
Novel benzenesulfonamides containing a dual triazole moiety with selective carbonic anhydrase inhibition and anticancer activity†
Aida
Buza
a,
Cüneyt
Türkeş
 *b,
Mustafa
Arslan
*c,
Yeliz
Demir
d,
Busra
Dincer
e,
Arleta Rifati
Nixha
*a and
Şükrü
Beydemir
f
*b,
Mustafa
Arslan
*c,
Yeliz
Demir
d,
Busra
Dincer
e,
Arleta Rifati
Nixha
*a and
Şükrü
Beydemir
f
aDepartment of Chemistry, Faculty of Mathematical and Natural Sciences, University of Prishtina, Prishtina 1000, Republic of Kosova. E-mail: arleta.rifati@uni-pr.edu
bDepartment of Biochemistry, Faculty of Pharmacy, Erzincan Binali Yıldırım University, Erzincan 24002, Turkey. E-mail: cuneyt.turkes@erzincan.edu.tr
cDepartment of Chemistry, Faculty of Sciences, Sakarya University, Sakarya 54187, Turkey. E-mail: marslan@sakarya.edu.tr
dDepartment of Pharmacy Services, Nihat Delibalta Göle Vocational High School, Ardahan University, Ardahan 75700, Turkey
eDepartment of Pharmacology, Faculty of Pharmacy, Ondokuz Mayıs University, Samsun 55020, Turkey
fDepartment of Biochemistry, Faculty of Pharmacy, Anadolu University, Eskişehir 26470, Turkey
First published on 4th October 2024
Abstract
A series of sulfonamides incorporating a 1,2,3-triazolyloxime substituted 1,2,3-triazolyl moiety were conceptualized and synthesized as human carbonic anhydrase (hCA) inhibitors. The synthesized small structures, denoted 7a through 7o, exhibited moderate inhibitory effects against the tumor-associated isoforms hCA IX and hCA XII compared to the well-known hCA inhibitor acetazolamide. In contrast, these molecules demonstrated higher potency and a diverse range of selectivity against the cytosolic isoforms hCA I and hCA II. Notably, the 4-hydroxyphenyl derivative (compound 7dversus cytosolic isoforms), the 4-acetylphenyl derivative (compound 7o), and the phenyl derivative (compound 7a) emerged as the most potent and selective inhibitors in this series, with inhibition constants (KI) of 47.1, 35.9, 170.0, and 149.9 nM, respectively, against hCA I, II, IX, and XII. Further cytotoxicity assays of compounds 7a–o against cancer cell lines Hep3B and A549, as well as normal cell line L929, were conducted to assess their selectivity towards malignant cells. Compounds 7d, 7g, and 7k exhibited selective cytotoxicity towards the Hep3B cell line, with reduced selectivity towards A549, whereas compound 7j demonstrated higher selectivity for the A549 cell line. Additionally, molecular docking studies were performed to elucidate the binding modes of these compounds within the active sites of hCAs, revealing crucial interactions that underpin their significant activity and selectivity for the tumor-specific isoforms.
1. Introduction
The progression of click chemistry has been meticulously aligned with pharmaceutical and medical research, facilitating the creation of extensive molecular libraries essential for drug discovery, thus establishing it as a crucial and transformative synthetic methodology. The successful synthesis of stable 1,2,3-triazolyl isosteres has significantly expanded their application in the development of bioactive molecule analogs.1 This renewed demand for innovative chemotherapeutic agents has spurred various research groups to focus on synthesizing triazole analogs. The significance of click chemistry lies in its rapid and efficient approach, capable of producing complex and high-purity drugs from simple, cost-effective starting materials.2 This methodology, which effectively links substrates to specific biomolecules, is highly valued in medicinal chemistry for overcoming practical synthesis limitations, enhancing throughput, and improving the quality of compound libraries.3 1,2,3-Triazole units synthesized via the metal-catalyzed alkyne–azide cycloaddition reaction have proven to be an exceptionally effective tool. Huisgen's 1,3-cycloaddition, involving the reaction between an azide and an alkyne to yield 1,2,3-triazole,4 represents a revolutionary advance in azole chemistry. Click chemistry, conceptualized by Sharpless, offers numerous advantages over Huisgen's 1,3-dipolar cycloaddition for synthesizing 1,2,3-triazole derivatives, including high reaction efficiency, mild conditions, remarkable chemo- and regioselectivities, and excellent functional group compatibility. Over the past decade, this approach has garnered significant attention for its utility in creating compounds with diverse applications, from pharmaceuticals to bioconjugation linkers.5 The 1H-1,2,3-triazole, produced through this versatile and selective click reaction, has become a preferred tool for synthetic and medicinal chemists due to its ability to mimic various functional groups and enhance targeted biological activities.Heterocyclic compounds, particularly the 1,2,3-triazole ring,6 have become increasingly vital in designing new medicinally important structures due to their moderate dipole character, hydrogen bonding capability, and stability.7 Furthermore, 1,4-substituted-1,2,3-triazole plays multiple roles in bioactive molecules: it acts as a key pharmacophore element forming hydrogen bonds or hydrophobic interactions, serves as a molecular scaffold maintaining the active conformation, and functions as a connecting group linking conjugated molecules or probes.
Cancer malignancy is a global public health concern that results in numerous deaths each year.8 Malignancy arises from many factors, including the proliferation of many mutations, uncontrolled proliferation of cancer cells, the formation of new blood vessels, and resistance to chemotherapy.9 Hypoxia in the solid tumor microenvironment is another important factor leading to malignancy.10 Cancer cells employ glycolysis and increase the expression of certain enzymes to survive in their surroundings and decrease the pH outside their cells.11 Of the sixteen kinds of carbonic anhydrases (CAs) found in human(h)s,12 two of them, hCA IX and hCA XII, are excessively produced in various types of solid (hypoxic) tumors that lack oxygen.13 These cancers include colon, breast, renal, lung, and ovarian carcinomas.14 The overexpression of hCA IX and XII isoforms is caused by the hypoxia-inducible factor (HIF-1α) cascade activation.15 Therefore, the hCA IX/XII isoforms have been firmly established as targets for anticancer drugs and markers for the prognosis,16 diagnosis,17 and treatment18 of many solid malignancies.19
CAs are Zn2+-based metalloenzymes20,21 in both prokaryotes22 and eukaryotes.23 These enzymes utilize zinc as a metal cofactor24 to facilitate the reversible conversion of carbon dioxide and bicarbonate ions.25 There are eight families of CAs,26 which are named α-CAs to ι-CAs.27 These families produce distinct variations of the enzyme.28 However, in humans, only the α-hCA isoform is expressed.29 Currently, 16 different α-hCAs have been recorded with the codes hCA I–IV, VA, VB, and VI–XV.30 They vary in terms of their placements, functional features, and kinetics.31 The transmembrane isoforms hCA IX and XII play a vital role in maintaining the balance of extracellular pH.32 Notably, most healthy tissues do not produce hCA IX, but its increased expression is associated with hypoxic cancer characteristics,33 which HIF-1α strongly induces,34 as mentioned above. The hCA XII isoform is overexpressed in several solid cancers.35 Hence, it is recommended that directing attention towards hCA IX and XII isoforms could be a feasible strategy for creating innovative anticancer drugs to treat hypoxic malignancies.36 Twelve catalytically active human α-CA isoforms have a conserved cone-shaped pocket at the active site's base.37 This pocket is regulated by three histidine residues (His94, His96, and His119) and a water molecule.38 The outer section of the active site has hydrophobic and hydrophilic areas with different levels of hydrophobicity and polarity across different CAs.39 In the catalytic cycle of CAs, the Zn2+ ion initially forms a coordination bond with the hydroxide ion.40 Subsequently, a chemical reaction occurs with a carbon dioxide molecule, resulting in the formation of bicarbonate.41 Thus, the distinguishing factor of human CA inhibitors (hCAIs) is the Zn2+-binding group that binds to the zinc ion in the active site.42 Designing molecules with high affinity and isoform-selectivity for CAs without inducing off-target effects is problematic due to the significant similarity among hCA structures, especially in the active regions.43
Sulfonamides and related bioisosteres are acknowledged as potent inhibitors of CAs.44 Many sulfonamide-containing drugs, like acetazolamide (AAZ), brinzolamide, celecoxib, ethoxzolamide, furosemide, indisulam, methazolamide, pazopanib, etc., are being clinically used for the treatment of hCA-related diseases45,46 (Fig. 1). Furthermore, compounds containing the 1,2,3-triazole ring have emerged in the last decade as potential pharmacological agents targeting different therapeutic areas, including hCA isoforms47–51 (Fig. 2). As stated in our previous studies, it has a high affinity for the hCA IX and XII isoforms with KIs of 437.2 and 338.9 nM, respectively.52,53 Nevertheless, due to the drug's ability to impede the specific hCA isoforms it targets, as well as other hCA isoforms present in many organs, prolonged usage of this medication may lead to adverse consequences.54 Among different strategies used to synthesize isoform selective hCAIs, the tail approach technique is among the most successful.55 This method involves appending differently substituted molecular moieties as tails to the aromatic or heterocyclic scaffolds of sulfonamides.56
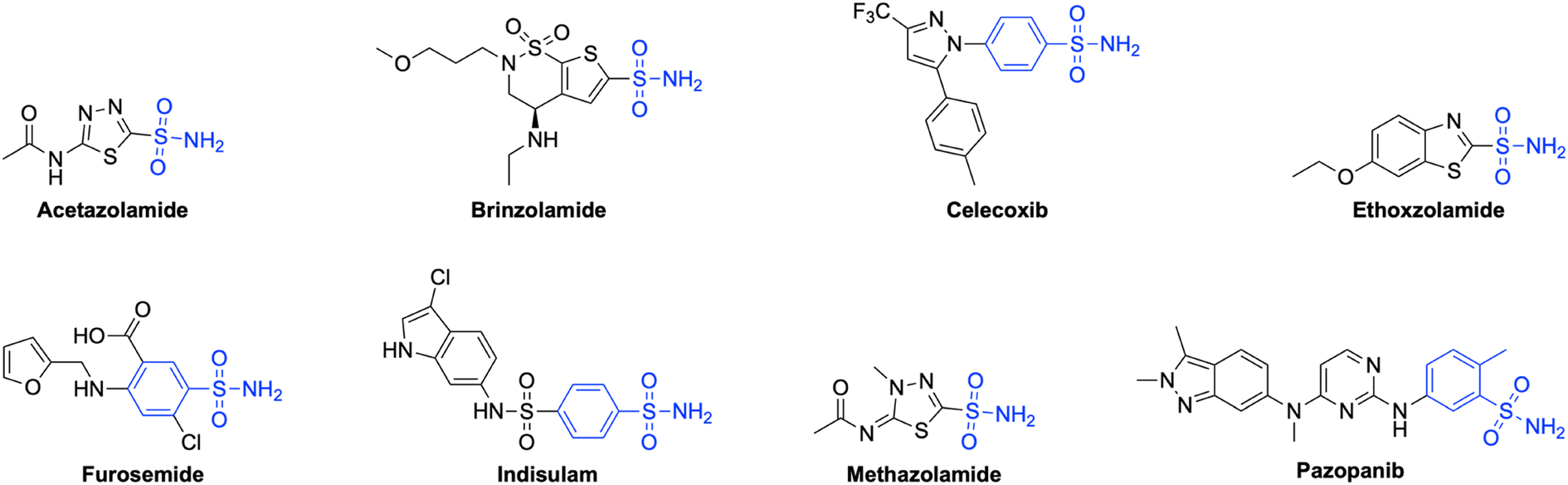 | ||
| Fig. 1 Some sulfonamide-containing drugs in clinical use as potential carbonic anhydrase inhibitors. | ||
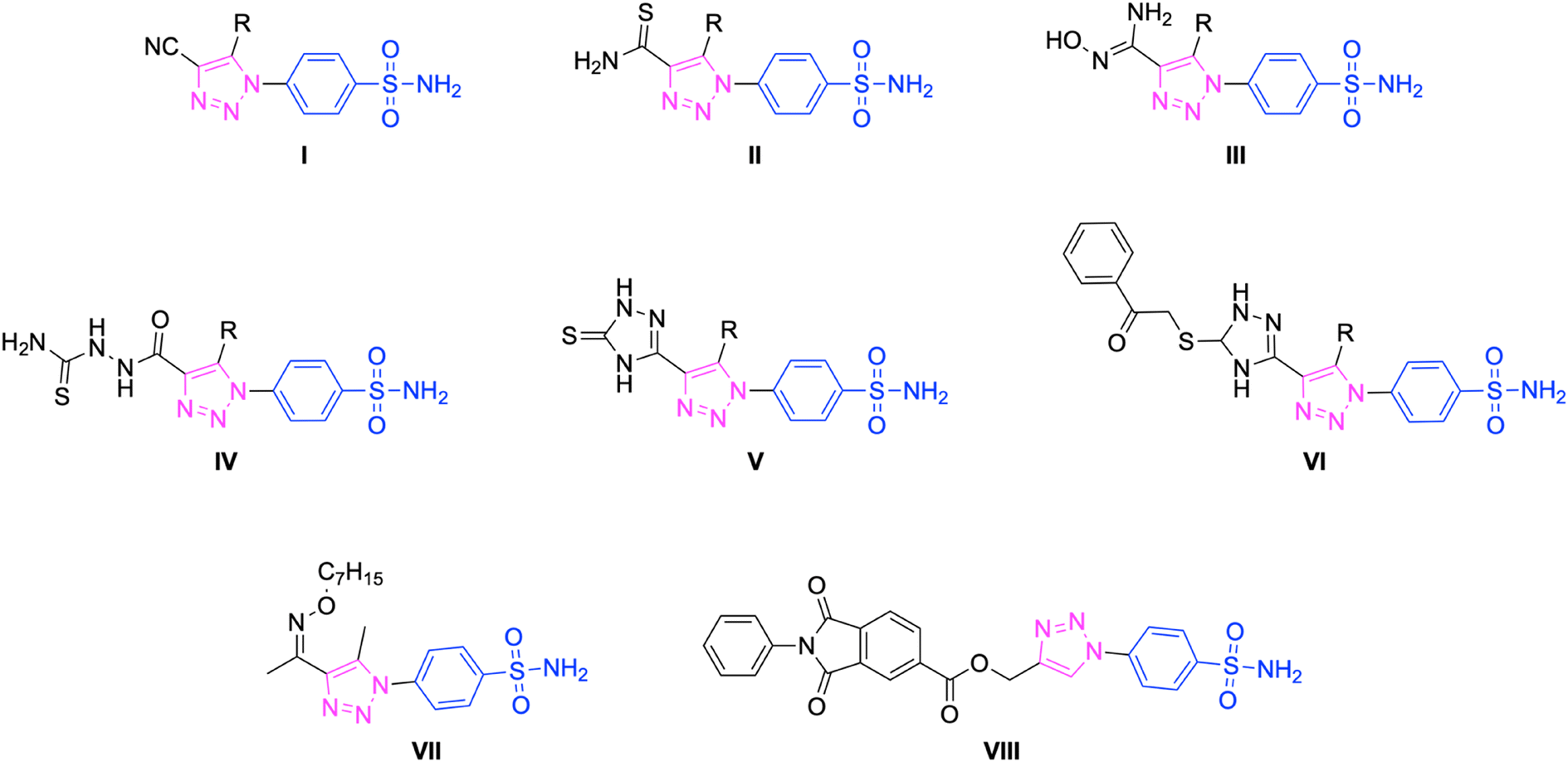 | ||
| Fig. 2 The chemical structure of some 1,2,3-triazole containing potential carbonic anhydrase inhibitors. | ||
In the last few years, our research group has made efforts to explore sulfonamides as inhibitors of carbonic anhydrase isoforms.57–60 Recently, there have been reports of research focused on developing new and specific inhibitors for hCA I, II, IX, and XII.61,62 For instance, like compound VII (KIs of 195.9 and 116.9 nM for hCA IX and XII, respectively)52 and compound VIII (KIs of 18.3 and 24.2 nM for hCA IX and XII, respectively)53 in our recent studies, these research studies combine benzenesulfonamide, a compound often used in conventional hCAIs, with a 1,2,3-triazole scaffold tail (as shown in Fig. 2).
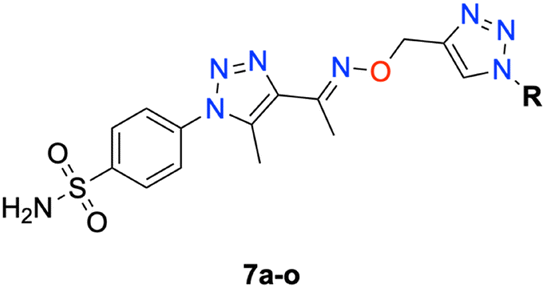
In this context, the tail technique has gained attraction as a strategy for designing inhibitors targeting hCA isoforms, utilizing the significant active site similarities. Typically, design studies have focused on the benzenesulfonamide scaffold,63 where diverse aryl or heterocyclic moieties are attached to the aromatic sulfonamide ring to interact selectively with the hydrophobic residues of these isoforms' active sites.64 The synthesized compounds, designated as 7a–o, feature a benzenesulfonamide zinc-binding group linked to a rigid triazole scaffold, optimizing the positioning towards the hydrophobic or hydrophilic rims of the active site. The 1,2,3-triazolyloxime moiety enhances the flexibility and hydrophilicity of the molecules, improving selectivity for hCA IX and hCA XII through interactions with specific residues in the hydrophilic region of the active site. Various elongated linkers were employed to fine-tune the inhibitory effect of the targeted structures on hCAs (Fig. 3).
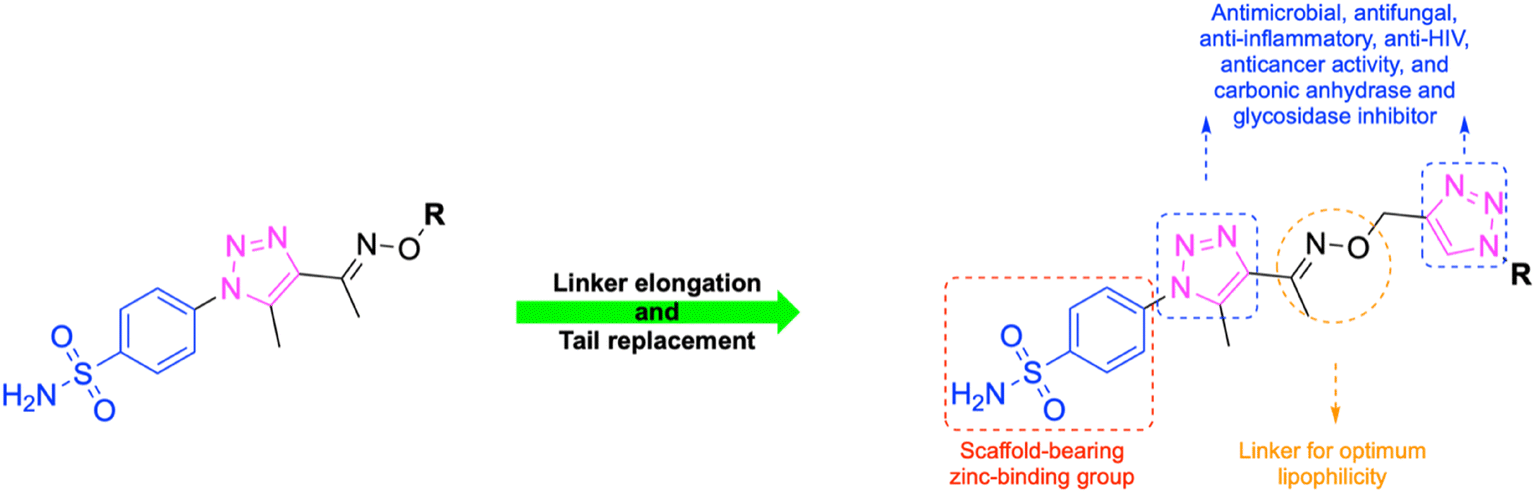 | ||
| Fig. 3 The design strategy of targeted small structure derivatives (7a–o). | ||
Motivated by the previously mentioned discoveries and continuing our current investigations, this study presents the creation of fifteen novel 1,2,3-triazolyloxime-substituted 1,2,3-triazolyl derivatives (7a–o) (Scheme 1). Specifically, to produce potent and specific inhibitors for the hCA IX and XII isoforms linked to tumors, we focus on sulfonamide-based hCAIs. In the previous study,52 the oxime ether-linked 1,4-disubstituted 1,2,3-triazole tail was replaced by the 1,2,3-triazolyl oxime moiety. That is, the linker has been elongated, and the tail has been changed, as illustrated in Fig. 3. Moreover, the targeted small structures were tested under in vitro conditions to see how well they stopped the growth of human hepatocellular carcinoma (Hep3B) and human lung carcinoma (A549) cell lines. We also conducted complementary molecular docking studies to clarify the expected in silico molecular binding interactions of the novel synthesized molecules (7a–o) within the active sites of the hCA isoforms.
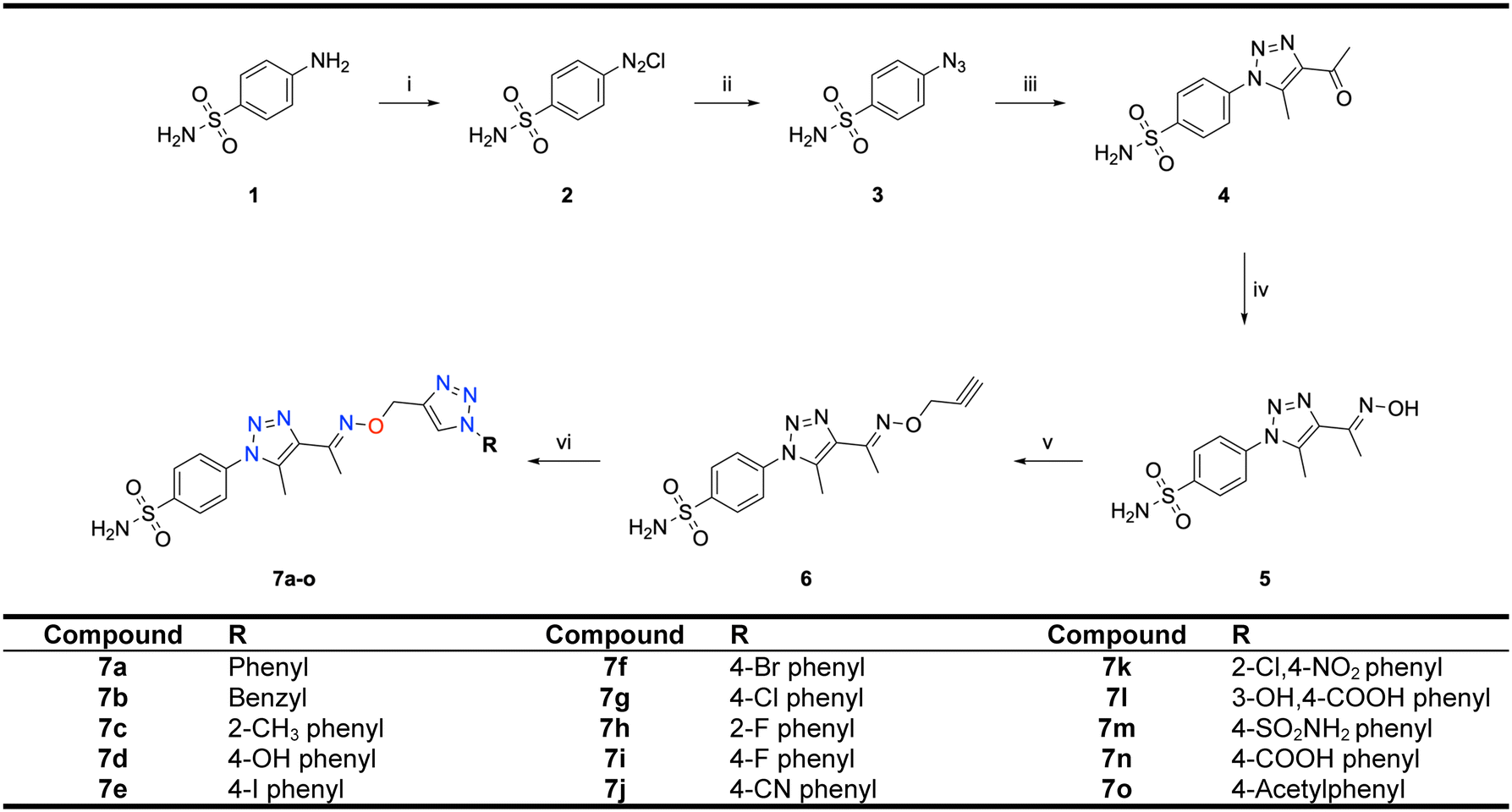 | ||
| Scheme 1 Synthetic pathway of novel 1,2,3-triazolyloxime substituted-1,2,3-triazolyl sulfonamide derivatives (7a–o). | ||
2. Experimental
2.1. Chemistry
All starting chemicals and solvents utilized in the research were procured from Sigma-Aldrich and employed without additional purification. The determination of melting points was conducted utilizing a Yanagimoto micro-melting point apparatus and values were left uncorrected. Infrared (FTIR) spectra were analyzed using a Shimadzu Prestige-21 (200 VCE) spectrometer in DMSO-d6. Acquisition of 1H and 13C NMR spectra was carried out with a VARIAN Infinity Plus at 300 and 75 Hz, respectively. The chemical shifts of 1H and 13C were calibrated against the internal deuterated solvent. Mass spectrometry was conducted on a 6200 series TOF/6500 series Q-TOF B.08.00 spectrometer.2.2. Synthesis of 4-azidobenzenesulfanilamide (3)
Sulfanilamide (1) (3 mmol, 0.517 g) was dissolved in an aqueous 6 M HCl solution and subsequently cooled to 0 °C. NaNO2 (3.6 mmol, 0.250 g), dissolved in cold water (in 15 mL), was then added dropwise to the mixture while maintaining the temperature at 0 °C, followed by continuous stirring for 30 minutes, resulting in the formation of sulfanilamide diazonium salt (2). Subsequently, sodium azide (NaN3, 4.4 mmol, 0.290 g) was introduced into the diazonium salt solution and stirred for an additional 2 hours at room temperature, yielding the corresponding azido derivative (3). The final product was washed with water, filtered, and dried, and then used in the subsequent step without further purification. The compound's purity was confirmed by 1H NMR, 13C NMR, and FTIR spectrophotometry.2.3. Synthesis of 1,2,3,-triazolebenzensulfonamide (4)
A solution containing azidobenzenesulfonamide (3) (1 mmol, 0.213 g) in N,N-dimethylformamide (DMF) was treated with acetylacetone (1.5 mmol, 0.150 g) in the presence of potassium carbonate (2 mmol, 0.276 g) as a primary catalyst at a temperature of 80 °C with continuous stirring for a duration of 5–6 hours. Upon completion of the reaction, the resulting mixture was subjected to extraction with ethyl acetate, followed by washing steps using 1 M NaOH solution, saturated NaHCO3 solution, and brine. The organic layer was subsequently dehydrated with magnesium sulfate (MgSO4), filtered, and concentrated under reduced pressure. The obtained product underwent purification via crystallization from chloroform (CHCl3). The final product, 1,2,3-triazolebenzenesulfonamide derivative (4), was isolated in a pure form. The compound was checked by 1H-, 13C-NMR, and FTIR spectrophotometry.2.4. Synthesis of compound 5
Compound 5 was synthesized following the literature.52 Hydroxylamine hydrochloride (3 mmol, 0.210 g) was introduced to azidobenzenesulfonamide (1 mmol, 0.213 g) (4), which was previously dissolved in dimethyl formaldehyde (DMF) with the addition of 2 mL triethylamine (Et3N) serving as a catalyst. The resulting mixture was stirred at 100 °C for 10 hours to produce the respective oxime derivative (5). Upon completion, the reaction mixture was allowed to cool to room temperature, followed by the addition of ice-cold water, filtration, and drying. Subsequently, the oxime derivative (5) was purified through crystallization using acetone–hexane as the solvent. The characterization of the compound was carried out via1H NMR, 13C NMR, and FTIR analysis.2.5. Synthesis of compound 6
Propargyl bromide (2 mmol, 0.238 g) was added to the oxime compound (2 mmol, 0.590 g) in the presence of dimethylsulfoxide (DMSO) as a solvent, NaOH as a base, and tetrabutylammonium bromide (TBAB) as a phase transfer catalyst to accelerate the reaction. The reaction mixture was stirred for 3 hours at room temperature. Then, propargylated oxime was obtained. Compound 6 was purified by crystallization from acetone–hexane. The compound was characterized by 1H NMR, 13C NMR, and FTIR.2.6. General method for the synthesis of targeted small compounds (7a–o)
The propargylated oximes (6) (1 mmol, 0.333 g) were treated with substituted aromatic azides (1 mmol) with different substituents. This reaction was carried out in the presence of DMF as a solvent and CuSO4 (1 mmol, 0250 g) as a catalyst in combination with a small amount of ascorbic acid (0.1 mmol), which played the role of reducing agent, and for 3 hours at room temperature. The reaction gave the desired products in reasonably good yields. The compounds (7a–o) shown in Scheme 1 were purified by crystallization from acetone–hexane and characterized by 1H-, 13C-NMR, QTOF LC/MS, and FTIR spectrophotometry, with detailed spectral data provided in the ESI.†M.p: 211 °C; yield: 95%; FTIR (cm−1, ν): 3344–3594 (N–H), 1597 (C
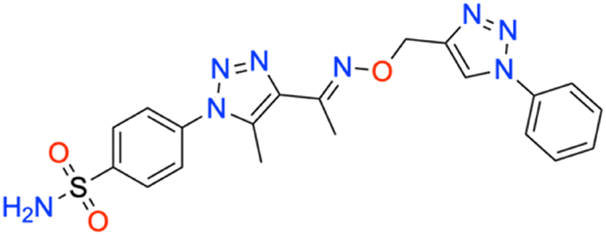
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1502 (CC), 1156 and 1328 (SO2); 1H NMR (DMSO-d6, 300 MHz): 8.54 (1H, s, CH), 8.027–8.051 (2H, d, –Ar–H), 7.82–7.85 (2H, d, –Ar–H), 7.405–7.465 (5H, d, –Ar–H), 7.59 (2H, s, –NH2), 5.326 (2H, s, –CH2), 2.46 (3H, s, –CH3), 2.126 (3H, s, –CH3); 13C NMR (DMSO-d6, 75 MHz): 151.3, 145.7, 137.3, 133.1 (2C), 130.3, 129.1 (3C), 127.89 (2C), 126.2 (2C), 123.1 (2C), 120.6 (2C), 67.5, 13.3, 11.3; MS (m/z) [M + H]+: calcd. for C20H20N8O3S: 454.1450, and found: 454.1478.
N), 1502 (CC), 1156 and 1328 (SO2); 1H NMR (DMSO-d6, 300 MHz): 8.54 (1H, s, CH), 8.027–8.051 (2H, d, –Ar–H), 7.82–7.85 (2H, d, –Ar–H), 7.405–7.465 (5H, d, –Ar–H), 7.59 (2H, s, –NH2), 5.326 (2H, s, –CH2), 2.46 (3H, s, –CH3), 2.126 (3H, s, –CH3); 13C NMR (DMSO-d6, 75 MHz): 151.3, 145.7, 137.3, 133.1 (2C), 130.3, 129.1 (3C), 127.89 (2C), 126.2 (2C), 123.1 (2C), 120.6 (2C), 67.5, 13.3, 11.3; MS (m/z) [M + H]+: calcd. for C20H20N8O3S: 454.1450, and found: 454.1478.
M.p: 134 °C; yield: 91%; FTIR (cm−1, ν): 3349–3267 (N–H), 1597 (C
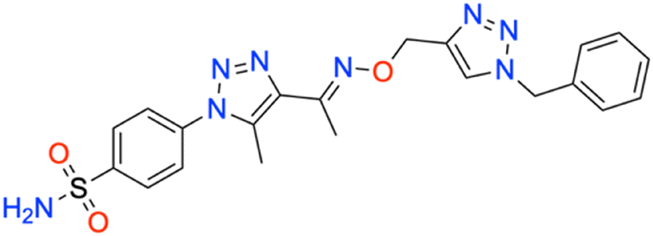 N), 1503 (CC), 1161 and 1130 (SO2); 1H NMR (DMSO-d6, 300 MHz): 8.22 (1H, s, CH), 8.03–8.057 (2H, d, –Ar–H), 7.816–7.845 (2H, d, –Ar–H), 7.615 (2H, s, –NH2), 7.289–7.331 (5H, d, –Ar–H), 5.578 (2H, s, –CH2), 5.20 (2H, s, –CH2), 2.482 (3H, s, –CH3), 2.367, (3H, s, –CH3); 13C NMR (DMSO-d6, 75 MHz): 154.6, 151.1, 141.1, 138.6 (2C), 136.7 (2C), 133.3 (2C), 129.4 (2C), 128.8 (2C), 127.8 (2C), 126.5 (2C), 67.7, 53.5, 13.4, 11.06; MS (m/z) [M + H]+: calcd. for C21H22N8O3S: 467.1608, and found: 467.1633.
N), 1503 (CC), 1161 and 1130 (SO2); 1H NMR (DMSO-d6, 300 MHz): 8.22 (1H, s, CH), 8.03–8.057 (2H, d, –Ar–H), 7.816–7.845 (2H, d, –Ar–H), 7.615 (2H, s, –NH2), 7.289–7.331 (5H, d, –Ar–H), 5.578 (2H, s, –CH2), 5.20 (2H, s, –CH2), 2.482 (3H, s, –CH3), 2.367, (3H, s, –CH3); 13C NMR (DMSO-d6, 75 MHz): 154.6, 151.1, 141.1, 138.6 (2C), 136.7 (2C), 133.3 (2C), 129.4 (2C), 128.8 (2C), 127.8 (2C), 126.5 (2C), 67.7, 53.5, 13.4, 11.06; MS (m/z) [M + H]+: calcd. for C21H22N8O3S: 467.1608, and found: 467.1633.
M.p: 193 °C; yield: 87%; FTIR (cm−1, ν): 3157–3319 (N–H), 3032 (
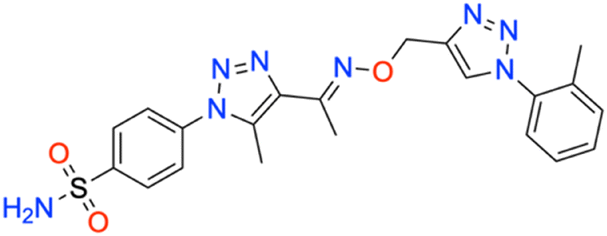 C–H), 1498 (CC), 1344 and 1165 (SO2); 1H NMR (DMSO-d6, 300 MHz): 8.56 (1H, s, CH), 8.053 (2H, d, –Ar–H), 7.871 (2H, d, –Ar–H), 7.587 (2H, s, –NH2), 7.478 (2H, d, –Ar–H), 7.419 (2H, t, –Ar–H), 5.340 (2H, s, –CH2), 2.498 (3H, s, –CH3), 2.361 (3H, s, –CH3), 2.141 (3H, s, –CH3); 13C NMR (DMSO-d6, 75 MHz): 151.3, 145.7, 144.3, 141.1, 138.6 (2C), 136.9, 133.7 (2C), 133.4, 132.1 (2C), 130.5, 127.8 (2C), 126.5 (2C), 67.6, 58.1, 13.5, 11.1; MS (m/z) [M + H]+: calcd. for C21H22N8O3S: 467.1609, and found: 467.1596.
C–H), 1498 (CC), 1344 and 1165 (SO2); 1H NMR (DMSO-d6, 300 MHz): 8.56 (1H, s, CH), 8.053 (2H, d, –Ar–H), 7.871 (2H, d, –Ar–H), 7.587 (2H, s, –NH2), 7.478 (2H, d, –Ar–H), 7.419 (2H, t, –Ar–H), 5.340 (2H, s, –CH2), 2.498 (3H, s, –CH3), 2.361 (3H, s, –CH3), 2.141 (3H, s, –CH3); 13C NMR (DMSO-d6, 75 MHz): 151.3, 145.7, 144.3, 141.1, 138.6 (2C), 136.9, 133.7 (2C), 133.4, 132.1 (2C), 130.5, 127.8 (2C), 126.5 (2C), 67.6, 58.1, 13.5, 11.1; MS (m/z) [M + H]+: calcd. for C21H22N8O3S: 467.1609, and found: 467.1596.
M.p: 109 °C; yield: 89%; FTIR (cm−1, ν): 3167 (O–H), 3257–3325 (N–H), 1596 (C
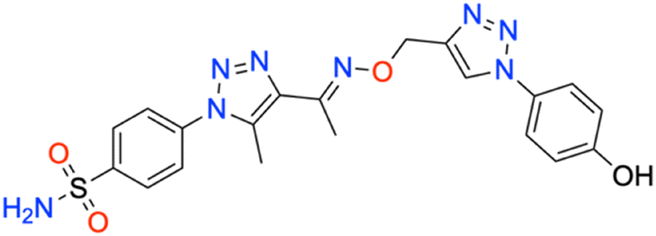 N), 1518 (CC aromatic), 1157 and 1334 (SO2); 1H NMR (DMSO-d6, 300 MHz): 10.08 (1H, s, –OH), 8.63 (1H, s, CH), 8.020–8.046 (2H, d, –Ar–H), 7.796–7.823 (2H, d, –Ar–H), 7.596–7.606 (2H, –Ar–H), 7.636 (2H, s, –NH2) 6.894–6.920 (2H, d, –Ar–H), 5.277 (2H, s, –CH2), 2.477 (3H, s, –CH3), 2.312 (3H, s, –CH3); 13C NMR (DMSO-d6, 75 MHz): 158.4, 151.3, 145.5, 138.6 (2C), 133.4 (2C), 129.4 (2C), 127.8 (2C), 126.5 (2C), 122.7 (2C), 116.7 (2C), 67.5, 13.4, 11.1; MS (m/z) [M + H]+: calcd. for C20H20N8O4S: 469.1401, and found: 469.1441.
N), 1518 (CC aromatic), 1157 and 1334 (SO2); 1H NMR (DMSO-d6, 300 MHz): 10.08 (1H, s, –OH), 8.63 (1H, s, CH), 8.020–8.046 (2H, d, –Ar–H), 7.796–7.823 (2H, d, –Ar–H), 7.596–7.606 (2H, –Ar–H), 7.636 (2H, s, –NH2) 6.894–6.920 (2H, d, –Ar–H), 5.277 (2H, s, –CH2), 2.477 (3H, s, –CH3), 2.312 (3H, s, –CH3); 13C NMR (DMSO-d6, 75 MHz): 158.4, 151.3, 145.5, 138.6 (2C), 133.4 (2C), 129.4 (2C), 127.8 (2C), 126.5 (2C), 122.7 (2C), 116.7 (2C), 67.5, 13.4, 11.1; MS (m/z) [M + H]+: calcd. for C20H20N8O4S: 469.1401, and found: 469.1441.
M.p: 242 °C; yield: 90%; FTIR (cm−1, ν): 3252–3339 (N–H), 1559 (C
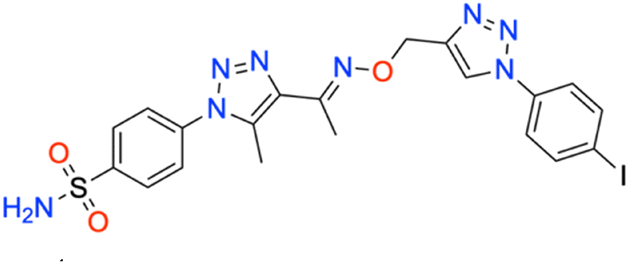 N), 1494 (CC), 1160 and 1332 (SO2), 578 (C–I); 1H NMR (DMSO-d6, 300 MHz): 8.878 (1H, s, CH), 8.017–8.053 (2H d, –Ar–H), 7.919–7.956 (2H, d, –Ar–H),7.814–7.851 (2H, d, –Ar–H), 7.699–7.736 (2H, d, –Ar–H), 7.59 (2H, s, –NH2), 5.304 (2H, s, –CH2), 2.455 (3H, s, –CH3), 2.331 (3H, s, –CH3); 13C NMR (DMSO-d6, 75 MHz): 151.4, 145.6, 145.5, 141.1, 139.3, 138.6, 136.9 (2C), 133.4 (2C), 127.8 (2C), 126.5 (2C), 122.6 (2C), 95.1, 67.5, 13.4, 11.3; MS (m/z) [M + H]+: calcd. for C20H19IN8O3S: 579.0418, and found: 579.0441.
N), 1494 (CC), 1160 and 1332 (SO2), 578 (C–I); 1H NMR (DMSO-d6, 300 MHz): 8.878 (1H, s, CH), 8.017–8.053 (2H d, –Ar–H), 7.919–7.956 (2H, d, –Ar–H),7.814–7.851 (2H, d, –Ar–H), 7.699–7.736 (2H, d, –Ar–H), 7.59 (2H, s, –NH2), 5.304 (2H, s, –CH2), 2.455 (3H, s, –CH3), 2.331 (3H, s, –CH3); 13C NMR (DMSO-d6, 75 MHz): 151.4, 145.6, 145.5, 141.1, 139.3, 138.6, 136.9 (2C), 133.4 (2C), 127.8 (2C), 126.5 (2C), 122.6 (2C), 95.1, 67.5, 13.4, 11.3; MS (m/z) [M + H]+: calcd. for C20H19IN8O3S: 579.0418, and found: 579.0441.
M.p: 251 °C; yield: 88%; FTIR (cm−1, ν): 3337–3251 (N–H), 1598 (C
 N), 1560 (CC), 1161 and 1336 (SO2); 1H NMR (DMSO-d6, 300 MHz): 8.83 (1H, s, CH), 8.017–8.159 (4H, d, –Ar–H), 7.814–7.843 (4H, d, –Ar–H), 7.588 (2H, s, –NH2), 5.328 (2H, s, –CH2), 2.525 (3H, s, –CH3), 2.341 (3H, s, –CH3); 13C NMR (DMSO-d6, 75 MHz): 151.4, 145.6, 141.1, 138.6 (2C), 136.4 (2C), 133.5 (2C), 127.8 (2C), 126.5 (2C), 123.4 (2C), 122.7, 122.1, 67.5, 13.4, 11.1; MS (m/z) [M + H]+: calcd. for C20H19BrN8O3S: 531.0556, and found: 531.0529.
N), 1560 (CC), 1161 and 1336 (SO2); 1H NMR (DMSO-d6, 300 MHz): 8.83 (1H, s, CH), 8.017–8.159 (4H, d, –Ar–H), 7.814–7.843 (4H, d, –Ar–H), 7.588 (2H, s, –NH2), 5.328 (2H, s, –CH2), 2.525 (3H, s, –CH3), 2.341 (3H, s, –CH3); 13C NMR (DMSO-d6, 75 MHz): 151.4, 145.6, 141.1, 138.6 (2C), 136.4 (2C), 133.5 (2C), 127.8 (2C), 126.5 (2C), 123.4 (2C), 122.7, 122.1, 67.5, 13.4, 11.1; MS (m/z) [M + H]+: calcd. for C20H19BrN8O3S: 531.0556, and found: 531.0529.
M.p: 237 °C; yield: 92%; FTIR (cm−1, ν): 3270–3368 (N–H), 1649 (C
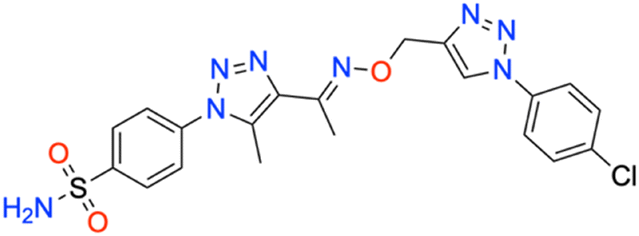 N), 1596 (CC), 1167 and 1500 (SO2), 630 (C–Cl); 1H NMR (DMSO-d6, 300 MHz): 8.92 (1H, s, CH), 8.045–8.073 (2H, d, –Ar–H), 7.949–7.978 (2H, d, –Ar–H), 7.844–7.872 (2H, d, –Ar–H), 7.665–7.694 (2H, d, –Ar–H), 7.619 (2H, s, –NH2), 5.338 (2H, s, –CH2), 2.486 (3H, s, –CH3), 2.362 (3H, s, –CH3); 13C NMR (DMSO-d6, 75 MHz): 151.4, 145.7, 141.1, 138.6 (2C), 136.1, 133.6, 133.4 (2C), 130.5 (2C), 127.8 (2C), 126.5 (2C), 122.5 (2C), 67.5, 13.4, 11.1; MS (m/z) [M + H]+: calcd. for C20H19ClN8O3S: 487.1062, and found: 488.1041.
N), 1596 (CC), 1167 and 1500 (SO2), 630 (C–Cl); 1H NMR (DMSO-d6, 300 MHz): 8.92 (1H, s, CH), 8.045–8.073 (2H, d, –Ar–H), 7.949–7.978 (2H, d, –Ar–H), 7.844–7.872 (2H, d, –Ar–H), 7.665–7.694 (2H, d, –Ar–H), 7.619 (2H, s, –NH2), 5.338 (2H, s, –CH2), 2.486 (3H, s, –CH3), 2.362 (3H, s, –CH3); 13C NMR (DMSO-d6, 75 MHz): 151.4, 145.7, 141.1, 138.6 (2C), 136.1, 133.6, 133.4 (2C), 130.5 (2C), 127.8 (2C), 126.5 (2C), 122.5 (2C), 67.5, 13.4, 11.1; MS (m/z) [M + H]+: calcd. for C20H19ClN8O3S: 487.1062, and found: 488.1041.
M.p: 232 °C; yield: 89%; FTIR (cm−1, ν): 3252–3338 (N–H), 1609 (C
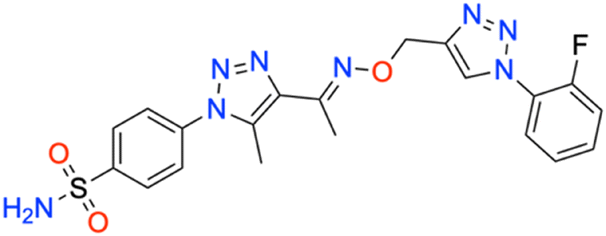 N), 1504 (CC), 1164 and 1344 (SO2), 1032 (C–F); 1H NMR (DMSO-d6, 300 MHz): 8.936 (1H, s, CH), 8.041–8.070 (2H, d, –Ar–H), 7.837–7.867 (2H, d, –Ar–H), 7.806 (2H, s, –NH2), 7.591–7.691 (2H, d, –Ar–H), 7.328–7.392 (2H, t, –Ar–H), 5.342 (2H, s, –CH2), 2.515 (3H, s, –CH3), 2.367 (3H, s, –CH3); 13C NMR (DMSO-d6, 75 MHz): 164.7, 161.5, 151.5, 145.6, 141.1, 138.4, 133.4, 132.6, 132.5, 127.8 (2C), 126.5 (2C), 123.5, 116.7, 115.9, 108.1, 67.5, 13.4, 11.1; MS (m/z) [M + H]+: calcd. for C20H19FN8O3S: 471.1353, and found: 471.1374.
N), 1504 (CC), 1164 and 1344 (SO2), 1032 (C–F); 1H NMR (DMSO-d6, 300 MHz): 8.936 (1H, s, CH), 8.041–8.070 (2H, d, –Ar–H), 7.837–7.867 (2H, d, –Ar–H), 7.806 (2H, s, –NH2), 7.591–7.691 (2H, d, –Ar–H), 7.328–7.392 (2H, t, –Ar–H), 5.342 (2H, s, –CH2), 2.515 (3H, s, –CH3), 2.367 (3H, s, –CH3); 13C NMR (DMSO-d6, 75 MHz): 164.7, 161.5, 151.5, 145.6, 141.1, 138.4, 133.4, 132.6, 132.5, 127.8 (2C), 126.5 (2C), 123.5, 116.7, 115.9, 108.1, 67.5, 13.4, 11.1; MS (m/z) [M + H]+: calcd. for C20H19FN8O3S: 471.1353, and found: 471.1374.
M.p: 232 °C; yield: 82%; FTIR (cm−1, ν): 3260–3344 (N–H), 1602 (C
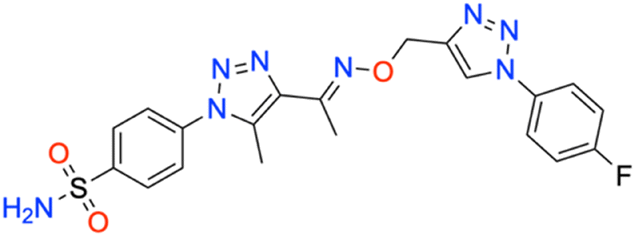 N), 1555 (CC), 1164 and 1335 (SO2), 1032 (C–F); 1H NMR (DMSO-d6, 300 MHz): 8.851 (1H, s, CH), 8.026–8.054 (2H, d, –Ar–H), 7.914–7.930 (2H, d, –Ar–H), 7.944–7.960 (2H, d, –Ar–H), 7.813–7.841 (2H, d, –Ar–H), 7.402 (2H, s, –NH2), 5.31 (2H, s, –CH2), 2.467 (3H, s, –CH3), 2.336 (3H, s, –CH3); 13C NMR (DMSO-d6, 75 MHz): 161.4, 151.3, 145.4, 141.1, 133.7 (2C), 133.4 (2C), 127.8 (2C), 126.4 (2C), 123.6 (2C), 123.1 (2C), 117.5, 67.4, 13.3, 11.1; MS (m/z) [M + H]+: calcd. for C20H19FN8O3S: 471.1358, and found: 471.1381.
N), 1555 (CC), 1164 and 1335 (SO2), 1032 (C–F); 1H NMR (DMSO-d6, 300 MHz): 8.851 (1H, s, CH), 8.026–8.054 (2H, d, –Ar–H), 7.914–7.930 (2H, d, –Ar–H), 7.944–7.960 (2H, d, –Ar–H), 7.813–7.841 (2H, d, –Ar–H), 7.402 (2H, s, –NH2), 5.31 (2H, s, –CH2), 2.467 (3H, s, –CH3), 2.336 (3H, s, –CH3); 13C NMR (DMSO-d6, 75 MHz): 161.4, 151.3, 145.4, 141.1, 133.7 (2C), 133.4 (2C), 127.8 (2C), 126.4 (2C), 123.6 (2C), 123.1 (2C), 117.5, 67.4, 13.3, 11.1; MS (m/z) [M + H]+: calcd. for C20H19FN8O3S: 471.1358, and found: 471.1381.
M.p: 265 °C; yield: 80%; FTIR (cm−1, ν): 3080–3291 (N–H), 2234 (C
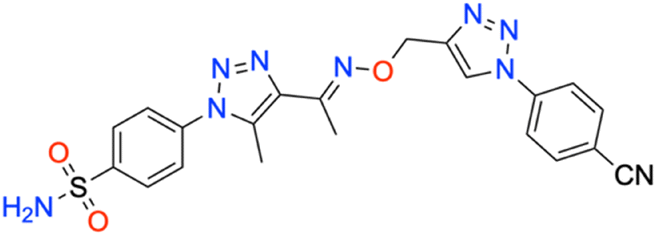
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), 1608 (CN), 1165 and 1345 (SO2); 1H NMR (DMSO-d6, 300 MHz): 8.997 (1H, s, CH), 8.083–8.121 (2H, d, –Ar–H), 8.042–8.053 (2H, d, –Ar–H), 7.998–8.019 (2H, d, –Ar–H), 7.804–7.832 (2H, d, –Ar–H), 7.587 (2H, s, –NH2), 5.325 (2H, s, –CH2), 2.486 (3H, s, –CH3), 2.334 (3H, s, –CH3); 13C NMR (DMSO-d6, 75 MHz): 151.5, 145.9, 145.6, 141.1, 140.1, 138.5, 134.9 (2C), 127.8 (2C), 126.5 (2C), 123.6 (2C), 121.2 (2C), 118.7, 111.7, 67.8, 13.4, 11.1; MS (m/z) [M + H]+: calcd. for C21H19N9O3S: 478.1404, and found: 478.1404.
N), 1608 (CN), 1165 and 1345 (SO2); 1H NMR (DMSO-d6, 300 MHz): 8.997 (1H, s, CH), 8.083–8.121 (2H, d, –Ar–H), 8.042–8.053 (2H, d, –Ar–H), 7.998–8.019 (2H, d, –Ar–H), 7.804–7.832 (2H, d, –Ar–H), 7.587 (2H, s, –NH2), 5.325 (2H, s, –CH2), 2.486 (3H, s, –CH3), 2.334 (3H, s, –CH3); 13C NMR (DMSO-d6, 75 MHz): 151.5, 145.9, 145.6, 141.1, 140.1, 138.5, 134.9 (2C), 127.8 (2C), 126.5 (2C), 123.6 (2C), 121.2 (2C), 118.7, 111.7, 67.8, 13.4, 11.1; MS (m/z) [M + H]+: calcd. for C21H19N9O3S: 478.1404, and found: 478.1404.
M.p: 105 °C; yield: 89%; FTIR (cm−1, ν): 3080–3291 (N–H), 1604 (C
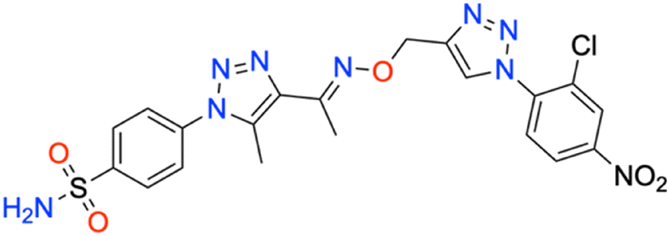 N), 1501 (CC), 1164 and 1341 (SO2), 1535 (C–NO2), 841 (C–Cl); 1H NMR (DMSO-d6, 300 MHz): 8.80 (1H, s, CH), 8.242–8.266 (2H, d, –Ar–H), 8.050–8.69 (2H, d, –Ar–H), 8.145 (1H, s, –Ar–H), 7.849–7.877 (2H, d, –Ar–H), 7.592 (2H, s, –NH2), 5.369 (2H, s, –CH2), 2.500 (3H, s, –CH3), 2.374 (3H, s, –CH3); 13C NMR (DMSO-d6, 75 MHz): 151.5, 145.7, 145.2, 143.3, 141.1, 139.1, 138.6, 133.5, 131.6 (2C), 130.8, 128.2, 127.9, 127.8 (2C), 126.5 (2C), 67.4, 13.5, 11.2; MS (m/z) [M + H]+: calcd. for C20H18ClN9O5S: 532.0911, and found: 532.0869.
N), 1501 (CC), 1164 and 1341 (SO2), 1535 (C–NO2), 841 (C–Cl); 1H NMR (DMSO-d6, 300 MHz): 8.80 (1H, s, CH), 8.242–8.266 (2H, d, –Ar–H), 8.050–8.69 (2H, d, –Ar–H), 8.145 (1H, s, –Ar–H), 7.849–7.877 (2H, d, –Ar–H), 7.592 (2H, s, –NH2), 5.369 (2H, s, –CH2), 2.500 (3H, s, –CH3), 2.374 (3H, s, –CH3); 13C NMR (DMSO-d6, 75 MHz): 151.5, 145.7, 145.2, 143.3, 141.1, 139.1, 138.6, 133.5, 131.6 (2C), 130.8, 128.2, 127.9, 127.8 (2C), 126.5 (2C), 67.4, 13.5, 11.2; MS (m/z) [M + H]+: calcd. for C20H18ClN9O5S: 532.0911, and found: 532.0869.
M.p: 257 °C; yield: 95%; FTIR (cm−1, ν): 3089–3193 (N–H), 1680 (C
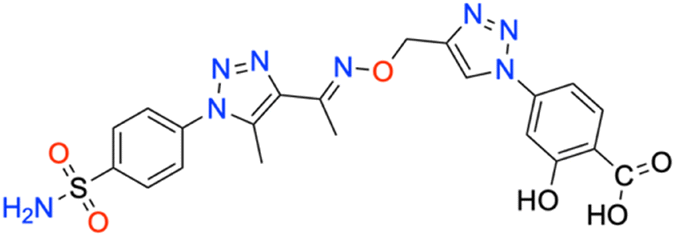 O) 1594 (CN), 1498 (CC), 1157 and 1330 (SO2); 1H NMR (DMSO-d6, 300 MHz): 11.3 (1H, s, –COOH), 10.06 (1H, s, –OH), 8.791 (1H, s, CH), 8.096–8.181 (2H, d, –Ar–H), 7.644–7.868 (2H, d, –Ar–H), 7.152–7.493 (3H, d, –Ar–H), 7.046 (2H, s, –NH2), 5.330 (2H, –CH2), 4.55 (3H, s, –CH3), 2.513 (3H, s, –CH3); 13C NMR (DMSO-d6, 75 MHz): 162.7, 151.5, 145.7, 144.5, 141.1, 139.2 (2C), 138.6 (2C), 128.2 (2C), 127.8 (2C), 126.5 (2C), 123.6 (2C), 121.1 (2C), 67.5, 13.5, 11.1; MS (m/z) [M + H]+: calcd. for C21H20N8O6S: 513.1297, and found: 513.1297.
O) 1594 (CN), 1498 (CC), 1157 and 1330 (SO2); 1H NMR (DMSO-d6, 300 MHz): 11.3 (1H, s, –COOH), 10.06 (1H, s, –OH), 8.791 (1H, s, CH), 8.096–8.181 (2H, d, –Ar–H), 7.644–7.868 (2H, d, –Ar–H), 7.152–7.493 (3H, d, –Ar–H), 7.046 (2H, s, –NH2), 5.330 (2H, –CH2), 4.55 (3H, s, –CH3), 2.513 (3H, s, –CH3); 13C NMR (DMSO-d6, 75 MHz): 162.7, 151.5, 145.7, 144.5, 141.1, 139.2 (2C), 138.6 (2C), 128.2 (2C), 127.8 (2C), 126.5 (2C), 123.6 (2C), 121.1 (2C), 67.5, 13.5, 11.1; MS (m/z) [M + H]+: calcd. for C21H20N8O6S: 513.1297, and found: 513.1297.
M.p: 257 °C; yield: 95%; FTIR (cm−1, ν): 3271–3370 (N–H), 1595 (C
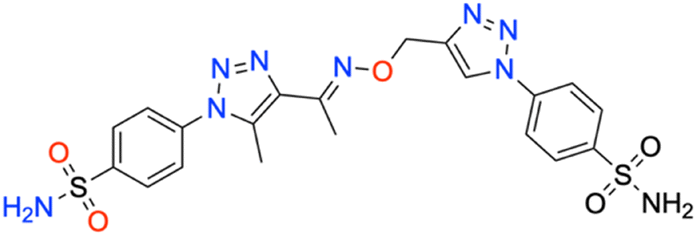 N), 1504 (CC), 1156 and 1325 (SO2); 1H NMR (DMSO-d6, 300 MHz): 8.996 (1H, s, CH), 8.16–8.133 (2H, d, –Ar–H), 8.037–8.066 (4H, d, –Ar–H), 7.842–7.869 (2 H, d, –Ar–H), 8.008 (4H, s, –NH2), 5.356 (2H, s, –CH2), 2.510 (3H, s, –CH3), 2.368 (3H, s, –CH3); 13C NMR (DMSO-d6, 75 MHz): 151.5, 145.7, 144.5, 141.1, 139.2, 138.6 (2C), 128.2 (2C), 127.8 (2C), 126.5 (2C), 123.6 (2C), 121.1 (2C), 67.5, 13.5, 11.1; MS (m/z) [M + H]+: calcd. for C20H21N9O5S2: 532.1176, and found: 532.1176.
N), 1504 (CC), 1156 and 1325 (SO2); 1H NMR (DMSO-d6, 300 MHz): 8.996 (1H, s, CH), 8.16–8.133 (2H, d, –Ar–H), 8.037–8.066 (4H, d, –Ar–H), 7.842–7.869 (2 H, d, –Ar–H), 8.008 (4H, s, –NH2), 5.356 (2H, s, –CH2), 2.510 (3H, s, –CH3), 2.368 (3H, s, –CH3); 13C NMR (DMSO-d6, 75 MHz): 151.5, 145.7, 144.5, 141.1, 139.2, 138.6 (2C), 128.2 (2C), 127.8 (2C), 126.5 (2C), 123.6 (2C), 121.1 (2C), 67.5, 13.5, 11.1; MS (m/z) [M + H]+: calcd. for C20H21N9O5S2: 532.1176, and found: 532.1176.
M.p: 277 °C; yield: 87%; FTIR (cm−1, ν): 2970 (O–H), 3290–3370 (N–H), 1679 (C
 O), 1608 (CN), 1506 (CC), 1156 and 1299 (SO2); 1H NMR (DMSO-d6, 300 MHz): 8.994 (1H, s, –COOH), 8.137 (1H, s, CH), 8.069–8.108 (4H, d, –Ar–H), 8.019–8.450 (2H, d, –Ar–H), 7.826–7.854 (2H, d, –Ar–H), 7.607 (2H, s, –NH2), 5.330 (2H, s, –CH2), 2.467 (3H, s, –CH3), 2.292 (3H, s, –CH3); 13C NMR (DMSO-d6, 75 MHz): 167.1, 151.5, 145.6, 140.1, 138.5, 133.5, 131.8 (2C), 131.2 (2C), 127.8 (2C), 126.5 (2C), 123.5 (2C), 120.5 (2C), 67.4, 13.4, 11.2; MS (m/z) [M + H]+: calcd. for C21H20N8O5S: 497.1350, and found: 497.1350.
O), 1608 (CN), 1506 (CC), 1156 and 1299 (SO2); 1H NMR (DMSO-d6, 300 MHz): 8.994 (1H, s, –COOH), 8.137 (1H, s, CH), 8.069–8.108 (4H, d, –Ar–H), 8.019–8.450 (2H, d, –Ar–H), 7.826–7.854 (2H, d, –Ar–H), 7.607 (2H, s, –NH2), 5.330 (2H, s, –CH2), 2.467 (3H, s, –CH3), 2.292 (3H, s, –CH3); 13C NMR (DMSO-d6, 75 MHz): 167.1, 151.5, 145.6, 140.1, 138.5, 133.5, 131.8 (2C), 131.2 (2C), 127.8 (2C), 126.5 (2C), 123.5 (2C), 120.5 (2C), 67.4, 13.4, 11.2; MS (m/z) [M + H]+: calcd. for C21H20N8O5S: 497.1350, and found: 497.1350.
M.p: 212 °C; yield: 87%; FTIR (cm−1, ν): 3241–3335 (N–H), 1739 (C
 O), 1677 (CN), 1603 (CC), 1159 and 1364 (SO2); 1H NMR (DMSO-d6, 300 MHz): 9.00 (1H, s, CH), 8.152–8.159 (2H, d, –Ar–H), 8.084–8.129 (2H, d, –Ar–H), 8.046–8.054 (2H, d, –Ar–H), 7.814–7.843 (2H, d, –Ar–H), 7.588 (2H, s, –NH2), 5.328 (2H, s, –CH2), 2.754 (3H, s, –CH3), 2.614 (3H, s, –CH3), 2.341 (3H, s, –CH3); 13C NMR (DMSO-d6, 75 MHz): 197.7, 151.5, 145.7, 145.6, 141.1, 140.2, 138.5, 137.1, 133.4 (2C), 130.7 (2C), 127.8 (2C), 126.5 (2C), 120.5 (2C), 67.4, 27.5, 13.4, 11.1; MS (m/z) [M + H]+: calcd. for C22H22N8O4S: 495.1557, and found: 495.1556.
O), 1677 (CN), 1603 (CC), 1159 and 1364 (SO2); 1H NMR (DMSO-d6, 300 MHz): 9.00 (1H, s, CH), 8.152–8.159 (2H, d, –Ar–H), 8.084–8.129 (2H, d, –Ar–H), 8.046–8.054 (2H, d, –Ar–H), 7.814–7.843 (2H, d, –Ar–H), 7.588 (2H, s, –NH2), 5.328 (2H, s, –CH2), 2.754 (3H, s, –CH3), 2.614 (3H, s, –CH3), 2.341 (3H, s, –CH3); 13C NMR (DMSO-d6, 75 MHz): 197.7, 151.5, 145.7, 145.6, 141.1, 140.2, 138.5, 137.1, 133.4 (2C), 130.7 (2C), 127.8 (2C), 126.5 (2C), 120.5 (2C), 67.4, 27.5, 13.4, 11.1; MS (m/z) [M + H]+: calcd. for C22H22N8O4S: 495.1557, and found: 495.1556.
2.7. Biological studies
2.8. Computational study
Determining the pharmacokinetic and physicochemical properties of designed compounds is crucial in developing new drug molecules. Therefore, to assess the drug-likeness of the targeted derivatives, measurement of in silico physicochemical properties, ADME predictions, and molecular docking analysis were conducted using the latest Schrödinger Small-Molecule Drug Discovery Suite 2024-2 iteration for Mac. The experimental models utilized X-ray crystallographic structures of hCA isoforms co-crystallized with native ligands. The following PDB IDs of hCAs (1AZM for I, 2.00 Å,723V2M for II, 1.47 Å,735FL4 for IX, 1.82 Å,74 and 1JD0 for XII, 1.50 Å (ref. 75)) were obtained from the RCSB Protein Data Bank. The Protein Preparation Wizard tool from the suite was utilized to prepare the enzyme structures for docking. The ChemDraw program for Mac (PerkinElmer) was used to sketch the structures of new 1,2,3-triazolyloxime substituted-1,2,3-triazolyl sulfonamide derivatives (7a–o). The LigPrep software76 of the same module was employed to optimize these sulfonamide derivatives (7a–o) at pH 7.4 ± 0.5 using the OPLS4 force field with Epik.77 Receptor grids were generated in the Maestro software by defining the active site residues defined by the SiteMap tool78 in the Receptor Grid Generation tool.79 Ligands were docked to hCAs using the extra precision method of the Glide application80,81 under default settings.82,83 Furthermore, the relative binding affinity in the VSGB energy model and OPLS force field was predicted using MM-GBSA84 for the hCA-complexes 1AZM, 3V2M, 3IAI, and 1JD0.2.9. Statistical study
The data analysis and visualization were conducted utilizing GraphPad Prism v10 software for Mac (La Jolla, CA, USA). The KI constants were ascertained using the SigmaPlot v12 module of Systat Software for Windows. To compare the fits of inhibition models, the extra sum-of-squares F test and the Akaike information criterion method were applied. The results are presented as mean ± SEM with 95% confidence intervals (p < 0.05).3. Results and discussion
3.1. Chemistry
In this paper, we successfully synthesized a series of 1,2,3-triazolyloxime substituted-1,2,3-triazolyl sulfonamide derivatives (7a–o) via a 1,3-dipolar cycloaddition reaction, commencing from benzenesulfonamide and completing the process in six steps (Scheme 1). In the first step of the reaction, 4-azidobenzenesulfanilamide was prepared starting from 4-aminobenzenesulfonamide in acidic medium at 0 °C followed by adding sodium azide to form azido derivatives of the benzenesulfonamide compound. Then, the azido compound is to diketone to synthesize the first triazole ring-containing compound. The oxime derivative of triazole containing sulfonamide was prepared under basic conditions with hydroxylamine hydrochloride in the fourth step. Then, the propargylated oxime derivative was synthesized using NaOH in DMSO and TBAB as a phase transfer catalyst. In the last step of the reaction, the targeted compounds were obtained by adding the prepared azido derivatives to the synthesized propargylated oxime derivative in DMF solvent with CuSO4 and ascorbic acid as catalysts. The synthesized compounds were characterized using FT-IR, 1H NMR, 13C NMR, and mass spectrometry, with detailed spectral data provided in the ESI.†Specifically, the 1H NMR spectra revealed resonances corresponding to the sulfanilamide NH2 and the CH proton on the benzenesulfonamide ring at approximately 7.60 ppm and between 7.80 and 8.00 ppm, respectively. Additionally, CH3 protons appeared around 2.1 and 2.5 ppm, while the methylene protons attached to the oxime–ether–triazole ring resonated at approximately 5.3 ppm. The triazole ring proton was observed at around 8.5–9.0 ppm. In the 13C NMR spectra, the carbon atom situated between oxygen and nitrogen resonated at approximately 67 ppm, and the carbon atom bonded to NOR via a double bond was observed around 150 ppm. Carbon atoms in the triazole ring are seen at around 130 and 140 ppm.
The FTIR spectra of compounds 7a–o exhibited absorption bands between 3210 and 3360 cm−1, corresponding to NH2 peaks. Consistent with literature reports,81 two distinct peaks associated with SO2 symmetric and asymmetric stretching were observed around 1350 and 1150 cm−1, respectively. Absorption peaks for CO, CN, and CC bonds appeared at approximately 1730, 1600, and 1520 cm−1, respectively. Aliphatic C–H absorption peaks were seen around 2990 cm−1. In compound 7j, the CN triple bond comes around 2234 cm−1. In compounds 7l and 7n, the COOH group has broad a peak between 2700 and 3500 cm−1. In compound 7k, N–O asymmetric and symmetric stretching were observed around 1535 and 1340 cm−1, respectively. These spectral analyses collectively confirm the structural integrity of the synthesized compounds.
3.2. Inhibitory effect of the targeted small structures
The newly synthesized target hCAIs, novel 1,2,3-triazolyloxime substituted-1,2,3-triazolyl sulfonamide derivatives 7a–o, underwent evaluation for their inhibitory activity on hCAs via Verpoorte's method, where it has been known that the zinc-based catalytic mechanism responsible for CO2 hydration also underlies the esterase activity of targeting four isoforms, hCA I, II, IX, and XII. The inhibitory potencies of these compounds were benchmarked against the standard drug AAZ. The structure–activity relationships derived from the inhibition data, as delineated in Tables 1 and 2 and Fig. 4, were subsequently determined.|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compounds | hCA I | hCA II | hCA IX | hCA XII | |||||
| ID | R | K I (nM) | R 2 | K I (nM) | R 2 | K I (nM) | R 2 | K I (nM) | R 2 |
| a The test results were expressed as means of triplicate assays ± SEM. b Acetazolamide. | |||||||||
| 7a | Phenyl | 47.7 ± 4.4 | 0.9875 | 45.8 ± 4.4 | 0.9882 | 465.6 ± 47.0 | 0.9890 | 149.9 ± 14.7 | 0.9871 |
| 7b | Benzyl | 73.5 ± 7.0 | 0.9877 | 42.7 ± 3.8 | 0.9887 | 302.5 ± 33.4 | 0.9873 | 349.4 ± 40.4 | 0.9875 |
| 7c | 2-CH3 phenyl | 82.8 ± 8.0 | 0.9870 | 93.2 ± 10.0 | 0.9873 | 383.2 ± 46.5 | 0.9884 | 309.0 ± 31.3 | 0.9872 |
| 7d | 4-OH phenyl | 47.1 ± 4.3 | 0.9878 | 35.9 ± 3.6 | 0.9874 | 434.7 ± 48.8 | 0.9867 | 416.1 ± 43.3 | 0.9881 |
| 7e | 4-I phenyl | 54.0 ± 5.1 | 0.9869 | 57.3 ± 5.2 | 0.9886 | 477.4 ± 58.2 | 0.9870 | 195.7 ± 22.5 | 0.9873 |
| 7f | 4-Br phenyl | 60.6 ± 5.4 | 0.9874 | 49.7 ± 4.7 | 0.9877 | 185.6 ± 18.8 | 0.9872 | 242.4 ± 28.7 | 0.9869 |
| 7g | 4-Cl phenyl | 85.1 ± 7.9 | 0.9876 | 83.5 ± 8.4 | 0.9870 | 306.4 ± 30.6 | 0.9886 | 336.4 ± 38.8 | 0.9863 |
| 7h | 2-F phenyl | 98.4 ± 9.2 | 0.9870 | 80.8 ± 7.7 | 0.9876 | 449.2 ± 49.2 | 0.9878 | 483.4 ± 55.2 | 0.9888 |
| 7i | 4-F phenyl | 118.4 ± 11.8 | 0.9871 | 103.6 ± 9.7 | 0.9883 | 552.3 ± 63.1 | 0.9882 | 385.6 ± 41.6 | 0.9866 |
| 7j | 4-CN phenyl | 184.2 ± 17.9 | 0.9888 | 164.6 ± 16.8 | 0.9877 | 459.4 ± 44.9 | 0.9892 | 360.9 ± 40.7 | 0.9871 |
| 7k | 2-Cl,4-NO2 phenyl | 109.0 ± 10.3 | 0.9879 | 137.0 ± 13.0 | 0.9883 | 260.8 ± 27.9 | 0.9875 | 330.9 ± 39.5 | 0.9874 |
| 7l | 3-OH,4-COOH phenyl | 196.7 ± 20.1 | 0.9874 | 198.3 ± 21.0 | 0.9882 | 689.2 ± 81.6 | 0.9880 | 503.5 ± 60.5 | 0.9863 |
| 7m | 4-SO2NH2 phenyl | 118.1 ± 11.1 | 0.9883 | 109.9 ± 10.7 | 0.9876 | 429.3 ± 47.3 | 0.9879 | 222.8 ± 22.6 | 0.9887 |
| 7n | 4-COOH phenyl | 208.1 ± 21.5 | 0.9876 | 202.8 ± 21.6 | 0.9872 | 359.6 ± 44.9 | 0.9873 | 509.9 ± 55.4 | 0.9864 |
| 7o | 4-Acetylphenyl | 181.2 ± 16.5 | 0.9888 | 240.8 ± 24.6 | 0.9871 | 170.0 ± 16.8 | 0.9871 | 426.9 ± 38.1 | 0.9896 |
| AAZb | — | 459.5 ± 23.5 | 0.9893 | 330.1 ± 21.2 | 0.9876 | 441.8 ± 23.3 | 0.9895 | 339.7 ± 21.2 | 0.9877 |
| Compounds ID | S I | |||
|---|---|---|---|---|
| hCA IX/I | hCA IX/II | hCA XII/I | hCA XII/II | |
| a Selectivity index. b Acetazolamide. | ||||
| 7a | 9.8 | 10.2 | 3.1 | 3.3 |
| 7b | 4.1 | 7.1 | 4.8 | 8.2 |
| 7c | 4.6 | 4.1 | 3.7 | 3.3 |
| 7d | 9.2 | 12.1 | 8.8 | 11.6 |
| 7e | 8.8 | 8.3 | 3.6 | 3.4 |
| 7f | 3.1 | 3.7 | 4.0 | 4.9 |
| 7g | 3.6 | 3.7 | 4.0 | 4.0 |
| 7h | 4.6 | 5.6 | 4.9 | 6.0 |
| 7i | 4.7 | 5.3 | 3.3 | 3.7 |
| 7j | 2.5 | 2.8 | 2.0 | 2.2 |
| 7k | 2.4 | 1.9 | 3.0 | 2.4 |
| 7l | 3.5 | 3.5 | 2.6 | 2.5 |
| 7m | 3.6 | 3.9 | 1.9 | 2.0 |
| 7n | 1.7 | 1.8 | 2.5 | 2.5 |
| 7o | 0.9 | 0.7 | 2.4 | 1.8 |
| AAZb | 1.0 | 1.3 | 0.7 | 1.0 |
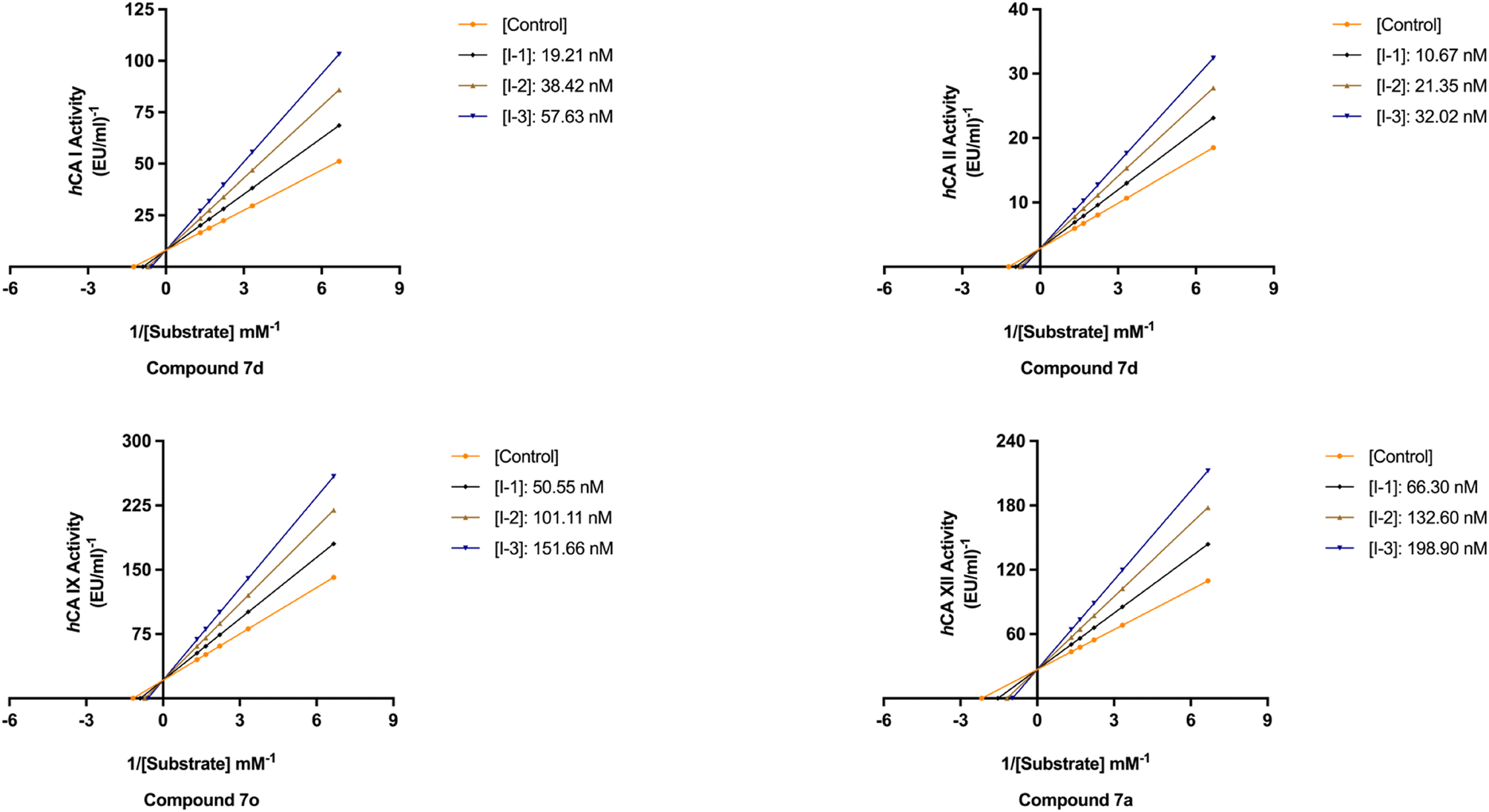 | ||
| Fig. 4 In vitro inhibition effects of the most potent inhibitors, 7d against hCA I (top left panel) and hCA II (top right panel), 7o against hCA IX (bottom left panel), and 7a against hCA XII (bottom right panel). | ||
All the compounds profiled for the hCA I isoform showed quite varied KI constants, and all were higher when compared with that of 7d (KI of 47.1 ± 4.3 nM) and were found to be better inhibitors in comparison to the reference drug AAZ (KI of 459.5 ± 23.5 nM) (Table 1). Specifically, the introduction within the phenyl moiety of 7a (KI of 47.7 ± 4.4 nM) of some functional groups, such as –4F in 7i, –4CN in 7j, 2-Cl,4-NO2 in 7k, 3-OH,4-COOH in 7l, 4-SO2NH2 in 7m, 4-COOH in 7n, and 4-acetyl in 7o, determined a sensible rise of the three-digit KI constants down to the high nanomolar range (KIs of 118.4 ± 11.8 nM, 184.2 ± 17.9 nM, 109.0 ± 10.3 nM, 196.7 ± 20.1 nM, 118.1 ± 11.1 nM, 208.1 ± 21.5 nM, and 181.2 ± 16.5 nM, respectively). In contrast, the –I, –Br, –Cl, and –F halogens at the 2nd or 4th position were slightly beneficial for the inhibition potency when compared with compound 7a (KIs of 54.0 ± 5.1 nM, 60.6 ± 5.4 nM, 85.1 ± 7.9 nM, and 98.4 ± 9.2 nM for 7e, 7f, 7g, and 7h, respectively). The bulky benzyl moiety in the para-position, as in compound 7b, lowly increased the KI constant to 73.5 ± 7.0 nM, thus making it comparable with the 2-methylphenyl-substituted derivative 7c (KI of 82.8 ± 8.0 nM). Interestingly, replacing the fluorine group from the 2nd position, as in compound 7h (KI of 98.4 ± 9.2 nM), to the 4th position (7i, KI of 118.4 ± 11.8 nM) reduced the inhibitory effect.
As for hCA II, substituting the phenyl in compound 7a with a hydroxy group to afford compound 7d reduced the KI value (KIs of 45.8 ± 4.4 nM and 35.9 ± 3.6 nM for 7a and 7d, respectively) as compared to the clinically used drug, AAZ (KI of 330.1 ± 21.2 nM). The iodo (7e), bromo (7f), chloro (7g), and fluoro (7h and 7i) phenyl derivatives were less effective than the unsubstituted phenyl 7a (Table 1). As expected, the bulky para-benzyl derivative 7b was a potent nanomolar inhibitor with a KI of 42.7 ± 3.8 nM. Similarly, as in hCA I, derivatives 7i, 7j, 7k, 7l, 7m, 7n, and 7o exhibited a noticeable increase towards the high nanomolar range with three-digit KI constants (KIs of 103.6 ± 9.7 nM, 164.6 ± 16.8 nM, 137.0 ± 13.0 nM, 198.3 ± 21.0 nM, 109.9 ± 10.7 nM, 202.8 ± 21.6 nM, and 240.8 ± 24.6 nM, respectively). Again, the switch of the fluorine from the ortho- to para-position (i.e., from compounds 7h to 7i) allowed the reduction of the compound's hCA II inhibition potency from 80.8 ± 7.7 nM to 103.6 ± 9.7 nM. The kinetic trend of 3-hydroxy-4-carboxyphenyl derivative 7l (KI of 198.3 ± 21.0 nM) towards hCA II was partially similar to that of hCA I (KI of 196.7 ± 20.1 nM). It was even more effective than its 4-carboxyphenyl substituted counterpart, compound 7n, against both hCA I and II isoforms (KIs of 208.1 ± 21.5 nM and 202.8 ± 21.6 nM, respectively).
Compared with the cytosolic isoforms I and II, compounds 7a–o showed a flatter kinetic profile for hCA IX. For instance, the hydroxy 7d, the fluoro 7h, the cyano 7j, and the sulfamoyl derivative 7m were found to be equipotent hCA IX inhibitors (i.e., KIs of 434.7 ± 48.8 nM, 449.2 ± 49.2 nM, 459.4 ± 44.9 nM, and 429.3 ± 47.3 nM, respectively). Similarly, the 4-acetylphenyl (7o) and the 4-bromophenyl (7f) derivatives were both potent nanomolar inhibitors (KIs of 170.0 ± 16.8 nM and 185.6 ± 18.8 nM, respectively) in comparison to reference drug AAZ (KI of 441.8 ± 23.3 nM). Quite interestingly, the introduction of bromo (i.e., 7f) instead of the iodo, chloro, and fluoro atoms in compounds 7e, 7g, and 7h or 7i strongly increased the KI constant (KIs of 477.4 ± 58.2 nM, 306.4 ± 30.6 nM, 449.2 ± 49.2, and 552.3 ± 63.1 nM, respectively). Also, the 2-chloro-4-nitrophenyl derivative 7k effectively inhibited hCA IX (KI of 260.8 ± 27.9 nM). The halogen effect for compounds 7e and 7h was almost undetectable (KIs of 477.4 ± 58.2 nM and 449.2 ± 49.2 nM, respectively). In contrast, the kinetics was slightly affected by the position of fluorine since the ortho-fluoro 7h was more effective than its para-substituted counterpart 7i (KIs of 449.2 ± 49.2 nM and 552.3 ± 63.1 nM, respectively). Finally, the 3-hydroxy-4-carboxyphenyl derivative 7l, the weakest inhibitor within the compound series, elicited a KI of 689.2 ± 81.6 nM against hCA IX (Table 1).
As for the other tumor-associated isoform XII, the iodo-containing derivative 7e was a slightly weaker inhibitor than its phenyl progenitor counterpart 7a (KIs of 149.9 ± 14.7 nM and 195.7 ± 22.5 nM for 7a and 7e, respectively), but both were potent inhibitors compared to AAZ (KI of 339.7 ± 21.2 nM). Interestingly, replacing the phenyl moiety in compound 7a with the benzyl ring, as in compound 7b, increased the KI constant by 2.3-fold (KIs of 149.9 ± 14.7 nM and 349.4 ± 40.4 nM, respectively). Moreover, such constants were almost superimposable to the bromo derivative 7f (KI of 242.4 ± 28.7 nM). As for the remaining compounds, higher KI constants were obtained. The kinetic trend of chloro derivatives 7g and 7k (KIs of 336.4 ± 38.8 and 330.9 ± 39.5 nM, respectively) against hCA XII was partially similar. Significant reduction of the inhibition potency was observed for the 4-hydroxyphenyl derivative 7d (KI of 416.1 ± 43.3 nM), for the 2-fluorophenyl derivative 7h (KI of 483.4 ± 55.2 nM), for the 4-fluorophenyl derivative 7i (KI of 385.6 ± 41.6 nM), and for the 4-cyanophenyl derivative 7j (KI of 360.9 ± 40.7 nM). Interestingly, 7d (KI of 416.1 ± 43.3 nM) showed an inhibitory profile against hCA XII almost similar to that of the acetylphenyl derivative 7o (KI of 426.9 ± 38.1 nM). Among the compounds, carboxyphenyl derivatives 7l and 7n were the least effective in inhibiting hCA XII (KIs of 503.5 ± 60.5 nM and 509.9 ± 55.4, respectively).
The selectivity indexes (SIs) of 1,2,3-triazolyloxime substituted-1,2,3-triazolyl sulfonamide derivatives 7a–o for the ubiquitous and physiologically relevant hCA I and II isoforms, relative to the tumor-associated isoforms hCA IX and XII, are presented in Table 2. Since the experimental inhibitory activities in Table 2 accounted for compound 7d (SIs of 9.2, 12.1, 8.8, and 11.6 for hCA IX/I, IX/II, XII/I, and XII/II, respectively) being a highly selective inhibitor for the physiological isoforms hCA I and II, we consider investigating its binding modes on such enzymes (i.e., both I/II and IX/XII) by molecular docking simulations.
3.3. Cytotoxicity of the targeted small molecules
The upregulation of hCA IX and XII isoforms has been extensively documented across various tumor types. Given the significant association between hCAs and cancer, these enzymes present compelling targets for cancer prevention and therapy. To elucidate the molecular mechanisms of novel 1,2,3-triazolyloxime substituted-1,2,3-triazolyl sulfonamide derivatives (7a–o) and confirm their interaction with hCAs, the synthesized compounds were subjected to in vitro anti-proliferative activity screening against distinct cell lines: Hep3B and A549 cells. Furthermore, a normal cell line (mouse fibroblast, L929) was also included for comparative analysis. The results expressed as IC50s were benchmarked with a well-established anticancer agent, 5-fluorouracil (5-FU). Compounds 7m–o exhibited notable anti-proliferative activities against both cancer cell lines, with IC50 values ranging from 5.5 ± 1.2 μM to 6.9 ± 0.9 μM (for Hep3B) and 5.9 ± 0.8 μM to 9.1 ± 0.3 μM (for A549). Notably, compound 7m (IC50s of 5.5 ± 1.2 μM versus Hep3B and 6.0 ± 0.4 μM versus A549), featuring a sulfamoyl substituent on the para position of the phenyl group, demonstrated intriguing anti-proliferative effects, affirming the essential role of the sulfamoyl moiety in conferring biological activity, particularly anticancer impact, as per established postulates. Moreover, compounds 7f (IC50 of 2.9 ± 0.6 μM) and 7m (IC50 of 8.2 ± 3.4 μM) displayed minimal cytotoxicity towards the L929 cell line, as illustrated in Table 3.| Compounds ID | IC50a (μM) | S I | |||
|---|---|---|---|---|---|
| L929b | Hep3Bc | A549d | Hep3B/A549 | A549/Hep3B | |
| a IC50 values are the concentration of novel 1,2,3-triazolyloxime substituted-1,2,3-triazolyl sulfonamides (7a–o) that reduces the cell viability to 50%, measured at 24 hours. The test results were expressed as the mean of triplicate assays. b Mouse fibroblast cell line. c Human hepatocellular carcinoma cell line. d Human lung carcinoma cell line. e Selectivity index. f 5-Fluorouracil. g Not determined. | |||||
| 7a | 241.4 ± 0.2 | NDg | NDg | NDg | NDg |
| 7b | 32.3 ± 0.2 | 54.6 ± 9.9 | 94.2 ± 0.1 | 0.6 | 1.7 |
| 7c | 31.1 ± 0.1 | 22.4 ± 0.1 | 38.8 ± 0.4 | 0.6 | 1.7 |
| 7d | NDg | 132.7 ± 0.1 | 21.1 ± 0.3 | 6.3 | 0.2 |
| 7e | 48.8 ± 0.1 | 72.9 ± 0.1 | 29.4 ± 0.1 | 2.5 | 0.4 |
| 7f | 2.9 ± 0.6 | 47.1 ± 0.2 | 14.2 ± 0.1 | 3.3 | 0.3 |
| 7g | 28.0 ± 0.2 | 140.5 ± 0.7 | 9.1 ± 0.2 | 15.4 | 0.1 |
| 7h | 53.9 ± 0.1 | 35.9 ± 0.1 | 11.5 ± 0.1 | 3.1 | 0.3 |
| 7i | 62.9 ± 0.1 | 53.4 ± 0.1 | 13.6 ± 0.1 | 3.9 | 0.3 |
| 7j | 349.3 ± 5.5 | 14.5 ± 0.1 | 31.2 ± 0.1 | 0.5 | 2.2 |
| 7k | NDg | 117.4 ± 0.1 | 17.5 ± 0.3 | 6.7 | 0.1 |
| 7l | 99.0 ± 4.1 | 7.4 ± 0.2 | 10.8 ± 0.1 | 0.7 | 1.5 |
| 7m | 8.2 ± 3.4 | 5.5 ± 1.2 | 6.0 ± 0.4 | 0.9 | 1.1 |
| 7n | NDg | 6.9 ± 0.9 | 5.9 ± 0.8 | 1.2 | 0.9 |
| 7o | 48.7 ± 0.1 | 5.7 ± 1.0 | 9.1 ± 0.3 | 0.6 | 1.6 |
| 5-FUf | 14.0 ± 0.1 | 60.6 ± 0.1 | 100.3 ± 0.1 | — | — |
The tested compounds were more effective toward A549 cells than Hep3B cells, and compounds 7d, 7g, and 7k, in particular, revealed high selectivity with values of 6.3, 15.4, and 6.7, respectively. Compared to the anticancer drug 5-FU (IC50 of 14.0 ± 0.1 μM), known for its elevated toxicity on cancer and normal cell lines, all the compounds (except for 7f and 7m), particularly compounds 7a (17.2-fold with IC50 of 241.4 ± 0.2 μM) and 7j (25-fold with IC50 of 349.3 ± 5.5 μM), demonstrated lower cytotoxicity against L929 mouse fibroblasts (Table 3). Collectively, these findings position novel synthesized 1,2,3-triazolyloxime substituted-1,2,3-triazolyl sulfonamide derivatives (7a–o) as promising hCAIs and anticancer agents, underscored by their potent anti-proliferative effects and favorable selectivity profiles. Due to these promising characteristics, compounds 7a–o have been prioritized for further investigation to gain deeper insights into their mechanism of action.
3.4. Computational analysis of the targeted small compounds
Structural analysis reveals that, despite differences in subcellular localization, hCA isoforms display a remarkable degree of architectural similarity, a reflection of their high sequence homology.85 A hallmark of this structural conservation is the presence of a central twisted β-sheet flanked by helices and additional β-strands.86 The active site, housed within a large, conical cavity approximately 12 Å wide and 13 Å deep, extends from the protein's surface to its core, where a catalytic zinc ion is positioned at the base.87 This Zn2+ ion engages in tetrahedral coordination with three conserved histidine residues and a water molecule or hydroxide ion, which acts as a ligand.88 The Zn2+-bound water molecule/hydroxide ion is further stabilized by a complex hydrogen-bond network, notably involving a conserved threonine residue (Thr199) and two water molecules.89 One of these water molecules, referred to as the deep water, resides within a hydrophobic pocket defined by conserved residues at positions 121, 143, 198, and 209, while the other occupies a more hydrophilic region near the entrance of the active site.90 Across all hCA isoforms, the active site is compartmentalized into two distinct regions: a hydrophobic domain and a hydrophilic domain, each contributing uniquely to enzyme functionality.91Herein, the in vitro carbonic anhydrase KI results indicated that compounds 7d (for hCA I and hCA II), 7o (for hCA IX), and 7a (for hCA XII) were the most effective hCAIs among these series compared to the reference AAZ. The self-docking validation of the employed in silico docking protocol confirmed its suitability, as evidenced by the low RMSD values (<1.0 Å) between the native co-crystallized ligands, and the re-docked poses in hCAs. Furthermore, the native ligand's ability to reproduce all significant interactions with the active site hot spots in hCAs further validated the protocol's appropriateness.
As demonstrated in Fig. 5–8, the interaction of the synthesized agents with the active sites of hCA isoforms revealed a consistent accommodation of the sulfonamide moiety, which was deeply embedded and interacted with the zinc ion, as well as forming hydrogen bonds with key residues such as Thr199 and Thr200. Furthermore, the triazole moiety engaged in hydrogen bonding with specific residues like Phe91 and Tyr204 in hCA I, while in hCA II, it formed hydrogen bonds and pi–pi stacking interactions with Gln92 and Phe131. In the case of hCA IX, a hydrogen bond was formed with Gln92. Additionally, the phenyl moieties exhibited hydrophobic interactions with Trp5 and His94 in the hCA XII isoform. The newly synthesized compounds displayed a diverse range of binding affinities and selectivities across different hCA isoforms, attributed to distinct structural modifications that imparted varied steric and electronic properties. This study provides valuable insights into the binding mechanisms of 1,2,3-triazolyloxime substituted-1,2,3-triazolyl sulfonamides (7a–o) within hCAs, highlighting their potential as selective therapeutic agents for conditions associated with carbonic anhydrase dysfunction.
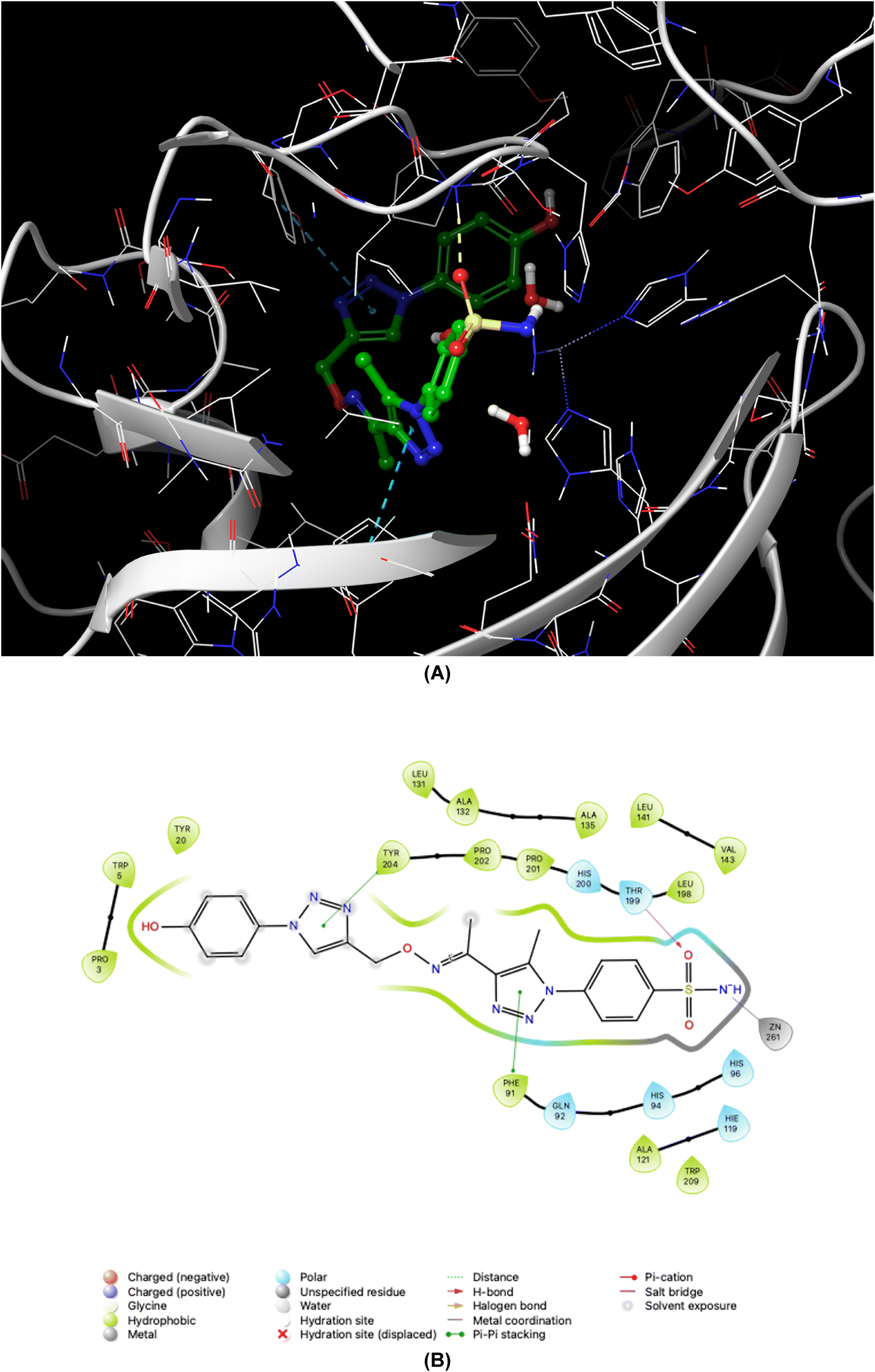 | ||
| Fig. 5 Binding orientations resulting from docking of the carbonic anhydrase I isoform (PDB ID 1AZM) with 4-{4-[1-{[(1-(4-hydroxyphenyl)-1H-1,2,3-triazol-4-yl]methoxy}imino)ethyl]-5-methyl-1H-1,2,3-triazol-1-yl}benzenesulfonamide (7d). (A) and (B) 3D and 2D interaction diagrams of 1AZM with compound 7d, respectively. | ||
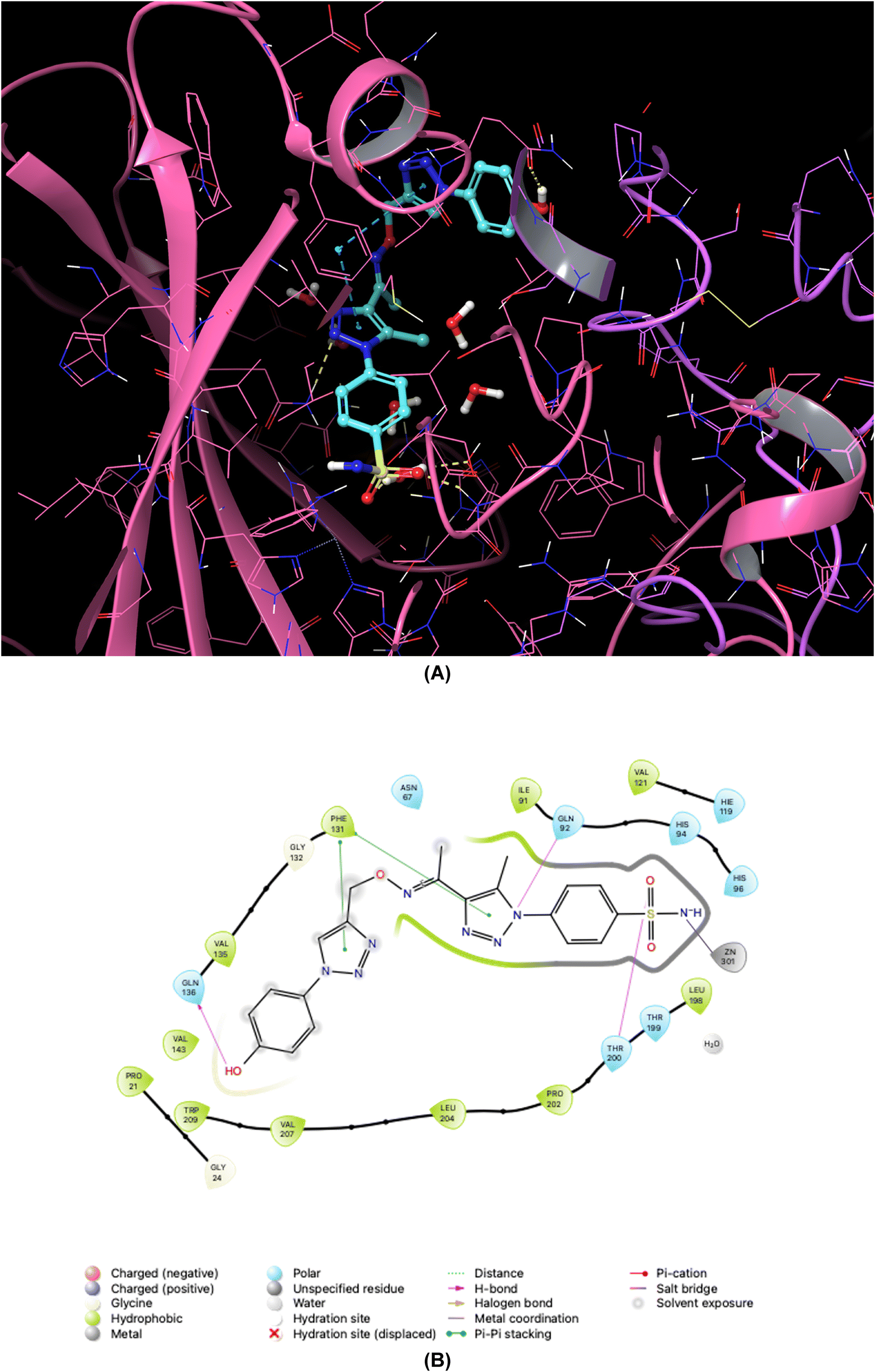 | ||
| Fig. 6 Binding orientations resulting from docking of the carbonic anhydrase II isoform (PDB ID 3V2M) with 4-{4-[1-{[(1-(4-hydroxyphenyl)-1H-1,2,3-triazol-4-yl]methoxy}imino)ethyl]-5-methyl-1H-1,2,3-triazol-1-yl}benzenesulfonamide (7d). (A) and (B) 3D and 2D interaction diagrams of 3V2M with compound 7d, respectively. | ||
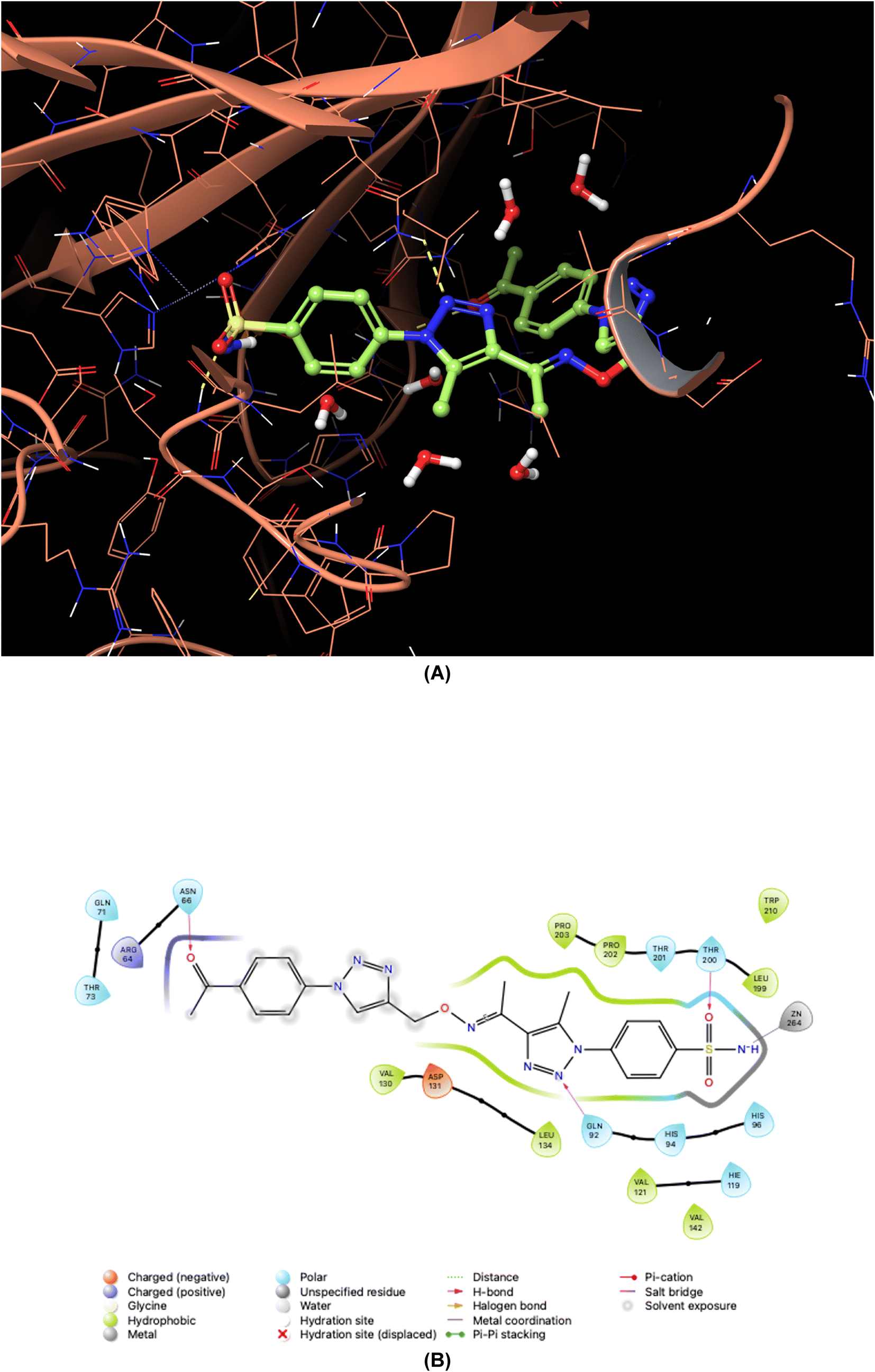 | ||
| Fig. 7 Binding orientations resulting from docking of the carbonic anhydrase IX isoform (PDB ID 5FL4) with 4-{4-[1-({[1-(4-acetylphenyl)-1H-1,2,3-triazol-4-yl]methoxy}imino)ethyl]-5-methyl-1H-1,2,3-triazol-1-yl}benzenesulfonamide (7o). (A) and (B) 3D and 2D interaction diagrams of 5FL4 with compound 7o, respectively. | ||
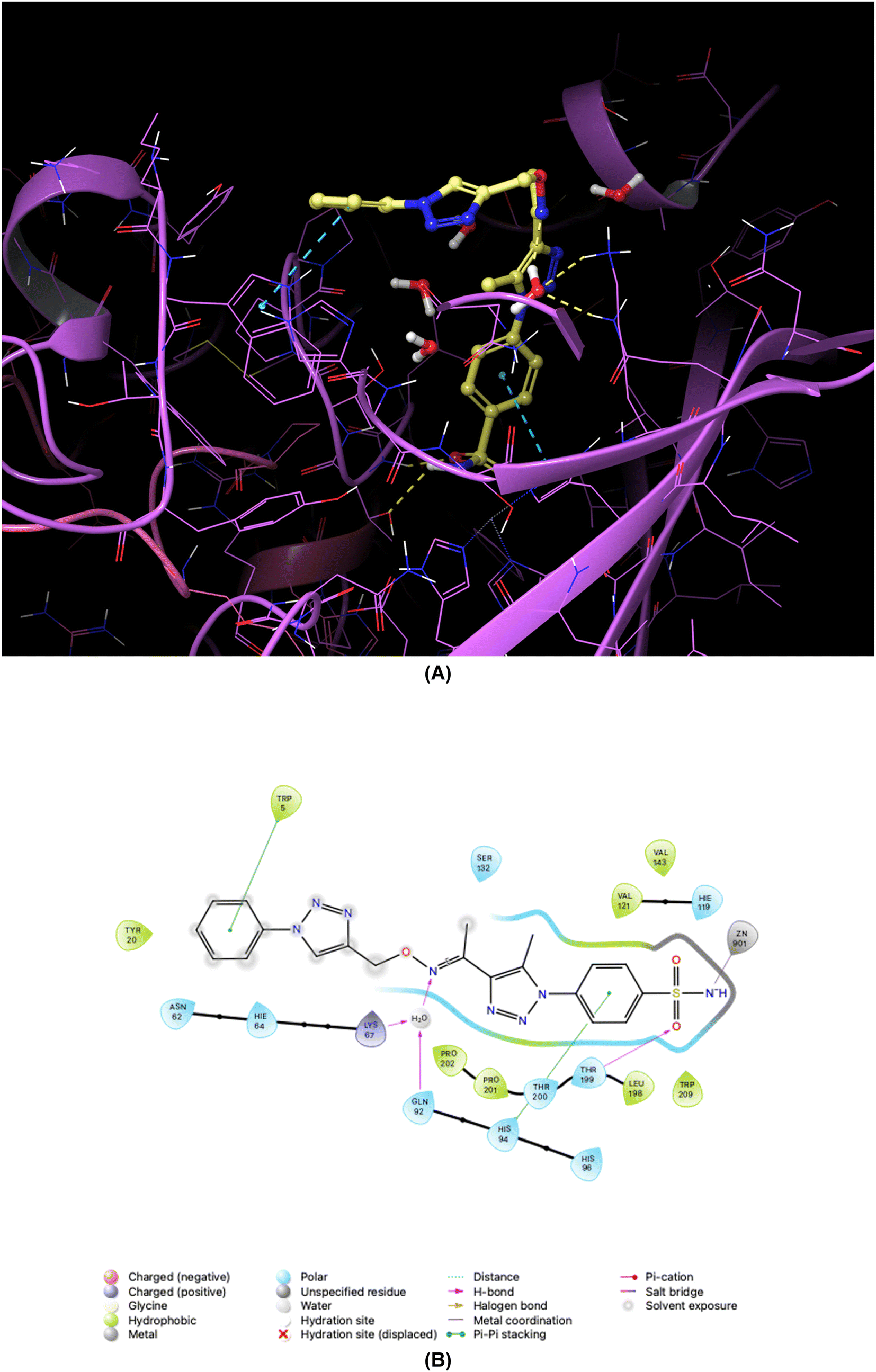 | ||
| Fig. 8 Binding orientations resulting from docking of the carbonic anhydrase XII isoform (PDB ID 1JD0) with 4-[5-methyl-4-(1-{[(4-phenyl-1H-1,2,3-triazol-4-yl)methoxy]imino}ethyl)-1H-1,2,3-triazol-1-yl]benzenesulfonamide (7a). (A) and (B) 3D and 2D interaction diagrams of 1JD0 with compound 7a, respectively. | ||
The 3D structures of the synthesized compounds (70a–o), which were optimized using the LigPrep tool, were subjected to ADME analysis through both the QikProp module of the Schrödinger Suite and the SwissADME platform.92,93 The computed physicochemical and pharmacokinetic descriptors, as outlined in Tables S1–S4,† reveal that all the target compounds conform to well-established drug-likeness frameworks, including Lipinski's Rule of Five (Pfizer),94 Jorgensen's parameters (Melinta),95 Egan's criteria (Pharmacia),96 Ghose's filter (Amgen),97 Muegge's algorithm (Bayer),98 and Veber's rules (GSK).99 Furthermore, favorable bioavailability scores (Abbot)100 and the absence of PAINS (pan assay interference compounds)101 confirm their potential as viable therapeutic agents. Of particular note, compounds 7d, 7o, and 7a, each exhibiting significant inhibitory activity, displayed exceptional drug-likeness and pharmacokinetic profiles, surpassing the performance of the standard reference inhibitor AAZ (Fig. 9 and S1†).
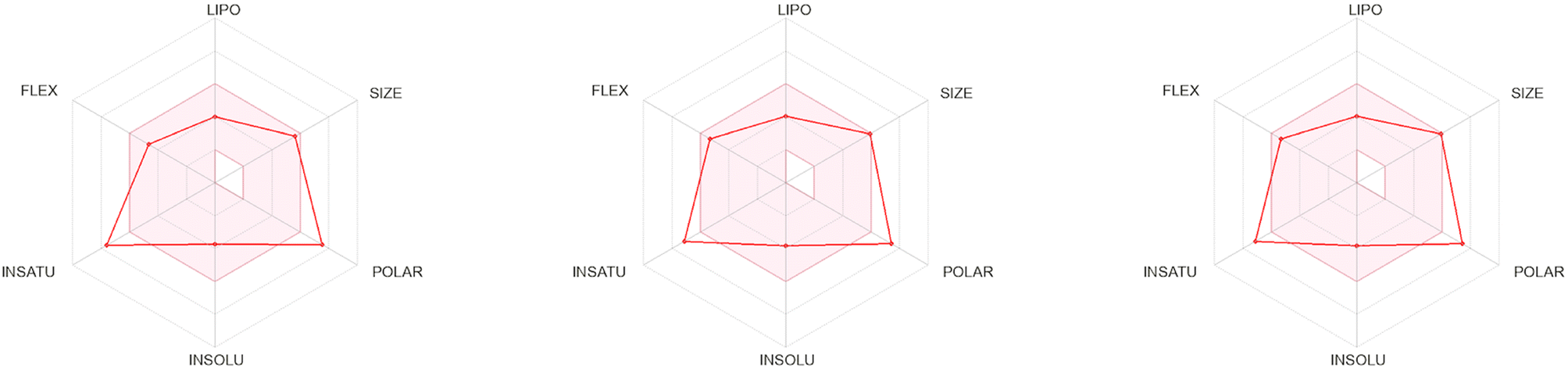 | ||
| Fig. 9 Diagrams showing ‘drug-likeness’ descriptors for this series' most potent inhibitors, 7d against hCA I and hCA II (left), 7o against hCA IX (middle), and 7a against hCA XII (right). The red-colored zone has been delineated as an optimal physicochemical region for improving oral bioavailability, encompassing parameters such as lipophilicity (LIPO), molecular weight (SIZE), insolubility (INSOLU), saturation (INSATU), and molecular flexibility (FLEX). | ||
4. Conclusion
In conclusion, a novel series of sulfonamide derivatives (7a–o) incorporating a 1,2,3-triazolyloxime substituted 1,2,3-triazolyl framework was synthesized and evaluated for their inhibitory potential against isoforms hCA I, II, IX, and XII. The inhibitory activities of these synthesized compounds were tested against the cytosolic hCA isoforms I (KIs of 47.1–208.1 nM) and II (KIs of 35.9–240.8 nM), as well as the transmembrane tumor-associated isoforms IX (KIs ranging between 170.0 nM and 689.2 nM) and XII (KIs ranging from 222.8 nM to 509.9 nM). The majority of the compounds demonstrated significant inhibitory effects on all isoforms compared to the well-known hCAI AAZ (KI of 459.5 nM, 330.1 nM, 441.8 nM, and 339.7 nM against hCA isoforms, I, II, IX, and XII, respectively). Among them, compounds 7d (for hCA I and II), 7o (for hCA IX), and 7a (for hCA XII) emerged as the most potent inhibitors. Further, an in vitro assessment of the cytotoxic properties of these potent derivatives was conducted on various cell lines, including cancer cell lines Hep3B (IC50s of 5.5–140.5 μM) and A549 (IC50s of 5.9–94.2 μM), as well as normal cell line L929 (IC50s of 2.9–349.3 μM). Notably, these compounds showed the highest selectivity towards Hep3B cells, followed by A549 cells. Molecular docking studies within the active sites of hCA isoforms I (PDB ID 1AZM), II (PDB ID 3V2M), IX (PDB ID 5FL4), and XII (PDB ID 1JD0) indicated that these compounds engage in crucial interactions essential for receptor binding, corroborating their potent and selective inhibition effects. The inhibition profiles and structure–activity relationship insights detailed in this study underscore the promising potential of the reported derivatives. These compounds, incorporating both sulfanilamide and triazole moieties, demonstrate enhanced potency and selectivity. These findings suggest that the strategic modification of such scaffolds can inform the rational design of next-generation inhibitors, potentially yielding pharmacologically relevant candidates for further therapeutic development.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
A. B.: formal analysis and investigation. C. T.: conceptualization, formal analysis, investigation, methodology, validation, and writing. M. A.: conceptualization, formal analysis, investigation, methodology, validation, and writing. Y. D.: formal analysis and investigation. B. D.: formal analysis and investigation. A. R. N.: conceptualization and methodology. Ş. B.: conceptualization, funding acquisition, and methodology.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the Research Fund of Anadolu University (grant number 2102S003).References
- L. da S. M. Forezi, C. G. Lima, A. A. Amaral, P. G. Ferreira, M. C. B. de Souza, A. C. Cunha, F. de C. da Silva and V. F. Ferreira, Bioactive 1, 2, 3-Triazoles: An Account on their Synthesis, Structural Diversity and Biological Applications, Chem. Rec., 2021, 21, 2782–2807, DOI:10.1002/tcr.202000185.
- X. Jiang, X. Hao, L. Jing, G. Wu, D. Kang, X. Liu and P. Zhan, Recent applications of click chemistry in drug discovery, Expert Opin. Drug Discovery, 2019, 14, 779–789, DOI:10.1080/17460441.2019.1614910.
- J. Totobenazara and A. J. Burke, New click-chemistry methods for 1,2,3-triazoles synthesis: recent advances and applications, Tetrahedron Lett., 2015, 56, 2853–2859, DOI:10.1016/j.tetlet.2015.03.136.
- J. John, J. Thomas and W. Dehaen, Organocatalytic routes toward substituted 1,2,3-triazoles, Chem. Commun., 2015, 51, 10797–10806, 10.1039/C5CC02319J.
- S. Kumar, B. Sharma, V. Mehra and V. Kumar, Recent accomplishments on the synthetic/biological facets of pharmacologically active 1H-1,2,3-triazoles, Eur. J. Med. Chem., 2021, 212, 113069, DOI:10.1016/j.ejmech.2020.113069.
- M.-X. Pu, H.-Y. Guo, Z.-S. Quan, X. Li and Q.-K. Shen, Application of the Mannich reaction in the structural modification of natural products, J. Enzyme Inhib. Med. Chem., 2023, 38, 2235095, DOI:10.1080/14756366.2023.2235095.
- A. Kumar, K. Siwach, T. Rom, R. Kumar, A. Angeli, A. Kumar Paul, C. T. Supuran and P. K. Sharma, Tail-approach based design and synthesis of Arylthiazolylhydrazono-1,2,3-triazoles incorporating sulfanilamide and metanilamide as human carbonic anhydrase I, II, IV and IX inhibitors, Bioorg. Chem., 2022, 123, 105764, DOI:10.1016/j.bioorg.2022.105764.
- O. Gaidai, P. Yan and Y. Xing, Future world cancer death rate prediction, Sci. Rep., 2023, 13, 303, DOI:10.1038/s41598-023-27547-x.
- U. Anand, A. Dey, A. K. S. Chandel, R. Sanyal, A. Mishra, D. K. Pandey, V. De Falco, A. Upadhyay, R. Kandimalla, A. Chaudhary, J. K. Dhanjal, S. Dewanjee, J. Vallamkondu and J. M. Pérez de la Lastra, Cancer chemotherapy and beyond: Current status, drug candidates, associated risks and progress in targeted therapeutics, Genes Dis., 2023, 10, 1367–1401, DOI:10.1016/j.gendis.2022.02.007.
- M. A. Khan, Z. A. Khan, F. Shoeb, G. Fatima, R. H. Khan and M. M. Khan, Role of de novo lipogenesis in inflammation and insulin resistance in Alzheimer's disease, Int. J. Biol. Macromol., 2023, 242, 124859, DOI:10.1016/j.ijbiomac.2023.124859.
- P. Swietach, What is pH regulation, and why do cancer cells need it?, Cancer Metastasis Rev., 2019, 38, 5–15, DOI:10.1007/s10555-018-09778-x.
- C. T. Supuran, Novel Carbonic Anhydrase Inhibitors, Future Med. Chem., 2021, 13, 1935–1937, DOI:10.4155/fmc-2021-0222.
- C. T. Supuran, Experimental Carbonic Anhydrase Inhibitors for the Treatment of Hypoxic Tumors, J. Exp. Pharmacol., 2020, 12, 603–617, DOI:10.2147/JEP.S265620.
- C. T. Supuran, Carbonic anhydrase inhibitors as emerging agents for the treatment and imaging of hypoxic tumors, Expert Opin. Invest. Drugs, 2018, 27, 963–970, DOI:10.1080/13543784.2018.1548608.
- R. Ronca and C. T. Supuran, Carbonic anhydrase IX: An atypical target for innovative therapies in cancer, Biochim. Biophys. Acta, Rev. Cancer, 2024, 1879, 189120, DOI:10.1016/j.bbcan.2024.189120.
- C. T. Supuran, V. Alterio, A. Di Fiore, K. D'Ambrosio, F. Carta, S. M. Monti and G. De Simone, Inhibition of carbonic anhydrase IX targets primary tumors, metastases, and cancer stem cells: three for the price of one, Med. Res. Rev., 2018, 38, 1799–1836, DOI:10.1002/med.21497.
- J. Y. Winum, M. Rami, A. Scozzafava, J. L. Montero and C. Supuran, Carbonic anhydrase IX: a new druggable target for the design of antitumor agents, Med. Res. Rev., 2008, 28, 445–463, DOI:10.1002/med.20112.
- M. Krasavin, S. Kalinin, T. Sharonova and C. T. Supuran, Inhibitory activity against carbonic anhydrase IX and XII as a candidate selection criterion in the development of new anticancer agents, J. Enzyme Inhib. Med. Chem., 2020, 35, 1555–1561, DOI:10.1080/14756366.2020.1801674.
- D. Dar'in, G. Kantin, S. Kalinin, T. Sharonova, A. Bunev, G. I. Ostapenko, A. Nocentini, V. Sharoyko, C. T. Supuran and M. Krasavin, Investigation of 3-sulfamoyl coumarins against cancer-related IX and XII isoforms of human carbonic anhydrase as well as cancer cells leads to the discovery of 2-oxo-2H-benzo[h]chromene-3-sulfonamide – A new caspase-activating proapoptotic agent, Eur. J. Med. Chem., 2021, 222, 113589, DOI:10.1016/j.ejmech.2021.113589.
- S. Burmaoğlu, E. Dilek, A. O. Yılmaz and C. T. Supuran, Synthesis of two phloroglucinol derivatives with cinnamyl moieties as inhibitors of the carbonic anhydrase isozymes I and II: an in vitro study, J. Enzyme Inhib. Med. Chem., 2016, 31, 208–212, DOI:10.1080/14756366.2016.1181626.
- Y. Thakur, M. Tripathi, B. Verma, R. Khilari, R. Agrawal, Likheshwari, M. Khursheed Siddiqi, R. Pande, E. Mohapatra and R. H. Khan, Interaction of cobalt(II) and copper(II) hydroxamates with polyriboadenylic acid: An insight into RNA based drug designing, Nucleosides, Nucleotides Nucleic Acids, 2019, 38, 481–508, DOI:10.1080/15257770.2018.1562074.
- F. S. Tokalı, Z. Alım and Ü. Yırtıcı, Carboxylate- and Sulfonate-Containing Quinazolin-4(3H)-one Rings: Synthesis, Characterization, and Carbonic Anhydrase I–II and Acetylcholinesterase Inhibition Properties, ChemistrySelect, 2023, 8, e202204191, DOI:10.1002/slct.202204191.
- P. Taslimi, C. Caglayan, V. Farzaliyev, O. Nabiyev, A. Sujayev, F. Turkan, R. Kaya and İ. Gulçin, Synthesis and discovery of potent carbonic anhydrase, acetylcholinesterase, butyrylcholinesterase, and α-glycosidase enzymes inhibitors: The novel N,N′-bis-cyanomethylamine and alkoxymethylamine derivatives, J. Biochem. Mol. Toxicol., 2018, 32, e22042, DOI:10.1002/jbt.22042.
- A. Nocentini, C. T. Supuran and C. Capasso, An overview on the recently discovered iota-carbonic anhydrases, J. Enzyme Inhib. Med. Chem., 2021, 36, 1988–1995, DOI:10.1080/14756366.2021.1972995.
- Y. Hirakawa, M. Senda, K. Fukuda, H. Y. Yu, M. Ishida, M. Taira, K. Kinbara and T. Senda, Characterization of a novel type of carbonic anhydrase that acts without metal cofactors, BMC Biol., 2021, 19, 105, DOI:10.1186/s12915-021-01039-8.
- M. Gümüş, Ş. N. Babacan, Y. Demir, Y. Sert, İ. Koca and İ. Gülçin, Discovery of sulfadrug–pyrrole conjugates as carbonic anhydrase and acetylcholinesterase inhibitors, Arch. Pharm., 2022, 355, 2100242, DOI:10.1002/ardp.202100242.
- İ. Gülçin, B. Trofimov, R. Kaya, P. Taslimi, L. Sobenina, E. Schmidt, O. Petrova, S. Malysheva, N. Gusarova, V. Farzaliyev, A. Sujayev, S. Alwasel and C. T. Supuran, Synthesis of nitrogen, phosphorus, selenium and sulfur-containing heterocyclic compounds – Determination of their carbonic anhydrase, acetylcholinesterase, butyrylcholinesterase and α-glycosidase inhibition properties, Bioorg. Chem., 2020, 103, 104171, DOI:10.1016/j.bioorg.2020.104171.
- A. E. Elsawi, M. M. Elbadawi, A. Nocentini, H. Almahli, S. Giovannuzzi, M. Shaldam, R. Salem, T. M. Ibrahim, H. A. Abdel-Aziz, C. T. Supuran and W. M. Eldehna, 1,5-Diaryl-1,2,4-triazole Ureas as New SLC-0111 Analogues Endowed with Dual Carbonic Anhydrase and VEGFR-2 Inhibitory Activities, J. Med. Chem., 2023, 66, 10558–10578, DOI:10.1021/acs.jmedchem.3c00721.
- F. Carta, A. Maresca, A. S. Covarrubias, S. L. Mowbray, T. A. Jones and C. T. Supuran, Carbonic anhydrase inhibitors. Characterization and inhibition studies of the most active β-carbonic anhydrase from Mycobacterium tuberculosis, Rv3588c, Bioorg. Med. Chem. Lett., 2009, 19, 6649–6654, DOI:10.1016/j.bmcl.2009.10.009.
- S. Akocak and C. T. Supuran, Activation of α-, β-, γ- δ-, ζ- and η- class of carbonic anhydrases with amines and amino acids: a review, J. Enzyme Inhib. Med. Chem., 2019, 34, 1652–1659, DOI:10.1080/14756366.2019.1664501.
- C. T. Supuran, Structure and function of carbonic anhydrases, Biochem. J., 2016, 473, 2023–2032, DOI:10.1042/BCJ20160115.
- E. Berrino and C. T. Supuran, Novel approaches for designing drugs that interfere with pH regulation, Expert Opin. Drug Discovery, 2019, 14, 231–248, DOI:10.1080/17460441.2019.1567488.
- G. De Simone, R. M. Vitale, A. Di Fiore, C. Pedone, A. Scozzafava, J.-L. Montero, J.-Y. Winum and C. T. Supuran, Carbonic Anhydrase Inhibitors:
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Hypoxia-Activatable Sulfonamides Incorporating Disulfide Bonds that Target the Tumor-Associated Isoform IX, J. Med. Chem., 2006, 49, 5544–5551, DOI:10.1021/jm060531j.
Hypoxia-Activatable Sulfonamides Incorporating Disulfide Bonds that Target the Tumor-Associated Isoform IX, J. Med. Chem., 2006, 49, 5544–5551, DOI:10.1021/jm060531j. - Z. Ditte, P. Ditte, M. Labudova, V. Simko, F. Iuliano, M. Zatovicova, L. Csaderova, S. Pastorekova and J. Pastorek, Carnosine inhibits carbonic anhydrase IX-mediated extracellular acidosis and suppresses growth of HeLa tumor xenografts, BMC Cancer, 2014, 14, 358, DOI:10.1186/1471-2407-14-358.
- H. I. Gul, C. Yamali, H. Sakagami, A. Angeli, J. Leitans, A. Kazaks, K. Tars, D. O. Ozgun and C. T. Supuran, New anticancer drug candidates sulfonamides as selective hCA IX or hCA XII inhibitors, Bioorg. Chem., 2018, 77, 411–419, DOI:10.1016/j.bioorg.2018.01.021.
- C. T. Supuran, Targeting carbonic anhydrases for the management of hypoxic metastatic tumors, Expert Opin. Ther. Pat., 2023, 33, 701–720, DOI:10.1080/13543776.2023.2245971.
- C. T. Supuran, Advances in structure-based drug discovery of carbonic anhydrase inhibitors, Expert Opin. Drug Discovery, 2017, 12, 61–88, DOI:10.1080/17460441.2017.1253677.
- N. Bijari, S. Ghobadi, H. Mahdiuni, R. Khodarahmi and S. A. Ghadami, Spectroscopic and molecular modeling studies on binding of dorzolamide to bovine and human carbonic anhydrase II, Int. J. Biol. Macromol., 2015, 80, 189–199, DOI:10.1016/j.ijbiomac.2015.06.028.
- R. P. Tanpure, B. Ren, T. S. Peat, L. F. Bornaghi, D. Vullo, C. T. Supuran and S.-A. Poulsen, Carbonic Anhydrase Inhibitors with Dual-Tail Moieties To Match the Hydrophobic and Hydrophilic Halves of the Carbonic Anhydrase Active Site, J. Med. Chem., 2015, 58, 1494–1501, DOI:10.1021/jm501798g.
- J. K. Kim, C. Lee, S. W. Lim, A. Adhikari, J. T. Andring, R. McKenna, C.-M. Ghim and C. U. Kim, Elucidating the role of metal ions in carbonic anhydrase catalysis, Nat. Commun., 2020, 11, 4557, DOI:10.1038/s41467-020-18425-5.
- H. Imtaiyaz, B. Shajee, A. Waheed, F. Ahmad and W. S. Sly, Structure, function and applications of carbonic anhydrase isozymes, Bioorg. Med. Chem., 2013, 21, 1570–1582, DOI:10.1016/j.bmc.2012.04.044.
- C. T. Supuran, How many carbonic anhydrase inhibition mechanisms exist?, J. Enzyme Inhib. Med. Chem., 2016, 31, 345–360, DOI:10.3109/14756366.2015.1122001.
- M. A. Said, W. M. Eldehna, A. Nocentini, A. Bonardi, S. H. Fahim, S. Bua, D. H. Soliman, H. A. Abdel-Aziz, P. Gratteri, S. M. Abou-Seri and C. T. Supuran, Synthesis, biological and molecular dynamics investigations with a series of triazolopyrimidine/triazole-based benzenesulfonamides as novel carbonic anhydrase inhibitors, Eur. J. Med. Chem., 2020, 185, 111843, DOI:10.1016/j.ejmech.2019.111843.
- L. Alaei, R. Khodarahmi, V. Sheikh-Hasani, N. Sheibani and A. A. Moosavi-Movahedi, Mechanistic investigation of sulfonamide ligands as human carbonic anhydrase II inhibitors, Int. J. Biol. Macromol., 2018, 120, 1198–1207, DOI:10.1016/j.ijbiomac.2018.08.186.
- F. P. Busardò, A. F. Lo Faro, A. Sirignano, R. Giorgetti and J. Carlier, In silico, in vitro, and in vivo human metabolism of acetazolamide, a carbonic anhydrase inhibitor and common “diuretic and masking agent” in doping, Arch. Toxicol., 2022, 96, 1989–2001, DOI:10.1007/s00204-022-03289-z.
- L. Vats, V. Sharma, A. Angeli, R. Kumar, C. T. Supuran and P. K. Sharma, Synthesis of novel 4-functionalized 1,5-diaryl-1,2,3-triazoles containing benzenesulfonamide moiety as carbonic anhydrase I, II, IV and IX inhibitors, Eur. J. Med. Chem., 2018, 150, 678–686, DOI:10.1016/j.ejmech.2018.03.030.
- Y.-C. Duan, Y.-C. Ma, E. Zhang, X.-J. Shi, M.-M. Wang, X.-W. Ye and H.-M. Liu, Design and synthesis of novel 1, 2, 3-triazole-dithiocarbamate hybrids as potential anticancer agents, Eur. J. Med. Chem., 2013, 62, 11–19, DOI:10.1016/j.ejmech.2012.12.046.
- R. Kant, V. Singh, G. Nath, S. K. Awasthi and A. Agarwal, Design, synthesis and biological evaluation of ciprofloxacin tethered bis-1, 2, 3-triazole conjugates as potent antibacterial agents, Eur. J. Med. Chem., 2016, 124, 218–228, DOI:10.1016/j.ejmech.2016.08.031.
- P. Yadav, K. Lal, A. Kumar, S. K. Guru, S. Jaglan and S. Bhushan, Green synthesis and anticancer potential of chalcone linked-1, 2, 3-triazoles, Eur. J. Med. Chem., 2017, 126, 944–953, DOI:10.1016/j.ejmech.2016.11.030.
- S. Gatadi, J. Gour, M. Shukla, G. Kaul, S. Das, A. Dasgupta, S. Malasala, R. S. Borra, Y. Madhavi and S. Chopra, Synthesis of 1, 2, 3-triazole linked 4 (3H)-Quinazolinones as potent antibacterial agents against multidrug-resistant Staphylococcus aureus, Eur. J. Med. Chem., 2018, 157, 1056–1067, DOI:10.1016/j.ejmech.2018.08.070.
- M. Allam, A. Bhavani, A. Mudiraj, N. Ranjan, M. Thippana and P. P. Babu, Synthesis of pyrazolo [3, 4-d] pyrimidin-4 (5H)-ones tethered to 1, 2, 3-triazoles and their evaluation as potential anticancer agents, Eur. J. Med. Chem., 2018, 156, 43–52, DOI:10.1016/j.ejmech.2018.06.055.
- A. Buza, C. Türkeş, M. Arslan, Y. Demir, B. Dincer, A. R. Nixha and Ş. Beydemir, Discovery of novel benzenesulfonamides incorporating 1,2,3-triazole scaffold as carbonic anhydrase I, II, IX, and XII inhibitors, Int. J. Biol. Macromol., 2023, 239, 124232, DOI:10.1016/j.ijbiomac.2023.124232.
- C. Kakakhan, C. Türkeş, Ö. Güleç, Y. Demir, M. Arslan, G. Özkemahlı and Ş. Beydemir, Exploration of 1,2,3-triazole linked benzenesulfonamide derivatives as isoform selective inhibitors of human carbonic anhydrase, Bioorg. Med. Chem., 2023, 77, 117111, DOI:10.1016/j.bmc.2022.117111.
- A. A. Shukralla, E. Dolan and N. Delanty, Acetazolamide: Old drug, new evidence?, Epilepsia Open, 2022, 7, 378–392, DOI:10.1002/epi4.12619.
- A. Bonardi, S. Bua, J. Combs, C. Lomelino, J. Andring, S. M. Osman, A. Toti, L. Di Cesare Mannelli, P. Gratteri, C. Ghelardini, R. McKenna, A. Nocentini and C. T. Supuran, The three-tails approach as a new strategy to improve selectivity of action of sulphonamide inhibitors against tumour-associated carbonic anhydrase IX and XII, J. Enzyme Inhib. Med. Chem., 2022, 37, 930–939, DOI:10.1080/14756366.2022.2053526.
- A. Kumar, K. Siwach, C. T. Supuran and P. K. Sharma, A decade of tail-approach based design of selective as well as potent tumor associated carbonic anhydrase inhibitors, Bioorg. Chem., 2022, 126, 105920, DOI:10.1016/j.bioorg.2022.105920.
- N. Lolak, S. Akocak, M. Durgun, H. E. Duran, A. Necip, C. Türkeş, M. Işık and Ş. Beydemir, Novel bis-ureido-substituted sulfaguanidines and sulfisoxazoles as carbonic anhydrase and acetylcholinesterase inhibitors, Mol. Diversity, 2023, 27, 1735–1749, DOI:10.1007/s11030-022-10527-0.
- Ö. Güleç, C. Türkeş, M. Arslan, Y. Demir, B. Dincer, A. Ece, Ö. İ. Küfrevioğlu and Ş. Beydemir, Bioactivity, cytotoxicity, and molecular modeling studies of novel sulfonamides as dual inhibitors of carbonic anhydrases and acetylcholinesterase, J. Mol. Liq., 2024, 410, 125558, DOI:10.1016/j.molliq.2024.125558.
- Ö. Güleç, C. Türkeş, M. Arslan, Y. Demir, B. Dincer, A. Ece and Ş. Beydemir, Novel beta-lactam substituted benzenesulfonamides: in vitro enzyme inhibition, cytotoxic activity and in silico interactions, J. Biomol. Struct. Dyn., 2024, 42, 6359–6377, DOI:10.1080/07391102.2023.2240889.
- Ö. Güleç, C. Türkeş, M. Arslan, M. Işık, Y. Demir, H. E. Duran, M. Fırat, Ö. İ. Küfrevioğlu and Ş. Beydemir, Dynamics of small molecule-enzyme interactions: Novel benzenesulfonamides as multi-target agents endowed with inhibitory effects against some metabolic enzymes, Arch. Biochem. Biophys., 2024, 759, 110099, DOI:10.1016/j.abb.2024.110099.
- V. Sharma, R. Kumar, A. Angeli, C. T. Supuran and P. K. Sharma, Benzenesulfonamides with trisubstituted triazole motif as selective carbonic anhydrase I, II, IV, and IX inhibitors, Arch. Pharm., 2023, 356, 2200391, DOI:10.1002/ardp.202200391.
- L. Vats, R. Kumar, S. Bua, A. Nocentini, P. Gratteri, C. T. Supuran and P. K. Sharma, Continued exploration and tail approach synthesis of benzenesulfonamides containing triazole and dual triazole moieties as carbonic anhydrase I, II, IV and IX inhibitors, Eur. J. Med. Chem., 2019, 183, 111698, DOI:10.1016/j.ejmech.2019.111698.
- M. M. Elbadawi, W. M. Eldehna, A. Nocentini, W. R. Somaa, S. T. Al-Rashood, E. B. Elkaeed, M. A. El Hassab, H. A. Abdel-Aziz, C. T. Supuran and M. Fares, Development of 4-((3-oxo-3-phenylpropyl)amino)benzenesulfonamide derivatives utilizing tail/dual-tail approaches as novel carbonic anhydrase inhibitors, Eur. J. Med. Chem., 2022, 238, 114412, DOI:10.1016/j.ejmech.2022.114412.
- A. Bonardi, A. Nocentini, S. Bua, J. Combs, C. Lomelino, J. Andring, L. Lucarini, S. Sgambellone, E. Masini, R. McKenna, P. Gratteri and C. T. Supuran, Sulfonamide Inhibitors of Human Carbonic Anhydrases Designed through a Three-Tails Approach: Improving Ligand/Isoform Matching and Selectivity of Action, J. Med. Chem., 2020, 63, 7422–7444, DOI:10.1021/acs.jmedchem.0c00733.
- A. Innocenti, A. Scozzafava, S. Parkkila, L. Puccetti, G. De Simone and C. T. Supuran, Investigations of the esterase, phosphatase, and sulfatase activities of the cytosolic mammalian carbonic anhydrase isoforms I, II, and XIII with 4-nitrophenyl esters as substrates, Bioorg. Med. Chem. Lett., 2008, 18, 2267–2271, DOI:10.1016/j.bmcl.2008.03.012.
- S. M. Gould and D. S. Tawfik, Directed Evolution of the Promiscuous Esterase Activity of Carbonic Anhydrase II, Biochemistry, 2005, 44, 5444–5452, DOI:10.1021/bi0475471.
- J. A. Verpoorte, S. Mehta and J. T. Edsall, Esterase Activities of Human Carbonic Anhydrases B and C, J. Biol. Chem., 1967, 242, 4221–4229, DOI:10.1016/S0021-9258(18)95800-X.
- C. Türkeş, Carbonic anhydrase inhibition by antiviral drugs in vitro and in silico, J. Mol. Recognit., 2023, 36, e3063, DOI:10.1002/jmr.3063.
- F. Topal, Inhibition profiles of Voriconazole against acetylcholinesterase, α-glycosidase, and human carbonic anhydrase I and II isoenzymes, J. Biochem. Mol. Toxicol., 2019, 33, e22385, DOI:10.1002/jbt.22385.
- H. Lineweaver and D. Burk, The Determination of Enzyme Dissociation Constants, J. Am. Chem. Soc., 1934, 56, 658–666, DOI:10.1021/ja01318a036.
- T. Mosmann, Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays, J. Immunol. Methods, 1983, 65, 55–63, DOI:10.1016/0022-1759(83)90303-4.
- S. Chakravarty and K. K. Kannan, Drug-Protein Interactions: Refined Structures of Three Sulfonamide Drug Complexes of Human Carbonic Anhydrase I Enzyme, J. Mol. Biol., 1994, 243, 298–309, DOI:10.1006/jmbi.1994.1655.
- M. Aggarwal, C. D. Boone, B. Kondeti, C. Tu, D. N. Silverman and R. McKenna, Effects of cryoprotectants on the structure and thermostability of the human carbonic anhydrase II-acetazolamide complex, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 860–865, DOI:10.1107/S0907444913002771.
- J. Leitans, A. Kazaks, A. Balode, J. Ivanova, R. Zalubovskis, C. T. Supuran and K. Tars, Efficient Expression and Crystallization System of Cancer-Associated Carbonic Anhydrase Isoform IX, J. Med. Chem., 2015, 58, 9004–9009, DOI:10.1021/acs.jmedchem.5b01343.
- D. A. Whittington, A. Waheed, B. Ulmasov, G. N. Shah, J. H. Grubb, W. S. Sly and D. W. Christianson, Crystal structure of the dimeric extracellular domain of human carbonic anhydrase XII, a bitopic membrane protein overexpressed in certain cancer tumor cells, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 9545–9550, DOI:10.1073/pnas.161301298.
- Ö. Güleç, C. Türkeş, M. Arslan, Y. Demir, B. Dincer, A. Ece, Ö. İrfan Küfrevioğlu and Ş. Beydemir, Novel spiroindoline derivatives targeting aldose reductase against diabetic complications: Bioactivity, cytotoxicity, and molecular modeling studies, Bioorg. Chem., 2024, 145, 107221, DOI:10.1016/j.bioorg.2024.107221.
- J. C. Shelley, A. Cholleti, L. L. Frye, J. R. Greenwood, M. R. Timlin and M. Uchimaya, Epik: a software program for pKaprediction and protonation state generation for drug-like molecules, J. Comput.-Aided Mol. Des., 2007, 21, 681–691, DOI:10.1007/s10822-007-9133-z.
- T. A. Halgren, Identifying and Characterizing Binding Sites and Assessing Druggability, J. Chem. Inf. Model., 2009, 49, 377–389, DOI:10.1021/ci800324m.
- C. Türkeş, A. Ö. Kesebir, Y. Demir, Ö. İ. Küfrevioğlu and Ş. Beydemir, Calcium Channel Blockers: The Effect of Glutathione S-Transferase Enzyme Activity and Molecular Docking Studies, ChemistrySelect, 2021, 6, 11137–11143, DOI:10.1002/slct.202103100.
- C. Türkeş, Y. Demir and Ş. Beydemir, Infection Medications: Assessment In-Vitro Glutathione S-Transferase Inhibition and Molecular Docking Study, ChemistrySelect, 2021, 6, 11915–11924, DOI:10.1002/slct.202103197.
- C. Türkeş, Aldose reductase with quinolone antibiotics interaction: In vitro and in silico approach of its relationship with diabetic complications, Arch. Biochem. Biophys., 2024, 761, 110161, DOI:10.1016/j.abb.2024.110161.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P. Francis and P. S. Shenkin, Glide:A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy, J. Med. Chem., 2004, 47, 1739–1749, DOI:10.1021/jm0306430.
- T. A. Halgren, R. B. Murphy, R. A. Friesner, H. S. Beard, L. L. Frye, W. T. Pollard and J. L. Banks, Glide:A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening, J. Med. Chem., 2004, 47, 1750–1759, DOI:10.1021/jm030644s.
- G. Barreiro, C. R. W. Guimarães, I. Tubert-Brohman, T. M. Lyons, J. Tirado-Rives and W. L. Jorgensen, Search for Non-Nucleoside Inhibitors of HIV-1 Reverse Transcriptase Using Chemical Similarity, Molecular Docking, and MM-GB/SA Scoring, J. Chem. Inf. Model., 2007, 47, 2416–2428, DOI:10.1021/ci700271z.
- C. T. Supuran, Exploring the multiple binding modes of inhibitors to carbonic anhydrases for novel drug discovery, Expert Opin. Drug Discovery, 2020, 15, 671–686, DOI:10.1080/17460441.2020.1743676.
- M. Aggarwal, C. D. Boone, B. Kondeti and R. McKenna, Structural annotation of human carbonic anhydrases, J. Enzyme Inhib. Med. Chem., 2013, 28, 267–277, DOI:10.3109/14756366.2012.737323.
- V. Alterio, A. Di Fiore, K. D'Ambrosio, C. T. Supuran and G. De Simone, Multiple Binding Modes of Inhibitors to Carbonic Anhydrases: How to Design Specific Drugs Targeting 15 Different Isoforms?, Chem. Rev., 2012, 112, 4421–4468, DOI:10.1021/cr200176r.
- C. Keum, M.-C. Kim and S.-Y. Lee, Effects of transition metal ions on the catalytic activity of carbonic anhydrase mimics, J. Mol. Catal. A: Chem., 2015, 408, 69–74, DOI:10.1016/j.molcata.2015.07.006.
- A. S. Lipton, R. W. Heck and P. D. Ellis, Zinc Solid-State NMR Spectroscopy of Human Carbonic Anhydrase:Implications for the Enzymatic Mechanism, J. Am. Chem. Soc., 2004, 126, 4735–4739, DOI:10.1021/ja0305609.
- C. D. Boone, M. Pinard, R. McKenna and D. Silverman, Catalytic Mechanism of α-Class Carbonic Anhydrases: CO2 Hydration and Proton Transfer, in Carbonic Anhydrase: Mechanism, Regulation, Links to Disease, and Industrial Applications, ed. S. C. Frost and R. McKenna, Springer Netherlands, Dordrecht, 2014, pp. 31–52 Search PubMed.
- Z. Hou, B. Lin, Y. Bao, H.-n. Yan, M. Zhang, X.-w. Chang, X.-x. Zhang, Z.-j. Wang, G.-f. Wei, M.-s. Cheng, Y. Liu and C. Guo, Dual-tail approach to discovery of novel carbonic anhydrase IX inhibitors by simultaneously matching the hydrophobic and hydrophilic halves of the active site, Eur. J. Med. Chem., 2017, 132, 1–10, DOI:10.1016/j.ejmech.2017.03.023.
- A. Daina, O. Michielin and V. Zoete, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 7, 42717, DOI:10.1038/srep42717.
- H. E. Duran and Ş. Beydemir, Naphthoquinones and anthraquinones: Exploring their impact on acetylcholinesterase enzyme activity, Biotechnol. Appl. Biochem., 2024, 71, 1079–1093, DOI:10.1002/bab.2599.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Delivery Rev., 1997, 23, 3–25, DOI:10.1016/S0169-409X(96)00423-1.
- E. M. Duffy and W. L. Jorgensen, Prediction of Properties from Simulations:Free Energies of Solvation in Hexadecane, Octanol, and Water, J. Am. Chem. Soc., 2000, 122, 2878–2888, DOI:10.1021/ja993663t.
- W. J. Egan, K. M. Merz and J. J. Baldwin, Prediction of Drug Absorption Using Multivariate Statistics, J. Med. Chem., 2000, 43, 3867–3877, DOI:10.1021/jm000292e.
- A. K. Ghose, V. N. Viswanadhan and J. J. Wendoloski, A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases, J. Comb. Chem., 1999, 1, 55–68, DOI:10.1021/cc9800071.
- I. Muegge, S. L. Heald and D. Brittelli, Simple Selection Criteria for Drug-like Chemical Matter, J. Med. Chem., 2001, 44, 1841–1846, DOI:10.1021/jm015507e.
- D. F. Veber, S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, Molecular Properties That Influence the Oral Bioavailability of Drug Candidates, J. Med. Chem., 2002, 45, 2615–2623, DOI:10.1021/jm020017n.
- Y. C. Martin, A Bioavailability Score, J. Med. Chem., 2005, 48, 3164–3170, DOI:10.1021/jm0492002.
- J. B. Baell and G. A. Holloway, New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays, J. Med. Chem., 2010, 53, 2719–2740, DOI:10.1021/jm901137j.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00617h |
| This journal is © The Royal Society of Chemistry 2025 |