cm−1, hydroxyl groups at 3600–2700 cm−1, and OH groups of physiosorbed water at 1638 cm−1. CC vibration of the imidazolium cation appears at 1575 cm−1, while the PO frequency of the phosphate group resonates at 1170 cm−1. The broader band at 1105 cm−1 is assignable to P–O–Ti stretching vibration, thereby confirming the covalent anchoring of the phosphate anion to the TiO2 surface.55 Materials with long alkyl chains show weak C–H stretching modes at 2853–2919 cm−1, indicating the retention of PIL's organic motifs within the TiO2 framework (Fig. S5, ESI†).
The solid-state 31P MAS NMR spectrum of various PILn@TiO2 exhibits a well resolved single signal ranging from approximately −0.04 to −1.32 ppm, attributed to the mineral phase bridged by a phosphate anion through the Ti–O–P(O)–O linker (Fig. 1b and Fig. S6, ESI†). This notable shift toward negative values, from 2.87 ppm for PIL1 to −0.04 ppm for PIL1@TiO2, can be attributed to the formation of at least one Ti–O–P bond.56 The variation in phosphorus chemical shifts observed with different PILn is associated with the nature of the counter-cation. When the positive charge of the cation decreases due to its inductive donor groups (such as butyl), the negative charge of its phosphate counter-anion becomes more significant, leading to enhanced shielding of the phosphorus. This suggests that the sol–gel process induces condensation between the titanium alkoxide and the alkoxy groups of the phosphate component of the ionic liquid, while maintaining the anionic character stabilized by its counter-cation. The cation remains intact, as confirmed by 1H and 13C NMR spectroscopy.
Surprisingly, the solid-state 1H MAS NMR spectrum of PIL1@TiO2 reveals intense and well-defined peaks corresponding to the protons of the 1,3-dimethylimidazolium cation, alongside a broad peak at 5.44 ppm attributed to the Ti–OH hydroxyl protons on the TiO2 surface (Fig. 1b). The signal at 8.86 ppm corresponds to the proton in position 2 of the imidazole ring, and the peak at 7.35 ppm represents the two hydrogens in positions 4 and 5 due to molecular symmetry. Additionally, the protons of the methyl group attached to nitrogen appear at 3.92 ppm. The integral intensities correlate perfectly with the number of protons present in the cationic part of the PIL1. Therefore, the absence of the methoxy group peak of the phosphate anion indicates their hydrolysis and release during the sol–gel process. 1H MAS NMR spectra of PIL2@TiO2 and PIL3@TiO2 show identical peaks corresponding to 1-butyl-3-methylimidazolium with their integration indicating that they exclusively correspond to the protons of the cation (Fig. 1b and Fig. S7, ESI†). This further confirms the removal of butoxy groups from the anion during the sol–gel process. This phenomenon also occurs in PIL4@TiO2, where all the peaks correspond exclusively to the carbon proton of the butylic groups attached to the nitrogen in the ring (Fig. S7, ESI†). The noticeably narrow signals observed for these ionogels reflect a pronounced dynamicity of the solid framework and suggest that the ionic character of the network remains dominant in the solid state, despite the significant network reticulation occurring during sol–gel polymerization.42
The solid-state 13C MAS NMR of PIL1@TiO2 exhibits characteristic signals of the 1,3-dimethylimidazolium cation, with carbon resonances of N–CH–N, N–CHCH–N, and N–CH3 observed at approximately 137, 122, and 35 ppm, respectively (Fig. 1b). The carbon peak from the anionic part of the starting methyl phosphate, expected around 53 ppm, vanishes. Similarly, in the case of ionogels with other PILn, the presence of cationic components was also noticed (Fig. S8, ESI†). These spectra collectively demonstrate the absence of peaks corresponding to carbons from the anionic part, consolidating its consumption and the creation of new P–O–Ti bonds during mineralization, while preserving the structure of the cation and its ionic interaction with the phosphate anion.
Thermogravimetric analysis was conducted to assess the thermal stability of these hybrid ionogels. Nearly the same weight degradation of 4% could be observed until 200 °C for all PILn@TiO2 due to the evaporation of water, alcohols, and physically adsorbed species. Subsequently, a much steeper slope takes place above 230 °C due to the decomposition of 1,3-dialkylimidazolium, with mass loss increasing with the cation's size (Fig. S9, ESI†). Therefore, ionogels with the same cation exhibit similar thermal behavior. Moreover, compared to TiO2 prepared under similar conditions, the weight loss beyond 230 °C in PIL1@TiO2 matches the calculated mass of the 1,3-dimethylimidazolium cation, suggesting its complete incorporation inside of the network (Fig. S9, ESI†).
