
Multiple H-bonds induced mechanically robust vat photopolymerization 3D printing poly(urethane–urea) elastomers†
Xingxing
Yang
ab,
Changcheng
Bai
b,
Bin
Zhu‡
ab,
Jiayu
Wu
b,
Mingyang
Wang
a,
Yixian
Wang
a,
Tao
Wu
ac,
Desheng
Liu
 *b,
Pan
Jiang
*b and
Xiaolong
Wang
*ab
*b,
Pan
Jiang
*b and
Xiaolong
Wang
*ab
aSchool of Chemistry and Chemical Engineering/State Key Laboratory Incubation Base for Green Processing of Chemical Engineering, Shihezi University, Shihezi 832003, P. R. China
bState Key Laboratory of Solid Lubrication, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou 730000, P. R. China. E-mail: pan.jiang@ijm.fr; liudesheng@licp.cas.cn; wangxl@licp.cas.cn
cShandong Laboratory of Advanced Materials and Green Manufacturing at Yantai, Yantai 264006, P. R. China
First published on 29th October 2024
Abstract
Vat photopolymerization (VP) elastomers show great promise across various fields, yet they face significant challenges in achieving adequate mechanical strength, elasticity, and durability due to incomplete polymerization and weak interfacial bonding between printed layers. In this study, we introduce high-performance poly(urethane–urea) elastomers (PUEs) utilizing a dual cross-linked network (DCN) strategy compatible with VP 3D printing. This innovative approach enhances mechanical properties by incorporating multiple hydrogen-bonded urethane and urea groups. The presence of multiple hydrogen bonds facilitates energy dissipation under external mechanical stress and improves interfacial interlocking, while the covalent cross-linked network provides stability and flexibility during deformation. The resulting elastomer exhibits a tensile strength of 28.30 ± 1.10 MPa, a recovery strain of approximately 300%, and a fracture energy of 22.90 ± 4.20 kJ m−2. As a proof of concept, we demonstrate the rapid fabrication of 3D-printed stents with intricate architectures, outstanding load-bearing capabilities, and excellent biocompatibility. This strategy not only paves the way for the development of mechanically robust, complex-structured PUEs but also broadens their application scope in engineering and biomedical fields.
New conceptsHere, we propose a “dual cross-linked network” design concept to address the problem of poor compatibility between mechanical properties and processability of printable elastomeric materials, while balancing rapid fabrication and biocompatibility. By preparing a photosensitive poly(urethane–urea) oligomer containing urethane and urea groups, acrylate groups can undergo UV-triggered free radical polymerization to form a covalently cross-linked network during 3D printing. The urethane and urea groups within the elastomer can form a second physically cross-linked network through multiple hydrogen bonds, enhancing interactions between polymer chains and adhesion between printed layers. The HMDI-PUU2000 elastomer exhibited a tensile strength of up to 28.30 ± 1.10 MPa, toughness of up to 57.90 ± 4.20 MJ m−3, and elongation at break of up to 755 ± 37%. In addition, the HMDI-PUU2000 elastomer exhibited good biocompatibility and hemocompatibility because no organic dyes were used in the photosensitive resin preparation. Combined with 3D printing technology, this provides an opportunity to manufacture personalized medical products such as vascular stents and heart valves. |
Introduction
Three-dimensional (3D) printing of polyurethane elastomers is commonly applied in the fields of biomedical engineering,1 soft robotics,2,3 actuators,4–6 sensors,7,8 and flexible electronic devices.9–11 This is due to its notable benefits, such as personalized design capabilities, and low cost.12 Currently, several 3D printing technologies have been developed for fabricating structured elastomeric parts, such as direct ink writing (DIW),13–15 fused deposition modeling (FDM),12,16 and vat photopolymerization (VP) 3D printing.17,18 While DIW and FDM technologies have certain advantages in printing poly(urethane–urea) elastomers (PUEs) in terms of mechanical properties and biocompatibility, the limited printing resolution and speed make it difficult to achieve complex structured device manufacturing.19 Compared with the others, VP 3D printing technology is being widely used in rapid manufacturing of 3D complex geometries due to its high printing resolution, fast printing speed, and desirable surface quality and free-forming ability.8,14 In order to achieve optimal photopolymerization performance and manufacturing quality, elastomer inks must be highly photosensitive to the specific wavelength of light used in the printing system to ensure the rapid photocuring process and high resolution. Second, the viscosity of 3D printing elastomer inks must be low enough to spread evenly for high quality rapid manufacturing. Most importantly, the 3D printed elastomers should also have strong interlayer bonding to maintain the structural integrity and enhance the mechanical properties.20,21 Although PUE flexible actuators, soft robots, and organ models with complex structures have been fabricated through VP 3D printing technology, the intractable issue is the insufficient mechanical properties of the photosensitive soft materials after shaping (breaking strength<20 MPa, elongation at break<400%).6,9,22,23 Therefore, it is desirable to develop high-performance polyurethane elastomers for VP 3D printing to broaden their practical applications in various scenarios.At present, mixing aliphatic polyurethane diacrylate with functional diluents is an alternative strategy to prepare photosensitive resin for VP 3D printing of high-performance polyurethane elastomers.2,18,24 For example, Wu14,25 and colleagues prepared photosensitive inks by mixing commercial polyurethane acrylate resin with a diluent, and successfully printed intricate elastic parts with good mechanical properties. Besides, Huang26 and colleagues synthesized polyurethane acrylate oligomers to formulate a photosensitive ink utilizing 3D printing to manufacture structured elastic parts with certain mechanical properties. Despite enormous efforts from researchers in preparing photosensitive ink using either commercial polyurethane acrylate resin or synthetic polyurethane acrylate oligomers, the resulting elastic parts still exhibit significant limitations in terms of mechanical performance and biocompatibility. Firstly, VP 3D printing requires rapid ultraviolet (UV)-triggered free radical polymerization within a few seconds for each layer. The fast reaction process leads to incomplete crosslinking networks with relatively short polymer chains, which fail in further extension and repair of polymer chains or networks even under postprocessing. In contrast, traditional thermoplastic PUEs commonly need a long curable time to form complete long-chain polymer networks.6,26–31 As a result, VP 3D printing of elastomers exhibits poor mechanical properties and structure stability. Secondly, introducing absorptive dyes could improve printing resolution, but these additives would cause potential biological toxicity of the printed objects, limiting their application as biomedical polymer materials.23,32,33 Hence, there is an urgent need to design and develop novel photosensitive resin for VP 3D printing technology to integrate high mechanical properties and complex structures manufacturing toward promising applications.
In this study, we propose a strategy to improve the mechanical properties and structure manufacturing of photosensitive elastomers based on a dual cross-linked network (DCN) strategy. Specifically, a photosensitive poly(urethane–urea) oligomer containing urethane and urea groups was synthesized. The grafted acrylate groups could form a covalently crosslinked network by UV triggered free radical polymerization during VP 3D printing. After further annealing treatment, the urethane and urea groups in the PU elastomer could form a second physical crosslinking network via multiple hydrogen bonds to enhance interactions between polymer chains. In this case, multiple hydrogen bonds, as dynamic sacrificial units, could quickly dissociate and then dissipate energy originated from external mechanical stimuli, while stable and robust covalent networks endow elastomers with excellent stability and flexibility. The resulting DCN elastomer could efficiently reduce the deterioration of the mechanical properties caused by rapid VP. The well-designed elastomer exhibits high crack resistance (22.85 kJ m−2) and toughness (57.93 MJ m−3). Furthermore, multiple hydrogen-bonding PUEs exhibit good biocompatibility. As a proof of concept, we successfully printed a high precision stent structure, providing an alternative to functional biomedical devices.
Results and discussion
Design of poly(urethane–urea) elastomers with DCN structures for 3D printing
Fig. 1a shows the molecular design of poly(urethane–urea) acrylate photosensitive resins for VP 3D printing of high-performance elastomers. It is widely recognized that the molecular weight of poly(urethane–urea) acrylate blocking oligomers is crucial to the tensile strength and elongation at break of elastomers. In this case, to further evaluate correlation between molecular weight and mechanical properties, PTMEG of varying molecular weights (Mn = 1000 and 2000 g mol−1) and a chain extender (PEA) were chosen as reactants to react with HMDI. Subsequently, the remaining –NCO functional groups reacted with HPA to impart UV-curable reactivity (Fig. S1, ESI†). The reaction was monitored in detail by Fourier transform infrared spectroscopy (FTIR) (Fig. S2 and S3, ESI†). The specific peaks at 1660 and 812 cm−1 were attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and the out-of-plane bending vibration absorption of C–H, respectively. FTIR spectra indicated the complete reaction of HPA with –NCO. Furthermore, the characteristic absorption peak of the –NCO groups disappeared at 2265 cm−1, which further confirmed the formation of the oligomer HMDI-PUU. In addition, the chemical structure of HMDI-PUU was confirmed by 1H NMR characteristic peaks H1 and H2 at 5.60–6.30 ppm of –CCH2 (Fig. S4 and S5, ESI†). The number average molecular weights (Mn) of HMDI-PUU oligomers were further determined by gel permeation chromatography (GPC). The Mn values of HMDI-PUU1000 and HMDI-PUU2000 were 8294 g mol−1 and 16998 g mol−1, respectively (Table S1, ESI†). The increase in Mn mainly originated from the PTMEG molecular weight. The corresponding polydispersity indices (PDI) for two oligomers were 3.30 and 2.00, respectively. The photosensitive resins only contained acrylate blocking oligomers and photoinitiators without any other additives, which were directly adapted for digital light processing 3D printing in high resolution with complex microstructure lattice structures (Fig. 1b and c). Subsequently, Fig. 1d shows the optimal curing and double cross-linked network structure for photosensitive resins. The acrylate double bonds underwent a free radical-initiated polymerization reaction, forming a covalently crosslinked network. In addition, the polymer networks also contained urethane and urea groups, which could further form a mass of multiple hydrogen bonding interaction sites to tight polymer chains. Annealing can remove the extra solvent and further strengthen hydrogen bonding interactions between polymer chains, resulting in the formation of a robust noncovalent dynamic network based on hydrogen bonds. In this system, covalent bonds contributed to strong mechanical properties because of their durability, whereas hydrogen bonds exhibited dynamic interactions that can efficiently dissipate energy and recovery. It is anticipated that the double cross-linked network structure would significantly enhance the performance of VP-printed elastomers.
C and the out-of-plane bending vibration absorption of C–H, respectively. FTIR spectra indicated the complete reaction of HPA with –NCO. Furthermore, the characteristic absorption peak of the –NCO groups disappeared at 2265 cm−1, which further confirmed the formation of the oligomer HMDI-PUU. In addition, the chemical structure of HMDI-PUU was confirmed by 1H NMR characteristic peaks H1 and H2 at 5.60–6.30 ppm of –CCH2 (Fig. S4 and S5, ESI†). The number average molecular weights (Mn) of HMDI-PUU oligomers were further determined by gel permeation chromatography (GPC). The Mn values of HMDI-PUU1000 and HMDI-PUU2000 were 8294 g mol−1 and 16998 g mol−1, respectively (Table S1, ESI†). The increase in Mn mainly originated from the PTMEG molecular weight. The corresponding polydispersity indices (PDI) for two oligomers were 3.30 and 2.00, respectively. The photosensitive resins only contained acrylate blocking oligomers and photoinitiators without any other additives, which were directly adapted for digital light processing 3D printing in high resolution with complex microstructure lattice structures (Fig. 1b and c). Subsequently, Fig. 1d shows the optimal curing and double cross-linked network structure for photosensitive resins. The acrylate double bonds underwent a free radical-initiated polymerization reaction, forming a covalently crosslinked network. In addition, the polymer networks also contained urethane and urea groups, which could further form a mass of multiple hydrogen bonding interaction sites to tight polymer chains. Annealing can remove the extra solvent and further strengthen hydrogen bonding interactions between polymer chains, resulting in the formation of a robust noncovalent dynamic network based on hydrogen bonds. In this system, covalent bonds contributed to strong mechanical properties because of their durability, whereas hydrogen bonds exhibited dynamic interactions that can efficiently dissipate energy and recovery. It is anticipated that the double cross-linked network structure would significantly enhance the performance of VP-printed elastomers.
 | ||
| Fig. 1 Photosensitive ink composition and printing. (a) Preparation of poly(urethane–urea) acrylate blocking oligomers and the chemical structure of the photopolymer resin. (b) Schematic illustrations of the bottom-up VP 3D printing. (c) A 3D-printed lattice structure. Scale bars represent 10 mm. (d) Schematic diagram of the curing and dual cross-linked networks (covalent cross-linking networks and hydrogen bonding interactions) of the vat photopolymerization-printed HMDI-PUU elastomers. | ||
Prior to the implementation of VP 3D printing, the initial viscosity, storage modulus, and double bond conversion of the photosensitive resins need to be optimized to match the VP 3D printing process. As shown in Fig. S6 (ESI†), both photosensitive resins showed a Newtonian fluid state at room temperature. The viscosity increased from 1.20 Pa s (HMDI-PUU1000) to 2.10 Pa s (HMDI-PUU2000) at a shear rate of 10 s−1, which was attributed to enhance physical entanglement caused by an increase of molecular weight. Notably, the viscosity of these resins was suitable for VP 3D printing to fabricate a high-resolution structure. The photopolymerization rheological behaviors of the specimens were examined by in situ photorheological tests (Fig. 2a). After 25 s UV irradiation, the storge moduli (G′) and loss moduli (G′′) of all specimens rapidly increased, and the corresponding gelation points (the cross points of G′ and G′′ curves) appeared after ∼16 s UV exposure. When the G′ value exceeded G′′, the resin transitioned from a liquid-like to solid-like state (Fig. 2b). This means the formation of a covalently cross-linked network. Moreover, an ATR-FTIR instrument combined with UV irradiation was employed to monitor the transformation of double bonds.6,34 With an extended exposure time of UV irradiation, the intensity of CC–H at a characteristic peak of 812 cm−1 gradually decreased (Fig. S7and S8, ESI†), which suggested that the reaction of acrylate groups of oligomers was triggered by photoinduced free radical polymerization. After ∼18 seconds of UV exposure, the 812 cm−1 peak gradually disappeared. The corresponding double bond conversion is approximately 98% in the samples (Fig. 2c). The abovementioned results indicated that the proposed photosensitive resins possessed good rheology behaviors and favorable photopolymerization dynamics for adapting VP 3D printing.
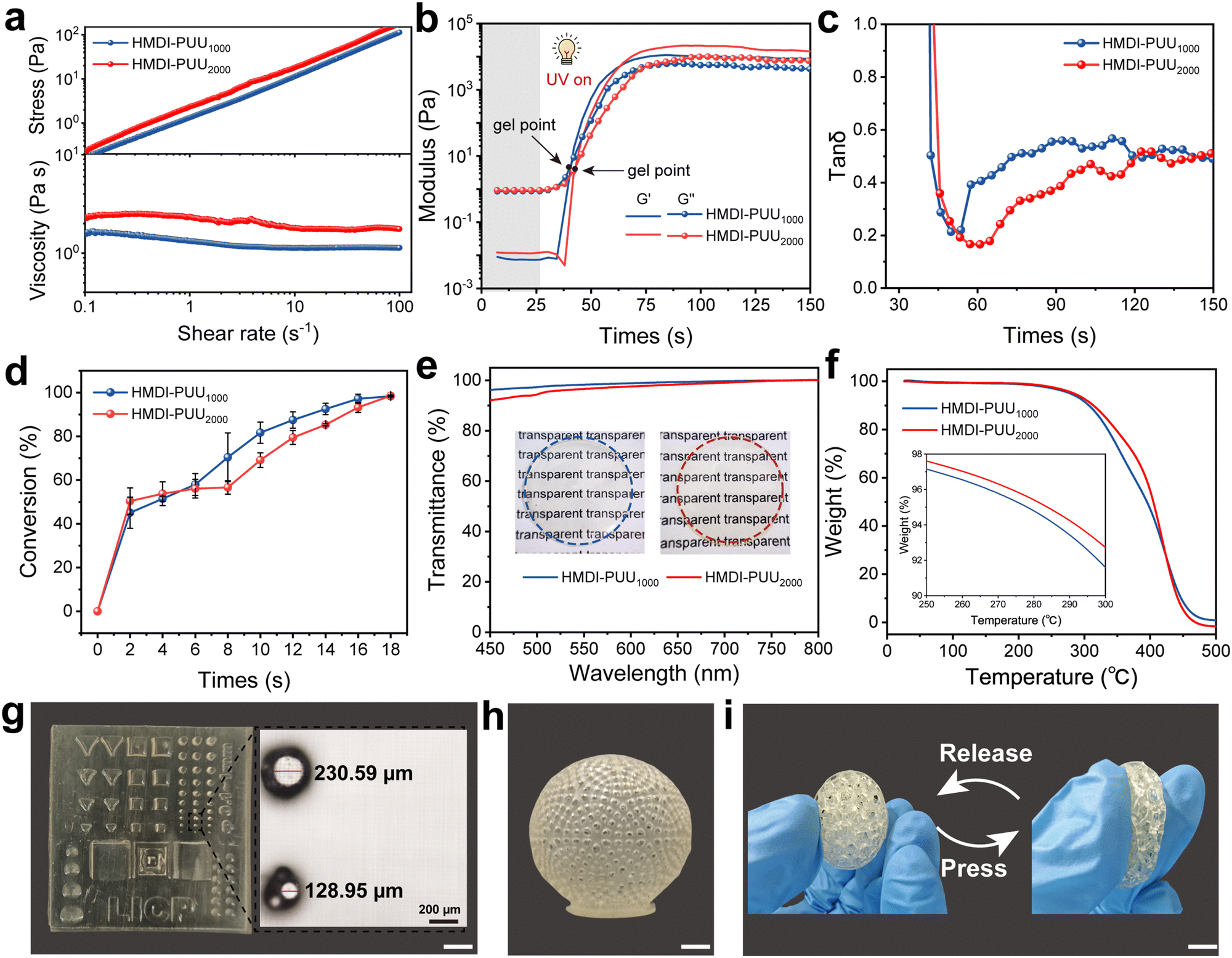 | ||
Fig. 2 Photosensitive ink rheological properties and model manufacturing. (a) Photopolymerization kinetics of the photosensitive resins. (b) Tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ of the photosensitive resins, derived from the photorheological measurements. (c) Double bond conversion UV irradiation time. (d) UV-vis transmission spectra of the HMDI-PUU1000 and HMDI-PUU2000 elastomers. (e) TGA curves of the HMDI-PUU1000 and HMDI-PUU2000 elastomers. (f) The performance of ultraviolet (UV)-cured samples and 3D printed samples. (g) A 3D-printed cylindrical structure and corresponding photograph show a diameter of 128.95 μm. Scale bars represent 10 mm. (h) Physical photographs of printed hollow 3D structures. Scale bars represent 10 mm. (i) Physical photographs of the spherical lattice structure elastomer pressing and releasing process. Scale bars represent 10 mm. δ of the photosensitive resins, derived from the photorheological measurements. (c) Double bond conversion UV irradiation time. (d) UV-vis transmission spectra of the HMDI-PUU1000 and HMDI-PUU2000 elastomers. (e) TGA curves of the HMDI-PUU1000 and HMDI-PUU2000 elastomers. (f) The performance of ultraviolet (UV)-cured samples and 3D printed samples. (g) A 3D-printed cylindrical structure and corresponding photograph show a diameter of 128.95 μm. Scale bars represent 10 mm. (h) Physical photographs of printed hollow 3D structures. Scale bars represent 10 mm. (i) Physical photographs of the spherical lattice structure elastomer pressing and releasing process. Scale bars represent 10 mm. | ||
After UV curing and thermal treatment, the HMDI-PUU1000 and HMDI-PUU2000 samples exhibited good transparency, as shown in Fig. 2d. When the thickness of the sample films was 500 μm, the average transmittance at room temperature was greater than 90% in the visible light wavelength range (450–800 nm). The thermal properties of the prepared elastomers were further studied by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) (Fig. 2e and Fig. S9, ESI†). It can be found that the glass transition temperature (Tg) of the HMDI-PUU1000 and HMDI-PUU2000 samples was approximately 5 °C while the thermal decomposition temperature was about 250 °C. The thermal resistance of the prepared elastomers can be attributed to the formation of robust covalent networks. Furthermore, there is little performance difference between the UV-cured film samples and the 3D-printed samples (Fig. 2f). This indicates that the poly(urethane–urea) acrylate photosensitive resins, through the interaction of multiple hydrogen bonds during the printing process, effectively overcomes the issue of weak interfacial bonding between layers. This allows the 3D-printed elastomers, constructed through layer-by-layer stacking, to maintain excellent mechanical properties (Fig. S10, ESI†). To reflect the advantages of vat photopolymerization printing in the manufacture of sophisticated and high-precision structures, as shown in Fig. 2g, the photopolymer resins enable the high printing resolution (128.95 μm) with a cylindrical structure. Various complex structures, including 3D hollow spheres and spherical lattice were successfully printed using HMDI-PU2000, demonstrating the good printability of the ink (Fig. 2h and i). The elastomer with a spherical lattice was soft and elastic, deformed rapidly when pressed, and recovered its original shape after release (Fig. 2i). Furthermore, the printed structures exhibited uniform shrinkage in dimensions following solvent removal, with no unexpected geometric distortions were observed. This was confirmed by statistical dimensional analysis (Fig. S11 and S12, ESI†).
Given the thermal responsiveness of hydrogen bonds,35,36 temperature-dependent FTIR spectroscopy is utilized to evaluate the interactions of multiple hydrogen bonds between urethane and urea groups (Fig. 3a). According to two-dimensional correlation FTIR spectroscopy (2D COS) analysis, the peak changes predominantly occurred in the ranges of 1800–1600 cm−1 (Fig. 3b and c). For hydrogen bonds associated with CO groups, the weaker hydrogen bonds at 1559 and 1542 cm−1 are more sensitive to temperature changes than the stronger hydrogen bonds at 1651 and 1629 cm−1, resulting in preferential cleavage of the weaker bonds, further suggesting that the N–H groups are involved in the formation of hydrogen bonding.
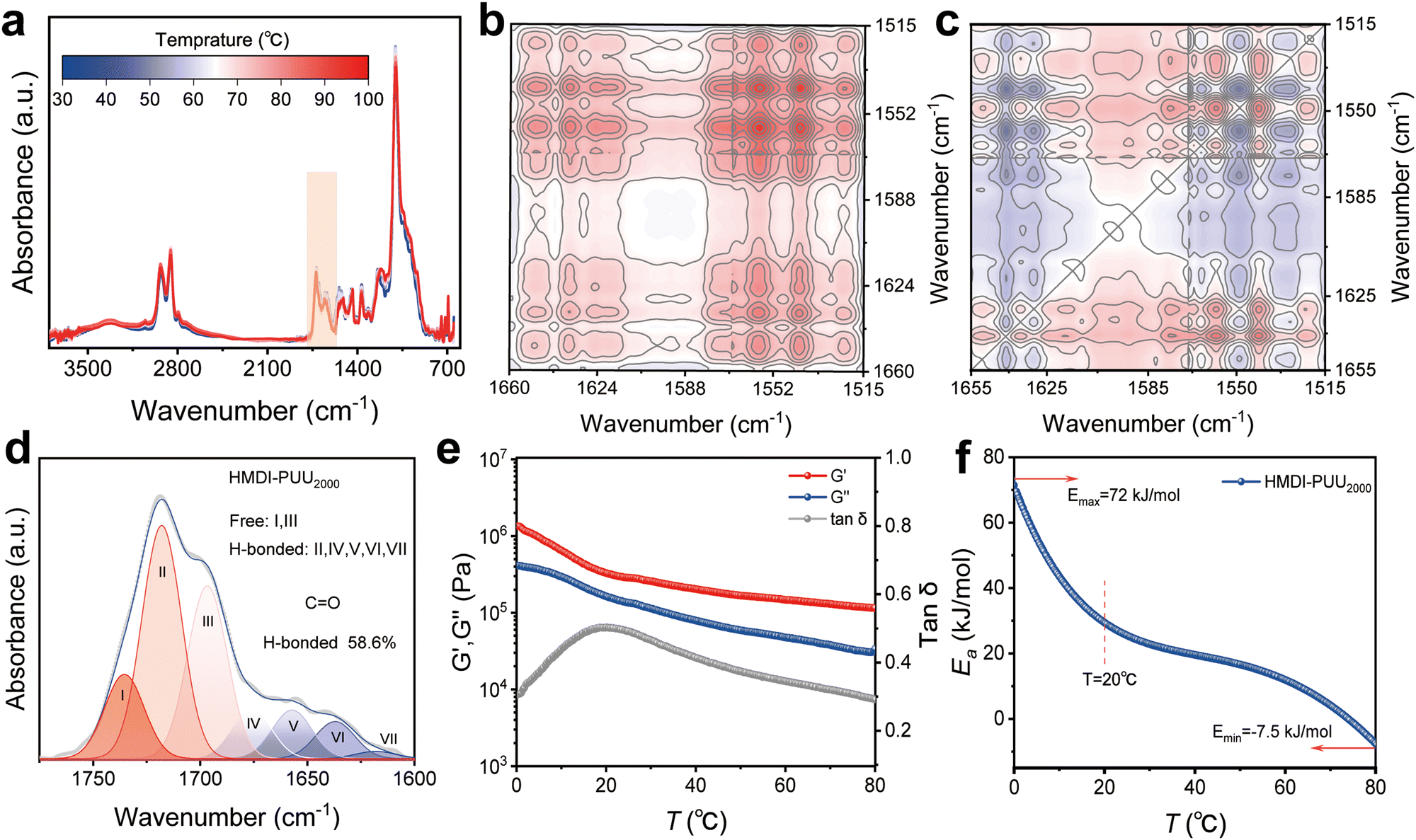 | ||
| Fig. 3 Elucidation of multiple hydrogen bond interactions in HMDI-PUU2000. (a) In situ FTIR spectra of HMDI-PUU2000 over a temperature range of 30 to 100 °C. (b) and (c) Synchronous and asynchronous 2D COS spectra of HMDI-PUU2000 in the 1665–1510 cm−1 band. (d) FTIR spectrum of HMDI-PUU2000 in the CO stretching vibration region. (e) Storage modulus, loss modulus, and loss angle of HMDI-PUU2000 as a function of temperature. (f) Ea − T curve of HMDI-PUU2000. | ||
The above results indicate that there are multiple hydrogen bonding interactions in the elastomer, but quantitative analysis is lacking. To this end, the CO absorption band in the range of 1800–1600 cm−1 was deconvoluted into 7 subpeaks (Fig. 3D). The deconvoluted subpeaks were assigned to the free groups and hydrogen-bonded CO groups in urethane (NHC(O)), urea ((NH)2C(O)), and amide (NHC(O)) functionalities.37–39 Quantitative analysis showed that the CO content of hydrogen bonds in the elastomer was as high as 58.6%, which further confirmed that the urethane and urea groups were conducive to the formation of denser hydrogen bonds.
Fig. 3e illustrates the temperature dependence of the linear viscoelastic storage modulus (G′), loss modulus (G′′), and loss tangent (tanδ) of the elastomer at an angular frequency of 10 rad s−1. As the temperature increases from 0 °C to 80 °C (with G′ > G′′), the values of G′ and G′′ gradually decrease, indicating the transition of the amorphous phase from the glassy state to the rubbery state.39,40 Additionally, the temperature point corresponding to the maximum peak of tanδ is not the glass transition temperature. When tanδ reaches its maximum at about 20 °C, it indicates that the polymer chains in the elastomer undergo a rheological transition from a solid-like state to a liquid-like state at this temperature, which is defined as the softening temperature (Ts). The maximum peak of tanδ indicates that the material exhibits the strongest energy dissipation capability at that temperature, which means that the elastomer material has optimal energy dissipation performance at room temperature. The lnαT value of the elastomer can be found to vary significantly over the temperature range of 0 to 20 °C (Fig. S13, ESI†). The corresponding activation energy (Ea) of the elastomer reaches its maximum value of about 72 kJ mol−1 at a temperature of approximately 0 °C. With the increase of temperature, the hydrogen bonds in the elastomer are gradually dissociated, leading to the decrease in Ea of the elastomer, which is consistent with the results of variable temperature FTIR spectroscopy. In conclusion, it is anticipated that the performance of elastomeric materials could be improved by designing the molecular structures of polymers that interact with hydrogen bonds.
Mechanical properties of 3D-printed elastomers
The tensile mechanical properties of the 3D printed elastomers were measured by using a universal testing machine with a stretching speed of 50 mm min−1. Fig. 4a shows the tensile stress–strain curves of the HMDI-PUU2000 samples. It could be observed that the elastomers exhibited classic elastomeric behavior without obvious yielding phenomena and plastic deformation during elongation. As shown in Table S2, (ESI†) the molecular weight of the soft segment PTMEG (Mn = 1000 and 2000 g mol−1) could efficiently govern the mechanical properties of 3D printed PUEs. Compared with HMDI-PUU1000, which has relatively short soft segments, HMDI-PUU2000 exhibited excellent mechanical performance, with a maximum stress of 28.30 ± 1.10 MPa at a high strain of 755 ± 37%. Interestingly, the printed elastomers showed J-shaped stress–strain curves, which indicated strain-dependent mechanical responses.41 In the unstretched state, the polymer chains were in a random folded state. The movement and internal friction between polymer chains along the stretched direction caused a slight increase in stress at small strain. With the increase in strain, the breakage of hydrogen bonding networks could dissipate large external mechanical stimuli, which resulted in the rapid increase of stress. Upon further stretching, highly oriented covalent cross-linked networks induced a strain hardening effect to reinforce the load-bearing ability of elastomers, leading to a significant increase in stress before complete fracture. From the calculated toughness and Young’ s moduli in Fig. 4b, HMDI-PUU2000 indicated a toughness up to 57.90 ± 4.20 MJ m−3 and Young’ s moduli up to 3.52 ± 0.43 MPa. Although HMDI-PUU have the same bi-hard segment structure, the HMDI-PUU2000 elastomer features a higher molecular weight soft segment. For the polyether polyurethane the high molecular weight soft segment could result in better ductility and flexibility during elongation. In addition, the longer soft segment in the polyurethane is conducive to disperse stress concentration when subjected to external forces. As a result, HMDI-PUU2000 exhibits better mechanical performance than HMDI-PUU1000. The above research results show that satisfactory mechanical performance is attributed to the synergistic double network, in which the covalent network structure was further tightened by a wide range of multiple hydrogen bonds. Compared with the currently reported and commercially available VP printing elastomers, our HMDI-PUU2000 elastomers showed good tensile strength42–47 (Fig. 4c). | ||
| Fig. 4 Mechanical properties of the HMDI-PUU1000 and HMDI-PUU2000 elastomers. (a) Stress–strain curves of the HMDI-PUU1000 and HMDI-PUU2000 elastomer. (b) Toughness and Young’ s moduli of the HMDI-PUU2000 elastomer. (c) Comparison of the tensile strength and strain among the photocuring elastomers (PE) and several of the literature-reported PUEs (non-photocuring elastomers: NPE). (d) Successive cyclic loading–unloading tensile curves of the HMDI-PUU2000 at different strains. (e) Tensile loading–unloading 100 cycle tests for the HMDI-PUU2000. (f) Cyclic loading–unloading tensile curves of the HMDI-PUU2000 at different times. (g) Time-dependent recovery of hysteresis and stress, as well as residual strain. (h) Photographs of the notched HMDI-PUU2000 sample that is elongated to different strains. Scale bars represent 10 mm. (i) Typical engineering stress–strain curves of unnotched and notched HMDI-PUU2000. | ||
Subsequently, the resilience of the 3D-printed HMDI-PUU2000 elastomer was further assessed through loading–unloading cycle tests at 100% to 300% strain as illustrated in Fig. 4d. The results showed that the hysteresis loop increases gradually with the increase in strain during distortion. This could be ascribed to the energy dissipation originated from the disruption of hydrogen bonds at low strains (<200%), and the deformation of the covalent cross-linking network. Furthermore, the energy dissipation increases from 0.26 MJ m−3 to 1.58 MJ m−3, and the corresponding energy loss coefficient increases from 36.60% to 44.50% (Fig. S14, ESI†). The above results indicated that the high elasticity and resilience of VP 3D-printed HMDI-PUU2000 elastomers lay in the DCN structure. To further evaluate the fatigue resistance, as illustrated in Fig. 4e, 100 consecutive tensile loading–unloading tests were conducted at a constant strain of 300%. The hysteresis loop area of the subsequent loading–unloading curves dramatically reduced compared to the initially larger hysteresis loop. And meanwhile, the dissipated energy also reduced from 1.59 MJ m−3 to 0.45 MJ m−3 after 100 cycles, and the corresponding energy loss coefficient decreased from 44.70% to 38.30% (Fig. S15, ESI†). These results further indicated that the elastomer exhibited favorable mechanical stability and fatigue resistance. Fig. 4f displays the cyclic tensile curves of an HMDI-PUU2000 sample after being loaded at 300% strain and then relaxed for different times. It can be seen that the disruption of the hydrogen bonding network caused by the mechanical force leads to a large energy dissipation of the elastomer. As the relaxation time increased from 1 min to 60 min, the strength almost returns to their original state but they still exhibited a residual strain of 40%. Fig. 4g illustrates the calculation of dissipation recovery based on the data presented in Fig. 4f, in which a 60 min rest resulted in 93.55% stress recovery and 78.28% toughness recovery. The photographs in Fig. S16 (ESI†) showed that the HMDI-PUU2000 elastomer was stretched to 3 times its original length and almost recovered to its original shape after 10 min relaxation. These results demonstrate the excellent elastic recovery performance of DCN elastomers, which was mainly attributed to the existence of dynamic hydrogen bonding networks. In this system, multiple hydrogen bonds could restore mechanical properties by reconstruction and dissipating the energy of external mechanical stimuli (Fig. S17, ESI†). The fracture energy was measured with a notched sample and the Greensmith method was employed to quantitatively evaluate crack tolerances. As illustrated in Fig. 4h, the HMDI-PUU2000 elastomer with a 1 mm pre-damaged notch reached 4.5 times its initial length. The corresponding ultimate engineering stress and breaking elongation were 1.90 MPa and 482.96%, respectively. It was found that the fracture energy of the HMDI-PUU2000 elastomer was up to 22.9 kJ m−2, which was much higher than that of natural rubber (<10 kJ m−2) (Fig. 4i). It is worth mentioning that the pre-notched HMDI-PUU2000 elastomer crack only gradually grew along the longitudinal direction instead of random expansion (Fig. S18, ESI†). The high crack resistance of HMDI-PUU2000 elastomers originated from the synergistic enhancement of dynamic hydrogen bonding networks and covalent crosslinking networks, which enables the stick-slip motion of polymer chains under external stresses. As a result, tthe stress concentration at the crack tip could efficiently propagate throughout the entire polymer network, impeding the lateral propagation of the crack to achieve excellent tear resistance.16,48
Various 3D objects with complex metamaterial structures have been printed with HMDI-PUU2000. Photographs of these structures are shown in Fig. 5a and b. To investigate the hysteresis behavior of the printed HMDI-PUU2000 elastomer in detail, compression experiments with different strains were performed on the third printed metamaterial structure. In addition, compression experiments with different strains are executed to further investigate the hysteresis behavior of HMDI-PUU2000 elastomers. Fig. 5c displays that the HMDI-PUU2000 elastomer exhibited good compressive resilience even at large compression deformations (80%) without breakage. Various strains were applied ranging from 10% to 80%, followed by a continuous compression cycle without any waiting time (Fig. 5d). The printed metamaterial structure rapidly regained its original shape, demonstrating exceptional elasticity for the metamaterial structure. As the strain increases from 10% to 80%, the compressive stress also increases from 0.02 to 0.54 MPa. At low levels of strain, the metamaterial structures with hollowed spaces allowed for deformation without breakage. At low levels of strain, the metamaterial structures with hollowed spaces allowed for deformation without significant resistance, leading to a hysteresis loop area of relatively small size and residual strains ranging from 4.20% to 8.44% (Fig. S19, ESI†). As the deformation exceeded 40%, compression was applied to the solid part of the metamaterial structure, resulting in a rapid increase in the energy dissipation and residual strain which significantly increased to 0.03 MJ m−3 and 17.75%, respectively (Fig. S19, ESI†). These results indicate the remarkable resilience of the metamaterial structure. In addition, 10 consecutive compression cycles were performed at a constant strain of 80% to evaluate the durability of HMDI-PUU2000 elastomers (Fig. 5e). It can be found that the dissipated energy gradually decreases and stabilizes at approximately 0.03 MJ m−3 (Fig. S20, ESI†). The maximum compressive stress remains at around 0.60 MPa and meanwhile, the corresponding residual strain increases slightly from 12.80% to 21.82%, during the whole test (Fig. S20, ESI†). In this process, the dynamic hydrogen bonding network achieved a balance between rupture and reconstruction while the stability of the covalent cross-linking network could rapidly recover once external forces were removed. These results further confirm that DCN tactics are an effective approach to improve the mechanical properties of VP 3D-printed elastomers, such as strength, resilience, and durability.
 | ||
| Fig. 5 Characteristics and resilience properties of the HMDI-PUU2000 elastomers. (a) 3D model of complex metamaterial structure models. (b) Physical photographs of various 3D printed metamaterial structures. The scale bars correspond to 10 mm. (c) Photographs of the 3D-printed HMDI-PUU2000 elastomer under compression and releasing at 80% strain deformation. (d) Cyclic loading–unloading compressive stress curves at different strains from 10% to 80%. Scale bars represent 10 mm. (e) Compressive stress–strain curves at 80% strain for 10 cycles. | ||
Biocompatibility of the 3D-printed elastomers
Cardiovascular stenosis and occlusion are among the most prevalent cardiovascular diseases (Fig. 6a).49 Currently, stent intervention therapy is the most effective approach. Polymer stents exhibit better biocompatibility compared to metal stents.50,51 Biomedical polyurethane is a polymer material with excellent biocompatibility and durability, which can effectively minimize immune rejection and inflammatory responses when used in the body for a long time.52,53 In addition, its superior elasticity and flexibility facilitate free expansion of stent implants within the blood vessel to adapt to complex vascular environments.54 The surface of polyurethane can easily introduce drug coatings or other functional molecules through physical or chemical methods to further reduce the risk of thrombosis.55 The inherent low protein adsorption and good hemocompatibility of polyurethanes also minimize the potential for platelet adhesion formation.53 These advantages make it ideal for the construction of cardiovascular stent implants. Given the significant advantages of VP 3D printed polyurethane in terms of mechanical properties and rapid structural manufacturing, it is easy to fabricate stents that can adapt to varying vessel sizes as a concept validation of polyurethane stents (Fig. 6b). Additionally, we verified the compressive load capacity and load stability of support structures of different sizes. The results indicate that polyurethane stents maintain good load-bearing capacity and mechanical stability even after being immersed in 37 °C PBS buffer for three days (Fig. 6c and Fig. S21, ESI†). To confirm the cytotoxicity of the prepared polyurethane stent it was further evaluated by using a cell counting kit-8 (CCK-8) and live/dead staining assay. As shown in Fig. 6d and e the cell viability of the HMDI-PUU2000 elastomer was found to be higher than 80%. And meanwhile, the fluorescence microscopy images based on live/dead staining further revealed that the cells are morphologically healthy and had beneficial proliferation activity, indicating that the elastomer possessed excellent biocompatibility.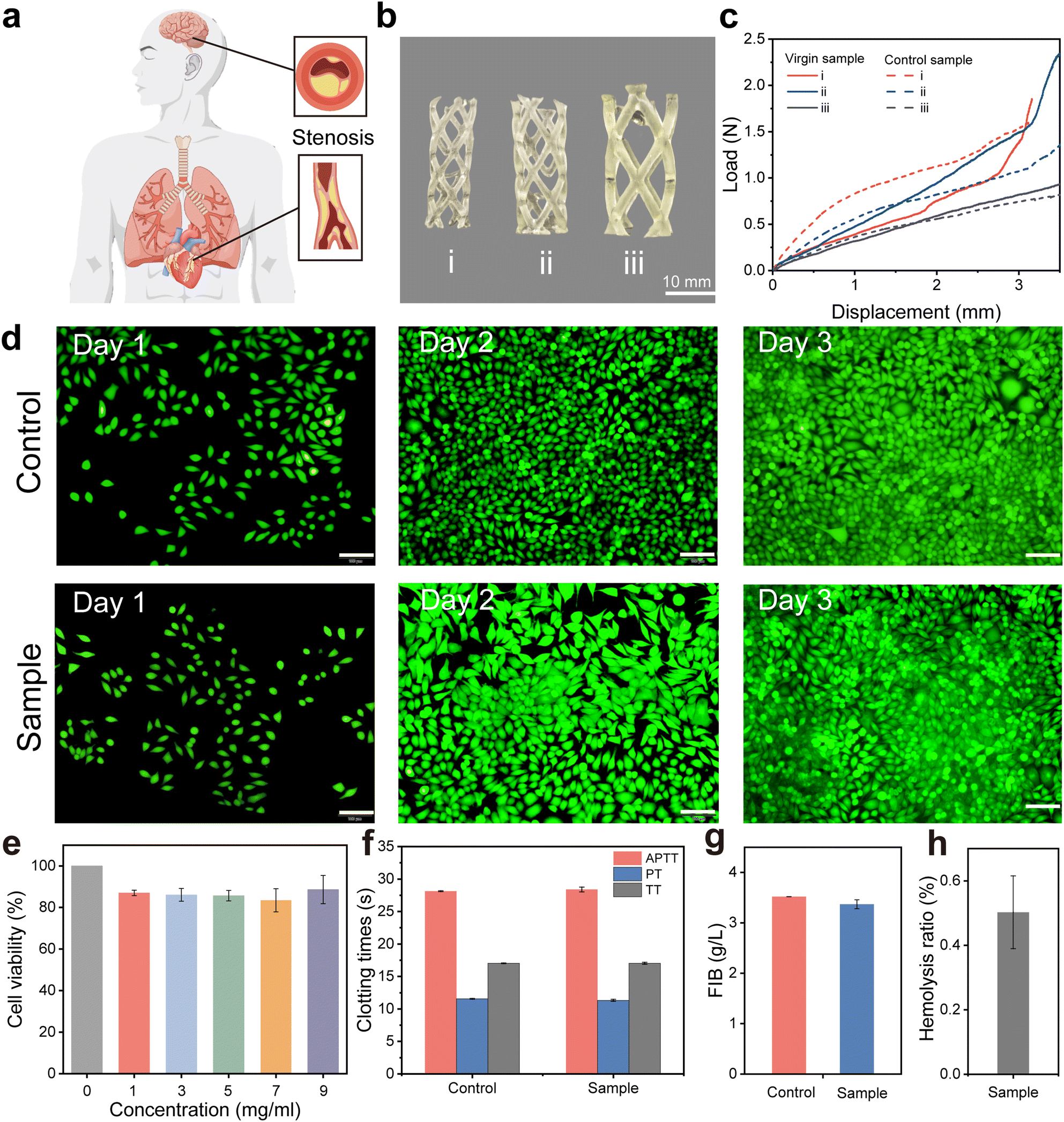 | ||
| Fig. 6 Mechanical properties and biocompatibility of polyurethane stents. (a) Illustration of stenotic lesions in different organs of the human body. (b) Physical photographs of different sizes stents. Scale bars represent 1 cm. (c) Load-displacement curves of stents with different sizes (control sample: 3 days at 37 °C in PBS buffer). (d) Live and dead cell staining. Scale bars represent 500 μm. (e) CCK-8 assay of cell viability. Error bars represent the standard deviation (n = 3). (f)–(h) Values of four coagulation indices, APTT, TT, PT, and FIB, for the HMDI-PUU2000 elastomer. Error bars represent the standard deviation (n = 3). | ||
Subsequently, the hemocompatibility of polyurethane elastomers was further estimated by using four indicators of blood coagulation, namely activated partial thromboplastin time (APTT), thrombin time (TT), prothrombin time (PT), and fibrinogen (FIB). As revealed in Fig. 6f and g the anticoagulant performance of polyurethane elastomers exhibits an APTT of 28.40 ± 0.36 s, PT of 11.33 ± 0.15 s, TT of 17.3 ± 0.15 s, and FIB of 3.37 ± 0.09 mg ml−1. The results demonstrated that the anticoagulant properties of the 3D printed elastomer were much better than that of platelet-poor plasma (PPP). Moreover, the elastomers displayed a hemolysis rate (HR) of 0.50 ± 0.11, which also met the reference HR standard of 5% in biomedical fields (Fig. 6h and Fig. S22, ESI†). The above-mentioned results indicated that the VP 3D-printed polyurethane elastomers have beneficial biocompatibility and hemocompatibility, serving as a promising candidate biomaterial for biomedical applications. The proposed 3D printable polyurethane elastomers are expected to provide the possibility of rapid manufacturing of complex medical devices for biomedical applications.
Conclusions
In conclusion, we have successfully prepared mechanically robust vat photopolymerization 3D-printed elastomers through the synergistic effect of dynamic hydrogen bonding and covalent crosslinked networks. Compared with the 3D-printed elastomers reported to date, the proposed poly(urethane–urea) elastomers possessed better mechanical properties, such as tensile strength, resilience, fatigue resistance, and tear resistance. Due to its favorable rheological behaviors and high photopolymerization rate, furthermore, various elastomers 3D objects with sophisticated and high-precision features, such as metamaterial structures, cylinders, and tapered arrays, were also realized by vat photopolymerization 3D printing. What is more, the as-fabricated PUEs also displayed beneficial biocompatibility and hemocompatibility, which could meet the requirements of implantable biomaterials. As a proof of concept, the cardiovascular stents with high-precision and exceptional mechanical performance were easily manufactured. It is believed that VP 3D printing of PUEs with high performance and flexible structure manufacturing would broaden their application in biomedicine and other engineering fields.Animal cell experiments
All animal cell procedures were approved by the Animal Experimentation Ethics Committee of Yanxuan Biotechnology (Hangzhou) Co., Ltd (permission number SYXK (Zhejiang)2020-0043), and all procedures were performed in strict accordance with the Guide for the Care and Use of Laboratory Animals and the Regulation of Animal Protection Committee.Human blood experiments
The human blood experiments involved in this experiment were conducted at Yanxuan Biotechnology (Hangzhou) Co., Ltd and approved by the Experimental Animal Ethics Committee of Yanxuan Biotechnology (Hangzhou) Co., Ltd (permission number SYXK (Zhejiang)2020-0043)Data availability
The data used to support the findings of this study are available from the corresponding author upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support from the National Key Research and Development Program of China (2022YFB4600101), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0470303), the National Natural Science Foundation of China (52175201, 52205228, and 51935012), the Research Program of Science and Technology Department of Gansu Province (22JR5RA107, 22JR5RA093, and 24JRRA044), the Shandong Provincial Natural Science Foundation (ZR2023OE090), the Major Program (ZYFZFX-2) and the Cooperation Foundation for Young Scholars (HZJJ23-02) of the Lanzhou Institute of Chemical Physics, CAS, the Western Light Project, CAS (xbzg-zdsys-202007), and the Taishan Scholars Program. The authors would like to thank Minglong Wang from Shiyanjia Lab (www.shiyanjia.com) for the hemocompatible analysis and SCI-GO (www.sci-go.com) for the thermal analysis measurement.Notes and references
- H. Goodarzi Hosseinabadi, A. Biswas, A. Bhusal, A. Yousefinejad, A. Lall, W.-H. Zimmermann, A. K. Miri and L. Ionov, Small, 2024, 20, 2306387 CrossRef CAS PubMed.
- D. K. Patel, A. H. Sakhaei, M. Layani, B. Zhang, Q. Ge and S. Magdassi, Adv. Mater., 2017, 29, 1606000 CrossRef.
- J. Mauriello, R. Maury, Y. Guillaneuf and D. Gigmes, Adv. Mater. Technol., 2023, 8, 2300366 CrossRef CAS.
- B. Zhang, H. Li, J. Cheng, H. Ye, A. H. Sakhaei, C. Yuan, P. Rao, Y.-F. Zhang, Z. Chen, R. Wang, X. He, J. Liu, R. Xiao, S. Qu and Q. Ge, Adv. Mater., 2021, 33, 2101298 CrossRef CAS.
- J. Cheng, R. Wang, Z. Sun, Q. Liu, X. He, H. Li, H. Ye, X. Yang, X. Wei, Z. Li, B. Jian, W. Deng and Q. Ge, Nat. Commun., 2022, 13, 7931 CrossRef CAS.
- X. Huang, S. Peng, L. Zheng, D. Zhuo, L. Wu and Z. Weng, Adv. Mater., 2023, 35, 2304430 CrossRef CAS.
- H. Wu, X. Luo, C. Wang, Q. Jin, Y. Li, F. Guo, W. Guo and Y. Long, Colloids Surf., A, 2024, 685, 133248 CrossRef CAS.
- Y.-C. M. Wu, G. Chyr, H. Park, A. Makar-Limanov, Y. Shi, J. M. DeSimone and Z. Bao, Chem. Sci., 2013, 14, 12535–12540 RSC.
- S. Peng, Y. Li, L. Wu, J. Zhong, Z. Weng, L. Zheng, Z. Yang and J.-T. Miao, ACS Appl. Mater. Interfaces, 2020, 12, 6479–6488 CrossRef CAS PubMed.
- S. Peng, N. Thirunavukkarasu, J. Chen, X. Zheng, C. Long, X. Huang, Z. Weng, L. Zheng, H. Wang, X. Peng and L. Wu, Chem. Eng. J., 2023, 463, 142312 CrossRef CAS.
- J. Shi, Z. Wang, T. Zheng, X. Liu, B. Guo and J. Xu, Mater. Horiz., 2022, 9, 3070–3077 RSC.
- J. Herzberger, J. M. Sirrine, C. B. Williams and T. E. Long, Prog. Polym. Sci., 2019, 97, 101144 CrossRef CAS.
- Y. Yao, B. Liu, Z. Xu, J. Yang and W. Liu, Mater. Horiz., 2021, 8, 2742–2749 RSC.
- Z. Weng, X. Huang, S. Peng, L. Zheng and L. Wu, Nat. Commun., 2023, 14, 4303 CrossRef CAS PubMed.
- S. Zhang, Q. Gao, Y. Zhang, X. Sheng, Z. Miao, J. Qin, G. Zhang and X. Shi, Addit. Manuf., 2023, 76, 103770 CAS.
- R. Guo, Q. Zhang, Y. Wu, H. Chen, Y. Liu, J. Wang, X. Duan, Q. Chen, Z. Ge and Y. Zhang, Adv. Mater., 2023, 35, 2212130 CrossRef CAS.
- H. Lai, X. Peng, L. Li, D. Zhu and P. Xiao, Polym. Sci., 2022, 128, 101529 CAS.
- J. L. Rapp, M. A. Borden, V. Bhat, A. Sarabia and F. A. Leibfarth, ACS Polym. Au, 2024, 4, 120–127 CrossRef CAS.
- Y. Chen, Z. Yu, H. Oguzlu, J. Jiang, M. Cho, M. Karaaslan, S. Renneckar and F. Jiang, Addit. Manuf., 2021, 46, 102107 CAS.
- A. Al Rashid, W. Ahmed, M. Y. Khalid and M. Koç, Addit. Manuf., 2021, 47, 102279 CAS.
- X. Huang, S. Peng, L. Zheng, D. Zhuo, L. Wu and Z. Weng, Adv. Mater., 2023, 35, 2304430 CrossRef CAS.
- Y. Wu, M. Fei, T. Chen, C. Li, T. Fu, R. Qiu and W. Liu, Addit. Manuf., 2021, 47, 102268 CAS.
- G. Ding, X. Li, M. Sun, Y. He, F. Zhao, T. Wu, J. Wang, S. Ren, W. Shi, L. Xu, X. Hu, W. Huang, R. Yu and Y. Ao, Chem. Eng. J., 2022, 431, 133861 CrossRef CAS.
- S. Peng, Z. Wang, J. Lin, J. Miao, L. Zheng, Z. Yang, Z. Weng and L. Wu, Adv. Funct. Mater., 2020, 31, 2008729 CrossRef.
- N. Thirunavukkarasu, J. Gao, S. Peng, A. Laroui, L. Wu and Z. Weng, Addit. Manuf., 2023, 66, 103451 Search PubMed.
- X. Li, R. Yu, Y. He, Y. Zhang, X. Yang, X. Zhao and W. Huang, ACS Macro Lett., 2019, 8, 1511–1516 CrossRef CAS.
- W. Wang, S. Sun, S. Hu, B. Yang, S. He, R. Wang and L. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 3324–3333 CrossRef CAS.
- L. Zhao, R. Yu, Y. He, M. Zhang, F. Tian, L. Wang, Y. Zhao and W. Huang, Addit. Manuf., 2024, 79, 103904 CAS.
- H. Zhang, D. Lu, S. Yang, Y. He, H. Zhang and J. Bao, Polymer, 2024, 292, 126657 CrossRef CAS.
- O. Bliah, S. Joe, R. Reinberg, A. B. Nardin, L. Beccai and S. Magdassi, Mater. Horiz., 2023, 10, 4976–4985 RSC.
- J. Ye, G. Lin, Z. Lin, H. Deng, J. Huang, H. Xiang, M. Z. Rong and M. Q. Zhang, Macro Mater. Eng., 2022, 307, 2100874 CrossRef CAS.
- W.-B. Lim, J.-H. Bae, M.-J. Seo, J.-G. Min, J.-H. Lee, S. H. Kim, Y.-S. Jung and P. Huh, Addit. Manuf., 2022, 51, 102625 CAS.
- Y. Bao, N. Paunović and J. Leroux, Adv. Funct. Mater., 2022, 32, 2109864 CrossRef CAS.
- P. Jiang, Y. Zhang, X. Mu, D. Liu, Y. Liu, R. Guo, Z. Ji, X. Wang and X. Wang, Adv. Mater. Technol., 2022, 7, 2101288 CrossRef CAS.
- I. Noda, Appl. Spectrosc., 1993, 47, 1329–1336 CrossRef CAS.
- K. Mantala and D. Crespy, Macromolecules, 2023, 56, 7332–7343 CrossRef CAS.
- Z. Li, Y.-L. Zhu, W. Niu, X. Yang, Z. Jiang, Z.-Y. Lu, X. Liu and J. Sun, Adv. Mater., 2021, 33, 2101498 CrossRef CAS.
- Y. W. Wen, M. Li, L. F. Fan, M. Z. Rong and M. Q. Zhang, Adv. Mater., 2024, 36, 2406574 CrossRef CAS.
- L. Wang, K. Zhang, X. Zhang, Y. Tan, L. Guo, Y. Xia and X. Wang, Adv. Mater., 2024, 36, 2311758 CrossRef CAS.
- J. Wu, Z. Zhang, Z. Wu, D. Liu, X. Yang, Y. Wang, X. Jia, X. Xu, P. Jiang and X. Wang, Adv. Funct. Mater., 2022, 33, 2210395 CrossRef.
- J. Chen, Y. Gao, L. Shi, W. Yu, Z. Sun, Y. Zhou, S. Liu, H. Mao, D. Zhang, T. Lu, Q. Chen, D. Yu and S. Ding, Nat. Commun., 2022, 13, 4868 CrossRef CAS PubMed.
- Z. Fang, H. Mu, Z. Sun, K. Zhang, A. Zhang, J. Chen, N. Zheng, Q. Zhao, X. Yang, F. Liu, J. Wu and T. Xie, Nature, 2024, 631, 783–788 CrossRef CAS.
- H. Wu, H. Wang, M. Luo, Z. Yuan, Y. Chen, B. Jin, W. Wu, B. Ye, H. Zhang and J. Wu, Mater. Horiz., 2024, 11, 1548–1559 RSC.
- X. Huang, S. Peng, L. Zheng, D. Zhuo, L. Wu and Z. Weng, Adv. Mater., 2023, 35, 2304430 CAS.
- C. B. Cooper, S. Nikzad, H. Yan, Y. Ochiai, J.-C. Lai, Z. Yu, G. Chen, J. Kang and Z. Bao, ACS Cent. Sci., 2021, 7, 1657–1667 CrossRef CAS PubMed.
- J. Chen, Z. Wang, B. Yao, Y. Geng, C. Wang, J. Xu, T. Chen, J. Jing and J. Fu, Adv. Mater., 2024, 36, 2401178 CAS.
- J. Jing, B. Yao, W. Sun, J. Chen, J. Xu and J. Fu, Angew. Chem., Int. Ed., 2024, e202410 Search PubMed.
- Z. Li, Y.-L. Zhu, W. Niu, X. Yang, Z. Jiang, Z.-Y. Lu, X. Liu and J. Sun, Adv. Mater., 2021, 33, 2101498 CAS.
- P. Unger, A. Powers, E. Le Nezet, E. Lacasse-Rioux, X. Galloo and M.-A. Clavel, J. Am. Cardiol., 2024, 83, 1109–1119 CrossRef PubMed.
- L. Ning, S. Zanella, M. L. Tomov, M. S. Amoli, L. Jin, B. Hwang, M. Saadeh, H. Chen, S. Neelakantan, L. P. Dasi, R. Avazmohammadi, M. Mahmoudi, H. D. Bauser-Heaton and V. Serpooshan, Adv. Sci., 2024, 10, 2400476 CrossRef.
- M. Li, M. Jiang, Y. Gao, Y. Zheng, Z. Liu, C. Zhou, T. Huang, X. Gu, A. Li, J. Fang and X. Ji, Bioact. Mater., 2022, 11, 140–153 CAS.
- X. Guo, J. Liang, Z. Wang, J. Qin, Q. Zhang, S. Zhu, K. Zhang and H. Zhu, Adv. Mater., 2023, 35, 2210092 CrossRef CAS.
- H. Wang, T. Li, J. Li, R. Zhao, A. Ding and F.-J. Xu, Prog. Polym. Sci., 2024, 151, 101803 CrossRef CAS.
- Z. Hou, Y. Wu, C. Xu, S. Reghu, Z. Shang, J. Chen, D. Pranantyo, K. Marimuth, P. P. De, O. T. Ng, K. Pethe, E.-T. Kang, P. Li and M. B. Chan-Park, ACS Cent. Sci., 2020, 6, 2031–2045 CrossRef CAS PubMed.
- Z. Zhang, L. Wang, J. Liu, H. Yu, X. Zhang, J. Yin, S. Luan and H. Shi, Small, 2023, 19, 2304 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mh01191k |
| ‡ Current address: College of Chemistry and Environmental Engineering, Shenzhen University, Shenzhen 518060, P. R. China. |
| This journal is © The Royal Society of Chemistry 2025 |