
Modulating polybasic character of galactose-based glycosylated antitumor ether lipids for enhanced cytotoxic response†
Rajat
Arora
 a,
Ayan
Mukherjee
a,
Gilbert
Arthur
b,
Mark W.
Nachtigal
bcd and
Frank
Schweizer
*ae
a,
Ayan
Mukherjee
a,
Gilbert
Arthur
b,
Mark W.
Nachtigal
bcd and
Frank
Schweizer
*ae
aDepartment of Chemistry, Faculty of Science, University of Manitoba, Winnipeg, Manitoba R3T 2N2, Canada. E-mail: frank.schweizer@umanitoba.ca
bDepartment of Biochemistry and Medical Genetics, University of Manitoba, Winnipeg, Manitoba R3E 0J9, Canada
cDepartment of Obstetrics, Gynecology and Reproductive Sciences, University of Manitoba, Winnipeg, Manitoba R3E 0J9, Canada
dPaul Albrechtsen Research Institute, CancerCare Manitoba, Winnipeg, Manitoba R3E 0V9, Canada
eDepartment of Medical Microbiology and Infectious Diseases, University of Manitoba, Winnipeg, Manitoba R3E 0J9, Canada
First published on 14th October 2024
Abstract
We describe the structure–activity relationship studies of galactose-based glycosylated antitumor ether lipids (GAELs) by installing amine groups at different positions of galactose and the glycerol backbone. Different dibasic and tribasic analogues of galacto-GAELs were synthesized and tested against a panel of human epithelial cancer cell lines. A β-anomeric triamino galactose scaffold, was the most active compound of the series and displayed CC50 in the range of 2.6 ± 0.2 μM to 6.5 ± 0.1 μM against various epithelial cancer cell lines. This compound exhibited superior activity to kill cancer cells than cisplatin. The hit GAEL compound did not induce caspase activation and therefore, the cell-killing effect does not occur due to caspase-mediated apoptosis. This observation is in line with the previously reported GAEL prototypes.
1. Introduction
The American Cancer Society estimates over two million people will be affected by cancer in 2024.1 Drug resistance and cancer recurrence are obstacles that limit the complete eradication of the disease. Together, they pose a major clinical challenge to many conventional cytotoxic drugs employed in cancer treatment. Prolonged exposure of pro-apoptotic agents like doxorubicin and cisplatin, results in cancer cells acquiring resistance to apoptosis allowing survival of the cells and tumor progression.2–5 Furthermore, growing evidence in the literature indicates cancer stem cells may be responsible for tumor metastasis, relapse, and resistance to both chemo- and radiation therapy.6,7 Therefore, it is pertinent to explore non-apoptotic drug candidates that can eliminate drug-resistant cancer cells and cancer stem cells.A subclass of antitumor ether lipids (AELs) known as glycosylated antitumor ether lipids (GAELs) are synthetically prepared novel anticancer compounds demonstrated to kill a variety of human epithelial cancer cells8–18via an apoptosis-independent pathway with the formation of cytoplasmic vacuoles closely resembling cellular responses to methuosis.8,19,20 GAELs possess the ability to effectively kill chemosensitive and chemoresistant high-grade serous ovarian cancer (HGSC) cells and patient samples.8,21 In addition, GAEL analogues disintegrate cancer stem cell (CSC) spheroids resulting in complete loss of cell viability in BT-474 cells.11
Structure–activity relationship studies indicate that the cationic charge of GAELs may significantly influence their cytotoxic activity.13,21 The impact of cationicity among many cationic amphiphilic drugs (CADs) inducing non-apoptotic lysosomal-dependent cell death in cancer cells has been discussed.22–24 There are indications that the cancer-specific toxicity of CADs could be due to the electrostatic attraction between CADs and the negatively charged surface of cancer cells22 as a consequence of altered cellular metabolic activity and elevated glycolysis.25,26 The characteristic amphiphilic nature of CADs bearing a hydrophobic chain and a polar head group renders them lysosomotropic. With the propensity to protonate at physiological or at neutral pH, CADs can freely diffuse to acidic lysosomes and incorporate in luminal membranes to induce events that trigger lysosome membrane permeabilization which further leads to plasma membrane rupture.22,24 If the cell-killing effect of GAELs either partially or completely relies on a lysosomal-based pathway like other CADs, the identification of the optimal GAEL cationicity in the molecular structure will be crucial to identify a potent scaffold for further development.
The prototypic GAEL, β-Gln B (Fig. 1) with a primary amine group at the C-2 position of the glucose sugar scaffold exhibited 10-fold higher cytotoxic activity than the analogous D-gluco-GAEL A (Fig. 1) without the amine group.12 Subsequently, diamino GAEL C (Fig. 1) bearing amine groups at the C-2 and C-6 positions of the glucose sugar enhanced the cytotoxic effect by 2–3-fold depending on the cell line11 in comparison with the monoamino GAEL B. In contrast, a diglycosylated GAEL D (Fig. 1), prepared by fusing two glucose sugars each bearing an amine group, resulted in a significant loss of activity.15 Collectively, these observations point out that modulation of the cationic charge should be restricted to a single sugar scaffold to attain optimization of the cationic charge in the GAEL molecular structure. Consequently, the effect of a third amine group attached with a long carbon chain linker in gluco-GAEL E, introduced by amphiphilic modulation of the second amine group at the C-6 position of β-diamino gluco-GAEL C resulted in 2–3-fold higher cytotoxic effect than C against a panel of human epithelial cancer cell lines.9 No studies have been conducted on the cytotoxic effect of having three amine groups directly on the sugar scaffold in GAELs. Furthermore, the effect of mono- and diamine substitution has been mainly investigated in glucose-based GAELs9–11,15 and whether this effect applies to other sugar moieties in GAELs has not been explored.
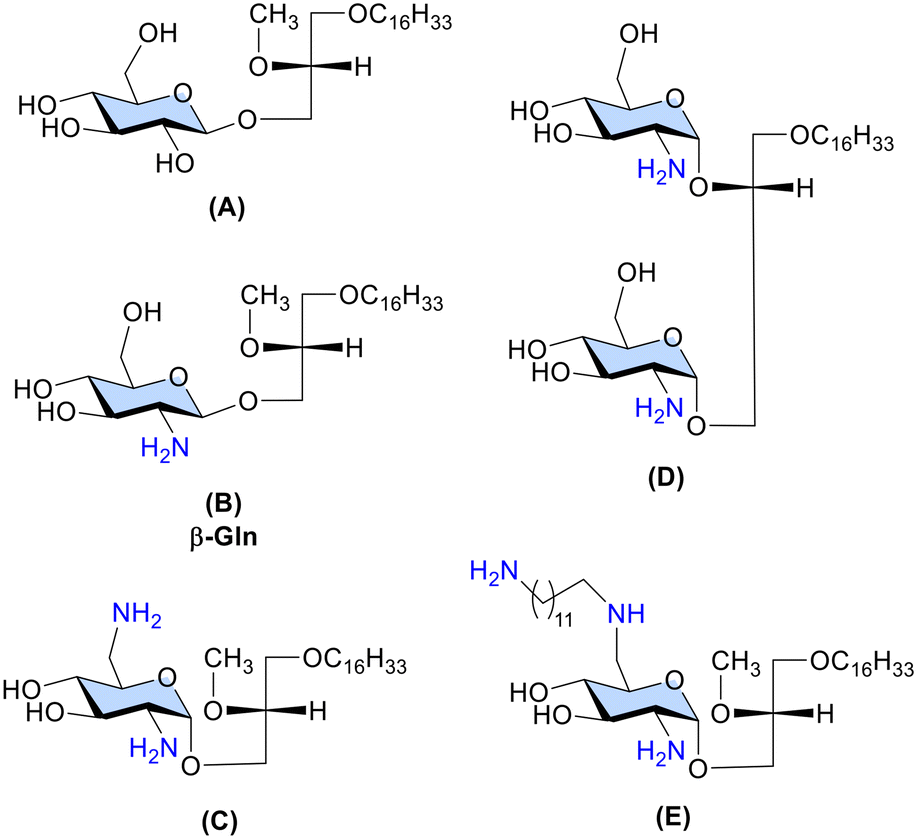
We report here the synthesis of different dibasic and tribasic α- and β-anomeric galactose-based GAEL analogues (Fig. 2) and investigation of their effect on the cell viability of several cancer cell lines including drug-resistant human epithelial cancer cell lines. In addition, we performed a caspase inhibition assay to validate the caspase independent non-apoptotic cell-killing effect of the most active GAEL compound of the series.
 | ||
| Fig. 2 Molecular structures of previously synthesized reference GAEL F (ref. 14) and dibasic (9a–b, 14a–b) and tribasic (17a–b, 21a–b) galactose-based GAELs synthesized and examined in this study. Compounds a and b denote alpha and beta anomers, respectively. | ||
2. Results and discussion
2.1 Chemistry
To study the effect of the number and position of the amine groups on galactose-based GAELs and the effect of glycosidic linkage on cytotoxicity, we synthesized eight structural GAEL analogues (Fig. 2). | ||
Scheme 1 Synthesis of GAELs 9a and 9b. Reagents and conditions: (i) as reported,14 73%; (ii) MeONa, MeOH, 1 h, rt, 71%; (iii) TsCl, pyridine, DMAP, 0 °C to rt, 18 h, 51%; (iv) sodium azide, DMF, 70 °C, 24 h, 71%; (v) Ac2O, DMAP, pyridine, rt, 18 h, 70%; (vi) AgOTf, NIS, DCM, rt, 3 h, (α![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :β/2:1) 7a-34%, 7b-17%; (vii) MeONa, MeOH, 30 min, 8a-58%, 8b-81%; (viii) 1 M trimethyl phosphine solution in THF, THF:H2O (1:4), 2 h, rt, 9a-72%, 9b-79%; abbreviations: 4-dimethylaminopyridine (DMAP), dichloromethane (DCM), 2,2,N,N-dimethylformamide (DMF), room temperature (rt) 23 °C, N-iodosuccinimide (NIS), tetrahydrofuran (THF). :β/2:1) 7a-34%, 7b-17%; (vii) MeONa, MeOH, 30 min, 8a-58%, 8b-81%; (viii) 1 M trimethyl phosphine solution in THF, THF:H2O (1:4), 2 h, rt, 9a-72%, 9b-79%; abbreviations: 4-dimethylaminopyridine (DMAP), dichloromethane (DCM), 2,2,N,N-dimethylformamide (DMF), room temperature (rt) 23 °C, N-iodosuccinimide (NIS), tetrahydrofuran (THF). | ||
The synthesis of 2,6-diamino-galacto-configured GAELs begins from commercially available D-galactosamine 1 which was converted to a thioglycoside donor 2 as described before.14 To obtain a 2,6-diazido compound, acetyl groups on 2 were deprotected using catalytic sodium methoxide in methanol to produce deprotected compound 3. Subsequently, the primary C-6 hydroxy group on 3 was selectively activated as tosylate using toluene sulphonyl chloride to generate compound 4 which underwent azidation with sodium azide to yield diazide 5. The remaining hydroxy groups at C-3 and C-4 were protected with acetic anhydride in pyridine which afforded fully protected glycosyl donor 6. The glycosyl donor 6 was reacted with a commercially available lipid alcohol L1 (Scheme 1) using N-iodosuccinimide (NIS)/silver trifluoromethyl sulfonate promoter combination to afford glycolipid anomers 7a and 7b in 34% and 17% isolated yield, respectively. Subsequent deprotection of acetyl groups using sodium methoxide in methanol produced 8a and 8b, which underwent reduction of azide group by trimethyl phosphine in THF/water (1:4) to yield the final dibasic glycolipid 9a (72%) and 9b (79%).
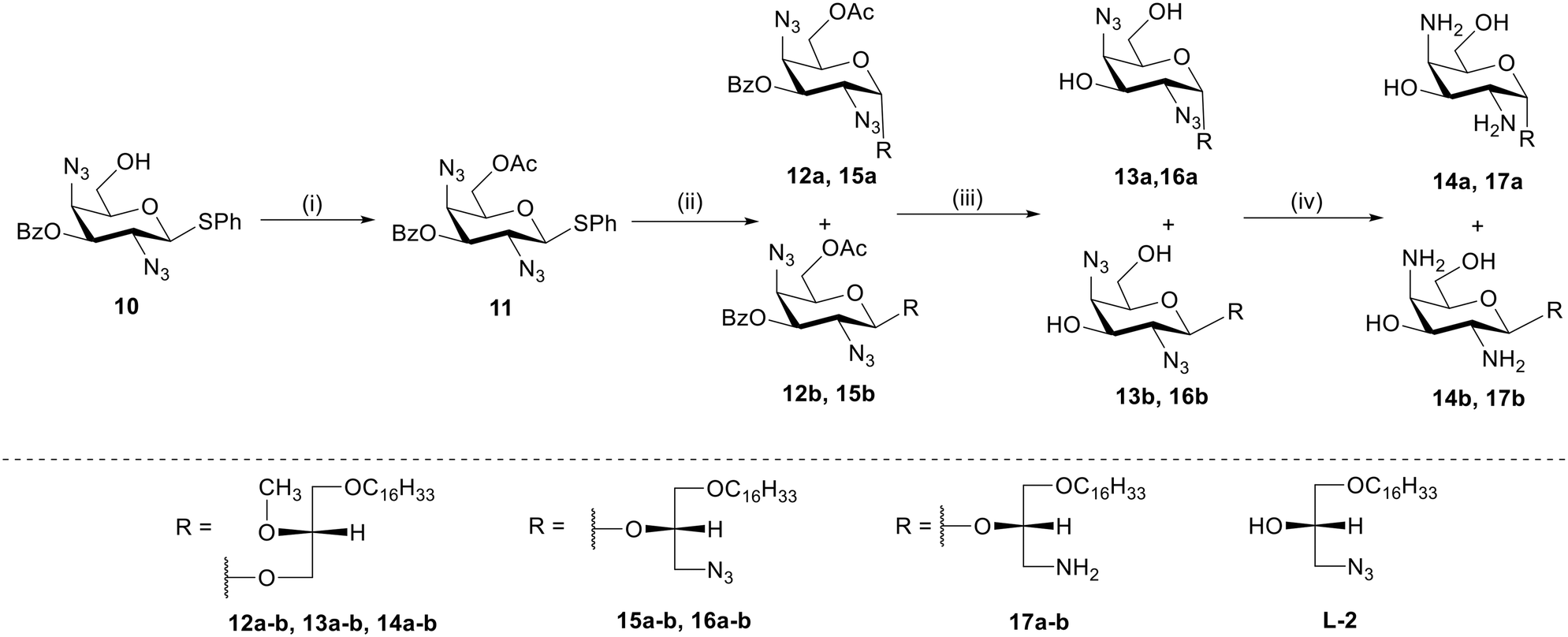 | ||
| Scheme 2 Synthesis of GAELs 14a, 14b, 17a, and 17b. Reagents and conditions: (i) acetic anhydride, DMAP, pyridine, 0 °C to rt, 3 h, 70%.; (ii) respective L1/L2 alcohol, NIS, AgOTf, dry DCM, 0 °C to rt, 3 h, (α:β/1:3) 12a-15%, 12b-56%, (α:β/3:1) 15a-42%, 15b-15%; (iii) NaOMe, MeOH, rt, 3 h, 13a-92%, 13b-94%, 16a-96%, 16b-92%; (iv) 1 M P(CH3)3 solution in THF, THF/water, rt, 5 h, 14a-91%, 14b-93%, 17a-80%, 17b-83%. Abbreviations: 4-dimethylaminopyridine (DMAP), dichloromethane (DCM), room temperature (rt) 23 °C, N-iodosuccinimide (NIS), tetrahydrofuran (THF). | ||
:1 ratio to give 15a (42%) and 15b (15%) purified by column chromatography. Compounds 15a and 15b underwent a reaction with catalytic sodium methoxide for deprotection of acetyl and benzoyl group to yield 16a and 16b. Finally, the azide group reduction was carried out using trimethyl phosphine in 1 M THF that yielded the target compounds, 17a (80%) and 17b (83%).
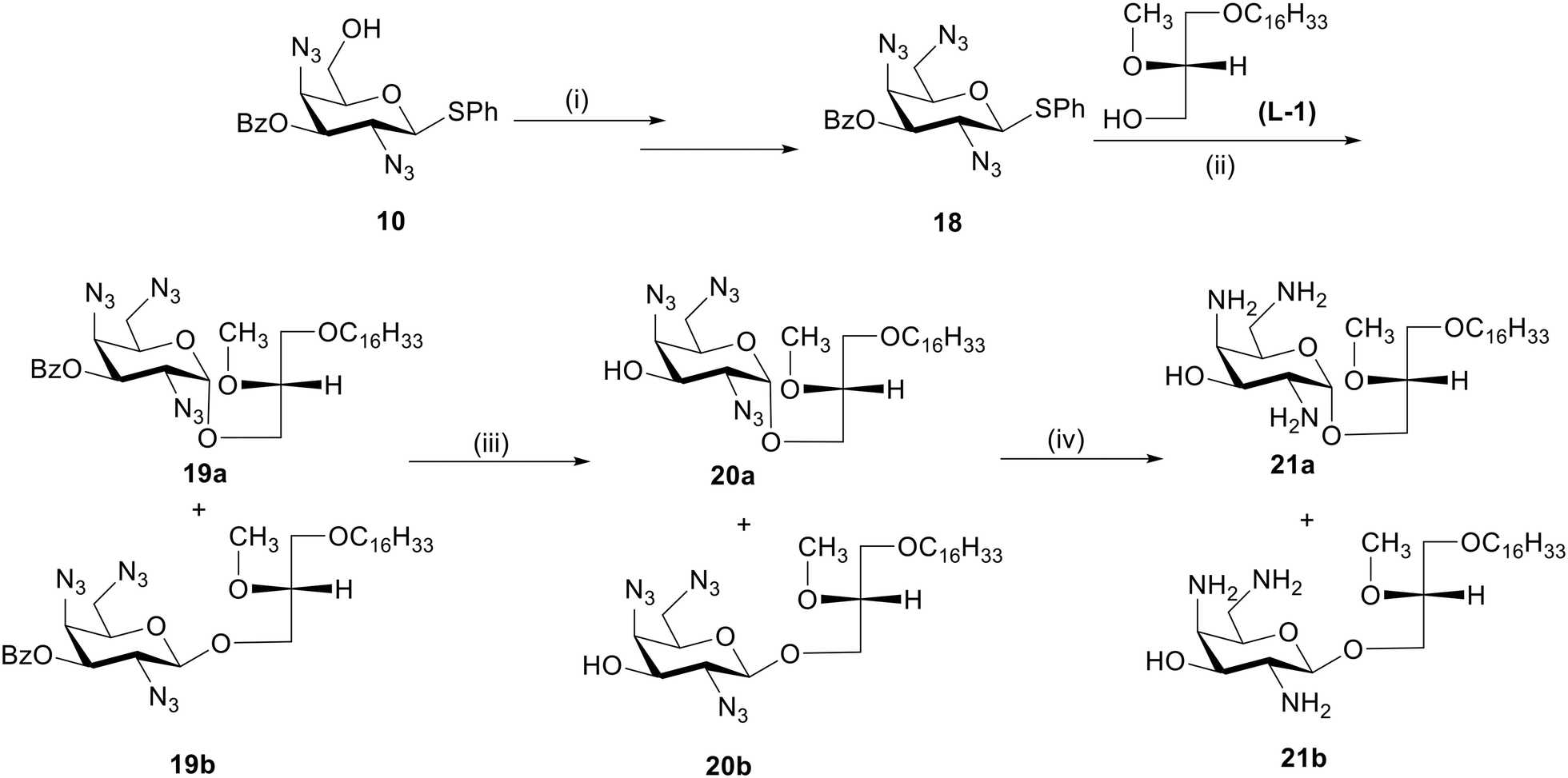 | ||
| Scheme 3 Synthesis of GAELs 21a and 21b. Reagents and conditions: (i) as reported,27 84%; (ii) L1 alcohol, NIS, AgOTf, dry DCM, 0 °C to rt, 3 h, (α:β/1:3) 19a-17%, 19b-45%; (iii) NaOMe, MeOH, rt, 3 h, 20a-90%, 20b-92%; (iv) 1 M P(CH3)3 solution in THF, THF/water, 60 °C, 12 h, 21a-93%, 21b-93%; abbreviations: dichloromethane (DCM), room temperature (rt) 23 °C, N-iodosuccinimide (NIS), tetrahydrofuran (THF). | ||
2.2 Biological evaluation
 | ||
| Fig. 3 Effect of 48 hour incubation of target GAEL compounds 9a–b, 14a–b, 17a–b, 21a–b, cisplatin, and doxorubicin on the viability of a panel of human epithelial cancer cells from the pancreas (MIAPaCa-2), breast (BT-474, JIMT-1), and prostate (DU-145) assessed by using PrestoBlue cell viability assay. Dots and error bars indicate the mean and standard deviation obtained from two independent experiments with five wells per concentration for each experiment. | ||
| Compound code | DU-145 | MIAPaCa-2 | JIMT-1 | BT-474 |
|---|---|---|---|---|
| 9a | 6.6 ± 0.3 | 5.5 ± 0.3 | 4.9 ± 0.2 | 6.9 ± 0.5 |
| 9b | 7.5 ± 0.1 | 6.6 ± 0.2 | 5.7 ± 0.1 | 6.4 ± 0.8 |
| 14a | 6.5 ± 0.4 | 6.3 ± 0.3 | 5.0 ± 0.3 | >10 |
| 14b | 7.8 ± 0.6 | 6.6 ± 0.3 | 5.8 ± 0.5 | >10 |
| 17a | 8.1 ± 0.9 | 8.2 ± 0.2 | 6.3 ± 0.1 | 7.4 ± 1.0 |
| 17b | 7.4 ± 0.5 | >10 | 8.9 ± 0.2 | >10 |
| 21a | 5.3 ± 0.3 | 6.7 ± 0.3 | 5.4 ± 0.2 | 8.0 ± 0.9 |
| 21b | 4.5 ± 0.4 | 3.3 ± 0.1 | 3.8 ± 0.2 | 5.6 ± 0.3 |
| Reference GAEL | 10.0 ± 0.3 | 10.9 ± 0.5 | 9.9 ± 0.3 | >20 |
| Cisplatin | 6.5 ± 0.3 | >10 | 7.4 ± 1.6 | >10 |
Comparison of the activity of compounds 9a and 9b, α- and β-anomers of 2,6-diamino galacto-GAEL, respectively, with the reference monobasic GAEL revealed both dibasic compounds showed about 1.5 to 2-fold higher cytotoxic effect than the monobasic reference GAEL against DU-145, MIAPaCa-2, and JIMT-1 cell lines. In BT-474 cells, the cytotoxic effects of 9a and 9b were greater than 2-fold relative to the activity with the reference compound. Both 9a and 9b displayed comparable CC50 against DU-145 (9a 6.6 ± 0.3 μM, 9b 7.5 ± 0.1 μM) and BT-474 cells (9a 6.9 ± 0.5 μM, 9b 6.4 ± 0.8 μM) whereas a statistical t-test analysis between 9a and 9b revealed that 9a displayed significantly lower CC50 than 9b against MIAPaCa-2 (9a 5.5 ± 0.3 μM, 9b 6.6 ± 0.2 μM) (P < 0.05) and JIMT-1 cells (9a 4.9 ± 0.2 μM, 9b 5.7 ± 0.1 μM) (P < 0.05). This indicates that the effect of anomeric configuration is cell-line specific. The anomeric configuration of 2,6-diamino galacto-GAELs does not play a significant role in the cytotoxic activity of DU-145 and BT-474 cells while the α-anomeric 2,6-diamino galacto-GAEL (9a) is more active than the corresponding β-isomer (9b) against MIAPaCa-2 and JIMT-1 cells. This observation is largely in line with previous reports on glucose-based GAELs where the nature of the glycosidic linkage was shown to affect the cytotoxic action in GAELs, with the α-isomer either displaying similar10 or slightly higher activity than the β-anomer.11,13,14
To investigate the effect of the position of the amine group on the sugar on cytotoxicity, α- and β-anomers of compounds with amine groups installed at the C-2 and C-4 positions of the galactose sugar of 14a and 14b were synthesized and evaluated for cytotoxicity (Table 1). Comparison of the results of 14a with 14b revealed no significant differences in their activity against the cell lines tested. Thus, the anomeric configuration of the C-2, and C-4 compounds (14a, 14b) did not impact the activity, a result similar to that observed for the C-2, and C-6 compounds (9a, 9b) against DU-145 and BT-474 cells.
The activities of 14a were next compared with 9a, and 14b with 9b as shown in Table 1, to assess the impact of having the second amine at the C-4 position as opposed to C-6 of the galactose moiety. The results revealed no significant differences in the CC50 values obtained between 14a and 9a or 14b and 9b for DU-145, MIAPaCa-2, and JIMT-1 cells. In contrast, the effect of the compounds on BT-474 cells revealed significant differences in the activities of 2,6-diamino and 2,4-diamino GAELs. 9a and 9b had CC50 6.9 ± 0.5 μM, and 6.4 ± 0.8 μM, respectively, while 14a and 14b both had CC50 >10 μM (P < 0.01). Thus, the positioning of the second amine group at the C-6 position resulted in significantly higher activity relative to the C-4 for BT-474 cells suggesting a cell-specific effect.
To study the cytotoxic effect of three amine groups placed directly on the C-2, C-4, and C-6 position of the galactose sugar, α- and β-anomeric 21a and 21b respectively, were synthesized. The triamino α-anomeric compound 21a exhibited a slightly higher activity (CC50 5.3 ± 0.3 μM) than the diamino α-anomeric versions 9a (6.6 ± 0.3 μM) and 14a (6.5 ± 0.4 μM) against DU-145 cells whereas 21a was either comparable or slightly less active than the dibasic compounds against the remaining cell lines. In contrast, the corresponding β-anomer of this tribasic GAEL, 21b, exhibited potent cytotoxic activity against all four cell lines, with CC50 ranging from 3.3 ± 0.1 μM to 5.6 ± 0.3 μM. As cationicity is expected to play a major role in the GAEL action, the tribasic GAEL 21b consistently displayed more potent cytotoxicity than the monobasic reference GAEL as well as the dibasic GAELs (9a–b, 14a–b). Depending on the cell line, GAEL 21b exhibited about 2.2 to 3.5-fold enhanced cytotoxic activity in contrast to the monobasic reference GAEL. Additionally, compound 21b also showed a better cytotoxic response than cisplatin on the four tested human epithelial cancer cell lines (Fig. 3). While doxorubicin rapidly reduced the cell viability at lower concentrations than 21b, it did not achieve complete cell death up to 10 μM whereas GAEL 21b displayed no viable cells at concentrations between 6.0–10.0 μM across the panel of human epithelial cancer cell lines (Fig. 3). The α-anomer 21a also resulted in lower CC50 than cisplatin and complete cell death of JIMT-1 and DU-145 cells (Fig. 3).
Since the tribasic 21b with three amine groups on the galactose proved to be the most active GAEL, we investigated whether there was a requirement for the presence of the amines on the sugar or whether it was just a function of increased basicity. Therefore, compounds 17a and 17b were synthesized and the results of the cytotoxicity studies revealed that α and β analogues, 17a and 17b, were less active than the corresponding tribasic GAELs 21a and 21b with the amine groups attached directly to the sugar. Indeed, 17a and 17b had similar activity or were less active than the parent dibasic GAELs 14a and 14b. The results above reinforce the significance of amine groups on the galactose sugar and requirement of the methoxy group at the sn-2 position of the glycerol backbone for optimum activity.
With 21b identified as the most active compound of the series tested against the panel of epithelial cancer cells used in our screening, we investigated its cytotoxicity against additional drug-resistant cancer cell lines. These included triple-negative breast cancer cell line MDA-MB-231, tubo-ovarian, high-grade serous carcinoma (HGSC) cell lines including OVCAR-3 (resistant to clinically relevant concentrations of adriamycin, melphalan, cisplatin),28 cisplatin-resistant COV362,29 carboplatin-resistant HEYC2,30 multidrug-resistant liver cancer cell line HepG2,31 and gemcitabine-resistant pancreatic cancer cell line BxPC3.32 The results presented in Fig. 4 and Table 2 show that after 48 h incubation of cells with compound 21b eliminated 50% of the cell population of OVCAR-3 and BxPC3 cells at 2.6 ± 0.2 μM and 2.6 ± 0.1 μM, respectively, relative to percent of the untreated control (Table 2). At 6 μM, 21b resulted in the complete elimination of OVCAR-3 and BxPC3 as well as HepG2 cells (21b CC50 3.6 ± 0.5 μM), rendering them the most sensitive cell lines among the series followed by COV362 (21b CC50 3.1 ± 0.4 μM) in which case complete cell death was observed at 8 μM. The least sensitive cell lines were HEYC2 (21b CC50 5.5 ± 0.2 μM) and MDA-MB-231 (21b CC50 6.5 ± 0.1 μM). However, incubation with 10 μM 21b resulted in complete cell death of HEYC2 and MDA-MB-231 cells. Overall, 21b proved to be effective against human epithelial cancer cell lines that are resistant to conventional anticancer drugs.
 | ||
| Fig. 4 Effect of 21b on the viability of a panel of epithelial cancer cells from the breast (MDA-MB-231), ovary (OVCAR-3, COV362, HEYC2), liver (HepG2), and pancreas (BxPC3) assessed by using PrestoBlue cell viability assay. Dots and error bars indicate the mean and standard deviation from two independent experiments with five wells per concentration for each experiment. Asterisks (*) represent a significant difference from vehicle-treated cells analysed by one-way ANOVA analysis on GraphPad Prism version 10.2.1 [****<0.0001 (P-value)]. | ||
| Classification | Cell line | CC50(μM) |
|---|---|---|
| Breast (triple negative) | MDA-MB-231 | 6.5 ± 0.1 |
| Ovarian | HEYC2 | 5.5 ± 0.2 |
| COV362 | 3.1 ± 0.4 | |
| OVCAR-3 | 2.6 ± 0.2 | |
| Liver | HepG2 | 3.6 ± 0.5 |
| Pancreas | BxPC3 | 2.6 ± 0.1 |
 | ||
| Fig. 5 Pan-caspase inhibition of DU-145 cells on treatment with varying concentrations of cisplatin and 21b, with and without MX1013 caspase inhibitor (70 μM). Asterisks (*) represent significant and “ns” indicates “not significant” difference in drug response with and without inhibitor analysed by two-way ANOVA analysis on GraphPad Prism version 10.2.1 [ns not significant, *<0.05, **<0.01, ***<0.001, ****<0.0001 (P-value)]. | ||
3. Conclusion
We report for the first-time multi-step syntheses of galactose-based dibasic and tribasic GAEL analogues with the amine groups attached directly to the sugar and the glycerol backbone. Cytotoxic studies revealed increasing the number of amines on the GAEL sugar scaffold enhanced the activity of the compound against drug resistant human epithelial cancer cell lines relative to the monoamine analogue. The presence of amine groups on C-2 and C-6 position of the galactose sugar yielded similar or greater activity than compounds with the amines at the C-2 and C-4 positions. The tribasic β-anomeric galacto-GAEL 21b, the first GAEL synthesised with three amine groups directly on the sugar was the most active compound synthesized with about 2-to 4-fold increase in cytotoxicity relative to the monobasic reference galacto-GAEL and effectively killed drug-resistant pancreatic cancer, BxPC3, and HGSC cell lines, OVCAR-3 and COV362. Cell death induced by 21b was independent of caspase activation unlike conventionally used anticancer drugs. Gal-GAELs are therefore similar to Glu-GAELs in the modulation of activity by basicity and caspase independent mechanism of cell death. Thus, Gal-GAELs are a viable alternative for further development of GAELs into clinically useful compounds. Subsequent investigation of potent Gal-GAEL will be directed towards testing with 3D spheroids and cancer stem cells along with combination studies with apoptosis-inducing anticancer agents.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization: RA, GA and FS; investigation: RA (synthesis, characterization, and biological experiments); methodology: RA, AM and FS (synthetic scheme), RA and GA (biological experiments); data curation: RA, supervision: AM, FS, GA; writing – original draft: RA, GA, FS; writing – review and editing: RA, GA, FS, MN.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) in the form of a discovery grant (2018-06047). RA thanks the University of Manitoba for a University of Manitoba Graduate Fellowship (UMGF). Frozen stocks of cell lines originally obtained from ATCC.References
- D. S. Dizon and A. H. Kamal, Cancer Statistics 2024: All Hands on Deck, Ca-Cancer J. Clin., 2024, 74(1), 8–9, DOI:10.3322/CAAC.21824.
- R. Ranasinghe, M. L. Mathai and A. Zulli, Cisplatin for Cancer Therapy and Overcoming Chemoresistance, Heliyon, 2022, 8(9), e10608, DOI:10.1016/J.HELIYON.2022.E10608.
- C. Christowitz, T. Davis, A. Isaacs, G. Van Niekerk, S. Hattingh and A. M. Engelbrecht, Mechanisms of Doxorubicin-Induced Drug Resistance and Drug Resistant Tumour Growth in a Murine Breast Tumour Model, BMC Cancer, 2019, 19(1) DOI:10.1186/S12885-019-5939-Z.
- J. Cox and S. Weinman, Mechanisms of Doxorubicin Resistance in Hepatocellular Carcinoma, Hepatic Oncol., 2016, 3(1), 57, DOI:10.2217/HEP.15.41.
- D. W. Shen, L. M. Pouliot, M. D. Hall and M. M. Gottesman, Cisplatin Resistance: A Cellular Self-Defense Mechanism Resulting from Multiple Epigenetic and Genetic Changes, Pharmacol. Rev., 2012, 64(3), 706, DOI:10.1124/PR.111.005637.
- A. Lyakhovich and M. E. Lleonart, Bypassing Mechanisms of Mitochondria-Mediated Cancer Stem Cells Resistance to Chemo- and Radiotherapy, Oxid. Med. Cell. Longevity, 2016, 2016 DOI:10.1155/2016/1716341.
- M. Cojoc, K. Mäbert, M. H. Muders and A. Dubrovska, A Role for Cancer Stem Cells in Therapy Resistance: Cellular and Molecular Mechanisms, Semin. Cancer Biol., 2015, 31, 16–27, DOI:10.1016/J.SEMCANCER.2014.06.004.
- M. W. Nachtigal, P. Musaphir, S. Dhiman, A. D. Altman, F. Schweizer and G. Arthur, Cytotoxic Capacity of a Novel Glycosylated Antitumor Ether Lipid in Chemotherapy-Resistant High Grade Serous Ovarian Cancer in Vitro and in Vivo, Transl. Oncol., 2021, 14(11) DOI:10.1016/J.TRANON.2021.101203.
- T. Idowu, P. Samadder, G. Arthur and F. Schweizer, Amphiphilic Modulation of Glycosylated Antitumor Ether Lipids Results in a Potent Triamino Scaffold against Epithelial Cancer Cell Lines and BT474 Cancer Stem Cells, J. Med. Chem., 2017, 60(23), 9724–9738, DOI:10.1021/ACS.JMEDCHEM.7B01198.
- M. Ogunsina, P. Samadder, T. Idowu, G. Arthur and F. Schweizer, Replacing D-Glucosamine with Its l-Enantiomer in Glycosylated Antitumor Ether Lipids (GAELs) Retains Cytotoxic Effects against Epithelial Cancer Cells and Cancer Stem Cells, J. Med. Chem., 2017, 60(5), 2142–2147, DOI:10.1021/ACS.JMEDCHEM.6B01773.
- M. Ogunsina, P. Samadder, T. Idowu, G. Arthur and F. Schweizer, Design, Synthesis and Evaluation of Cytotoxic Properties of Bisamino Glucosylated Antitumor Ether Lipids against Cancer Cells and Cancer Stem Cells, MedChemComm, 2016, 7(11), 2100–2110, 10.1039/C6MD00328A.
- G. Arthur, F. Schweizer and M. Ogunsina, Synthetic Glycosylated Ether Glycerolipids as Anticancer Agents, RSC Drug Discovery Ser., 2015, 2015(43), 151–179, 10.1039/9781849739993-00151.
- Y. Xu, M. Ogunsina, P. Samadder, G. Arthur and F. Schweizer, Structure-Activity Relationships of Glucosamine-Derived Glycerolipids: The Role of the Anomeric Linkage, the Cationic Charge and the Glycero Moiety on the Antitumor Activity, ChemMedChem, 2013, 8(3), 511–520, DOI:10.1002/CMDC.201200489.
- P. Samadder, Y. Xu, F. Schweizer and G. Arthur, Cytotoxic Properties of D-Gluco-, D-Galacto- and D-Manno-Configured 2-Amino-2-Deoxy-Glycerolipids against Epithelial Cancer Cell Lines and BT-474 Breast Cancer Stem Cells, Eur. J. Med. Chem., 2014, 78, 225–235, DOI:10.1016/J.EJMECH.2014.03.057.
- M. Ogunsina, H. Pan, P. Samadder, G. Arthur and F. Schweizer, Structure Activity Relationships of N-Linked and Diglycosylated Glucosamine-Based Antitumor Glycerolipids, Molecules, 2013, 18(12), 15288–15304, DOI:10.3390/MOLECULES181215288.
- M. Ogunsina, P. Samadder, T. Idowu, M. Nachtigal, F. Schweizer and G. Arthur, Syntheses of L-rhamnose-linked Amino Glycerolipids and Their Cytotoxic Activities against Human Cancer Cells, Molecules, 2020, 25(3) DOI:10.3390/MOLECULES25030566.
- T. Idowu, P. Samadder, G. Arthur and F. Schweizer, Design, Synthesis and Antitumor Properties of Glycosylated Antitumor Ether Lipid (GAEL)-Chlorambucil-Hybrids, Chem. Phys. Lipids, 2016, 194, 139–148, DOI:10.1016/J.CHEMPHYSLIP.2015.07.003.
- R. K. Erukulla, X. Zhou, P. Samadder, G. Arthur and R. Bittman, Synthesis and Evaluation of the Antiproliferative Effects of 1-O-Hexadecyl-2-O-Methyl-3-O-(2′-Acetamido-2′-Deoxy-Beta-D-Glucopyranosyl)-Sn-Glycerol and 1-O-Hexadecyl-2-O-Methyl-3-0-(2′-Amino-2′-Deoxy-Beta-D-Glucopyranosyl)-Sn-Glycerol on Epithelial Cancer Cell Growth, J. Med. Chem., 1996, 39(7), 1545–1548, DOI:10.1021/JM950928F.
- G. Arthur and R. Bittman, Glycosylated Antitumor Ether Lipids: Activity and Mechanism of Action, Anti-Cancer Agents Med. Chem., 2014, 14(4), 592–606, DOI:10.2174/1871520614666140309231144.
- P. Samadder, R. Bittman, H. S. Byun and G. Arthur, A Glycosylated Antitumor Ether Lipid Kills Cells via Paraptosis-like Cell Death, Biochem. Cell Biol., 2009, 87(2), 401–414, DOI:10.1139/O08-147.
- M. W. Nachtigal, A. D. Altman, R. Arora, F. Schweizer and G. Arthur, The Potential of Novel Lipid Agents for the Treatment of Chemotherapy-Resistant Human Epithelial Ovarian Cancer, Cancers, 2022, 14(14) DOI:10.3390/CANCERS14143318.
- A. M. Ellegaard, P. Bach and M. Jäättelä, Targeting Cancer Lysosomes with Good Old Cationic Amphiphilic Drugs, Rev. Physiol., Biochem. Pharmacol., 2023, 185, 107–152, DOI:10.1007/112_2020_56.
- R. S. Funk and J. P. Krise, Cationic Amphiphilic Drugs Cause a Marked Expansion of Apparent Lysosomal Volume: Implications for an Intracellular Distribution-Based Drug Interaction, Mol. Pharmaceutics, 2012, 9(5), 1384–1395, DOI:10.1021/MP200641E.
- A. Anand, B. Liu, J. D. Giacobini, K. Maeda, M. Rohde and M. Jaattela, Cell Death Induced by Cationic Amphiphilic Drugs Depends on Lysosomal Ca2+ Release and Cyclic AMP, Mol. Cancer Ther., 2019, 18(9), 1602–1614, DOI:10.1158/1535-7163.MCT-18-1406.
- O. Warburg, On the Origin of Cancer Cells, Science, 1956, 123(3191), 309–314, DOI:10.1126/SCIENCE.123.3191.309/ASSET/A8D38B53-799F-4009-AAD3-E77CEF33D301/ASSETS/SCIENCE.123.3191.309.FP.PNG.
- N. Hammoudi, K. B. R. Ahmed, C. Garcia-Prieto and P. Huang, Metabolic Alterations in Cancer Cells and Therapeutic Implications, Chin. J. Cancer, 2011, 30(8), 508, DOI:10.5732/CJC.011.10267.
- A. Mukherjee, D. Ramirez, R. Arora, G. Arthur and F. Schweizer, Amphiphilic Tribasic Galactosamines Potentiate Rifampicin in Gram-Negative Bacteria at Low Mg++/Ca++concentrations, Bioorg. Med. Chem. Lett., 2023, 129371, DOI:10.1016/J.BMCL.2023.129371.
- T. C. Hamilton, R. C. Young, W. M. McKoy, K. R. Grotzinger, J. A. Green, E. W. Chu, J. Whang-Peng, A. M. Rogan, W. R. Green and R. F. Ozols, Characterization of a Human Ovarian Carcinoma Cell Line (NIH:OVCAR-3)1 with Androgen and Estrogen Receptors, Cancer Res., 1983, 43(11), 5379–5389 CAS.
- J. Haley, S. Tomar, N. Pulliam, S. Xiong, S. M. Perkins, A. R. Karpf, S. Mitra, K. P. Nephew, A. K. Mitra, J. Haley, S. Tomar, N. Pulliam, S. Xiong, S. M. Perkins, A. R. Karpf, S. Mitra, K. P. Nephew and A. K. Mitra, Functional Characterization of a Panel of High-Grade Serous Ovarian Cancer Cell Lines as Representative Experimental Models of the Disease, Onco Targets Ther, 2016, 7(22), 32810–32820, DOI:10.18632/ONCOTARGET.9053.
- C. Guo, C. Song, J. Zhang, Y. Gao, Y. Qi, Z. Zhao and C. Yuan, Revisiting Chemoresistance in Ovarian Cancer: Mechanism, Biomarkers, and Precision Medicine, Genes Dis., 2022, 9(3), 668, DOI:10.1016/J.GENDIS.2020.11.017.
- Y. R. Lei, X. L. He, J. Li and C. F. Mo, Drug Resistance in Hepatocellular Carcinoma: Theoretical Basis and Therapeutic Aspects, Front. Biosci.-Landmark, 2024, 29(2), 52, DOI:10.31083/J.FBL2902052.
- A. Arlt, A. Gehrz, S. Müerköster, J. Vorndamm, M. L. Kruse, U. R. Fölsch and H. Schäfer, Role of NF-KB and Akt/PI3K in the Resistance of Pancreatic Carcinoma Cell Lines against Gemcitabine-Induced Cell Death, Oncogene, 2003, 22(21), 3243–3251, DOI:10.1038/sj.onc.1206390.
- L. Jahreiss, M. Renna, R. Bittman, G. Arthur and D. C. Rubinsztein, 1-O-Hexadecyl-2-O-Methyl-3-O-(2′-Acetamido-2′-Deoxy-∃-D-Glucopyranosyl)-Sn-Glycerol (Gln) Induces Cell Death with More Autophagosomes Which Is Autophagy-Independent, Autophagy, 2009, 5(6), 835–846, DOI:10.4161/AUTO.9120.
- A. I. Moraya, J. L. Ali, P. Samadder, L. Liang, L. C. Morrison, T. E. Werbowetski-Ogilvie, M. Ogunsina, F. Schweizer, G. Arthur and M. W. Nachtigal, Novel Glycolipid Agents for Killing Cisplatin-Resistant Human Epithelial Ovarian Cancer Cells, J. Exp. Clin. Cancer Res., 2017, 36(1) DOI:10.1186/S13046-017-0538-9.
- W. Yang, J. Guastella, J. C. Huang, Y. Wang, L. Zhang, D. Xue, M. Tran, R. Woodward, S. Kasibhatla, B. Tseng, J. Drewe and S. X. Cai, MX1013, a Dipeptide Caspase Inhibitor with Potent in Vivo Antiapoptotic Activity, Br. J. Pharmacol., 2003, 140(2), 402–412, DOI:10.1038/SJ.BJP.0705450.
- S. Dasari and P. Bernard Tchounwou, Cisplatin in Cancer Therapy: Molecular Mechanisms of Action, Eur. J. Pharmacol., 2014, 740, 364, DOI:10.1016/J.EJPHAR.2014.07.025.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00662c |
| This journal is © The Royal Society of Chemistry 2025 |