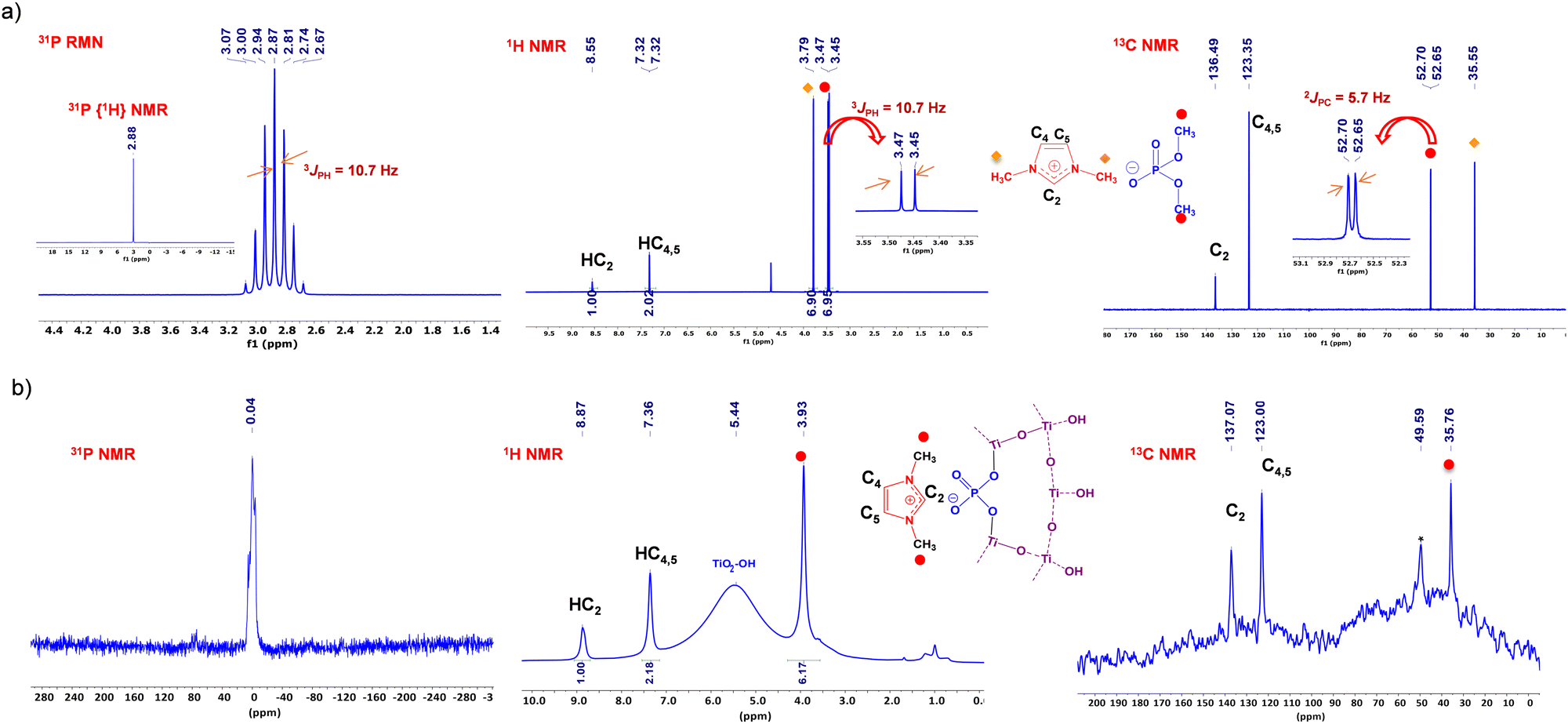
X-ray diffraction (XRD) analysis of PILn@TiO2 shows exclusively crystalline peaks of the anatase phase at 25.2, 37.8, 47.8, 53.9, 62.5, 69.2 and 74.8 corresponding respectively to the (101), (004), (200), (105), (213), (116) and (215) planes (Fig. 2a). The average crystallite size estimated from Scherrer's formula is 5 nm. However, the absence of polyphosphate or layered titanate phosphate peaks indicates that phosphorus is well dispersed in the composite and that TiO2 is sufficiently doped with phosphorus.57–60 Additionally, another peak was observed at 18° for the PIL4@TiO2 ionogel, which could be attributed to the packing of imidazolium ring moieties. This peak may indeed result from the large size of the cation, whose long alkyl chains could facilitate the formation of a certain organic periodicity, reminiscent of the one seen in metal–organic frameworks. For the smaller ionic liquid, this peak was observed at 17.9° when the PIL1 content was increased in the PIL1@TiO2 to a molar ratio of 1:5 (LIP1:Ti(OiPr)4) (Fig. S10, ESI†). Raman spectroscopy confirms the formation of anatase phase in PILn@TiO2, with distinct vibrational modes observed at 144, 396, 509, and 634 cm−1 corresponding to Eg, B1g (O–Ti–O bending), A1g (Ti–O stretching), and Eg (Ti–O stretching), respectively (Fig. 2b).61
 | ||
| Fig. 2 (a) XRD patterns and (b) Raman spectra of PILn@TiO2. | ||
Nitrogen adsorption–desorption of PILn@TiO2 shows an isotherm profile of type IV, with a hysteresis loop typical of mesoporous materials (Fig. S11, ESI†). While a surfactant-free design was chosen here for its simplicity and cost-effectiveness, PILn@TiO2 displayed a consistent specific surface area of 200 m2 g−1. The specific surface area is slightly higher for hybrid ionogels with asymmetric cationic and larger PILs (Table S1, ESI†).
Therefore, both the size and volume of the pores increase with the size of the anion, suggesting that the inorganic burst initially nucleates and then condenses with the anionic phosphate near to the surface. It is reasonable to assume the critical role of PILn serving as a structure-directing agent and confining medium to grow and crystallize discrete mesoporous anatase nanoparticles.62,63
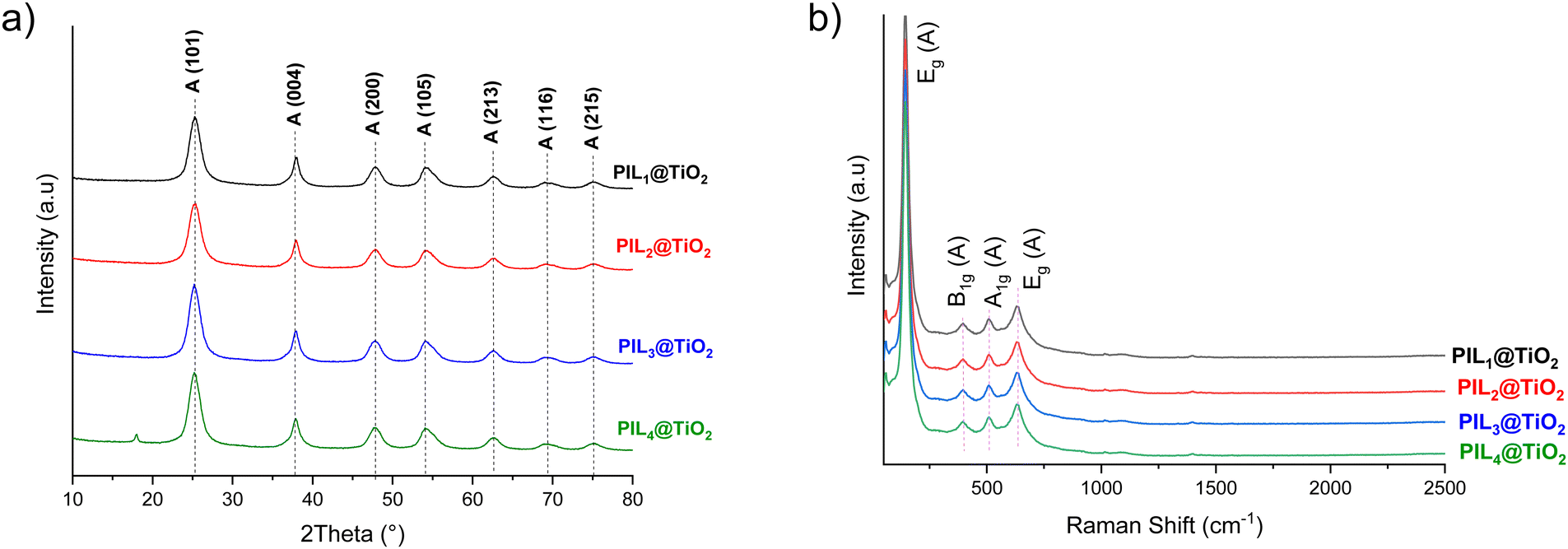
X-ray photoelectron spectroscopy (XPS) analysis of PIL1@TiO2 reveals the presence of Ti, O, P, N, and C, consistent with the expected chemical composition of the final ionogel (Fig. 3a). The C 1s spectrum can be deconvoluted into three different species, namely C–H, CH3–N and carbons from the imidazolium ring N–CC–N and NC–N components, with binding energies centered at 284.8, 286.3, and 288.6 eV, respectively.64 In the nitrogen region, the peak at 401.3 eV corresponds to the nitrogen (N+) in the imidazolium cation, while the smaller peak at 398.7 eV is attributed to the non-ionized nitrogen in the ring.65 These results indicate that the imidazolium cation retains its structural integrity and remains cationic. For oxygen species, the O 1s spectrum displays signals at 529.8 eV and 533.0 eV for oxygen in the oxide lattice (Ti–O–Ti) and on the surface (Ti–OH), respectively.66 The peak at 531.5 eV is associated with P–O–Ti and PO.67 In the phosphorus region, the P 2p peak can be deconvoluted into two main components located at 133.3 and 134.1 eV assigned to the Ti–O–P bond and phosphate species, respectively.68,69 The absence of a peak at 133.1 eV corresponding to the P–O–C bond indicates that the two methoxyl groups of PIL1 have been hydrolyzed and condensed with the TiO2 network.69 The high-resolution Ti 2p spectrum was deconvoluted into peaks at 458.5 eV (Ti 2p3/2) and 462.2 eV (Ti 2p1/2), confirming Ti–O bonds in the Ti4+ state. The negative shift of Ti 2p3/2 from the typical 459.3 eV in TiO2 is attributed to phosphorus doping and oxygen defects.70
 | ||
| Fig. 3 (a) XPS spectra and (b) SEM image/elemental analysis mapping by SEM-EDX of N, P, O and Ti of PIL1@TiO2. | ||
Scanning electron microscopy (SEM) images of PIL1@TiO2 reveal the formation of aggregates of spherical nanoparticles approximately 100 nm in size (Fig. 3b). The uniform size of the primary particles and the absence of large crystals indicate a controlled sol–gel polymerization, corroborating our assumptions that PIL1 plays a pivotal role in directing the nucleation and growth of the anatase mineral phase. Energy-dispersive X-ray spectroscopy (EDX) mapping of the elemental composition of both organic and inorganic phases reveals a uniform distribution of phosphorus and titanium throughout the microstructure. This provides convincing evidence for the intimate hybridization of PIL1 and TiO2 within the network, without phase separation or segregation (Fig. 3b).
Cation exchange on PIL1@TiO2
PIL1@TiO2 was next subjected to cation exchange. This route could provide a route for expanding the molecular diversity of the dynamic ionogels and further tailoring its properties. The exchange was executed using two reagents (NaBH4 and NH4Cl) to replace 1,3-dimethylimidazolium cations in PIL1@TiO2 with Na+ and NH4+, resulting, respectively, in the formation of Na+@P-TiO2 and NH4+@P-TiO2 (Scheme 1c).The exchange reaction was performed in ethanol, where PIL1@TiO2 was dispersed. A stoichiometric amount of the exchanging agent relative to 1,3-dimethylimidazolium was added. After stirring for 4 hours, the mixture was filtered and thoroughly washed with ethanol, and the solid was dried at 60 °C. Cation exchange was monitored by FTIR analysis, focusing on the CC band of the imidazolium ring at approximately 1573 cm−1. The absence of this signature for NH4+@P-TiO2 indicates complete exchange of the 1,3-dimethylimidazolium. In contrast, for Na+@P-TiO2, the sharp band transformed into a broad band (Fig. S12, ESI†). Solid-state 31P MAS NMR spectroscopy of Na+@P-TiO2 and NH4+@P-TiO2 showed broad signals at δ = −0.17 ppm and δ = −1.07 ppm (Fig. S13, ESI†). These peaks indicate the intactness of the phosphate moiety after the exchange reaction, with the slight chemical shift difference attributed to the nature of the counter-cation. Solid-state 13C MAS NMR confirms the disappearance of the characteristic peaks of the 1,3-dimethylimidazolium cation (Fig. S14, ESI†), demonstrating effective exchange with both sodium Na+ and ammonium NH4+ cations.
The thermal weight degradation profile of Na+@P-TiO2 was compared to that of PIL1@TiO2 (Fig. 4). The significant mass loss observed in the original ionogel above 230 °C was absent in the inorganic material, with the mass difference between the two materials from 500 °C onward corresponding to the 4% mass of the 1,3-dimethylimidazolium cation present before the exchange. Similarly, cation exchange with NH4Cl shifted the degradation onset from 260 °C in PIL1@TiO2 to 410 °C in NH4+@P-TiO2, demonstrating enhanced thermal stability of the exchanged materials compared to the original ionogels.
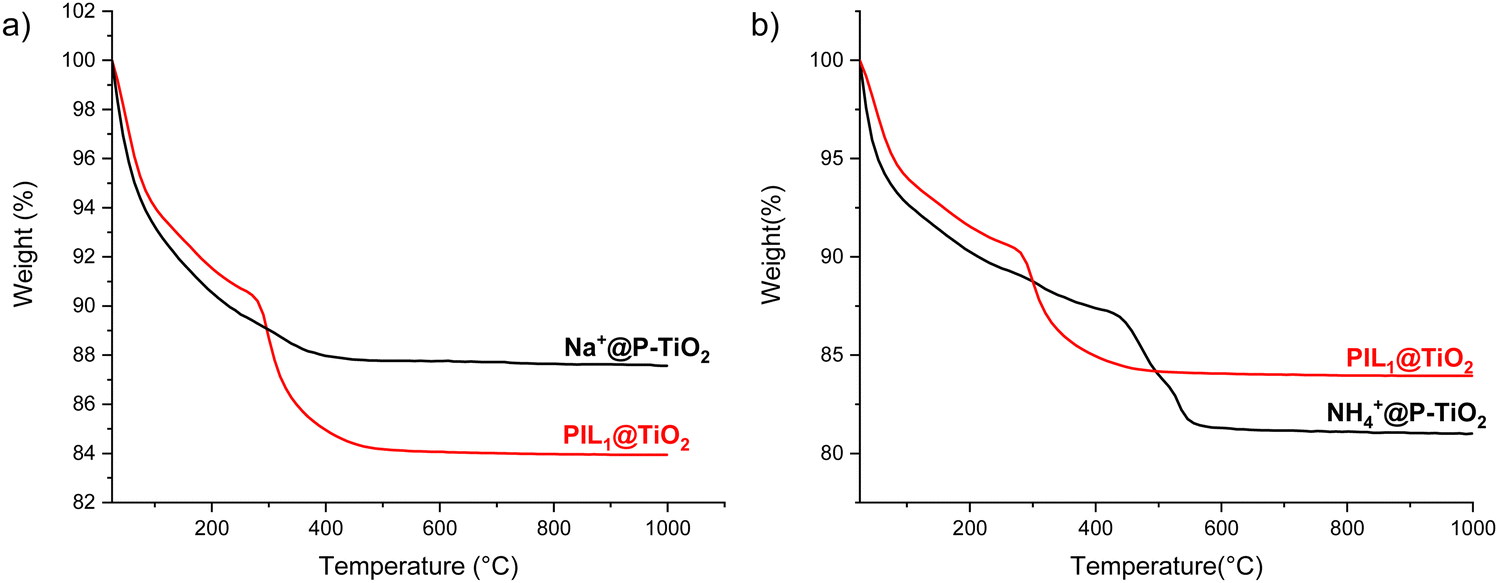 | ||
| Fig. 4 TGA analysis of (a) Na+@P-TiO2 and (b) NH4+@P-TiO2 compared with the starting material PILn@TiO2. | ||
Nitrogen adsorption–desorption isotherms of both materials show a hysteresis loop characteristic of open mesoporous structures (Fig. S15, ESI†). Na+@P-TiO2 displays a specific surface area of 194 m2 g−1 and an average pore size of 5 nm, indicating a slight shrinkage of the network during exchange. NH4+@P-TiO2 exhibits a specific surface area of 254 m2 g−1, which is 56 m2 g−1 greater than that of the parent ionogel. After cation exchange, both materials retain the crystalline anatase phase, as confirmed by XRD spectra with well-resolved peaks and an average crystallite size of about 5 nm (Fig. S16, ESI†). Raman spectroscopy further supports these findings by revealing the characteristic anatase modes consistent with the XRD data (Fig. S17, ESI†).
Removal of organic pollutants and metal ions from water
Ionic liquids display great affinity for chemical and metal pollutants, but are difficult to recover from the medium because of their inherent solubility.71 To address this issue, supported ionic liquids including ionogels present a scalable alternative. PILn@TiO2 materials were consequently tested for the removal of cationic methylene blue (MB) dye and tetracycline antibiotic (TC), and two metal ions namely Fe(III) and Au(III) from aqueous solutions.Fig. 5 and Fig. S18, and S19 (ESI†) illustrate the removal efficiency (%) and adsorption capacity (mg g−1) of MB, TC, Fe(III) and Au(III) using both unmodified TiO2 and PILn@TiO2. Pristine TiO2 enables only 20% removal of methylene blue, with a low adsorption capacity of 6 × 10−2 mg g−1, contrasting with the enhanced performance reached using PILn@TiO2 (Fig. 5a and Fig. S20, ESI†). The highest removal efficiency (99%) was recorded for PIL1@TiO2 (99%) and an adsorption capacity of 32 × 10−2 mg g−1.
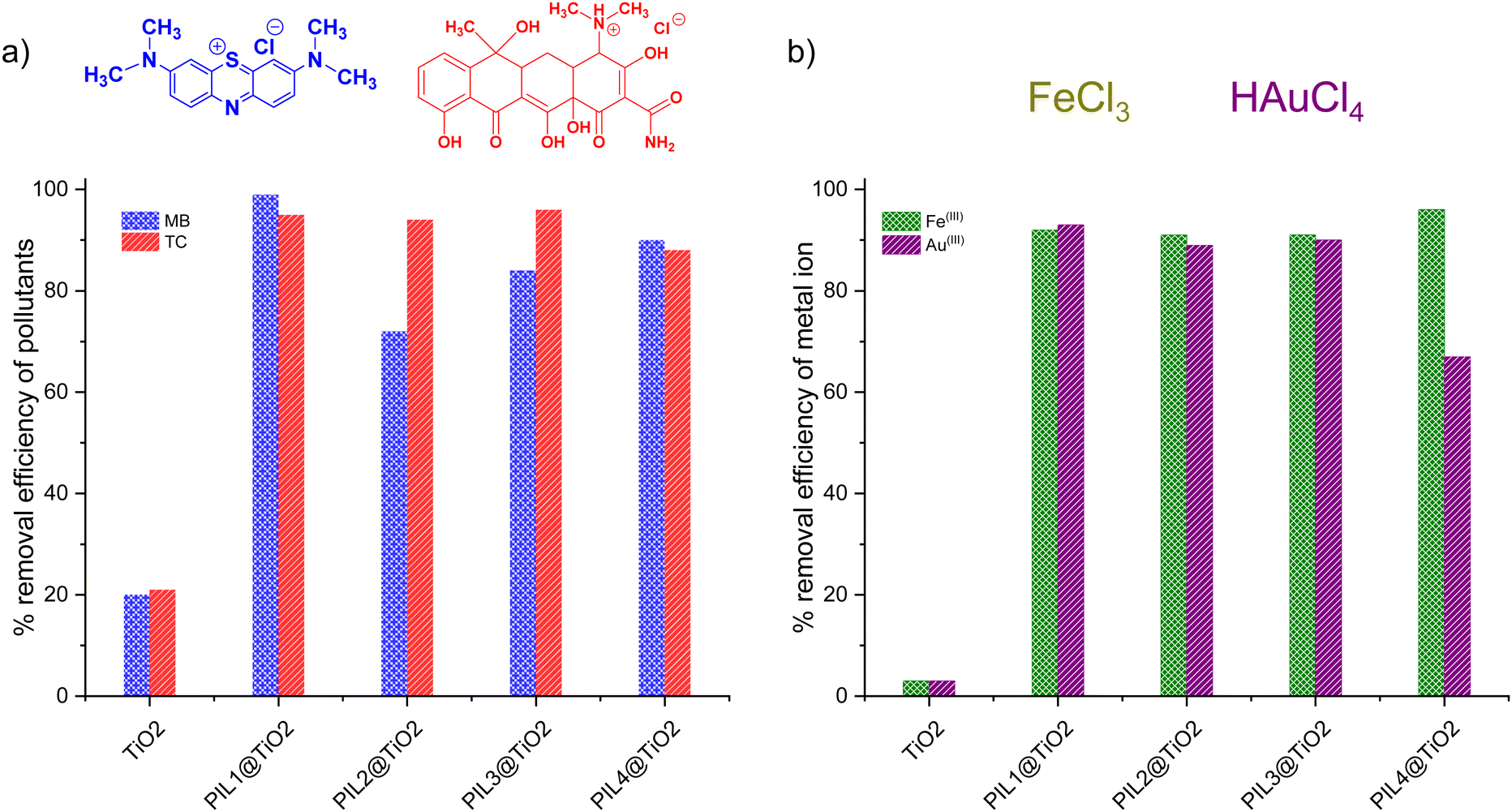 | ||
| Fig. 5 Removal of (a) methylene blue (MB) and tetracycline (TC), and (b) removal of Fe(III) and Au(III) using TiO2 and PILn@TiO2 materials. | ||
A similar trend was also observed for the uptake of tetracycline, with unmodified TiO2 being able to remove only 21%, with an adsorption capacity of 9.33 mg g−1. Comparatively, the removal efficiency attained by PILn@TiO2 exceeds 88%, with adsorption capacities greater than 39 mg g−1. PIL3@TiO2 stands as the most effective, with a removal efficiency of 96% and an adsorption capacity of 42.61 mg g−1.
Ionogels also offer substantial potential for metal extraction due to the immobilization of active chemical groups, which leads to a marked increase in the binding capacity of materials for a wide range of metal ions.72 In our study, unmodified TiO2 demonstrated marginal uptake of 3% for Fe(III) removal, with an adsorption capacity of 0.15 mg g−1. In contrast, regardless of their composition, PILn@TiO2 displayed nearly quantitative removal efficiency that exceeds 91%, with PIL4@TiO2 being the best scavenger (96% removal and an adsorption capacity of 4.77 mg g−1). Unmodified TiO2 was also ineffective for Au(III) uptake, enabling only 3% removal and an adsorption capacity of 0.85 mg g−1. PILn@TiO2 showed spectacular improvements, with PIL1@TiO2 achieving the highest removal efficiency (93%) and an adsorption capacity of 25.9 mg g−1. Notably, during the adsorption process, Au(III) was spontaneously reduced to Au(0) nanoparticles, as evidenced by the color variation of PILn@TiO2 powders from white to dark purple,73–75 after filtration and drying of the adsorbents (Fig. S21, ESI†). Overall, these results highlight the interesting performance of PILn@TiO2 compared to unmodified TiO2, with the resulting ionogels being competitive compared to standard absorbents (Table S2, ESI†). Besides, the reducing ability of these ionogels as evidenced for gold, and their high-surface area open new channels of possibilities in the field of heterogeneous (photo)catalysis.
Conclusions
In summary, this study focused on the straightforward preparation of a set of dialkylphosphate-containing ionic liquids, using a catalyst-free, solvent-free, single-step synthesis. Next, the co-condensation of these ionic building-blocks with titanium alkoxide resulted in the formation of novel titanium oxide entrapping imidazolium phosphate ionic liquids. These dynamic hybrid ionogels exhibit several distinctive features, including the formation of stable P–O–Ti covalent bonds, while retaining the ionic character and the mobility of organic moieties as shown by solid-state 1H and 13C NMR of the imidazolium cation, an open porous network formed under surfactant-free conditions, and a crystalline framework built from discrete anatase nanoparticles. The potential for cation exchange within the iono-materials was demonstrated, affecting thermal and structural properties. Additionally, the hybrid ionogels showed a remarkable ability to remove chemical pollutants and metal ions from water, showcasing their environmental relevance.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by UEMF.References
- E. Kuhlmann, S. Himmler, H. Giebelhaus and P. Wasserscheid, Green Chem., 2007, 9, 233–242 RSC.
- T. Endo, S. Yoshida and Y. Kimura, Cryst. Growth Des., 2020, 20, 6267–6271 CrossRef CAS.
- P. D. Vu, A. J. Boydston and C. W. Bielawski, Green Chem., 2007, 9, 1158–1159 RSC.
- M. Musiał, E. Zorebski, K. Malarz, M. Kuczak, A. Mrozek-Wilczkiewicz, J. Jacquemin and M. Dzida, ACS Sustainable Chem. Eng., 2021, 9, 7649–7657 CrossRef.
- K. D. Weaver, H. J. Kim, J. Sun, D. R. MacFarlane and G. D. Elliott, Green Chem., 2010, 12, 507–513 RSC.
- W.-T. Wang, J. Zhu, X.-L. Wang, Y. Huang and Y.-Z. Wang, J. Macromol. Sci. Chem., Part B, 2010, 49, 528–541 CrossRef CAS.
- M. Feng, X. Lu, L. Wang, J. Zhang, S. Yang, C. Shi, Q. Zhou and S. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 11990–11998 CAS.
- L. Wang, Y. Nie, X. Zhang, S. Zeng, S. Zhang and S. Zheng, Chem. Eng. Technol., 2016, 39, 979–986 CrossRef CAS.
- J.-i Horinaka, A. Okamoto and T. Takigawa, Int. J. Biol. Macromol., 2016, 91, 789–793 CrossRef CAS PubMed.
- M. A. Martins, U. Domanska, B. Schröder, J. A. Coutinho and S. P. Pinho, ACS Sustainable Chem. Eng., 2016, 4, 548–556 CrossRef CAS.
- C. Kornpointner, A. S. Martinez, M. Schnürch, H. Halbwirth and K. Bica-Schröder, Green Chem., 2021, 23, 10079–10089 RSC.
- K. Desai, S. Dharaskar, J. Pandya, S. Shinde and V. Vakharia, Process Saf. Environ. Prot., 2022, 166, 512–523 CrossRef CAS.
- H. Gao, S. Zeng, X. Liu, Y. Nie, X. Zhang and S. Zhang, RSC Adv., 2015, 5, 30234–30238 RSC.
- Y. Chen, F. Mutelet and J.-N. Jaubert, J. Chem. Eng. Data, 2014, 59, 603–612 CrossRef CAS.
- M. Smiglak, J. M. Pringle, X. Lu, L. Han, S. Zhang, H. Gao, D. R. Macfarlane and R. D. Rogers, Chem. Commun., 2014, 50, 9228–9250 RSC.
- M. Moniruzzaman, N. Kamiya and M. Goto, J. Colloid Interface Sci., 2010, 352, 136–142 CrossRef CAS.
- X. Han, Y. Wang and Q. Li, J. Chem. Eng. Data, 2022, 67, 2418–2425 CrossRef CAS.
- Z. Zhang, X. Zhao, Y. Wang, Y. Ma and G. Li, Sep. Purif. Technol., 2022, 287, 120491 CrossRef CAS.
- K. Yue and G. Zhou, J. Mol. Liq., 2022, 348, 118404 CrossRef CAS.
- F. Chen, L. Zhang, Z. Liu and G. Yu, Ind. Eng. Chem. Res., 2020, 59, 13271–13282 CrossRef CAS.
- A. Zicmanis and L. Anteina, Tetrahedron Lett., 2014, 55, 2027–2028 CrossRef CAS.
- Z. I. Ishak, N. A. Sairi, Y. Alias, M. K. T. Aroua and R. Yusoff, Chem. Eng. J., 2016, 297, 128–138 CrossRef CAS.
- T. Ståhlberg, M. G. Sørensen and A. Riisager, Green Chem., 2010, 12, 321–325 RSC.
- S. Zhang, L. Dias Goncalves, H. Lefebvre, M. Tessier, B. Rousseau and A. Fradet, ACS Macro Lett., 2012, 1, 1079–1082 CrossRef CAS PubMed.
- M. A. Redouane, N. Khiri-Meribout, S. Benzerka and A. Debache, Heterocycl. Commun., 2019, 25, 167–179 CAS.
- M. Y. Machado and R. Dorta, Synthesis, 2005, 2473–2475 CAS.
- J. Vitz, T. Erdmenger, C. Haensch and U. S. Schubert, Green Chem., 2009, 11, 417–424 RSC.
- M. T. Clough, J. A. Griffith, O. Kuzmina and T. Welton, Green Chem., 2016, 18, 3758–3766 RSC.
- S. Bahrani, S. Raeissi and M. Sarshar, Bioresour. Technol., 2015, 185, 411–415 CrossRef CAS.
- D. Silva and E. Bogel-Łukasik, Green Chem., 2017, 19, 4048–4060 RSC.
- A. K. Tripathi, Mater. Today Energy, 2021, 20, 100643 CrossRef CAS.
- H. Hu, J. Li and X. Ji, Chem. – Eur. J., 2024, 30, e202302826 CrossRef CAS PubMed.
- N. Gao, Y. Yang, Z. Wang, X. Guo, S. Jiang, J. Li, Y. Hu, Z. Liu and C. Xu, Chem. Rev., 2023, 124, 27–123 CrossRef PubMed.
- S. Zhang, J. Zhang, Y. Zhang and Y. Deng, Chem. Rev., 2017, 117, 6755–6833 CrossRef CAS PubMed.
- M. Dong, K. Zhang, X. Wan, S. Wang, S. Fan, Z. Ye, Y. Wang, Y. Yan and X. Peng, Small, 2022, 18, 2108026 CrossRef CAS PubMed.
- M.-A. Néouze, J. Le Bideau, P. Gaveau, S. Bellayer and A. Vioux, Chem. Mater., 2006, 18, 3931–3936 CrossRef.
- E. H. Lahrar, A. Belhboub, P. Simon and C. Merlet, ACS Appl. Mater. Interfaces, 2019, 12, 1789–1798 CrossRef PubMed.
- Y. Chen, Z. Chang, Y. Liu, X. Wan, T. Wang, Z. Zhou and G. Li, Eur. Polym. J., 2024, 210, 112992 CrossRef CAS.
- B. Xin and J. Hao, Chem. Soc. Rev., 2014, 43, 7171–7187 RSC.
- Q. Dou, L. Liu, B. Yang, J. Lang and X. Yan, Nat. Commun., 2017, 8, 2188 CrossRef PubMed.
- M. J. Park, J. K. Lee, B. S. Lee, Y.-W. Lee, I. S. Choi and S.-G. Lee, Chem. Mater., 2006, 18, 1546–1551 CrossRef CAS.
- R. Göbel, P. Hesemann, J. Weber, E. Möller, A. Friedrich, S. Beuermann and A. Taubert, Phys. Chem. Chem. Phys., 2009, 11, 3653–3662 RSC.
- B. Gadenne, P. Hesemann and J. J. Moreau, Chem. Commun., 2004, 1768–1769 RSC.
- N. Abdou, B. Alonso, N. Brun, P. Landois, A. Taubert, P. Hesemann and A. Mehdi, Mater. Chem. Front., 2022, 6, 939–947 RSC.
- N. Abdou, B. Alonso, N. Brun, S. Devautour-Vinot, M. Paillet, P. Landois, A. Mehdi and P. Hesemann, J. Phys. Chem. C, 2022, 126, 20937–20945 CrossRef CAS.
- N. Abdou, P. Dieudonné-George, N. Brun, A. Mehdi and P. Hesemann, Phys. Chem. Chem. Phys., 2022, 24, 21853–21862 RSC.
- J. Le Bideau, L. Viau and A. Vioux, Chem. Soc. Rev., 2011, 40, 907–925 RSC.
- J. Le Bideau, P. Gaveau, S. Bellayer, M.-A. Neouze and A. Vioux, Phys. Chem. Chem. Phys., 2007, 9, 5419–5422 RSC.
- K. Lunstroot, K. Driesen, P. Nockemann, C. Görller-Walrand, K. Binnemans, S. Bellayer, J. Le Bideau and A. Vioux, Chem. Mater., 2006, 18, 5711–5715 CrossRef CAS.
- A. El Kadib, P. Hesemann, K. Molvinger, J. Brandner, C. Biolley, P. Gaveau, J. J. Moreau and D. Brunel, J. Am. Chem. Soc., 2009, 131, 2882–2892 CrossRef CAS PubMed.
- A. Dutta, D. K. Mishra, D. Kundu, U. Mahanta, S. P. Jiang, D. S. Silvester and T. Banerjee, Ind. Eng. Chem. Res., 2022, 61, 8763–8774 CrossRef CAS.
- X. Li, Z. Zhang, L. Yang, K. Tachibana and S.-I. Hirano, J. Power Sources, 2015, 293, 831–834 CrossRef CAS.
- Z. Zhao, Y. Yin, X. Jin, G. Zhang, L.-M. Wang, Y. D. Liu and H. J. Choi, ACS Appl. Nano Mater., 2021, 4, 12382–12392 CrossRef CAS.
- R. Streck and A. J. Barnes, Spectrochim. Acta, Part A, 1999, 55, 1049–1057 CrossRef.
- T.-Y. Ma, L. Liu, Q.-F. Deng, X.-Z. Lin and Z.-Y. Yuan, Chem. Commun., 2011, 47, 6015–6017 RSC.
- A. I. Bortun, L. Bortun, A. Clearfield, M. A. Villa-García, J. R. García and J. Rodríguez, J. Mater. Res., 1996, 11, 2490–2498 CrossRef CAS.
- Y. Brahmi, N. Katir, J. A. M. Agullo, A. Primo, M. Bousmina, J. P. Majoral, H. Garcia and A. El Kadib, Dalton Trans., 2015, 44, 15544–15556 RSC.
- Y. Brahmi, N. Katir, A. Hameau, A. Essoumhi, E. M. Essassi, A.-M. Caminade, M. Bousmina, J.-P. Majoral and A. El Kadib, Chem. Commun., 2011, 47, 8626–8628 RSC.
- M. Boundor, F. Semerci, N. Katir, S. Royer and A. El Kadib, J. Energy Storage, 2024, 79, 110119 CrossRef.
- Y. Brahmi, N. Katir, M. Ianchuk, V. Colliere, E. M. Essassi, A. Ouali, A.-M. Caminade, M. Bousmina, J. P. Majoral and A. El Kadib, Nanoscale, 2013, 5, 2850–2856 RSC.
- W. Ma, Z. Lu and M. Zhang, Appl. Phys. A: Mater. Sci. Process., 1998, 66, 621–627 CrossRef CAS.
- N. Katir, Y. Brahmi, J. P. Majoral, M. Bousmina and A. El Kadib, Chem. Commun., 2015, 51, 17716–17719 RSC.
- Y. Zhou and M. Antonietti, J. Am. Chem. Soc., 2003, 125, 14960–14961 CrossRef CAS PubMed.
- J. Y. Lee, N. Y. Kim, D. Y. Shin, H.-Y. Park, S.-S. Lee, S. Joon Kwon, D.-H. Lim, K. W. Bong, J. G. Son and J. Y. Kim, J. Nanopart. Res., 2017, 19, 98 CrossRef.
- L. Cen, K. Neoh and E.-T. Kang, Adv. Mater., 2005, 17, 1656–1661 CrossRef CAS.
- L. Azizova, D. Morgan, J. Rowlands, E. Brousseau, T. Kulik, B. Palianytsia, J. P. Mansell, J. Birchall, T. Wilkinson and A. Sloan, Appl. Surf. Sci., 2022, 604, 154462 CrossRef CAS.
- G. Mani, D. M. Johnson, D. Marton, V. L. Dougherty, M. D. Feldman, D. Patel, A. A. Ayon and C. M. Agrawal, Langmuir, 2008, 24, 6774–6784 CrossRef CAS PubMed.
- J. Song, Z. Yu, M. L. Gordin, S. Hu, R. Yi, D. Tang, T. Walter, M. Regula, D. Choi and X. Li, Nano Lett., 2014, 14, 6329–6335 CrossRef CAS PubMed.
- X. Wu, K. Gong, G. Zhao, W. Lou, X. Wang and W. Liu, RSC Adv., 2018, 8, 4595–4603 RSC.
- S. Wang, J. Lian, W. Zheng and Q. Jiang, Appl. Surf. Sci., 2012, 263, 260–265 CrossRef CAS.
- C. Rizzo, S. Marullo, P. R. Campodonico, I. Pibiri, N. T. Dintcheva, R. Noto, D. Millan and F. D’Anna, ACS Sustainable Chem. Eng., 2018, 6, 12453–12462 CrossRef CAS.
- S. Boudesocque, L. Viau, H. Nouali and L. Dupont, Sep. Purif. Technol., 2023, 322, 124285 CrossRef CAS.
- V. Amendola, R. Pilot, M. Frasconi, O. M. Maragò and M. A. Iatì, J. Phys. Condens. Matter., 2017, 29, 203002 CrossRef PubMed.
- M. Boundor, B. Bielska, N. Katir, N. Wronska, K. Lisowska, M. Bryszewska, K. Miłowska and A. El Kadib, ACS Appl. Polym. Mater., 2023, 5, 9952–9963 CrossRef CAS.
- M. Boundor, N. Katir, S. Royer and A. El Kadib, ACS Appl. Nano Mater., 2025, 8, 639–648 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Apparatus and analytical procedures; experimental details; infrared spectra; supplementary liquid and solid NMR spectra; N2 adsorption/desorption isotherms and textural parameters; supplementary XRD and Raman spectra. See DOI: https://doi.org/10.1039/d4ma01073f |
| This journal is © The Royal Society of Chemistry 2025 |