
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible-light-driven hydrogen evolution using calcium titanate codoped with aluminium, antimony, magnesium, and rhodium, loaded with a platinum (core)–chromia (shell) cocatalyst†
Tomoya
Ota
a,
Ryota
Tomizawa
b,
Tomoya
Nagano
b,
Koji
Hayashi
b and
Shigeru
Ikeda
 *ac
*ac
aDepartment of Chemistry, Konan University, 9-1 Okamoto, Higashinada-ku, Kobe, Hyogo 658-8501, Japan. E-mail: s-ikeda@konan-u.ac.jp
bCarbon Neutral Development Division, Toyota Motor Corporation, 1200 Mishuku, Susono, Shizuoka 410-1193, Japan
cInstitute for Energy Conversion Materials, Konan University, 9-1 Okamoto, Higashinada-ku, Kobe, Hyogo 658-8501, Japan
First published on 24th April 2025
Abstract
Calcium titanate codoped with Al3+, Sb5+, Mg2+, and Rh (Rh3+ or Rh4+), prepared via the flux method, showed efficient visible-light-driven H2 evolution using methanol as a hole scavenger. Loading Pt (core)–Cr2O3 (shell) nanoparticles as a cocatalyst enhanced the performance, with an apparent quantum yield of 7.1% being achieved at 420 nm.
Sunlight-induced water splitting using mixed-oxide semiconductor photocatalysts has gained attention for its potential in hydrogen (H2) production, an alternative energy source for fossil fuels.1 Efficient utilization of sunlight requires photocatalysts with narrow band gaps to absorb visible light. However, many mixed-oxide semiconductors have a deep valence band maximum (VBM) because valence bands of such mixed-oxide semiconductors consist of oxygen 2p orbitals,2 making it challenging to achieve a sufficient reduction potential for H2 evolution. One approach is to hybridize oxygen 2p orbitals with other atomic orbitals, creating a shallower VBM. Materials such as oxynitrides,3–5 oxysulfides,6,7 and oxyhalides8,9 have demonstrated visible-light-driven H2 production.
Another approach to provide photocatalytic activity under visible-light irradiation is doping of a transition metal cation to form a mid-gap state (or an impurity level) in the forbidden band of a wide-gap semiconductor compound. Although such a concept used to be unpromising because it was thought that such an energy state would facilitate recombination of photoexcited carriers,10 several reports have shown that visible light-driven photocatalytic reactions are induced over transition metal-doped photocatalysts based on wide-gap host compounds including SrTiO3,11–20 BaTiO3,21 NaTaO3,22 and ZnGa2O4.23 Among them, SrTiO3 is a promising host compound for photocatalytic H2 evolution under visible light. Since the first example of doping trivalent rhodium ion (Rh3+) to substitute the B site (Ti4+) in the crystal lattice of SrTiO3 (SrTiO3:Rh),11 several photocatalytic materials using various kinds of dopants have been reported by Kudo et al.12–16,20 Further improvements were achieved by applying a codoping concept that is charge compensation of a doped cation by a codopant, such as pentavalent antimony ion (Sb5+) at the Ti4+ site (SrTiO3:Sb,Rh)12,13 or tetravalent lanthanum ion (La3+) at the Sr2+ site (SrTiO3:La,Rh).19,20 These strategies stabilize the trivalent state of the doped rhodium, leading to significant enhancement of photocatalytic H2 evolution activity.
Calcium titanate (CaTiO3), a perovskite compound with a band gap of 3.5 eV,24 has potential as a host material for photocatalysts, but there has been limited success in the use of CaTiO3 compared to SrTiO3. Compared to SrTiO3, the wider band gap of CaTiO3, along with its structural properties of CaTiO3, make it suitable for transition-metal doping. However, there have been few reports on the application of CaTiO3 as the host material, and high photocatalytic activity has not been achieved.25 In this study, visible-light-responsive CaTiO3:Rh photocatalysts were developed with codoping of Al3+, Sb5+, and Mg2+ at the Ti4+ site (CaTiO3:Al,Sb,Mg,Rh). In addition, a cocatalyst of Pt (core)–Cr2O3 (shell) nanoparticles was developed to further boost H2 evolution efficiency. The optimized CaTiO3:Al,Sb,Mg,Rh system achieved impressive apparent quantum yields (AQYs) of 8.0% at 400 nm and 7.1% at 420 nm in a sacrificial H2 evolution.
We synthesized powder samples of CaTiO3 doped with Al3+ (CaTiO3:Al), CaTiO3:Al codoped with Rh (Rh3+ or Rh4+) (CaTiO3:Al,Rh), CaTiO3:Al,Rh codoped with Sb5+ (CaTiO3:Al,Sb,Rh), and CaTiO3:Al,Sb,Rh codoped with Mg2+ (CaTiO3:Al,Sb,Mg,Rh) using the flux method in molten CaCl2 (details in ESI†). All of the samples contained Al3+ derived from the alumina crucible used during the flux treatment, while the other dopants were intentionally introduced during synthesis. Powder XRD analyses revealed that all samples exhibited orthorhombic CaTiO3 perovskite structures (ICSB 1000022) with no detectable impurities (Fig. 1a), suggesting successful incorporation of dopants into the crystalline lattice of the host CaTiO3. The ionic radii of the dopants used (Rh3+: 66.5 pm (or Rh4+: 60.0 pm), Al3+: 53.5 pm, Sb5+: 60.0 pm, and Mg2+: 72.0 pm) are significantly smaller than the ionic radius of A site Ca2+ (134 pm) but comparable to the B-site Ti4+ radius (60.5 pm). Thus, these dopants should occupy the B-site positions. Furthermore, no appreciable shifts in the diffraction peaks of CaTiO3 were observed, indicating negligible lattice expansion or contraction due to doping.
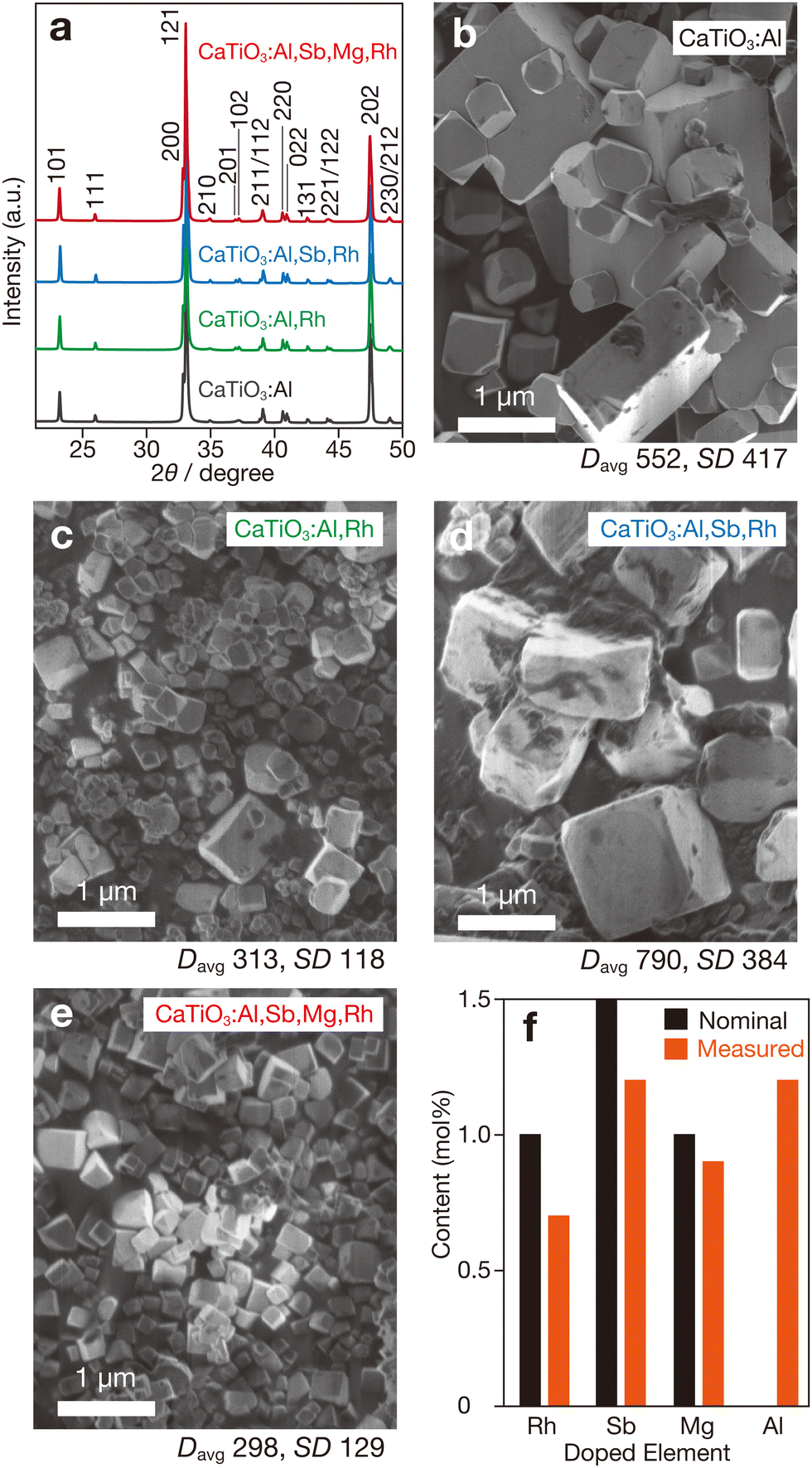 | ||
| Fig. 1 Structural characterizations of doped CaTiO3 samples. (a) XRD patterns and SEM images of (b) CaTiO3:Al, (c) CaTiO3:Al,Rh, (d) CaTiO3:Al,Sb,Rh and (e) CaTiO3:Al,Sb,Mg,Rh, (f) nominal and measured amounts of doped elements in CaTiO3:Al,Sb,Mg,Rh. Davg and SD values in panels (b)–(e) denote average diameter and standard deviation in nanometre units, respectively. | ||
In contrast to the crystallographic results, the morphological characteristics of samples with different compositions showed notable variations. The CaTiO3:Al sample consisted of cubic or rectangular particles with relatively large sizes (Fig. 1b). The exposed surfaces were likely facets oriented along the {100} and {001} directions, suggesting that each particle would be in a single-crystalline state. For the CaTiO3:Al,Rh sample, a significant reduction in particle size was observed (Fig. 1c), indicating that rhodium doping suppressed particle growth. Measurements of average particle size (Davg) and standard deviation (SD) (Fig. S1, ESI†) revealed reductions of more than half in both parameters compared to the CaTiO3:Al sample. Conversely, substantial particle growth was observed in the antimony-codoped sample (CaTiO3:Al,Sb,Rh), with a larger Davg than that of CaTiO3:Al, as shown in Fig. 1d. On the other hand, the sample further codoped with magnesium (CaTiO3:Al,Sb,Mg,Rh) exhibited smaller particles with relatively uniform size distributions (Fig. 1e). Although the underlying mechanisms remain unclear, these results suggest that particle size depends on the valency of the dopant. The high-valent Sb5+ cations promoted particle growth, whereas the low-valent Mg2+ cation appeared to suppress particle growth. ICP measurements (Fig. 1f) confirmed that the intentionally added dopants were present in amounts exceeding half of their respective nominal contents, while the Al3+ component, leached from the alumina crucible, was present in comparable quantities.
