
Development, biological evaluation, and molecular modelling of some benzene-sulfonamide derivatives as protein tyrosine phosphatase-1B inhibitors for managing diabetes mellitus and associated metabolic disorders†
Nagat
Ghareb
a,
Khaled M.
Darwish
 bc,
Mohamed S.
Nafie
de,
Ranwa
Elrayess
f,
Noha M.
Abourobe
g,
Shaimaa A.
Fattah
h,
Reem M.
Hazem
i,
Eman T.
Mehanna
h and
Ranza
Elrayess
*aj
bc,
Mohamed S.
Nafie
de,
Ranwa
Elrayess
f,
Noha M.
Abourobe
g,
Shaimaa A.
Fattah
h,
Reem M.
Hazem
i,
Eman T.
Mehanna
h and
Ranza
Elrayess
*aj
aDepartment of Pharmaceutical Organic Chemistry, Faculty of Pharmacy, Suez Canal University, Ismailia 41522, Egypt. E-mail: Ranza.el-rayes@pharm.suez.edu.eg; Fax: +20 064 3230741; Tel: +20 102 548 8849
bDepartment of Medicinal Chemistry, Faculty of Pharmacy, Suez Canal University, Ismailia 41522, Egypt
cDepartment of Medicinal Chemistry, Faculty of Pharmacy, Galala University, New Galala 43713, Egypt
dDepartment of Chemistry, College of Sciences, University of Sharjah, Sharjah, 27272, United Arab Emirates
eDepartment of Chemistry, Faculty of Science, Suez Canal University, Ismailia 41522, Egypt
fDepartment of Zoology, Faculty of Science, Suez Canal University, Ismailia 41522, Egypt
gDepartment of Pharmaceutics, Faculty of Pharmacy, Suez Canal University, Ismailia 41522, Egypt
hDepartment of Biochemistry, Faculty of Pharmacy, Suez Canal University, Ismailia 41522, Egypt
iDepartment of Pharmacology and Toxicology, Faculty of Pharmacy, Suez Canal University, Ismailia 41522, Egypt
jPharmaceutical Organic Chemistry Department, College of Pharmacy, Al-Ayen University, Dhi-Qar 64001, Iraq
First published on 23rd October 2024
Abstract
Exploring new inhibitors with good bioavailability and high selectivity for managing type 2 diabetes mellitus (T2DM) and its associated complications is a major challenge for research, academia, and the pharmaceutical industry. Protein tyrosine phosphatase-1B (PTP1B) arose as an important negative regulator in insulin signaling pathways associated with metabolic disorders, including T2DM and obesity. Novel neutral compounds with a benzene-sulfonamide scaffold were designed and synthesized based on structural- and ligand-based drug design strategies for fragment growth. Promising hits against PTP1B were identified through in vitro enzymology inhibition assay. Mechanistic aspects of the compound's different inhibition activities were rigorously investigated through molecular docking coupled with explicit dynamics simulations. Four identified hits, 3c, 8, 10a, and 11, with sub-micromolar PTP-1B IC50 and significant predicted pharmacokinetic and pharmacodynamic parameters, were further biologically evaluated for their anti-diabetic, anti-obesity, anti-inflammatory, and anti-oxidant effects in a high-fat diet (HFD) + streptozotocin (STZ)-induced T2DM rat model. All these hit compounds exhibited a significant anti-diabetic and anti-obesity effect and a significant efficacy in reducing oxidative stress and increasing anti-oxidant enzymes while reducing inflammatory markers. Improving compound potency was further highlighted by improving the pharmacokinetic profile of the most active compound, 10a, through nano formulation. Compound 10a nano formulation showed the most promising anti-diabetic and anti-obesity effects and a remarkable histopathological improvement in all organs studied.
1. Introduction
The global prevalence of obesity and type 2 diabetes mellitus (T2DM) is steadily increasing due to modern nutrition and hereditary factors. Better blood glucose levels, lower insulin resistance, fewer diabetes complications, and a reduction in cardiovascular disease risk factors have been associated with moderate weight loss.1 The treatment strategy for lowering blood glucose levels should therefore include techniques that support weight management goals, with semaglutide and tirzepatide currently showing the greatest efficacy in weight reduction among medications approved for glycemic control.2 Insulin has the advantage of being effective in situations where other agents are ineffective, and it should be included in any combination regimen when hyperglycemia is severe, especially when catabolic features (hypertriglyceridemia, ketosis) are present.3 However, long-term insulin injections are very uncomfortable and difficult for patients, and the efficacy of oral hypoglycemic drugs is often inadequate.4 Besides variable therapeutic efficiency, current hypoglycemic agents have severe side effects such as hypoglycemia, weight gain, risk of liver disease, and other complications.5 There is a significant link between high blood glucose levels, oxidative stress caused by hyperglycemia, inflammation and the onset and progression of T2DM. Numerous studies have shown that chronic low-grade inflammation is associated with an increased risk of developing T2DM. This subclinical inflammation also contributes to insulin resistance and is associated with features of metabolic syndrome, including high blood glucose. Oxidative stress promotes the production of inflammatory mediators, and inflammation in turn increases the production of reactive oxygen species.6 Additionally, oxidative stress and inflammation are significantly associated with the pathophysiology of micro- and macrovascular T2DM complications, and so drugs targeting these important metabolic pathways have been developed to treat diabetic complications. Only a limited number of anti-inflammatory/oxidative drugs, including gabapentin, pregabalin, and duloxetine, have been approved by the FDA.7Human protein tyrosine phosphatase-1B (PTP1B) arose as a promising target for the management of T2DM and associated metabolic disorders like obesity through boosting insulin resistance.7 PTP1B is an intracellular PTP that negatively regulates insulin and leptin signaling8 and has been shown to play a role in inflammatory responses.9 Inhibition of PTP1B has been considered beneficial for interfering with the dephosphorylation-driven inhibition of actually active insulin receptor kinases and insulin receptor substrate-1 within the leptin signaling pathway.10,11 PTP1B inhibitors can help regulate insulin and plasma glucose levels while improving insulin resistance without causing hypoglycemia.12 PTP1B is highly expressed in tissues affected by diabetes complications. Deletion of PTP1B in mice led to an improvement in the lipid profile, particularly in the liver, and provided some protection against liver inflammation. Heart failure is common among diabetes patients, and studies have shown that deletion of PTP1B specifically in endothelial cells of mice restores nitric oxide production, prevents endothelial dysfunction and reduces heart failure. In the context of diabetic nephropathy, inhibition of PTP1B helps to protect fibroblasts from dying during acute kidney injury.13
In the course of research into diabetes pathogenesis, numerous drugs targeting mechanisms and pathways have been identified and serve as pragmatic inspiration for the development of new, more effective synthetic compounds for the treatment of T2DM. Currently, α-amylase, α-glucosidase inhibitors14–17 and PTP1B inhibitors are at the forefront of T2DM drug research and development. Numerous synthetic PTP1B inhibitors have been described, including aminobenzoic acid, vanadium complexes, benzofuran benzothiophene biphenyls, and thiazolidinediones. Thiazolidinediones (TZDs), also known as glitazones, are molecules that have the same molecular scaffold (2,4-TZDs31).18 They are able to improve glycemic control in type 2 diabetics by reducing insulin resistance in both the liver and peripheral tissues and improving insulin sensitivity in target tissues.19–21 Unfortunately, the in vivo activity showed insufficient efficiency due to the low cellular permeability of hydrophilic small molecule inhibitors, which have an increased affinity for the active binding site. This led to the imminent development of an effective PTP1B inhibitor.22 The development of small molecule drugs for PTP1B (from synthetic or plant sources) could have a positive impact on bioavailability in the treatment of T2DM mellitus.23 A previous study from our laboratory has shown that benzene-sulfonamide derivatives have promising antidiabetic activity by inhibiting PTP1B, resulting in a significant reduction in fasting blood glucose (FBG) and insulin resistance compared to the obese control group.24
The focus of the present study is to modify further the chemical space of benzene-sulfonamide compounds for developing novel analogs based on combined structural- and ligand-based drug design strategies. Adopting the fragment growing strategy, the current study aimed to develop novel-tailed scaffolds linked to the benzene-sulfonamide heads to serve as anti-diabetic, anti-inflammatory, and anti-oxidant agents with improved pharmacodynamic and pharmacokinetic profiles. Synthesized compounds were screened in vitro for their efficacy in targeting PTP1B, and their predicted affinity towards PTP1B enzyme through molecular docking coupled with dynamics studies was investigated. Furthermore, the compounds' pharmacokinetic profiles were predicted through comprehensive computational ADME/Tox profiling. Promising synthesized compounds were further in vivo investigated for their anti-diabetic, anti-inflammatory, and anti-oxidant properties in a high-fat diet (HFD) + streptozotocin (STZ)-induced diabetic rat model to postulate promising clinical candidates for future research.
2. Scientific rationale for compound design development
Developing relevant candidate inhibitors for the PTP1B target has been challenged by the adequacy of ligand/pocket binding while maintaining a relevant safety and absorption profile.12 The high-charge density active site imposes difficulties against ligand binding due to high solvation entropy as well as poor cell membrane penetration for highly hydrophilic ligands. On the other hand, the conserved pocket residues across the PTPase family would raise potential off-target concerns.12,25 In this regard, the authors adopted a combined structure- and ligand-approach for designing their target compounds taking into account the advent of target-pocket selective binding as well as the key pharmacophoric features of actually reported PTP1B inhibitors. Adopting such a strategy would highly guarantee safety, pharmacodynamic selectivity, and a relevant pharmacokinetic profile for the synthesized compounds owing to the knowledge of pocket topology and characteristic ligand-based scaffolds.Adopting structure-based insights was proceeded through exploring the target pocket. The catalytic site of PTP1B, generally called the P-loop (His214-to-Arg221), is the most accessible pocket on the target surface, comprising the central binding site for the target's substrate (Fig. 1A). Down to the P-loop bottom site, the conserved Cys215 is settled together with a horseshoe-like orientation of neighboring amino acid mainchains.26 Both the conserved Arg221 and amides permit polar/hydrogen bonding with the substrate's phosphate for stabilizing the transition state.27 Substrate binding triggers PTP1B conformational changes being culminated through WPD loop closure as it comprises the acidic conserved residue, Asp181, servicing as a generalized acid/base for the enzyme catalysis. Two surface/solvent exposed amino acids, Arg47 and Tyr46, were recognized to contribute to substrate specificity and so targeting these residues via small ligands could reduce the substrate's affinity.27 Beside the catalytic P-loop (a.k.a. site A), additional secondary binding sites (namely sites B, C, and D) were recognized (Fig. 1A). Notably, site B (13 × 20 × 4 Å) is generally at the cleft area between the frontiers of the PTR loop and Q loop. First identified by Puius et al., this second aryl phosphate (Tyr-P; site B) is found vicinal to the P-loop and harbors several non-PTPase conserved residues; Arg24, Cys32, Phe52, Arg254, and Met258, critical for target selectivity.28 Site C is quite wide, flat, and fully solvent exposed, whereas site D is narrow, partially shielded from solvent, and very close to the P-loop site.29 Critical residues for PTP1B selectivity in sites C (Leu41, Tyr46, Arg47, Asp48) and site D (Lys116, Leu119, Lys120, Asp181, Ser216) were also identified.30 Among all site D residues, Lys120 has been identified as a determinant for PTP1B selectivity owing to its unique orientation and spatial position.29,30 Reported strategies for addressing selectivity/safety challenges have focused on developing extended molecules that are able to accommodate both the active catalytic site and the neighboring less conserved clefts of PTPase surface topology.25,28 Thus, we aimed to doubly target both P-loop/Tyr-P (sites A and B) with our designed compounds that would provide the advent of improved potencies down to nM ranges as well as PTP1B selectivity.25,31 Additionally, incorporating balanced hydrophilic/lipophilic scaffolds in our compounds has also been proposed for accommodating the polar pocket while exhibiting good cell penetration and overall kinetic profiles.32
 | ||
| Fig. 1 Rationalized design of our presented synthesized compounds as PTP1B inhibitors. (A) Structural-based insights from exploring the PTP1B target binding site. Surface representation showing the main catalytic loop (P-loop) as well as the secondary catalytically active loops; PTR, Q, WPD, and E loops in color codes. Zoomed in image illustrates the canonical substrate binding site (site A) as well as neighboring less/non-conserved PTPase sites; site B (Tyr-P), site C, and site D. Critical residues for PTP1B selectivity are shown as lines, colored with regard to their location at the enzyme's loops, and labelled according to their sequence within the protein structure. (B) Scientific approach for the compound's architectural design was further guided through ligand-based insights based on mapping the common pharmacophoric features of clinical phase investigated and literature reported PTP1B clinical and pre-clinical inhibitors. | ||
Moving towards the ligand-based insights, the structure features of several PTP1B inhibitors reaching pre-clinical or clinical phases were thoroughly revised. One of the earliest clinically tested PTP1B inhibitors, ertiprotafib, developed by Wyeth mimicking phosphotyrosine monocarboxylic acid derivatives, has reached phase-II trials for T2DM management.33 The tricyclic thiophene-based inhibitor exhibited relevant binding towards the A/B subpockets of the PTP1B binding site.34 Developed by Japan Tobacco Company, JTT-551 is another small molecule PTP1B inhibitor possessing promising preclinical results on diabetic and obesity mice models.35 This thiazole-based small molecule showed great selectivity towards PTP1B over the T-cell protein tyrosine phosphatase (Ki = 0.22 ± 0.04 versus 9.30 ± 0.40 μM, respectively) exhibiting anti-diabetic activity without mice weight changes.36 Another small molecule showing great preclinical data is DPM-1001 which is a potent selective non-competitive PTP1B inhibitor with a high oral bioavailability profile.37 Employing a non-charged polyamine steroid conjugate skeleton, DPM-1001 exhibited mixed action through binding to the C-terminal domain of PTP1B while chelating copper potentiating its inhibitory activity.38,39 Several other PTP1B small molecule inhibitors have been reported in the literature employing sulphur-containing heterocyclic rings as the relevant head group for PTP1B polar pocket binding. CID11786814, CID5327048, and CID11786814 showed preferential binding to the A/B/C subpockets of the target site while being capable of being actively transported into hepatocytes circumventing the poor membrane permeability of most PTP1B inhibitors.40,41 Thoroughly revising these inhibitors' chemical architectures revealed that these small molecules generally promoted three canonical pharmacophoric features: i) polar functionality (anionic or more preferably less ionizable) on linear/cyclic scaffold, ii) elongated cyclic trunk, and iii) terminal extended tail decorated with hydrogen binding and hydrophobic fragments being directed towards these less conserved clefts (Fig. 1B).
Our compounds were designed to cover both strategies, where our head ring scaffolds, including thiazole, oxazole, and other related analogs, were decorated with balanced ratios of polar and lipophilic functionalities. This attempt would satisfy the hydrophilic nature of the PTP1B catalytic site while limiting solvation penalties and poor kinetic profiles contributing to the compound's pharmacological activity. Further, these warhead scaffolds were meant to provide relevant anchoring at the site D polar subpocket. The elongated architecture of our synthesized compounds would permit multi-site accommodation for addressing selectivity/off-target concerns such as reaching site B across site A.25 The terminal N-heterocyclic sulphonamide group of our compounds was reasoned to provide significant binding affinity at selectivity site B. It is worth noting that the presence of the sulfonamide moiety as the compound's linker was rationalized for introducing relevant structural flexibility at the tail end, which would permit a favored direction for the terminal scaffold towards the less conserved sub-pocket, site B.
3. Experimental
3.1 Chemistry
The synthetic protocol for the desired compounds 2–11 is illustrated in Schemes 1 and 2. The synthetic procedure and spectral data of compounds 2a–c, 5–10 and 11 were reported previously.24,42,43 | ||
Scheme 1 Reagents and conditions: (a) Ar/R–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CX, DMF (dry), reflux, 24 h, yield: 55–70%; (b) ethyl bromoacetate, Na. Acetate, EtOH (HPLC), reflux, 12 h, yield: 67–77%; (c) ethyl 2-chloropropionate, Na. Acetate, EtOH (HPLC), reflux, 12 h, yield: 30–62%. CX, DMF (dry), reflux, 24 h, yield: 55–70%; (b) ethyl bromoacetate, Na. Acetate, EtOH (HPLC), reflux, 12 h, yield: 67–77%; (c) ethyl 2-chloropropionate, Na. Acetate, EtOH (HPLC), reflux, 12 h, yield: 30–62%. | ||
 | ||
Scheme 2 Reagents and conditions: (a) 2-cholroacetylchloride, TEA, DMF (dry), stirring, 4 h, yield: 80%; (b) NH4SCN, EtOH (HPLC), reflux, 10 h, yield: 67%; (c) ethyl acetoacetate, DMF: pyridine (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.01, dry), reflux, 4 h, yield: 56%; (d) ethyl cyanoacetate, DMF: pyridine (1:0.01, dry), reflux, 6 h, yield: 53%; (e) Ar/R–NCX, ACN (HPLC), r.t, 14 h, yield: 33–50%; (f) N–Ar/R piperazine, ACN (HPLC), r.t, 12 h, yield: 44–52%; (g) 2-(bromomethyl)-1H-benzo[d]imidazole, KI, MeOH (HPLC), reflux, 10 h, KOH, stirring, 3 h, yield: 50%. :0.01, dry), reflux, 4 h, yield: 56%; (d) ethyl cyanoacetate, DMF: pyridine (1:0.01, dry), reflux, 6 h, yield: 53%; (e) Ar/R–NCX, ACN (HPLC), r.t, 14 h, yield: 33–50%; (f) N–Ar/R piperazine, ACN (HPLC), r.t, 12 h, yield: 44–52%; (g) 2-(bromomethyl)-1H-benzo[d]imidazole, KI, MeOH (HPLC), reflux, 10 h, KOH, stirring, 3 h, yield: 50%. | ||
General procedure for synthesis of target compounds 3a–c and 4a–c. A solution of the corresponding thioureido-N-(pyrimidin-2-yl)benzene sulfonamide (2a–c) derivatives (0.01 mol) in ethanol (HPLC) (50 mL) was refluxed with ethyl bromoacetate or ethyl-2-chloropropionate (0.01 mol) and sodium acetate (0.02 mol) for 12 h. The reaction mixture was then filtered while hot concentrated and allowed to cool. The product obtained was washed with water and recrystallized from ethanol.44
(Z)-4-(4-Oxo-3-phenylthiazolidin-2-ylideneamino)-N-(pyrimidin-2-yl)benzene sulfonamide (3a). Grey powder m.p. = 160–163 °C, (yield 77%). IR (cm−1): 694 (C–S), 1153(SO2 Sym), 1373(SO2 Asym), 1539 (C
N), 1593 (CN), 1635 (CN), 1724 (CO), 3356 (NH). Mass spectrum: m/z (%): 425 (M+). 1H-NMR (400 MHz, DMSO-d6) δ (ppm) 8.49–6.55 (m, Ar–H, 12H), 6.03 (s, NH, 1H), 4.17 (s, CH2, 2H). 13C-NMR (101 MHz, DMSO-d6) δ (ppm) 165.7, 165.4, 158.7, 157.1 (2C), 142.5, 135.8, 135.1, 129.4 (2C), 129.3, 129.3 (2C), 118.9 (2C), 116.1 (2C), 113.1, 37.5. Anal. calcd for C19H15N5O3S2:C, 53.63; H, 3.55; N, 16.46; S, 15.07. Found: C, 53.28; H, 3.58; N, 16.71; S, 15.22 (Fig. S1 and S2†).
(Z)-4-(4-Oxo-3-phenyloxazolidin-2-ylideneamino)-N-(pyrimidin-2-yl)benzene sulfonamide (3b). White powder m.p. = 200–203 °C, (yield 67%). IR (cm−1): 1153(SO2 Sym), 1230 (C–O–C), 1315 (SO2 Asym), 1543 (C
N), 1597 (CN), 1651 (CN), 1747 (CO), 3464 (NH). Mass spectrum: m/z (%): 409 (M+). 1H-NMR (400 MHz, DMSO-d6) δ (ppm) 9.08–6.54 (m, Ar–H, 12H), 5.75 (s, NH, 1H), 4.33 (s, CH2, 2H). 13C-NMR (101 MHz, DMSO-d6) δ (ppm) 173.5, 159.5 (2C), 157.9, 154.0, 153.4, 141.6, 136.5, 131.0 (2C), 130.4 (2C), 130.2, 122.5 (2C), 116.9 (2C), 113.8, 33.9. Anal. calcd. for C19H15N5O4S: C, 55.74; H, 3.69; N, 17.11; S, 7.83. Found: C, 55.52; H, 3.31; N, 17.38; S, 7.64 (Fig. S3 and S4†).
(Z)-4-(3-Ethyl-4-oxothiazolidin-2-ylideneamino)-N-(pyrimidin-2-yl)benzene sulfonamide (3c). Brownish red powder m.p. = 160–162 °C, (yield 72%). IR (cm−1): 694 (C–S), 1153(SO2 Sym), 1338 (SO2 Asym), 1577 (C
N), 1593(CN), 1616 (CN), 1728 (CO), 3363 (NH). Mass spectrum: m/z (%): 377 (M+). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 8.45–6.93 (m, Ar–H, 7H), 6.01 (s, NH, 1H), 3.50 (s, CH2, 2H), 3.46 (q, CH2 ethyl, 2H, J = 8 Hz), 1.12 (t, CH3 ethyl, 3H, J = 8 Hz). 13C-NMR (101 MHz, DMSO-d6) δ (ppm) 172.1, 170.8, 157.9 (2C), 139.9, 134.0, 128.7, 119.6 (2C), 116.6 (2C), 112.7, 36.3, 31.3, 12.5. Anal. calcd. for C15H15N5O3S2: C, 47.73; H, 4.01; N, 18.55; S, 16.99. Found: C, 47.46; H, 4.06; N, 18.39; S, 16.57 (Fig. S5 and S6†).
(Z)-4-(5-Methyl-4-oxo-3-phenylthiazolidin-2-ylideneamino)-N-(pyrimidin-2-yl)benzene sulfonamide (4a). Brown powder m.p. = 190–193 °C, (yield 62%). Mass spectrum: m/z (%): 439 (M+). 1H-NMR (400 MHz, DMSO-d6) δ (ppm) 8.60–6.67 (m, Ar–H, 12H), 6.14 (s, NH, 1H), 4.60 (q, CH, 1H, J = 8 Hz), 1.44 (d, CH3, 3H, J = 8 Hz). 13C-NMR (101 MHz, DMSO-d6) δ (ppm) 161.4, 161.2, 160.9 (2C), 151.7, 147.3, 146.3, 130.9, 128.9 (2C), 128.6 (2C), 128.6 (2C), 128.5 (2C), 116.6, 107.9, 21.1, 19.4. Anal. calcd. for C20H17N5O3S2: C, 54.65; H, 3.90; N, 15.93; S, 14.59. Found: C, 54.82; H, 3.39; N, 16.07; S, 14.76 (Fig. S7 and S8†).
(Z)-4-(5-Methyl-4-oxo-3-phenyloxazolidin-2-ylideneamino)-N-(pyrimidin-2-yl)benzene sulfonamide (4b). White powder m.p. = 260–263 °C, (yield 52%). IR (cm−1): 1153(SO2 Sym), 1234 (C–O–C), 1323 (SO2 Asym), 1543 (C
N), 1593 (CN), 1643 (CN), 1708 (CO), 3425 (NH). Mass spectrum: m/z (%): 423 (M+). 1H-NMR (400 MHz, DMSO-d6) δ (ppm) 7.97–7.18 (m, Ar–H, 12H), 5.96 (s, NH, 1H), 4.32 (q, CH, 1H, J = 4 Hz), 1.68 (d, CH3, 3H, J = 8 Hz). 13C-NMR (101 MHz, DMSO-d6) δ (ppm) 159.3, 158.5 (2C), 157.9, 152.9, 149.9, 141.1, 140.5, 129.9 (2C), 128.7 (2C), 126.2, 124.2, 122.3 (2C), 115.1 (2C), 22.4, 19.9. Anal. calcd. for C20H17N5O4S: C, 56.73; H, 4.05; N, 16.54; S, 7.57. Found: C, 56.38; H, 4.16; N, 16.79; S, 7.71 (Fig. S9 and S10†).
(Z)-4-(3-Ethyl-5-methyl-4-oxothiazolidin-2-ylideneamino)-N-(pyrimidin-2-yl)benzene sulfonamide (4c). Yellow powder m.p. = 202–203 °C, (yield 30%). IR (cm−1): 694 (C–S), 1157 (SO2 Sym), 1315 (SO2 Asym), 1500 (C
N), 1589 (CN), 1624 (CN), 1674 (CO), 3360 (NH). Mass spectrum: m/z (%): 391 (M+). 1H-NMR (400 MHz, DMSO-d6) δ (ppm) 8.41–6.88 (m, Ar–H, 7H), 3.45 (q, CH, 1H, J = 8 Hz), 3.35 (q, CH2, 2H, J = 8 Hz), 1.13–1.03 (m, 2CH3, 6H). 13C-NMR (100 MHz, DMSO-d6) δ (ppm) 180.2, 158.6, 158.4 (2C), 153.3, 143.4, 130.1, 128.4 (2C), 121.3 (2C), 115.7, 38.9, 21.4, 18.7, 14.1. Anal. calcd. for C16H17N5O3S2: C, 49.09; H, 4.38; N, 17.89; S, 16.38. Found: C, 49.38; H, 4.69; N, 17.56; S, 16.55 (Fig. S11 and S12†).
3.2 In vitro PTP1B inhibition assay
In accordance with the manufacturer's protocol, the CycLex®Protein Tyrosine Phosphatase PTP1B Fluorometric Assay Kit was used to evaluate the inhibitory activity of the compounds under test on PTP1B. Each well of the 96-well microplate was promptly filled with a mixture of 40 μL of reaction buffer and 5 μL of target compounds or pioglitazone, with varying concentrations achieved by successive dilutions. The reaction mixture was then incubated at room temperature for 15 minutes after adding 5 μL of a human recombinant PTP1B enzyme solution (20 m units per μL). The following steps were taken after incubation: 20 μL of development buffer and 5 μL of fluorophospho substrate (20 μM) (×10). Each well was supplemented with 25 μL of stop solution following 15 minutes of incubation at room temperature. Following excitation at 482–502 nm, the fluorescence was checked at emission between 510 and 530 nm.24
3.3 Molecular docking coupled with dynamics simulations
Compounds were constructed, PTP1B protein (PDB = 2QBP) was prepared, and the docking protocol was done via AutoDock Vina V.1.2.0 (Scripps Research, La Jolla, CA, United States) as per reported studies.45,46 The binding site was defined based on the co-crystallized ligand and also refined to include key small molecule binding residues. Docking was proceeded under Vina Forcefield and Lamarckian_Genetics with the biological target center as the docking box center.45,46 Global search exhaustiveness of 100 kcal mol−1 and poses' maximum energy differences of 3 kcal mol−1 were set.47 Visualizing poses and compound–PTP1B binding interactions was performed via PyMol V2.0.6 (Schrödinger, NY, USA).48Docking poses were considered as reference complexes for explicit molecular dynamics using GROMACS-2019 under CHARMM-General forcefield and CHARMM36m forcefields for compounds and PTP1B, respectively.49,50 Complexes were solvated in TIP3P cubes with periodic boundaries at 10 Å marginal distances.51 Standard ionization at pH 7.4 was set for PTP1B's residues and the whole model was neutralized via chloride/potassium ions.52 Systems were minimized at steep descent (5 ps),53 and then double equilibrated at ensembles NVT (303 K) then NPT (303 K; one atmospheric_Pressure) at 100 ps each.54 Molecular dynamics were measured for 100 ns under NPT at particle-mesh Ewald to compute long-range electrostatics.55 LINCS was used at 2 fs time steps to model covalent bonding.56 The Verlet cut-off scheme was truncated at 10 Å to estimate Coulomb's and van der Waals non-bonded interactions.57 Compound–PTP1B binding-free energies were estimated via MM_PBSA calculations as per a reported study.58
3.4 Nano formulation
A pre-formulation screening of oils and surfactants was performed to investigate crucial factors regarding the produced nanoparticles. Since these nanoparticles were designed to be nano emulsions, these factors were set to be the self-emulsification ability of the oil–surfactant mixtures alongside the drug's solubility in these blends. In the preliminary screening, we investigated the following combinations: (oleic acid/Tween 20), (olive oil/Tween 20), (castor oil/Tween 20), (oleic acid/Tween 80), (olive oil/Tween 80), and (castor oil/Tween 80) in gradients of oil to surfactant ratio starting from 80:20 up to 50:50 (Table S1†).
The constituents were accurately weighed and vortexed, after which they were inspected for any phase separation. The predetermined drug dose was then loaded to formulae that showed no phase separation. The emulsification ability and grading were done as reported.59 Besides, the particle size and polydispersity index were determined according to ref. 60–63.
3.5 In vivo biological evaluation
The experimental protocol of this study was approved by the Ethics Committee of the Faculty of Pharmacy, Suez Canal University (202211RA2). The animals were maintained and used in accordance with the criteria of the National Research Council (2011). Fifty-six male albino rats weighing 150 ± 17 g purchased from the Egyptian Organization for Biological Products and Vaccines (Cairo, Egypt) were used for the present study. The animals were kept at room temperature (25 ± 5 °C) for seven days to acclimatize and had unrestricted access to a standard balanced diet and tap water on a 12 hour light–dark cycle.To calculate the liver index [(liver weight/body weight) × 100], the livers of the rats were removed, washed with ice-cold saline solution and weighed. To determine the adipose tissue index (adipose tissue weight/body weight) × (100), the white visceral (including epididymal) and subcutaneous adipose tissues were also removed, washed and weighed. A portion of the adipose tissue was stored at −80 °C to be used later for quantitative real-time PCR (qRT-PCR) assessments.
| HOMA-IR = fasting insulin level (μIU ml−1)/405 × FDG (mg dL−1), |
| QUICKI = 1/[log fasting insulin level (μU ml−1) + log FBG (mg dL−1)]. |
Calorimetric analysis was used to determine the serum levels of liver enzymes alanine aminotransferase (ALT) and aspartate aminotransferase (AST) and the renal function parameters creatinine and urea (Diamond, Egypt). In addition, the serum lipid profile was determined through the measurement of triglycerides (TG), total cholesterol (TC), low-density lipoprotein cholesterol (LDL-C) and high-density lipoprotein cholesterol (HDL-C) (Biodiagnostic, Egypt).
ELISA kits (MyBioSource, San Diego, CA, USA) were used to measure the serum levels of glutathione (GSH) (MBS724319), superoxide dismutase (SOD) (MBS036924), and catalase (CAT) (MBS726781). In addition, rat ELISA kits (MyBioSource, San Diego, CA, USA) were used to determine the levels of malondialdehyde (MDA) (Cat. No. MBS738685), tumor necrosis factor-alpha (TNF-α) (Cat. No. MBS2507393), interleukin-6 (IL-6) (Cat. No. MBS726707) and interleukin-1β (IL-1β) (Cat. No. MBS175967) in serum.
Serum leptin and adiponectin levels were measured using ELISA. In addition, their gene expression levels were measured in adipose tissue using the GoTaq® 1-Step RT-qPCR System (Promega, Madison, WI, USA). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an endogenous control. The primers and annealing temperatures are listed in Table S2.† For the experiment, 4 μL RNA template, 0.4 μL GoScriptTM RT mix for 1-step RT-qPCR, 1 μL forward and reverse primers, 10 μL GoTaq® qPCR master mix, 0.31 μL additional CXR reference dye and 3.29 μL nuclease-free water were mixed. The PCR reaction cycle included reverse transcription at 37 °C for 15 minutes, reverse transcriptase enzyme inactivation at 95 °C for 10 minutes, and 40 cycles of denaturation at 95 °C for 10 seconds, annealing for 30 seconds, and extension at 72 °C for 30 seconds. The StepOnePlus™ real-time PCR thermal cycler (Applied Biosystems, Waltham, MA, USA) was used for all real-time PCR experiments. ΔΔCt and fold change were determined,
4. Results and discussion
4.1 Chemistry
Synthesis of our compounds started using a commercially available chemical synthon, the marketed FDA-approved anti-bacterial agent, sulfadiazine, 4-amino-N-(pyrimidine-2-yl)benzene-sulfonamide 1. The nucleophilic reaction between our starting compound and appropriate isothiocyanate or phenyl isocyanate derivatives provided the first intermediates 2a–c as presented in Scheme 1. The IR spectra of the synthesized intermediates showed (N-H) bands in the range 3109–3444 cm−1. Moreover, the IR spectra of compounds 2a and 2c showed characteristic bands corresponding to their respective (CS) at 1095 and 1087 cm−1, respectively, while compound 2b showed its characteristic (CO) band at 1708 cm−1. It is worth noting that these furnished intermediates 2a–c were designed to harbor the chemically versatile urea/thiourea arms, permitting further structure derivatization and ring formation at the last step of the chemical reaction. The latter approach was highly rationalized for cutting-down chemical synthetic steps, making it cost-effective and time-saving. Further, chemical reactions and ring closure between 2a–c and either ethyl bromoacetate or ethyl-2-chloropropionate provided the target final chemical compounds 3a–c and 4a–c with their imine linker connecting the decorated hydrogen-bond warheads with the compounds' trunk regions. The IR spectra of the synthesized compounds showed the absorption bands of (CO) in the range 1747–1674 cm−1 and characteristic bands corresponding to (N–H) within the range of 3464–3356 cm−1. The obtained structures of the extra-methylated compounds 4a, 4b, and 4c were confirmed through the presence of the up-field doublet of CH3 at 1H-NMR (δH) 1.44 ppm, 1.68 ppm, and 1.13 ppm, respectively (ESI†). The quartet of the CH of oxazolidinone or thiazolidinone appeared at 1H-NMR (δH) 4.60 ppm, 4.32 ppm, and 3.45 ppm, respectively. On the other hand, the respective 1H-NMR spectra of 3a, 3b, and 3c showed an up-field singlet signal corresponding to CH2 of oxazolidinone or thiazolidinone at (δH) 4.17 ppm, 4.33 ppm, and 3.50 ppm, respectively. Additionally, the 1H-NMR spectra of the N-ethyl substituted thiazole-based compounds 3c and 4c showed the up-field quartet of CH2 (ethyl) at (δH) 3.46 ppm and 3.35 ppm, whereas the up-field triplet of CH3 (ethyl) was observed at 1.12 ppm and 1.03 ppm, respectively. In addition, the 13C-NMR spectra of the synthesized compounds 3a, 3b, and 3c showed the characteristic aliphatic signal of CH2 of oxazolidinone or thiazolidinone at (δC) 37.5, 33.9, and 36.2 ppm, respectively. On the other hand, the 13C-NMR spectra of compounds 4a, 4b, and 4c showed the aliphatic CH of oxazolidinone or thiazolidinone at (δC) 21.1, 22.4, and 21.4 ppm and also CH3 appeared at (δC) 19.4, 19.9 and 18.7 ppm, respectively. The C13-NMR spectra of compounds 3c and 4c showed signals belonging to the ethyl group at (δC) 12.5 (CH3), 31.3 (CH2), and 14.1 (CH3), 38.9 (CH2), respectively.
For exploring different spacer sizes connecting the decorated hydrogen-bond warheads with the compounds' trunk regions, several compounds were synthesized. N-Acylation of the sulfadiazine starting compound with 2-chloroacetyl chloride in the presence of triethylamine and DMF afforded 2-chloro-N-(4-(N-pyrimidin-2-ylsulfamoyl)phenyl) acetamide 5 as another chemical synthesis versatile intermediate. Within the latter reaction, triethylamine was used to scavenge the released hydrogen chloride during the reaction and the structural conformation was determined through the obtained IR spectra showing the characteristic absorption band of the amidic carbonyl (CO) at 1681 cm−1. The terminal chloroacetamide arm in intermediate 5 permitted the straightforward synthesis of several heterocyclic five-membered hydrogen-bond warheads 6–9 being decorated with lipo/polar balanced functionalities (Scheme 2). This attempt was achieved through a coupling reaction of chloroacetamide 5 with ammonium thiocyanate to give the 2-thiocyanatoacetamide intermediate, which was rapidly cyclized into 2-imino-4-oxothiazolidine final compound 6. The IR spectra of this compound showed two bands at 3194–3271 cm−1 corresponding to its respective NH groups. On the other hand, cyclization of chloroacetamide 5 with ethyl acetoacetate in dimethylformamide and pyridine led to the final acetyl-substituted pyrrolidone-based compound 7. The IR-spectra of 7 confirmed the presence of carbonyl groups from the characteristic absorption bands at 1712 cm−1. Regarding the synthesis of another pyrrolidone-based compound 8, the initial alkylation of chloroacetamide 5 with ethyl cyanoacetate gave the dihydropyrrole intermediate followed by intramolecular cyclization, which oxidized the intermediate under the reaction conditions to yield the target compound. Notably, the IR spectra of this novel pyrrole derivative exhibited a nitrile band (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) at 2194 cm−1 as well as two bands at 1662 and 1693 cm−1 due to carbonyl (CO). Finally, coupling 5 with appropriate isothiocyanate or phenyl isocyanate in the presence of pyridine, followed by nucleophilic substitution of chlorine by the sulfur or oxygen atom of isothiocyanate or isocyanate, resulted in the formation of thiazolidine, oxazolidine, and oxothiazolidine rings of the N-(pyrimidin-2yl) benzene sulfonamide derivatives 9a, 9b and 9c. The 1H-NMR spectra of these compounds exhibited a singlet signal of CH2 of thiazolidine and oxazolidine rings at δH 3.51 ppm, while as the ethyl group of compound 9c exhibited a triplet signal of CH3 at δH 1.30 ppm and a quartet signal of its CH2 at δH 3.20 ppm.
N) at 2194 cm−1 as well as two bands at 1662 and 1693 cm−1 due to carbonyl (CO). Finally, coupling 5 with appropriate isothiocyanate or phenyl isocyanate in the presence of pyridine, followed by nucleophilic substitution of chlorine by the sulfur or oxygen atom of isothiocyanate or isocyanate, resulted in the formation of thiazolidine, oxazolidine, and oxothiazolidine rings of the N-(pyrimidin-2yl) benzene sulfonamide derivatives 9a, 9b and 9c. The 1H-NMR spectra of these compounds exhibited a singlet signal of CH2 of thiazolidine and oxazolidine rings at δH 3.51 ppm, while as the ethyl group of compound 9c exhibited a triplet signal of CH3 at δH 1.30 ppm and a quartet signal of its CH2 at δH 3.20 ppm.
It is worth noting that all the above acetamido-furnished compounds possessed a zero-atom sized spacer, which would allow us to investigate the impact of spacer contraction on hypoglycemic bioactivity and PTP1B target pocket accommodation. Moving towards the opposite direction of compound derivatization and development, expansion of the spacer into two- and even three-sized atoms was then conducted to obtain the optimum structural topology being required for preferential biological activity and target pocket accommodation. Heating chloroacetamide 5 under refluxing conditions with N-substituted piperazine in acetonitrile solvent led to the formation of the substituted piperazin-1-yl)-N-(4-(N-pyrimidin-2-ylsulfamoyl) phenyl) acetamide compounds 10a and 10b with their elongated three-atom acetamido spacer (Scheme 2). The 1H-NMR spectra were characterized by multiplet signals at δH 2.16–3.60 ppm for (CH2)4 of the piperazine ring.
Through a different synthetic pathway, the starting compound sulfadiazine, 4-amino-N-(pyrimidine-2-yl)benzene sulfonamide 1, was refluxed with 2-(bromomethyl)-1H-benzo[d]imidazole affording final compound 11 incorporating a two-atom sized spacer (Scheme 2). Confirming the afforded compound, the IR spectrum showed three (N–H) bands at 3255, 3356, and 3421 cm−1, whereas the corresponding 1H-NMR spectrum illustrated the characteristic up-field singlet corresponding to CH2 at δH 4.25 ppm. For compounds 10a, b, and 11, an elongated bigger-sized topology was assigned as compared to the other synthesized compounds while also inheriting greater structure flexibility owing to their respective rotatable acetamido or amino methylene spacers. Such structural advantages were rationally designed to permit deeper anchoring and better lodging of synthesized compounds at the PTP1B binding site while adopting favored conformations and structural maneuvers that could minimize possible steric clashes with the receptor site.
4.2 In vitro PTP1B inhibition assay
The investigated compounds were tested against protein tyrosine phosphatase PTP1B in comparison with pioglitazone, as summarized in Table 1. Interestingly, compounds 3c, 4c, 7, 8, and 10a exhibited potent PTB1B inhibition with IC50 values down to the sub-micromolar levels and percentages of inhibition above 84% compared to the positive control market drug, pioglitazone (IC50 = 0.65 μM; 85% inhibition, Fig. S22†). In the meantime, compounds 4a, 4b, 6, and 10b exhibited moderate protein inhibition with IC50 values ranging from 1.89 μM to 8.97 μM. In contrast, the rest of the synthesized compounds exhibited modest-to-poor protein inhibition values.| Compound | % inhibition (@ 10 μM) | PTP1B IC50 (μM) |
|---|---|---|
| IC50 values were expressed as mean ± SD of three independent trials and were calculated from the non-linear regression dose–response curve of viability percentage at four concentrations (0.01, 0.1, 1, 10 μM) at a time interval of 30 min. NA = not active. | ||
| 3a | 56.30 ± 2.90 | 11.13 ± 0.10 |
| 3b | 19.00 ± 0.57 | NA |
| 3c | 87.00 ± 3.00 | 0.62 ± 0.01 |
| 4a | 81.20 ± 2.80 | 1.89 ± 0.10 |
| 4b | 80.30 ± 2.90 | 1.95 ± 0.20 |
| 4c | 84.00 ± 3.10 | 0.71 ± 0.01 |
| 6 | 71.60 ± 2.80 | 8.97 ± 0.79 |
| 7 | 84.10 ± 2.60 | 0.83 ± 0.01 |
| 8 | 84.90 ± 2.80 | 0.72 ± 0.04 |
| 9a | 51.40 ± 1.80 | 13.98 ± 1.10 |
| 9b | 18.90 ± 0.80 | NA |
| 9c | 59.70 ± 2.10 | 16.64 ± 0.70 |
| 10a | 90.10 ± 3.40 | 0.57 ± 0.02 |
| 10b | 61.40 ± 2.80 | 3.67 ± 0.90 |
| 11 | 86.90 ± 3.10 | 0.68 ± 0.05 |
| Pioglitazone | 85.00 ± 3.15 | 0.65 ± 0.01 |
Comprehensive investigation of the structure–activity relationship (SAR) has been considered as a crucial part of pharmacology for guiding future lead optimization and development. Exploring the compounds' structure diversity and its correlation with their in vitro PTP1B inhibition activity revealed interesting insights. Firstly, lengthy compounds such as 10a and 11 exhibited more favourable activity profiles (sub-micromolar range: IC50 = 0.57 ± 0.02 and 0.68 ± 0.05, respectively), which could be attributed to their ability to occupy multiple PTP1B sites. This was also obvious for much more extended compounds incorporating a spacer group (imine, acetamide, or methylamino) between the polar warhead and the compound's trunk part. The latter could partially explain the relatively better activity values for 3c and 4c (IC50 = 0.62 ± 0.01 and 0.71 ± 0.01) as compared to corresponding close analogs, 7 and 8 (IC50 = 0.83 ± 0.01 and 0.72 ± 0.04). However, correlating better PTP1B inhibition activity to just lengthy structures would be an oversimplification of the SAR analysis since structurally extended compounds, like 10b, provided moderate activity at the micromolar concentration. This observation would confirm the impact of the polar warhead type, bulkiness, and the substituent's nature and orientation on the compounds' activity profiles. This brought us to a second SAR insight where activity is highly dependent on the steric size and bulkiness of the polar warhead. For the 5-membered isosteric heterocyclic polar warheads (thiazole, oxazole, and pyrroline-based compounds), there is a great activity shift from nanomolar concentration up to micromolar ones following introduction of bulky substituents (phenyl moiety). This was obvious for the comparative activity profiles of 3c and 4c in relation to their bulkier close analogs 3a, b and 4a, b or even a distant warhead analogue (piperazine) 10b, where all harbor the phenyl substituent on their warhead groups.
This would bring us to the third SAR insight where activity on PTP1B has been highly dependent on the nature of the compounds' polar warheads. The activity of compounds against PTP1B is infavour to the warhead groups of high hydrogen bond potentiality as being decorated with several hydrogen bond donors and/or acceptors for relevant interactions at the hydrophilic site D. That is why the well decorated polar warheaded compounds of the thiazole, oxazole, and pyrroline series depicted nanomolar range activities, being pretty obvious with their highly polar less steric members, 3c, 4c, 7, and 8. Despite the fact that compound 6 possesses a polar decorated warhead, it depicts moderate activity (IC50 = 8.97 ± 0.79) as compared to its chemical class members, 7 and 8 (IC50 = nanomolar concentrations). This could be due to the higher hydrogen bond potentiality of the pyrrolidione rings (7 and 8) over that of the 4-oxo-thiazol-2-imine since oxygen is more electrophilic than the NH group. Further, compounds 7 and 8 possess extra hydrogen bonding arms (acetyl and cyano moieties) capable of mediating more polar networks with the target pocket site D. Although the third SAR insight seems solid and well validated, its application is not straightforward for full explanation of the comparative activity profile. It is worth noting that an extra methyl substitution was found beneficial for 4a as compared to its congener, 3a (IC50 = 1.89 ± 0.10 versus 11.13 ± 0.10). This observation could confirm the favourable impact of not just the polar well decorated warhead, but also the balanced lipophilic/polar nature of these head groups for a compound's activity. Generally, having a balanced lipophilic/polar characteristic for a compound would be greatly favorable for its pharmacodynamics/target binding (lower solvation entropy) and pharmacokinetic profiles (better dissolution and membrane passage), impacting the compound's overall activity.69 Finally, validation of all these SAR insights has been highlighted with the applied positive reference control, pioglitazone, exhibiting potent activity and pharmacophoric features in good agreement with SAR insights. The reference compound exhibits a polar thiazolidinedione head with one carbon spacer (methylene bridge) between the warhead and phenyl trunk which was correlated to its nanomolar activity profile (IC50 = 0.65 ± 0.01). The presence of the flexible ethoxy spacer connecting its trunk with the heterocyclic tail group was suggested to be beneficial for providing extra length and flexible maneuver capability for the compound to reach the PTP1B selectivity pocket (site B).
4.3 Computational screening analysis
 | ||
| Fig. 2 Predicted poses of tested compounds at PTP1B catalytic sites and subsites. (A) Overlaid docked synthesized ligands (magenta lines) and co-crystallized ligand (CID11786814; blue sticks) at surface representation of the PTP1B active pocket; PDB = 2QBP. (B–H) Predicted poses of synthesized hits (magenta sticks); (B) 3a, (C) 9a, (D) 4a, (E) 9c, (F) 3c, (G) 8, and (H) 10a at the PTP1B pocket. Only amino acids within a 5 Å radius from the inbound ligand are shown in lines, colored in regard to their corresponding locations at structural loops, and finally labelled in protein sequence numbers. Predicted polar/hydrogen interactions are displayed in black-dashed lines. | ||
For exploring the different activity profiles of the synthesized compounds, comprehensive evaluation of the compound's binding mode was conducted. Compound 3a is an example of a sterically hindered head having its thiazolone ring settled at the opening of the P-loop near the site D interface. This orientation predicted polar interactions for the thiazolone ring with site C Tyr42 (bond length/angle; 3.2 Å/125°), site D Lys120 (3.4 Å/111°), and Asp181 (3.1 Å/121°) of the WPD loop (Fig. 2B). Close proximity towards P-loop Arg221 and Ser216 sidechains suggested relatively weak H-bonding (>3.4 Å) or even water-bridge polar interaction with the ligand's oxo group. The thiazolone ring was further stabilized through hydrophobic T-shaped stacking with WPD loop residue Phe182 (4.6 Å) contributing to the 3a–target complex stability. Stacking with Phe182 was reported to be beneficial for stabilizing bis-(p-phosphophenyl) methane-based PTP1B inhibitors with comparable affinity (Ki = 16 μM).28 Owing to its steric bulkiness, 3a's N-substituted phenyl group was settled facing the site D interface. Concerning the tail part of 3a, the sulfonamide linker and terminal pyrimidine ring depicted H-bonds with site B Arg24 (1.9 Å/158°) and Gln262 (3.4 Å/143°), respectively, in addition to van der Waal attractions for the heterocyclic ring towards Met258, Gly259, and Leu260. Reported data showed that polar interaction with Q-loop Gln262 assisted in improving target selectivity for bis-nitrophenols scaffolds.70
3a's comparable orientation was also predicted for 9a, yet with quite more retracted orientation for its head ring owing to its larger steric-hindered imine phenyl moiety (Fig. 2C). Fewer polar interaction networks were depicted for 9a since its thiazolone head managed to adopt H-bonds with the P-loop Ala216 mainchain (3.2 Å/128°) and WPD Asp181 (2.8 Å/132°) while lacking relevant contacts with the site D Lys120 residue and site C Tyr46. Additionally, improper orientation of its terminal pyrimidine ring made it lose its polar contact with site b Arg24. Instead, polar interaction with PTR-loop/site C Asp48 (3.3 Å/121°) and pi–cation interaction with Arg24 (4.6 Å) managed to compensate for and provide stabilization of the tail scaffold at selectivity site B. A similar kind of compensatory interaction with Lys120 (4.7 Å) was observed to stabilize the imine phenyl moiety at site D through pi–cation interaction. Both predicted 4SD and 15SD orientations were correlated to moderate docking scores of −5.576 and −5.177 kcal mol−1, respectively, being successfully translated into their moderate two-digit micromolar activities. An improved docking score was assigned to 4a (−6.117 kcal mol−1), the methyl-substituted analogue of 3a. Despite depicting the close steric-hindered orientation of its phenyl moiety, the extra methyl arm at the ligand's thiazolone head ring showed insertion within the P-loop site, which seems to provide a gripping point for the ligand's anchoring at the PTP1B catalytic site (Fig. 2D). The latter orientation showed proper anchoring of the tail group at site B, mediating closeness and polar contacts with Arg24 (1.8 Å/168°) and Gln262 (3.4 Å/145°) as well as strong contacts with Asp181 (3.1 Å/128°) and Tyr46 (3.3 Å/121°) by the head scaffold.
On the other hand, the oxazole ring bioisosteres/congeners of 3a and 9a, which are 3b and 9b, respectively, were assigned with quite low negative docking scores −4.620 kcal mol−1 and −4.948 kcal mol−1. This could be due to for the different electronegativity between the sulphur and oxygen atoms within the five-membered system. Pagliai et al. showed through ab initio molecular dynamics, Raman spectra, and density functional theory analysis for thiazole and oxazole rings that higher electronegativity is concentrated at the oxygen atom rather than sulphur.71 Moreover, the sulphur atom within this five-membered system even adopted a positive charge characteristic as compared to its oxygen congener. This could explain why 9a in our results could mediate polar contact with Asp181 at the WPD loop domain. In this regard, more electrostatic charges would be concentrated at the nitrogen and oxo group within the thiazolone rings as compared to the oxazolone one in 3b and 9b. Since the oxo group at both 3a and 9a was predicted to be important for anchoring ligands at the PTP1B pocket, it was suggested that this compromised electronegativity would impact their binding affinity towards polar residues, resulting in lower fold-inhibition activities (<20%). This could also partially explain why 9c furnished a moderate docking score (−4.576 kcal mol−1) correlating to micromolar inhibition activity despite having less sterically hindered ring substitution (ethylamine moiety). Harboring an oxo-ring isostere in the thiazole ring could have compromised the polar network of 9c despite its deep anchoring at the P loop site (Fig. 2E).
Moving towards less sterically hindered ring derivatives, compounds like 3c and 4c (−7.371 and −7.217 kcal mol−1, respectively) predicted deeper anchoring at the catalytic P-loop site with more extended polar networks with their H-bonding warheads. This was obvious for 3c (Fig. 2F) where its thiazolone ring depicted a salt bridge with P-loop Arg221 (1.9 Å / 128° and 2.8 Å/127°), catalytic nucleophile Cys215 (3.6 Å/119°), and WPD loop Asp181 (2.9 Å/129°). The ligand's hetero-ring was further stabilized through T-shaped pi–pi packing with Phe182 (4.0 Å). At the other end, the ligand's tail showed optimum closeness/orientation towards site B Arg24 furnishing relevant H-bonding (2.3 Å/135°) while maintaining H–pi attraction with Asp262 (3.1 Å). All of which would be successfully translated into tight PTP1B catalytic site blocking relevant for the high inhibitory activity profile obtained at nano-molar concentrations. Comparable P loop-oriented deep-anchoring was also depicted for 8 and 7, which was correlated to their high negative docking scores (−7.484 and −6.965 kcal mol−1, respectively). Exemplary for compound 8, the dioxo pyrrole ring head mediated salt bridges with P loop Arg221 main and sidechains (2.5 Å/149° and 2.1 Å/139°), as well as polar interactions with Asp181 (2.4 Å/127°) and Tyr46 (3.0 Å/119°) (Fig. 2G). The benzene was quite sandwiched between Phe182 (5.0 Å) and Tyr46 (3.9 Å) through T-shaped and displaced pi–pi stacking, respectively. Interestingly, the ligand's terminal tail depicted preferential direction towards site C rather than site B which could be related to the above hydrophobic sandwiching and singularly depicted polar contact with the Asp48 mainchain (3.4 Å/147°). This caused us to believe that 8 and 7 are better site AC inhibitors. Despite the fact site C is quite undruggable, flat and solvent exposed, potent selective site AC PTP1B was introduced, relying mainly on developed salt bridges with lining residues.29,30
Regarding our lengthy and structure-extended compounds, ligands showed relevant docking poses reaching out across sites A and D of the PTP1B site. Compound 10a (−7.842 kcal mol−1) depicted deep anchoring with its substituted piperazine ring at the P-loop site. Furnishing extended H-bonds with lining residues via its ternary aliphatic amine, the ligand's head was tightly anchored at the P-loop site (Fig. 2H). Polar contacts with Arg221 side and mainchains (3.0 Å/150° and 2.9 Å/132°) and WPD-loop Asp181 (2.3 Å/144°) were depicted at optimum parameters. At the compound's terminal, the pyrimidine ring showed relevant anchoring at site B reaching far more than any other investigated compounds. Double H-bonding with Arg24 (2.7 Å/121° and 3.4 Å/123°) and polar contact with Asp48 (3.3 Å/118°), in addition to pi–cation hydrophobic interaction with Arg24 (3.8 Å), were predicted to contribute to site B–ligand stability as well as presenting 10a as a better site AB inhibitor. Methyl substitution was suggested to be beneficial to increase the electrophilicity of piperazine nitrogen for H-bonding while maintaining low steric-hindrance against P-loop-oriented anchoring for being small in size. In contrast, phenyl substitution on the piperazine head ring of compound 10b was considered detrimental for binding at the P-loop owing to increased steric resistance. In brief, optimum PTP1B inhibition activity could be correlated with the ability of binding ligands to achieve deep anchoring at the P-loop via furnished complex polar binding with lining and/or vicinal residues, including Lys120, Asp181, and Tyr46. On the other hand, the ability of the ligand to extend back deep towards site B is considered relevant for excellent site AB binding and selectivity. However, short ligands with salt bridging at the P-loop and favored direction towards site C are also considered relevant for PTP1B inhibition activity and selectivity profiles.
Docking poses were validated through the redocking procedure for the co-crystallized ligand as it illustrated a low-value RMSD of 1.176 Å for aligned co-crystallize/redocked poses (redocked ligand energy score = −8.318 kcal mol−1). Obtaining redocked RMSD with values less than 2.0 Å generally highlights that the adopted docking procedure is valid where furnished docking poses and their corresponding binding energies would be of relevant biological significance.49,72–75 Additionally, docking findings were further confirmed as the docked ligands managed to replicate canonical binding modes as well as adopt comparable patterns of residue-oriented interactions in relation to the literature small molecules reported with actual or even potential PTP1B inhibition activity.76–79
Notably, simulated proteins depicted typical thermodynamics where carbon-alpha RMSDs were elevated at initial run times due to system relaxation. Subsequently, RMSDs were leveled-off at average values for >50% of the simulation runs. It worth noting that RMSDs of holo proteins (ligand bounded) were of minimal fluctuations and lower averages than those of unliganded/apo enzymes (2.04 ± 0.15 Å versus 2.41 ± 0.37 Å) (Fig. 3A). Therefore, it was inferred that PTP1B enzyme gained higher compactness and favored stability following compound binding/interactions. Different RMSD tones among holo proteins illustrated limited fluctuations/greater stability for 10a (1.96 ± 0.17 Å) and the co-crystallized ligand (1.98 ± 0.25 Å). In contrast, slightly higher fluctuations were assigned for 3c and even more for the 8-bound PTP1B proteins (2.01 ± 0.45 Å and 2.26 ± 0.64 Å, respectively). Despite different protein RMSD tones across the simulation window, all proteins managed to converge around a mean RMSD of almost 2.27 Å at the end of the simulations. Such behavior has been reported to be relevant for proteins' stability/convergence and the validity of the adopted molecular dynamics protocols requiring no further simulation time extensions.
 | ||
| Fig. 3 Thermodynamic stability analysis of the simulated compounds' ligands bounded to PTP1B proteins. (A) Carbon-alpha RMSD of PTP1B protein; (B) ligands' RMSDs, in relation to simulation time (ns). (C) Overlaid compound/PTP1B extracted frames at simulation beginning and ending for 10a (upper left), 3c (upper right), 8 (lower left), and the co-crystallized molecule (upper right quadrant). Simulated molecules (sticks) and in complex PTP1B proteins (cartoon) are in green and red colors corresponding to zero and 100 ns extracted frames. Residues at 4 Å from the in complex compounds are shown in gray surface representation. | ||
Concerning the sole ligand's RMSDs, limited fluctuations and steady tones were demonstrated for 10a (3.71 ± 0.46 Å) as compared to other ligands (3.69 ± 0.87 Å for 3c, and 4.68 ± 0.94 Å for 8), even the co-crystallized ligand (3.94 ± 1.05 Å), Fig. 3B. Despite limited fluctuations at the initial times, ligand 3c depicted lower value RMSD trajectories, only second to 10a. Increased RMSD tones were depicted for 8 and co-crystallized ligands beyond 30 ns or 10 ns time frames, respectively, inferring significant orientation shift at the binding pocket. For the co-crystallized ligand, an abrupt RMSD rise (from 1.46 to 2.65 Å) was detected around 12 ns. Nevertheless, the co-crystallized ligand rapidly attained its equilibration plateau around an average RMSD value till the end of the simulation, except for limited fluctuations at 80 ns. It is worth noting that the depicted ligand RMSDs never exceeded 3-fold those of corresponding bounded enzymes which confirms ligand-pocket accommodation, ligand–enzyme complex stability, and successful protein convergence.84,85
Time evolution of the compound–PTP1B complex conformations/orientations was monitored over overlaid extracted frames at the start and end of dynamics runs (Fig. 3C). Limited orientation/conformation alterations were assigned with 10a at the dynamic run end. The ligand maintained its elongated anchoring at the target's site B, while being more deeply anchored at the catalytic site A cleft. On the other hand, limited shift was depicted for 3c's ligand terminal ring at site B, yet the compound's polar head ring was maintained at deep anchoring at catalytic site A. The latter dynamic behavior could explain the limited RMSD fluctuations observed with the ligand across the time frames. Interestingly, more terminal scaffold drift was seen with compound 8 at the simulation end run. This ligand showed more directed orientation towards the target's site C while keeping its polar head with relevant anchoring at site A. Such observed drift could be due to the higher RMSD trajectories obtained for 8 as compared to any other simulated compound. Conformational analysis for compound 8 would further highlight the ligand's preferential inhibition activity as a site AC inhibitor against the PTP1B target. Finally, the co-crystallized ligand showed stable orientation at the target's sites A and B, however the ligand adopted a different conformation for its polar ring head. The ligand's thiophene ring showed relevant rotation around its axis depicting a different conformation in relation to its initial architecture at the simulation beginning. This caused quite a shift for the ring's double substituted carboxylate arms at the P-loop site. The 2-carboxylic acid group was shifted towards the solvent side when resting at the site D interface; yet significant head anchoring and strong polar contacts with site A and D lining residues were maintained by the ring's oxyacetic acid arm. The latter observed conformational changes could explain the abrupt RMSD rise around 12 ns for the simulated co-crystallized ligand. It is worth noting that examining the WPD-loop conformation at the end of simulation runs depicted a stable conformation for the target's inactive state where the WPD-loop maintained its closed folding over the P-loop site.
Further stability analysis was highlighted through RMS trajectory monitoring where the protein's flexibility/immobility profiles were dissected down to their comprising amino acids.86 Generally, RMSF analysis permits an understanding of residue-wise dynamic behaviors at the enzyme's active site/vicinal loops and would aid in pinpointing the key residues for the ligand's anchoring.87,88 In concordance with RMSD findings, higher flexibility/mobility was depicted for the apo PTP1B target protein as compared to the holo states (Fig. 4). This would infer the compounds' positive impacts on PTP1B stability being extended beyond the canonical binding pocket impacting the distant regions. RMSF analysis further illustrated the typical protein's dynamic behaviors where the far carboxy terminal residues were inherited with higher immobility as compared to core amino acids which was reported to be typical of PTP1B thermodynamics and B-factors.89 Notably, core residue ranges across Leu59-to-Tyr66 and Gln127-to-Leu140 showed significant flexibility (RMSFs reaching 2.16 Å) due to reported low intermolecular bindings and reduced secondary structure compactness.90–92 In contrast, the illustrated immobility profiles for the N-terminus and vicinal amino acids indicate the compound-related stability for the residues within proximity towards the protein's catalytic loops and Tyr-P cleft.
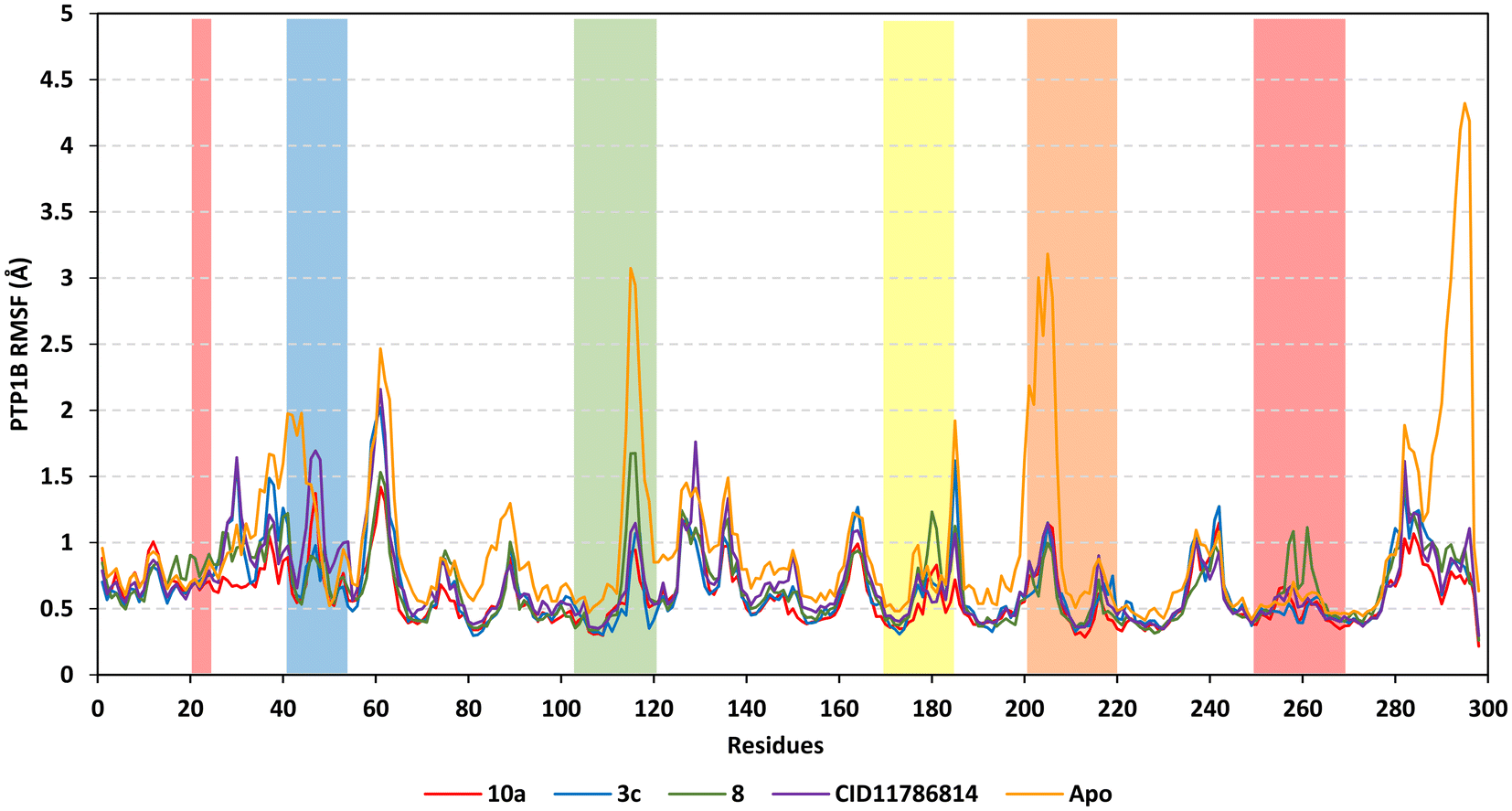 | ||
| Fig. 4 Analysis of RMSFs for PTP1B protein across the entire molecular dynamics runs. Residue-wise flexibility contributions of the target protein in complex to 10a, 3c, 8, or the co-crystallized ligand are represented as compared to the apo/unliganded state. Secondary structural loops and main catalytic sites are color highlighted in residue ranges: 40–52 (PTR-loop; blue), 110–121 (E-loop; green), 176–186 (WPD-loop; yellow), 214–221 (catalytic P-loop; orange), 20–27/254–269 (Q-loop and Tyr-P cleft; red). | ||
Concerning the different PTP1B loop stability, the Q-loop followed by the catalytic P-loop was assigned with the most immobility and stability profiles for all bounded proteins as compared to different protein regions. In contrast, both the PTR-loop and E-loop depicted higher mobility profiles as compared to the catalytic P-loop and even other catalytic-associated loops, the WPD. The latter infers the significant role of site A (P-loop) and site B (Q-loop) residues in the preferential ligand's stability and pocket accommodation. Additionally, the limited mobility profile for the WPD-loop confirmed the retainment of the loop's inactive conformation across the whole molecular dynamics simulation providing tightness for the ligand's head ring anchoring at the P-loop site. Notably, compound 8 was the only ligand that showed the highest fluctuating protein residues at the Q-loop/Tyr-P cleft (residue range Gly259-to-Leu267) as well as the lowest RMSF tones (high stability) at the PTR-loop. This could be due to the preferentially depicted binding of 8 at site C (embedded at PTR-loop) rather than site B (at Q-loop/Tyr-P cleft) throughout the previously described docking studies. This could further highlight the preferentiality of compound 8 as a site AC inhibitor. In this regard, all highlighted residue ranges were considered important for the compound's stability and pocket binding as well as being in good concordance with the above-described residue-directed docked findings.
Final analysis was performed to estimate the free binding energies via molecular mechanics-Poisson Boltzmann surface area (MM_PBSA) calculations in order to understand the nature of compound–PTP1B binding, estimate affinity magnitudes, and individual energy-based contribution for key pocket amino acids.93 MM_PBSA calculation is heavily used being as accurate as free-energy perturbations, yet at much reduced computational expense.58 The estimated total binding energy of compound 10a came just next to the co-crystallized ligand exhibiting close binding affinities (−61.77 ± 21.32 and −69.16 ± 30.50 kJ mol−1, respectively) as shown in (Fig. S23A†). Relatively lower negative-value free binding energies were estimated for 3c (−54.72 ± 26.90 kJ mol−1) and the site AC binding 8 (−50.53 ± 5.54 kJ mol−1). The latter's preferential affinity for 10a and the co-crystallized ligand confirms the advent of the compound's elongated topology for reaching across site A and site B as well. This was in good agreement with findings in the literature showing preferential binding and higher potency/selectivity correlated to site AB inhibitors.29
Dissecting the total free-binding energy to its constituting energy terms showed that the four simulated ligand–target complexes exhibited superior van der Waal energy contributions over electrostatic potentials. The hydrophobic potential preferentiality was reaching more than 4-fold for the synthesized compounds and just double for the co-crystallized ligand. The electrostatic potential was higher for the co-crystallized ligand (−89.70 ± 25.62 kJ mol−1) as compared to the synthesized compounds (more than the double) owing to its ionizable negatively charged free carboxylate moieties. However, this was associated with much greater solvation polar penalties (231.40 ± 21.20 kJ mol−1) that would compromise the co-crystallized ligand anchoring since binding is a solvent-displacement procedure. Additionally, the higher aromatic functionalities at the co-crystallized ligand could impose relevant solvation penalties for displacing high-ordered water molecules at the protein's non-polar surface. Nevertheless, the compound's deep anchoring towards the more lipophilic Tyr-P cleft can partially compensate for the predicted solvation entropy by furnishing high van der Waals energies and so high total-free binding energy. Similarly, the higher van der Waal energies for 10a as compared to other synthesized compounds further highlight the advent of elongated structural topology to anchor the terminal tail scaffold at the more hydrophobic site B and Tyr-P cleft.
Thus, the above-depicted patterns highlight the beneficial strategy of introducing polar/less ionizable functionalities at our synthesized compounds' warhead ring. In these terms, we could manage to satisfy the high-charged density of the P-loop while maintaining favorable binding at the catalytic site with minimized solvation penalty and suggested improved kinetics. Additionally, introducing balanced hydrophobic/polar functionality could be considered the next step of lead optimization, where extra branched/extended non-polar scaffolds capable of achieving further extension down to the Tyr-P site are added. The scaffold should also be decorated via hydrophilic/polar substitutions for fulfilling the limited polar residues (e.g. Arg24, Arg258, Gln262) lining the surface sub-pockets. Exemplary among such moieties are the tetrazole ring and bioisosteres of the carboxylate chemical group.
Per residue, energy contributions for the simulated target proteins showed coherent findings to those obtained with RMSF analysis. Interestingly, a wide range of Q-loop/Tyr-P cleft, WPD-loop, and catalytic P-loop residues were of higher negative energy contributions as compared to those at the PTR and E-loop (Fig. S23B†). Residue-wise energy contributions were at their highest values for 10a and the co-crystallized ligand at the Q-loop/Tyr-P cleft. In contrast, compound 8 showed preferential higher negative energy contributions for residues at the PTR-loop confirming the ligand's binding preferentiality towards sites AC. Adopting ≥−2.00 kJ mol−1 cut-off for significant energy contributions, residues like Arg24 (−3.80 to −6.05 kJ mol−1), Met258 (−2.14 to −5.27 kJ mol−1), Gln262 (−4.10 to −5.49 kJ mol−1) at Q-loop/Tyr-P cleft; Asp181 (−9.59 to −10.36 kJ mol−1), and Phe182 (−2.24 to −5.67 kJ mol−1) at the WPD-loop; Ile219 (−2.63 to −6.38 kJ mol−1) and Arg221 (−2.37 to −5.86 kJ mol−1) at the P-loop were depicted important for 10a, 3c, and co-crystallized ligand binding. On the other hand, those residues were of low negative energies or even positive repulsive ones in the case of the 8 bounded complex. Nevertheless, higher negative attractive energies were depicted with Tyr46 (−9.15 kJ mol−1) and Asp48 (−13.19 kJ mol−1) at the PTR-loop and Lys120 (−3.64 kJ mol−1) at the E-loop for 8. It is worth mentioning that the mixed hydrophobic/polar nature of most binding-relevant amino acids illustrated the advent of introducing balanced functionalities within the compound structures. Finally, the co-crystallized ligand was assigned with high positive repulsive energy contributions, mostly recognized with Q-loop vicinal residue Asp29 (6.34 kJ mol−1), PTR-loop Asp48 (9.75 kJ mol−1), and Q-loop Arg254 (18.10 kJ mol−1). This further highlights the impact of the ligand's extended aromaticity as well as ionizable war head in mediating unfavored polar solvation penalties against its binding to the target site.
4.4 Nano-formulation and bioactivity data
Interest in nanotechnology has increased following evidence of its effectiveness in improving the pharmacokinetic profile of drugs. This involves improving solubility, dissolution rate, and stability, and most importantly, regulating the permeability of the drug through absorption in the membranes, resulting in lower drug dosage.91 Owing to the above predicted in silico ADMET/pharmacokinetic profiles, it was assumed that nano formulation for the top-active compound would be beneficial and worthy of further investigation. The oils screened for their ability to emulsify and solubilize the required dose of the active compound were primarily chosen for their edibility. Therefore, they will be administered orally with great ease. The formula of choice was oleic acid and Tween 80 in a composition ratio of 70:30, and it displayed a uniform droplet size distribution of grade A (clear with slight bluish)59 as the polydispersity index was 0.181 and the mean particle size was around 135.2 nm (Fig. S24†).Further, conducting screening of the dispersion by transmission electron microscopy (TEM) verified the particle size range and confirmed the spherical droplet shape formation of the nano dispersions (Fig. S25†). The decision originated after inspecting all screened formulae for any phase separation, drug precipitation, and the formula that would produce nanometric dispersion. The chosen formula fulfilled all these requirements. The reduced particle size of the dispersion modulates the drug absorption and bioavailability by increasing the interfacial area for drug release.94 Furthermore, the polydispersity index expresses the degree of homogeneity of the dispersion with values that range from zero to one. The lower the value, the more favorable, indicating the uniformity of the dispersion.94
4.5 In vivo investigation for antidiabetic and anti-obesity activities
Identified hits, out of the computational studies, 3c, 8, 10a and 11 were then pursued for their in vivo activity in hampering T2DM-linked hyperglycemic status, obesity, and disease-associated pathogenesis, including oxidative stress and inflammation. A group of rats was also treated with a prepared 10a nano-formulation. HFD + STZ-induced T2DM rats showed a significant increase in FBG, plasma insulin levels, insulin resistance, % body weight gain, adipose tissue index, TG (tri-glyceride), TC (total cholesterol), and LDL-c (low density lipoprotein cholesterol), and a significant decrease in insulin sensitivity (indicated by QUICKI) and HDL-c (high density lipoprotein cholesterol) compared to the control group (p < 0.001) (Tables 2 and 3). Oral administration of all the synthetic compounds resulted in a significant decrease in FBG, insulin resistance index (HOMA-IR), and% body weight gain compared with the control (HFD + STZ) rats. Moreover, all the administered synthetic compounds, except compound 8, significantly decreased the adipose tissue index, TG, TC and LDL-c, but increased HDL-c compared to the HFD+STZ control group (Table 3). Interestingly, the rats receiving 10a (NPs) had the lowest FBG, HOMA-IR, plasma insulin, TG, TC, and LDL-c levels and the highest HDL-c levels (Tables 2 and 3). In an earlier study, the efficacy of benzene-sulfonamide nanoparticles as antidiabetics was demonstrated.92| Groups | FBG (mg dL−1) | Insulin (μIU mL−1) | HOMA-IR | QUICKI |
|---|---|---|---|---|
| Data are expressed as mean ± SD and analyzed using one-way ANOVA followed by Bonferroni's post hoc test (n = 6–8).a Significantly different at p < 0.05 vs. normal.b Vs. control (HFD + STZ).c Vs.3c.d Vs.8.e Vs.10a.f Vs.10a (NPs). NPs = nanoparticles; FBG = fasting blood glucose; HOMA-IR = homeostasis model assessment-insulin resistance; QUICKI = quantitative insulin sensitivity check index. | ||||
| Normal | 99.0 ± 9.5 | 6.7 ± 0.7 | 1.6 ± 0.2 | 0.35 + 0.03 |
| Control (HFD + STZ) | 290.3 ± 30.2a | 22.4 ± 2.3a | 16.0 ± 2.3a | 0.26 ± 0.02a |
| Compound 3c | 193.1 ± 20.3a,b | 12.3 ± 1.4a,b | 5.9 ± 0.9a,b | 0.30 ± 0.03a,b |
| Compound 8 | 223.3 ± 25.5a,b,c | 18.3 ± 2.0a,b,c | 10.1 ± 1.4a,b,c | 0.28 ± 0.03a,b,c |
| Compound 10a | 185.9 ± 20.1a,b,d | 9.8 ± 1.1a,b,d | 4.5 ± 0.6a,b,d | 0.31 ± 0.03a,b,d |
| Compound 10a (NPs) | 174.3 ± 18.3a,b,c,d | 8.7 ± 0.9a,b,c,d | 3.7 ± 0.5a,b,c,d | 0.31 ± 0.03a,b,d |
| Compound 11 | 217.9 ± 22.4a,b,c,e,f | 14.5 ± 1.5a,b,d,e,f | 7.8 ± 1.1a,b,c,d,e,f | 0.29 ± 0.03a,b,d,e,f |
| Groups | Increase in body weight (%) | Adipose tissue index (%) | TG (mg dL−1) | TC (mg dL−1) | LDL-c (mg dL−1) | HDL-c (mg dL−1) |
|---|---|---|---|---|---|---|
| Data are expressed as mean ± SD and analyzed using one-way ANOVA followed by Bonferroni's post hoc test (n = 6–8).a Significantly different at p < 0.05 vs. normal.b Vs. control (HFD + STZ).c Vs.3c.d Vs.8.e Vs.10a.f Vs.10a (NPs). NPs = nanoparticles; TG = triglycerides; TC = total cholesterol; LDL-c = low density lipoprotein-cholesterol; HDL-c = high density lipoprotein-cholesterol. | ||||||
| Normal | 10.5 ± 2.2 | 2.1 ± 0.2 | 72 ± 8 | 59 ± 6 | 46 ± 4 | 51 ± 5 |
| Control (HFD + STZ) | 89.1 ± 10.1a | 5.5 ± 0.5a | 136 ± 15a | 104 ± 12a | 84 ± 9a | 22 ± 2a |
| Compound 3c | 43.6 ± 5.5a,b | 4.2 ± 0.4a,b | 92 ± 10a,b | 83 ± 8a,b | 56 ± 6b | 39 ± 4b |
| Compound 8 | 58.3 ± 7.2a,b,c | 5.1 ± 0.4a,c | 124 ± 13a,c | 97 ± 10a | 84 ± 8a,c | 23 ± 3a,c |
| Compound 10a | 40.2 ± 4.2a,b,d | 3.9 ± 0.3a,b,d | 90 ± 9a,b,d | 67 ± 7b,c,d | 51 ± 5b,d | 47 ± 4b,c,d |
| Compound 10a (NPs) | 36.2 ± 3.8a,b,c,d | 3.4 ± 0.3a,b,c,d | 76 ± 8b,c,d,e | 67 ± 6b,c,d | 46 ± 5b,d | 47 ± 4b,c,d |
| Compound 11 | 54.3 ± 6.7a,b,c,e,f | 4.7 ± 0.5a,b,e,f | 98 ± 11a,b,d,f | 83 ± 9a,b,e,f | 63 ± 7a,b,d,f | 38 ± 4a,b,d,e,f |
The results of the present study showed that HFD+STZ significantly increased the serum levels of liver enzymes alanine aminotransferase (ALT) and aspartate aminotransferase (AST) as well as the serum levels of renal function markers urea and creatinine in the experimental rats compared with the normal group (p < 0.001). All the compounds 3c, 8, 10a, 10a (NPs) and 11 significantly decreased ALT, AST, and serum urea levels compared to the control group. Only the rats treated with 10a (NPs) showed a significant decrease in serum creatinine levels compared to the control group. In addition, 10a (NPs) showed the strongest protective effect, indicated by the levels of ALT, serum creatinine, and urea which is not significantly different compared with the normal levels (p < 0.001) (Table S3†).
The anti-oxidant effect of the synthetic compounds 3c, 8, 10a, 10a (NPS) and 11 was demonstrated by correcting the HFD + STZ-induced oxidative stress in rats: a significant increase in glutathione (GSH), superoxide dismutase (SOD) and catalase (CAT) serum levels and a decrease in malondialdehyde (MDA) serum levels compared to the normal group. Both the 10a and 10a (NPs) treated groups had the lowest CAT levels compared to the other groups. Again, the nano formula of the 10a treatment had the highest GSH and SOD, and the lowest MDA levels compared to the other groups. In addition, only the 10a (NPs) treatment brought all anti-oxidant markers to normal levels (p > 0.001), indicating the maximum anti-oxidant effect (Fig. 5).
 | ||
| Fig. 5 Anti-oxidant effect of synthetic compounds on serum levels of (A) CAT, (B) GSH, (C) SOD, and (D) MDA (n = 8). Data are expressed as mean ± SD and analyzed using one-way ANOVA followed by Bonferroni's post hoc test (n = 6–8). aSignificantly different at p < 0.05 vs. normal, bvs. control (HFD+STZ), cvs.3c, dvs.8, evs.10a, fvs.10a (NPs). GSH; glutathione, SOD; superoxide dismutase, CAT; catalase; MDA; malondialdehyde. | ||
To evaluate the anti-inflammatory effect of the synthetic compounds, NF-κB was measured in all the experimental animals together with the pro-inflammatory cytokines tumor necrosis factor-alpha (TNF)-α, interleukin (IL)-1β, and IL-6. HFD and administration of STZ apparently elicited an inflammatory response in the experimental rats, as evidenced by a significant increase in serum levels of NF-κB, TNF-α, IL-1β, and IL-6 compared to normal levels. Serum levels of all the inflammatory markers were significantly decreased by the administration of 3c, 8, 10a, 10a (NPs), and 11 compared to the control group. 10a (NPs) showed the strongest anti-inflammatory effect by significantly lowering all inflammatory markers compared to all other groups (p < 0.001). It is worth mentioning that the levels of NF-κB, TNF-α, and IL-1β in the serum of the rats treated with 10a (NPs) were not significantly different from the normal group (p > 0.001) (Fig. 6).
 | ||
| Fig. 6 Anti-inflammatory effect of synthetic compounds on serum levels of (A) NF-κB, (B) TNF-α, (C) IL-1β, and (D) IL-6 (n = 8). Data are expressed as mean ± SD and analyzed using one-way ANOVA followed by Bonferroni's post hoc test (n = 6–8). aSignificantly different at p < 0.05 vs. normal, bvs. control (HFD+STZ), cvs.3c, dvs.8, evs.10a, fvs.10a (NPs). NF-κB; nuclear factor- κB, TNF-α; tumor necrosis factor-α, IL; interleukin. | ||
Levels of leptin and adiponectin were determined in the serum, along with their gene expression levels in adipose tissue (Fig. 7). As expected, serum leptin levels were increased after treatment with HFD+STZ, and its adipose tissue expression was upregulated, while serum adiponectin levels decreased, and its expression in adipose tissue was downregulated compared to the normal group. In all treated groups, a significant decrease in serum leptin levels and a significant downregulation of its expression in adipose tissue were observed compared to the untreated control group. Treatment with 10a and 10a (NPs) resulted in significantly lower serum leptin levels and lower leptin expression in adipose tissue than in the other treatment groups. In all treated groups, an upregulation of adiponectin in adipose tissue was observed, along with an increase in its serum levels. Adiponectin serum levels and adiponectin expression in adipose tissue of rats treated with 10a and 10a (NPs) were not significantly different from those of the normal group (p < 0.001) (Fig. 7).
 | ||
| Fig. 7 Effect of synthetic compounds on (A) leptin serum levels, (B) leptin gene expression in adipose tissue, (C) adiponectin serum levels, and (D) adiponectin gene expression in adipose tissue (n = 8). Data are expressed as mean ± SD and analyzed using one-way ANOVA followed by Bonferroni's post hoc test (n = 6–8). aSignificantly different at p < 0.05 vs. normal, bvs. control (HFD + STZ), cvs.3c, dvs.8, evs.10a, fvs.10a (NPs). | ||
In obese people with T2DM, adipose tissue triggers chronic inflammation, which in turn plays a key role in increasing insulin resistance.95 Activation of the NF-κB inflammatory pathway is associated with obesity. Ultimately, this leads to the upregulation of many NF-κB target genes [such as IL-6, TNF-α, and IL-1β], which exacerbates the progression of insulin resistance.96 IL-1β affects adipose tissue insulin sensitivity by inhibiting insulin signaling and causing adipocytes to reduce insulin-stimulated glucose uptake and lipogenesis.97 Similarly, IL-6 has been hypothesized to play a role in the development of obesity and T2DM-induced insulin resistance.97 Adipocytokines, such as leptin and adiponectin, are associated with both insulin sensitivity and obesity-related inflammation and are closely linked to T2DM. Fluctuations in adipocytokines such as leptin and adiponectin are associated with both insulin sensitivity and obesity-related inflammation and are closely linked to T2DM. Higher serum concentrations of leptin and lower serum concentrations of adiponectin are associated with T2DM and obesity. Inflammation promotes leptin resistance,98 which leads to metabolic abnormalities99 and a positive correlation with insulin resistance.100 In contrast, adiponectin promotes insulin sensitization by reducing hepatic glucose synthesis while increasing insulin sensitivity in the liver.101 In addition, obesity-induced adipose tissue remodeling generates a variety of signals, such as hypoxia, that can trigger inflammatory response.102 Moreover, insulin resistance is associated with increased oxidative stress and lipid peroxidation,103 which is reflected in increased MDA levels, reduced glutathione metabolism, and reduced levels of the anti-oxidant enzymes SOD and CAT.104
Specific knockouts have shown that PTP1B regulates body weight, adiposity, and leptin activity. PTP1B-deficient mice exhibited lower weight and higher energy expenditure. In addition, suppression of PTP1B decreased hypothalamic AMP-activated protein kinase (AMPK) activity in peripheral tissues, leading to changes in the expression of genes that promote leanness and energy expenditure.8 Deletion of PTP1B in mice reduced the production of the pro-inflammatory markers TNF- α, IL-6, and IL-1β, highlighting its function in inflammation.105 In addition, a previous study with one of the new PTP1B showed that MDA levels decreased dramatically while SOD activity increased, suggesting that PTP1B can reduce oxidative stress.106 In addition, the study by Swarbrick et al.107 investigated the effects of antisense oligonucleotides (ISIS 113715) on the inhibition of PTP1B in monkeys, where adiponectin levels increased, and insulin sensitivity was improved by ISIS 113715.107 As a result, the PTP pathway was elucidated and is now thought to be a crucial mediator in the control of these metabolic pathways. In an STZ-induced diabetic rat model, benzene-sulfonamide derivatives, as selective inhibitors of PTP1B, improved oral glucose tolerance and insulin resistance by restoring insulin levels and normalizing the serum lipid profile. In addition, other experiments demonstrated the anti-adipogenic properties of this compound and its inhibition of insulin-induced lipid accumulation.108
Consistent with previous reports,109–111 all the synthetic compounds that were in vivo investigated in the current work showed antidiabetic and anti-obesity effects as evidenced by the reduction in FBG, insulin levels, HOMA-IR, percentage increase in body weight, and adipose tissue index after oral administration. In addition, the current results demonstrated the effect of the synthetic benzene-sulfonamide compounds on the lipid profile, which showed lower TG, TC, and LDL-c levels and higher HDL-c levels compared to the untreated rats. The results of the present study showed that oral administration of all the compounds resulted in a decrease in leptin and an increase in adiponectin serum levels, along with decreased leptin gene expression and increased adiponectin expression in adipose tissue.
 | ||
| Fig. 8 Results of hepatic tissue histopathological examination. (A) Hepatic central area and (B) hepatic portal area of different experimental groups showing: a) the normal group with typical hepatic architecture; b) the HFD + STZ-control group showed that the liver lost its normal structure with fatty degeneration of hepatocytes, congestion of portal veins, infiltration of inflammatory cells and bile duct hyperplasia; c) the 3c-treated group showed moderate improvement in hepatocyte fatty degeneration but evidence of portal vein congestion and lymphatic infiltration was still recorded; d) the 8-treated group showed no obvious change with fatty degeneration of hepatocytes, congestion of portal veins, infiltration of inflammatory cells and bile duct hyperplasia; e) the 10a-treated group showed moderate improvement in hepatocyte fatty degeneration but still experienced portal vein congestion and lymphatic infiltration; f) the 10a (NPs)-treated group showed restored hepatic architecture with no evidence of histopathological changes; g) the 11-treated group showed no obvious change with fatty degeneration of hepatocytes, congestion of portal veins, infiltration of inflammatory cells and bile duct hyperplasia. (cv) central vein, (hc) hepatocytes, (s) sinusoids, (fd) fatty degeneration, (hpv) hepatic portal vein, (ha) hepatic artery, (b) bile duct, (lyi) lymphatic infiltration, (*) portal vein congestion and (arrow) bile duct hyperplasia. | ||
The microscopy damage score for the liver for all experimental groups is illustrated in (Fig. S26†). The hepatocyte fatty degeneration score for all treated groups was significantly increased (p < 0.001) compared with the normal group except for the 10a (NPs)-treated group, which showed no difference, while all the groups showed a significant decrease (p < 0.001) of fatty degeneration when compared with the ‘HFD + STZ’-control group. Regarding the cellular infiltration, portal vein congestion and bile duct hyperplasia scoring, all the groups showed a significant increase (p < 0.001) except for the 10a (NPs)-treated group, which exhibited no difference when compared to the normal group. However, 3c, 10a, and 10a (NPs)-treated groups' scores were significantly decreased (p < 0.001), while groups 8 and 11 showed no significant difference when compared with the ‘HFD + STZ’-control group.
The microscopy examination of the kidney showed that the kidney of the normal group showed normal appearance of the renal cortex and medulla (Fig. S27A and B†). In contrast, the kidney of the HFD + STZ-control group displayed several changes in the renal cortical architecture, including hypertrophy of glomerulus, infiltration of inflammatory cells, and hemorrhage, while the medullary region showed signs of tubular fatty changes. Treatment with compounds 8 and 11 showed no obvious amelioration compared to the HFD + STZ-control group, whereas 3c, 10a and 10a (NPs)-treated groups showed completely restored renal cortical architecture with no evidence of histopathological changes (Fig. S27A and B†).
Kidney cortical damage score was significantly increased (p < 0.001) in all treated groups except for 10a and 10a (NPs)- treated groups, which showed no significant difference when compared with the normal group. However, the cortical kidney damage score of 3c, 10a, and 10a (NPs)-treated groups was significantly decreased (p < 0.001) when compared with the ‘HFD + STZ’-control group (Fig. S27C†).
The adipose tissue examination showed a general increase in adipocyte size in the HFD + STZ control group with mild blood vessel congestions but no signs of inflammation were recorded (Fig. 9A). Adipocyte diameter revealed a significant increase (p < 0.001) in the 8, 11 and HFD + STZ-control groups compared to the ‘normal’ group, while 3c, 10a and 10a (NPs)-treated groups showed no significant difference. Adipocyte diameter showed a significant decrease (p < 0.001) in all experimental group when compared to the HFD + STZ-control group (Fig. 9B).
 | ||
| Fig. 9 White subcutaneous adipose tissue examination for all experimental groups. (A) H&E-stained adipose tissue showing: a) the normal group with regular uniformed adipocytes; b) the HFD + STZ control group showed mild congestion of blood vessels and increased size of adipocytes; c) the 3c-treated group with normal adipose tissue; d) the 8-treated group with mild blood vessel congestion; e & f) 10a and 10a (NPs)-treated groups respectively with normal adipose tissue appearance; g) the 11-treated group with mild blood vessel congestion. No signs of inflammation were recorded in all treated groups, (arrow) blood vessel congestion. (B) Adipocyte diameter (μ) represented as mean ± SD and analyzed using one-way ANOVA followed by Bonferroni's post hoc test (n = 6–8). aSignificantly different at p < 0.05 vs. normal, bvs. control (HFD + STZ), cvs.3c, dvs.8, evs.10a, fvs.10a (NPs). | ||
5. Conclusions
Molecular modeling provided great insights regarding the compound's different affinity towards the biological target as well as the direction for improvement. Superior PTP1B inhibition activity was highlighted with extended structures as seen with 10a, being capable of reaching across sites A and B while maintaining deep anchoring at site catalytic loop-P. However, the ability of the compound to anchor at site AC was relevant for high ligand–target stability and profound inhibition activity, as seen with 8. In contrast, bulky and improper group substitution on the polar ring head would impact steric hinderance and compromised the ligand's binding. Further, this paper highlighted a successful approach for obtaining balanced hydrophilic/lipophilic compounds with potential target inhibition activity even down to nanomolar concentration ranges while mitigating lead development challenges. The latter absorption-related parameters highlighted the potentiality for future structural development and optimization. Structure expansion with polarly decorated hydrophobic fragments (e.g., triazole ring scaffold) would further favor the compound's dynamics and ligand–target binding as described with the above molecular modeling studies. On the other hand, adopting a site-specific targeted drug delivery nano system has also been highlighted as beneficial for improving 10a's potency through predicted boosted pharmacokinetics. Selected synthetic compounds were in vivo investigated in a rat model of HFD + STZ-induced diabetes, where the antidiabetic, anti-obesity, anti-oxidant, and anti-inflammatory properties of the synthetic compounds were elucidated both biochemically and histopathologically. Interestingly, the nano-formulated 10a exhibited the most potent effects. Further study is still needed to fine-tune the ligand's interactions within the enzyme active site leading to improvement of the activity, selectivity, and hence the safety of these leads. Future lead optimization and development work will be conducted altering the trunk and tail parts of the compounds through structural modifications and fine tuning approaches.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
This work was carried out in collaboration among all authors. The authors designed the study, wrote the protocol, managed the analysis of the study, performed the statistical analysis, wrote the first draft of the manuscript, and managed the literature searches. All authors read and approved the final manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.References
- D. Dhirani, A. Shahid and H. Mumtaz, A new kind of diabetes medication approved by the FDA: is there hope for obesity?, Int. J. Surg., 2023, 109(2), 81–82 CrossRef PubMed.
- J. P. Frias, et al., Tirzepatide versus Semaglutide Once Weekly in Patients with Type 2 Diabetes, N. Engl. J. Med., 2021, 385(6), 503–515 CrossRef CAS.
- A. Babu, et al., Safe and simple emergency department discharge therapy for patients with type 2 diabetes mellitus and severe hyperglycemia, Endocr. Pract., 2009, 15(7), 696–704 CrossRef PubMed.
- W. Zou, Type 2 Diabetes Mellitus and Protein-Tyrosine Phosphatase 1B1, J. Diabetes Metab. Disord. Control, 2016, 3(8), 180–183 Search PubMed.
- M. Bodmer, et al., Metformin, sulfonylureas, or other antidiabetes drugs and the risk of lactic acidosis or hypoglycemia: a nested case-control analysis, Diabetes Care, 2008, 31(11), 2086–2091 Search PubMed.
- O. O. Oguntibeju, Type 2 diabetes mellitus, oxidative stress and inflammation: examining the links, Int. J. Physiol., Pathophysiol. Pharmacol., 2019, 11(3), 45–63 CAS.
- T. Behl, et al., Exploring protein tyrosine phosphatases (PTP) and PTP-1B inhibitors in management of diabetes mellitus, Biomed. Pharmacother., 2022, 153, 113405 CrossRef CAS.
- K. K. Bence, et al., Neuronal PTP1B regulates body weight, adiposity and leptin action, Nat. Med., 2006, 12(8), 917–924 CrossRef CAS PubMed.
- P. A. Thiebaut, et al., Role of protein tyrosine phosphatase 1B in cardiovascular diseases, J. Mol. Cell. Cardiol., 2016, 101, 50–57 CrossRef CAS PubMed.
- B. L. Seely, et al., Protein tyrosine phosphatase 1B interacts with the activated insulin receptor, Diabetes, 1996, 45(10), 1379–1385 CrossRef CAS PubMed.
- B. J. Goldstein, et al., Tyrosine dephosphorylation and deactivation of insulin receptor substrate-1 by protein-tyrosine phosphatase 1B. Possible facilitation by the formation of a ternary complex with the Grb2 adaptor protein, J. Biol. Chem., 2000, 275(6), 4283–4289 CrossRef CAS.
- G. Liu, Protein tyrosine phosphatase 1B inhibition: opportunities and challenges, Curr. Med. Chem., 2003, 10(15), 1407–1421 CrossRef CAS PubMed.
- M. Teimouri, et al., The role of protein tyrosine phosphatase 1B (PTP1B) in the pathogenesis of type 2 diabetes mellitus and its complications, J. Physiol. Biochem., 2022, 78(2), 307–322 CrossRef CAS.
- M. Feng, et al., Synthesis, anti-α-glucosidase activity, inhibition interaction, and anti-diabetic activity of novel cryptolepine derivatives, J. Mol. Struct., 2024, 1310, 138311 CrossRef CAS.
- B. Liang, et al., Novel thiosemicarbazide-based beta-carboline derivatives as alpha-glucosidase inhibitors: Synthesis and biological evaluation, Eur. J. Med. Chem., 2024, 275, 116595 CrossRef CAS.
- C. Hu, et al., Synthesis and biological evaluation of indole derivatives containing thiazolidine-2,4-dione as alpha-glucosidase inhibitors with antidiabetic activity, Eur. J. Med. Chem., 2024, 264, 115957 CrossRef CAS.
- X. T. Xu, et al., Synthesis and biological evaluation of coumarin derivatives as alpha-glucosidase inhibitors, Eur. J. Med. Chem., 2020, 189, 112013 CrossRef.
- H. Koyama, et al., 5-Aryl thiazolidine-2,4-diones as selective PPARgamma agonists, Bioorg. Med. Chem. Lett., 2003, 13(10), 1801–1804 Search PubMed.
- S. Mudaliar and R. R. Henry, New oral therapies for type 2 diabetes mellitus: The glitazones or insulin sensitizers, Annu. Rev. Med., 2001, 52, 239–257 Search PubMed.
- M. Li, et al., Identification of 1,3,4-Thiadiazolyl-Containing Thiazolidine-2,4-dione Derivatives as Novel PTP1B Inhibitors with Antidiabetic Activity, J. Med. Chem., 2024, 67(10), 8406–8419 Search PubMed.
- Y. Zheng, et al., New chromone derivatives bearing thiazolidine-2,4-dione moiety as potent PTP1B inhibitors: Synthesis and biological activity evaluation, Bioorg. Chem., 2024, 143, 106985 Search PubMed.
- M. Verma, et al., Protein tyrosine phosphatase 1B inhibitors as antidiabetic agents - A brief review, Bioorg. Chem., 2017, 70, 267–283 CrossRef CAS PubMed.
- Z. Q. Liu, et al., Fumosorinone, a novel PTP1B inhibitor, activates insulin signaling in insulin-resistance HepG2 cells and shows anti-diabetic effect in diabetic KKAy mice, Toxicol. Appl. Pharmacol., 2015, 285(1), 61–70 CAS.
- N. Ghareb, et al., Toward a treatment of diabesity: Rational design, synthesis and biological evaluation of benzene-sulfonamide derivatives as a new class of PTP-1B inhibitors, Bioorg. Chem., 2019, 86, 322–338 CAS.
- Z. Y. Zhang, Protein tyrosine phosphatases: structure and function, substrate specificity, and inhibitor development, Annu. Rev. Pharmacol. Toxicol., 2002, 42, 209–234 CAS.
- D. Barford, A. J. Flint and N. K. Tonks, Crystal structure of human protein tyrosine phosphatase 1B, Science, 1994, 263(5152), 1397–1404 CrossRef CAS.
- Z. Y. Zhang, Protein-tyrosine phosphatases: biological function, structural characteristics, and mechanism of catalysis, Crit. Rev. Biochem. Mol. Biol., 1998, 33(1), 1–52 Search PubMed.
- Y. A. Puius, et al., Identification of a second aryl phosphate-binding site in protein-tyrosine phosphatase 1B: a paradigm for inhibitor design, Proc. Natl. Acad. Sci. U. S. A., 1997, 94(25), 13420–13425 CAS.
- R. Liu, et al., Human Protein Tyrosine Phosphatase 1B (PTP1B): From Structure to Clinical Inhibitor Perspectives, Int. J. Mol. Sci., 2022, 23(13), 7027 CAS.
- X. Li, L. Wang and D. Shi, The design strategy of selective PTP1B inhibitors over TCPTP, Bioorg. Med. Chem., 2016, 24(16), 3343–3352 CAS.
- K. Shen, et al., Acquisition of a specific and potent PTP1B inhibitor from a novel combinatorial library and screening procedure, J. Biol. Chem., 2001, 276(50), 47311–47319 CAS.
- S. Zhang and Z. Y. Zhang, PTP1B as a drug target: recent developments in PTP1B inhibitor discovery, Drug Discovery Today, 2007, 12(9–10), 373–381 CrossRef CAS PubMed.
- Z. Y. Zhang and S. Y. Lee, PTP1B inhibitors as potential therapeutics in the treatment of type 2 diabetes and obesity, Expert Opin. Invest. Drugs, 2003, 12(2), 223–233 CrossRef CAS PubMed.
- G. S. Kumar, R. Page and W. Peti, The mode of action of the Protein tyrosine phosphatase 1B inhibitor Ertiprotafib, PLoS One, 2020, 15(10), e0240044 CrossRef CAS PubMed.
- S. Fukuda, et al., Pharmacological profiles of a novel protein tyrosine phosphatase 1B inhibitor, JTT-551, Diabetes, Obes. Metab., 2010, 12(4), 299–306 CrossRef CAS.
- G. Liu, et al., Selective protein tyrosine phosphatase 1B inhibitors: targeting the second phosphotyrosine binding site with non-carboxylic acid-containing ligands, J. Med. Chem., 2003, 46(16), 3437–3440 CrossRef CAS PubMed.
- N. Krishnan, et al., A potent, selective, and orally bioavailable inhibitor of the protein-tyrosine phosphatase PTP1B improves insulin and leptin signaling in animal models, J. Biol. Chem., 2018, 293(5), 1517–1525 CrossRef CAS PubMed.
- N. Krishnan, et al., DPM-1001 decreased copper levels and ameliorated deficits in a mouse model of Wilson's disease, Genes Dev., 2018, 32(13–14), 944–952 CrossRef CAS.
- Q. Wang, et al., Potent inhibition of protein tyrosine phosphatase 1B by copper complexes: implications for copper toxicity in biological systems, Chem. Commun., 2010, 46(20), 3547–3549 RSC.
- A. F. Moretto, et al., Bicyclic and tricyclic thiophenes as protein tyrosine phosphatase 1B inhibitors, Bioorg. Med. Chem., 2006, 14(7), 2162–2177 CrossRef CAS PubMed.
- D. P. Wilson, et al., Structure-based optimization of protein tyrosine phosphatase 1B inhibitors: from the active site to the second phosphotyrosine binding site, J. Med. Chem., 2007, 50(19), 4681–4698 CrossRef CAS.
- T. Joutsamo, et al., Compounds Useful For Treatment Or Prevention Of Disease Mediated By Alpha-2b-Adrenoceptor, U.S. Pat., 6767909, 2003 Search PubMed.
- N. G. Ghareb and M. S. Elgawish, Design and Synthesis of some Azoles Incorporating Sulphadiazine Derivatives as Protein Tyrosine Phosphatase-1B inhibitors, Rec. Pharm. Biomedical Sci., 2019, 3(2), 81–90 Search PubMed.
- H. M. Faidallah, K. A. Khan and A. M. Asiri, Synthesis and biological evaluation of new 3-trifluoromethylpyrazolesulfonyl-urea and thiourea derivatives as antidiabetic and antimicrobial agents, J. Fluorine Chem., 2011, 132(2), 131–137 CrossRef CAS.
- J. Eberhardt, et al., AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings, J. Chem. Inf. Model., 2021, 61(8), 3891–3898 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J. Comput. Chem., 2010, 31(2), 455–461 CrossRef CAS.
- Q. Xue, et al., Evaluation of the binding performance of flavonoids to estrogen receptor alpha by Autodock, Autodock Vina and Surflex-Dock, Ecotoxicol. Environ. Saf., 2022, 233, 113323 CAS.
- The PyMOL Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC, https://pymol.sourceforge.net/faq.html#:~:text=external%20GUI%20window%3F-,How%20does%20one%20cite% Search PubMed; Impact, New York, NY, Schrödinger, LLC, 2016 Search PubMed; Prime, New York, NY, Schrödinger, LLC, 2016 Search PubMed.
- S. S. Elhady, et al., Molecular Docking and Dynamics Simulation Study of Hyrtios erectus Isolated Scalarane Sesterterpenes as Potential SARS-CoV-2 Dual Target Inhibitors, Biology, 2021, 10(5), 389 CAS.
- S. Páll, et al., Tackling Exascale Software Challenges in Molecular Dynamics Simulations with GROMACS, in Solving Software Challenges for Exascale, Springer International Publishing, Cham, 2015 Search PubMed.
- A. H. Saleh, et al., Deciphering the molecular basis of the kappa opioid receptor selectivity: A Molecular Dynamics study, J. Mol. Graphics Modell., 2021, 106, 107940 CrossRef CAS PubMed.
- G. A. Ross, et al., Biomolecular Simulations under Realistic Macroscopic Salt Conditions, J. Phys. Chem. B, 2018, 122(21), 5466–5486 CAS.
- A. A. Zaki, et al., Calendulaglycoside A Showing Potential Activity Against SARS-CoV-2 Main Protease: Molecular Docking, Molecular Dynamics, and SAR Studies, J. Tradit. Complementary Med., 2021, 12(1), 16–34 Search PubMed.
- S. C. Tuble, J. Anwar and J. D. Gale, An Approach to Developing a Force Field for Molecular Simulation of Martensitic Phase Transitions between Phases with Subtle Differences in Energy and Structure, J. Am. Chem. Soc., 2004, 126(1), 396–405 CAS.
- T. Darden, D. York and L. Pedersen, Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems, J. Chem. Phys., 1993, 98(12), 10089–10092 CrossRef CAS.
- B. Hess, et al., LINCS: A linear constraint solver for molecular simulations, J. Comput. Chem., 1997, 18(12), 1463–1472 CrossRef CAS.
- S. Páll and B. Hess, A flexible algorithm for calculating pair interactions on SIMD architectures, Comput. Phys. Commun., 2013, 184(12), 2641–2650 CrossRef.
- R. Kumari, R. Kumar and A. Lynn, g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations, J. Chem. Inf. Model., 2014, 54(7), 1951–1962 CrossRef CAS PubMed.
- Y. Zhao, et al., Self-nanoemulsifying drug delivery system (SNEDDS) for oral delivery of Zedoary essential oil: formulation and bioavailability studies, Int. J. Pharm., 2010, 383(1–2), 170–177 CrossRef CAS.
- S. Beg, et al., Solid self-nanoemulsifying systems of olmesartan medoxomil: Formulation development, micromeritic characterization, in vitro and in vivo evaluation, Powder Technol., 2016, 294, 93–104 CrossRef CAS.
- R. N. Dash, H. Mohammed and T. Humaira, Design, optimization, and evaluation of ezetimibe solid supersaturatable self-nanoemulsifying drug delivery for enhanced solubility and dissolution, J. Pharm. Invest., 2016, 46(2), 153–168 CrossRef CAS.
- A. A. Date and M. S. Nagarsenker, Design and evaluation of self-nanoemulsifying drug delivery systems (SNEDDS) for cefpodoxime proxetil, Int. J. Pharm., 2007, 329(1–2), 166–172 CrossRef CAS.
- R. Kahn, Consensus Development Conference on Insulin Resistance, 5-6 November 1997, American Diabetes Association, Diabetes Care, 1998, 21(2), 310–314 CrossRef PubMed.
- J. M. Park, et al., Postprandial hypoglycemic effect of mulberry leaf in Goto-Kakizaki rats and counterpart control Wistar rats, Nutr. Res. Pract., 2009, 3(4), 272–278 CrossRef CAS PubMed.
- A. Katz, et al., Quantitative insulin sensitivity check index: a simple, accurate method for assessing insulin sensitivity in humans, J. Clin. Endocrinol. Metab., 2000, 85(7), 2402–2410 CrossRef CAS PubMed.
- R. D. Cardiff, C. H. Miller and R. J. Munn, Manual hematoxylin and eosin staining of mouse tissue sections, Cold Spring Harb. Protoc., 2014, 2014(6), 655–658 Search PubMed.
- B. A. Neuschwander-Tetri and S. H. Caldwell, Nonalcoholic steatohepatitis: summary of an AASLD Single Topic Conference, Hepatology, 2003, 37(5), 1202–1219 CrossRef.
- P. J. Medeiros, et al., Effect of sildenafil in renal ischemia/reperfusion injury in rats, Acta Cir. Bras., 2010, 25(6), 490–495 CrossRef.
- J. D. Baggot, Pharmacokinetic-pharmacodynamic relationship, Ann. Rech. Vet., 1990, 21(Suppl 1), 29s–40s CAS.
- X. Chen, et al., Virtual Screening of Novel and Selective Inhibitors of Protein Tyrosine Phosphatase 1B over T-Cell Protein Tyrosine Phosphatase Using a Bidentate Inhibition Strategy, J. Chem. Inf. Model., 2018, 58(4), 837–847 CAS.
- M. Pagliai, et al., Competitive Solvation and Chemisorption in Silver Colloidal Suspensions, Prog. Colloid Polym. Sci., 2012, 139, 39–44 CAS.
- M. Kontoyianni, L. M. McClellan and G. S. Sokol, Evaluation of Docking Performance: Comparative Data on Docking Algorithms, J. Med. Chem., 2004, 47(3), 558–565 CAS.
- S. O. Albuquerque, et al., Biological evaluation and molecular modeling of peptidomimetic compounds as inhibitors for O-GlcNAc transferase (OGT), Eur. J. Pharm. Sci., 2020, 154, 105510 CAS.
- A. S. de Souza, et al., 3-Acyltetramic acids as a novel class of inhibitors for human kallikreins 5 and 7, Bioorg. Med. Chem. Lett., 2019, 29(9), 1094–1098 CAS.
- A. M. El-Naggar, et al., Design, synthesis, and SAR studies of novel 4-methoxyphenyl pyrazole and pyrimidine derivatives as potential dual tyrosine kinase inhibitors targeting both EGFR and VEGFR-2, Bioorg. Chem., 2022, 123, 105770 CAS.
- W. S. Liu, et al., Design, synthesis, biological evaluation and molecular dynamics studies of 4-thiazolinone derivatives as protein tyrosine phosphatase 1B (PTP1B) inhibitors, J. Biomol. Struct. Dyn., 2020, 38(13), 3814–3824 CAS.
- M. K. Mahapatra, et al., In silico modelling and molecular dynamics simulation studies of thiazolidine based PTP1B inhibitors, J. Biomol. Struct. Dyn., 2018, 36(5), 1195–1211 CAS.
- B. Manasa, et al., Virtual structure-based docking, WaterMap, and molecular dynamics guided identification of the potential natural compounds as inhibitors of protein-tyrosine phosphatase 1B, J. Mol. Struct., 2021, 1226, 129396 CAS.
- J. Cai, L. Zhao and W. Tao, Potent protein tyrosine phosphatase 1B (PTP1B) inhibiting constituents from Anoectochilus chapaensis and molecular docking studies, Pharm. Biol., 2015, 53(7), 1030–1034 CrossRef CAS PubMed.
- A. A. Zaki, et al., Calendulaglycoside A showing potential activity against SARS-CoV-2 main protease: Molecular docking, molecular dynamics, and SAR studies, J. Tradit. Complementary Med., 2022, 12(1), 16–34 CrossRef CAS.
- M. A. Soltan, et al., In Silico Designing of a Multitope Vaccine against Rhizopus microsporus with Potential Activity against Other Mucormycosis Causing Fungi, Cell, 2021, 10(11), 3014 CrossRef CAS.
- M. Arnittali, A. N. Rissanou and V. Harmandaris, Structure Of Biomolecules Through Molecular Dynamics Simulations, Procedia Comput. Sci., 2019, 156, 69–78 CrossRef.
- K. Liu, E. Watanabe and H. Kokubo, Exploring the stability of ligand binding modes to proteins by molecular dynamics simulations, J. Comput.-Aided Mol. Des., 2017, 31(2), 201–211 CrossRef CAS.
- A. Manandhar, et al., Targeting SARS-CoV-2 M3CLpro by HCV NS3/4a Inhibitors: In Silico Modeling and In Vitro Screening, J. Chem. Inf. Model., 2021, 61(2), 1020–1032 CrossRef CAS.
- A. J. Almalki, et al., Computational and Biological Evaluation of β-Adrenoreceptor Blockers as Promising Bacterial Anti-Virulence Agents, Pharmaceuticals, 2022, 15(2), 110 CrossRef CAS PubMed.
- N. C. Benson and V. Daggett, A comparison of multiscale methods for the analysis of molecular dynamics simulations, J. Phys. Chem. B, 2012, 116(29), 8722–8731 CrossRef CAS.
- W. Singh, et al., Conformational Dynamics, Ligand Binding and Effects of Mutations in NirE an S-Adenosyl-L-Methionine Dependent Methyltransferase, Sci. Rep., 2016, 6, 20107 CrossRef CAS PubMed.
- J. F. Fatriansyah, et al., Molecular docking and dynamics studies on propolis sulabiroin-A as a potential inhibitor of SARS-CoV-2, J. King Saud Univ., Sci., 2022, 34(1), 101707 CrossRef.
- M. V. Reddy, et al., X-ray structure of PTP1B in complex with a new PTP1B inhibitor, Protein Pept. Lett., 2014, 21(1), 90–93 CrossRef PubMed.
- J. P. Sun, et al., Crystal structure of PTP1B complexed with a potent and selective bidentate inhibitor, J. Biol. Chem., 2003, 278, 12406–12414 CrossRef CAS.
- G. Scapin, et al., The structural basis for the selectivity of benzotriazole inhibitors of PTP1B, Biochemistry, 2003, 42(39), 11451–11459 CrossRef CAS.
- H. Zhao, et al., Isoxazole carboxylic acids as protein tyrosine phosphatase 1B (PTP1B) inhibitors, Bioorg. Med. Chem. Lett., 2004, 14(22), 5543–5546 CrossRef CAS PubMed.
- C. N. Cavasotto, Binding Free Energy Calculation Using Quantum Mechanics Aimed for Drug Lead Optimization, Methods Mol. Biol., 2020, 2114, 257–268 CrossRef CAS.
- A. Nasr, A. Gardouh and M. Ghorab, Novel Solid Self-Nanoemulsifying Drug Delivery System (S-SNEDDS) for Oral Delivery of Olmesartan Medoxomil: Design, Formulation, Pharmacokinetic and Bioavailability Evaluation, Pharmaceutics, 2016, 8(3), 20 CrossRef.
- F. Zatterale, et al., Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes, Front. Physiol., 2019, 10, 1607 CrossRef.
- G. Panahi, et al., High glucose induces inflammatory responses in HepG2 cells via the oxidative stress-mediated activation of NF-kappaB, and MAPK pathways in HepG2 cells, Arch. Physiol. Biochem., 2018, 124(5), 468–474 CrossRef CAS PubMed.
- B. Feve and J. P. Bastard, The role of interleukins in insulin resistance and type 2 diabetes mellitus, Nat. Rev. Endocrinol., 2009, 5(6), 305–311 CrossRef CAS PubMed.
- A. Perez-Perez, et al., Role of Leptin in Inflammation and Vice Versa, Int. J. Mol. Sci., 2020, 21(16), 5887 CrossRef CAS.
- O. Gruzdeva, et al., Leptin resistance: underlying mechanisms and diagnosis, Diabetes, Metab. Syndr. Obes.: Targets Ther., 2019, 12, 191–198 CrossRef CAS PubMed.
- I. Osegbe, H. Okpara and E. Azinge, Relationship between serum leptin and insulin resistance among obese Nigerian women, Ann. Afr. Med., 2016, 15(1), 14–19 CrossRef PubMed.
- W. Liu, et al., Serum leptin, resistin, and adiponectin levels in obese and non-obese patients with newly diagnosed type 2 diabetes mellitus: A population-based study, Medicine, 2020, 99(6), e19052 CrossRef.
- S. M. Reilly and A. R. Saltiel, Adapting to obesity with adipose tissue inflammation, Nat. Rev. Endocrinol., 2017, 13(11), 633–643 CrossRef CAS.
- S. C. Shabalala, et al., Detrimental Effects of Lipid Peroxidation in Type 2 Diabetes: Exploring the Neutralizing Influence of Antioxidants, Antioxidants, 2022, 11(10), 2071 CrossRef CAS.
- U. Asmat, K. Abad and K. Ismail, Diabetes mellitus and oxidative stress-A concise review, Saudi Pharm. J., 2016, 24(5), 547–553 CrossRef PubMed.
- E. Delile, et al., Reduced Insulin Resistance Contributes to the Beneficial Effect of Protein Tyrosine Phosphatase-1B Deletion in a Mouse Model of Sepsis, Shock, 2017, 48(3), 355–363 CrossRef CAS PubMed.
- D. Li, et al., A Novel PTP1B Inhibitor-Phosphate of Polymannuronic Acid Ameliorates Insulin Resistance by Regulating IRS-1/Akt Signaling, Int. J. Mol. Sci., 2021, 22(23), 12693 CrossRef CAS.
- M. M. Swarbrick, et al., Inhibition of protein tyrosine phosphatase-1B with antisense oligonucleotides improves insulin sensitivity and increases adiponectin concentrations in monkeys, Endocrinology, 2009, 150(4), 1670–1679 CrossRef CAS.
- V. M. Balaramnavar, et al., Identification of novel PTP1B inhibitors by pharmacophore based virtual screening, scaffold hopping and docking, Eur. J. Med. Chem., 2014, 87, 578–594 CrossRef CAS PubMed.
- S. I. Attah, et al., Pro-Gly based dipeptide containing sulphonamide functionality, their antidiabetic, antioxidant, and anti-inflammatory activities. Synthesis, characterization and computational studies, J. Mol. Struct., 2022, 1264, 133280 Search PubMed.
- V. Dayma, et al., Synthesis, antidiabetic, antioxidant and anti-inflammatory activities of novel hydroxytriazenes based on sulpha drugs, Heliyon, 2020, 6(8), e04787 Search PubMed.
- N. Hosseinzadeh, et al., Synthesis and antidiabetic evaluation of benzenesulfonamide derivatives, Iran. J. Pharm. Res., 2013, 12(2), 325–330 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00594e |
| This journal is © The Royal Society of Chemistry 2025 |