
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHuman microbiome derived synthetic antimicrobial peptides with activity against Gram-negative, Gram-positive, and antibiotic resistant bacteria†
Walaa K.
Mousa‡
*acd,
Ashif Y.
Shaikh‡
b,
Rose
Ghemrawi
ac,
Mohammed
Aldulaimi
 b,
Aya
Al Ali
ac,
Nour
Sammani
ac,
Mostafa
Khair
e,
Mohamed I.
Helal
f,
Farah
Al-Marzooq
g and
Emilia
Oueis
*bh
b,
Aya
Al Ali
ac,
Nour
Sammani
ac,
Mostafa
Khair
e,
Mohamed I.
Helal
f,
Farah
Al-Marzooq
g and
Emilia
Oueis
*bh
aCollege of Pharmacy, Al Ain University, PO BOX 64141, Abu Dhabi, United Arab Emirates. E-mail: walaa.mousa@aau.ac.ae
bDepartment of chemistry, Khalifa University of Science and Technology, PO BOX 127788, Abu Dhabi, United Arab Emirates. E-mail: emilia.oueis@ku.ac.ae
cAAU Health and Biomedical Research Center, Al Ain University, PO BOX 112612, Abu Dhabi, United Arab Emirates
dCollege of Pharmacy, Mansoura University, Mansoura 35516, Egypt
eCore Technology Platforms, New York University Abu Dhabi, PO BOX 127788, United Arab Emirates
fElectron Microscopy Core Labs, Khalifa University of Science and Technology, PO BOX 127788, Abu Dhabi, United Arab Emirates
gDepartment of Medical Microbiology and Immunology, College of Medicine and Health Sciences, UAE University, P.O. Box 15551, Al Ain, United Arab Emirates
hHealthcare Engineering Innovation Group, Khalifa University of Science and Technology, PO BOX 127788, Abu Dhabi, United Arab Emirates
First published on 28th October 2024
Abstract
The prevalence of antibacterial resistance has become one of the major health threats of modern times, requiring the development of novel antibacterials. Antimicrobial peptides are a promising source of antibiotic candidates, mostly requiring further optimization to enhance druggability. In this study, a series of new antimicrobial peptides derived from lactomodulin, a human microbiome natural peptide, was designed, synthesized, and biologically evaluated. Within the most active region of the parent peptide, linear peptide LM6 with the sequence LSKISGGIGPLVIPV-NH2 and its cyclic derivatives LM13a and LM13b showed strong antibacterial activity against Gram-positive bacteria, including resistant strains, and Gram-negative bacteria. The peptides were found to have a rapid onset of bactericidal activity and transmission electron microscopy clearly shows the disintegration of the cell membrane, suggesting a membrane-targeting mode of action.
Introduction
The high occurrence of antibiotic resistance in bacterial infections is a serious global health problem, contributing to financial burden, increased morbidity, and the death of millions worldwide.1–3 Hence, there is an urgent need for novel antibiotics capable of targeting difficult to treat infections. One natural source of potential drugs that can be explored for antibiotics discovery is the human microbiome.4–6 Indeed, there has been mounting evidence regarding the role that the human microbiota plays in invading pathogens, among others.7,8 Specifically, antimicrobial peptides (AMPs), innate to all living organisms, have been shown to have a dual action as antimicrobials and modulators of the host immune system.9,10 Natural AMPs and their derivatives show great potential as promising antibiotic candidates, notably because of their ability to penetrate bacterial cell membranes. As such, many have already been marketed as antibiotics (i.e. daptomycin, polymyxin B, and colistin), while others are in the clinical pipeline.11,12 Microbiota-derived AMPs have been found to have 35 to 57 amino acids on average, with the most common producers in the gut being Bacillus and Lactobacillus.13 In our previous work, we identified lactomodulin (LM), a class IIb bacteriocin AMP that we isolated from a strain of Lacticaseibacillus rhamnosus (ATCC 7469).14 LM is a 52 amino acid long peptide belonging to the lactobin A/cerein 7B family. Our investigation into the activity of LM revealed that the peptide possesses a dual antibacterial and anti-inflammatory activity, including against resistant strains, while showing low cytotoxicity on human cell lines. In this work, we set out to find the shorter sequences derived from LM that conserve the antibacterial activity, setting the groundwork for further modifications and fine-tuning of the structural and physicochemical properties. AMPs with longer sequences generally have unfavourable properties that hinder their therapeutic applications, including stability and cost. As such, it is important to determine the essential regions within the peptide that have functionality, or in this case antibacterial effect. The identification of these key regions is the first step in the design and development of potent antibacterial peptides with desirable properties through chemical modifications. Using systemic and rational design, a first series of nine peptides (LM1–LM9) derived from LM was assessed, followed by another set of five peptides (LM10–LM13) based on LM6. Compared with the parent peptide, the best analogues showed similar activity against Gram-positive pathogens, while showing additional, albite lower, Gram-negative activity in vitro, leading to the identification of the core key regions responsible for the antibacterial activity of LM. Killing kinetics combined with transmission electron microscopy imaging (TEM) suggested that these antimicrobial peptides might target the cell membrane resulting in its disruption in a fast-killing mode. However, this action is associated with a considerable cytotoxicity on human cell lines which needs to be addressed in the next design stage to improve the peptides' therapeutic potential.Results and discussion
Peptide design
A library of nine short peptides derived from LM was designed and synthesized with the aim to pinpoint the pharmacophore region within the parent peptide that would retain the antibacterial activity. As such two methodologies were used generating two separate series. The first series included five peptides that were systematically derived from the primary sequence of LM by splicing it into one 12-mer (LM1) and four 10-mer (LM2–LM5) peptides (Fig. 1). The rationale behind this strategy was to cover all parts of the original AMP, regardless of any predictions. The second series consisted of four derivatives that were rationally designed through the analysis of the LM sequence, by examining and evaluating the antimicrobial prediction scores, toxicity, and secondary structure (LM6–LM9, Fig. 1 and Table 1). In silico AMP prediction tools have proven beneficial to AMP discovery and design, saving in time and cost. CAMPR3, a curated database of known AMPs, offers an AMP prediction tool based on four different prediction models: discriminant analysis (DA), support