
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLazertinib: breaking the mold of third-generation EGFR inhibitors
Kishan B.
Patel
a and
David E.
Heppner
 *abc
*abc
aDepartment of Chemistry, The State University of New York at Buffalo, Natural Sciences Complex, Buffalo, NY 14260, USA. E-mail: davidhep@buffalo.edu
bJacobs School of Medicine and Biomedical Sciences, Department of Structural Biology, The State University of New York at Buffalo, Buffalo, NY, USA
cDepartment of Pharmacology and Therapeutics, Roswell Park Comprehensive Cancer Center, Buffalo, NY, USA
First published on 7th January 2025
Abstract
Small molecules targeting activating mutations within the epidermal growth factor receptor (EGFR) are efficacious anticancer agents, particularly in non-small cell lung cancer (NSCLC). Among these, lazertinib, a third-generation tyrosine kinase inhibitor (TKI), has recently gained FDA approval for use in combination with amivantamab, a dual EGFR/MET-targeting monoclonal antibody. This review delves into the discovery and development of lazertinib underscoring the improvements in medicinal chemistry properties, especially in comparison with osimertinib. Analysis of its structure–activity relationships (SAR), as outlined in the patent literature, reveals the structural diversity explored enroute to the candidate molecule. The resulting structure of lazertinib is distinguished among other TKIs due to the combination of the hydrophobic phenyl and hydrophilic amine substituents on the pyrazole. The structural basis for the selectivity against the T790M mutation is enabled by the substituted pyrazole moiety, which facilitates both van der Waals and H-bonding interactions with the EGFR kinase domain. Insights from this case study offer lessons that can inform the future design of kinase inhibitors with improved safety and efficacy profiles for cancer treatment and other diseases.
 Kishan Patel | Kishan Patel received his BPharm degree from CHARUSAT, Anand, in 2017 and his MPharm degree from The Maharaja Sayajirao University, Vadodara, in 2019. He gained industrial experience in drug discovery and process chemistry while working as a senior research associate at Piramal Pharma Solutions, Ahmedabad and as an R&D officer at CTX lifesciences, Surat. In 2021, Kishan joined Dr. Heppner's group as a medicinal chemistry Ph.D. student where he works on the synthesis and development of small-molecule inhibitors targeting protein kinases and other disease-relevant targets. |
 David Heppner | David Heppner is the J. Solo Assistant Professor of Medicinal Chemistry in the Department of Chemistry at The State University of New York at Buffalo. He obtained his B.S. in Chemistry from the University of Minnesota and Ph.D. in Chemistry with Edward I. Solomon from Stanford University. He acquired training in biomedical research as an NIH postdoctoral fellow with Albert van der Vliet at the University of Vermont and subsequently in medicinal chemistry, cancer biology, and structural biology as a postdoctoral research fellow with Michael J. Eck at the Dana-Farber Cancer Institute and Harvard Medical School. The Heppner Lab applies a structural perspective to drug development and focuses on developing novel small molecules relevant to several diseases. |
Introduction
The emergence of drug resistance in treating cancer presents drug hunters with “moving target” obstacles in the discovery and development of effective therapies.1,2 The past few decades of drug development efforts in targeting mutant EGFR non-small cell lung cancer (NSCLC) have proven to be such a challenge resulting in a diverse arsenal of therapies.3–5 Initially, the observation that NSCLC patients harboring EGFR activating mutations responded well to small-molecule tyrosine kinase inhibitors (TKIs) laid the foundation for advancements in targeted therapies for diverse cancers.6,7 The subsequent gatekeeper point mutation T790M, which strongly resembles a resistance mechanism in BCR-ABL1 treatment by imatinib,8 provided the incentive to shift from reversible-binding TKIs to mutant-selective irreversible-covalent inhibitors.9,10 More recently, despite having been shown to lead to significant lengthening of progression-free survival (PFS),11–13 drug discovery has pivoted on account of the acquired tertiary C797S mutation,14,15 which renders irreversible inhibitors unable to form their potency-enabling covalent bond, thereby, shifting the focus towards advanced reversible binding ATP-competitive as well as ATP-non-competitive allosteric scaffolds, and even combinations thereof.16–22On August 19, 2024, the US Food and Drug Administration (FDA) approved a new regiment for NSCLC patients: the combination of a novel TKI lazertinib (Lazcluze, Leclaza, GNS-1480, YH25448, JNJ-73841937)23 with the bispecific EGFR/c-MET antibody amivantamab (MARIPOSA, NCT04487080).24,25 Amivantamab (Rybrevant) is a monoclonal antibody originally developed by Janssen (now Johnson & Johnson Innovation Medicine), which was granted this accelerated approval in 2021 and full FDA approval in March for exon 20 insertion activating EGFR mutations in combination with carboplatin and pemetrexed (CHRYSALIS, NCT02609776).26 Combination antibody and TKI regimens have been studied appreciably with respect to the EGFR antibody cetuximab (Erbitux), which has been known since 1988, and previously utilized in EGFR overexpressing head and neck as well as colorectal cancers,27 although discontinued due to poorer efficacy compared to cisplatin.28,29 Nevertheless, the recent success of lazertinib in combination with amivantamab represents a meaningful advancement in mutant EGFR NSCLC therapy showing a PFS of approximately two years.30
This combination therapy is the most recent FDA approval for a disease where the most common treatment consists of a TKI monotherapy. The most effective of which to date has been osimertinib (AZD9291),31–33 developed on the basis of the tool compounds WZ4002,34 was the first third-generation TKI to reach the clinic for drug-resistant T790M-positive NSCLC35 and subsequently treatment-naive NSCLC patients.13 Despite these clinical advancements, osimertinib still has room for improvement due to several factors that contribute to a dose-limiting toxicity profile, such as its toxic metabolite AZ5014.36–38 Accordingly, lazertinib was developed out of the deliberate effort to improve on osimertinib, and led to a drug with superior medicinal chemistry behavior enabling more effective targeting of mutant EGFR as well as equivalent blood–brain barrier penetrations and activity against brain metastasis.38
The main objective of this review is to showcase the pathway toward the development of lazertinib and how this compound results in improvements to its medicinal chemistry profile through a structural perspective. Examination of the X-ray cocrystal structures, irreversible inhibitor kinetic values, and cellular data provide a structural basis for the superior T790M-selectivity of lazertinib.39,40 Off-target effects are also a key aspect of this story including how the alternative structure of the pyrazole in lazertinib eliminates the risk of forming an active metabolite associated with dose-limiting toxicities (AZ5014) and diminished activity against HER2.38,39 Additionally, since limited is established regarding the discovery and optimization of the lazertinib molecule, we have presented a review of cellular structure–activity relationships (SAR) that provide the structures and properties of candidates produced eventually on the path to providing lazertinib.41 Taken together, this accumulated overview provides several insights pertinent to the design of kinase inhibitors and a perspective into the complex nature underlying the ability to produce more effective anticancer agents.
Chemical structures
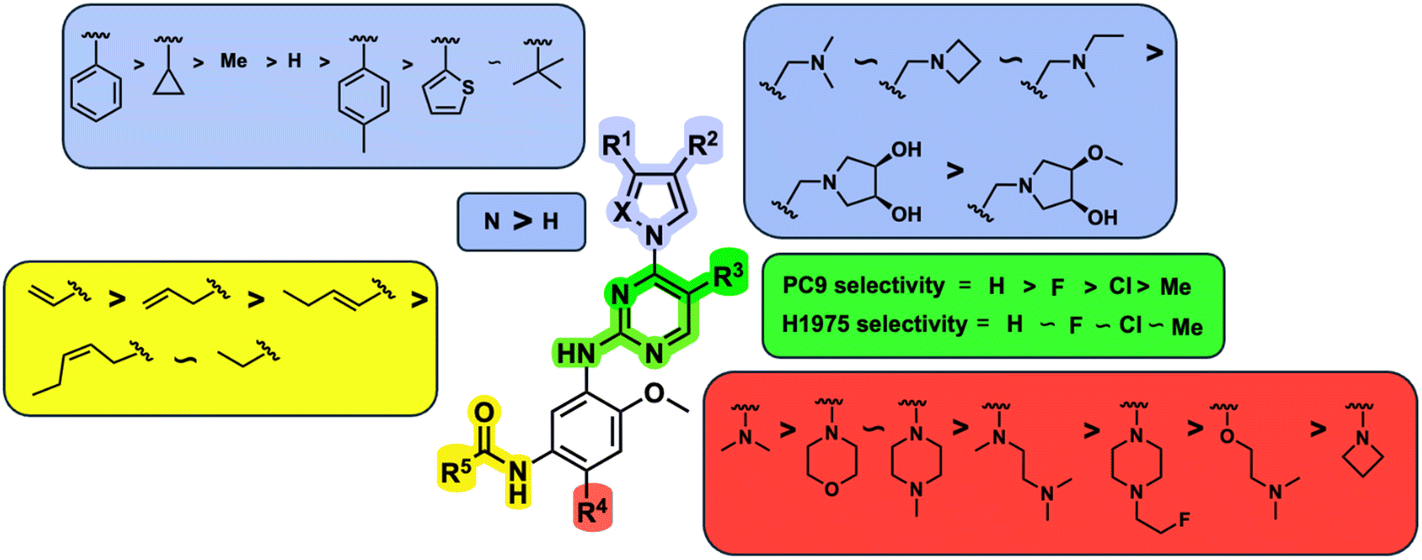
An examination of the chemical structure of lazertinib and osimertinib reveals generally consistent structural framework with the most significant differences being the functional groups attached to the aminopyrimidine (Fig. 1, purple). For osimertinib, this group is an N-methylindole while at the same position lazertinib contains a more elaborated substituted pyrazole with an N,N-dimethylmethyleneamine (methyleneamine) and a phenyl ring. The two drugs are otherwise practically identical with the only exception being the solubilization group (Fig. 1, red). The aminopyrimidine (Fig. 1, green) is a frequently occurring motif in kinase inhibitors that bind the conserved hinge region backbone, and the acrylamide Michael acceptor provides the ability for these two molecules to form their potency-enabling covalent bonds to C797 (Fig. 1, yellow). An important element in both drugs, as will become evident in the discussion of the structural basis for their activity, is the single bond (N–C for lazertinib and C–C for osimertinib) that allows for the distinct groups to freely rotate.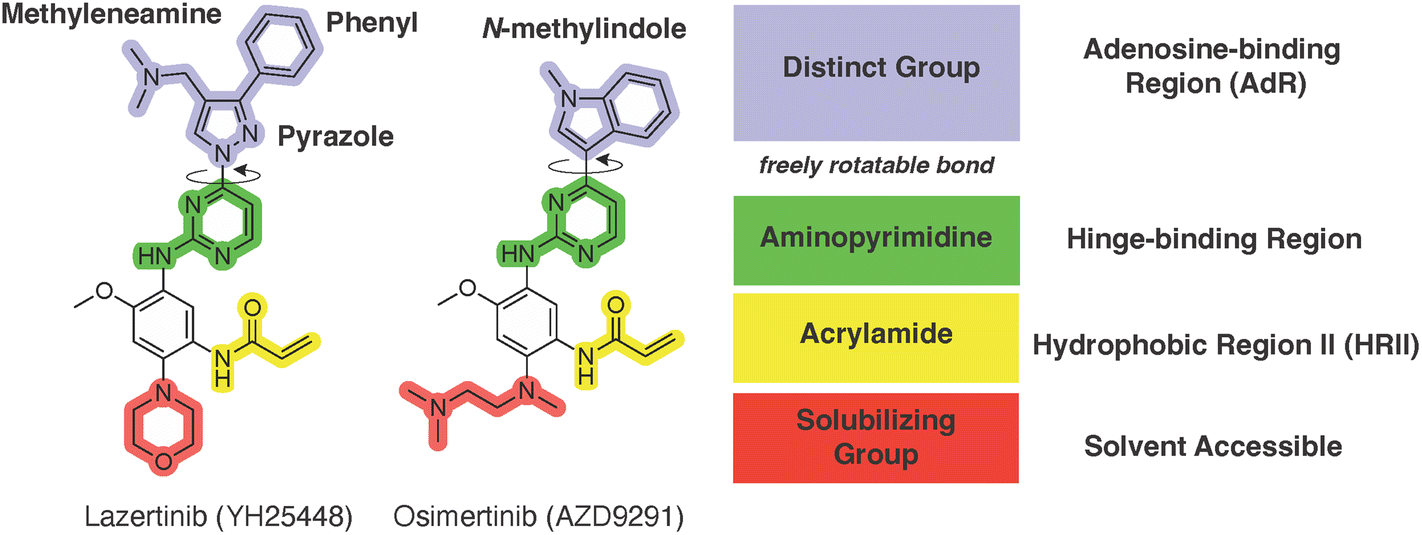 | ||
| Fig. 1 The chemical structures of lazertinib (YH25448) and osimertinib (AZD9291) along with accompanying labels of similar and distinct groups along with the location of their binding within the EGFR kinase domain. | ||
Synthesis of lazertinib
A gram-scale, convergent synthesis of lazertinib is available from the patent US20200207750A1 by the Yuhan Corp. and summarized in Scheme 1 including percent yields in the caption.42 For simplicity, we have adopted compound numbering in this section relevant only to the discussion of the synthesis of lazertinib (compounds 1–8) as this was reported in a patent distinct from the SAR that will be covered in the subsequent section.41 The synthesis begins with a nucleophilic aromatic substitution (SNAr) reaction between 3-phenyl-1H-pyrazole-4-carbaldehyde and 4-chloro-2-(methylthio)pyrimidine, yielding intermediate 1. This is followed by ammonium heptamolybdate tetrahydrate-catalyzed oxidation of the S-methyl group using aqueous hydrogen peroxide, resulting in the first key intermediate 2. The second key intermediate 7, is prepared via a sequential five-step process starting with an SNAr reaction between 4-fluoro-2-methoxy-5-nitroaniline and morpholine to form intermediate 3. This is succeeded by formic acid-mediated formylation of the aniline nitrogen and reduction of the nitro group using ammonium formate and Pd/C, producing intermediates 4 and 5, respectively. Intermediate 5 undergoes a one-pot, two-step reaction involving amine acylation with 3-chloropropionyl chloride followed by base-mediated elimination, forming the acrylamide intermediate 7. In the penultimate step, intermediates 2 and 7 are condensed to yield intermediate 8, which undergoes STAB-mediated reductive elimination to produce lazertinib.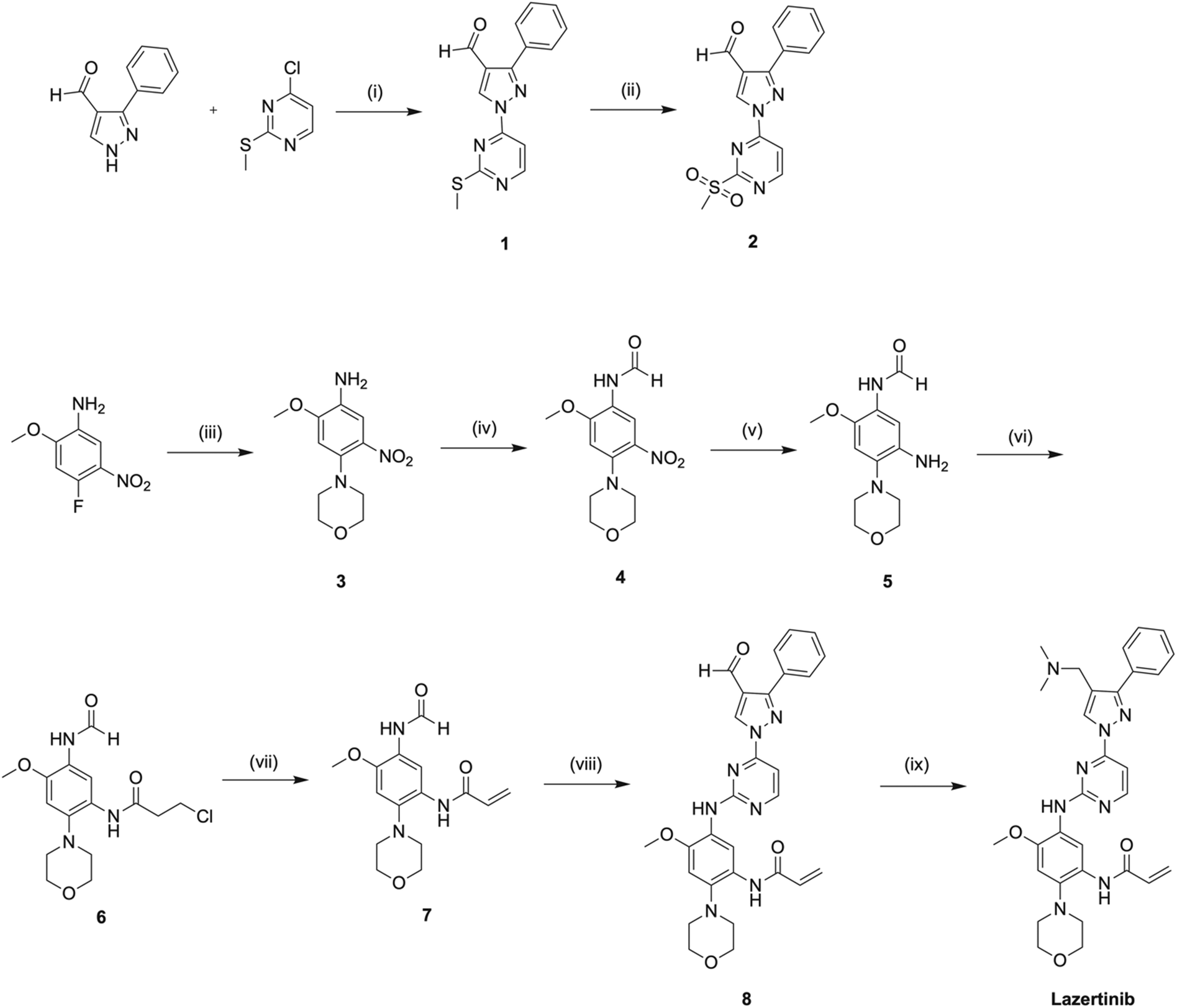 | ||
| Scheme 1 A gram-scale preparation of lazertinib based on US20200207750A1 Yuhan Corp. patent.42 Reagents and conditions are as follows: (i) K2CO3, DMF, 50 °C, 2 h, 90%; (ii) 35% HOOH (aq), (NH4)6Mo7O24·4H2O, EtOH, rt, 2 h, 84%; (iii) morpholine, DIPEA, MeCN, rf, 4 h, 96%; (iv) HCO2H, AcOH, THF, rt, 1 h, 91%; (v) NH4HCO2, 10% Pd/C, EtOH, THF, 40 °C, 2 h, 68%; (vi) 3-chloropropionyl chloride, NaHCO3, MeCN, rt, 0.5 h; (vii) Et3N, n-PrOH, rf, 3 h, 99% over last 2 steps; (viii) 2, t-BuONa, THF, rt, 1 h, 2 N NaOH (aq), rt, 15 h, 56%; (ix) Me2NH·HCl, DIPEA, DMA, rt, 1 h, STAB, rt, 1 h, 92%. | ||
Structure–activity relationships in mutant NSCLC cell line models
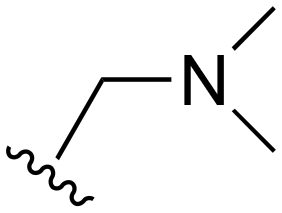
While a report detailing the discovery and development of the lazertinib molecule is lacking from the peer-review literature, insights into structural activity relationships (SAR) are available in cellular viability experiments from the original composition of matter patent from Yuhan Corp. WO2016060443A2.41 For consistency with the patent, we have elected to discuss these molecules using the same compound numbering system. The potency of various molecules was gauged against two prevalent activating mutations in NSCLC, namely the H1975 that harbors the first-generation resistant L858R/T790M double mutation and PC9 that contains the E746-A750 exon19del mutation, alongside H2073, which is derived from lung adenocarcinoma and contains wild type (WT) EGFR. The comparison of viability between these mutant and WT cell lines allows for the assessment of mutant selectivity within the SAR. Overall, the set of 45 compounds feature a central aminopyrimidine ring and various substituents such as the acrylamide consistent with many third-generation TKIs. Additionally, the vast majority of molecules tested include a substituted pyrazole ring on the pyrimidine (purple, Fig. 1), which exhibits over 10 times greater potency and selectivity for PC9 and H1975 mutant cell lines compared to a matched pyrrole group (19vs.48).41 It should be noted that we have not exhaustively presented all of the data contained within the Yuhan Corp. patent, however this cell-based experiment and subset of structures are most informative with respect to the SAR of lazertinib.The substituents at the R1 position of the pyrazole ring are found to be important for enabling selectivity profile of these inhibitors (Table 1). As the patent presents ranges of viability EC50 values (the patent reports IC50 values, which are best reserved for enzymatic inhibition assays), we discuss approximate fold changes in antiproliferative effects based on the ranges provided.41 Replacing a methyl group (14 & 46) with a cyclopropyl moiety (55 & 74) at R1 position enhances selectivity for the H1975 mutant cell line over WT by 10-fold. Substituting the cyclopropyl with a phenyl ring (73 & 78) further increases the potency and selectivity for H1975 by another 10-fold. As a result, replacing the methyl with a phenyl ring results in a 100-fold increase in potency and selectivity for H1975 mutant. However, substituting a methyl group at the para position of the phenyl ring (82) leads to a 10-fold reduction in potency and selectivity for both PC9 and H1975 cells. When compared to an unsubstituted R1 position (R1 = H, 32), the methyl group (76) results in a modest 2-fold increase in mutant potency and selectivity over WT. On the other hand, replacing the methyl with cyclopropyl group (123) significantly enhances both potency and selectivity for both mutant cell lines by over 10-folds. The substitution of phenyl ring at R1 with thiophene (118), t-Bu (86) and isopropyl (93) makes them active against WT, which results in loss of selectivity for both mutants. However, the methyl substitution at R3 (124) in comparison to 93 renders the compound inactive against WT EGFR, thus restoring its selectivity profile for both mutant cell lines. Overall, these cellular results indicate that a phenyl ring at R1 is preferred for enabling desired potency and selectivity against these cell line models.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | EGFR wild type | EGFR mutants | Selectivity over wild type | ||
| H2073 (nM) | PC9 (nM) | H1975 (nM) | H2073/PC9 (fold) | H2073/H1975 (fold) | ||||
| 14 | Me |
|
H | >1000 | <20 | 20–200 | >200 | 20–100 |
| 32 | H |
|
Me | >1000 | 201–1000 | 20–200 | 20–100 | 20–100 |
| 46 | Me |
|
H | >1000 | <20 | 20–200 | >200 | 101–200 |
| 55 |
|
|
H | >1000 | <20 | 20–200 | >200 | 101–200 |
| 73 (Lazertinib) | Ph |
|
H | >1000 | <20 | <20 | >200 | >200 |
| 74 |
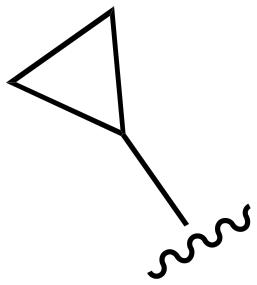
|
|
H | >1000 | <20 | <20 | >200 | >200 |
| 76 | Me |
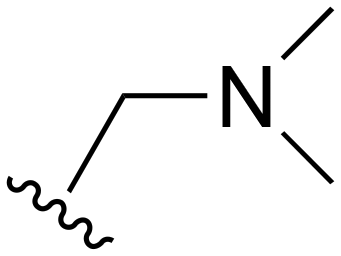
|
Me | >1000 | 20–200 | 20–200 | 101–200 | 101–200 |
| 78 | Ph |

|
H | >1000 | <20 | <20 | >200 | >200 |
| 82 |
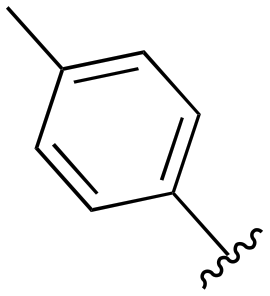
|
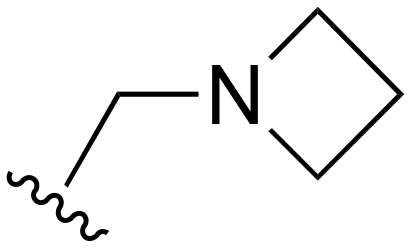
|
H | >1000 | 20–200 | 20–200 | 101–200 | 101–200 |
| 86 | t-Bu |
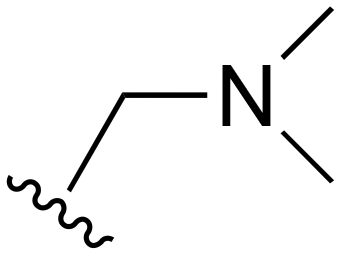
|
H | 201–1000 | <20 | <20 | 20–200 | 20–200 |
| 93 |
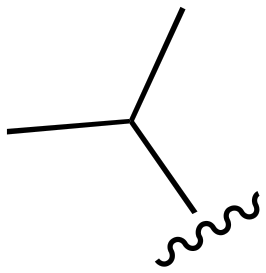
|
|
H | 201–1000 | <20 | <20 | 20–200 | 20–200 |
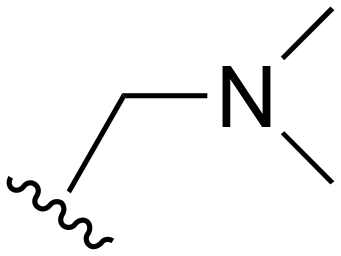
| 118 |
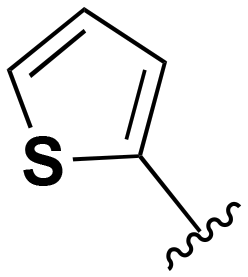
|
|
H | 201–1000 | <20 | <20 | 20–200 | 20–200 |
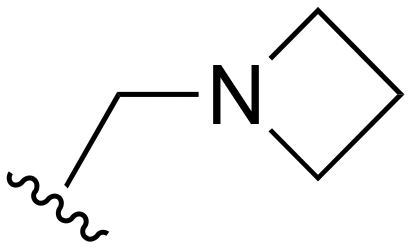
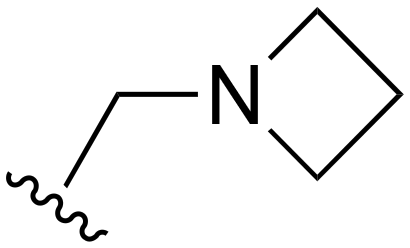
| 123 |
|
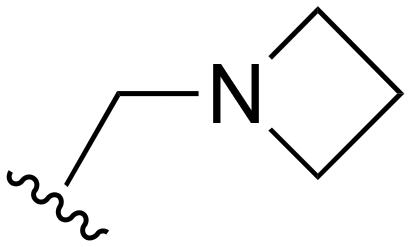
|
H | >1000 | <20 | <20 | >200 | >200 |
| 124 |
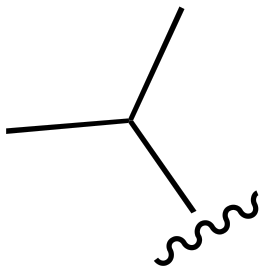
|

|
Me | >1000 | <20 | <20 | >200 | >200 |
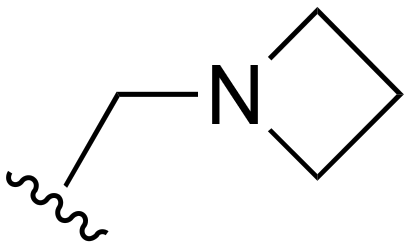
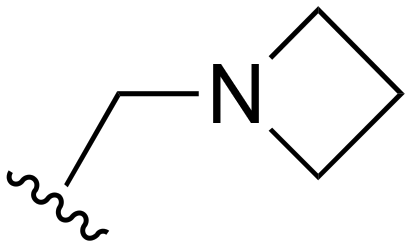
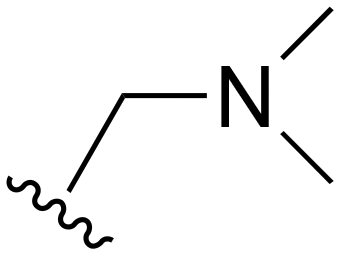
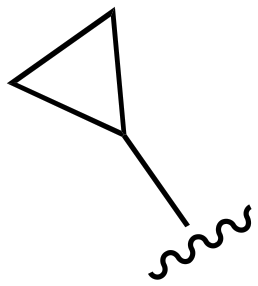
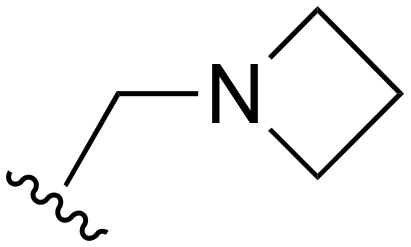
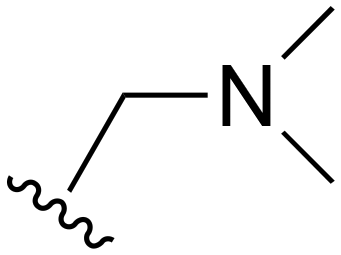
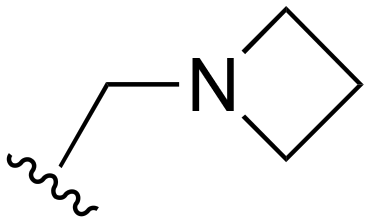
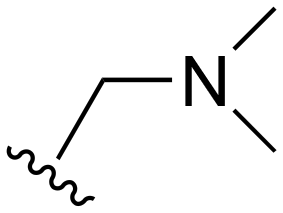
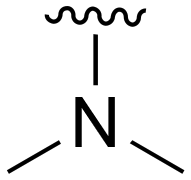
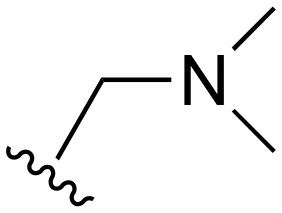
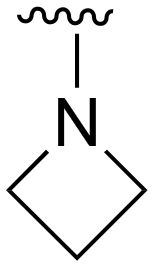
The other substitution on the pyrazole ring (R2) was also found to be important for achieving the desired activity profile of these inhibitors (Tables 1 and 2). These substituents span diverse tertiary aliphatic amines, which are protonated at physiological pH making these groups positively charged and capable of acting as H-bond donors. Given different groups at R1 (cyclopropyl, phenyl), maximal potency and mutant selectivity is generally observed when R2 is substituted with the N,N-dimethylmethyleneamine (methyleneamine), such as for 73 (lazertinib), 74, 78, 122. Generally, potency diminishes to an extent when R2 is a methyleneazetidine and the R1 position is a smaller methyl or cyclopropyl group 14, 55, 76, except for 123, which shows maximal potency and selectivity on both mutants. Additionally, a singular example of R2 = N-ethyl-N-methylmethanmine with R1 = phenyl also exhibits maximal potency and selectivity showing limited impacts of longer aliphatic groups on the amine. Furthermore, substituting R2 with a more elaborative methyleneazetidine containing an alcohol at C3-position (19) enhances the potency and selectivity for PC9 over WT EGFR by more than 10-fold (compared to 32). The R2 substitution of methylpyrrolidine-3,4-diol (25) increases the potency for both PC9 and H1975 by over 10-fold. This can be counteracted by methylation of the alcohol, which results in 10-fold loss in potency for both mutant cell lines. These results indicate that more simple tertiary amines are most productive at R2, and less so for the more elaborate pyrrolidines, with most maximally active and selective compounds containing R2 = methyleneamine.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | EGFR wild type | EGFR mutants | Selectivity over wild type | ||
| H2073 (nM) | PC9 (nM) | H1975 (nM) | H2073/PC9 (fold) | H2073/H1975 (fold) | ||||
| 19 | H |
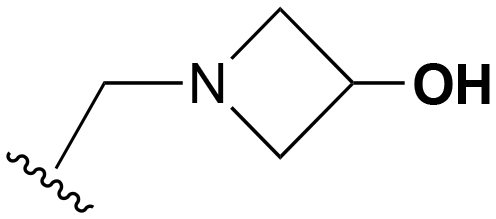
|
Me | >1000 | <20 | 20–200 | 100–200 | 20–100 |
| 25 | Me |
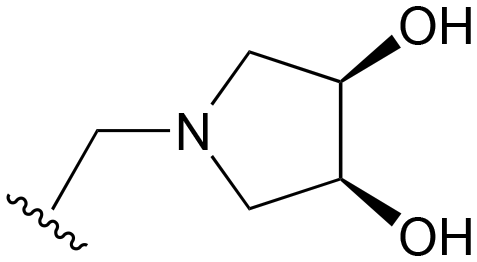
|
Me | >1000 | <20 | <20 | >200 | 101–200 |
| 26 | Me |
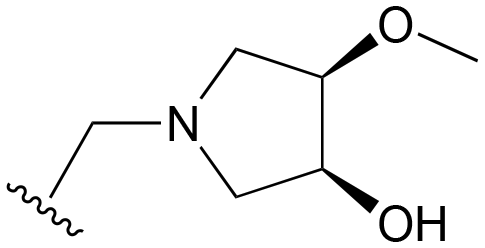
|
Me | >1000 | 20–200 | 20–200 | 20–100 | 101–200 |
| 75 | Me |
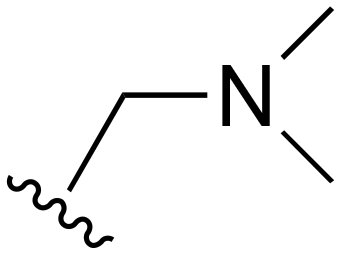
|
Me | >1000 | 20–200 | 20–200 | 101–200 | 101–200 |
| 92 | Ph |
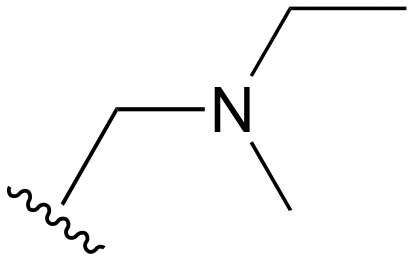
|
H | >1000 | <20 | <20 | >200 | >200 |
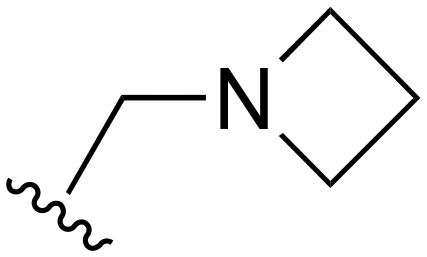
| 122 |
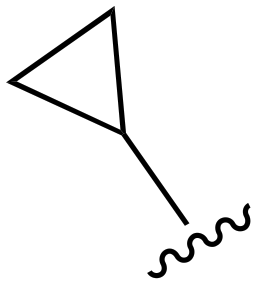
|
|
Me | >1000 | <20 | <20 | >200 | >200 |
The substitution of R3 position (Tables 1–3) with H, Cl and F or methyl in compounds that have R1 = methyl (14, 46, 50, 62, 75 & 76) results in a notable decrease in potency and selectivity against PC9 cell line associated with an increase in the size of the substituents. Specifically, compounds with R3 = H as substituent are 10 times more potent and twice as selective than compounds with R3 = F, Cl and methyl substitutions. These effects are unique to PC9 cells while these molecules exhibit no appreciable effects on potency against the H1975 mutant indicating that the R3 position differently impacts targeting of L858R/T790M and exon19del mutations. No other substitutions appear to show this disconnect. Interestingly, a different trend is observed when the R1 position is substituted with cyclopropyl (55, 75, 122 & 123) or isopropyl groups (93 & 124). In these cases, the methyl substituent at R3 shows ∼10-fold improved potency and selectivity for H1975 mutant over WT EGFR. These results indicate that alternative functional groups at R3 can have divergent effects on these two prevalent NSCLC mutations and showcase a potential avenue for discriminative inhibition of L858R/T790M over exon19del mutations.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | EGFR wild type | EGFR mutants | Selectivity over wild type | ||
| H2073 (nM) | PC9 (nM) | H1975 (nM) | H2073/PC9 (fold) | H2073/H1975 (fold) | ||||
| 50 | Me |

|
Cl | >1000 | 20–200 | 20–200 | 101–200 | >200 |
| 62 | Me |
|
F | >1000 | 20–200 | 20–200 | >200 | >200 |
| 76 | Me |
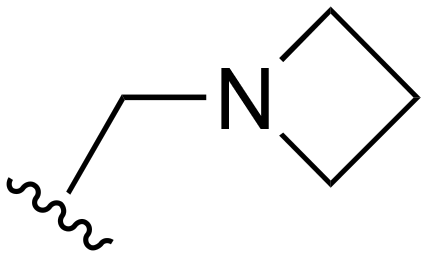
|
Me | >1000 | 20–200 | 20–200 | 101–200 | 101–200 |
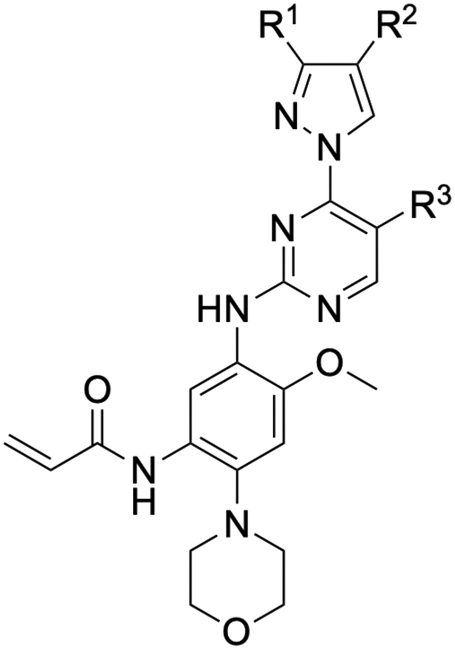
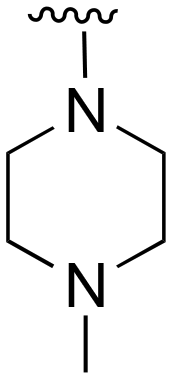
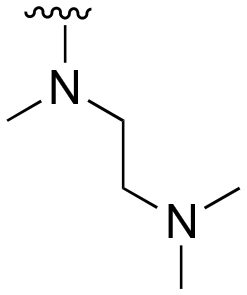
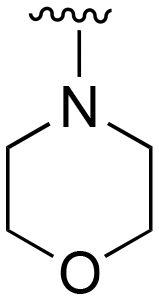
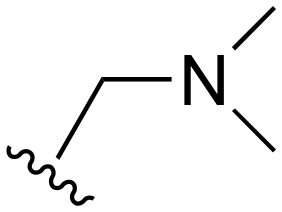
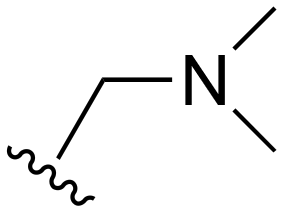
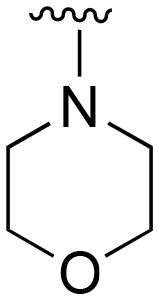
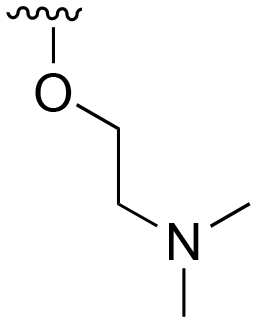
The R4 substituents comprise various solubility-enabling groups located in the solvent exposed region, which provide opportunities for optimizing pharmacokinetic properties. Compounds with R4 = morpholine are generally less potent than R4 = N-methylpiperazine or N,N-dimethylamine (71). For example, substituting morpholine (32) with N-methylpiperazine (36) enhances PC9 potency by 10-fold. On the other hand, replacing a morpholine (46) with a N,N-dimethylamine (71) increases the potency and selectivity for H1975 by over 10-fold. Interestingly, changing a morpholine (73, lazertinib) to an azetidine group (84) reduces selectivity for PC9 and H1975 selectivity by more than 10-fold, as the azetidine makes the compound more active against WT EGFR. The 2-methoxy-N,N-dimethylethan-1-amine (106) leads to a significant reduction in potency and selectivity for both mutants. Compound 86 with morpholine group at R4 position is found exhibit enhanced WT EGFR activity when this is seen to not be the case for when R4 = 2-methoxy-N,N-dimethylethan-1-amine (100). This contrasting trend in WT activity appears to be uniquely dependent on the substitution of t-Bu group at R1 (86, 100) and is not observed in any of the previously discussed molecules showcasing a potential liability for diminished mutant selectivity in this series. Overall, the various solubilization groups exhibit differential impacts to potency and mutant selectivity demonstrating soft SAR at this position as expected based on the location of these groups in the solvent accessible HRII region of the kinase domain. The rationale for the morpholine at R4 in lazertinib given these observations is unclear, to our knowledge, but we suspect other pharmacokinetic factors may have directed for retaining this group in the lead compound.
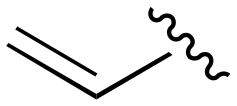
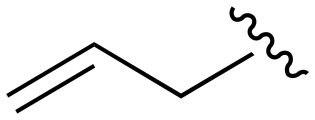
The Michael acceptor “warhead” substituents at R5 position are critical for the effectiveness of these compounds as third-generation EGFR TKIs react to form covalent bonds with C797.43,44 Generally, alterations to this group as surveyed in the patent are detrimental to potency and selectivity. Specifically, saturating the acrylamide alkene of lazertinib (157) results in a complete loss of potency in both PC9 and H1975 cell lines as expected and proves lazertinib is made effective by its ability to form covalent bonds to EGFR. Adding a methylene spacer (154) to the acrylamide (73) causes a 10-fold decrease in potency in the PC9 and over 50-fold in H1975 cell lines. Furthermore, adding an ethyl group at the end of the acrylamide (155) causes a dramatic loss in potency in both the mutant cell lines. Lastly, adding a methylene spacer (156) results in complete loss of potency.
In summary, through a review of data available in the Yuhan Corp. patent,41 the various molecules and their impact on cellular antiproliferative activity provide a perspective into the development of lazertinib (Tables 1–5, Fig. 2). The cellular SAR demonstrates that maximal potency and mutant selectivity is achieved when R1 = phenyl out of a variety of hydrophobic groups while simple tertiary amines are effective at the R2 position. The substituents at R1 and R2 highlighted in this experiment indicate that the investigators likely focused their optimization of the substituted pyrazole deliberately to contain distinctive hydrophobic and hydrophilic polar substituents, which have been shown to be critically necessary for enabling T790M selectivity,39 as will be mentioned in upcoming sections. Interestingly, substituents at R3 exhibit differential antiproliferative effects on PC9 and H1975 where no substitution appears ideal for inhibition of both exon19del and L858R/T790M EGFR. Aside from variations in the solubilization groups at R4, the rest of the compound strongly resembles the chemical structures of other third-generation TKIs as consistent with their conserved irreversible C797-targeting mechanism. Overall, the cellular SAR showcases the most impactful functional groups incorporated into lazertinib (73) that enable the desired potent and mutant selective cellular activity of this scaffold positioning this molecule for advanced biological testing.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | EGFR wild type | EGFR mutants | Selectivity over wild type | ||
| H2073 (nM) | PC9 (nM) | H1975 (nM) | H2073/PC9 (fold) | H2073/H1975 (fold) | |||||
| 32 | H |
|
Me |
|
>1000 | 201–1000 | 20–200 | 20–100 | 20–100 |
| 36 | H |
|
Me |
|
>1000 | 20–200 | 20–200 | 20–100 | 20–100 |
| 42 | H |
|
Me |
|
>1000 | 201–1000 | 20–200 | 20–100 | >200 |
| 46 | Me |
|
H |
|
>1000 | <20 | 20–200 | >200 | 101–200 |
| 71 | Me |
|
H |
|
>1000 | <20 | <20 | >200 | >200 |
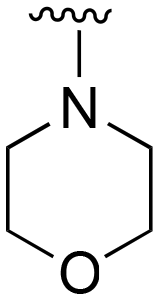
| 73 (Lazertinib) | Ph |
|
H |
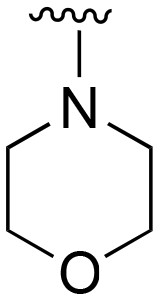
|
>1000 | <20 | <20 | >200 | >200 |
| 74 |
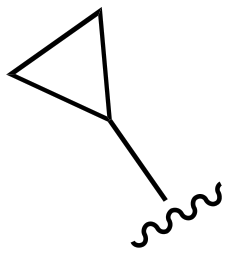
|
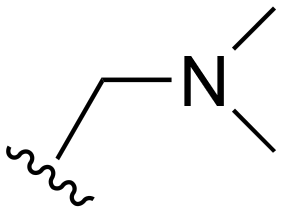
|
H |
|
>1000 | <20 | <20 | >200 | >200 |
| 75 | Me |
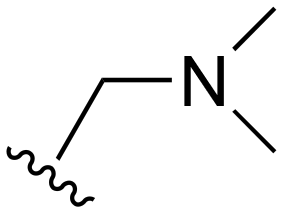
|
Me |
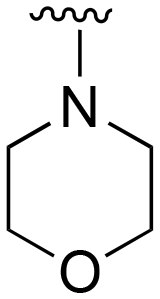
|
>1000 | 20–200 | 20–200 | 101–200 | 101–200 |
| 79 | Me |
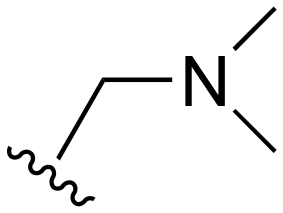
|
H |
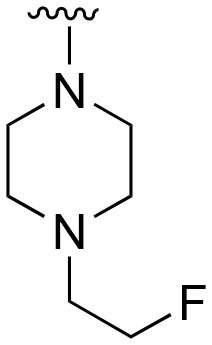
|
>1000 | 20–200 | 20–200 | 101–200 | 101–200 |
| 84 | Ph |
|
H |
|
20–200 | <20 | <20 | <20 | <20 |
| 86 | t-Bu |
|
H |
|
201–1000 | <20 | <20 | 20–200 | 20–200 |
| 100 | t-Bu |
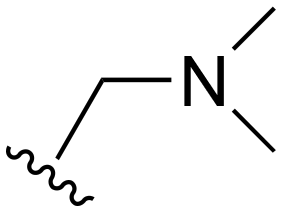
|
H |
|
>1000 | 20–200 | 20–200 | 101–200 | 101–200 |
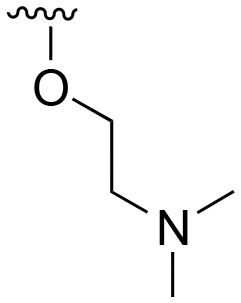
| 106 |
|
|
H |
|
>1000 | 20–200 | 20–200 | 101–200 | 101–200 |
|
|
||||||
|---|---|---|---|---|---|---|
| Compound | R5 | EGFR wild type | EGFR mutants | Selectivity over wild type | ||
| H2073 (nM) | PC9 (nM) | H1975 (nM) | H2073/PC9 (fold) | H2073/H1975 (fold) | ||
| 73 (Lazertinib) |
|
>1000 | <20 | <20 | >200 | >200 |
| 154 |
|
>1000 | 20–200 | 201–1000 | <20 | <20 |
| 155 |
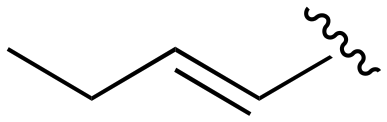
|
>1000 | 201–1000 | 201–1000 | <20 | <20 |
| 156 |
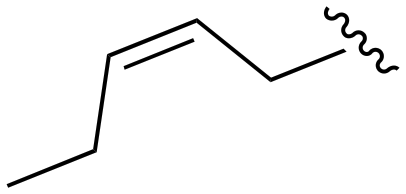
|
>1000 | >1000 | >1000 | <20 | <20 |
| 157 |
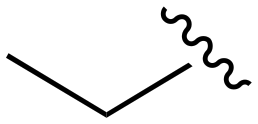
|
>1000 | >1000 | >1000 | <20 | <20 |
 | ||
| Fig. 2 Structure–activity relationships (SAR) generated in the development of lazertinib through cellular viability experiments against PC9 and H1975 cells. | ||
Pre-clinical assessments of the functional profile of lazertinib vs. osimertinib
At the time lazertinib was first disclosed in the peer-review literature, the compound had advanced into clinical trials for NSCLC patients with previously first-generation TKI treatment with disease-progressed T790M-positive NSCLC (LASER201, NCT03046992).38 In general, lazertinib was shown to be superior to osimetinib with respect to both activity and safety in addition to a variety of other important drug profile parameters (Table 6). In a diverse set of mutant EGFR cell lines and mice xenografts, lazertinib was observed to be superiorly effective at suppressing active EGFR and tumor regressions in vivo, respectively. As brain metastasis is a common route to disease progression in NSCLC patients, lazertinib was found to exhibit meaningful blood–brain barrier penetration and comparable impacts to antitumor brain metastasis to osimertinib. Overall, these activity studies show that lazertinib was a promising agent showing improved efficacy in mutant EGFR NSCLC-relevant biological settings.38| Parameters | Lazertinib | Osimertinib | Ref. |
|---|---|---|---|
| Physicochemical properties | |||
| Molecular weight | 554.66 g mol−1 | 499.63 g mol−1 | 45 |
Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D7.4 D7.4 |
3.9 ± 0.1 | 3.2 ± 0.2 | |
| Polar surface area (PSA) | 100 Å2 | 79 Å2 | |
| Hydrogen bond donors | 2 | 2 | |
| In vitro efficacy and permeability | |||
|---|---|---|---|
| Ba/F3 calls expressing mutant and WT EGFR and HER2 | Cellular antiproliferative activities – EC50 (nM) | ||
| Wild type | 722.7 | 519.1 | 38 |
| Del19 | 3.3 | 3.5 | 38 |
| L858R | 3.9 | 3.6 | 38 |
| 4.8 ± 2 | 3.2 ± 0.3 | 39 | |
| Del19/T790M | 4.9 | 3.4 | 38 |
| L858R/T790M | 5.7 | 4.3 | 38 |
| 7.4 ± 1 | 14 ± 4 | 39 | |
| HER2 | >100 | 34 ± 8 | 39 |
| Cell permeability (Caco2 assay) | 17 ± 7 | 10 ± 4 | 45 |
| pH 7.4/7.4 Paa (10−6 cm s−1) | |||
| In vivo parameters | |||
|---|---|---|---|
| Drug exposure in mouse brain (Kp,uu,brain)a | 0.29 | 0.39 | 38 |
| Drug exposure in healthy rat brain (Kp,uu,brain)a | 0.087 | 0.21 | 45 |
| Drug exposure in cynomolgus macaque brain (KP) | 0.97 | 4.18 ± 2.49 | 45 |
| Tumor growth suppression in H1975 (L858R/T790M) xenograft (3 mg kg−1, QD, 13 days period) | 86.85% | 7.24% | 38 |
| Tumor growth suppression in YHIM-1009 (exon19del) xenograft (25 mg kg−1, QD, 20 days period) | 87.5% | 83.6% | 38 |
| Survival rate in H1975-tumor bearing mice (10 mg kg−1, QD) | 124 days | 65 days | 38 |
| Pharmacokinetic parameters (human) | ||||
|---|---|---|---|---|
| Lazertinib | Ref. | Osimertinib | Ref. | |
| a Unbound brain-to-plasma partition coefficient.46 | ||||
| Oral dose (QD) | 240 mg | 47 | 80 mg | 48 |
| Oral bioavailability (human) | — | — | 70% | 48 |
| Volume of distribution | 4264 L | 47 | 986 L | 49 |
| 1216 L | 48 | |||
| Protein plasma binding (PPB) | 99.1–99.7% | 23, 44 | 95% | 47, 48 |
| Peak plasma concentration (Cmax) | 517.15 (ng mL−1) | 50 | 550.4 (nmol L−1) | 48, 51 |
| Time to peak drug concentration (Tmax) | 2–4 h | 47 | 4 h | 48 |
| 6 h | 49 | |||
| Half-life (T1/2) | 64.7 h | 47 | 55 h | 49 |
| Time to steady state | 15 days | 47 | 22 days | 49 |
| Mode of excretion | Bile (60%) | 23 | Fecal (68%) | 49 |
| Fecal (24%) | Urinary (14%) | |||
The clear standout improvement for lazertinib when compared to osimertinib were elements of the safety and tolerability.38 In many respects, this has allowed for lazertinib to be administered at higher doses to osimertinib, 240 mg vs. 80 mg QD, respectively, however their pharmacokinetic profiles are otherwise fairly similar (Table 6). Osimertinib is highly efficacious but is well associated with toxic liabilities. An established source of adverse events are caused by the susceptibility of the N-methylindole of osimertinib to CYP-dependent oxidative dealkylation to give AZ5104, an active and toxic metabolite due to elevated potency against WT EGFR (Fig. 3).36,37,52,53 This substituent is completely replaced with the substituted pyrazole in lazertinib resulting in a distinct and improved toxicity profile. This study also reported that lazertinib was less potent than osimertinib against HER2, which is highly similar with respect to sequence similarity to the WT kinase domain and considered a source of potential off-target complications (e.g., osimertinib is ∼3-fold more potent against L858R/T790M EGFR versus HER2 compared to ∼18-fold more selective for lazertinib).38 The investigators specifically assessed the potential for both drugs to cause cutaneous toxicities in skin, which are not life-threatening but contribute to the tolerability of these anticancer agents. In these respects, lazertinib dosing was associated with reduced skin inflammation in vivo compared to osimertinib showcasing a wider therapeutic window. This pre-clinical evaluation demonstrates lazertinib as a best-in-class third-generation EGFR TKI and motivates deeper understanding into the molecular nature governing the functional differences between lazertinib and osimertinib.
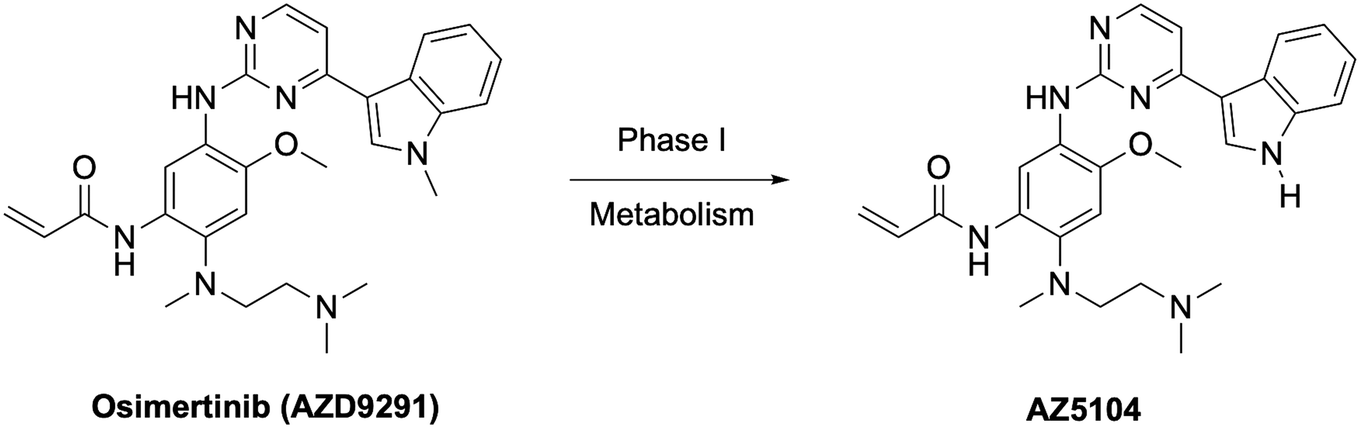 | ||
| Fig. 3 AZ5104 is a phase I metabolite of osimertinib and associated with dose-limiting adverse events. | ||
Time-dependent inhibition properties of lazertinib against WT and mutant EGFR
Since third-generation TKIs are irreversible EGFR inhibitors, proper measurements of their biochemical activity rely on time-dependent measurements. In this respect, it is impossible to assess the potency and selectivity of an irreversible TKI on the basis of IC50 measurements since these values fluctuate based on the incubation times of the assays and changes in rates of enzyme inactivation.40,54,55 First, it is important to appreciate that these compounds operate through a two-step irreversible covalent mechanism (Fig. 4) that involves initial reversible binding to EGFR followed (first step) by permanent inactivation through covalent bond formation to C797 (second step).40,55 For a particular covalent inhibitor, the potency can be assessed through the second-order rate constant (kinact/KI) known as the inactivation efficiency that reflects how quickly the free enzyme (E) is irreversibly inactivated to form the E–I complex.54–56kinact/KI is the most ideal value for determining irreversible inhibitor potency as it contains all the rate constants that govern target inhibition.56 The first-order rate constant kinact in the numerator defines the maximum rate of inactivation achieved at an infinite concentration of an inhibitor and KI (the inactivation constant) is the concentration of the inhibitor where the rate constant of inactivation is equal to 1/2kinact. KI contains the rate constants kon, koff, and kinact and can be used to estimate the relative impact of reversible binding (kon, koff) on kinact/KI. A variety of activity assay formats have been developed to measure kinact/KI, kinact, and KI values, such as progress-curve analysis (PCA), time-dependent IC50 value determination, stopped-flow rapid mixing and others, to construct structure-kinetic relationships (SKR) for irreversible covalent inhibitors.40,54,57–59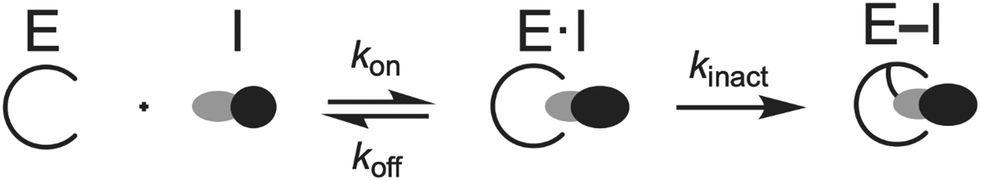 | ||
| Fig. 4 Two-step mechanism that governs the activity of irreversible inhibitors. “E” generally refers to any enzyme but in this context is the EGFR kinase domain. “E·I” indicates the enzyme–inhibitor complex formed from fully intermolecular interactions and “E–I” stands for the completely inactivated enzyme–inhibitor complex after the covalent bond is formed. Reproduced from ref. 55 with permission from the American Chemical Society, copyright 2024. | ||
Our group set out to characterize the time-dependent inhibition of lazertinib compared to osimertinib to provide activity values to generate a structural basis for the potency and selectivity of these TKIs.39 These results were found in good agreement with the earlier study38 indicating that lazertinib exhibited a greater degree of potency for L858R/T790M compared to WT, which was superior to osimertinib. Additionally, it was also showed that lazertinib was limitedly potent against WT HER2, which was corroborated with potency data from human cancer cell lines and Ba/F3 cells.39 An alternative liquid handling protocol with 100% DMSO in the setup of the PCA assays, while final concentrations were identical to earlier experiments (1% DMSO), were found to result in elevated values of kinact/KI.40 The exact origins of this change in potency are still unknown, however we speculate it could be due to differences in solubility or other solution-phase behavior as the elevated kinact/KI values also tracked with reduction in KI, which is dependent on inhibitor concentration.40 Despite this complication, we have elected to showcase the kinetic values from this most recent work as the best representations of the potency and mutant selectivity of lazertinib and osimertinib (Table 7).
| Lazertinib (YH25448) | Osimertinib (AZD9291) | |||||
|---|---|---|---|---|---|---|
| k inact × 10−3 (s−1) | K I app (nM) | (kinact/KI)app × 105 (M−1 s−1) | k inact × 10−3 (s−1) | K I app (nM) | (kinact/KI)app × 105 (M−1 s−1) | |
| WT | 6.51 ± 0.62 | 72.1 ± 8.5 | 0.903 ± 0.026 | 7.20 ± 0.46 | 34.4 ± 3.3 | 2.09 ± 0.10 |
| L858R | 5.88 ± 0.34 | 4.68 ± 0.33 | 12.6 ± 0.3 | 6.36 ± 0.29 | 4.47 ± 0.3 | 14.2 ± 0.40 |
| L858R/T790M | 5.20 ± 0.23 | 14.6 ± 0.8 | 3.56 ± 0.05 | 7.60 ± 0.61 | 16.4 ± 1.7 | 4.63 ± 0.14 |
A head-to-head comparison to irreversible covalent inhibitor kinetics shows that lazertinib and osimertinib are nearly equivalently potent against L858R/T790M, however lazertinib is significantly less potent against WT (Table 7).40 The apparent difference in WT potency is observed to be due to a higher KI values for lazertinib consistent with weaker reversible binding affinity to WT compared to L858R/T790M. Both compounds exhibit elevated kinact/KI and lower KI against L858R consistent with the universally weaker ATP binding affinity for this mutation.60 These observations indicate that both lazertinib and osimertinib are potent and selective inhibitors of L858R/T790M, however lazertinib is superior due to the ∼4-fold lower kinact/KI against WT. These results indicate that the improved selectivity against T790M is mainly due to differences in the irreversible binding properties of these drugs (first-step, Fig. 4) as kinact values are virtually identical for both compounds and all EGFR enzymes (Table 7).
Structural basis for selectivity and potency of lazertinib
As mentioned earlier, lazertinib and osimertinib differ most significantly with respect to the distinct group that binds the adenosine-binding region (Fig. 1). The key result from irreversible inhibitor kinetic values (Table 7) indicates that reduced binding strength of lazertinib to WT EGFR accounts for the greater degree of T790M selectivity and likely pinpoints that the substituted pyrazole to be responsible for this effect. To fully appreciate the structural basis for the potency and mutant selectivity of lazertinib, a direct comparison of several EGFR X-ray cocrystal structures is required.For a number of years, the precise structural basis for the T790M selectivity of osimertinib had been unknown as only a single cocrystal structure had been known in the literature with this drug in complex with WT EGFR featuring osimertinib bound without the covalent bond formed to C797.61 A major advancement in our understanding was reported by Yun and Shaw through a combination of molecular dynamic (MD) simulations and X-ray cocrystal structures from both soaking and cocrystallization strategies.62 This work revealed that the N-methylindole of osimertinib is observed in two distinct conformations depending on the gatekeeper residue (790) being either a Thr (as in WT) or methionine (as in T790M). As shown in the X-ray cocrystal structure of osimertinib bound to WT, the N-methylindole is located with the benzene ring facing “away” from the gatekeeper T790 (Fig. 5A). Additionally, the T790 alcohol is involved in H-bonding with a water network that is maintained through interactions with nearby conserved polar residues K745, D855, and T854. Alternatively, an X-ray structure obtained through cocrystallization of osimertinib and EGFR (T790M) showed a “flipped” conformation of the N-methylindole now with the benzene ring involved in van der Waals interactions with the T790M methionine and complete loss of the bound water network (Fig. 5B). Computational simulations support the observations in cocrystal structures and reinforce the conclusion that the N-methylindole adopts different conformations depending on the gatekeeper mutation. The T790M-selectivity of osimertinib can be readily credited to the formation of productive van der Waals allowed by the “flipped” conformation of the N-methylindole,62 and is consistent with the trends in kinact/KI and KI values for WT and L858R/T790M.40,55,58
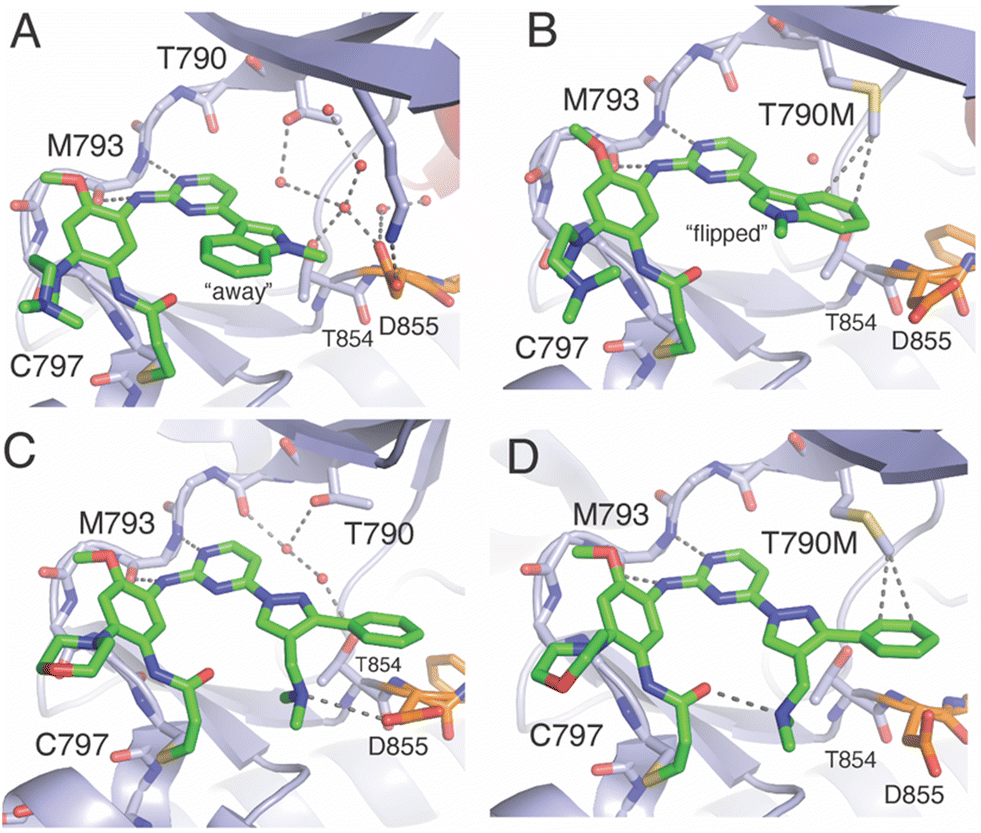 | ||
| Fig. 5 Comparison of the binding modes of osimertinib and lazertinib when bound to EGFR as WT or the T790M gatekeeper mutation. A) X-ray cocrystal structure of osimertinib in complex with WT EGFR through soaking. This binding mode shows the N-methylindole rotated “away” from the gatekeeper T790 (PDB ID 6JXT). B) The binding mode of osimertinib and T790M mutant EGFR kinase domain obtained through cocrystallization (PDB ID 6JX0). C) Binding mode of lazertinib in complex with WT EGFR obtained through soaking (PDB ID 7UKV). D) Binding mode of lazertinib and T790M/V948R obtained through soaking (PDB ID 7UKW). | ||
X-ray cocrystal structures determined by our group feature lazertinib in complex with both WT and T790M/V948R EGFR.39 The WT EGFR kinase domain reliably crystallizes in the αC-helix “in” active conformation while the V948R mutation confers the ability for the kinase to crystallize into the αC-helix “out” inactive, as observed previously.63–65 As expected, the binding mode of lazertinib shares much in common with osimertinib such as the anchoring of the aminopyrimidine hinge-binding group and the covalent bond to C797 (Fig. 5C). The substituted pyrazole moiety is located in the adenosine-binding region with the phenyl ring bound toward the gatekeeper and methyleneamine positioned toward the solvent accessible HRII donating a H-bond to the carboxylic acid side chain of D855. The water network previously seen in the osimertinib WT structure (Fig. 5A) is disrupted potentially due to the rearrangement of the D855 to H-bond with lazertinib as well as the placement of the hydrophobic phenyl ring near these resides. The T790M/V948R cocrystal structure shows lazertinib bound in an identical orientation with the phenyl moiety involved in van der Waals interactions with T790M and the methyleneamine producing an intramolecular H-bond to the carbonyl of the acrylamide moiety that has formed the covalent bond to C797 (Fig. 5D). The observation of the van der Waals interactions with T790M are comparable to those seen in the osimertinib X-ray cocrystal structure (Fig. 5B) indicating that these two drugs share similar features that enable T790M selectivity, namely constructive hydrophobic interactions via aromatic benzene/phenyl rings and consistent with results from kinetic assays (Table 7).62
The relatively lower potency and reversible binding against WT for lazertinib is of considerable interest given that this attribute is likely responsible for the improved functional profile. In this respect, weaker reversible binding can be attributed to the substituted pyrazole of lazertinib and was shown to enable maximal potency and mutant selectivity in the cell-based SAR (Tables 1–5). The methyleneamine is most likely not the direct source of this weaker binding as it is found making a H-bond to D855, which would presumably lead to stronger reversible binding. Alternatively, the phenyl ring of lazertinib is in a region of WT EGFR where a polar water network is seen within the WT osimertinib structure. The hydrophobic phenyl ring is opposed, to an extent, by the polar nature of this region (Fig. 5C), which is likely that such an interaction is preferred for osimertinib owing to the more stable “away” positioning of the hydrophobic benzene of the N-methylindole (Fig. 5A). The local polarity of this region of the kinase domain is altered significantly by the presence of the hydrophobic T790M methionine where and both lazertinib and osimertinib are productively anchored to this pocket through non-polar interactions to the T790M gatekeeper residue (Fig. 5B and D). This rationale also explains the diminished inhibition of HER2 by lazertinib where this closely related ErbB-family kinase shares a practically identical ATP binding sites, including the T798 gatekeeper residue as well as the other conserved polar residues analogous to K745, T854, and D855 in EGFR.66
A reasonable question given this explanation would concern the ability for lazertinib to rotate the methyleneamine and phenyl akin to the N-methylindole. This would appear likely given the hydrophilic nature of the gatekeeper/HRI region in WT EGFR; however, this binding mode is not observed in our WT cocrystal structure. It should be noted that a rotated lazertinib pyrazole is observed in a distinct chain in the T790M/V948R cocrystal structure,39 however, the location of the positively charged amine near the catalytic lysine is likely far more repulsive than the phenyl as observed in the WT structure. Instead, it is most likely that the unique methyleneamine of lazertinib rigidifies the pyrazole into the conformation through productive intramolecular H-bonding with the acrylamide group as observed in the X-ray cocrystal structure (Fig. 5D) or the D855 carboxyl side chain as seen in WT (Fig. 5C). The relatively unproductive binding of the lazertinib phenyl to WT is not contradicted by kinetic results for L858R since this kinase domain mutation leads to stronger TKI binding due to intrinsically reduced ATP binding affinities, which is also the case for exon19del mutations, and uniformly results in tighter TKI reversible binding (KI values in Table 7).9,60,67
Clinical assessments
TKIs have long been the focus of clinical investigations, especially as potent agents for targeted therapies in mutant EGFR non-small cell lung cancer (NSCLC). Lazertinib was initially approved in the Republic of Korea for patients with T790M-positive mutations following first-generation TKI treatment.23 Later, it gained FDA approval for front-line therapy, specifically in combination with the dual-targeting antibody, amivantamab.25 The growing use of combination therapies has led to numerous clinical trials involving lazertinib, with 39 trials listed as of now (https://www.clinicaltrials.gov).47The first clinical evaluation of lazertinib as monotherapy was the LASER201 trial (NCT03046992, phase I/II), which demonstrated that the drug had a tolerable safety profile and promising clinical activity in patients with T790M-positive NSCLC who had progressed on or after EGFR TKI therapy.50 (A phase I/II trial is a combined study that assesses safety and tolerability within a patient population to quickly evaluate clinical efficacy). A subsequent report showed that lazertinib exhibited durable antitumor activity, including against brain metastases.68 To evaluate whether lazertinib was superior to the first-generation TKI gefitinib, the LASER301 (NCT04248829) trial demonstrated significant efficacy improvements with lazertinib in the first-line treatment of EGFR-mutated advanced NSCLC, alongside a manageable safety profile.69 These findings support that lazertinib is an effective agent for both untreated mutant NSCLC and second-line treatment for T790M-positive resistant patients.
The CHRYSALIS study (NCT02609776, phase I) aimed to evaluate the effectiveness of combining the dual EGFR/c-MET antibody amivantamab with lazertinib in both treatment-naive and osimertinib-relapsed settings. Early results indicated that the combination of amivantamab and lazertinib had favorable safety profiles, similar to use as single agents.70 The combination was further assessed in osimertinib-relapsed EGFR-mutant NSCLC, where progression-free survival (PFS) rates were comparable to platinum-based chemotherapy. Additionally, clinical response was associated with high expressions of EGFR and/or MET.70 A related study (CHRYSALIS-2, phase I/Ib, NCT04077463) explored the combination's efficacy in rarer, atypical EGFR mutations (e.g., exon 20 insertions, S768I, L861Q, G719X), showing durable antitumor activity for both untreated patients and those who had progressed after treatment with the second-generation inhibitor afatinib.71
Given the prominence of osimertinib as the first-line therapy for mutant EGFR NSCLC,13 subsequent trials were designed to compare the combination of lazertinib and amivantamab to osimertinib monotherapy. Results from the MARIPOSA phase III trial (NCT04487080) demonstrated that the lazertinib–amivantamab combination was superior to osimertinib, extending PFS to approximately 24 months.26,72 Lazertinib monotherapy (240 mg daily) resulted in a longer PFS compared to osimertinib (80 mg daily), though still not as effective as the combination. A related trial (MARIPOSA-2, NCT04988295) explored amivantamab plus chemotherapy, with or without lazertinib, in EGFR-mutant advanced NSCLC after progression on osimertinib. Both the amivantamab-chemotherapy and amivantamab–lazertinib-chemotherapy groups showed improved PFS compared to chemotherapy alone.73 More recently, assessments of subcutaneous versus intravenous amivantamab administration in combination with lazertinib is also being assessed in a phase III trial (PALOMA-3, NCT05388669) in attempt to provide greater convenience of amavantamab administration for NSCLC patients.74
Conclusions and outlook
The emergence of lazertinib as an approved drug has reshaped the landscape of targeted therapies and provided unique molecular insights into how third-generation inhibitors can be designed to become more effective and safer agents for mutant EGFR NSCLC. The binding modes and the resulting structural insights, informed by time-dependent kinetic values, show how the pyrazole group of lazertinib, substituted with a hydrophobic phenyl and a hydrophilic H-bond-donating amine weakens binding to WT EGFR while maintaining potent inhibition of L858R and the T790M gatekeeper mutation. A persistent challenge is highlighted by the fact that lazertinib, like other third-generation TKIs, is vulnerable to the C797S resistance mutation, which impairs the formation of the covalent bond critical for potency.14,15 Indeed, the cell-based SAR in the Yuhan Corp. patent also demonstrate that alterations to the acrylamide warhead dramatically impact antiproliferative activity (Table 5).41 The development of agents capable of overcoming this C797S limitation while maintaining effectiveness against prior targets remains an unmet need.5,16 Additionally, modern treatments for mutant EGFR NSCLC appear to be made more effective due to their ability to anticipate future resistance as exemplified by the combination of lazertinib and amivantamab as a superiorly effective “one-two punch” against diverse resistance mechanisms, particularly “gatekeeper” T790M acquired resistance and c-MET amplification. As such, monotherapies may become less common and increasingly replaced with combinations potentially made of blends of diverse agents spanning small molecules as well as biologics and cell-based therapies.Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support from startup funds from The State University of New York, the National Center for Advancing Translational Sciences of the National Institutes of Health under award number UL1TR001412-08 (BTC K Scholar Award), and the National Institutes of General Medical Sciences R35GM155353-01.References
- N. Vasan, J. Baselga and D. M. Hyman, Nature, 2019, 575, 299–309 CrossRef CAS PubMed.
- G. Housman, S. Byler, S. Heerboth, K. Lapinska, M. Longacre, N. Snyder and S. Sarkar, Cancers, 2014, 6, 1769–1792 CrossRef PubMed.
- W. Pao and J. Chmielecki, Nat. Rev. Cancer, 2010, 10, 760–774 CrossRef CAS PubMed.
- D. E. Heppner and M. J. Eck, Protein Sci., 2021, 30, 1535–1553 CrossRef CAS PubMed.
- L. Xu, B. Xu, J. Wang, Y. Gao, X. He, T. Xie and X.-Y. Ye, Eur. J. Med. Chem., 2023, 245, 114900 CrossRef CAS PubMed.
- J. G. Paez, P. A. Jänne, J. C. Lee, S. Tracy, H. Greulich, S. Gabriel, P. Herman, F. J. Kaye, N. Lindeman, T. J. Boggon, K. Naoki, H. Sasaki, Y. Fujii, M. J. Eck, W. R. Sellers, B. E. Johnson and M. Meyerson, Science, 2004, 304, 1497–1500 CrossRef CAS PubMed.
- T. J. Lynch, D. W. Bell, R. Sordella, S. Gurubhagavatula, R. A. Okimoto, B. W. Brannigan, P. L. Harris, S. M. Haserlat, J. G. Supko, F. G. Haluska, D. N. Louis, D. C. Christiani, J. Settleman and D. A. Haber, N. Engl. J. Med., 2004, 350, 2129–2139 CrossRef CAS PubMed.
- A. Quintás-Cardama and J. Cortes, Clin. Cancer Res., 2008, 14, 4392–4399 CrossRef PubMed.
- C.-H. Yun, K. E. Mengwasser, A. V. Toms, M. S. Woo, H. Greulich, K.-K. Wong, M. Meyerson and M. J. Eck, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2070–2075 CrossRef CAS PubMed.
- S. Kobayashi, T. J. Boggon, T. Dayaram, P. A. Jänne, O. Kocher, M. Meyerson, B. E. Johnson, M. J. Eck, D. G. Tenen and B. Halmos, N. Engl. J. Med., 2005, 352, 786–792 CrossRef CAS PubMed.
- R. S. Herbst, D. Morgensztern and C. Boshoff, Nature, 2018, 553, 446–454 CrossRef CAS PubMed.
- S. S. Ramalingam, J. Vansteenkiste, D. Planchard, B. C. Cho, J. E. Gray, Y. Ohe, C. Zhou, T. Reungwetwattana, Y. Cheng, B. Chewaskulyong, R. Shah, M. Cobo, K. H. Lee, P. Cheema, M. Tiseo, T. John, M.-C. Lin, F. Imamura, T. Kurata, A. Todd, R. Hodge, M. Saggese, Y. Rukazenkov and J.-C. Soria, N. Engl. J. Med., 2020, 382, 41–50 CrossRef CAS PubMed.
- J.-C. Soria, Y. Ohe, J. Vansteenkiste, T. Reungwetwattana, B. Chewaskulyong, K. H. Lee, A. Dechaphunkul, F. Imamura, N. Nogami and T. Kurata, N. Engl. J. Med., 2018, 378, 113–125 CrossRef CAS PubMed.
- K. S. Thress, C. P. Paweletz, E. Felip, B. C. Cho, D. Stetson, B. Dougherty, Z. Lai, A. Markovets, A. Vivancos and Y. Kuang, Nat. Med., 2015, 21, 560–562 CrossRef CAS PubMed.
- S. Park, B. M. Ku, H. A. Jung, J.-M. Sun, J. S. Ahn, S.-H. Lee, K. Park and M.-J. Ahn, Cancer Res. Treat., 2020, 52, 1288–1290 Search PubMed.
- H.-Y. Zhao, X.-X. Xi, M. Xin and S.-Q. Zhang, Bioorg. Chem., 2022, 128, 106057 CrossRef CAS PubMed.
- D. E. Heppner, M. Günther, F. Wittlinger, S. A. Laufer and M. J. Eck, J. Med. Chem., 2020, 63, 4293–4305 CrossRef CAS PubMed.
- T. Damghani, F. Wittlinger, T. S. Beyett, M. J. Eck, S. A. Laufer and D. E. Heppner, in Methods in Enzymology, ed. J. P. Richard and G. R. Moran, Academic Press, 2023, vol. 685, pp. 171–198 Search PubMed.
- Y. Jia, C.-H. Yun, E. Park, D. Ercan, M. Manuia, J. Juarez, C. Xu, K. Rhee, T. Chen and H. Zhang, Nature, 2016, 534, 129–132 CrossRef CAS PubMed.
- C. To, J. Jang, T. Chen, E. Park, M. Mushajiang, D. J. De Clercq, M. Xu, S. Wang, M. D. Cameron and D. E. Heppner, Cancer Discovery, 2019, 9, 926–943 CrossRef CAS PubMed.
- T. S. Beyett, C. To, D. E. Heppner, J. K. Rana, A. M. Schmoker, J. Jang, D. J. De Clercq, G. Gomez, D. A. Scott and N. S. Gray, Nat. Commun., 2022, 13, 2530 CrossRef CAS PubMed.
- J. Niggenaber, L. Heyden, T. Grabe, M. P. Müller, J. Lategahn and D. Rauh, ACS Med. Chem. Lett., 2020, 11, 2484–2490 CrossRef CAS PubMed.
- S. Dhillon, Drugs, 2021, 81, 1107–1113 CrossRef CAS PubMed.
- B. C. Cho, E. Felip, H. Hayashi, M. Thomas, S. Lu, B. Besse, T. Sun, M. Martinez, S. N. Sethi and S. M. Shreeve, Future Oncol., 2022, 18, 639–647 CrossRef CAS PubMed.
- U. S. FDA, FDA approves lazertinib with amivantamab-vmjw for non-small lung cancer, https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-lazertinib-amivantamab-vmjw-non-small-lung-cancer.
- C. Zhou, K.-J. Tang, B. C. Cho, B. Liu, L. Paz-Ares, S. Cheng, S. Kitazono, M. Thiagarajan, J. W. Goldman, J. K. Sabari, R. E. Sanborn, A. S. Mansfield, J.-Y. Hung, M. Boyer, S. Popat, J. M. Dias, E. Felip, M. Majem, M. Gumus, S.-W. Kim, A. Ono, J. Xie, A. Bhattacharya, T. Agrawal, R. E. Knob, S. M. Shreevelauch, K. Park and N. Girard, N. Engl. J. Med., 2023, 389, 2039–2051 CrossRef CAS PubMed.
- Y.-H. Xie, Y.-X. Chen and J.-Y. Fang, Signal Transduction Targeted Ther., 2020, 5, 22 CrossRef CAS PubMed.
- S. M. Magrini, M. Buglione, R. Corvò, L. Pirtoli, F. Paiar, P. Ponticelli, A. Petrucci, A. Bacigalupo, M. Crociani, L. Lastrucci, S. Vecchio, P. Bonomo, N. Pasinetti, L. Triggiani, R. Cavagnini, L. Costa, S. Tonoli, M. Maddalo and S. Grisanti, J. Clin. Oncol., 2016, 34, 427–435 CrossRef CAS PubMed.
- J. M. Bauml, R. Vinnakota, Y.-H. Anna Park, S. E. Bates, T. Fojo, C. Aggarwal, J. Di Stefano, C. Knepley, S. Limaye, R. Mamtani, J. Wisnivesky, N. Damjanov, C. J. Langer, R. B. Cohen and K. Sigel, Cancer, 2019, 125, 406–415 CrossRef CAS PubMed.
- Key Reference: B. C. Cho, S. Lu, E. Felip, A. I. Spira, N. Girard, J.-S. Lee, S.-H. Lee, Y. Ostapenko, P. Danchaivijitr, B. Liu, A. Alip, E. Korbenfeld, J. M. Dias, B. Besse, K.-H. Lee, H. Xiong, S.-H. How, Y. Cheng, G.-C. Chang, H. Yoshioka, J. C.-H. Yang, M. Thomas, D. Nguyen, S.-H. I. Ou, S. Mukhedkar, K. Prabhash, M. D'Arcangelo, J. Alatorre-Alexander, J. C. V. Limón, S. Alves, D. Stroyakovskiy, M. Peregudova, M. A. N. Şendur, O. Yazici, R. Califano, V. G. Calderón, F. D. Marinis, A. Passaro, S.-W. Kim, S. M. Gadgeel, J. Xie, T. Sun, M. Martinez, M. Ennis, E. Fennema, M. Daksh, D. Millington, I. Leconte, R. Iwasawa, P. Lorenzini, M. Baig, S. Shah, J. M. Bauml, S. M. Shreeve, S. Sethi, R. E. Knoblauch and H. Hayashi, N. Engl. J. Med., 2024, 391, 1486–1498 CrossRef CAS PubMed.
- D. A. Cross, S. E. Ashton, S. Ghiorghiu, C. Eberlein, C. A. Nebhan, P. J. Spitzler, J. P. Orme, M. R. V. Finlay, R. A. Ward and M. J. Mellor, Cancer Discovery, 2014, 4, 1046–1061 CrossRef CAS PubMed.
- M. R. V. Finlay, M. Anderton, S. Ashton, P. Ballard, P. A. Bethel, M. R. Box, R. H. Bradbury, S. J. Brown, S. Butterworth, A. Campbell, C. Chorley, N. Colclough, D. A. E. Cross, G. S. Currie, M. Grist, L. Hassall, G. B. Hill, D. James, M. James, P. Kemmitt, T. Klinowska, G. Lamont, S. G. Lamont, N. Martin, H. L. McFarland, M. J. Mellor, J. P. Orme, D. Perkins, P. Perkins, G. Richmond, P. Smith, R. A. Ward, M. J. Waring, D. Whittaker, S. Wells and G. L. Wrigley, J. Med. Chem., 2014, 57, 8249–8267 CrossRef CAS PubMed.
- F. Wittlinger and S. A. Laufer, Expert Opin. Drug Discovery, 2021, 16, 1091–1103 CrossRef CAS PubMed.
- W. Zhou, D. Ercan, L. Chen, C.-H. Yun, D. Li, M. Capelletti, A. B. Cortot, L. Chirieac, R. E. Iacob, R. Padera, J. R. Engen, K.-K. Wong, M. J. Eck, N. S. Gray and P. A. Jänne, Nature, 2009, 462, 1070–1074 CrossRef CAS PubMed.
- P. A. Jänne, J. C.-H. Yang, D.-W. Kim, D. Planchard, Y. Ohe, S. S. Ramalingam, M.-J. Ahn, S.-W. Kim, W.-C. Su, L. Horn, D. Haggstrom, E. Felip, J.-H. Kim, P. Frewer, M. Cantarini, K. H. Brown, P. A. Dickinson, S. Ghiorghiu and M. Ranson, N. Engl. J. Med., 2015, 372, 1689–1699 CrossRef PubMed.
- E. Ishikawa, Y. Yokoyama, H. Chishima, H. Kasai, O. Kuniyoshi, M. Kimura, J. Hakamata, H. Nakada, N. Suehiro and N. Nakaya, Invest. New Drugs, 2023, 41, 122–133 CrossRef CAS PubMed.
- T. Okuno, M. Hongo, R. Tanino, Y. Tsubata and T. Isobe, in C110. Surfing the Innovation Wave in San Diego: Cutting-Edge Lung Cancer Biomarkers, Diagnostics and Treatments, American Thoracic Society, 2024, p. A6889 Search PubMed.
- Key Reference: J. Yun, M. H. Hong, S.-Y. Kim, C.-W. Park, S. Kim, M. R. Yun, H. N. Kang, K.-H. Pyo, S. S. Lee, J. S. Koh, H.-J. Song, D. K. Kim, Y.-S. Lee, S.-W. Oh, S. Choi, H. R. Kim and B. C. Cho, Clin. Cancer Res., 2019, 25, 2575–2587 CrossRef CAS PubMed.
- Key Reference: D. E. Heppner, F. Wittlinger, T. S. Beyett, T. Shaurova, D. A. Urul, B. Buckley, C. D. Pham, I. K. Schaeffner, B. Yang, B. C. Ogboo, E. W. May, E. M. Schaefer, M. J. Eck, S. A. Laufer and P. A. Hershberger, ACS Med. Chem. Lett., 2022, 13, 1856–1863 CrossRef CAS PubMed.
- Key Reference: K. W. Hoyt, D. A. Urul, B. C. Ogboo, F. Wittlinger, S. A. Laufer, E. M. Schaefer, E. W. May and D. E. Heppner, J. Med. Chem., 2024, 67, 2–16 CrossRef CAS PubMed.
- B.-C. Suh, P. D. Salgaonkar, J. Lee, J. S. Koh, H.-J. Song, I. Y. Lee, J. Lee, D. S. Jung, J.-H. Kim and S.-W. Kim, US Pat., US20160102076A1, 2016 Search PubMed.
- S.-H. Oh, J.-H. Khoo, J.-C. Lim, D.-B. Lee, J.-A. Lee, J.-S. Lii, H. Ju, W.-S. Shin and S.-S. Joen, US Pat., US20200207750A1, 2020 Search PubMed.
- L. Hillebrand, X. J. Liang, R. A. M. Serafim and M. Gehringer, J. Med. Chem., 2024, 67, 7668–7758 CrossRef CAS PubMed.
- M. Gehringer and S. A. Laufer, J. Med. Chem., 2019, 62, 5673–5724 CrossRef CAS PubMed.
- N. Colclough, K. Chen, P. Johnström, N. Strittmatter, Y. Yan, G. L. Wrigley, M. Schou, R. Goodwin, K. Varnäs, S. J. Adua, M. Zhao, D. X. Nguyen, G. Maglennon, P. Barton, J. Atkinson, L. Zhang, A. Janefeldt, J. Wilson, A. Smith, A. Takano, R. Arakawa, M. Kondrashov, J. Malmquist, E. Revunov, A. Vazquez-Romero, M. M. Moein, A. D. Windhorst, N. A. Karp, M. R. V. Finlay, R. A. Ward, J. W. T. Yates, P. D. Smith, L. Farde, Z. Cheng and D. A. E. Cross, Clin. Cancer Res., 2021, 27, 189–201 CrossRef CAS PubMed.
- I. Loryan, A. Reichel, B. Feng, C. Bundgaard, C. Shaffer, C. Kalvass, D. Bednarczyk, D. Morrison, D. Lesuisse, E. Hoppe, G. C. Terstappen, H. Fischer, L. Di, N. Colclough, S. Summerfield, S. T. Buckley, T. S. Maurer and M. Fridén, Pharm. Res., 2022, 39, 1321–1341 CrossRef CAS PubMed.
- A. Shaji and A. Sharma, Cancer Research, Statistics, and Treatment, 2024, 7(2), 234–240 CrossRef.
- C. Bordet, V. Dongay, Y. Zelmat, J. Mazieres and F. Despas, J. Clin. Pharm. Ther., 2024, 2024, 5437090 CrossRef.
- S. L. Greig, Drugs, 2016, 76, 263–273 CrossRef CAS PubMed.
- Key Reference: M.-J. Ahn, J.-Y. Han, K. H. Lee, S.-W. Kim, D.-W. Kim, Y.-G. Lee, E. K. Cho, J.-H. Kim, G.-W. Lee and J.-S. Lee, Lancet Oncol., 2019, 20, 1681–1690 CrossRef CAS PubMed.
- H. Zhao, J. Cao, J. Chang, Z. Zhang, L. Yang, J. Wang, M. Cantarini and L. Zhang, J. Clin. Pharmacol., 2018, 58, 504–513 CrossRef CAS PubMed.
- J. W. T. Yates, S. Ashton, D. Cross, M. J. Mellor, S. J. Powell and P. Ballard, Mol. Cancer Ther., 2016, 15, 2378–2387 CrossRef CAS PubMed.
- Y. Meng, B. Yu, H. Huang, Y. Peng, E. Li, Y. Yao, C. Song, W. Yu, K. Zhu, K. Wang, D. Yi, J. Du and J. Chang, J. Med. Chem., 2021, 64, 925–937 CrossRef CAS PubMed.
- E. Mons, S. Roet, R. Q. Kim and M. P. Mulder, Curr. Protoc., 2022, 2, e419 CrossRef CAS PubMed.
- D. E. Heppner, B. C. Ogboo, D. A. Urul, E. W. May, E. M. Schaefer, A. S. Murkin and M. Gehringer, J. Med. Chem., 2024, 67, 14693–14696 CrossRef CAS PubMed.
- R. A. Copeland, Evaluation of enzyme inhibitors in drug discovery: a guide for medicinal chemists and pharmacologists, John Wiley & Sons, 2013 Search PubMed.
- L. K. Mader and J. W. Keillor, ACS Med. Chem. Lett., 2024, 15, 731–738 CrossRef CAS PubMed.
- X. Zhai, R. A. Ward, P. Doig and A. Argyrou, Biochemistry, 2020, 59, 1428–1441 CrossRef CAS PubMed.
- B.-F. Krippendorff, R. Neuhaus, P. Lienau, A. Reichel and W. Huisinga, SLAS Discovery, 2009, 14, 913–923 CrossRef CAS PubMed.
- M. J. Eck and C.-H. Yun, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 559–566 CrossRef CAS PubMed.
- Y. Yosaatmadja, S. Silva, J. M. Dickson, A. V. Patterson, J. B. Smaill, J. U. Flanagan, M. J. McKeage and C. J. Squire, J. Struct. Biol., 2015, 192, 539–544 CrossRef CAS PubMed.
- X.-E. Yan, P. Ayaz, S.-J. Zhu, P. Zhao, L. Liang, C. H. Zhang, Y.-C. Wu, J.-L. Li, H. G. Choi, X. Huang, Y. Shan, D. E. Shaw and C.-H. Yun, J. Med. Chem., 2020, 63, 8502–8511 CrossRef CAS PubMed.
- D. J. H. De Clercq, D. E. Heppner, C. To, J. Jang, E. Park, C.-H. Yun, M. Mushajiang, B. H. Shin, T. W. Gero, D. A. Scott, P. A. Jänne, M. J. Eck and N. S. Gray, ACS Med. Chem. Lett., 2019, 10, 1549–1553 CrossRef CAS PubMed.
- F. Wittlinger, D. E. Heppner, C. To, M. Günther, B. H. Shin, J. K. Rana, A. M. Schmoker, T. S. Beyett, L. M. Berger, B.-T. Berger, N. Bauer, J. D. Vasta, C. R. Corona, M. B. Robers, S. Knapp, P. A. Jänne, M. J. Eck and S. A. Laufer, J. Med. Chem., 2022, 65, 1370–1383 CrossRef CAS PubMed.
- F. Wittlinger, B. C. Ogboo, E. Shevchenko, T. Damghani, C. D. Pham, I. K. Schaeffner, B. T. Oligny, S. P. Chitnis, T. S. Beyett, A. Rasch, B. Buckley, D. A. Urul, T. Shaurova, E. W. May, E. M. Schaefer, M. J. Eck, P. A. Hershberger, A. Poso, S. A. Laufer and D. E. Heppner, Commun. Chem., 2024, 7, 38 CrossRef CAS PubMed.
- K. Aertgeerts, R. Skene, J. Yano, B.-C. Sang, H. Zou, G. Snell, A. Jennings, K. Iwamoto, N. Habuka, A. Hirokawa, T. Ishikawa, T. Tanaka, H. Miki, Y. Ohta and S. Sogabe, J. Biol. Chem., 2011, 286, 18756–18765 CrossRef CAS PubMed.
- C.-H. Yun, T. J. Boggon, Y. Li, M. S. Woo, H. Greulich, M. Meyerson and M. J. Eck, Cancer Cell, 2007, 11, 217–227 CrossRef CAS PubMed.
- B. C. Cho, J.-Y. Han, S.-W. Kim, K. H. Lee, E. K. Cho, Y.-G. Lee, D.-W. Kim, J.-H. Kim, G.-W. Lee and J.-S. Lee, J. Thorac. Oncol., 2022, 17, 558–567 CrossRef CAS PubMed.
- B. C. Cho, M.-J. Ahn, J. H. Kang, R. A. Soo, T. Reungwetwattana, J. C.-H. Yang, I. Cicin, D.-W. Kim, Y.-L. Wu and S. Lu, J. Clin. Oncol., 2023, 41, 4208–4217 CrossRef CAS PubMed.
- Key Reference: B. C. Cho, D.-W. Kim, A. I. Spira, J. E. Gomez, E. B. Haura, S.-W. Kim, R. E. Sanborn, E. K. Cho, K. H. Lee, A. Minchom, J.-S. Lee, J.-Y. Han, M. Nagasaka, J. K. Sabari, S.-H. I. Ou, P. Lorenzini, J. M. Bauml, J. C. Curtin, A. Roshak, G. Gao, J. Xie, M. Thayu, R. E. Knoblauch and K. Park, Nat. Med., 2023, 29, 2577–2585 CrossRef CAS PubMed.
- C. A. Shu, K. Goto, B. C. Cho, F. Griesinger, J. C.-H. Yang, E. Felip, J. Xie, J. Chen, J. Mahoney and M. Thayu, J. Clin. Oncol., 2021, 39 DOI:10.1200/JCO.2021.39.15_suppl.TPS9132.
- B. C. Cho, E. Felip, H. Hayashi, M. Thomas, S. Lu, B. Besse, T. Sun, M. Martinez, S. N. Sethi, S. M. Shreeve and A. I. Spira, Future Oncol., 2022, 18, 639–647 CrossRef CAS PubMed.
- A. Passaro, J. Wang, Y. Wang, S. H. Lee, B. Melosky, J. Y. Shih, J. Wang, K. Azuma, O. Juan-Vidal, M. Cobo, E. Felip, N. Girard, A. B. Cortot, R. Califano, F. Cappuzzo, S. Owen, S. Popat, J. L. Tan, J. Salinas, P. Tomasini, R. D. Gentzler, W. N. William Jr, K. L. Reckamp, T. Takahashi, S. Ganguly, D. M. Kowalski, A. Bearz, M. MacKean, P. Barala, A. B. Bourla, A. Girvin, J. Greger, D. Millington, M. Withelder, J. Xie, T. Sun, S. Shah, B. Diorio, R. E. Knoblauch, J. M. Bauml, R. G. Campelo and B. C. Cho, Ann. Oncol., 2024, 35, 77–90 CrossRef CAS PubMed.
- N. B. Leighl, H. Akamatsu, S. M. Lim, Y. Cheng, A. R. Minchom, M. E. Marmarelis, R. E. Sanborn, J. C.-H. Yang, B. Liu, T. John, B. Massutí, A. I. Spira, S.-H. Lee, J. Wang, J. Li, C. Liu, S. Novello, M. Kondo, M. Tamiya, E. Korbenfeld, M. Moskovitz, J.-Y. Han, M. Alexander, R. Joshi, E. Felip, P. J. Voon, P. Danchaivijitr, P.-C. Hsu, F. J. S. M. Cruz, T. Wehler, L. Greillier, E. Teixeira, D. Nguyen, J. K. Sabari, A. Qin, D. Kowalski, M. A. N. Şendur, J. Xie, D. Ghosh, A. Alhadab, N. Haddish-Berhane, P. L. Clemens, P. Lorenzini, R. B. Verheijen, M. Gamil, J. M. Bauml, M. Baig, A. Passaro, H. Akamatsu, M. Alexander, A. Bleckmann, F. Cappuzzo, Y. Cheng, B. C. Cho, T. Cil, A. Cortot, P. Danchaivijitr, T.-O. Emde, D. Erdem, E. Felip, F. Estevinho, M. L. Ferreira, F. F. D. Silva, M. D. R. G. Campelo, L. Greillier, A. Greystoke, J.-Y. Han, P.-C. Hsu, J.-Y. Hung, M. Ji, T. John, R. Joshi, Y.-C. Kim, M. Kondo, E. Korbenfeld, D. Kowalski, S.-H. Lee, N. Leighl, J. Li, S.-H. Lin, B. Liu, C. Liu, J. S.-H. Low, M. E. Marmarelis, B. Massutí, A. R. Minchom, S. Moore, M. Moskovitz, A. Nagrial, D. Nguyen, S. Novello, Y. Ohe, M. Özgüroğlu, O. Ozyilkan, A. Passaro, N. Peled, N. Prasongsook, A. Qin, E. F. Ramos, J. K. Sabari, J. Salinas, R. E. Sanborn, M. A. N. Sendur, F. J. S. M. Cruz, A. I. Spira, T. Suksombooncharoen, M. Tamiya, J. L. Tan, E. Teixeira, R. Tota, D. Urban, A. Vergnenègre, P. J. Voon, V. Wainsztein, J. Wang, T. Wehler, J. C.-H. Yang, H. Yoshioka, A. Zer, Y. Zhao and B. Zurawski, J. Clin. Oncol., 2024, 42, 3593–3605 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |