
Synthesis and evaluation of tetrahydropyrrolo[1,2-a]quinolin-1(2H)-ones as new tubulin polymerization inhibitors†
Mikhail N.
Anisimov‡
 ab,
Maksim A.
Boichenko‡
c,
Vitaly V.
Shorokhov
c,
Julia N.
Borzunova
a,
Marina
Janibekova
d,
Vadim V.
Mustyatsa
bd,
Ilya A.
Lifshits
a,
Andrey Yu.
Plodukhin
c,
Ivan A.
Andreev
e,
Nina K.
Ratmanova
e,
Sergey S.
Zhokhov
c,
Elena A.
Tarasenko
c,
Daria A.
Ipatova
c,
Alexander R.
Pisarev
f,
Ivan A.
Vorobjev
dgh,
Igor V.
Trushkov
*i,
Olga A.
Ivanova
*c and
Nikita B.
Gudimchuk
*ab
ab,
Maksim A.
Boichenko‡
c,
Vitaly V.
Shorokhov
c,
Julia N.
Borzunova
a,
Marina
Janibekova
d,
Vadim V.
Mustyatsa
bd,
Ilya A.
Lifshits
a,
Andrey Yu.
Plodukhin
c,
Ivan A.
Andreev
e,
Nina K.
Ratmanova
e,
Sergey S.
Zhokhov
c,
Elena A.
Tarasenko
c,
Daria A.
Ipatova
c,
Alexander R.
Pisarev
f,
Ivan A.
Vorobjev
dgh,
Igor V.
Trushkov
*i,
Olga A.
Ivanova
*c and
Nikita B.
Gudimchuk
*ab
aDepartment of Physics, M.V. Lomonosov Moscow State University, Moscow, 119991, Russia. E-mail: ngudimch@gmail.com
bCenter for theoretical problems of physicochemical pharmacology, Moscow, 109029, Russia
cDepartment of Chemistry, M.V. Lomonosov Moscow State University, Moscow, 119991, Russia. E-mail: iv@kinet.chem.msu.ru
dNational Laboratory Astana, Astana, 010000, Kazakhstan
eDmitry Rogachev National Medical Research Center of Pediatric Hematology, Oncology and Immunology, Moscow, 117997, Russia
fFaculty of Biology and Biotechnologies, Higher School of Economics, Moscow 117418, Russia
gDepartment of Biology, School of Sciences and Humanities, Nazarbayev University, Astana, 010000, Kazakhstan
hDepartment of Biology, M.V. Lomonosov Moscow State University, Moscow, 119991, Russia
iN.D. Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, Moscow, 119991, Russia. E-mail: trush@ioc.ac.ru
First published on 15th October 2024
Abstract
Here we explored new 1,5-disubstituted pyrrolidin-2-ones 1, 2 and 5-aryl-3,3a,4,5-tetrahydropyrrolo[1,2-a]quinoline-1(2H)-ones 3 as inhibitors of tubulin polymerization. We evaluated their effects on microtubule dynamics in vitro and on the proliferation of A549 cells, using flow cytometry-based cell cycle analysis. The results were verified with phase-contrast microscopy in three cancer cell lines: A549, HeLa and MCF-7. Guided by molecular modeling of the interactions between tubulin and the most active of the identified compounds, we designed, synthesized, and tested the 3-hydroxyphenyl-substituted compound 3c. This compound was further shown to bind to the colchicine site of tubulin and reduce microtubule growth rates in vitro. Moreover, compound 3c arrested division of the A549 cells in the low micromolar range (IC50 = 5.9 μM) and exhibited cytotoxicity against four different cell lines in the MTT assay for cell proliferation. Our findings demonstrate that 5-aryltetrahydropyrrolo[1,2-a]quinoline-1(2H)-one is a promising scaffold for the development of novel tubulin polymerization inhibitors.
Introduction
Microtubules are dynamic cytoskeletal structures containing thirteen protofilaments formed by α- and β-tubulin dimers. These polymers are essential for numerous cellular processes, including cell division.1 Individual microtubules switch from growth to shortening and reverse both in vitro and in cells, a remarkable nonequilibrium behavior known as dynamic instability.2 The dynamic instability of microtubules is significantly accelerated during mitosis.3 Numerous drugs have been shown to inhibit the microtubule dynamics and thus inhibit cell division,4,5 leading to cell arrest at the metaphase often followed by apoptosis.6 Some of these microtubule-targeting cytostatic drugs have been widely used for cancer treatment for several decades. The first clinically approved class of anticancer chemotherapy drugs was vinca alkaloids.7 These agents bind at the interface between tubulin dimers, inhibiting microtubule dynamics.8 In addition to the vinca-binding site, at least five other binding pockets have been characterized on the α,β-tubulin heterodimer to date5 and more recently several new pockets have been identified.9 The two best characterized of these are the taxane site, located on the luminal side of the β-tubulin subunit,10 and the colchicine site, located in the intradimer interface of tubulin.11 Taxane site binders, such as paclitaxel and some of its analogues, were clinically approved in the 1990s and have been widely and successfully used to treat various types of cancers.12 The colchicine-binding site, named after a long-known drug derived from the plant Colchicum autumnale, is a very attractive and not yet clinically exploited anti-cancer target. Colchicine itself is a potent inhibitor of microtubule assembly, which has been applied in medicine for over a thousand years. It is currently used to treat gout, Behcet's disease, pericarditis and familial Mediterranean fever due to its anti-inflammatory and anti-vascular effects.13 Due to its high toxicity against normal cells, colchicine is not approved for the cancer therapy.14 However, development of its analogs with reduced toxicity, which can serve as anticancer agents, is of great interest. Like in the case of vinca alkaloids, vinblastine and vincristine, which are structurally very similar but are known to be effective against different types of cancer and have substantially different levels of toxicity,7 other colchicine site binders may be less toxic than colchicine, but still effective against cancer. Potential advantages of colchicine site binders over taxanes and vinca alkaloids include their ability to overcome multidrug resistance, act as vascular-disrupting agents, and their structural simplicity, which ensures the relative ease of their synthesis.15Numerous ligands targeting the colchicine site on tubulin have been developed, and some of them are promising candidates for chemotherapy in clinical trials.16 Diverse colchicine site binders contain two electron-rich (hetero)aromatic rings, predominantly polyoxygenated ones, connected by different spacer chains and rings.15–24 Some well-known examples of such compounds are shown in Fig. 1a.
 | ||
| Fig. 1 Structures of colchicine and some other colchicine site binders as well as the subjects of this study. IC50 values in panel (b) are based on the cytotoxicity (MTT) assay with HEK-293 cells, while the IC50 value in panel (c) is derived from the flow cytometry-based cell cycle assay with A549 cells, as detailed below. | ||
In accordance with this structural motif, we previously found that tetracyclic compounds containing a 5-aryl-1-benzylpyrrolidin-2-one moiety demonstrate variable cytotoxicity against a range of cancer cells.25 On the other hand, we also demonstrated that completely different compounds, polyoxygenated 1-arylindane derivatives and their heterocyclic analogues, also exhibited toxicity against some tumor cells.26 The general formulas for these two series of compounds are also given in Fig. 1b. Our ongoing interest in biologically active nitrogen-containing heterocycles and activated small ring systems in drug design led us to explore the anticancer properties of a set of novel 1,5-disubstituted pyrrolidin-2-ones 1, 2 and related pyrrolidinone based compounds 3 (Fig. 1c). Pyrrolidones are readily available synthetically and the designed synthetic procedures allow a straightforward fine-tuning of substituents to influence the structure–activity relationship.27–30 Moreover, pyrrolidine derivatives are well-known as effective anti-proliferative agents.31 Thus, we decided to test them as novel tubulin polymerization inhibitors targeting the colchicine site.