Photocatalytic activity of doped CaTiO3 samples for water reduction in the presence of methanol as a sacrificial hole scavenger under visible-light irradiation (>400 nm) was evaluated. The samples were loaded with a nanoparticulate platinum (Pt) cocatalyst by using the photodeposition method (details in ESI†). Since the Pt was found to be the most effective of the known cocatalysts for the SrTiO3-based doped photocatalytic system11–13,19,20 (data not shown), the effect of the Pt cocatalyst was focused in this study. Fig. 2a shows the amounts of H2 evolved as a function of photoirradiation time for these photocatalysts. The CaTiO3:Al-based photocatalyst showed almost no H2 evolution due to its lack of photoabsorption in the visible light region (see below). In contrast, photocatalysts containing rhodium exhibited visible light-driven H2 evolution. Compared to the CaTiO3:Al,Rh-based photocatalyst, codoping with Sb5+ significantly increased the H2 evolution rate. The activity was further enhanced in the CaTiO3:Al,Sb,Mg,Rh-based photocatalyst.
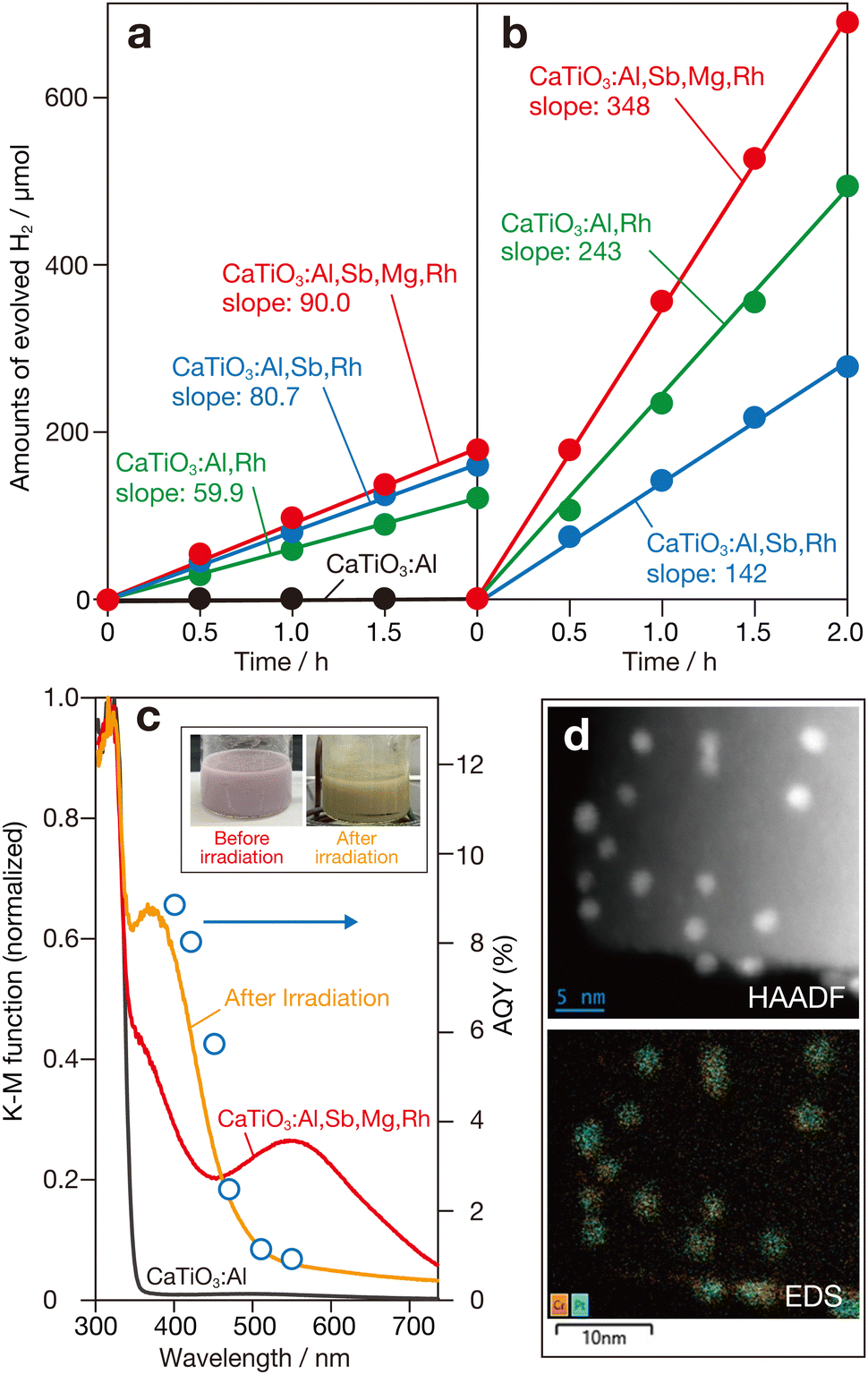 | ||
| Fig. 2 Amount of evolved H2 as a function of photoirradiation (>400 nm) duration from aqueous methanol solutions containing doped CaTiO3 samples loaded with (a) Pt and (b) Pt (core)–Cr2O3 (shell) (Pt/Cr2O3) cocatalysts. Slope values denote the rate of H2 evolution (μmol h−1). (c) DR spectra of CaTiO3:Al, CaTiO3:Al,Sb,Mg,Rh, and a suspension of Pt/Cr2O3-loaded CaTiO3:Al,Sb,Mg,Rh after photoirradiation. Dependence of AQYs for the sacrificial H2 evolution on wavelength of incident photons is also plotted. The inset shows photographs of a suspension containing CaTiO3:Al,Sb,Mg,Rh (labelled Before irradiation) and that loaded with Pt/Cr2O3 cocatalyst obtained after photodeposition process (labelled after irradiation). (d) HAADF scanning TEM (upper) and EDS elemental mapping (lower) images of loaded Pt/Cr2O3 cocatalyst. | ||
Instead of conventional Pt cocatalysts, a Pt (core)–chromia (shell) (Pt/Cr2O3) cocatalyst was used for the sacrificial H2 evolution system. This cocatalyst was prepared by successive reductive photodeposition of aqueous precursors, similar to the method used for Rh (core)–Cr2O3 (shell) (Rh/Cr2O3) cocatalysts, which are known to be effective in several photocatalysts for overall water splitting.5,26,27Fig. 2b shows typical time courses of H2 evolution for Pt/Cr2O3-loaded CaTiO3:Al,Rh, CaTiO3:Al,Sb,Rh, and CaTiO3:Al,Sb,Mg,Rh photocatalysts. Compared to photocatalysts with conventional Pt cocatalysts, significant improvements in H2 evolution were observed for the photocatalysts. Notably, the CaTiO3:Al,Sb,Mg,Rh-based photocatalyst exhibited a substantial increase in the H2 production rate. The activity was continued for a further 2 runs without any appreciable degradation (Fig. S2, ESI†). The apparent quantum yields (AQYs) of this photocatalyst were 8.0% at 400 nm and 7.1% at 420 nm. To the best of our knowledge, these results represent the first demonstration of significantly high photocatalytic performance in an Rh-doped photocatalytic system using a host semiconductor material other than SrTiO3.
Fig. 2c shows diffuse reflection (DR) spectra of CaTiO3:Al and CaTiO3:Al,Sb,Mg,Rh samples. The CaTiO3:Al sample exhibited fundamental photoabsorption in the ultraviolet region, with a photoabsorption onset at 355 nm. From this onset, the band-gap energy was estimated to be approximately 3.5 eV, being consistent with the previously reported value for undoped CaTiO3.24 In contrast, the CaTiO3:Al,Sb,Mg,Rh sample exhibited additional photoabsorption extending from the ultraviolet region into the visible light region. This additional absorption can be divided into two components: a shoulder component near the band-gap absorption of CaTiO3, centered around 390 nm and a broad absorption band in the visible region, centered around 560 nm. Based on results of previous studies on Rh-doped photocatalysts using SrTiO3,11,13 these two absorption bands are attributed to transitions related to trivalent rhodium ion (Rh3+) and tetravalent rhodium ion (Rh4+). The Rh3+ dopant acts as an electron donor, with its absorption band corresponding to transitions from the donor level introduced by Rh3+ to the conduction band, which facilitates water reduction to H2. In contrast, the Rh4+ dopant forms a deep acceptor level that can negatively impact photocatalytic H2 evolution by promoting recombination of photogenerated charge carriers. Based on the charge compensation mechanism, codoping with pentavalent antimony ion (Sb5+) at the B site of CaTiO3 (Ti4+) stabilizes the trivalent state of the rhodium component at the B site. In contrast, doping with low-valent aluminium and magnesium ions (Al3+ and Mg2+) at the B site increases the proportion of Rh4+ species to compensate for the charge imbalance caused by these ions. As a result, the DR spectrum of the CaTiO3:Al,Sb,Mg,Rh sample showed noticeable absorption attributed to the Rh4+-derived deep acceptor band, whereas the Rh3+-related transition band was not significantly enhanced. The greyish-purple appearance of the sample further indicates the dominant presence of the Rh4+ component in the material.
As observed in the photocatalytic H2 evolution under visible-light irradiation over Rh-doped SrTiO3 photocatalysts,11,12 the color of the suspension changed to yellow during the photodeposition process of Pt or Pt/Cr2O3 cocatalysts (inset of Fig. 2c), though the color changed back to the original color upon exposure to air due probably to the above-mentioned charge imbalance of B site cation. Compared to the DR spectrum of the bare CaTiO3:Al,Sb,Mg,Rh sample, the DR spectrum of the suspension obtained after 3 h of light irradiation with a Xe lamp (>400 nm) and measured under argon (Ar) (details in ESI†) showed an increase in the Rh3+-related band and a corresponding decrease in the Rh4+-related band, as depicted in Fig. 2c. This color change suggests the occurrence of reductive activation of doped rhodium species in the sample. Furthermore, the plots of apparent quantum yields (AQYs) as a function of incident photon wavelength were consistent with the photoabsorption characteristics of the yellow-colored suspension, reinforcing this interpretation. Since the suspensions containing CaTiO3:Al,Rh and CaTiO3:Al,Sb,Rh exhibited nearly the same yellow color after photoirradiation (Fig. S3, ESI†), it is considered that similar photoactivation occurred in these samples as in the CaTiO3:Al,Sb,Mg,Rh sample. Although detailed quantitative analyses have not yet been conducted, the highest activity observed for the CaTiO3:Al,Sb,Mg,Rh-based photocatalysts is likely due to significant suppression of particle growth (Fig. 1), as well as stabilization of trivalent rhodium species (Rh3+) induced by co-doping with pentavalent antimony ions (Sb5+).
To investigate the structural properties of the Pt/Cr2O3 cocatalyst, TEM-EDS measurements were performed using the Pt/Cr2O3-loaded CaTiO3:Al,Sb,Mg,Rh sample recovered after the sacrificial H2 evolution reaction. A low-magnification secondary electron (SE) scanning image revealed homogeneous deposition of nanoparticles across the entire surface of the rectangular CaTiO3:Al,Sb,Mg,Rh particles (Fig. S4, ESI†). Bright-field (BF) and high-angle annular dark field (HAADF) scanning TEM images confirmed the presence of Pt in the observed nanoparticles (Fig. S4, ESI† and Fig. 2d). As shown in the same figures, corresponding EDS elemental mapping images showed overlapping signals of Pt and chromium (Cr) at the locations of the nanoparticles. Since Pt and Cr2O3 were sequentially deposited by photoreduction of their respective precursor ions (i.e., PtCl42− and CrO42−), these results confirm the formation of Cr2O3 layers over the initially deposited Pt nanoparticles, i.e., the creation of Pt (core)–Cr2O3 (shell) cocatalysts resulted in significant enhancement of H2 evolution.
For the Rh/Cr2O3 cocatalyst used in several photocatalysts for overall water splitting, the Cr2O3 surface layer is known to prevent O2 from reaching the surface of the inner Rh nanoparticle core, effectively suppressing the backward reaction of water splitting.28 A similar effect of the Cr2O3 layer on Pt cocatalysts has also been proposed.5 Since the current system involves a sacrificial reaction using methanol as a hole scavenger, the suppression of the reverse reaction in overall water splitting cannot fully account for the observed phenomenon. According to recent studies on the electrocatalytic properties of Rh/Cr2O3 nanoparticles supported on conductive substrates,29 the Cr2O3 layer may facilitate proton (H+) access to the Pt core surface while inhibiting the approach of other reagents and ions. If the Cr2O3 shell indeed has such effects, there may be potential to enhance catalytic activity by optimizing its thickness. This is being considered for future investigation.
In this study, we explored the potential of CaTiO3 as a host material for Rh-doped photocatalysts used in H2 evolution under visible-light irradiation. Significant performance enhancement was achieved by using Rh-doped CaTiO3 codoped with Al3+, Sb5+, and Mg2+ cations, loaded with the Pt/Cr2O3 cocatalyst: the use of the photocatalyst resulted in a high AQY of 7.1% at 420 nm among the doped photocatalytic systems. Although the perovskite CaTiO3 compound has not been extensively studied as a host material, it demonstrates considerable potential for further activity enhancement.
Author contributions
T. O. and S. I. directed and led the research. T. O. performed structural analyses and photocatalytic experiments. R. T., T. N., and K. H. performed TEM measurements and analyses. All the authors discussed the results. T. O. and S. I. wrote the manuscript.Data availability
Experimental details and the data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was partly supported by JSPS KAKENHI Grant No. 23H02074 and JSPS Bilateral Program No. JPJSBP120247415. We gratefully acknowledge the helpful discussions with Professor Akihiko Kudo (Tokyo University of Science) and Professor Hideki Kato (Tohoku University).Notes and references
-
(a) B. A. Pinaud, J. D. Benck, L. C. Seitz, A. J. Forman, Z. Chen, T. G. Deutsch, B. D. James, K. N. Baum, G. N. Baum, S. Ardo, H. Wang, E. Miller and T. F. Jaramillo, Energy Environ. Sci., 2013, 6, 1983 RSC
; (b) D. M. Fabian, S. Hu, N. Singh, F. A. Houle, T. Hisatomi, K. Domen, F. E. Osterloh and S. Ardo, Energy Environ. Sci., 2015, 8, 2825 RSC
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC
- W.-J. Chun, A. Ishikawa, H. Fujisawa, T. Takata, J. N. Kondo, M. Hara, M. Kawai, Y. Matsumoto and K. Domen, J. Phys. Chem. B, 2003, 107, 1798 CrossRef CAS
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS PubMed
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, Angew. Chem., Int. Ed., 2006, 45, 7806 CrossRef CAS PubMed
- A. Ishikawa, T. Takata, J. N. Kondo, M. Hara, H. Kobayashi and K. Domen, J. Am. Chem. Soc., 2002, 124, 13547 CrossRef CAS PubMed
- Q. Wang, M. Nakabayashi, T. Hisatomi, S. Sun, S. Akiyama, Z. Wang, Z. Pan, X. Xiao, T. Watanabe, T. Yamada, N. Shibata, T. Takata and K. Domen, Nat. Mater., 2019, 18, 827 CrossRef CAS PubMed
- H. Fujito, H. Kunioku, D. Kato, H. Suzuki, M. Higashi, H. Kageyama and R. Abe, J. Am. Chem. Soc., 2016, 138, 2082 CrossRef CAS PubMed
- X. Tao, Y. Zhao, L. Mu, S. Wang, R. Li and C. Li, Adv. Energy Mater., 2018, 8, 1701392 CrossRef
- S. Ikeda, N. Sugiyama, B. Pal, G. Marcí, L. Palmisano, H. Noguchi, K. Uosaki and B. Ohtani, Phys. Chem. Chem. Phys., 2001, 3, 267 RSC
- R. Konta, T. Ishii, H. Kato and A. Kudo, J. Phys. Chem. B, 2004, 108, 8992 CrossRef CAS
-
(a) Y. Sasaki, H. Nemoto, K. Saito and A. Kudo, J. Phys. Chem. C, 2009, 113, 17536 CrossRef CAS
- K. Furuhashi, Q. Jia, A. Kudo and H. Onishi, J. Phys. Chem. C, 2013, 117, 19101 CrossRef CAS
- R. Niishiro, S. Tanaka and A. Kudo, Appl. Catal., B, 2014, 150–151, 187 CrossRef CAS
- S. Suzuki, H. Matsumoto, A. Iwase and A. Kudo, Chem. Commun., 2018, 54, 10606 RSC
- T. Ishii, H. Kato and A. Kudo, J. Photochem. Photobiol. A, 2004, 163, 181 CrossRef CAS
- P. Reunchan, S. Ouyang, N. Umezawa, H. Xu, Y. Zhang and J. Ye, J. Mater. Chem. A, 2013, 1, 422117 RSC
- K. Sayama, K. Mukasa, R. Abe, Y. Abe and H. Arakawa, Chem. Commun., 2001, 2416 RSC
- Q. Wang, T. Hisatomi, S. S. K. Ma, Y. Li and K. Domen, Chem. Mater., 2014, 26, 4144 CrossRef CAS
- Q. Wang, T. Hisatomi, Q. Jia, H. Tokudome, M. Zhong, C. Wang, Z. Pan, T. Takata, M. Nakabayashi, N. Shibata, Y. Li, I. D. Sharp, A. Kudo, T. Yamada and K. Domen, Nat. Mater., 2016, 15, 611 CrossRef CAS PubMed
- K. Maeda, ACS Appl. Mater. Interfaces, 2014, 6, 2167 CrossRef CAS PubMed
- X. Zhou, J. Shi and C. Li, J. Phys. Chem. C, 2011, 115, 8305 CrossRef CAS
- N. Kumagai, L. Ni and H. Irie, Chem. Commun., 2011, 47, 1884 RSC
- T. Kimijima, K. Kanie, M. Nakaya and A. Muramatsu, CrystEngComm, 2014, 16, 5591 RSC
-
(a) S. Nishimoto, M. Matsuda and M. Miyake, Chem. Lett., 2006, 35, 308 CrossRef CAS
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411 CrossRef CAS PubMed
- S. Ikeda, R. Okamoto, A. Kimura, Y. Nakayasu, A. Yamakata, R. Tomizawa, T. Masuda and K. Nakatani, Sustainable Energy Fuels, 2024, 8, 202 RSC
- M. Yoshida, K. Takanabe, K. Maeda, A. Ishikawa, J. Kubota, Y. Sakata, Y. Ikezawa and K. Domen, J. Phys. Chem. C, 2009, 113, 10151 CrossRef CAS
- T. Higashi, K. Seki, Y. Sasaki, Y. Pihosh, V. Nandal, M. Nakabayashi, N. Shibata and K. Domen, Chem. – Eur. J., 2023, 29, e20220458 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ma00098j |
| This journal is © The Royal Society of Chemistry 2025 |