vector network (SVM), artificial neural network (ANN), and random forest (RF).15 The latter has been shown to be the most accurate in a recent study.16 As the aim was to generate shorter bioactive peptides, four chain length peptides (7, 10, 12, and 15-mers) were generated through the prediction tool. By overlapping the prediction results based on all four algorithms and four lengths, it was apparent that most peptides were concentrated within the K3-L30 region of LM. All the peptides were predicted to be not toxic using the ToxinPred tool based on the SVM (Swiss-Prot) and motif based method.17 The helical secondary structure is considered as an important and prominent feature of AMPs and shorter sequences within the helical region of LM would be prioritized. However, the secondary structure of LM is not known. When we used the different structure prediction tools, AlphaFold, RoseTTAFold2, and Quark for the prediction of the secondary structure of the LM peptide, there was a convergence in the results, which showed the mostly helical structure of LM (Fig. S1†). This indicates that most predicted sequences were likely to come from helical regions within LM.18–21 Based on these results, three overlapping peptides (LM6–LM8) within the K3-L30 region with high AMP probability prediction were shortlisted for testing. LM6, a 15-mer with the sequence LSKISGGIGPLVIPV (position 9–23), along with its shorter derivatives, was one of the few peptides predicted by all four algorithms to have AMP character with high probability, occupying the centre of the AMP region. LM7, a 12-mer with the sequence IGPLVIPVAAIL (position 16–27), and its different variations have high scores in the RF algorithm and nicely overlap with LM6. LM8, a 13-mer with the sequence KLNEVELSKISGG (position 3–15), overlaps with LM6 on the N-terminal side and completes the AMP predicted region. The C-terminal region of LM has low AMP probability throughout all algorithms, but the sequence HADELVAGVKQ (LM9) was added to the series regardless, especially as it was predicted by PEP-FOLD3 (Fig. S1†) to have an α-helical structure.22 Along with it, LM1, LM3, LM5, and LM8 all showed predicted helicity as well. LM7 is mostly a random coil, except for a four amino acid stretch (PVAA) that is helical. LM6 is predicted to be a random coil, while the shorter version peptide LM2 is predicted to have a β-hairpin structure with a two-residue turn (GP). Some physicochemical properties were theoretically determined using the Database of Antimicrobial Activity and Structure of Peptides (DBAASP v3.0) to assess the hydrophobicity, amphiphilicity, and toxicity of the shortlisted compounds, using the Eisenberg and Weiss hydrophobicity scale.23,24 The parent peptide LM is negatively charged overall with a −2 charge, while the majority of known antimicrobial peptides are cationic. Anionic AMPs bind to metal ions and form salt bridges with the negatively charged bacterial membrane, allowing the AMP to penetrate through.25 Positively charged AMPs on the other hand interact directly with the bacterial membrane. The synthesized peptides of the first library are mostly cationic (+1 or +2), except for peptides LM4 (−2) and LM9 (0). The peptides were synthesized as their C-terminal amide, neutralizing the carboxylic acid and securing an additional positive charge for the linear peptide. At the same time, hydrophobicity is an important characteristic of peptides, which is essential for their interaction with the hydrophobic core of bacterial membranes, and has been shown to correlate with their antibacterial activity.26,27 An increased hydrophobicity however has been linked to hemotoxicity (red blood cell destruction). Hence, amphipathicity or amphiphilicity is important, and can be measured through the normalized hydrophobic moment. The normalized hydrophobicity of all truncated peptides and the parent peptide ranges from −0.86 to 0.23 and the normalized hydrophobic moment from 0.10 to 0.52 (Table 1). The values are within the expected ranges of known AMPs. The amphiphilicity index indicates the helical structural stability at the membrane–water interface. A value of zero shows no stability, while a higher value indicates a higher stability.28 Three peptides showed an amphiphilicity index of zero, LM2, LM3, and LM7, while all the others showed a certain stability with the highest value being for LM5. In vitro aggregation propensity is a useful prediction as it is associated with lower antimicrobial activity, decreased physical stability, and increased cytotoxicity of the peptide.29 Peptide LM3 showed a high value, similar to the parent peptide LM (690 and 580, respectively), LM9 showed a low propensity to in vitro aggregation, and all the other peptides are not predicted to aggregate.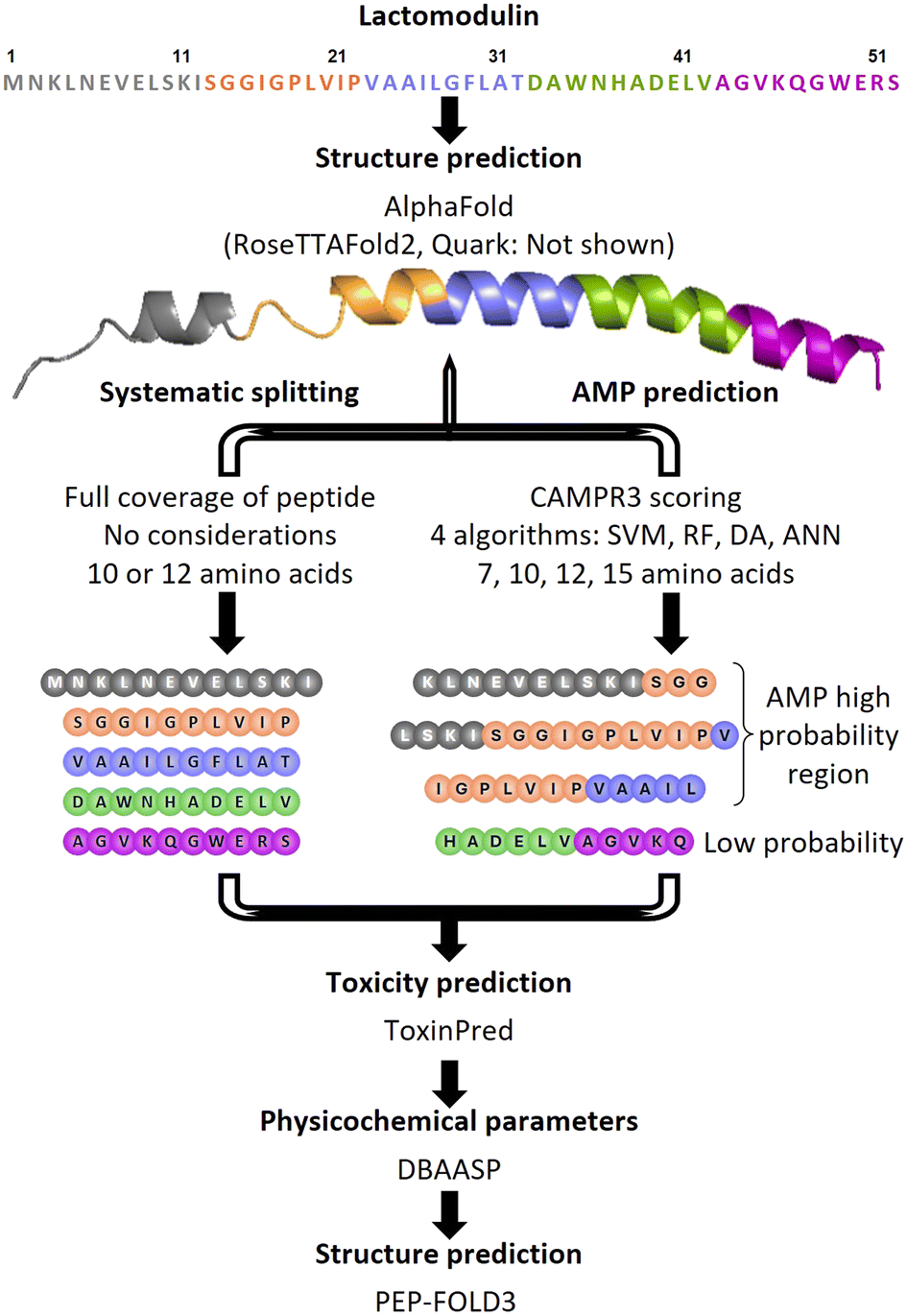 | ||
| Fig. 1 Summary of the overall workflow used for the design stage. | ||
| Peptides | Characterization | Computed physicochemical properties | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Code | Sequence | Puritya (%) | t R (min) | Theoretical [M + H]+ | Measuredc [M + H]+ | q | P/Ne | <H>d | <mH>d | A indexd | In vitro aggregationd |
| a Purity as determined by analytical RP-HPLC with UV absorption at 220 nm. b Retention time determined by analytical RP-HPLC. c Protonated mass confirmed by high resolution mass spectrometry. d Theoretical values of the physicochemical parameters determined by DBAASP q: net charge, <H>: normalized hydrophobicity, <mH>: normalized hydrophobic moment, A index: amphiphilicity index. e Ratio of polar (including glycine) to non-polar amino acids. | |||||||||||
| LM | MNKLNEVELSKISGGIGPLVIPVAAILGFLATDAWNHADELVAGVKQGWERS | −2 | 0.93 | −0.22 | 0.23 | 0.68 | 690.00 | ||||
| LM1 | MNKLNEVELSKI-NH2 | 76 | 11.81 | 1416.7888 | 1416.7736 | +1 | 1.40 | 0.08 | 0.24 | 0.82 | 0 |
| LM2 | SGGIGPLVIP-NH2 | 99 | 13.82 | 908.5569 | 908.5530 | +1 | 0.67 | −0.64 | 0.10 | 0 | 0 |
| LM3 | VAAILGFLAT-NH2 | 96 | 14.90 | 974.6039 | 974.6000 | +1 | 0.25 | −0.81 | 0.22 | 0 | 589.22 |
| LM4 | DAWNHADELV-NH2 | 97 | 15.95 | 1168.5387 | 1168.5317 | −2 | 1.00 | −0.05 | 0.52 | 0.96 | 0 |
| LM5 | AGVKQGWERS-NH2 | 99 | 6.41 | 1116.5909 | 1116.5793 | +2 | 2.33 | 0.23 | 0.40 | 1.56 | 0 |
| LM6 | LSKISGGIGPLVIPV-NH2 | 98 | 14.51 | 1449.9045 | 1449.9154 | +2 | 0.67 | −0.55 | 0.34 | 0.24 | 0 |
| LM7 | IGPLVIPVAAIL-NH2 | 97 | 15.83 | 1174.7927 | 1174.7838 | +1 | 0.09 | −0.86 | 0.26 | 0 | 342.26 |
| LM8 | KLNEVELSKISGG-NH2 | 98 | 14.98 | 1372.7800 | 1372.7774 | +1 | 2.25 | 0.01 | 0.17 | 0.76 | 0 |
| LM9 | HADELVAGVKQ-NH2 | 95 | 9.41 | 1165.6329 | 1165.6275 | 0 | 1.20 | −0.05 | 0.40 | 0.69 | 18.52 |
| LM10 | LSKISGGIGP-NH2 | 96 | 13.63 | 927.5627 | 927.5567 | +2 | 1.50 | −0.35 | 0.39 | 0.37 | 0 |
| LM11 | KISGGIGPLV-NH2 | 97 | 17.32 | 939.5991 | 939.5936 | +2 | 1.00 | −0.48 | 0.41 | 0.37 | 0 |
| LM12 | LSKISG-NH2 | 95 | 9.94 | 603.3830 | 603.3814 | +2 | 2.00 | −0.18 | 0.46 | 0.61 | 0 |
| LM13a | Cyc(LSKISGGIGPLVIPv) | 95 | 17.76 | 1431.8934 | 1431.8874 | +1 | 0.67 | ||||
| LM13b | Cyc(LSKISGGIGPLVIPV) | 95 | 18.35 | 1431.8934 | 1431.8874 | +1 | 0.67 | ||||
Biological evaluation
To assess whether the synthetized peptides retained the antibiotic activity of the parent compound, we first performed an agar diffusion on a panel of human pathogens, with peptide concentration ranging from 1 to 10 μM. All the peptides showed some level of antibiotic activity. We then measured their corresponding minimum inhibitory concentration (MIC) values (Table 2), as the average of five replicates each, against a collection of Gram-positive and Gram-negative pathogens. The parent peptide LM has shown good antibacterial activity against Gram-positive pathogens Staphylococcus aureus ATCC 25923 (hereafter referred to as SA; 0.8 μM MIC), Clostridium difficile ATCC 43602 (hereafter referred to as C. diff; 1.1 μM MIC), and Listeria monocytogenes ATCC 15313 (hereafter referred to as Lm; 1.2 μM MIC), as well as the resistant strains: methicillin resistant Staphylococcus aureus BAA 2313 (hereafter referred to as MRSA; 1.4 μM MIC) and vancomycin resistant Enterococcus faecium BAA 2317 (hereafter referred to as VRE; 1.2 μM MIC). It also showed low activity against the Gram-negative Escherichia coli ATCC 33694 (hereafter referred to as E. coli; 1.2 μM MIC), while no activity was observed at the highest tested concentration against the other Gram-negatives Pseudomonas aeruginosa BAA 1744 and Salmonella enterica ATCC 13314 (hereafter referred to Pa and Sen, respectively). The least active peptide of the first series is LM3, followed by LM1. LM3 is only active against SA with an MIC of 6.5 μM. It has a high hydrophobicity and a high propensity for in vitro aggregation. LM4 is the only peptide with a total negative charge and has the highest normalized hydrophobic moment with a high amphiphilicity index. In terms of activity however, the peptide showed MICs only against SA and MRSA, at 2.6 μM and 3.5 μM, respectively. LM2 showed activity against the Gram-positive pathogens SA, C. diff, and Lm with acceptable MIC values compared to LM, that is 2.1 μM, 1.6 μM, and 1.2 μM, respectively. However, it showed no activity at the tested concentration against the drug resistant strains. On the other hand, it showed activity against E. coli with an MIC of 8.9 μM in the same range as the parent peptide. More interestingly, LM2 showed a new activity against the Gram-negative strain Pa, with an MIC of 9.5 μM. Its physicochemical properties show a normalized hydrophobicity and normalized hydrophobic moment of −0.64 and 0.10, respectively, with an amphiphilicity index of zero, indicating an instability of the helical structure at the membrane–water interface. The last peptide of the systemic splicing is LM5 at the C-terminal of LM. It is positively charged with a +2 charge, has a high ratio of polar amino acids, and has the highest hydrophobicity of the family. It also shows high stability of its helical structure. LM5 showed good MIC values within the same range as the parent peptide against all Gram-positive pathogens (MIC SA = 1.2 μM, C. diff. = 2.1 μM, Lm = 1.8 μM), including resistant strains (MIC MRSA = 1.4 μM and VRE = 1.9 μM), as well as E. coli (MIC = 7.8 μM). Looking at the correlation between the antibacterial activity of the peptides and their physicochemical properties, it seems non-existent. The peptides coming from the different regions within LM are very different in terms of their primary sequence, hence making the comparison of their properties quite difficult, especially that the predicted values are well within the range of what is expected for AMPs. Regardless, just by focusing on the bioactivity profile of these systematically truncated peptides, we could identify two potential areas that can be further exploited and that showed the better activities, LM2 and LM5.| Peptide | MICs (μM) | Pro-inflammatory biomarker reduction (%) | EC50 (μM) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gram-positive bacteria | Gram-positive resistant strains | Gram-negative bacteria | ||||||||||||
| S. aureus ATCC 25923 | C. difficile ATCC 43602 | L. monocytogenes ATCC 15313 | S. aureus MRSA BAA 2313 | E. faecium VRE BAA 2317 | E. coli ATCC 33694 | P. aeruginosa BAA 1744 | S. enterica ATCC 13314 | IL-8 | IL-6 | IL-1B | TNF alpha | H292 | A549 | |
| NA: not observed within the tested concentrations. NT: not tested on these strains. | ||||||||||||||
| LM | 0.8 | 1.1 | 1.2 | 1.4 | 1.2 | 7.5 | NA | NA | 95 | 90 | 91 | 80 | — | — |
| LM1 | 6.2 | NA | 3.1 | NA | NA | NA | NA | NA | 1 | 2 | 0 | 0 | — | — |
| LM2 | 2.1 | 1.6 | 1.2 | NA | NA | 8.9 | 9.5 | NA | 6 | 6 | 20 | 0 | 2.97 | 6.35 |
| LM3 | 6.5 | NA | NA | NA | NA | NA | NA | NA | 0 | 0 | 0 | 0 | — | — |
| LM4 | 2.6 | NA | NA | 3.5 | NA | NA | NA | NA | 61 | 50 | 19 | 0 | — | — |
| LM5 | 1.2 | 2.1 | 1.8 | 1.4 | 1.9 | 7.8 | NA | NA | 18 | 12 | 18 | 0 | 2.28 | 11.11 |
| LM6 | 0.9 | 0.9 | 2.3 | 1.7 | 1.3 | 9.2 | 9.3 | NA | 10 | 0 | 7 | 10 | 2.80 | 3.69 |
| LM7 | 1.5 | 1.8 | 3.6 | 1.8 | 1.5 | NA | NA | NA | 18 | 25 | 27 | 25 | — | — |
| LM8 | 2.3 | 2.6 | 2.6 | 2.9 | 3.2 | NA | NA | NA | 17 | 5 | 8 | 0 | — | — |
| LM9 | 1.3 | 1.0 | 1.1 | 2.5 | 3.1 | NA | NA | NA | 12 | 32 | 33 | 40 | 2.72 | 11.71 |
| LM10 | 2.4 | 4.1 | 3.7 | NA | NA | NA | NA | NA | 0 | 0 | 0 | 0 | — | — |
| LM11 | 5.1 | NA | NA | NA | NA | NA | NA | NA | 0 | 0 | 0 | 0 | — | — |
| LM12 | 3.9 | 4.6 | 5.1 | NA | NA | NA | NA | NA | 0 | 0 | 0 | 0 | — | — |
| LM13a | 0.8 | 0.9 | 1.5 | 1.7 | 1.5 | 8.2 | 8.3 | NA | 14 | 9 | 4 | 15 | 2.07 | 8.50 |
| LM13b | 0.7 | 0.9 | 1.5 | 1.6 | 1.8 | 8 | 8.5 | NA | 10 | 4 | 13 | 5 | 2.55 | 4.73 |
| Amoxycillin | 5 | 5 | 5 | 35 | 35 | NT | NT | NT | ||||||
| Fidaxomicin | NT | 1 | 1 | NT | 2 | NT | NT | NT | ||||||
| Ciprofloxacin | 2 | NT | 2 | 2 | 4 | 2 | 2 | 2 | ||||||
The activity of the rationally designed peptides by looking mostly at their AMP prediction also correlates with these results. Very similar in primary structure to LM1, LM8 has most of the amino acids of LM1 except MN from the N-terminal with the additional SGG at the C-terminal. Most of its predicted physicochemical properties are similar or within the same range as LM1. The only difference is the ratio of polar to non-polar residue. But conversely, LM8 has a much better activity profile against all Gram-positive pathogens with MICs in the range of 2.3 μM to 3.2 μM, including resistant strains (2.9 μM and 3.2 μM). However, LM8 was the weakest compound within this second series. LM7 is a hybrid between LM2 and LM3 and has shown improvement in its MIC value compared to them against SA (1.5 μM), while recovering the activity against the resistant strains MRSA (1.8 μM) and VRE (1.5 μM). It was less active nonetheless against C. diff. (1.8 μM) and Lm (3.6 μM). However, it remained inactive against the Gram-negative pathogens. LM7 is highly hydrophobic and is predicted to aggregate in vitro. LM6, the peptide at the core of the AMP prediction region, showed a very good activity profile, almost matching the parent peptide activity with only 15 amino acids. The MIC against SA is 0.9 μM and 1.7 μM against MRSA, 0.9 μM against C. diff., 2.3 μM against Lm, and 1.3 μM against VRE. The Gram-negative E. coli activity is also conserved with an MIC of 9.2 μM, while a new Pa activity was also introduced with 9.3 μM MIC. Most peptides performed better than amoxycillin against the Gram-positive bacteria, and in particular activity was observed against resistant strains. Similar activity to fidaxomicin was observed for the most active peptides (LM6 and LM9) against C. diff, while the others were slightly less active. More specifically, the activity of LM6 is superior to both ciprofloxacin and amoxycillin in inhibiting Gram-positive bacteria while comparable to fidaxomicin in killing C. difficile (Table 2). However, it is about 5 times less active than ciprofloxacin against the Gram-negative bacteria. LM6 is the best peptide of the series and the library. When looking at its sequence, it is an extension of LM2 with an additional 4 amino acids at the N-terminal and one at the C-terminal. As LM2 gave good bioactivity, it is not surprising that LM6 has even surpassed it. LM6 has an additional positive charge, a higher hydrophobicity, and amphiphilicity. The second best, LM9, is a hybrid between LM4 and LM5. It is a neutral peptide with low hydrophobicity, and high polar amino acid content. LM9 was specifically chosen as it had some of the lowest scores for the AMP prediction. Nonetheless, it showed a very good activity profile against all Gram-positive pathogens, but with higher MIC values compared to LM5 for the resistant strains, with 1.3 μM, 1.0 μM, 1.1 μM, 2.5 μM, and 3.1 μM against SA, C. diff., Lm, MRSA, and VRE, respectively. LM9 however remained inactive against the Gram-negative pathogens.
To verify whether the determined MIC kills the entire bacterial population, we performed the minimal bactericidal concentration (MBC) experiment for all tested peptides against all the pathogens. It showed that the peptides indeed killed the bacteria at the observed MIC values, as no bacterial growth was observed after 48 hours of incubation at 37 °C, confirming that the MIC and MBC values were the same, and indicating a bactericidal mode of action.
Using this methodology, we were able to determine two main regions within the LM peptide that contribute most to the antibacterial activity, represented by the peptides LM6 and LM9. Both peptides come from the second series obtained through AMP predictions. However, LM9 and other peptides around that region had very low scores, probably because of the high proportion of polar amino acids, and would have been dismissed otherwise. Nonetheless, the other three AMP peptides with high AMP prediction scores all showed good MIC activities against the Gram-positive pathogens, including the resistant strains. Within the first series, the systemic splicing also led to the same general two regions with peptides LM2 and LM5. More importantly, both methods were convergent in pinpointing the core region with antibacterial activity. We also evaluated the peptides for their anti-inflammatory activity based on the percentage reduction in multiple proinflammatory biomarkers' (IL-8, IL-6, IL-1B, and TNF-alpha) concentration in Caco-2 cell lines inoculated with the inflammatory-inducing pathogen C. difficile ATCC 43602. LM has shown excellent reduction in all four biomarkers, with 95% reduction in IL-8 concentration, 90% for IL6, 91% for IL-1B, and 80% for TNF alpha. This anti-inflammatory level of activity however could not be replicated in any of the shorter peptides. LM4 showed some anti-inflammatory activity with 61% reduction in IL-8, 50% reduction in IL-6, and 19% reduction in IL-1B, while TNF alpha concentration was not affected. The LM9 peptide, which contains part of LM4, reduced all four biomarkers IL-8, IL-6, IL-1B, and TNF-alpha by 12%, 32%, 33%, and 40%, respectively. LM5, on the other hand, which completes the C-terminal of the peptide, has induced a lower response with 18%, 12%, and 18% reduction of the respective interleukins, but none against TNF alpha. LM7 also showed some moderate activity between 18 and 27% reduction of all four cytokines. LM2, LM6, and LM8 induced the reduction of some cytokines by less than 20%, while LM1 and LM3 had virtually no effect. Despite not having a single peptide that has retained the full anti-inflammatory activity of the parent peptide, these results indicate that the C-terminal of the peptide is one of the key regions involved in the observed activity. Having a dual mode of action to target both the bacterial pathogen and the associated inflammation is of great interest in the development of novel therapeutics, specifically targeting complicated infections leading to sepsis.30
Follow-up series
After analysis of the obtained results, and the key regions identified within the peptide sequence, LM6 was considered the best of the series, given its low micromolar MIC values against the Gram-positive pathogens and the resistant strains comparable to those of the parent peptide. But more importantly, it retained the Gram-negative activity against E. coli and showed new activity against Pa within the same range of the MIC as Lm. Three more peptides were considered by further truncating the sequence of LM6, given the activity profile of LM2. Two peptides, LM10 and LM11, have ten amino acids including the lysine residue, compared to LM2, thus having a +2 charge. The third peptide is a hexamer of the N-terminal side of LM6 with a charge of +2. LM2 remained the best truncated derivative of LM6, as the other three also showed no activity against the tested resistant strains, nor the Gram-negative pathogens. LM10 showed an MIC value of 2.4 μM against SA, similar to LM2, but higher MIC values against C. diff. and Lm. LM11 showed activity exclusively against SA with a higher MIC of 5.1 μM. Surprisingly, the shorter LM12 peptide still showed a moderate Gram-positive activity with MICs of 3.9 μM, 4.6 μM, and 5.1 μM against SA, C. diff. and Lm. The calculated physicochemical properties of the peptides differ slightly but are all within the same range or direction. As LM6 remains the best performer peptide, we then synthesized and tested its macrocyclized derivatives. Cyclic peptides have been shown to have better stability in vivo in general, while sometimes being more active caused by a more rigid structure and fewer conformations in solution, facilitating their interaction with the biological target.31–33 The macrocyclization reaction with PyBop gave two separable products (ML13a and ML13b), the major of which (LM13a) contains the epimerized valine residue at the C-terminal point of macrocyclization, as verified by Marfey's analysis (Table S2 and Fig. S2†).34 No attempts were conducted at this stage to optimize the macrocyclization reaction and both macrocycles were tested. The presence of D-amino acids is another chemical strategy that is used to increase the stability of peptides.35 Both cyclic peptides show mostly similar MIC values and a slight gain in activity compared to the linear peptide LM6 against most pathogens tested, making them the best performers of this first round of design. The cytotoxicity of the best antibiotic compounds was evaluated against two human lung cancer cell lines H292 and A549. Although favourable in silico predictions were obtained, toxicity was observed and was more pronounced against the H292 cell line compared to A549. Despite the low therapeutic index of these peptides (ranging from 1 to 10 depending on the peptide and cell line), there is ample room for further improvement and proper optimization of these molecules. Decreasing the overall lipophilicity of the peptides and increasing the net positive charge and amphipathicity are strategies that have been previously successful in reducing the toxicity of AMP analogues.36–38 More advanced modifications entail the introduction of peptoids, non-natural amino acids, and peptidomimetic scaffolds within the peptidic structure.36,39–42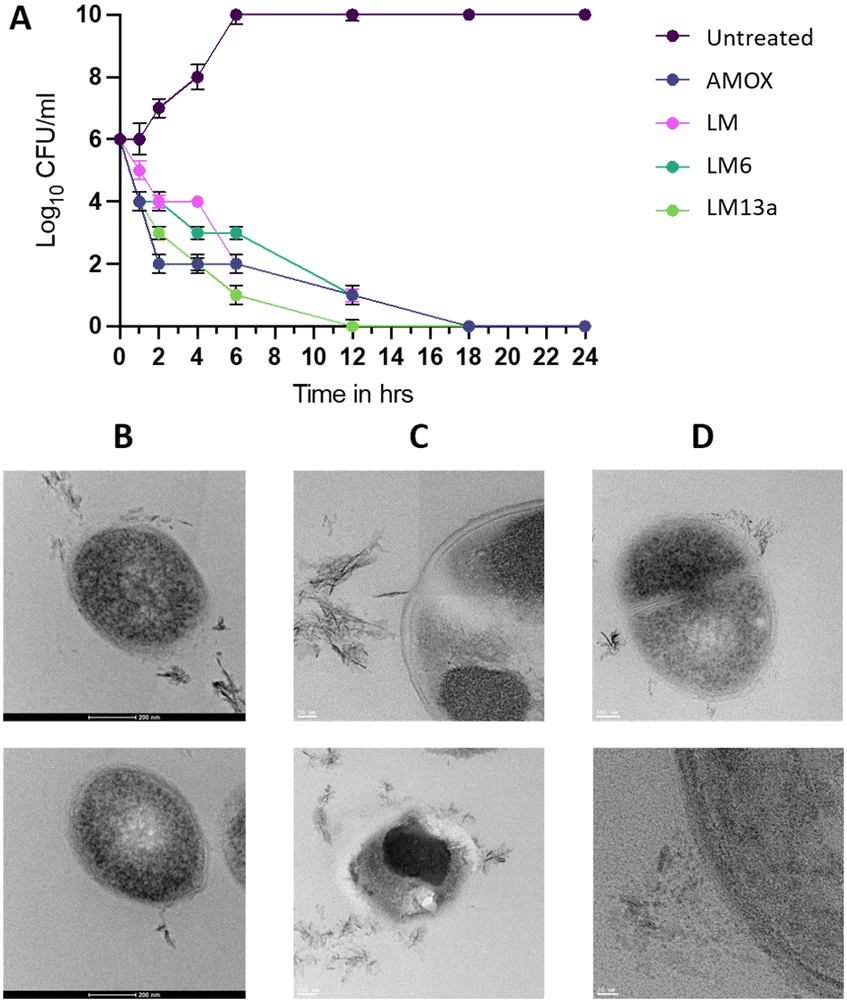 | ||
| Fig. 2 Preliminary investigation into the possible mode of action of the designed peptide derivatives. A: Time-killing graph of S. aureus in the presence of the parent peptide LM, the best of series peptide LM6, its cyclic derivative LM13a, and the positive control antibiotic amoxicillin (Amox). B–D: TEM micrographs demonstrate the intact cell membrane of growing S. aureus cells (B) compared to a ruptured cell membrane (top) or complete disintegration and leakage (bottom) observed when the cells are treated with LM6 (C) and a partially disintegrated cell membrane with clear extracellular protrusions when cells are treated with the cyclic derivative LM13a (D). | ||
Conclusions
Starting from lactomodulin, a 53-amino acid long peptide isolated from the gut microbiota with promising antibacterial and anti-inflammatory activities, we set out to determine the key core regions that would conserve the activity. As such, two series of nine peptides were determined based on systematic truncation of the parent peptide or AMP prediction calculations. The synthesized peptides ranging from 10–15 amino acid residues were evaluated for their antibacterial and anti-inflammatory activities. The two methods were convergent in identifying the best two regions within the peptide that conserved the antibacterial activity against Gram-positive pathogens and resistant strains spanning LM2/LM6 and LM5/LM9. The Gram-negative activity against E. coli was conserved for LM2 and LM6, in addition to the activity against P. aeruginosa. The anti-inflammatory activity however was very low for most peptides, with the best being peptides LM4, LM7, and LM9, showing anywhere between 18 and 61% reduction in some cytokine levels. The best of the series peptide LM6 was further truncated, but the shorter peptides showed a significant decrease in activity. Finally, LM6 was macrocyclized head-to-tail to generate two cyclic peptides LM13a and LM13b, where the former has an epimerized valine residue. The cyclic peptides proved to be the best performers, with similar or better activities against all the bacterial strains compared to LM6. The activity of LM6 and its cyclic derivatives against Gram-positive bacterial strains is comparable or superior to some commercial bactericidal antibiotics tested in this study. The best six antibacterial peptides were assessed against two human cancer cell lines but showed toxicity and low therapeutic index. Further optimization in the next round of design to improve the peptides' properties is considered. The LM6 and LM13a peptides were found to be bactericidal at their MIC concentration using a time-kill curve and were shown to have a membrane-targeting mode of action by transmission electron microscopy. Bacterial cells of S. aureus treated with either of the compounds have shown clear signs of membrane damage and intracellular content leakage. This study has identified LM6 as an ideal starting point for the generation of lactomodulin-inspired antibacterial peptides through further optimizations. Despite not having a conserved anti-inflammatory activity, other regions within the peptide have shown promise for the development of peptides with dual antibacterial and anti-inflammatory activities. Further investigations around the optimization of the identified peptides and a detailed study of their antibacterial properties and mechanism of action, as well as their potential for selecting resistance are currently underway.Experimental
Materials and methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000).
:1 DMF:DCM for 5 min before use and then washed with DMF. Fmoc-protected amino acid building block (5.0 equiv.) coupling was performed using a double coupling strategy at room temperature for 0.5 h each coupling with intermittent shaking every 2 min. The first coupling uses DIC (0.5 M in DMF; 5.0 equiv.) and OxymaPure® (2.0 M in DMF; 5.0 equiv.) and the second uses HBTU (0.48 M in DMF; 5.0 equiv.) and DIPEA (2 M in NMP; 5.0 equiv.). Fmoc deprotection was performed in two steps with 40% (v/v) piperidine in DMF at rt for 3 min and 20% (v/v) piperidine in DMF at rt for 9 min, with shaking, followed by washing with DMF. Cleavage was performed off-line by treating the resin-bound peptide with 2 mL of a TFA/H2O/TIPS mixture (95:2.5:2.5; v/v) two times for 1 h each. The combined eluates were kept at rt for 2–3 h. The solution was transferred into a round-bottom flask and co-evaporated with toluene. The addition of cold diethyl ether resulted in the precipitation of the crude peptide. Crude peptides were purified by semi-preparative RP-HPLC using a linear gradient of 0–40% ACN over 20 min with a flow rate of 20 mL min−1. The purity and identity of the peptides were verified by analytical HPLC and LC-MS HRMS analysis, respectively. The purified peptides were freeze-dried and stored at −20 °C until biological testing.
:2) in DCM (3 mL) to quench unreacted CTC for 0.5 h at rt. After washing, the initial linear peptide was then completed on the automated Syro I (Biotage) peptide synthesizer. The procedure for coupling/deprotection cycles is the same as the one described for the other linear peptides. After complete synthesis, the resin was washed with two cycles of successive DMF and DCM washing. Full cleavage of the side-chain fully-protected peptide was achieved by adding 30% HFIP in DCM and shaking at room temperature over 1 h. The crude peptide (80 mg) was obtained after evaporation and was directly used for the cyclization reaction. The crude peptide was dissolved in 40 mL DMF, and DIPEA (50 μL, 6 equiv.) was added to the solution. Then PyBOP (80 mg, 3 equiv.) was added and the reaction mixture stirred overnight at rt. Progress of the macrocyclization reaction was monitored by LCMS. DMF was removed under reduced pressure and the crude macrocyclic peptide was obtained by addition of 20 mL MeCN:H2O (1:1) followed by centrifugation. The final deprotection was then done by treating the crude macrocycle with the TFA/H2O (97.5:2.5) solution for 3 h while shaking. Progress of the deprotection was also monitored by LCMS. TFA was co-evaporated with toluene and cold diethyl ether was added to precipitate the peptide. The crude was then purified by semi-preparative RP-HPLC using a linear gradient of 0–100% MeCN over 25 min with a flow rate of 20 mL min−1. Two macrocyclic products, LM13a (major) and LM13b (minor), with the same mass were observed and separated. The purified peptides were freeze-dried and stored at −20 °C until biological testing.
000).
:1 DMF:DCM for 5 min before use and then washed with DMF. Fmoc-protected amino acid building block (5.0 equiv.) coupling was performed using a double coupling strategy at room temperature for 0.5 h each coupling with intermittent shaking every 2 min. The first coupling uses DIC (0.5 M in DMF; 5.0 equiv.) and OxymaPure® (2.0 M in DMF; 5.0 equiv.) and the second uses HBTU (0.48 M in DMF; 5.0 equiv.) and DIPEA (2 M in NMP; 5.0 equiv.). Fmoc deprotection was performed in two steps with 40% (v/v) piperidine in DMF at rt for 3 min and 20% (v/v) piperidine in DMF at rt for 9 min, with shaking, followed by washing with DMF. Cleavage was performed off-line by treating the resin-bound peptide with 2 mL of a TFA/H2O/TIPS mixture (95:2.5:2.5; v/v) two times for 1 h each. The combined eluates were kept at rt for 2–3 h. The solution was transferred into a round-bottom flask and co-evaporated with toluene. The addition of cold diethyl ether resulted in the precipitation of the crude peptide. Crude peptides were purified by semi-preparative RP-HPLC using a linear gradient of 0–40% ACN over 20 min with a flow rate of 20 mL min−1. The purity and identity of the peptides were verified by analytical HPLC and LC-MS HRMS analysis, respectively. The purified peptides were freeze-dried and stored at −20 °C until biological testing.
:2) in DCM (3 mL) to quench unreacted CTC for 0.5 h at rt. After washing, the initial linear peptide was then completed on the automated Syro I (Biotage) peptide synthesizer. The procedure for coupling/deprotection cycles is the same as the one described for the other linear peptides. After complete synthesis, the resin was washed with two cycles of successive DMF and DCM washing. Full cleavage of the side-chain fully-protected peptide was achieved by adding 30% HFIP in DCM and shaking at room temperature over 1 h. The crude peptide (80 mg) was obtained after evaporation and was directly used for the cyclization reaction. The crude peptide was dissolved in 40 mL DMF, and DIPEA (50 μL, 6 equiv.) was added to the solution. Then PyBOP (80 mg, 3 equiv.) was added and the reaction mixture stirred overnight at rt. Progress of the macrocyclization reaction was monitored by LCMS. DMF was removed under reduced pressure and the crude macrocyclic peptide was obtained by addition of 20 mL MeCN:H2O (1:1) followed by centrifugation. The final deprotection was then done by treating the crude macrocycle with the TFA/H2O (97.5:2.5) solution for 3 h while shaking. Progress of the deprotection was also monitored by LCMS. TFA was co-evaporated with toluene and cold diethyl ether was added to precipitate the peptide. The crude was then purified by semi-preparative RP-HPLC using a linear gradient of 0–100% MeCN over 25 min with a flow rate of 20 mL min−1. Two macrocyclic products, LM13a (major) and LM13b (minor), with the same mass were observed and separated. The purified peptides were freeze-dried and stored at −20 °C until biological testing.
All the peptides had a purity >95%, except for LM1 at 76% after two purification rounds. Total yields of the linear peptides were approximately 10–38% based on the resin loading (for each compound, the yield is stated in Table S1†). The analytical reversed-phase HPLC chromatograms and ESI mass spectra for purified peptides are presented in Fig. S2−S15.†
:10000 ratio, following the McFarland Standards. Subsequently, 196 μL of this inoculated medium was dispensed into each well, followed by the addition of 4 μL of various serial dilutions of each peptide into the respective wells. The control wells were filled with 196 μL of non-inoculated medium and 4 μL of DMSO, the solvent used to dissolve the peptides. Fidaxomicin, amoxicillin, and ciprofloxacin served as positive controls. Plates were then incubated aerobically or anaerobically, depending on the growth requirements of each pathogen, at 37 °C. Following a 24-hour incubation period, the OD600 of each well was measured using a microplate reader. Subsequently, the MIC, defined as the lowest peptide concentration resulting in 100% growth inhibition, was determined for each pathogen. Each concentration was tested in duplicate, and the entire assay was independently repeated five times. The percentage of growth inhibition was calculated using the following equation:| % of inhibition = 1 − [(OD600 test − OD600 blank medium)/(OD600 pathogen only − OD600 blank medium)] × 100 |
000 cells per well. Overall, the experimental design involved treatment of the cell lines challenged with an inflammation-inducing pathogen with each peptide then measurement of the released pro-inflammatory cytokines in the culture supernatant using commercially available ELISA kits. The pathogen C. difficile was used as a model to induce inflammation. Briefly, C difficile was cultured overnight in meat broth medium at 37 °C under anaerobic conditions. The culture was then subjected to centrifugation at 12000 × g for 2 minutes, followed by two washes with PBS buffer and suspension in meat broth. Subsequently, 1 μL of 0.5 OD600 culture was used for each 300 μL of culture medium in every well of the 12-well plate. After a 2-hour incubation period, 1 μL of each peptide was added to achieve a final concentration equivalent to the MIC. Negative control experiments included untreated cells or cells treated with DMSO, while positive controls consisted of cells treated with the pathogen only. The duration of incubation was determined through prior optimization experiments and referenced from previous reports regarding the optimal timing for cytokine release and measurement in Caco-2 cells. To quantify cytokine release in Caco-2 cells following co-incubation, an enzyme-linked immunosorbent assay (ELISA) was employed for IL8, IL-6, IL-1β, and TNF-α in the collected supernatant. Commercial kits from Invitrogen, Carlsbad, CA, USA (catalog numbers BMS204-3, BMS213HS, KHC001, and BMS223-4 for IL8, IL-6, IL-1β, and TNFα, respectively) were utilized for this purpose. Cytokine levels were determined following the provided protocol. Each analysis was conducted in three independent replicates.
000 cells per well (200 μL per well), treated with a serial dilution of each peptide at concentrations of 50 μM, 10 μM, 1 μM, 0.1 μM, and 0.01 μM, and incubated in a CO2 incubator for 24 hours. Following the removal of the supernatant and washing twice with PBS1X, 20 μL of MTT solution was added to each well and incubated for 3 hours. Subsequently, the absorbance intensity was measured using spectrophotometry with a microplate reader at an absorbance wavelength of 570 nm (Thermo Fisher Scientific, Waltham, MA, USA). DMSO was used as the negative control. EC50 values were calculated using OD values (triplicate) normalized to blank (average of six) and an online software program (AAT Bioquest EC50 Calculator).46
:30) in 40 ml of LB broth and shaken continuously for 2 hours until the OD600 reached approximately 0.5. Subsequently, the tested compounds were added at their minimum inhibitory concentration (MIC) and incubated at 37 °C. Cells were then centrifuged at 4 °C to prevent cell damage, followed by washing and resuspension in 0.1 M phosphate-buffered saline (PBS; pH 7.2). The fixed cells were preserved overnight using a fixative solution comprising 4% formaldehyde, 1.25% glutaraldehyde, 4% sucrose, 0.01 M CaCl2, and 0.075% ruthenium red in PBS. Following preservation, the fixed cells underwent a series of washes in PBS buffer and were post-fixed for 1 hour with 1% osmium tetroxide in PBS supplemented with 0.075% ruthenium red, followed by further washing. Dehydration was achieved through successive ethanol treatments (30%, 50%, 70%, 80%, 90%, 95%, and 100%), each lasting 15 minutes, ultimately culminating in treatment with propylene oxide. The cells were then infiltrated with a mixture of propylene oxide and Agar 100 epoxy resin in ratios of 1:1, 1:2, and 1:3, and subsequently polymerized at 65 °C for 24 hours. The resulting blocks were trimmed, and ultrathin sections (95 nm) with gold coloration were cut using an EM UC7 Ultracuts ultramicrotome from Leica (Vienna, Austria). Ultrathin sections were collected on 200 mesh copper grids, air-dried, and then contrasted with uranyl acetate and lead citrate. The grids were subsequently examined using a transmission electron microscope, with images captured at varying magnifications using the Titan system at 300 kV. The entire experiment was repeated in duplicate.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
E. O. and W. M. conceptualized, designed, directed, supervised, acquired funding, and coordinated the study. A. S. and M. A. performed the synthesis, purification, and analysis of the peptides; W. M. conducted the time-kill and ELISA assays; W. M. and A. A. conducted the antimicrobial assays; R. G., N. S. and M. K. conducted, and analysed data for cell-line based assays; F. A. and W. M. prepared the TEM samples and M. I. H. ran the TEM experiments; F. A. analysed the TEM results; E. O. and W. M. wrote the original draft. All authors contributed to editing and revising and have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support provided by Khalifa University of Science and Technology for starting grant to E. O. (FSU-2022-019) and internal grant from Al Ain University to W. M. (Ph2023-4-103). The authors would like to thank Mr. Mohan Rommala from the ACBC core lab at KU.Notes and references
- H. Fongang, A. T. Mbaveng and V. Kuete, in Advances in Botanical Research, Elsevier, 2023, vol. 106, pp. 1–20 Search PubMed.
- C. J. L. Murray, K. S. Ikuta, F. Sharara, L. Swetschinski, G. Robles Aguilar, A. Gray, C. Han, C. Bisignano, P. Rao, E. Wool, S. C. Johnson, A. J. Browne, M. G. Chipeta, F. Fell, S. Hackett, G. Haines-Woodhouse, B. H. K. Hamadani, E. A. P. Kumaran, B. McManigal, S. Achalapong, R. Agarwal, S. Akech, S. Albertson, J. Amuasi, J. Andrews, A. Aravkin, E. Ashley, F.-X. Babin, F. Bailey, S. Baker, B. Basnyat, A. Bekker, R. Bender, J. A. Berkley, A. Bethou, J. Bielicki, S. Boonkasidecha, J. Bukosia, C. Carvalheiro, C. Castañeda-Orjuela, V. Chansamouth, S. Chaurasia, S. Chiurchiù, F. Chowdhury, R. Clotaire Donatien, A. J. Cook, B. Cooper, T. R. Cressey, E. Criollo-Mora, M. Cunningham, S. Darboe, N. P. J. Day, M. De Luca, K. Dokova, A. Dramowski, S. J. Dunachie, T. Duong Bich, T. Eckmanns, D. Eibach, A. Emami, N. Feasey, N. Fisher-Pearson, K. Forrest, C. Garcia, D. Garrett, P. Gastmeier, A. Z. Giref, R. C. Greer, V. Gupta, S. Haller, A. Haselbeck, S. I. Hay, M. Holm, S. Hopkins, Y. Hsia, K. C. Iregbu, J. Jacobs, D. Jarovsky, F. Javanmardi, A. W. J. Jenney, M. Khorana, S. Khusuwan, N. Kissoon, E. Kobeissi, T. Kostyanev, F. Krapp, R. Krumkamp, A. Kumar, H. H. Kyu, C. Lim, K. Lim, D. Limmathurotsakul, M. J. Loftus, M. Lunn, J. Ma, A. Manoharan, F. Marks, J. May, M. Mayxay, N. Mturi, T. Munera-Huertas, P. Musicha, L. A. Musila, M. M. Mussi-Pinhata, R. N. Naidu, T. Nakamura, R. Nanavati, S. Nangia, P. Newton, C. Ngoun, A. Novotney, D. Nwakanma, C. W. Obiero, T. J. Ochoa, A. Olivas-Martinez, P. Olliaro, E. Ooko, E. Ortiz-Brizuela, P. Ounchanum, G. D. Pak, J. L. Paredes, A. Y. Peleg, C. Perrone, T. Phe, K. Phommasone, N. Plakkal, A. Ponce-de-Leon, M. Raad, T. Ramdin, S. Rattanavong, A. Riddell, T. Roberts, J. V. Robotham, A. Roca, V. D. Rosenthal, K. E. Rudd, N. Russell, H. S. Sader, W. Saengchan, J. Schnall, J. A. G. Scott, S. Seekaew, M. Sharland, M. Shivamallappa, J. Sifuentes-Osornio, A. J. Simpson, N. Steenkeste, A. J. Stewardson, T. Stoeva, N. Tasak, A. Thaiprakong, G. Thwaites, C. Tigoi, C. Turner, P. Turner, H. R. Van Doorn, S. Velaphi, A. Vongpradith, M. Vongsouvath, H. Vu, T. Walsh, J. L. Walson, S. Waner, T. Wangrangsimakul, P. Wannapinij, T. Wozniak, T. E. M. W. Young Sharma, K. C. Yu, P. Zheng, B. Sartorius, A. D. Lopez, A. Stergachis, C. Moore, C. Dolecek and M. Naghavi, Lancet, 2022, 399, 629–655 CrossRef CAS.
- G. S. Tillotson and S. H. Zinner, Expert Rev. Anti-infect. Ther., 2017, 15, 663–676 CrossRef CAS.
- M. S. Donia, P. Cimermancic, C. J. Schulze, L. C. Wieland Brown, J. Martin, M. Mitreva, J. Clardy, R. G. Linington and M. A. Fischbach, Cell, 2014, 158, 1402–1414 CrossRef CAS PubMed.
- C. D. Santos-Júnior, M. D. T. Torres, Y. Duan, Á. Rodríguez Del Río, T. S. B. Schmidt, H. Chong, A. Fullam, M. Kuhn, C. Zhu, A. Houseman, J. Somborski, A. Vines, X.-M. Zhao, P. Bork, J. Huerta-Cepas, C. De La Fuente-Nunez and L. P. Coelho, Cell, 2024, 187, 3761–3778 CrossRef.
- M. R. Wilson, L. Zha and E. P. Balskus, J. Biol. Chem., 2017, 292, 8546–8552 CrossRef CAS.
- H.-Y. Cheng, M.-X. Ning, D.-K. Chen and W.-T. Ma, Front. Immunol., 2019, 10, 607 CrossRef CAS PubMed.
- K. Hou, Z.-X. Wu, X.-Y. Chen, J.-Q. Wang, D. Zhang, C. Xiao, D. Zhu, J. B. Koya, L. Wei, J. Li and Z.-S. Chen, Signal Transduction Targeted Ther., 2022, 7, 135 CrossRef.
- M. Mahlapuu, J. Håkansson, L. Ringstad and C. Björn, Front. Cell. Infect. Microbiol., 2016, 6, 194 Search PubMed.
- J. Xuan, W. Feng, J. Wang, R. Wang, B. Zhang, L. Bo, Z.-S. Chen, H. Yang and L. Sun, Drug Resistance Updates, 2023, 68, 100954 CrossRef CAS PubMed.