Results and discussion
Chemistry
We hypothesized that the 1,5-diaryl- or 1,5-aryl(styryl)pyrrolidin-2-ones 1, 2 and tetrahydropyrrolo[1,2-a]quinoline-1(2H)-ones 3 may be a good starting point for the search for novel colchicine site binders. We synthesized these compounds starting from donor–acceptor cyclopropanes 4 (see the ESI† for the details) and anilines. To synthesize 1,5-diarylpyrrolidin-2-ones 1, we used a highly efficient and practical method recently developed by some of us (Scheme 1).30 This method includes Ni(ClO4)2-catalyzed ring opening of readily available donor–acceptor cyclopropanes 4 with the appropriate aniline affording γ-aminoesters followed by lactamization at reflux in toluene in the presence of acetic acid. Removal of the ester group in the intermediate 2-oxopyrrolidine-3-carboxylates was carried out by alkaline hydrolysis and decarboxylation at reflux in toluene in a one-pot mode. 1,5-Diarylpyrrolidin-2-ones 1a,b,d–f were obtained for the first time and fully characterized by 1H and 13C NMR spectroscopy, IR spectroscopy and high-resolution mass spectrometry. | ||
| Scheme 1 Synthesis of 1,5-diarylpyrrolidin-2-ones 1a–f from donor–acceptor cyclopropanes 4. Compounds 1 were obtained as racemic mixture. Reagent and conditions: (a) Ar′NH2, Ni(ClO4)2·6H2O, DCE, r.t.; (b) AcOH, PhMe, reflux; (c) NaOH, EtOH/H2O, r.t.; (d) PhMe, reflux. | ||
Using the same reaction sequence, from 2-styrylcyclopropane-1,1-dicarboxylates 4 we synthesized 1-aryl-5-styrylpyrrolidin-2-ones 2a–d, the treatment of which with polyphosphoric acid (PPA) produced 5-aryl-3,3a,4,5-tetrahydropyrrolo[1,2-a]quinolin-1(2H)-ones 3 (Scheme 2, conditions (a)–(e)).
 | ||
Scheme 2 Synthesis of 1-aryl-5-styrylpyrrolidin-2-ones 2 and 5-aryl-tetrahydropyrrolo[1,2-a]quinolin-1(2H)-ones 3. Compounds 2 and 3 were obtained as racemic mixtures. Reagents and conditions: (a) Ar′NH2, Ni(ClO4)2·6H2O, DCE, r.t.; (b) AcOH, toluene, reflux; (c) NaOH, EtOH/H2O, r.t.; (d) toluene, reflux; (e) PPA, 100 °C; (f) PhNH2, Y(OTf)3, DCE, r.t.; (g) AcOH, 1,4-dioxane, reflux; (h) NaOH, EtOH/H2O, r.t.; (i) PhCl/dioxane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), reflux; (j) MsOH/DCM (1:3), 40 °C. :1), reflux; (j) MsOH/DCM (1:3), 40 °C. | ||
Compounds 2e and 3c, which contain an unprotected OH group in one of the aromatic fragments, were prepared in a similar manner using a slightly modified synthetic procedure (Scheme 2, conditions (f)–(j)). It is worth noting that the reaction proceeds diastereoselectively and only one diastereomer was formed. Using 1H–1H NOESY spectroscopy32 and comparing spectra for pyrroloquinolines 3 with spectral data for the cognate compounds,33cis arrangement was assigned to the hydrogens at the stereogenic centers.
Evaluation of the effects of synthesized compounds on tubulin polymerization in vitro
We used an in vitro differential interference contrast (DIC) microscopy assay34,35 to characterize microtubule dynamics in the presence of compounds 1, 2, 3 (Fig. 2a). Tests of 2a and 3a showed a considerable suppression of the growth rate of microtubules (Fig. 2b and c). The frequency of microtubule catastrophes also decreased, but the effect was statistically significant only for compounds 2a and 3a. Their effect was similar to that of microtubule destabilizers, such as nocodazole, but distinct from that of stabilizers, such as paclitaxel (Fig. 2b–d). | ||
| Fig. 2 Effects of compounds 1, 2, 3 on dynamics of individual microtubules. a) Schematics of in vitro assay. b) Representative kymographs of microtubule dynamics in the presence of the tested compounds. Red dotted lines mark the boundaries between the stable microtubule seeds and the dynamic extensions of the microtubules. Nocodazole (Noc.) and paclitaxel (Pac.) were added at 0.5 μM and 0.02 μM concentrations as positive controls. All other compounds except 1f were added at a concentration of 100 μM. Compound 1f was added at a concentration of 50 μM because of its poor water solubility at 100 μM. Control samples 1 and 2 show microtubule dynamics in the absence of added compounds at the beginning and end of the experiment to ensure that normal microtubule dynamics is fully restored after the added compounds are washed away from the microscope flow chamber after each test. c and d) Quantification of microtubule growth rate and catastrophe frequency. For growth rate, each point is a measurement from one microtubule growth cycle. Each dot corresponds to a mean value from an independent experiment, based on analysis of N = 7–22 microtubules (34–60 microtubules per each tested compound); the short horizontal bars show the mean values, error bars show s.e.m. Dotted lines separate different experimental conditions. Both controls correspond to measurements in the presence of 1% DMSO in the chamber. Statistical significance was assessed using an unpaired t-test with Welch correction. ns – not significant, * – p-value (two-tailed) < 0.05, **** – p-value (two-tailed) < 0.0001. | ||
Compounds 1c, 2c, 2d and 3b exhibited a smaller, although statistically significant, effects on the microtubule growth rates, while the other tested compounds had no effect on microtubule dynamics in vitro (Fig. 2b–d). Although the 3,4,5-trimethoxyaryl group (known as the A-ring of combretastatin A-4) is optimal to maintain potent activity of many colchicine site binders,36 compounds 1a,b did not suppress either microtubule growth rate or catastrophe frequency.
Evaluation of the tubulin polymerization inhibitors from series 2 and 3 in the cancer cell culture
Inhibitors of microtubule dynamics are expected to arrest cells in mitosis, possibly further promoting apoptosis.37 To test whether this is true for compounds 2 and 3, we examined their effects on the proliferation of human cancer cells. First, we analyzed the effect of compounds 2 and 3 on cultured lung adenocarcinoma cells, A549, using the flow cytometry-based propidium iodide (PI) cell cycle assay (Fig. 3). In the presence of compound 2a, accumulation of cells in G2/M stage was observed at 116 μM and more prominent – at 200 μM. For compounds 2c and 2d, no significant G2/M accumulation was observed at maximum concentrations. Compound 3a induced G2/M accumulation at concentrations above 22 μM and more prominently – at 66 μM. For compound 3b, no significant G2/M accumulation was observed at 66 μM, the maximum concentration we tested. | ||
| Fig. 3 Analysis of the effects of compounds 2 and 3 on the A549 cell cycle. a) Two examples of DNA content curves used for cell cycle analysis (see Fig. S1† for additional information). The distribution of propidium iodide (PI) fluorescence intensity, assessed by flow cytometry, is displayed along with linear gates set to determine G0/G1, S, and G2/M cell cycle populations for the DMSO control and compound 3a at a concentration of 66 μM. b–f) Quantification of the percentages of A549 cells in different phases of the cell cycle after treatment with the tested compounds for 24 hours. Nocodazole (Noc.) is included as a positive control. Data are presented as mean ± s.d. from 2–3 independent experiments. The statistical significance of the deviation in the percentage of cells in the G2/M phase from the DMSO control (0 μM) was analyzed using Dunnett's multiple comparisons test. ns, not significant; **p < 0.01; ****p < 0.0001 (two-tailed). | ||
We further confirmed mitotic arrest in the presence of 2a and 3a by microscopic analysis of A549, MCF-7 and HeLa cell lines (Fig. 4). Treatment of cells with 1 μM nocodazole was used as a positive control. Compounds 2a and 3a induced an increase in the mitotic index in all cell lines in the same way as nocodazole, starting at concentrations above 200 μM and 50 μM, respectively.
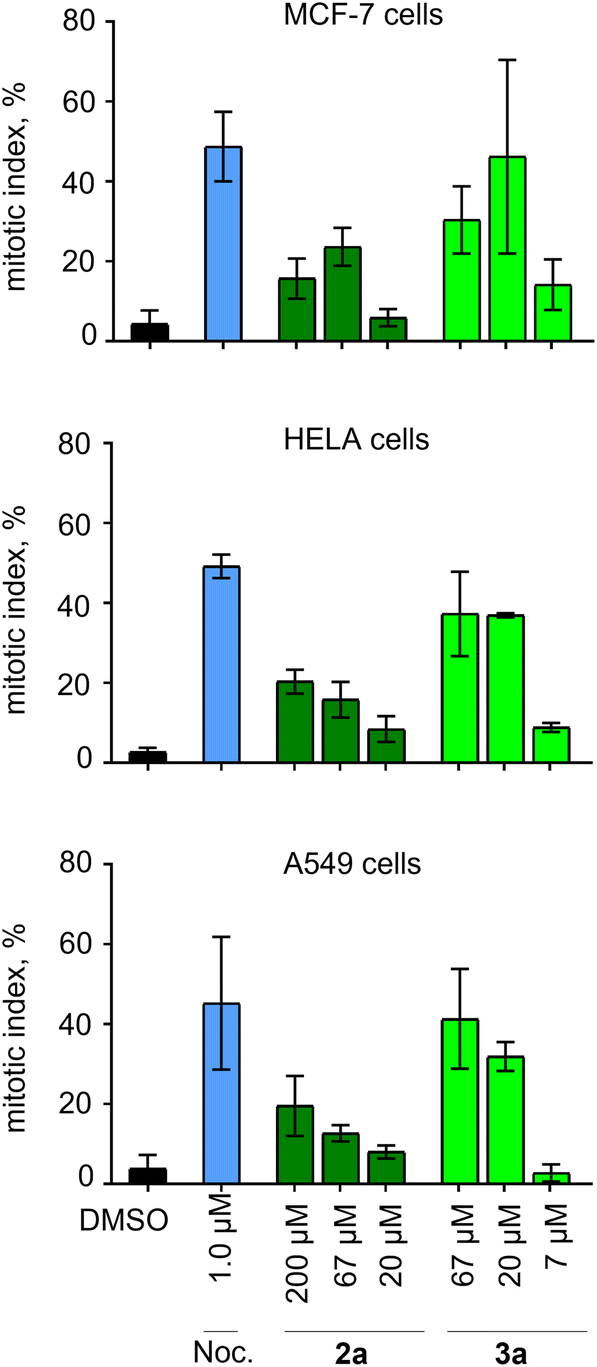 | ||
| Fig. 4 Evaluation of effects of compounds 2a and 3a based on phase contrast microscopy of live cells. Percentage of cells in mitosis after 12 h incubation with DMSO, nocodazole, or one of the tested drugs in 3 cell types. Mean ± standard deviations are shown for each concentration, based on four technical replicates. | ||
Molecular docking and prediction of compounds with enhanced affinity
We used a molecular docking approach to identify the potential binding mechanism of compounds 2a and 3a. The docking procedure requires the selection of the tubulin dimer structure to be used as the receptor for the ligand of interest. Available structures of tubulin from the Protein Data Bank (PDB), containing different ligands at the colchicine site, display slightly different conformations of the ligand-binding pocket.38 This flexibility of the binding site produces a problem for standard ‘rigid’ molecular docking methods, since the choice of tubulin structure can significantly affect the docking score and hence the ligand pose prediction. We found that this problem can be partially remedied using the ‘induced fit’ docking approach,39 incorporated into the Schrödinger software package, which can account for the flexibility of both ligand and receptor.40 However, even with this approach, the docking score alone is not a reliable predictor of the correct binding pose, as was confirmed by cross-docking of randomly selected colchicine site ligands into tubulin structures co-crystalized with other diverse ligands of the same site (Fig. S2a†).To overcome this problem, we performed a post-docking analysis using structural protein–ligand interaction fingerprints (SPLIF).41 SPLIF analysis allows for the quantitative evaluation of whether a compound's docking pose interacts with the protein target in a manner similar to any known ligand, and it can identify real poses that may have been penalized by a poor initial docking score. Indeed, using a test set of five protein–ligand structures from the PDB, which contained three well-established ligands: nocodazole, colchicine, combretastatin A-4, and two other randomly selected structures, we found that the combination of the induced fit docking and SPLIF similarity scores allowed us to select considerably more accurate poses of the test ligands within the colchicine site of tubulin, compared to the docking score alone (Fig. S2b and c†). Encouraged by this result, we applied the induced fit docking, enhanced by the SPLIF analysis, to predict the likely binding poses of compounds 2a and 3a (Fig. 5).
 | ||
| Fig. 5 Tubulin-binding modes of compounds 2a and 3a, predicted by the induced fit docking and SPLIF analysis. The predicted hydrogen bonds are shown with dashed lines. Hydrophobic interactions are shown as red arcs. The surface of β-tubulin (gray) is viewed from the side of the intradimer interface. a) Compound 2a bound to the colchicine site of tubulin (PDB ID 6BRY42). b) Compound 2a at the colchicine site of tubulin, viewed from a slightly different angle. c) (2E)-3-(3-hydroxy-4-methoxyphenyl)-1-(7-methoxy-2H-1,3-benzodioxol-5-yl)-2 methylprop-2-en-1-one from the PDB structure 5JVD43 is shown in brown together with compound 2a (yellow) in the colchicine site of tubulin. d) Compound 3a bound to the colchicine site of tubulin (PDB ID 6BRY). e) Compound 3a at the colchicine site of tubulin, viewed from a slightly different angle. f) Overlay of compounds 2a and 3a at the colchicine site of tubulin. | ||
The outcomes of molecular docking and SPLIF analysis indicate that the interactions of compounds 2a and 3a with tubulin are predominantly hydrophobic, as with most existing colchicine ligands. In addition, each of the compounds 2a and 3a are predicted to form a single hydrogen bond between the Asp-249 amino acid of β-tubulin and the oxygen of the pyrrolidone ring. The validity of these predictions is justified by the very similar binding patterns of previously characterized ligands, whose tubulin-bound X-ray structures are available from the PDB (Fig. 5c and S3a†). The higher binding ability of compound 3a compared to compound 2a is resulted from: a) fixation of the mutual arrangement of two aromatic groups in 3a, in contrast to that in 2a, wherein rotation around the bond between the C(5) atom of the pyrrolidone ring and the styryl moiety is possible; b) a more suitable dihedral angle between the two aromatic rings in 3a due to geometric constraints induced by the piperidine ring formation (Fig. 5f).
Based on molecular modelling results (Fig. 5 and S3†), we hypothesized that compound 3c, possessing an additional hydroxy group at the meta-position of exocyclic phenyl substituent, might allow the formation of an additional hydrogen bond (Fig. 6a). Induced fit docking of compound 3c confirmed this idea, identifying a possible binding pose of that compound, which is similar to the predicted pose of 3a, but enhanced with hydrogen bonds with Asn-347 and Lys-350 of β-tubulin (Fig. 6b–d and S3b†).
 | ||
| Fig. 6 Compound 3c and its predicted molecular pose. a) Structure of compound 3c. b) Compound 3c in the colchicine site of tubulin, as predicted by molecular docking (PDB ID 6BRY). The surface of β-tubulin (gray) is viewed from the side of the intradimer interface. c) The predicted hydrogen bonds between tubulin and compound 3c are shown with dashed lines. Hydrophobic interactions are shown as red arcs. d) Overlay of predicted poses of compounds 3a and 3c. | ||
Synthesis and evaluation of a modified compound 3c
Encouraged by that prediction, we synthesized compound 3c using the method described in Scheme 2. To verify the ability of compound 3c to target the colchicine site of tubulin, we used chlorpromazine as a more widely available probe, compared to many fluorescent colchicine analogues, such as 2-methoxy-5-(2′,3′,4′-trimethoxyphenyl)tropone (MTC).44,45 Chlorpromazine is a well-known fluorescent psychotropic drug that has been previously established as a reversible tubulin binder that displaces colchicine from its binding site on tubulin, as demonstrated through a combination of viscometry, [3H]colchicine binding studies, ultracentrifugation, and cell culture assays.46,47 We found that the fluorescence of chlorpromazine increased in a tubulin-concentration-dependent manner (Fig. 7), an enhancement of the fluorescence quantum yield typical of many fluorescent ligands upon binding to their protein targets.48 By measuring this fluorescence enhancement as an indicator of chlorpromazine–tubulin binding, we observed a clear displacement of chlorpromazine by compound 3c, which, while smaller in extent, was qualitatively similar to that caused by colchicine (Fig. 7). This validates our assumption that compound 3c and colchicine bind to the same site on tubulin. | ||
| Fig. 7 Exploration of the binding site of compound 3c using the chlorpromazine displacement assay. Ligands were added at a concentration of 100 μM. Data are presented as mean ± s.e.m. from three independent experiments. | ||
Using DIC microscopy in vitro assay, we visualized and quantified microtubule dynamics in the presence of compound 3c (Fig. 8a). At the concentration of 25 μM, this compound decreased the growth rate of microtubules by more than 50% and lowered the frequency of catastrophes (Fig. 8a and b).
 | ||
| Fig. 8 Effects of compound 3c on dynamics of individual microtubules in vitro and on mitosis of A549 cells. a) Representative kymographs of microtubule dynamics in the presence of the compound 3c at 25 μM concentration. Red dotted lines mark the boundaries between the stable microtubule seeds and the dynamic extensions of the microtubules. b) Quantification of microtubule growth rates and microtubule catastrophe frequencies in the presence or in the absence of compound 3c. c) Representative DNA content curves used for cell cycle analysis are shown (see Fig. S4† for additional information). Distributions of PI fluorescence intensity, assessed by flow cytometry, are displayed along with linear gates set to determine G0/G1, S, and G2/M cell cycle populations for examples of the DMSO control and compound 3c at a concentration of 64 μM. (d) Quantification of the percentages of A549 cells in different phases of the cell cycle after treatment with compound 3c for 24 hours. Nocodazole (Noc.) is included as a positive control. Data are presented as mean ± s.d. based on 2–3 independent experiments. The statistical significance of the deviation of the percentage of cells in the G2/M phase from the DMSO control (0 μM) was analyzed using Dunnett's multiple comparisons test. ns, not significant; *p < 0.05; ****p < 0.0001 (two-tailed). | ||
We further tested the effects of compound 3c on A549 cell proliferation using a flow cytometry-based cell cycle assay (Fig. 8c, d and S4†). Quantification of the percentage of mitotic cells demonstrated a clear concentration-dependent effect of this compound. The activity progressively increased from 2a to 3a to 3c, highlighting a distinct structure–activity relationship (Fig. 9).
 | ||
| Fig. 9 Percentage of mitotic cells in the population as a function of the concentration of the added compounds 2a, 3a or 3c. The upper grey zone indicates the mean ± standard deviation for A549 cells treated with 0.3 or 1 μM nocodazole, used as a positive control. The lower grey zone indicates the mean ± standard deviation for cells treated with DMSO, used as a negative control. Each dot is an average of 2–4 experiments, smoothed with a sliding average filter with a 3-point window. Solid lines are fits to the Hill's equation: Y = (Ymax − Y0)Xw/(Xw + IC50w) + Y0, where Ymax, Y0, w, IC50 are the fitting parameters. | ||
Consistently, the MTT assay for cell proliferation has demonstrated that compound 3c exhibited the highest cytotoxicity among the other tested compounds from series 2 and 3 on a panel of four cell lines: VA-13, MCF-7, HEK293T, and A549 (Fig. S5†). All tested substances demonstrated selective cytotoxicity toward the HEK293T cell line (IC50 = 4.1 ± 0.5 μM for compound 3c). This cell line is the most rapidly dividing in our set, and its highest susceptibility to the drugs supports the notion that the cytotoxicity of the tested drugs is primarily mediated by interference with the cell division process.
It is important to note that cytotoxicity observed in cell proliferation assays may not directly correlate with organismal toxicity. Ideally, mitostatic drugs, such as paclitaxel or nocodazole, induce mitotic arrest followed by apoptosis, rather than causing acute cell death. This mechanism reduces inflammation and damage to surrounding tissues, making mitostatics potentially less toxic at the organismal level compared to drugs that induce acute cell death through necrosis or other less regulated mechanisms. In our study, compound 3c exhibited mitostatic effects similar to nocodazole, in contrast to compounds 2a and 3a, which showed more acute cytotoxicity. This was evidenced by the similar plateau level of compound 3c and nocodazole (upper dotted red line), while the other compounds exhibited lower plateau levels (Fig. 9). It is conceivable that the meta-hydroxy group in the exocyclic phenyl substituent of compound 3c may play a role in reducing potential off-target interactions with other cellular receptors, thus enhancing the specificity of this compound for tubulin.
Conclusions
To summarize, in this work we report a synthesis of new 1,5-disubstituted pyrrolidin-2-ones 1, 2via a three-step sequence involving donor–acceptor cyclopropane ring opening with anilines as the key step. The 1-aryl-5-styrylpyrrolidin-2-ones 2 were further transformed into tricyclic ring systems 3 using an intramolecular Friedel–Crafts-type cyclization. To our knowledge, biological activity of these compounds, containing the indolizidine scaffold,49 has not been explored previously in any context. Here we examined the potential of these drugs as a new group of mitostatics. We establish 5-aryltetrahydropyrrolo[1,2-a]quinoline-1(2H)-ones 3, especially the hit compound 3c, as tubulin polymerization inhibitors, effectively suppressing division of cultured cancer cells.While a plethora of mitostatics with excellent antiproliferative activity have been developed previously, the search for new chemotypes among low molecular weight, synthetically accessible molecules remains crucial. The novel pyrrolo[1,2-a]quinoline skeleton, identified in this work, expands the known structure–activity relationship landscape for drugs targeting the colchicine site of tubulin. This skeleton can be easily modified due to its modular and simple assembly method. Moreover, the pyrrolidin-2-one and indolizidine motifs present in compounds 3 are known for their bioactive properties, reinforcing the potential of our hit compound as a promising candidate for further modification in the search for multitarget chemotherapeutics.
Experimental methods
Materials and reagents
Tubulin for all experiments was purified from bovine brains as described previously by three cycles of polymerization and depolymerization in a buffer with a high ionic strength.50 Labeling of the tubulin with 3-amino-3-deoxydigoxigenin hemisuccinamide succinimidyl ester (Invitrogen A-2952) was carried out using a previously published protocol.51 Bovine serum albumin was purchased from Sigma-Aldrich. Nocodazole (Sigma-Aldrich) was dissolved in DMSO at 10 mM concentration. Chlorpromazine (Sigma-Aldrich) and synthetic compounds 1, 2, 3 were dissolved in DMSO at 100 mM concentration. All small molecule compounds were aliquoted and stored at −20 °C; proteins and nucleotides were stored at −80 °C.In vitro assay for microtubule dynamics
Microtubule dynamics were investigated in vitro according to our previously developed protocols52–54 in a custom-designed flow chamber.55 The flow chamber was mounted on a Nikon Ti Eclipse microscope, equipped with an Andor iXon3 EMCCD camera and 1.49 NA TIRF 100× oil immersion objective. The solutions were pumped through the chamber by a syringe pump. Stabilized microtubule seeds were prepared by mixing 67 μM of unlabeled tubulin and 15 μM digoxigenin-labeled tubulin and 1 mM guanylyl-(α,β)-methylene-diphosphonate (GMPCPP, purchased from Jenna Biosciences) and incubating the mixture at 37 °C for 15 min. Unpolymerized fraction of tubulin was then removed by centrifugation at 25000g at 37 °C for 15 min. The stabilized seeds were diluted in BRB-80 buffer (80 mM K-PIPES, 1 mM MgCl2, 1 mM EGTA, pH 6.8) supplemented with 0.1 mM GMPCPP and stored at 37 °C for no more than 3 days. For observation of microtubule dynamics, a flow chamber with a silanized hydrophobic coverslip was assembled and incubated with a solution of anti-digoxigenin antibodies (Roche Applied Science, 11093274910) in BRB-80 buffer for 15 min. The coverslip was further blocked with 1% solution of Pluronic F-127 in BRB-80, followed by a wash with BRB-80 buffer. Stable GMPCPP microtubule seeds were then introduced into the microscopy chamber for 15–30 min. Unbound microtubule seeds were washed away by the BRB-80 buffer. Imaging Buffer, composed of the BRB-80 buffer, supplemented with 2 mM guanosine triphosphate (Sigma-Aldrich), 5 mg mL−1 bovine serum albumin, 0.08 mg mL−1 catalase, 0.1 mg mL−1 glucose oxidase, 12 mg mL−1 glucose, 1 mM dithiothreitol, 0.5% β-mercaptoethanol was freshly prepared and centrifuged 25000g at 37 °C for 15 min before each experiment. The polymerization mixture containing the imaging buffer, non-labeled tubulin at 12–18 μM concentration in the presence of either the working concentration of the tested compound, dissolved in DMSO, or equivalent concentration of DMSO (as a negative control) was pre-heated to 32 °C for 1 min and sequentially introduced into the flow chamber. Microtubule dynamics were imaged using DIC microscopy for 30 min at the temperature of 32 °C, controlled with an objective heater (Bioptechs). A motorized microscope XY-stage allowed cyclic data acquisition from 8 to 10 fields of view, collecting images of each field once per 10 seconds. This enabled us to acquire information about 7–22 microtubules on each independent day (34–60 microtubules per experimental condition). After imaging microtubules in the negative control (DMSO), the microscopy flow chamber was washed extensively by the imaging buffer and a new solution of tubulin with one of the test compounds was introduced. Up to 3 compounds could be tested sequentially in the same flow chamber, with intermittent washes between them. At the end of each experiment, we imaged the second negative control, which had an identical solution composition as the first negative control (12–18 μM tubulin and DMSO). This served as an intrinsic control to make sure that the parameters of microtubule dynamics were reproducible and the chamber did not degrade over time. This was always the case, based on N = 3 independent experiments.
Analysis of microtubule dynamics
Microtubule dynamics were processed in ImageJ software. Using a custom JavaScript, kymographs showing dependencies of microtubule length on time were built and analyzed to extract microtubule growth rates and catastrophe frequencies. Only faster growing ‘plus-ends’ of microtubules were analyzed. The growth rates of the plus-ends of microtubules were measured as the slopes on the kymographs, while the frequencies of catastrophes were defined as the number of growth-to-shortening transitions during microtubule growth recording. Microtubule growth rates were then pooled for each experimental condition. The second negative control was analyzed analogously to make sure that the sequence of experiments did not affect the conclusion. Statistical data analysis was performed with GraphPad Prism 8 software.Chlorpromazine displacement assay
The assay was performed using the Cary Eclipse fluorescence spectrometer (Agilent Technologies). Chlorpromazine fluorescence was excited at 440 nm and emission was detected at 495 nm. Tubulin was serially diluted from 9 μM to 18 nM using a two-fold dilution series in the BRB-80 buffer. The tested compounds were added to each sample at a final concentration of 100 μM. Prior to the measurements, the samples were incubated for 1 hour at room temperature. Each sample was first measured to establish the baseline chlorpromazine fluorescence. Next, a tested compound was added to the same sample, and the fluorescence of chlorpromazine was recorded.Live cell microscopy and flow cytometry for cell cycle progression quantification
Three cell lines (HeLa, A549, and MCF-7) were obtained from American Type Culture Collection (ATCC, VA, USA) and maintained according to a regular recommendation of ATCC in DMEM (ThermoFisher, Cat. # 11965092) or DMEM/F-12 (ThermoFisher Cat. # 21331020) supplemented with 10% FBS (ThermoFisher, Cat. # 10100147), supplemented with 5 mM of L-glutamine (Sigma, Cat. # G7513), and antibiotics penicillin–streptomycin (Sigma-Aldrich, Cat. # P4333).Microscopic observations and flow cytometry analysis were performed using previously developed protocols with small modifications.56 Briefly, cells were seeded into 24-well cell culture plates, then after 24–48 h the culture medium was exchanged to CO2 independent medium (ThermoFisher, Cat. # 18045088) with 10% FBS and 5 mM L-glutamine (Sigma, Cat. # G7513). Microtubule inhibitors were added, and cells were further incubated for 48 h in 37 °C in the heating incubator chamber, mounted on a Zeiss Cell Observer microscope (Carl Zeiss GmbH). For control cells, an equivalent amount of DMSO was added instead of the inhibitors. During the incubation, phase contrast microscopy images of four fields of view per culture plate well were recorded using an ORCA-Flash4.0 V2 camera (Hamamatsu Photonics, Hamamatsu, Japan) and a Plan-NEOFLUAR 20×/0.4 objective under ZEN software. The duration of the recording was 24 h with a 10-minute interval between the frames. 12 h after adding the drugs, the percentage of mitotic cells was determined using Fiji software.
Cell cycle analysis
Cells were seeded into 12-well plates. After 48 h growth, drugs were added and cells were further incubated for 24 h. DMSO at appropriate concentrations was added to each plate to control wells, as well as nocodazole as positive control. After the incubation, cells were detached from the plates, transferred to PBS and concentrated by centrifugation. Cells were then fixed with 70% ice-cold ethanol overnight, transferred into PBS, stained with 30 μg mL−1 of propidium iodide (PI) and the DNA content was measured using an Attune NxT flow cytometer (Thermo Fisher Scientific) at Ex. 488 nm/Em. 550–630 nm. The results obtained were analyzed using Flow Jo software (BD, Ashland, OR).MTT assay
Human breast cancer cell line MCF-7 was kindly provided by Dr D. Khochenkov, human lung adenocarcinoma cell line A549 was kindly provided by Dr. S. Dmitriev, immortalized human fibroblasts cell line VA-13 was kindly provided by Dr. M. Rubtsova, human embryonic kidney HEK293T cell line was kindly provided by Dr. E. Knyazhanskaya. MCF-7, VA-13, A549, and HEK293T cell lines were maintained in DMEM/F-12 (LLC Paneco, Russia) culture medium containing 10% fetal bovine serum (INTL Kang, China) and 50 μg mL−1 penicillin and 0.05 mg mL−1 streptomycin at 37 °C (Thermo Fisher Scientific, USA) in 5% CO2 in a humidified atmosphere. Genotypes of all four cell lines were validated by STR (LLC Gordiz, Russia). Cell cultures were tested for the absence of mycoplasma (MycoReport kit, Eurogen, Russia).Cytotoxicity was assessed using the MTT (3-(4,5-dimethylthiazol-2-yl)2,5-diphenyl tetrazolium bromide) assay.57 It was carried out on the Janus (PerkinElmer) automatic station. 5000 cells per well for VA-13, MCF-7 and 2500 cells per well for HEK293T, A549 cell lines were plated out in 140 μL of DMEM/F12 media (Paneco LLC, Russia) in a 96-well plate and incubated at 37 °C, 5% CO2 for 24 h before treatment. Then 11 μL of dilutions in media of all tested substances (all stock solutions were prepared in DMSO) were added. Each substance was applied at the final concentrations of 100–0.045 μM (eight dilutions), in triplicate. Then the cells were grown for 72 h. At the end of the incubation, we added MTT (Paneco LLC, Russia) into the media (up to 0.5 mg mL−1), incubated the cells for 1.5 h, followed by removing the media and addition of 120 μL of DMSO. The amount of MTT reduced by cells to its blue formazan derivative was measured photometrically at 565 nm using a plate reader VICTOR X5 (PerkinElmer) and normalized to the values for cells incubated without compounds. IC50 was calculated with “GraphPad Prism 5” software (GraphPad Software, Inc., San Diego, CA).
Molecular docking and SPLIF analysis
We used PyPDB python package58 to parse the PDB for any structures of tubulin with ligands at the colchicine site. A total of 143 structures of this kind were collected and 13 of them were removed from further analysis due to poor resolution (>3.5 Å). Three structures of well-established tubulin ligands, including colchicine (PDB id 4O2B), nocodazole (PDB id 5CA1), combretastatin A-4 (PDB id 5LYJ), and two structures of other randomly selected ligands (PDB ids: 5CB4 and 5XKH) were separated to form a test dataset. We used the clustering method, described earlier,59 to classify the remaining 125 structures into clusters (Fig. S2d†). PDB structures 5ITZ, 5XHC, 6BRY, 6PC4, and 7DBC were identified as the centroids of the five most populated clusters. We used these five structures to represent diverse but common receptor conformations of colchicine site ligands.3D structures of the most likely diastereomers of the five test ligands and the compounds 2a and 3a were prepared with the LigPrep module of the Schrödinger small molecule drug discovery suite and docked into the colchicine site of each of the selected receptor structures using the ‘induced fit’ docking approach39 with default settings of the Schrödinger software suite. Next, using the Open Drug Discovery Toolkit,60 we computed SPLIFs for all structures predicted by the induced fit docking and the 125 parsed tubulin–ligand structures. The SPLIF similarity scores were calculated between members of these two groups and used to range the predicted ligand poses. For compound 3c the ‘induced fit’ docking approach with default settings of the Schrödinger software suite was used to predict top-20 ligand poses for each of the two stereoisomers, which were then compared to predictions for compounds 2a and 3a, and the pose with the highest similarity and the largest number of hydrogen bonds was selected. Interactions between the ligands and tubulin were annotated with an automated protein–ligand interaction profiler tool.61 PyMOL program was used for visualization of the ligands in the colchicine site.
Data availability
Experimental methods, additional molecular docking data and NMR data (1H NMR and 13C NMR) of the synthesized compounds are available as ESI† (PDF).Author contributions
M. A. B., V. V. S., A. Yu. P., I. A. A., N. K. R., S. S. Z., E. A. T. performed chemical synthesis and analytical characterization of the synthesized compounds. M. N. A. and I. A. L. carried out in vitro experiments and analysis, J. N. B. performed molecular docking studies; M. J., V. V. M, D. A. I., and A. R. P. performed in vivo experiments with cell culture lines; N. B. G., I. A. V., I. V. T. and O. A. I. designed the study, analyzed data, wrote the manuscript, acquired funding; all authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Work with cell culture has been funded by the Grant number AP14869915 from the Science Committee of the Ministry of Science and Higher Education of the Republic of Kazakhstan. Chemical synthesis and analysis of the synthesized compounds was funded by the Russian Science Foundation (grant 21-73-20095 to O. A. I.). Development of the protocol for analysis of microtubule dynamics was supported by a fellowship from the Foundation for the Advancement of Theoretical Physics and Mathematics “BASIS” to M. N. A. In vitro tests of the effects of the chemical compounds and molecular modelling of drug–tubulin interactions were funded by a grant from the Russian Science Foundation 21-74-20035 to N. B. G. Molecular docking was carried out using the equipment of the shared research facilities of HPC computing resources at Lomonosov Moscow State University. Some of the optical filters for microscopy studies were purchased under the Development Program of Moscow State University (order No. 99 of February 6, 2023, contract No. 0183-44-2023). Flow cytometry and live-cell microscopy were performed using the equipment of the shared research facilities of Nazarbayev University. We thank Dr. D. A. Skvortsov for help with the MTT assay for cell proliferation.References
- N. B. Gudimchuk and J. R. McIntosh, Nat. Rev. Mol. Cell Biol., 2021, 22, 777–795 CrossRef CAS.
- T. Mitchison and M. Kirschner, Nature, 1984, 312, 237–242 CrossRef CAS.
- L. Wilson and M. A. Jordan, Chem. Biol., 1995, 2, 569–573 CrossRef CAS PubMed.
- M. A. Jordan and L. Wilson, Nat. Rev. Cancer, 2004, 4, 253–265 CrossRef CAS.
- M. O. Steinmetz and A. E. Prota, Trends Cell Biol., 2018, 28, 776–792 CrossRef CAS.
- C. Dumontet and M. A. Jordan, Nat. Rev. Drug Discovery, 2010, 9, 790–803 CrossRef CAS.
- R. L. Noble, Biochem. Cell Biol., 1990, 68, 1344–1351 CrossRef CAS.
- B. Gigant, C. Wang, R. B. G. Ravelli, F. Roussi, M. O. Steinmetz, P. A. Curmi, A. Sobel and M. Knossow, Nature, 2005, 435, 519–522 CrossRef CAS.
- T. Mühlethaler, D. Gioia, A. E. Prota, M. E. Sharpe, A. Cavalli and M. O. Steinmetz, Angew. Chem., Int. Ed., 2021, 60, 13331–13342 CrossRef.
- G. M. Alushin, G. C. Lander, E. H. Kellogg, R. Zhang, D. Baker and E. Nogales, Cell, 2014, 157, 1117–1129 CrossRef CAS.
- R. B. G. Ravelli, B. Gigant, P. A. Curmi, I. Jourdain, S. Lachkar, A. Sobel and M. Knossow, Nature, 2004, 428, 198–202 CrossRef CAS.
- C.-P. H. Yang and S. B. Horwitz, Int. J. Mol. Sci., 2017, 18, 1733 CrossRef.
- G. Liantinioti, A. A. Argyris, A. D. Protogerou and P. Vlachoyiannopoulos, Curr. Pharm. Des., 2018, 24, 690–694 CrossRef CAS.
- Z.-Y. Lin, C.-C. Wu, Y.-H. Chuang and W.-L. Chuang, Life Sci., 2013, 93, 323–328 CrossRef CAS.
- I. A. Gracheva, E. S. Shchegravina, H.-G. Schmalz, I. P. Beletskaya and A. Yu. Fedorov, J. Med. Chem., 2020, 63, 10618–10651 CrossRef CAS.
- E. C. McLoughlin and N. M. O'Boyle, Pharmaceuticals, 2020, 13, 8 CrossRef CAS.
- Z. Ma, X. Yan, L. Zhao, J. Zhou, W. Pang, Z. Kai and F. Wu, J. Agric. Food Chem., 2016, 64, 746–751 CrossRef CAS.
- L. Laraia, J. Stokes, A. Emery, G. J. McKenzie, A. R. Venkitaraman and D. R. Spring, ACS Med. Chem. Lett., 2014, 5, 598–603 CrossRef CAS PubMed.
- A. Kumari, S. S. Prassanawar and D. Panda, ACS Chem. Neurosci., 2023, 14, 19–34 CrossRef CAS PubMed.
- T. L. Nguyen, C. McGrath, A. R. Hermone, J. C. Burnett, D. W. Zaharevitz, B. W. Day, P. Wipf, E. Hamel and R. Gussio, J. Med. Chem., 2005, 48, 6107–6116 CrossRef CAS.
- C. Da, N. Telang, P. Barelli, X. Jia, J. T. Gupton, S. L. Mooberry and G. E. Kellogg, ACS Med. Chem. Lett., 2012, 3, 53–57 CrossRef CAS PubMed.
- A. Gangjee, Y. Zhao, L. Lin, S. Raghavan, E. G. Roberts, A. L. Risinger, E. Hamel and S. L. Mooberry, J. Med. Chem., 2010, 53, 8116–8128 CrossRef CAS.
- X.-F. Wang, F. Guan, E. Ohkoshi, W. Guo, L. Wang, D.-Q. Zhu, S.-B. Wang, L.-T. Wang, E. Hamel, D. Yang, L. Li, K. Qian, S. L. Morris-Natschke, S. Yuan, K.-H. Lee and L. Xie, J. Med. Chem., 2014, 57, 1390–1402 CrossRef CAS PubMed.
- P. Mowery, F. Banales Mejia, C. L. Franceschi, M. H. Kean, D. O. Kwansare, M. M. Lafferty, N. D. Neerukonda, C. E. Rolph, N. J. Truax and E. T. Pelkey, Bioorg. Med. Chem. Lett., 2017, 27, 191–195 CrossRef CAS PubMed.
- M. A. Boichenko, O. A. Ivanova, I. A. Andreev, A. O. Chagarovskiy, I. I. Levina, V. B. Rybakov, D. A. Skvortsov and I. V. Trushkov, Org. Chem. Front., 2018, 5, 2829–2834 RSC.
- O. A. Ivanova, E. M. Budynina, D. A. Skvortsov, M. Limoge, A. V. Bakin, A. O. Chagarovskiy, I. V. Trushkov and M. Ya. Melnikov, Chem. Commun., 2013, 49, 11482–11484 RSC.
- C.-H. Yeh, R. P. Korivi and C.-H. Cheng, Angew. Chem., Int. Ed., 2008, 47, 4892–4895 CrossRef CAS PubMed.
- Y. Ogiwara, T. Uchiyama and N. Sakai, Angew. Chem., Int. Ed., 2016, 55, 1864–1867 CrossRef CAS PubMed.
- N. Kise, Y. Hamada and T. Sakurai, Tetrahedron, 2017, 73, 1143–1156 CrossRef CAS.
- M. A. Boichenko, A. Yu. Plodukhin, V. V. Shorokhov, D. S. Lebedev, A. V. Filippova, S. S. Zhokhov, E. A. Tarasenko, V. B. Rybakov, I. V. Trushkov and O. A. Ivanova, Molecules, 2022, 27, 8468 CrossRef CAS PubMed.
- A. A. Bhat, I. Singh, N. Tandon and R. Tandon, Eur. J. Med. Chem., 2023, 246, 114954 CrossRef CAS PubMed.
- H. Doodhi, A. E. Prota, R. Rodríguez-García, H. Xiao, D. W. Custar, K. Bargsten, E. A. Katrukha, M. Hilbert, S. Hua, K. Jiang, I. Grigoriev, C.-P. H. Yang, D. Cox, S. B. Horwitz, L. C. Kapitein, A. Akhmanova and M. O. Steinmetz, Curr. Biol., 2016, 26, 1713–1721 CrossRef CAS PubMed.
- W. Zhang, L. Huang and J. Wang, Synthesis, 2006, 2006, 2053–2063 Search PubMed.
- R. J. Toso, M. A. Jordan, K. W. Farrell, B. Matsumoto and L. Wilson, Biochemistry, 1993, 32, 1285–1293 CrossRef CAS PubMed.
- W. B. Derry, L. Wilson and M. A. Jordan, Biochemistry, 1995, 34, 2203–2211 CAS.
- S. N. A. Bukhari, G. B. Kumar, H. M. Revankar and H.-L. Qin, Bioorg. Chem., 2017, 72, 130–147 CrossRef CAS PubMed.
- C. Dumontet and M. A. Jordan, Nat. Rev. Drug Discovery, 2010, 9, 790–803 CAS.
- J. Wang, D. D. Miller and W. Li, Drug Discovery Today, 2022, 27, 759–776 CAS.
- W. Sherman, T. Day, M. P. Jacobson, R. A. Friesner and R. Farid, J. Med. Chem., 2006, 49, 534–553 CAS.
- H. Zhong, L. M. Tran and J. L. Stang, J. Mol. Graphics Modell., 2009, 28, 336–346 Search PubMed.
- C. Da and D. Kireev, J. Chem. Inf. Model., 2014, 54, 2555–2561 CAS.
- S. Banerjee, K. E. Arnst, Y. Wang, G. Kumar, S. Deng, L. Yang, G. Li, J. Yang, S. W. White, W. Li and D. D. Miller, J. Med. Chem., 2018, 61, 1704–1718 CAS.
- M.-D. Canela, S. Noppen, O. Bueno, A. E. Prota, K. Bargsten, G. Sáez-Calvo, M.-L. Jimeno, M. Benkheil, D. Ribatti, S. Velázquez, M.-J. Camarasa, J. F. Díaz, M. O. Steinmetz, E.-M. Priego, M.-J. Pérez-Pérez and S. Liekens, Onco Targets Ther, 2017, 8, 14325–14342 Search PubMed.
- T. J. Fitzgerald, Biochem. Pharmacol., 1976, 25, 1383–1387 CrossRef CAS.
- V. Peyrot, D. Leynadier, M. Sarrazin, C. Briand, M. Menendez, J. Laynez and J. M. Andreu, Biochemistry, 1992, 31, 11125–11132 CrossRef CAS.
- N. D. Hinman and J. R. Cann, Mol. Pharmacol., 1976, 12, 769–777 CAS.
- M. Poffenbarger and G. M. Fuller, J. Neurochem., 1977, 28, 1167–1174 CrossRef CAS PubMed.
- B. Bhattacharyya, S. Kapoor and D. Panda, in Methods in Cell Biology, ed. L. Wilson and J. J. Correia, Academic Press, 2010, vol. 95, pp. 301–329 Search PubMed.
- N. K. Ratmanova, I. A. Andreev, A. V. Leontiev, D. Momotova, A. M. Novoselov, O. A. Ivanova and I. V. Trushkov, Tetrahedron, 2020, 76, 131031 CrossRef CAS.
- M. Castoldi and A. V. Popov, Protein Expression Purif., 2003, 32, 83–88 CrossRef CAS PubMed.
- A. Hyman, D. Drechsel, D. Kellogg, S. Salser, K. Sawin, P. Steffen, L. Wordeman and T. Mitchison, Methods Enzymol., 1991, 196, 478–485 CAS.
- A. E. Vartanova, A. Yu. Plodukhin, N. K. Ratmanova, I. A. Andreev, M. N. Anisimov, N. B. Gudimchuk, V. B. Rybakov, I. I. Levina, O. A. Ivanova, I. V. Trushkov and I. V. Alabugin, J. Am. Chem. Soc., 2021, 143, 13952–13961 CrossRef CAS.
- V. V. Alexandrova, M. N. Anisimov, A. V. Zaitsev, V. V. Mustyatsa, V. V. Popov, F. I. Ataullakhanov and N. B. Gudimchuk, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2208294119 CrossRef CAS.
- M. N. Anisimov, A. V. Korshunova, V. V. Popov and N. B. Gudimchuk, Eur. J. Cell Biol., 2023, 102, 151366 CrossRef CAS PubMed.
- V. A. Volkov, A. V. Zaytsev and E. L. Grishchuk, J. Visualized Exp., 2014, e51150 Search PubMed.
- M. Suleimenov, S. Bekbayev, M. Ten, N. Suleimenova, M. Tlegenova, A. Nurmagambetova, S. Kauanova and I. Vorobjev, Front. Pharmacol., 2022, 13, 933112 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- W. Gilpin, Bioinformatics, 2016, 32, 159–160 CrossRef CAS.
- D. Butina, J. Chem. Inf. Comput. Sci., 1999, 39, 747–750 CrossRef CAS.
- M. Wójcikowski, P. Zielenkiewicz and P. Siedlecki, Aust. J. Chem., 2015, 7, 26 Search PubMed.
- S. Salentin, S. Schreiber, V. J. Haupt, M. F. Adasme and M. Schroeder, Nucleic Acids Res., 2015, 43, W443–W447 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: ESI figures and legends, copies of NMR spectra, supporting references. See DOI: https://doi.org/10.1039/d4md00541d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |