
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNano-enabled dynamically responsive living acellular hydrogels†
Roya
Koshani
 a,
Sina
Kheirabadi
a and
Amir
Sheikhi
*abcde
a,
Sina
Kheirabadi
a and
Amir
Sheikhi
*abcde
aDepartment of Chemical Engineering, The Pennsylvania State University, University Park, PA 16802, USA. E-mail: sheikhi@psu.edu
bDepartment of Biomedical Engineering, The Pennsylvania State University, University Park, PA 16802, USA
cDepartment of Chemistry, The Pennsylvania State University, University Park, PA 16802, USA
dHuck Institutes of the Life Sciences, The Pennsylvania State University, University Park, PA 16802, USA
eDepartment of Neurosurgery, College of Medicine, The Pennsylvania State University, Hershey, PA 17033, USA
First published on 25th October 2024
Abstract
As a key building block of mammalian tissues, extracellular matrices (ECMs) stiffen under shear deformation and undergo cell-imparted healing after damage, features that regulate cell fate, communication, and survival. The shear-stiffening behavior is attributed to semi-flexible biopolymeric ECM networks. Inspired by the mechanical behavior of ECMs, we develop acellular nanocomposite living hydrogels (LivGels), comprising network-forming biopolymers and anisotropic hairy nanoparticle linkers that mimic the dynamic mechanical properties of living counterparts. We show that a bifunctional dynamic linker nanoparticle (nLinker), bearing semi-flexible aldehyde- and carboxylate-modified cellulose chains attached to rigid cellulose nanocrystals converts bulk hydrogels to ECM-like analogues via ionic and dynamic covalent hydrazone bonds. The nLinker not only enables the manipulation of nonlinear mechanics and stiffness within the biological window, but also imparts self-healing to the LivGels. This work is a step forward in designing living acellular soft materials with complex dynamic properties using bio-based nanotechnology.
New conceptsIt is non-trivial to convert conventional, static hydrogels to those that mimic the dynamic properties of extracellular matrices (ECMs) and tissues. The recent decade has witnessed the quest to engineer “living materials” that mimic the characteristic features of biological systems, such as the strain-stiffening behavior, anisotropy, and self-healing, driven by the need for innovations in biomedicine and beyond. Acellular living materials are an emerging class of engineered living materials, aiming to replicate the complexity of biological materials without using any living cells. Our research presents a new concept in converting static gels to those that partially mimic the ECM and tissue properties via anisotropic hairy nanoparticle linkers (nLinker). We develop novel acellular living hydrogels (LivGels) that are engineered using the nLinker and a biopolymer via tailoring polymer–polymer and polymer–nLinker dynamic interactions. Unlike conventional hydrogels, the LivGels, inspired by the structural organization of ECM networks and the healing potential of cells, enable shear-stiffening and self-healing traits. This conceptually novel design allows precise control of the nonlinear mechanics and stiffness of self-healing LivGels. The LivGels may open a new avenue for developing dynamic soft materials for regenerative engineering, tissue and disease models, organs-on-a-chip platforms, additive manufacturing, soft robotics, and active materials. |
1. Introduction
Engineering dynamically responsive hydrogels that mimic the mechanical and biological properties of tissues1–3 may enable living soft materials. The majority of native mammalian tissues comprise non-living extracellular matrices (ECMs) and living cell ensembles. The ECM dynamically responds to external physical stimuli and stiffens under shear.4,5 Accordingly, nonlinear shear-stiffening mechanics are crucial for tissue structural support and cell functions, such as adhesion, migration, and differentiation.6Tuning biopolymer concentration has been the main approach to tailor the nonlinear mechanics of ECM-mimetic soft materials,7–10 which may inevitably alter other hydrogel properties, including mechanical strength, swelling behavior, degradation rate, and biocompatibility. Additionally, synthetic hydrogels that mimic the mechanical properties of ECMs are undermined by the concerns associated with biocompatibility, immune response, and toxic degradation byproducts.11 As a mechanical–biological trait, self-healing is also highly desirable in fabricating living hydrogels to enable autonomous morphological and mechanical restorations following reoccurring deformation and defects.10
Developing multifunctional living acellular hydrogels with ECM-like mechanics and self-healing properties remains an ongoing challenge. Most living tissues have hierarchical and anisotropic structures, partially arising from the multiscale three-dimensional (3D) organization of ECM constituents.12,13 The isotropy of bulk polymeric hydrogels limits their ability to faithfully replicate the structural and mechanical features of ECMs. To address this shortcoming, nanoparticles have been included in 3D polymeric networks, yielding anisotropic nanocomposite hydrogels (NCH).14,15 Besides using anisotropic nanoparticles, nanoparticle–polymer crosslinking has also been tailored via non-covalent and/or covalent bond formation. Polymer–nanoparticle inclusions have widely been investigated in terms of microscale network dynamics and macroscale mechanical and rheological properties;16–18 however, a limited number of NCH with tunable ECM-mimetic mechanical characteristics, particularly strain-stiffening, exist. As an example, NCH with strain-stiffening and self-healing properties have been engineered via incorporating tannic acid-coated cellulose nanocrystals (CNC) into synthetic poly(vinyl alcohol)–borax dynamic networks.19 Other kinds of NCH with tunable strain-stiffening properties have been developed via manipulating the nature of polymer–nanoparticle crosslinks and density, albeit using synthetic polyisocyanide and anisotropic iron oxide nanorods.20,21
To the best of our knowledge, there has been no report of naturally derived NCH with anisotropic particles that mimic the dynamic characteristics of native ECM (i.e., strain-stiffening) or tissues (i.e., self-healing) and enable the control of these properties. Hairy CNC (HCNC) are a newly emerged member of the nanocellulose family, comprising a rod-like crystalline body with disordered cellulose chains (hairs) at both ends.22,23 The hairs contribute to the unique anisotropic surface chemistry and architecture. We hypothesize that chemically tailored HCNC, bearing a combination of carboxylate and aldehyde moieties on the hairs, can link polymer chains in NCH matrices via reversible ionic and covalent bonds, resulting in controlled strain-stiffening and self-healing properties. These bifunctional nanoparticle linkers are called nLinker.
Here, we aim to develop acellular nanocomposite living hydrogels (LivGels) with nonlinear mechanics and self-healing properties. Dihydrazide-modified alginate (DH-ALG) is synthesized as the matrix biopolymer to provide two types of active sites, i.e., carboxylate groups and hydrazide for ionic and dynamic covalent bond formation, respectively. To impart dynamic properties to bulk alginate hydrogels, we develop an anisotropic nLinker, bearing aldehyde and carboxylate groups. The network is engineered via varying the nLinker concentration and binding mechanisms (dynamic covalent bonding and/or ionic crosslinking). Structure–property relationships in the NCH are investigated to develop a phase diagram, identifying NCH compositions that yield LivGels.
2. Experimental section
2.1. Materials
Bleached, delignified softwood kraft pulp sheets were provided by Resolute Forest Products Inc., Canada, and used as the starting material for the nLinker synthesis. Medium viscosity (MV) alginic acid sodium salt from brown algae was obtained from MilliporeSigma, USA. Sodium (meta)periodate (NaIO4, >99%), sodium chlorite (NaClO2, 80%), sodium chloride (NaCl, >99.5%), hydrogen peroxide (H2O2, 30 wt%), sodium hydroxide (NaOH, ACS reagent, >97%), hydrochloric acid (HCl, ACS reagent, 37%), ethylene glycol (Reagent plus, >99%), hydroxylamine-hydrochloride (NH2OH·HCl, Reagent plus, 99%), adipic acid dihydrazide (ADH, NH2NHCO(CH2)4CONHNH2, ≥98%), calcium chloride dihydride (CaCl2·2H2O, ≥99%), poly-L-lysin (PLL, 0.1 w/v% in water), Whatman® filter papers (Grade 1, circles), and mineral oil (heavy) were purchased from MilliporeSigma, USA. AFM stainless steel disks and mica sheets (V1 grade) were obtained from SPI supplies (USA) and Ted Pella Inc. (USA), respectively. Deuterium oxide (D2O, 99.9%) was supplied by Cambridge Isotope Laboratories, Inc., USA. Anhydrous ethanol (200 proof) was purchased from KOPTEC, USA. All material synthesis and experiments were conducted using either ultrapure Milli-Q water (resistivity ∼18.2 MΩ cm at T = 25 °C) or deionized (DI) water, as specified.2.2. nLinker synthesis
The nLinker was prepared according to our previously established method for HCNC synthesis with minor modifications.24,25 The softwood pulp sheets (5 g) were torn into thin pieces with approximate dimensions of 2 cm × 2 cm, and pulp fragments were soaked in DI water (500 mL) while stirring with an overhead stirrer (SH-II-6C, Faithful Instrument CO., China) overnight. Afterwards, water was removed via vacuum filtration using a nylon cloth (pore size: 20 μm), and the collected wet pulps were suspended in a NaCl solution (325 mL, 1 M). Following 5 min mixing and thoroughly wrapping the beaker with aluminium foil, NaIO4 (6.60 g) was added to selectively oxidize the vicinal diols of cellulose to dialdehyde groups. The reaction mixture was stirred at ambient temperature for 42 h. Once completed, ethylene glycol (5 mL) was added and stirred for 10 min to deactivate unreacted NaIO4. The oxidized pulps (dialdehyde modified cellulose, DAMC) were then vacuum filtered and rinsed with DI water at least 5 times (250 mL each time) to remove residual chemicals and byproducts.To convert half of the DAMC aldehyde groups to carboxylate groups and isolate nLinker, DAMC was further oxidized with NaClO2. To this end, DAMC fibrils were suspended in DI water (250 mL, including the water in the wet DAMC), followed by adding NaClO2 (4.22 g) and NaCl (14.12 g). H2O2 (4.22 g) was then added dropwise within 2 min after which the mixture was stirred at 100 rpm for 12 h. The pH was adjusted to 5.0 ± 0.2 using a NaOH solution (0.5 M) during the reaction until pH remained almost constant at 5. The bifunctional fibrils were collected via centrifugation at 8000 × g for 20 min and rinsed with an ethanol solution in DI water (70 v/v%, 100 mL) four times. The fibrils were suspended in DI water (500 mL) and heated at 80 °C for 2 h, followed by centrifugation at 8000 × g for 20 min to remove the unfibrillated cellulose. The nLinker particles in the supernatant were precipitated via adding a nonsolvent (ethanol, volume ∼1.5 times the supernatant volume), collected by centrifugation at 8000 × g for 20 min, and purified via dialysis (Spectra/Por membranes, MW cutoff = 8–10 kDa) against DI water for 1 day. The nLinker concentration was increased to 5 wt% via evaporation at 40 °C under constant airflow for further use. The concentration was determined by oven-drying a known volume of nLinker dispersion at 60 °C, followed by weighing.
2.3. Matrix biopolymer (DH-ALG) synthesis
DH-ALG was synthesized via successive periodate oxidation and Schiff base reactions. A periodate-mediated oxidation reaction was conducted to functionalize alginate (ALG) with dialdehyde groups, yielding dialdehyde-modified alginate (DA-ALG, a reactive intermediate).26 DA-ALG synthesis was started by dispersing MV sodium ALG (5 g) in ethanol (50 mL), followed by adding DI water (200 mL). Ethanol, as a non-solvent, breaks up agglomerates, resulting in a homogenous distribution of ALG in water.27 Once fully dissolved, NaIO4 (5.7 g) was added to the ALG solution in an aluminium foil-wrapped beaker while stirring by the overhead stirrer (speed = 100 rpm) at room temperature. Given that NaIO4 is a photosensitive oxidizer, the reaction was performed in the dark.28 After 1 h, the reaction was quenched by adding ethylene glycol (5 mL) and stirring for 10 min. To purify the resulting DA-ALG, NaCl (5 g) was added to the solution to screen the negatively charged carboxylate groups, facilitating precipitation. The DA-ALG was subsequently rinsed with an ethanol solution in DI water (70 v/v%, 800 mL each time) three times, and the precipitated DA-ALG was dissolved in DI water (250 mL). Further purification was conducted via dialysis (Spectra/Por membranes, MW cutoff = 8–10 kDa) against DI water for 1 day. To prevent microbial contamination, DA-ALG was stored at 4 °C until further use.Next, the dialdehyde groups were reacted with hydrazide via a Schiff base reaction to form hydrazone bonds, yielding DH-ALG.29 ADH (0.9 g, ∼5 mmol) was dissolved in a DA-ALG solution (1 wt%, 100 mL, ∼5 mmol aldehyde), and a Schiff base reaction was carried out for 5 h at room temperature. The ADH amount was calculated based on a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of the aldehyde group:ADH. The resulting DH-ALG solution was then dialyzed (Spectra/Por membranes, MW cutoff = 8–10 kDa) against DI water for 12 h. DH-ALG was stored at 4 °C and used within 3 days to minimize hydrazone bond hydrolysis.30
:1 molar ratio of the aldehyde group:ADH. The resulting DH-ALG solution was then dialyzed (Spectra/Por membranes, MW cutoff = 8–10 kDa) against DI water for 12 h. DH-ALG was stored at 4 °C and used within 3 days to minimize hydrazone bond hydrolysis.30
2.4. NCH preparation
NCH were formed via (i) nLinker-mediated dynamic covalent crosslinking of DH-ALG or (ii) simultaneous nLinker-mediated and ionic crosslinking of DH-ALG. The nLinker-crosslinked NCH (1 mL) were prepared via mixing a DH-ALG (5 wt%) solution, the nLinker (5 wt%) dispersion, and Milli-Q water at varying ratios to yield a constant final DH-ALG concertation of 2 wt% and nLinker concentrations ranging from 1.25 to 2.5 wt%. The biopolymer–nLinker NCH were mixed using a positive displacement pipette (Microman E M1000E, Gilson, OH, USA) for ∼30 s and maintained at room temperature for 5 min prior to any characterization. To prepare the nLinker-ionically-crosslinked NCH, the nLinker (5 wt%, 10 mL) dispersion was mixed with 0.0875 mL, 0.175 mL, or 0.350 mL of a CaCl2 solution (5 M) to obtain a carboxylate:Ca2+ molar ratio of 1:0.5, 1:1, or 1:2, respectively, resulting in 4.95, 4.91, or 4.83 wt% of final nLinker concentrations. After 5 min of incubation, Ca2+-loaded nLinker dispersions were mixed with the DH-ALG solutions (5 wt%) to reach a total volume of 1 mL. The final DH-ALG concentration was maintained constant at 2 wt%, and the final nLinker concentration was varied (1.25, 1.75, 2, or 2.5 wt%) by adding Milli-Q water, similar to those in the nLinker-crosslinked NCH. Note that NCH with a 1:0.5, 1:1, or 1:2 molar ratio of carboxylate:Ca2+ are referred to as 9, 18, or 36 mM Ca2+, respectively. For the NCH preparation, Milli-Q water was used to dilute the nLinker dispersion when required.
2.5. Materials characterization
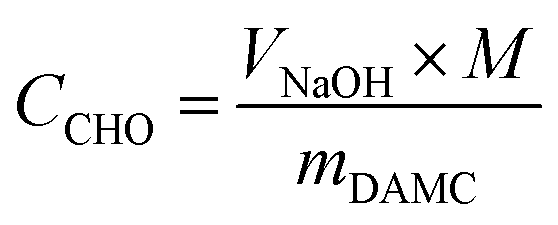 | (1) |
To measure the carboxylate group content, a known amount of nLinker or DH-ALG (20 mg dry mass) and NaCl solution (2 mL, 20 mM) were added to 100 mL of Milli-Q water. The pH of nLinker dispersion or DH-ALG solution was then decreased to 3.2 using a HCl solution (0.1 M). The dispersion was titrated by a NaOH solution (10 mM) up to pH ∼ 11 at a titration rate of 0.1 mL min−1. The carboxylate group content was calculated based on the volume of NaOH required to neutralize the weak acid (i.e., carboxylate groups) using eqn (2).
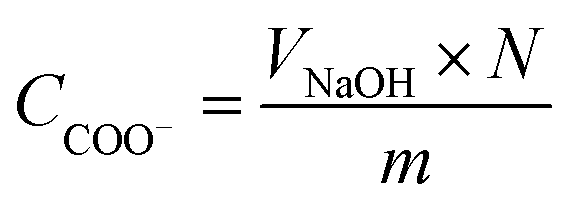 | (2) |
To determine ζ-potential, the electrophoretic mobility of nLinker was measured using a Nano ZS Zetasizer (Malvern instrument, UK) at room temperature and pH = 6.5. Before initiating the measurements, the nLinker dispersion was diluted to 0.1% w/v using Milli-Q water, followed by pipetting a desired volume (900 μL) into disposable folded capillary cells (Malvern, UK). To apply the effect of nLinker rod shape to the ζ-potential value, Oshima's mobility expression was used.34 Since the κa ∼ 1 (κ is the Debye–Hückel parameter, and a is the rod-like nLinker width, ∼5 nm, obtained from the AFM image analysis), the Henry's function f (κa) ∼ 0.5,35 and the following equation was used to calculate the ζ-potential using the electrophoretic mobility. Note that the ionic strength was calculated by considering the counter ions (Na+) of nLinker carboxylate groups.
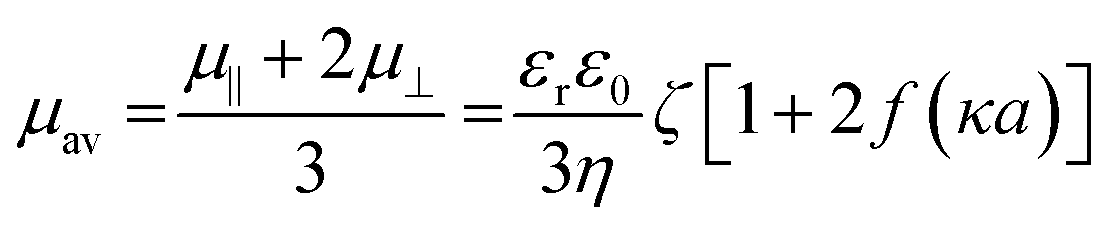 | (3) |
2.6. Rheological analyses
Rheological properties were measured using an AR-G2 rheometer (TA Instruments, DE, USA), equipped with a parallel plate geometry with an upper plate diameter of 20 mm and a truncation gap of 1000 μm. Freshly prepared NCH were sandwiched between two parallel plates. After trimming the excess hydrogel, the air-exposed area was covered with a mineral oil to prevent water evaporation from the gel during the experiments. To assess the strain-induced stiffening of NCH, an oscillatory amplitude sweep test was conducted at a constant frequency of 1 rad s−1 while varying the strain from 0.1 to 200%. To investigate the self-healing of NCH, recovery tests were conducted, wherein the dynamic moduli (i.e., storage modulus, G′, and loss modulus, G′′) were measured by applying alternating strain of 1% and 500% at time intervals of 100 s and a constant oscillation frequency (1 rad s−1). The time sweep test was performed on freshly prepared NCH at a constant frequency of 1 rad s−1 and strain of 0.1% for 15 min. Temperature was maintained at ∼20 °C in all measurements.2.7. Statistical analyses
Data were reported as the average of three independent samples (n = 3) ± standard deviation (SD), unless otherwise specified. GraphPad Prism (Version 9.4.1, MA, USA) was used for all statistical analyses. One-way (Fig. 4b, c and Fig. S4a, ESI†) or two-way (Fig. 4g, h and Fig. S4b, ESI†) analysis of variance (ANOVA), followed by the Tukey post hoc test was conducted to determine the statistical significance. The p-values <0.05 were considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).3. Results and discussion
3.1. LivGel design
LivGels are dynamically responsive bio-based NCH inspired by the mechanical responsiveness of ECM, specifically nonlinear strain-stiffening, as well as the self-healing capability of tissues. Fig. 1 presents the design rationale for LivGels, their building blocks, and strategies to control the nonlinear mechanical properties while imparting other functionalities, such as self-healing. Fig. 1a shows examples of human soft tissues and their main components, including ECM and cell ensembles, which have strain-stiffening and self-healing properties, respectively. LivGels are prepared via incorporating the bifunctional nLinker in reactive DH-ALG, prepared from semi-flexible ALG, as shown in Fig. 1b. ALG, the matrix biopolymer of LivGels, has a semi-flexible backbone with carboxylate groups and vicinal diols in repeating units,37,38 allowing for NaIO4-mediated selective oxidation and further functionalization with ADH (i.e., DH-ALG formation). The nLinker contains carboxylate and aldehyde groups, which are able to undergo Ca2+-mediated ionic crosslinking and Schiff base formation with the carboxylate and hydrazide groups of DH-ALG, respectively. The nLinker may enable not only dynamic covalent bonding (i.e., hydrazone bonds) with DH-ALG through a Schiff base reaction but also nanoparticle-polymer assembly via the Ca2+-mediated bridging of carboxylate groups in the DH-ALG matrix.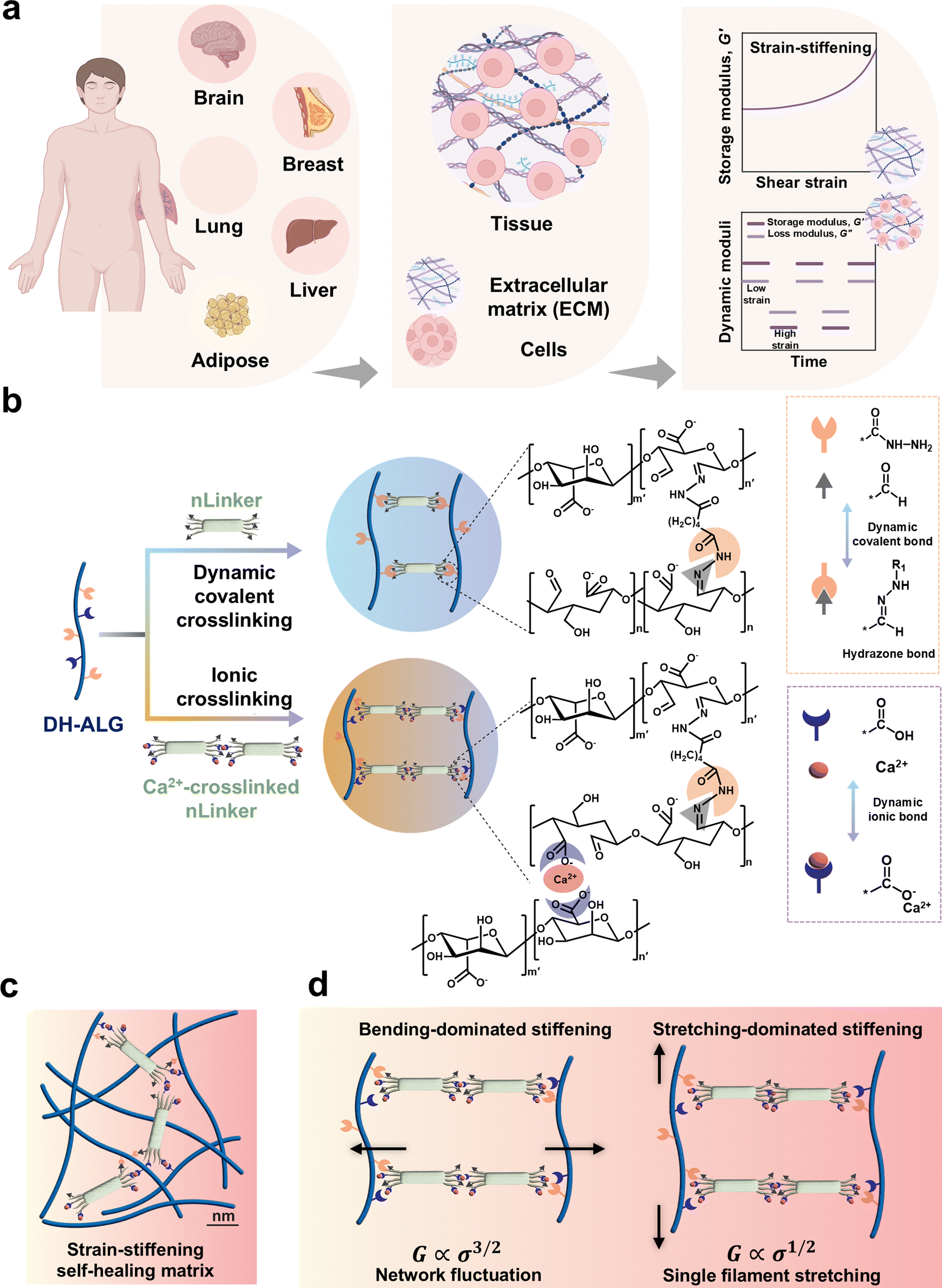 | ||
| Fig. 1 Design of dynamically responsive, self-healing LivGels. (a) Examples of human soft tissues, consisting of shear-stiffening ECM and cell ensembles conferring self-healing. (b) Materials, engineering pathways, and physicochemical interactions, designed to enable dynamically responsive LivGels. DH-ALG, a semi-flexible bifunctional biopolymer undergoes ionic crosslinking and dynamic covalent hydrazone bond formation with bifunctional nLinkers. (c) Proposed mechanisms governing the large deformation-induced stiffening of crosslinked LivGels, involving (i) pulling out of stress-path undulations on polymer–polymer and polymer–nLinker networks, which is bending dominated, and (ii) stress path reorientation of DH-ALG chains, which is dominated by stretching. Note that Ca2+-mediated DH-ALG/DH-ALG crosslinks are not shown in panels (b)–(d). | ||
The chemical modification of ALG is schematically shown in Fig. S1a (ESI†). The vicinal diols are converted to aldehyde groups via NaIO4-mediated oxidation, yielding DA-ALG (Fig. S1a-i, ESI†). Aldehyde groups are then converted to hydrazide via a Schiff base reaction, yielding DH-ALG (Fig. S1a-ii, ESI†). Hydrazides are commonly used for developing dynamic covalent bonds with aldehyde groups.39,40 ALG chemical modification is assessed via1H NMR spectroscopy, and the spectra of ALG, DA-ALG, and DH-ALG are shown in Fig. S1b (ESI†). In the DA-ALG spectrum, two discrete peaks at the chemical shifts of ∼5.35 ppm and 5.63 ppm were associated with the aldehyde groups in their hemiacetal forms.41 Aldehyde groups of DA-ALG (5.0 ± 0.2 mmol g−1, Fig. S1c, ESI†), are reacted with hydrazide to form DH-ALG. In the DH-ALG spectrum, peak (a) ranging from 7 to 8 ppm corresponds to the –NH–, originated from the Schiff base reaction of DA-ALG dialdehyde groups with the hydrazide. In addition, peaks (b) and (c) at 1.5 ppm and 2.25 ppm, respectively, are attributed to the –CH2– groups.42 Fig. S1d (ESI†) presents the content of remaining DH-ALG aldehyde groups after the reaction with hydrazide, which was only 0.4 ± 0.2 mmol g−1. Additionally, the carboxylate group content of DH-ALG is 3.2 ± 0.3 mmol g−1 (Fig. S1e, ESI†), which agrees with the previously reported values (e.g., 3 mmol g−1) for alginic acid.43
The schematic of DH-ALG/nLinker networks within the LivGel matrix is presented in Fig. 1c, indicating nLinker/nLinker and DH-ALG/nLinker crosslinks. Fig. 1d shows proposed mechanisms governing the strain-stiffening behavior of these crosslinked networks. In silico studies on random networks comprising crosslinked biopolymer chains have shown that a large-strain response is followed by the development of a stress path for axially stressed fibers and crosslinks.44 Strain-stiffening in the LivGels may result from two mechanisms: (i) pulling out of stress-path undulations on DH-ALG/DH-ALG and DH-ALG/nLinker conjunctions and (ii) the reorientation of stress path on DH-ALG. The pulling out of stress-path undulations is bending-dominated in which case shear modulus (G) is correlated with stress (σ) to the power of 3/2, mainly resulting from the entropic behavior of biopolymers. When the biopolymer chains are crosslinked within a network, the interplay between the entropic behavior of chains and the constraints imposed by the crosslinks (i.e., network fluctuations) may lead to nonlinear mechanical behaviors. As σ increases, the polymer network becomes stiffer because of the entropic effects of biopolymer chains. The reorientation of the stress path is stretch dominated in which case G is correlated with σ to the power of 1/2, originating from single filament stretching.44
3.2. nLinker synthesis and characterization
The nLinker, an integral component of LivGels responsible for imparting strain-stiffening and self-healing properties, was synthesized and characterized in terms of functional groups (specifically aldehyde and carboxylate groups), particle size, and morphology. Fig. 2a shows nLinker synthesis steps, involving the oxidation of cellulose fibrils and DAMC. Via the controlled partial oxidation of cellulose fibrils using NaIO4, DAMC was yielded and used as an intermediate precursor for nLinker synthesis. Fig. S2a (ESI†) shows a representative pH titration curve for DAMC fibrils. The NaOH amount required to neutralize the HCl released from the NH2OH·HCl-aldehyde reaction, is used to measure the aldehyde content (6.8 ± 0.3 mmol g−1).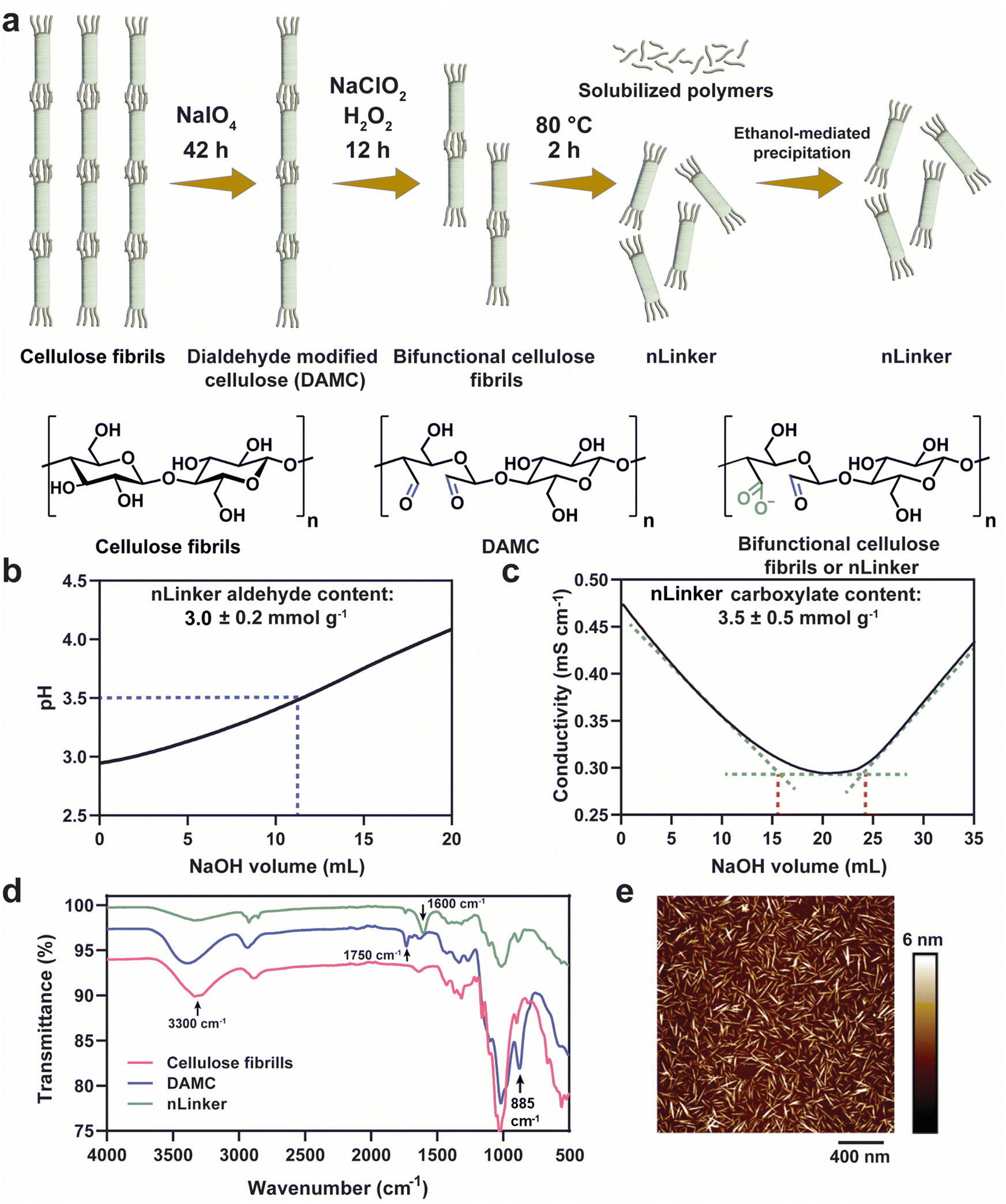 | ||
| Fig. 2 nLinker synthesis and characterization. (a) Schematic of nLinker synthesis via successive periodate-chlorite oxidation reactions, as well as the corresponding changes in cellulose chemical structure. (b) A representative aldehyde titration curve of nLinker. (c) A representative carboxylate titration curve of nLinker using a strong base (10 mM NaOH). (d) ATR-FTIR spectra of cellulose fibrils, DAMC, and nLinker. (e) An AFM image of the nLinker. | ||
After subjecting DAMC to a controlled chlorite oxidation reaction, whereby almost half of the aldehyde groups were transformed to carboxylate groups, the bifunctional nLinker was isolated following successive thermal treatment and ethanol-mediated precipitation. Fig. 2b and c show the representative titration curves of nLinker, bearing 3.0 ± 0.2 mmol g−1 aldehyde and 3.5 ± 0.5 mmol g−1 carboxylate groups, respectively. The coexistence of carboxylate and aldehyde groups enables concurrent physical (ionic) and chemical (dynamic covalent) bond formation with DH-ALG.
Fig. 2d shows the ATR-FTIR spectra of cellulose, DAMC fibrils, and the nLinker. In the DAMC spectrum, an increased intensity of the peak at ∼885 cm−1 compared with cellulose was correlated to the hemiacetal linkages, arising from the interactions of aldehyde groups with the neighboring hydroxyl groups.31 The small peak at ∼1750 cm−1 and broad peak at ∼3300 cm−1 were assigned to the stretching vibrations of carbonyl and hydroxyl groups, respectively. In the nLinker spectrum, the aldehyde and carboxylate groups were confirmed via the stretching vibrations of carbonyl groups at ∼1750 cm−1 and ∼1600 cm−1, respectively.
AFM images (see Fig. 2e as an example) showed that nLinker is a rod-shaped nanoparticle with a length of ∼103 ± 24 nm and a width of 5 ± 2 nm. The hydrodynamic equivalent size of nLinker was 132 ± 11 nm at pH ∼ 6 and ionic strength ∼3.5 mM, which was consistent with the previously reported values for bifunctional HCNC (∼108 nm) and trimmed anionic HCNC (∼140 nm, carboxylate content ∼3.4 mmol g−1).45,46 The ζ-potential of nLinker at the same condition was – 31 ± 6 mV, confirming the negatively charged carboxylate groups.
3.3. Mechanical characterization of LivGels
An overview of the strain-stiffening and self-healing properties of common gels in the literature is provided in Fig. 3a. Conventional gels comprising crosslinked, flexible polymers are typically inert to shear strain.47 However, gels made of semiflexible polymers, regardless of their biological or synthetic nature, may be shear-stiffening,7,8 albeit without self-healing. Similar to semiflexible polymer gels, ECM-mimetic analogs have been designed not only to undergo stiffening under strain,1,48 but also to enable other tissue-mimetic behaviors, such as anisotropic mechanics and shear-thinning. We aim to develop all-biopolymer-based LivGels that have both strain-stiffening and self-healing properties and enable the adjustment of stiffness and stiffening response.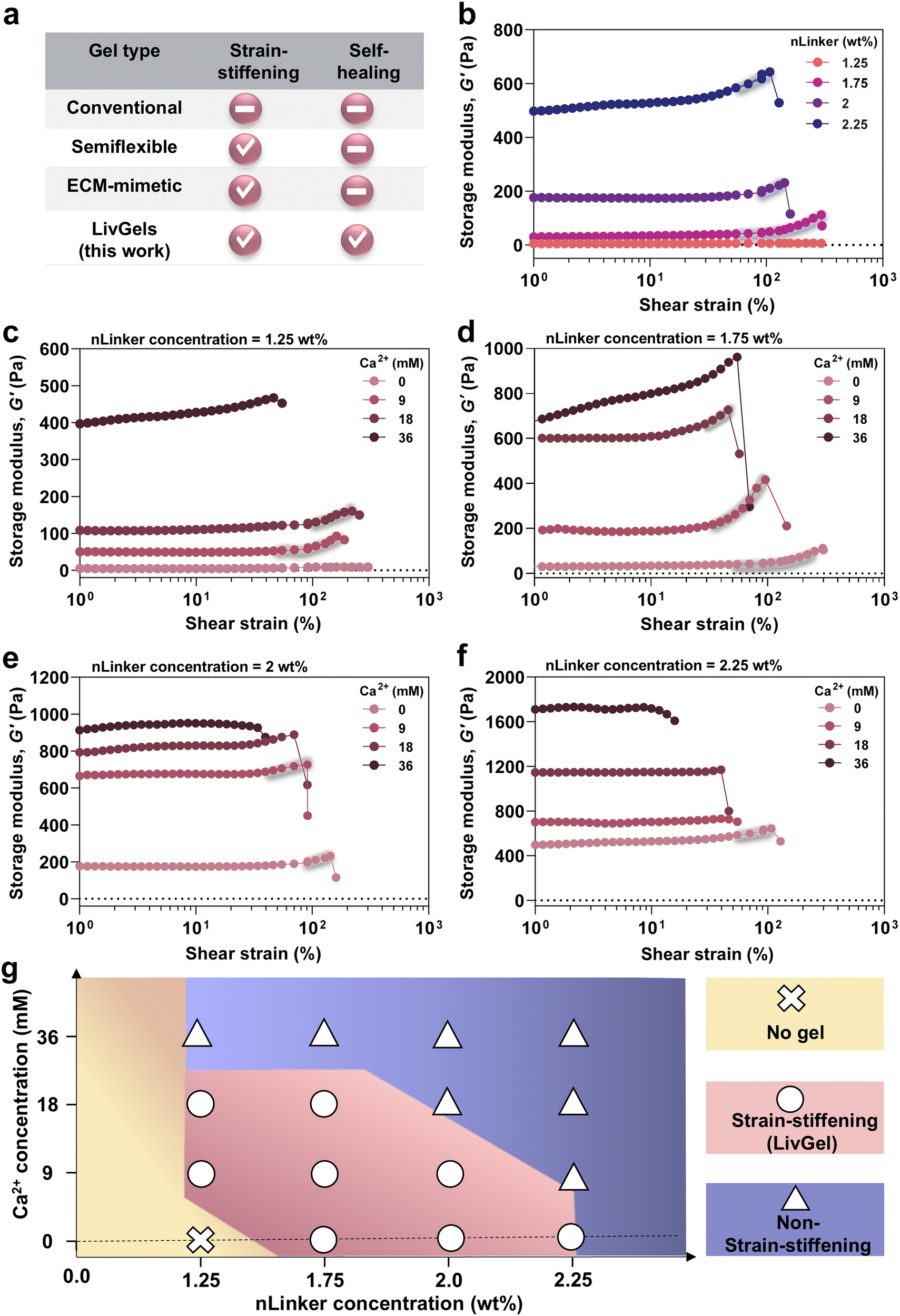 | ||
| Fig. 3 Strain-stiffening properties of LivGels are regulated by the nLinker and Ca2+-concentrations. (a) An overview of strain-stiffening and self-healing properties of conventional gels, comprising flexible chains, networks of semiflexible polymers, or ECM-mimetic biopolymer networks, compared with the LivGels. The LivGels mimic the ECM mechanics and are self-healing. Storage modulus (G′) versus oscillatory shear strain for the NCH, containing (b) varying Ca2+-free nLinker concentrations, or varying Ca2+ concentrations at a constant nLinker concentration ((c) 1.25 wt%, (d) 1.75 wt%, (e) 2 wt%, or (f) 2.25 wt%) and a constant frequency of 1 rad s−1. The matrix biopolymer (DH-ALG) concentration was 2 wt% in all the NCH. (g) The phase diagram of NCH behavior at varying nLinker and Ca2+ concentrations and a constant DH-ALG concentration (2 wt%). The LivGels are the NCH that exhibit a strain-stiffening behavior. | ||
Before conducting mechanical characterization, low-amplitude oscillatory rheology at a constant frequency of 1 rad s−1 and strain of 0.1% was performed on gels ∼5 min after preparation, to examine their rheological behavior over time, as shown in Fig. S3 (ESI†). G′ did not significantly change within 15 min after NCH formation at most of the experimented nLinker or Ca2+ concentrations, indicating no significant evolution in network formation. To investigate the effect of nLinker-mediated dynamic hydrazone bonds on the linear and nonlinear mechanical properties of NCH, oscillatory strain amplitude sweep tests were conducted. Fig. 3b shows the G′ of NCH versus shear strain at varying nLinker concentrations. At an nLinker concentration of 1.75 wt%, 2 wt%, or 2.25 wt%, the NCH underwent stiffening in the strain range of ∼56 ± 28% to 277 ± 54%, ∼66 ± 10% to 188 ± 88%, or ∼54 ± 1% to 95 ± 6% (n = 3), respectively, yielding LivGels. To determine the stiffening range of LivGels, we considered the stiffening onset as the strain point at which a 2% increase in G′ occurred. The G′ of LivGels, containing 1.75 wt%, 2 wt%, or 2.25 wt% nLinker, in the linear viscoelastic region (LVR) at a constant strain of 1%, was 30 ± 15 Pa, 165 ± 19 Pa, or 447 ± 119 Pa (n = 3), respectively, falling in the stiffness range of reconstituted ECM (G′ ∼ 30–1400 Pa). Type-1 collagen (G′ ∼ 30–1000 Pa), fibrin (G′ ∼ 300 Pa), and reconstituted basement membrane matrix (G′ ∼ 60–1400 Pa) are examples of reconstituted ECM.5
In the NCH, the nLinker form dynamic hydrazone bonds via a Schiff base reaction between the nLinker aldehyde and ALG-DH hydrazide groups. By increasing the nLinker concentration from 1.75 wt% to 2.25 wt% at a constant ALG-DH concentration (2 wt%), the G′ increased in the LVE region and NCH became stiffer as a result of the increased number of hydrazone bonds and nLinker-mediated network reinforcement. The mechanical properties of NCH strongly depend on the connectivity between polymer networks and particles.20 The NCH G′ within the LVR was used to estimate the average pore size (ξ) ≈ (G′/kBT)−1/3,49,50 where kB is the Boltzmann constant, and T is the absolute temperature. Fig. S4a (ESI†) shows that the NCH pore size monotonically decreased from 52 ± 6 nm to 21 ± 1 nm when the nLinker concentration increased from 1.75 wt% to 2.25 wt%, implying that the crosslinking density increased. Importantly, when the nLinker concentration was ≤1.25 wt%, no NCH was formed. The molar ratio of nLinker aldehyde groups to DH-ALG (2 wt%) hydrazide in the NCH containing 1.25 wt% nLinker was 1:0.4, which might not provide a sufficient number of dynamic hydrazone bonds for gel formation.
The effects of Ca2+-mediated ionic crosslinking along with the dynamic covalent bonds on the linear and nonlinear mechanics of NCH are presented in Fig. 3c–f. Note that 9 mM, 18 mM, or 36 mM of Ca2+ correspond to the saturation of ∼25%, ∼50%, or ∼100% of nLinker carboxylate groups, respectively. Fig. 3c shows the G′ and stiffening response of NCH, containing 1.25 wt% nLinker and varying Ca2+ concentrations. The nLinker (1.25 wt%) loaded with 9 mM or 18 mM of Ca2+ formed LivGels with a nonlinear stiffening behavior in the strain range of ∼59 ± 9% to 242 ± 33% or ∼91 ± 1% to 188 ± 28% (n = 3), respectively. However, NCH, containing the nLinker loaded with 36 mM of Ca2+ underwent stiffening over the whole strain range until the network was disrupted at a high strain. Increasing the Ca2+ concentration may shrink nLinker polymer chains via the neutralization of negatively charged carboxylates (stoichiometrically, at 36 mM, nLinker COO−:Ca2+ = 2:1 mol mol−1), resulting in an increase in crosslinking density and G′. In another study, the addition of salt to the ALG-chitosan complexes increased G′, which was attributed to the loss of water as the charges were screened by the counterions.51 Datapoints at which the gels undergo a significant drop in G′ have not been presented in our figures. By increasing the Ca2+ concentration from 0 to 36 mM, the NCH stiffness increased because of the additional ionic bond formation within the NCH network. The G′ of NCH, containing nLinker (1.25 wt%) with 9 mM or 18 mM of Ca2+, was 44 ± 6 Pa or 94 ± 20 Pa (within LVR, at strain ∼1%), respectively, mimicking the stiffness of type-1 collagen or reconstituted basement membrane matrix.5
Fig. 3d presents the G′ and stiffening of NCH, containing 1.75 wt% nLinker and varying Ca2+ concentrations. These gels, containing 9 or 18 mM of Ca2+, had a strain-stiffening behavior in the strain range of ∼58 ± 31% to 164 ± 62% or ∼31 ± 8% to 71 ± 26% (n = 3), respectively. The stiffness of LivGels, containing 1.75 wt% nLinker with 9 or 18 mM Ca2+, was 155 ± 43 Pa or 508 ± 138 Pa (at strain ∼1%, LVR), respectively, close to that of the reconstituted ECM.5 Besides increasing NCH stiffness by increasing the Ca2+ concentration (0 to 36 mM) at a constant nLinker concentration, increasing the Ca2+-loaded nLinker from 1.25 wt% to 1.75 wt% resulted in stiffer hydrogels, implying an increase in the crosslinking density.
The strain-stiffening mechanism of LivGels can be explained by stress localization at the hairy junction points where the majority of dynamic and ionic crosslinks exist. When the strain increases in NCH, stress is applied to the hairs. As a result, the nLinker can reorient locally along the strain direction, stretching the entire network and causing a stiffening response. Followed by nLinker reorientation, their extremely stiff crystalline body (Young's modulus ∼ 120–135 GPa52) may be connected to each other via the protruding hairs, forming semiflexible fibrils with finite bending stiffness. At an extremely large strain, some fraction of hairy junction points may break, initiating the yield (failure) process.
Fig. 3e and f show the G′ and stiffening response of NCH, containing 2 wt% and 2.25 wt% nLinker at varying Ca2+ concentrations, respectively. The NCH without Ca2+ (at 2 or 2.25 wt% nLinker) or with 9 mM of Ca2+ (at 2 wt% nLinker) had a stiffening behavior, shown in shades; however, at higher Ca2+ concentrations (18 and 36 mM), the NCH did not have a nonlinear response. At a constant Ca2+ concentration (9 mM), NCH with 2.25 wt% nLinker showed no stiffening, while the analogs with 2 wt% nLinker underwent ∼5% increase in G′, indicating that ionic crosslinking via nLinker further tailors the stiffening behavior. Additionally, the stiffness of LivGels, containing 2 wt% nLinker without Ca2+ (165 ± 19 Pa) or with 9 mM Ca2+ (592 ± 128 Pa), were in the range of reconstituted ECM. When the Ca2+ concentration was significantly high (36 mM), all the NCH containing 1.25 wt% to 2.25 wt% nLinker did not have any nonlinear response.
At any nLinker concentration, Ca2+ increased the NCH stiffness as a result of ionic crosslinking, reducing the nonlinear stiffening capability. At high Ca2+ concentrations (e.g., 18 mM for the NCH containing 2 or 2.25 wt% nLinker and 36 mM for all the NCH), the loss of strain-stiffening behavior in the gels may be a result of increased network connectivity beyond the isostatic threshold, rendering the otherwise semi-flexible networks stiff. This may impair network fluctuations through changes in the nLinker–nLinker or DH-ALG–nLinker connections along the strain direction. Thus, the deformation mechanism is mainly filament stretching (G ∝ σ1/2). In contrast, if the average connectivity is below the isostatic threshold, the network is floppy and the deformation is through changes in the angle between the rods (G ∝ σ3/2), allowing the alignment of rods along the strain direction.9,53,54
Fig. S4b (ESI†) shows the estimated NCH pore size at varying nLinker and Ca2+ concentrations. When the NCH, containing 1.25 wt% nLinker, were ionically crosslinked with 9 mM of Ca2+, the pore size was 43.5 ± 3 nm, which significantly decreased to 34 ± 2 nm at 18 mM of Ca2+ and to 22 ± 1 nm at 36 mM of Ca2+. In the NCH, containing 1.75 wt% nLinker, when the Ca2+ concentration increased from 0 to 9 mM, the pore size significantly decreased from 52 ± 6 nm to 27 ± 1 nm, and the pore size changes were insignificant by increasing the Ca2+ concentration from 18 mM (19 ± 1 nm) to 36 mM (18 ± 1 nm). The evolution of pore size suggests that the maximum crosslinking via ionic bonding in the NCH matrix was obtained at ∼18 mM of Ca2+. Stoichiometrically, 32 mM of Ca2+ is required for the saturation of all the carboxylate groups (3.2 ± 0.3 mmol g−1) of DH-ALG, considering that l mol of Ca2+ binds to 2 mol of COO−. However, not all the nLinker loaded with Ca2+ may be available for ionic interactions with DH-ALG carboxylate groups, likely as a result of Ca2+-mediated internal crosslinking of carboxylates within the nLinker. In the NCH containing 2 wt% nLinker (Fig. S4b, ESI†), the pore size significantly decreased from 28 ± 1 nm (Ca2+ concentration = 0) to 17 ± 1 nm at 9 mM of Ca2+, followed by no significant changes as the Ca2+ concentration was increased up to 36 mM. Similarly, increasing the Ca2+ concentration no longer affected the pore sizes of NCH, containing 2.25 wt% of nLinker. Fig. 3g shows the phase diagram of NCH, including strain-stiffening (LivGels) and non-strain-stiffening gels yielded at varying nLinker and Ca2+ concentrations. At a low (e.g., 1.25 wt%) nLinker concentration, no NCH was formed without Ca2+-mediated ionic crosslinking as a result of insufficient hydrazone bonds. As the Ca2+-free nLinker concentration increased up to 2.25 wt%, LivGels were formed. At any constant nLinker concentration ≤2 wt%, increasing Ca2+ concentration initially yielded LivGels, followed by non-strain-stiffening gels. At 2.25 wt% of nLinker, Ca2+ rendered the NCH non-strain-stiffening. Accordingly, the NCH are converted to LivGels at a specific concentration range of nLinker and Ca2+.
3.4. Quantitative analysis of strain-stiffening behavior in LivGels
To quantify stiffening, differential shear modulus, K′ = ∂σ/∂γ, where σ and γ are shear stress and strain, respectively, was calculated for the NCH, containing varying nLinker and Ca2+ concentrations. Fig. 4a shows K′ versus shear stress at a constant frequency for NCH, containing varying Ca2+-free nLinker concentrations. A linear region at low stress, followed by a nonlinear region at higher stress were observed at any nLinker concentration, except when nLinker concentration = 1.25 wt% at which no NCH was formed. In the lower stress region, K′ was equal to G′ in the LVR (G0), whereas in the high stress region, K′ had an exponential correlation with shear stress (K′ ∝ σm). The stiffening index (m) and critical stress (σc) are indicators of stiffening intensity and the onset of nonlinear region, respectively. When m is close to 0.5, the dominant stiffening mechanism is filament stretching; however as m approaches 1.5, the network fluctuation mainly regulates the stiffening responses.44 By conducting oscillatory amplitude sweep tests and extracting the exponents, we investigated how the LivGel components, specifically nLinker or Ca2+-loaded nLinker, and the corresponding networks affect the transition between the stiffening mechanisms (i.e., network fluctuation versus single filament stretching) and control the dominant power-law scaling behavior.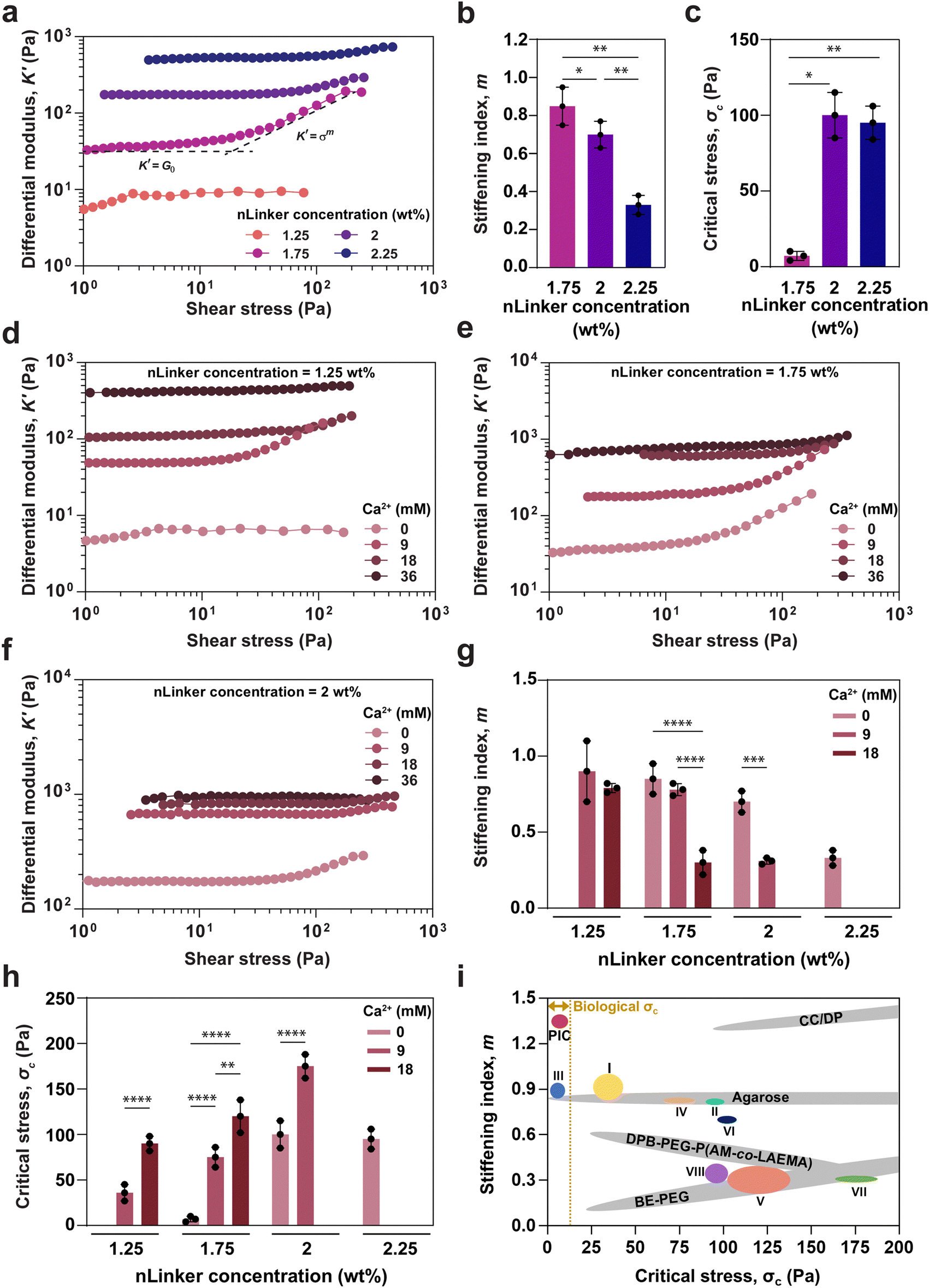 | ||
| Fig. 4 Quantitative analysis of strain-stiffening behavior in LivGels, containing varying nLinker and Ca2+ concentrations. (a) Differential modulus (K′) versus shear stress at a constant frequency of 1 rad s−1, (b) stiffening index (m), and (c) critical stress (σc) of LivGels, containing varying Ca2+-free nLinker concentrations. K′ versus oscillatory shear stress at a constant frequency of 1 rad s−1, measured for the LivGels, containing varying Ca2+ concentrations and a constant nLinker concentration of (d) 1.25 wt%, (e) 1.75 wt%, or (f) 2 wt%. (g) m and (h) σc of the LivGels at varying Ca2+ (0–18 mM)-loaded nLinker concentrations (1.25–2.25 wt%). The LivGels containing 36 mM of Ca2+ did not have strain-stiffening behavior (n =3, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). (i) m–σc profiles of biological and synthetic gels, compared with the LivGels, containing 1.25 wt% of nLinker with (I) 9 mM of Ca2+ or (II) 18 mM of Ca2+, LiveGels containing 1.75 wt% of nLinker with (III) 0 mM of Ca2+, (IV) 9 mM of Ca2+, or (V) 18 mM of Ca2+, LiveGels containing 2 wt% nLinker with (VI) 0 mM of Ca2+ or (VII) 9 mM of Ca2+, or (VIII) LiveGels containing 2.25 wt% of nLinker with 0 mM of Ca2+. PIC, DPB-PEG-P(AM-co-LAEMA), CC/DP, and BE-PEG stand for synthetic polyisocyanides, diphenylboronic acid-terminated telechelic poly(ethylene glycol)–poly(acrylamide-co-2-lactobionamidoethyl methacrylamide), O-carboxymethyl chitosan-dibenzaldehyde-terminated telechelic-ploy(ethylene glycol), and boronate ester-crosslinked 4-arm poly(ethylene glycol), respectively. Biological σc range ∼0.1–10 Pa.1,55 | ||
Based on the K′ plots (Fig. 4a), the m and σc values for LivGels containing varying concentrations of Ca2+-free nLinkers were obtained, which are presented in Fig. 4b and c, respectively. The m values for LivGels containing 1.75, 2, and 2.25 wt% of nLinker were 0.85 ± 0.10, 0.72 ± 0.07, and 0.30 ± 0.11, respectively, indicating a decrease in the stiffening extent by increasing the nLinker concentration. The m values imply that the dominant stiffening mechanism is a combination of filament stretching and network fluctuations at low nLinker concentrations, which shifts to more filament stretching by increasing the nLinker concentration (≥2 wt%). The σc values of LivGels, containing 1.75, 2, or 2.25 wt% nLinker were 7 ± 3, 100 ± 15, or 95 ± 11 Pa, respectively, showing a significant increase in σc, i.e., less nonlinear responsiveness, by increasing the nLinker concentration. The decrease in m and increase in σc may be associated with an increase in the stiffness (G0) by increasing the crosslinking density via the formation of hydrazone bonds and/or the reinforcing contribution of nLinker, rendering the LivGels less dynamically responsive to the applied strain. These results show that by changing the nLinker concentration, the nonlinear mechanical response of LivGels is readily tailored at a constant matrix biopolymer (DH-ALG) concentration (2 wt%).
To quantify the effect of Ca2+ on the strain-stiffening behavior of NCH, the K′ of LivGels, containing 2 wt% of DH-ALG and varying Ca2+ concentrations, was measured as a function of oscillatory shear stress at a constant nLinker concentration of 1.25, 1.75, 2, or 2.25 wt%, as presented in Fig. 4d–f, or Fig. S5 (ESI†), respectively. For the LivGels containing 1.25 wt% or 1.75 wt% of nLinker loaded with 9 or 18 mM Ca2+, or 1.75 wt% or 2 wt% of Ca2+-free nLinker, as well as the LivGel containing 2 wt% of nLinker loaded with 9 mM Ca2+, a linear region at low stress and a nonlinear region at higher stress were observed, which were consistent with the oscillatory strain sweep results (Fig. 3). For all the LivGels, G0 at low stress increased by increasing the Ca2+ concentration, confirming an increase in the ionic crosslinking density. The m and σc values of LivGels containing varying Ca2+ concentrations are shown in Fig. 4g and h, respectively. The m values for LivGels, containing 1.25 wt% nLinker loaded with 9 or 18 mM Ca2+ were ∼0.87 ± 0.18 or ∼0.73 ± 0.03, respectively. For these gels, by increasing the Ca2+ concentration, the σc value shifted significantly from 28 ± 11 Pa at 9 mM of Ca2+ to 90 ± 8 Pa at 18 mM of Ca2+. Similarly, in the LivGels containing 1.75 wt% or 2 wt% nLinker, by increasing the Ca2+ concentrations, m decreased and σc increased. This reduction in the mechanical responsiveness of LivGels may be attributed to an increase in stiffness as a result of dual dynamic hydrazone/ionic crosslinking and the increased network connectivity beyond the isostatic threshold, rendering the semi-flexible networks stiff. Accordingly, when the LivGels, containing 2.25 wt% of nLinker were loaded with Ca2+ (Fig. S5, ESI†), they did not have any strain-stiffening behavior. Additionally, the m values of LivGels, containing 1.75 wt% of nLinker, imply that the stiffening mechanism in the absence of Ca2+ (m = 0.85 ± 0.10) may be a result of filament stretching and network fluctuations, whereas by increasing the Ca2+ concentrations (e.g., m = 0.30 ± 0.08 at 18 mM), the stiffening may be regulated by filament stretching. A reason for this transition may be a high density of ionic crosslinking by which the local reorientation (bending) of nLinkers along the strain direction is restricted, and therefore the stiffening is governed via filament stretching.
Fig. 4i summarizes the stiffening parameters (m and σc) of biological and synthetic gels, as well as the LivGels. The LivGels, containing Ca2+-free nLinker (1.75 wt%) were engineered to resemble the strain-stiffening characteristics of biological gels (e.g., 0.5 Pa < σc < 10 Pa1,55). In the stress-stiffening region, m has been predicted to be 1.5 for a single semiflexible polymer, as observed in fibrin fibers55 and actin filaments.56 Among the developed hydrogels, synthetic polyisocyanide (PIC) gels have comparable strain-stiffening parameters with biological analogs. Synthetic boronate ester-crosslinked 4-arm poly(ethylene glycol) (BE-PEG) hydrogels have strain-stiffening properties with σc ranging from 20 Pa to 1000 Pa and m ranging from 0.1 to 0.5, regulated by the polymer concentration, temperature, and pH.57 Agarose hydrogels are a class of ECM-like biomaterials with strain-stiffening behavior because of their semi-flexible nature. The LivGels offer a broad range of m, spanning from ∼0.87 (9 mM Ca2+-loaded nLinker concentration = 1.25 wt%) to ∼0.25 (Ca2+-free nLinker concentration = 2.25 wt%), with σc ranging from ∼28 Pa to ∼157 Pa, adjusted by the crosslinking density or modes (i.e., hydrazone and ionic bonding). Diphenylboronic acid-terminated telechelic poly(ethylene glycol)–poly(acrylamide-co-2-lactobionamidoethyl methacrylamide) (DPB-PEG-P(AM-co-LAEMA))58 and O-carboxymethyl chitosan-dibenzaldehyde-terminated telechelic-ploy(ethylene glycol) (CC/DP)59 are other hydrogels with stiffening responses that are mechanically mimicked by the LivGels. Note that DPB-PEG-P(AM-co-LAEMA) and CC/DP gels do not recapitulate the stiffening properties, specifically σc, of biological gels. Overall, LivGels provide a novel bio-based platform that mimic the dynamic mechanical properties of ECM.
3.5. Qualitative and quantitative analyses of LivGel self-healing
The dynamic covalent hydrazone bonds and physical ionic interactions have been adopted extensively to develop self-healing gels.60 Accordingly, our LivGels were engineered using an aldehyde- and carboxylate-bearing nLinker to include reversible bonds that would enable self-healing behavior when mixed with DH-ALG. The self-healing capability of LivGels was assessed by strain amplitude measurements in which repeating cycles of low (1%) and high (500%) strains were used. Fig. 5a–d present the recovery tests in which the dynamic moduli of LivGels, containing varying nLinker concentrations loaded with 9 mM of Ca2+, are registered over time. The recovery tests of other NCH, containing varying nLinker and Ca2+ concentrations are shown in Fig. S6 (ESI†). These experiments were conducted to investigate how rapidly the LivGels recover their solid-like structure after the elimination of high strain. By applying strain of 500%, the LivGels had a fluid-like (viscous) behavior, reflected in G′ < G′′. Right after each cycle of high strain, the LivGel recovery was monitored at 1% strain and 1 rad s−1 angular frequency. All strain-stiffening LivGels, containing either Ca2+-free nLinker (formed via hydrazone bonds) or Ca2+-loaded nLinker (formed via both ionic interactions and hydrazone bonds), had excellent recovery within seconds. At an extremely large strain, a fraction of hairy junctions/crosslink points between nLinker/nLinker or nLinker/DH-ALG may break. These broken junction points, made of reversible bonds, reform upon strain reduction, healing the structure of LivGels. Fig. 5e shows a qualitative self-healing assessment of LivGels, containing Ca2+-free nLinker (2 wt%) or 9 mM Ca2+-loaded nLinker (2 wt%). The LivGels were prepared in star-shape molds, cut into two pieces, and healed in 10 min after juxtaposing the cut interfaces. The healed LivGels, containing Ca2+-free nLinker or Ca2+-loaded nLinker, were mechanically robust and preserved their integrity upon hanging.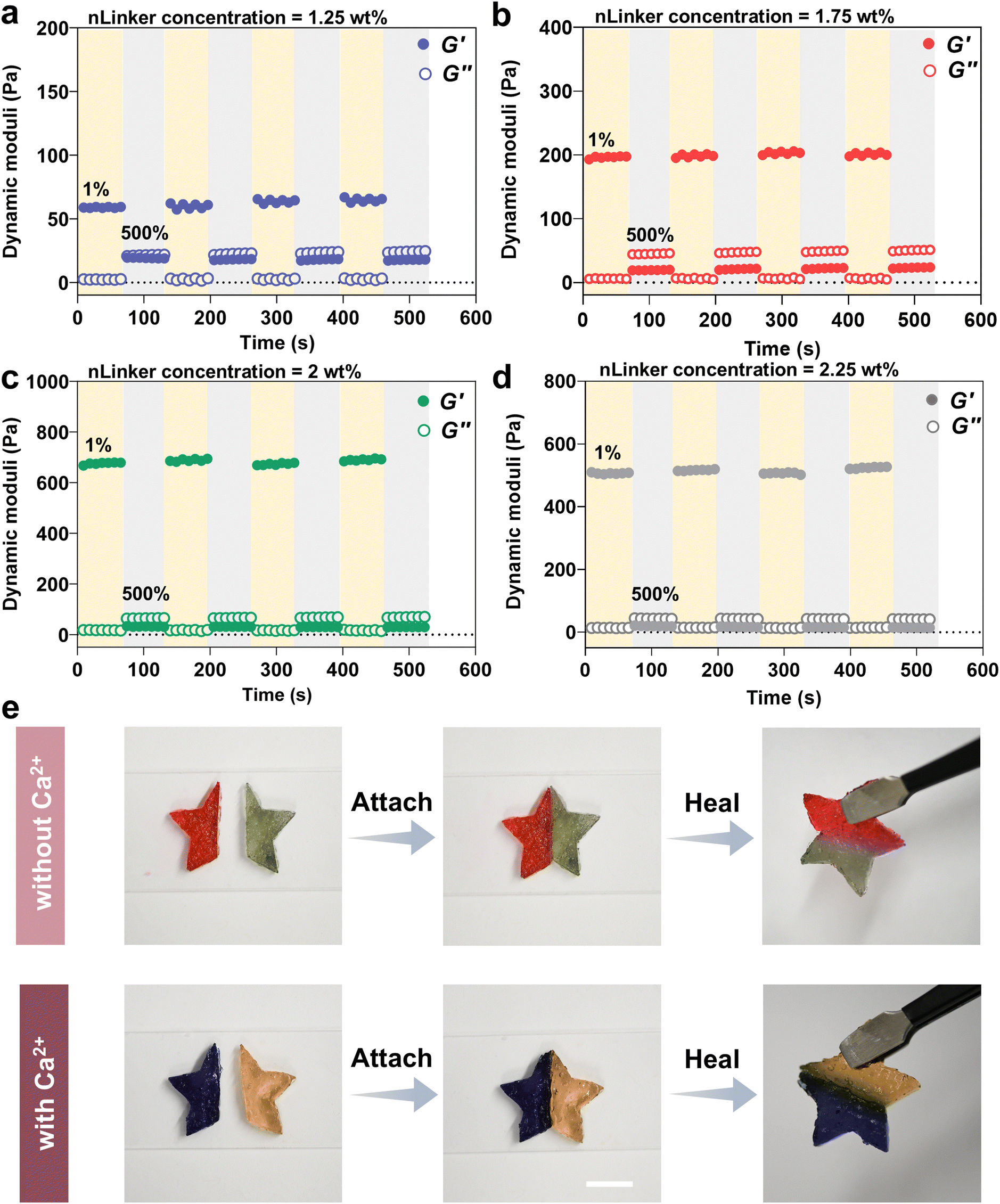 | ||
| Fig. 5 Self-healing of LivGels. Oscillatory shear strain amplitude tests at alternating low (1%)-high (500%) strain cycles, conducted on the LivGels, containing varying nLinker concentrations of (a) 1.25 wt%, (b) 1.75 wt%, or (c) 2 wt%, all loaded with 9 mM Ca2+, or (d) 2.25 wt% without Ca2+. (f) Representative photos, showing the self-healing behavior of LivGels, crosslinked with the nLinker (2 wt%) covalently or with Ca2+(9 mM)-loaded nLinker ionically and covalently (scale bar = 1 cm). | ||
4. Conclusions
Biopolymeric acellular nanocomposite living hydrogels, referred to as LivGels, were developed with nonlinear mechanical behavior and self-healing properties. The LivGel design was inspired by the shear-stiffening semi-flexible polymeric networks of natural ECM and cell-imparted healing after tissue damage. Via rationally tailoring the crosslinking of a network-forming biopolymer and anisotropic cellulose-based hairy nanoparticles (nLinker), conventional static gels were converted to dynamically responsive analogs using ionic and dynamic covalent hydrazone bonds. The anisotropic bifunctional nLinker, bearing aldehyde and carboxylate groups, enabled tailoring of the m, σc, and G′ of LivGels within the range of 0.30 to 0.87, 7 to 175 Pa, and 30 to 591 Pa, respectively, at a constant matrix biopolymer (DH-ALG) concentration. The strain-stiffening LivGels underwent rapid recovery under cyclic low (1%)–high (500%) strains. This work may pave the way for developing bio-based acellular living materials with complex dynamic attributes for a broad range of applications, including in vitro ECM models, tissue engineering, additive manufacturing, and soft robotics.Data availability
Data pertaining to this work are presented in the main manuscript and the ESI.† Further relevant data are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the support from Penn State, Dorothy Foehr Huck and J. Lloyd Huck Early Career Chair, the Convergence Center for Living Multifunctional Material Systems (LiMC2) and the Cluster of Excellence Living, Adaptive and Energy-autonomous Materials Systems (livMatS) Living Multifunctional Materials Collaborative Research Seed Grant Program, the Materials Research Institute (MRI), and the College of Engineering Materials Matter at the Human Level seed grants. Fig. 1a was created with https://BioRender.com.References
- M. Jaspers, M. Dennison, M. F. J. Mabesoone, F. C. MacKintosh, A. E. Rowan and P. H. J. Kouwer, Ultra-Responsive Soft Matter from Strain-Stiffening Hydrogels, Nat. Commun., 2014, 5(1), 5808, DOI:10.1038/ncomms6808.
- R. K. Das, V. Gocheva, R. Hammink, O. F. Zouani and A. E. Rowan, Stress-Stiffening-Mediated Stem-Cell Commitment Switch in Soft Responsive Hydrogels, Nat. Mater., 2016, 15(3), 318–325, DOI:10.1038/nmat4483.
- U. Blache, E. M. Ford, B. Ha, L. Rijns, O. Chaudhuri, P. Y. W. Dankers, A. M. Kloxin, J. G. Snedeker and E. Gentleman, Engineered Hydrogels for Mechanobiology, Nat. Rev. Methods Primers, 2022, 2(1), 98, DOI:10.1038/s43586-022-00179-7.
- Y. Fang, E. Han, X. X. Zhang, Y. Jiang, Y. Lin, J. Shi, J. Wu, L. Meng, X. Gao, P. J. Griffin, X. Xiao, H. M. Tsai, H. Zhou, X. Zuo, Q. Zhang, M. Chu, Q. Zhang, Y. Gao, L. K. Roth, R. Bleher, Z. Ma, Z. Jiang, J. Yue, C. M. Kao, C. T. Chen, A. Tokmakoff, J. Wang, H. M. Jaeger and B. Tian, Dynamic and Programmable Cellular-Scale Granules Tissue-like Materials, Matter, 2020, 2(4), 948–964, DOI:10.1016/j.matt.2020.01.008.
- O. Chaudhuri, J. Cooper-White, P. A. Janmey, D. J. Mooney and V. B. Shenoy, Effects of Extracellular Matrix Viscoelasticity on Cellular Behaviour, Nature, 2020, 584(7822), 535–546, DOI:10.1038/s41586-020-2612-2.
- C. Storm, J. F. Pastore, F. C. MacKintosh, T. C. Lubensky and P. A. Janmey, Nonlinear Elasticity in Biological Gels, Nature, 2005, 435(7039), 188–191, DOI:10.1038/nature03497.
- A. S. G. van Oosten, M. Vahabi, A. J. Licup, A. Sharma, P. A. Galie, F. C. MacKintosh and P. A. Janmey, Uncoupling Shear and Uniaxial Elastic Moduli of Semiflexible Biopolymer Networks: Compression-Softening and Stretch-Stiffening, Sci. Rep., 2016, 6, 19270, DOI:10.1038/srep19270.
- M. Vahabi, A. Sharma, A. J. Licup, A. S. G. van Oosten, P. A. Galie, P. A. Janmey and F. C. Mackintosh, Elasticity of Fibrous Networks under Uniaxial Prestress, Soft Matter, 2016, 12(22), 5050–5060, 10.1039/c6sm00606j.
- K. Bertula, L. Martikainen, P. Munne, S. Hietala, J. Klefström, O. Ikkala and Nonappa, Strain-Stiffening of Agarose Gels, ACS Macro Lett., 2019, 8(6), 670–675, DOI:10.1021/acsmacrolett.9b00258.
- B. Yan, J. Huang, L. Han, L. Gong, L. Li, J. N. Israelachvili and H. Zeng, Duplicating Dynamic Strain-Stiffening Behavior and Nanomechanics of Biological Tissues in a Synthetic Self-Healing Flexible Network Hydrogel, ACS Nano, 2017, 11(11), 11074–11081, DOI:10.1021/acsnano.7b05109.
- L. B. Nikolić, A. S. Zdravković, V. D. Nikolić and S. S. Ilić-Stojanović, Synthetic Hydrogels and Their Impact on Health and Environment, 2018, pp. 1–29 DOI:10.1007/978-3-319-76573-0_61-1.
- Z. Zhao, R. Fang, Q. Rong and M. Liu, Bioinspired Nanocomposite Hydrogels with Highly Ordered Structures, Adv. Mater., 2017, 29(45), 1703045, DOI:10.1002/adma.201703045.
- J. K. Mouw, G. Ou and V. M. Weaver, Extracellular Matrix Assembly: A Multiscale Deconstruction, Nat. Rev. Mol. Cell Biol., 2014, 15(12), 771–785, DOI:10.1038/nrm3902.
- H. Zhao, M. Liu, Y. Zhang, J. Yin and R. Pei, Nanocomposite Hydrogels for Tissue Engineering Applications, Nanoscale, 2020, 12(28), 14976–14995, 10.1039/d0nr03785k.
- A. Motealleh and N. S. Kehr, Nanocomposite Hydrogels and Their Applications in Tissue Engineering, Adv. Healthcare Mater., 2017, 6(1), 1600938, DOI:10.1002/adhm.201600938.
- V. Pryamitsyn and V. Ganesan, Mechanisms of Steady-Shear Rheology in Polymer-Nanoparticle Composites, J. Rheol., 2006, 50(5), 655–683, DOI:10.1122/1.2234483.
- N. Gao, G. Hou, J. Liu, J. Shen, Y. Gao, A. V. Lyulin and L. Zhang, Tailoring the Mechanical Properties of Polymer Nanocomposites: Via Interfacial Engineering, Phys. Chem. Chem. Phys., 2019, 21(34), 18714–18726, 10.1039/c9cp02948f.
- G. Schmidt and M. M. Malwitz, Properties of Polymer-Nanoparticle Composites, Curr. Opin. Colloid Interface Sci., 2003, 8(1), 103–108, DOI:10.1016/S1359-0294(03)00008-6.
- C. Shao, L. Meng, M. Wang, C. Cui, B. Wang, C. R. Han, F. Xu and J. Yang, Mimicking Dynamic Adhesiveness and Strain-Stiffening Behavior of Biological Tissues in Tough and Self-Healable Cellulose Nanocomposite Hydrogels, ACS Appl. Mater. Interfaces, 2019, 11(6), 5885–5895, DOI:10.1021/acsami.8b21588.
- W. Chen, J. Kumari, H. Yuan, F. Yang and P. H. J. Kouwer, Toward Tissue-like Material Properties: Inducing in Situ Adaptive Behavior in Fibrous Hydrogels, Adv. Mater., 2022, 34(37), 2202057, DOI:10.1002/adma.202202057.
- W. Chen and P. H. J. Kouwer, Combining Mechanical Tuneability with Function: Biomimetic Fibrous Hydrogels with Nanoparticle Crosslinkers, Adv. Funct. Mater., 2021, 31(47), 2105713, DOI:10.1002/adfm.202105713.
- T. G. M. van de Ven and A. Sheikhi, Hairy Cellulose Nanocrystalloids: A Novel Class of Nanocellulose, Nanoscale, 2016, 8(33), 15101–15114, 10.1039/C6NR01570K.
- A. Sheikhi and T. G. M. van de Ven, Colloidal Aspects of Janus-like Hairy Cellulose Nanocrystalloids, Curr. Opin. Colloid Interface Sci., 2017, 29, 21–31, DOI:10.1016/j.cocis.2017.02.001.
- H. Yang, A. Sheikhi and T. G. M. van de Ven, Reusable Green Aerogels from Cross-Linked Hairy Nanocrystalline Cellulose and Modified Chitosan for Dye Removal, Langmuir, 2016, 32(45), 11771–11779, DOI:10.1021/acs.langmuir.6b03084.
- M. Tavakolian, R. Koshani, N. Tufenkji and T. G. M. van de Ven, Antibacterial Pickering Emulsions Stabilized by Bifunctional Hairy Nanocellulose, J. Colloid Interface Sci., 2023, 643, 328–339, DOI:10.1016/j.jcis.2023.04.033.
- Y. Xu, C. Huang, L. Li, X. Yu, X. Wang, H. Peng, Z. Gu and Y. Wang, In Vitro Enzymatic Degradation of a Biological Tissue Fixed by Alginate Dialdehyde, Carbohydr. Polym., 2013, 95(1), 148–154, DOI:10.1016/j.carbpol.2013.03.021.
- E. Hermansson, E. Schuster, L. Lindgren, A. Altskär and A. Ström, Impact of Solvent Quality on the Network Strength and Structure of Alginate Gels, Carbohydr. Polym., 2016, 144, 289–296, DOI:10.1016/j.carbpol.2016.02.069.
- F. S. H. Head and G. Hughes, The Oxidation of Simple Organic Substances by Sodium Metaperiodate in Solutions Exposed to Daylight, J. Chem. Soc., 1952, 2046–2052 RSC.
- G. Janarthanan, J. H. Kim, I. Kim, C. Lee, E. J. Chung and I. Noh, Manufacturing of Self-Standing Multi-Layered 3D-Bioprinted Alginate-Hyaluronate Constructs by Controlling the Cross-Linking Mechanisms for Tissue Engineering Applications, Biofabrication, 2022, 14(3), 035013, DOI:10.1088/1758-5090/ac6c4c.
- K. Y. Lee, K. H. Bouhadir and D. J. Mooney, Degradation Behavior of Covalently Cross-Linked Poly(Aldehyde Guluronate) Hydrogels, Macromolecules, 2000, 33(1), 97–101, DOI:10.1021/ma991286z.
- U. J. Kim, S. Kuga, M. Wada, T. Okano and T. Kondo, Periodate Oxidation of Crystalline Cellulose, Biomacromolecules, 2000, 1(3), 488–492, DOI:10.1021/bm0000337.
- T. G. Mayerhöfer, S. Pahlow and J. Popp, The Bouguer–Beer–Lambert Law: Shining Light on the Obscure, Chemphyschem, 2020, 21(18), 2029–2046, DOI:10.1002/cphc.202000464.
- B. Huntington, M. L. Pitcher and A. Sheikhi, Nanoengineering the Redispersibility of Cellulose Nanocrystals, Biomacromolecules, 2023, 24(1), 43–56, DOI:10.1021/acs.biomac.2c00518.
- H. Ohshima, Approximate Analytic Expression for the Electrophoretic Mobility of Moderately Charged Cylindrical Colloidal Particles, Langmuir, 2015, 31(51), 13633–13638, DOI:10.1021/acs.langmuir.5b02969.
- H. Ohshima, Henry's Function for Electrophoresis of a Cylindrical Colloidal Particle, J. Colloid Interface Sci., 1996, 180(1), 299–301, DOI:10.1006/jcis.1996.0305.
- S. K. Bharti and R. Roy, Quantitative 1H NMR Spectroscopy, TrAC, Trends Anal. Chem., 2012, 35, 5–26, DOI:10.1016/j.trac.2012.02.007.
- K. Y. Lee and D. J. Mooney, Alginate: Properties and Biomedical Applications, Prog. Polym. Sci., 2012, 37(1), 106–126, DOI:10.1016/j.progpolymsci.2011.06.003.
- A. Dodero, S. Vicini, M. Alloisio and M. Castellano, Rheological Properties of Sodium Alginate Solutions in the Presence of Added Salt: An Application of Kulicke Equation, Rheol. Acta, 2020, 59(6), 365–374, DOI:10.1007/s00397-020-01206-8.
- A. Wilson, G. Gasparini and S. Matile, Functional Systems with Orthogonal Dynamic Covalent Bonds, Chem. Soc. Rev., 2014, 43(6), 1948–1962, 10.1039/c3cs60342c.
- Y. Zhang, Y. Qi, S. Ulrich, M. Barboiu and O. Ramström, Dynamic Covalent Polymers for Biomedical Applications, Mater. Chem. Front., 2020, 4(2), 489–506, 10.1039/c9qm00598f.
- S. K. Bajpai, M. Bajpai and F. F. Shah, Alginate Dialdehyde (AD)-Crosslinked Casein Films: Synthesis, Characterization and Water Absorption Behavior, Des. Monomers Polym., 2016, 19(5), 406–419, DOI:10.1080/15685551.2016.1169374.
- F. L. C. Morgan, J. Fernández-Pérez, L. Moroni and M. B. Baker, Tuning Hydrogels by Mixing Dynamic Cross-Linkers: Enabling Cell-Instructive Hydrogels and Advanced Bioinks, Adv. Healthcare Mater., 2022, 11(1), 2101576, DOI:10.1002/adhm.202101576.
- C. E. Pegg, G. H. Jones, T. J. Athauda, R. R. Ozer and J. M. Chalker, Facile Preparation of Ammonium Alginate-Derived Nanofibers Carrying Diverse Therapeutic Cargo, Chem. Commun., 2014, 50(2), 156–158, 10.1039/c3cc47232a.
- G. Žagar, P. R. Onck and E. Van Der Giessen, Two Fundamental Mechanisms Govern the Stiffening of Cross-Linked Networks, Biophys. J., 2015, 108(6), 1470–1479, DOI:10.1016/j.bpj.2015.02.015.
- R. Koshani and T. G. M. van de Ven, Electroacoustic Characterization of Trimmed Hairy Nanocelluloses, J. Colloid Interface Sci., 2020, 563, 252–260, DOI:10.1016/j.jcis.2019.12.034.
- S. L. Yeh, D. Alexander, N. Narasimhalu, R. Koshani and A. Sheikhi, Mussel-Inspired Nanocellulose Coating for Selective Neodymium Recovery, ACS Appl. Mater. Interfaces, 2023, 15(37), 44154–44166, DOI:10.1021/acsami.3c04512.
- M. Rubinstein and R. H. Colby, Polymer Physics, Oxford University Press, 2003, edn 1 Search PubMed.
- M. Jaspers, S. L. Vaessen, P. van Schayik, D. Voerman, A. E. Rowan and P. H. J. Kouwer, Nonlinear Mechanics of Hybrid Polymer Networks That Mimic the Complex Mechanical Environment of Cells, Nat. Commun., 2017, 8(1), 15478, DOI:10.1038/ncomms15478.
- C. F. Guimarães, L. Gasperini, A. P. Marques and R. L. Reis, The Stiffness of Living Tissues and Its Implications for Tissue Engineering, Nat. Rev. Mater., 2020, 5(5), 351–370, DOI:10.1038/s41578-019-0169-1.
- P.-G. De Gennes, Scaling Concepts in Polymer Physics, Cornell University Press, 1979 Search PubMed.
- A. Varadarajan, L. T. Kearney, J. K. Keum, A. K. Naskar and S. Kundu, Effects of Salt on Phase Behavior and Rheological Properties of Alginate-Chitosan Polyelectrolyte Complexes, Biomacromolecules, 2023, 24(6), 2730–2740, DOI:10.1021/acs.biomac.3c00171.
- M. Matsuo, C. Sawatari, Y. Iwai and F. Ozaki, Effect of Orientation Distribution and Crystallinity on the Measurement by X-Ray Diffraction of the Crystal Lattice Moduli of Cellulose i and Ii, Macromolecules, 1990, 23(13), 3266–3275, DOI:10.1021/ma00215a012.
- J. C. L. Maxwell, On the Calculation of the Equilibrium and Stiffness of Frames, London, Edinburgh Dublin Philos. Mag. J. Sci., 1864, 27(182), 294–299, DOI:10.1080/14786446408643668.
- J. Feng, H. Levine, X. Mao and L. M. Sander, Nonlinear Elasticity of Disordered Fiber Networks, Soft Matter, 2016, 12(5), 1419–1424, 10.1039/C5SM01856K.
- I. K. Piechocka, R. G. Bacabac, M. Potters, F. C. MacKintosh and G. H. Koenderink, Structural Hierarchy Governs Fibrin Gel Mechanics, Biophys. J., 2010, 98(10), 2281–2289, DOI:10.1016/j.bpj.2010.01.040.
- M. L. Gardel, J. H. Shin, F. C. MacKintosh, L. Mahadevan, P. Matsudaira and D. A. Weitz, Elastic Behavior of Cross-Linked and Bundled Actin Networks, Science, 2004, 304(5675), 1301–1305, DOI:10.1126/science.1095087.
- R. C. Ollier, Y. Xiang, A. M. Yacovelli and M. J. Webber, Biomimetic Strain-Stiffening in Fully Synthetic Dynamic-Covalent Hydrogel Networks, Chem. Sci., 2023, 14(18), 4796–4805, 10.1039/d3sc00011g.
- W. Wang, L. Xiang, D. Diaz-Dussan, J. Zhang, W. Yang, L. Gong, J. Chen, R. Narain and H. Zeng, Dynamic Flexible Hydrogel Network with Biological Tissue-like Self-Protective Functions, Chem. Mater., 2020, 32(24), 10545–10555, DOI:10.1021/acs.chemmater.0c03526.
- Y. Liu, S.-H. Lin, W.-T. Chuang, N.-T. Dai and S. Hsu, Biomimetic Strain-Stiffening in Chitosan Self-Healing Hydrogels, ACS Appl. Mater. Interfaces, 2022, 14(14), 16032–16046, DOI:10.1021/acsami.2c01720.
- X. Ding, L. Fan, L. Wang, M. Zhou, Y. Wang and Y. Zhao, Designing Self-Healing Hydrogels for Biomedical Applications, Mater. Horiz., 2023, 10(10), 3929–3947, 10.1039/d3mh00891f.
Footnote |
| † Electronic supplementary information (ESI) available: DH-ALG synthesis and characterization, aldehyde titration curve of DAMC, time sweep rheological measurements of NCH, LivGels pore size, differential modulus (K′) versus oscillatory shear stress for LivGels, containing 2.25 wt% nLinker, and strain amplitude tests of LivGels, containing varying nLinker and Ca2+ concentrations. See DOI: https://doi.org/10.1039/d4mh00922c |
| This journal is © The Royal Society of Chemistry 2025 |