- H. B. Koo and J. Seo, Pept. Sci., 2019, 111, e24122 CrossRef.
- G. S. Dijksteel, M. M. W. Ulrich, E. Middelkoop and B. K. H. L. Boekema, Front. Microbiol., 2021, 12, 616979 CrossRef.
- L. Gallardo-Becerra, M. Cervantes-Echeverría, F. Cornejo-Granados, L. E. Vazquez-Morado and A. Ochoa-Leyva, Microb. Ecol., 2024, 87, 8 CrossRef CAS.
- W. K. Mousa, R. Ghemrawi, T. Abu-Izneid, A. Ramadan and F. Al-Marzooq, IJMS, 2023, 24, 6901 CrossRef CAS.
- F. H. Waghu, R. S. Barai, P. Gurung and S. Idicula-Thomas, Nucleic Acids Res., 2016, 44, D1094–D1097 CrossRef CAS.
- M. N. Gabere and W. S. Noble, Bioinformatics, 2017, 33, 1921–1929 CrossRef CAS PubMed.
- S. Gupta, P. Kapoor, K. Chaudhary, A. Gautam, R. Kumar, Open Source Drug Discovery Consortium and G. P. S. Raghava, PLoS One, 2013, 8, e73957 CrossRef CAS.
- M. Baek, F. DiMaio, I. Anishchenko, J. Dauparas, S. Ovchinnikov, G. R. Lee, J. Wang, Q. Cong, L. N. Kinch, R. D. Schaeffer, C. Millán, H. Park, C. Adams, C. R. Glassman, A. DeGiovanni, J. H. Pereira, A. V. Rodrigues, A. A. van Dijk, A. C. Ebrecht, D. J. Opperman, T. Sagmeister, C. Buhlheller, T. Pavkov-Keller, M. K. Rathinaswamy, U. Dalwadi, C. K. Yip, J. E. Burke, K. C. Garcia, N. V. Grishin, P. D. Adams, R. J. Read and D. Baker, Science, 2021, 373, 871–876 CrossRef CAS.
- M. Baek, I. Anishchenko, I. R. Humphreys, Q. Cong, D. Baker and F. DiMaio, bioRxiv, 2023, preprint, DOI:10.1101/2023.05.24.542179.
- D. Xu and Y. Zhang, Proteins, 2012, 80, 1715–1735 CrossRef CAS PubMed.
- S. M. Mortuza, W. Zheng, C. Zhang, Y. Li, R. Pearce and Y. Zhang, Nat. Commun., 2021, 12, 5011 CrossRef CAS PubMed.
- A. Lamiable, P. Thévenet, J. Rey, M. Vavrusa, P. Derreumaux and P. Tufféry, Nucleic Acids Res., 2016, 44, W449–W454 CrossRef CAS PubMed.
- M. Pirtskhalava, A. A. Amstrong, M. Grigolava, M. Chubinidze, E. Alimbarashvili, B. Vishnepolsky, A. Gabrielian, A. Rosenthal, D. E. Hurt and M. Tartakovsky, Nucleic Acids Res., 2021, 49, D288–D297 CrossRef CAS.
- D. Eisenberg, R. M. Weiss, T. C. Terwilliger and W. Wilcox, Faraday Symp. Chem. Soc., 1982, 17, 109 RSC.
- Q.-Y. Zhang, Z.-B. Yan, Y.-M. Meng, X.-Y. Hong, G. Shao, J.-J. Ma, X.-R. Cheng, J. Liu, J. Kang and C.-Y. Fu, Mil. Med. Res., 2021, 8, 48 CAS.
- Y. Chen, M. T. Guarnieri, A. I. Vasil, M. L. Vasil, C. T. Mant and R. S. Hodges, Antimicrob. Agents Chemother., 2007, 51, 1398–1406 CrossRef CAS PubMed.
- I. A. Edwards, A. G. Elliott, A. M. Kavanagh, J. Zuegg, M. A. T. Blaskovich and M. A. Cooper, ACS Infect. Dis., 2016, 2, 442–450 CrossRef CAS.
- S. Mitaku, T. Hirokawa and T. Tsuji, Bioinformatics, 2002, 18, 608–616 CrossRef CAS.
- K. L. Zapadka, F. J. Becher, A. L. Gomes Dos Santos and S. E. Jackson, Interface Focus, 2017, 7, 20170030 CrossRef.
- Y. Acharya, K. K. Taneja and J. Haldar, RSC Med. Chem., 2023, 14, 1410–1428 RSC.
- H. Huang, J. Damjanovic, J. Miao and Y.-S. Lin, Phys. Chem. Chem. Phys., 2021, 23, 607–616 RSC.
- X. Ji, A. L. Nielsen and C. Heinis, Angew. Chem., 2024, 136, e202308251 CrossRef.
- E. Oueis, T. Klefisch, N. Zaburannyi, R. Garcia, A. Plaza and R. Müller, Org. Lett., 2019, 21, 5407–5412 CrossRef CAS.
- C. Bechtler and C. Lamers, RSC Med. Chem., 2021, 12, 1325–1351 RSC.
- Z. Y. Ong, N. Wiradharma and Y. Y. Yang, Adv. Drug Delivery Rev., 2014, 78, 28–45 CrossRef CAS.
- A. A. Robles-Loaiza, E. A. Pinos-Tamayo, B. Mendes, J. A. Ortega-Pila, C. Proaño-Bolaños, F. Plisson, C. Teixeira, P. Gomes and J. R. Almeida, Pharmaceuticals, 2022, 15, 323 CrossRef CAS PubMed.
- Y. Lyu, Y. Yang, X. Lyu, N. Dong and A. Shan, Sci. Rep., 2016, 6, 27258 CrossRef CAS PubMed.
- A. Hawrani, R. A. Howe, T. R. Walsh and C. E. Dempsey, J. Biol. Chem., 2008, 283, 18636–18645 CrossRef CAS PubMed.
- S. Jaber, V. Nemska, I. Iliev, E. Ivanova, T. Foteva, N. Georgieva, I. Givechev, E. Naydenova, V. Karadjova and D. Danalev, Molecules, 2021, 26, 7321 CrossRef CAS PubMed.
- G. N. Enninful, R. Kuppusamy, E. K. Tiburu, N. Kumar and M. D. P. Willcox, J. Pept. Sci., 2024, 30, e3560 CrossRef CAS PubMed.
- Y. M. Song, Y. Park, S. S. Lim, S.-T. Yang, E.-R. Woo, I.-S. Park, J. S. Lee, J. I. Kim, K.-S. Hahm, Y. Kim and S. Y. Shin, Biochemistry, 2005, 44, 12094–12106 CrossRef CAS.
- W. L. Zhu, K. Hahm and S. Y. Shin, J. Pept. Sci., 2007, 13, 529–535 CrossRef CAS.
- B. Zhou, G. Shetye, N. M. Wolf, S.-N. Chen, M. Qader, G. J. Ray, D. C. Lankin, S. Cho, J. Cheng, J.-W. Suh, S. G. Franzblau, J. B. McAlpine and G. F. Pauli, Org. Lett., 2022, 24, 7265–7270 CrossRef CAS PubMed.
- P. Parvekar, J. Palaskar, S. Metgud, R. Maria and S. Dutta, Biomater. Invest. Dent., 2020, 7, 105–109 CAS.
- M. Ghasemi, T. Turnbull, S. Sebastian and I. Kempson, IJMS, 2021, 22, 12827 CrossRef CAS.
- AAT Bioquest., AAT Bioquest, Inc., https://www.aatbio.com/tools/ec50-calculator, (accessed 30 April 2024).
- E. B. A. Adusei, R. K. Adosraku, J. Oppong-Kyekyeku, C. D. K. Amengor and Y. Jibira, J. Trop. Med., 2019, 2019, 1–8 CrossRef.
- J. Zahller and P. S. Stewart, Antimicrob. Agents Chemother., 2002, 46, 2679–2683 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00383g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |