
A porous network of boron-doped IrO2 nanoneedles with enhanced mass activity for acidic oxygen evolution reactions†
Fei
Hu
*a,
Peiyu
Huang
a,
Xu
Feng
a,
Changjian
Zhou
a,
Xinjuan
Zeng
 a,
Congcong
Liu
c,
Guangjin
Wang
*a,
Xiaowei
Yang
*b and
Huawen
Hu
*a
a,
Congcong
Liu
c,
Guangjin
Wang
*a,
Xiaowei
Yang
*b and
Huawen
Hu
*a
aSchool of Materials Science and Hydrogen Energy, Foshan University, Foshan, 528000, P.R. China. E-mail: mfhufei@fosu.edu.cn; wgj501@163.com; huawenhu@126.com
bSchool of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Shanghai 200240, P.R. China. E-mail: yangxw@sjtu.edu.cn
cFlexible Electronics Innovation Institute (FEII), Jiangxi Science and Technology Normal University, Nanchang 330013, P.R. China
First published on 1st November 2024
Abstract
While proton exchange membrane water electrolyzers (PEMWEs) are essential for realizing practical hydrogen production, the trade-off among activity, stability, and cost of state-of-the-art iridium (Ir)-based oxygen evolution reaction (OER) electrocatalysts for PEMWE implementation is still prohibitively challenging. Ir minimization coupled with mass activity improvement of Ir-based catalysts is a promising strategy to address this challenge. Here, we present a discovery demonstrating that boron doping facilitates the one-dimensional (1D) anisotropic growth of IrO2 crystals, as supported by both experimental and theoretical evidence. The synthesized porous network of ultralong boron-doped iridium oxide (B-IrO2) nanoneedles exhibits improved electronic conductivity and reduced charge transfer resistance, thereby increasing the number of active sites. As a result, B-IrO2 displays an ultrahigh OER mass activity of 3656.3 A gIr−1 with an Ir loading of 0.08 mgIr cm−2, which is 4.02 and 6.18 times higher than those of the un-doped IrO2 nanoneedle network (L-IrO2) and Adams IrO2 nanoparticles (A-IrO2), respectively. Density functional theory (DFT) calculations reveal that the B doping moderately increases the d-band center energy level and significantly lowers the free energy barrier for the conversion of *O to *OOH, thereby improving the intrinsic activity. On the other hand, the stability of B-IrO2 can be synchronously promoted, primarily attributed to the B-induced strengthening of the Ir bonds, which help resist electrochemical dissolution. More importantly, when the B-IrO2 catalysts are applied to the membrane electrode assembly for PEM water electrolysis (PEMWE), they generate a remarkable current density of up to 2.8 A cm−2 and maintain operation for at least 160 h at a current density of 1.0 A cm−2. This work provides new insights into promoting intrinsic activity and stability while minimizing the usage of noble-metal-based OER electrocatalysts for critical energy conversion and storage.
New conceptsIrO2-based materials are regarded as state-of-the-art electrocatalysts for sluggish oxygen evolution reactions. The improvement of the intrinsic activity and durability while maintaining a low usage for membrane electrode assembly fabrication in proton exchange membrane water electrolyzers (PEMWEs) is vital for bringing IrO2-based materials closer to real-world practical clean energy production. We present a discovery demonstrating that boron doping facilitates the one-dimensional (1D) anisotropic growth of IrO2 crystals. The synthesized porous network of ultralong boron-doped iridium oxide (B-IrO2) nanoneedles exhibits improved electronic conductivity and reduced charge transfer resistance, thereby increasing the number of active sites. While the morphology control to form a nanoneedle structure is favorable for enhancing the electrical conductivity and electrochemically active site number through elevating the concentration of active IrIII species and hydroxyl groups (OOH), B doping drives the unidirectional growth of IrO2 nanoneedles and moderately increases the d-band center energy level and significantly lowers the free energy barrier for the conversion of *O to *OOH, thereby improving the intrinsic activity. On the other hand, the stability of B-IrO2 can be synchronously promoted, primarily attributed to the B-induced strengthening of the Ir bonds, which help resist electrochemical dissolution. This new concept is highly expected to be generalized to many other noble- and non-noble-metal-based electrocatalysts for various critical energy conversion and storage applications. |
Introduction
Water electrolyzers’ installation capacitance is soaring for the decarbonization of modern society, as they can convert carbon-neutral renewable electricity into green hydrogen energy sources.1–3 With superiorities to their alkaline counterparts albeit with higher costs, proton exchange membrane water electrolyzers (PEMWEs) have attracted significant attention due to their fast response, low ohmic losses, ultra-high-purity hydrogen, and high power density, thus being more suitable and viable for coupling with intermittent renewable energy sources (e.g., solar, wind, tidal, and geothermal).4–6 In PEMWEs, protons discharged from water oxidation (oxygen evolution reaction, OER) migrate to the cathode through the proton exchange membrane for the subsequent hydrogen evolution reaction (HER). OER's complexity and requirements of multistep proton-coupled electron transfer processes relative to the HER result in its sluggish reaction kinetics. Therefore, it is of priority to develop PEMWE anodic OER electrocatalysts with high performance (including catalytic activity, corrosion resistance, and electrochemical stability) under harsh conditions (e.g., low pH and high oxidative potential).7,8IrO2 is the only reported state-of-the-art OER electrocatalyst with both high activity and stability for PEMWEs,9,10 which can be ascribed to its electronic structure, low resistivity, and excellent chemical stability.11,12 Although RuO2 may have higher activity compared with IrO2, RuO2 suffers from dissolution problems based on the lattice oxygen mechanism.13 To facilitate the scaling up of an IrO2-based electrolyzer using this expensive and scarce material in the Earth's crust,14 one needs to significantly reduce the IrO2 loading at the anode to below 0.1 mgIr cm−2, a notable decrease from the current range of 1–2 mgIr cm−2.15–17
While commonly used IrO2 nanoparticles tend to aggregate over time, leading to a gradual decline in catalytic activity and electrochemical stability,18 one-dimensional (1D) nanostructured electrocatalysts exhibit inherent anisotropic properties such as a larger aspect ratio, superior electrical conductivity,19 and enhanced stability.20–23 As a result, these 1D nanostructures can help minimize the use of iridium at the anode while maintaining high OER activity. For example, IrO2 nanoneedles with a length of ∼30 nm exhibit a mass activity of 51.6 A gIrO2−1, surpassing IrO2 nanoparticles with a mass activity of 15.5 A gIrO2−1 at 1.55 VRHE.23 Similarly, nanoribbon layered IrO2 with a length of around 22.53 μm demonstrates a mass activity of 2354.5 A gIr−1, which is 175.7 times higher than that of IrO2 nanoparticles (13.4 A gIr−1) at 1.5 VRHE.24 Therefore, the development of 1D nanostructured Ir-based materials is essential to help reduce the capital cost of PEMWEs while inheriting the superior electrochemical performance of Ir-based electrocatalysts.
Besides morphology tuning, many other strategies (e.g., multi-component incorporation and heteroatom doping) have been employed to modulate the local environment and electronic structure, regulate oxygen binding strength, and consequently enhance the Ir-based electrocatalyst performance.25 A wide spectrum of elements have been explored for Ir-based electrocatalyst doping, such as Pb,26 Ag,27 Sn,28 Mn,29 and B,30 especially the metal elements. However, the secondary metal introduced through doping may leach out and contaminate the Nafion membrane, thus causing the cell performance to deteriorate.31 In contrast, nonmetal element boron (B) doping provides a promising solution to address this challenge. Our group previously reported that the electron-deficient nature of B enables affinities to surface metal atoms, providing extra binding sites (such as metal–B–O bonds) and hence contributing to the superior OER activity.32,33 Meanwhile, the concomitantly enhanced electronic conductivity induced by B doping ultimately boosted OER activity and stability.32–36
Considering the favorable 1D nanostructure and B doping of Ir-based materials, we hypothesize that B doping of 1D nanostructured Ir-based electrocatalysts might give rise to high-performance Ir-based OER electrocatalysts with both activity and stability for large-scale OER applications in acidic environments. A scalable modified Adams fusion method was used to produce such a B-doped IrO2 nanostructure (in this case, a porous network of nanoneedles (B-IrO2)). The incorporation of boron species in the IrO2 catalyst promotes the controlled unidirectional growth of IrO2 nanoneedles. The reduced crystal boundaries decrease scattering and hence enhance the electronic conductivity. This adjustment also strikes a balance between the adsorption and desorption of oxygen intermediates, as evidenced by an increase in the d-band center energy level based on DFT calculations, leading to a notable enhancement in the intrinsic OER activity of the catalyst. When put to the test, the overpotentials required for OER operation at current densities of 40, 100, and 1000 mA cm−2 are measured at 233, 268, and 409 mV, respectively in a 0.5 M H2SO4 solution. Moreover, we integrate the B-IrO2 catalysts into a single-cell PEMWE, and water-splitting reactions necessitated voltages of 1.611, 1.998, and 2.679 for achieving current densities of 300, 1000, and 2500 mA cm−2, respectively. They generate a remarkable current density of up to 2.8 A cm−2 and maintain operation for at least 160 h at a current density of 1.0 A cm−2. The first efforts dedicated in this work to combine 1D morphology tuning and non-metal B doping of IrO2 open up an avenue to generate high-performance Ir-based electrocatalysts with well-balanced activity, stability, and cost.
Results and discussion
Synthesis and characterization of B-IrO2
The B-IrO2 materials were synthesized using a modified Adams fusion method,37,38 as depicted in Fig. 1. Initially, hexachloroiridate(IV) hydrate and NaNO3 were mixed in water, followed by the addition of cysteamine HCl as a structure-shaping agent,18,19 and boric acid (H3BO3) was subsequently introduced as the doping source. The resulting mixture was dried at 80 °C, ground, and then calcined at varying temperatures. The remaining metal salts were thoroughly washed with deionized (DI) water, and the product denoted as B-IrO2 was collected via centrifugation. For comparison, a pure IrO2 nanoneedle network (denoted as L-IrO2) was synthesized according to the procedures above except for that the B doping source was not added. Additionally, traditional Adams IrO2 nanoparticles (denoted as A-IrO2) were also prepared according to the same methodology except for that both B-doping and structure-shaping agents were not included. Cysteamine HCl has been tested as a favourable shaping agent for the anisotropic growth of IrO2, as illustrated in Fig. S1 in the ESI.†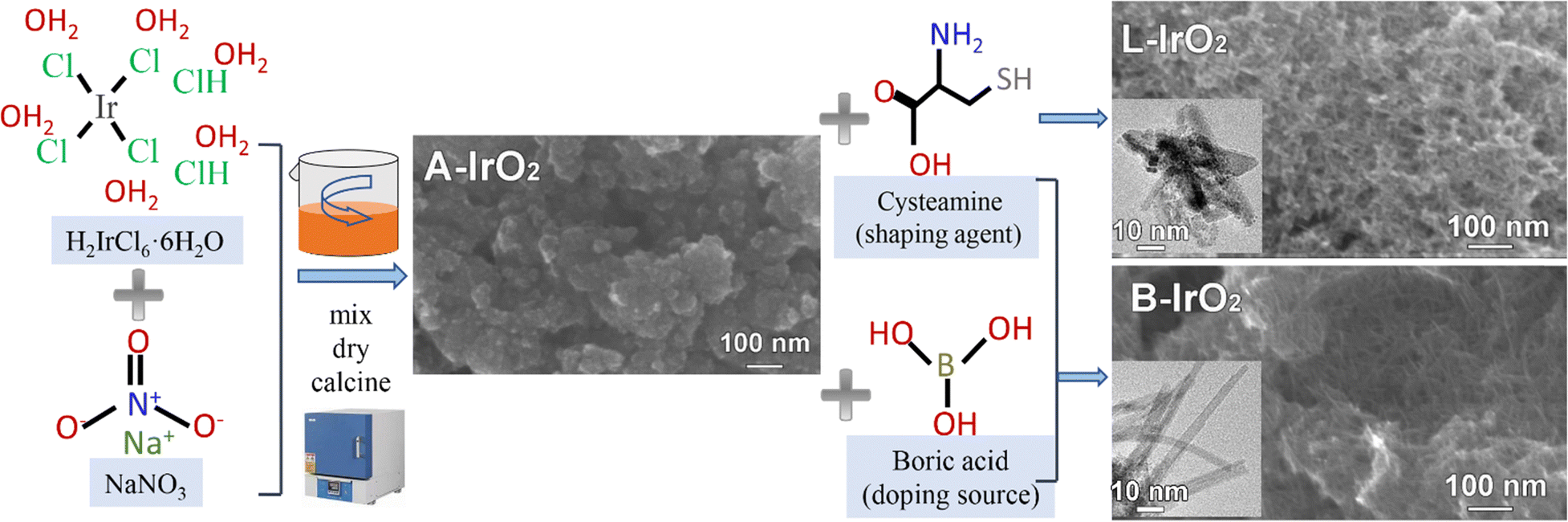 | ||
| Fig. 1 Schematic illustration for synthesizing B-IrO2 based on a modified Adams fusion method: the shaping agent and the boron sources were sequentially mixed, dried, and calcined to obtain B-IrO2. | ||
The scanning electron microscopy (SEM) images of B-IrO2 reveal a notable transformation in IrO2 morphology from an unshaped granular form (Fig. S2a, ESI†) to a porous network of 1D nanoneedles (Fig. S2b–d, ESI†) upon the addition of cysteamine HCl. The introduction of H3BO3 further enhances this transformation, facilitating the unidirectional growth of IrO2 into elongated and slender nanoneedle structures that intertwine with each other to form a porous network (Fig. S2b, ESI†). The resulting B-IrO2 nanoneedles exhibit entangled and slightly curved configurations, as corroborated by the transmission electron microscopy (TEM) image in Fig. 2a and Fig. S3 (ESI†). The brightest diffraction ring corresponding to the (101) plane observed in the selected area electron diffraction (SAED, inset of Fig. 2a) aligns with the X-ray diffraction (XRD) results shown in Fig. 2b, which can be indexed to the rutile IrO2 phase (PDF 88-0288) with a preference for the (101) plane orientation. The high-resolution TEM image in Fig. 2c unveils the ultrathin nature of IrO2, comprising approximately 5–7 layers of the IrO2 plane, with an interplanar spacing of the distinct lattice fringes calculated to be 0.32 nm, corresponding to the well-defined Ir(110) facet of the face-centered cubic (fcc) lattice.39 The cross-sectional diameter of a single needle is measured to be about 1.4 nm ± 0.3 nm, significantly slimmer compared to the un-doped IrO2 (L-IrO2, Fig. S2c and d, ESI†). The reduced crystal boundaries in the lattice planes of IrO2 nanoneedles contribute to decreased scattering, thereby enhancing charge transport and electronic conductivity.40–43
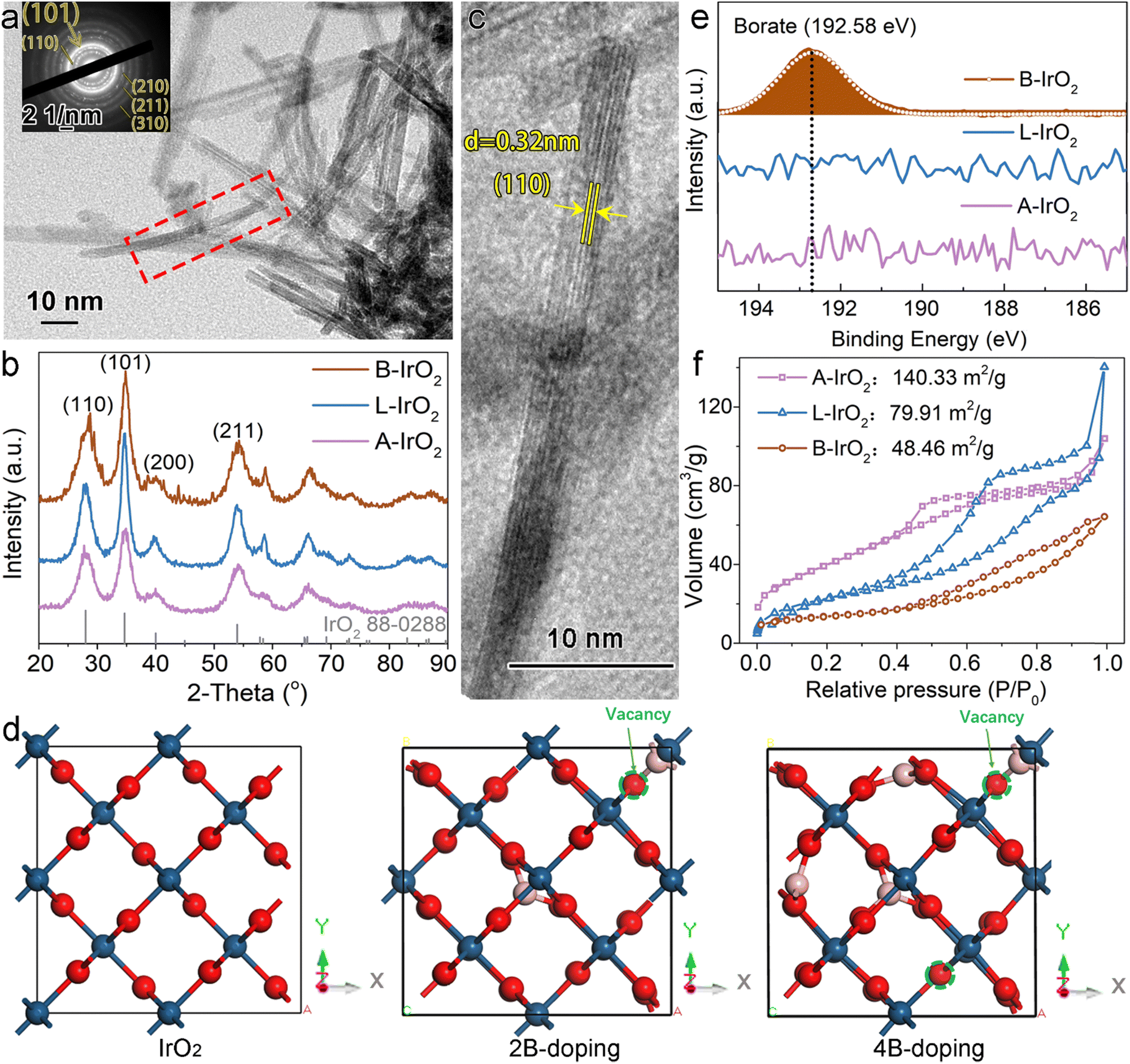 | ||
| Fig. 2 Morphology and structural analysis. (a) TEM image and SAED pattern of B-IrO2. (b) PXRD patterns of different IrO2-based samples, including B-IrO2, L-IrO2, and A-IrO2. (c) HRTEM image of B-IrO2. (d) The structure of IrO2 and B-IrO2 by doping two and four boron atoms based on DFT computations. Red, blue, and pink spheres represent oxygen, iridium, and boron atoms, respectively. (e) and (f) XPS spectra (e) and BET test results (f) of B-IrO2, L-IrO2, and A-IrO2. | ||
The unique elongated and curved morphologies of B-IrO2 reveal the beneficial impact of boron doping on the crystal growth of IrO2. By systematically varying the atomic ratio of B to Ir (B/Ir) in the precursors, we observe a morphologic transition in the IrO2 structure as the B/Ir increases from 0 to 3, 5, 8, 11, and 13, evolving from a short nanoneedle network to web-like wires, as illustrated in Fig. S4 (ESI†). In Fig. 2d and Fig. S5, S6 (ESI†), we simulate the incorporation of boron atoms into the IrO2 crystal, with the calculated lattice parameters presented in Table 1. Our calculated lattice parameters for IrO2 closely match those reported in other calculations44 and experiments,45 validating the reliability of our results. Upon doping with two boron atoms, all three axes (a, b, and c) shrink, resulting in a c/a(b) ratio of 0.706. When four boron atoms are incorporated, the c axis shrinks substantially compared to the a and b axes, yielding a c/a or c/b ratio of 0.682 or 0.681, respectively. More B doping gradually alters the crystal lattice to facilitate anisotropic growth. This observation signifies a notable unidirectional growth of IrO2 crystals. Therefore, the incorporation of boron atoms into the IrO2 crystal lattice drives anisotropic growth along specific crystallographic directions.
| Sample | Lattice parameters (Å) | Ref. | ||||
|---|---|---|---|---|---|---|
| a | b | c | c/a | c/b | ||
| Experimental | 9.018 | 9.018 | 6.322 | 0.701 | 0.701 | 45 |
| Theoretical calculation | 9.071 | 9.071 | 6.3898 | 0.704 | 0.704 | 44 |
| IrO2 | 9.00276 | 9.00276 | 6.34609 | 0.705 | 0.705 | This work |
| 2B doped IrO2 | 8.91708 | 8.91731 | 6.29410 | 0.706 | 0.706 | This work |
| 4B doped IrO2 | 8.93761 | 8.95544 | 6.09476 | 0.682 | 0.681 | This work |
| 6B doped IrO2 | 8.66720 | 8.86011 | 5.84001 | 0.674 | 0.659 | This work |
| 8B doped IrO2 | 8.64052 | 8.97547 | 5.50542 | 0.637 | 0.613 | This work |
Inductively coupled plasma optical emission spectroscopy (ICP-OES) is employed to determine the mass of Ir and B in the synthesized material. The analysis reveals the B/Ir molar ratio to be 1.26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Subsequently, X-ray photoelectron spectroscopy (XPS) was utilized to prove the local structure of boron species within the B-IrO2 sample (Fig. 2e). A distinct peak at ∼192.58 eV observed in the B 1s spectrum of B-IrO2 is indicative of boron-oxo species (B–O), in agreement with previous observations of borates.32,33,46,47 This result affirms the successful incorporation of boron species into the structure. Considering the atomic charge of B and Ir, it is plausible that B replaces Ir in the lattice. Subsequent measurements of B-IrO2 through nitrogen adsorption–desorption isotherms (Fig. 2f) reveal a Brunauer–Emmett–Teller (BET) specific surface area of approximately 48.46 m2 g−1 for B-IrO2, lower than the values recorded for L-IrO2 (79.91 m2 g−1) and A-IrO2 (140.33 m2 g−1). Similar N2 adsorption isotherms were previously obtained for IrO2-based granules24,30 and nanoneedles.23,24 The slender morphology of B-IrO2 with larger average pore sizes (Fig. S7, ESI†) and much smaller micropore volumes (Table S1, ESI†) results in reduced specific surface areas, as micropores are more effective in increasing the surface area.48,49
:1. Subsequently, X-ray photoelectron spectroscopy (XPS) was utilized to prove the local structure of boron species within the B-IrO2 sample (Fig. 2e). A distinct peak at ∼192.58 eV observed in the B 1s spectrum of B-IrO2 is indicative of boron-oxo species (B–O), in agreement with previous observations of borates.32,33,46,47 This result affirms the successful incorporation of boron species into the structure. Considering the atomic charge of B and Ir, it is plausible that B replaces Ir in the lattice. Subsequent measurements of B-IrO2 through nitrogen adsorption–desorption isotherms (Fig. 2f) reveal a Brunauer–Emmett–Teller (BET) specific surface area of approximately 48.46 m2 g−1 for B-IrO2, lower than the values recorded for L-IrO2 (79.91 m2 g−1) and A-IrO2 (140.33 m2 g−1). Similar N2 adsorption isotherms were previously obtained for IrO2-based granules24,30 and nanoneedles.23,24 The slender morphology of B-IrO2 with larger average pore sizes (Fig. S7, ESI†) and much smaller micropore volumes (Table S1, ESI†) results in reduced specific surface areas, as micropores are more effective in increasing the surface area.48,49
The chemical states of Ir and O in B-IrO2, L-IrO2, and A-IrO2 are further examined using XPS. Following the high-temperature Adams fusion process, all the IrO2 variants exhibit similar oxidic Ir characteristics, with no detectable metallic Ir. The oxidation states of Ir are identified as IrIV (4f 7/2) and IrIII (4f 5/2) species, along with their respective satellites, positioned at 61.7 eV (64.7 eV) and 62.5 eV (65.5 eV), as illustrated in Fig. 3a.33,50,51 The proportion of IrIII species is found to be 51.05%, 54.06%, and 56.91% for A-IrO2, L-IrO2, and B-IrO2, respectively. As IrIII species are recognized as precursors for active sites,52,53 the elevated concentration of IrIII species in B-IrO2 is indicative of enhanced activity for the OER,54 typically originating from vacancies on less crystalline IrO2 surfaces. The XPS spectra of the O 1s region in the IrO2 catalysts are analyzed and deconvoluted into three distinct peaks centered at ∼533.0, 529.7–530.1, and 531.1–531.9 eV, which can be assigned to adsorbed oxygen species (Oads), lattice oxygen species (Olattice), and surface hydroxyl O groups (OOH), respectively (Fig. 3b). The presence of OER intermediates (Ir–OH, Ir–OOH, and Ir–O species) on the active Ir oxide surface is linked to OOH. Notably, B-IrO2 exhibits a significantly higher fraction of OOH (47.39%) compared to L-IrO2 (28.50%). The increased presence of OOH in B-IrO2 underscores the improved catalytic activity of these materials.55
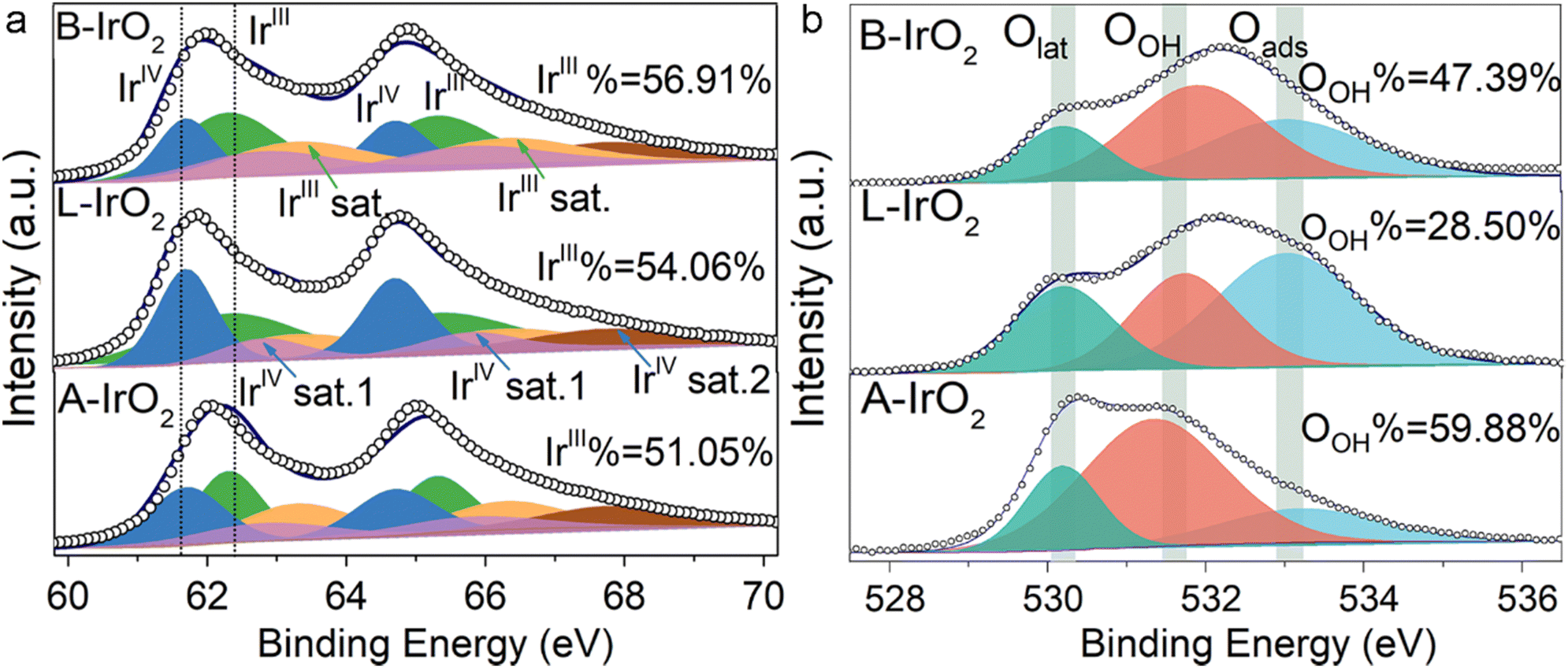 | ||
| Fig. 3 Electronic and atomic structure analysis. (a) and (b) Ir 4f (a) and O1 s (b) XPS spectra of A-IrO2, L-IrO2, and B-IrO2. | ||
It is worth noting that the oxygen peak of OOH is also significantly shifted, likely due to the presence of oxygen vacancies (Ov) formed in the B-doped IrO2. These vacancies are necessary for maintaining charge balance following B doping, which results in the oxygen peak moving to higher binding energies56–60 and promotes OOH formation as well.61–64 Thus, the generation of Ov is theoretically studied by DFT calculations, as presented in Fig. S5 and S6 (ESI†). To experimentally validate the presence of Ov in the B-IrO2 sample, we conducted additional electron paramagnetic resonance (EPR) tests, with the results included in Fig. S8 (ESI†).
Electrochemical measurements
Due to the sufficient slit pores and open spacing between the adjacent 1D B-IrO2 nanoneedles in the porous network, one can anticipate excellent electrochemical performance. The A-IrO2, L-IrO2, and B-IrO2 catalysts are sprayed onto carbon paper, maintaining an Ir loading of 0.08 mgIr cm−2. Their OER activity is evaluated in 0.5 mol L−1 H2SO4 using a standard three-electrode system. Linear sweep voltammetry (LSV) curves are generated at a scan rate of 5 mV s−1. The LSV curves in Fig. 4a reveal the superior activity of B-IrO2 that requires a low potential of 1.498 VRHE (corresponding to an overpotential of 268 mV) to achieve a current density of 100 mA cm−2, significantly outperforming L-IrO2 (1.582 VRHE), A-IrO2 (1.651 VRHE), and recently reported B-doped or incorporated IrO2-based electrocatalysts for the acidic OER, as listed in Table S2 (ESI†). To delve into the catalytic kinetics of the OER process, we examine the Tafel slopes of the IrO2 catalysts (Fig. 4b). B-IrO2 exhibits a Tafel slope of 88.5 mV dec−1, lower than those of L-IrO2 (103.5 mV dec−1) and A-IrO2 (121.2 mV dec−1), indicating enhanced OER kinetics for B-IrO2. Moreover, the exchange current densities (j0) of the catalysts are determined by extrapolating the Tafel plots. The j0 value is calculated to be −0.4149 mA cm−2 for B-IrO2, surpassing those of L-IrO2 (−1.227 mA cm−2) and A-IrO2 (−1.125 mA cm−2) and hence underscoring its superior intrinsic activity.65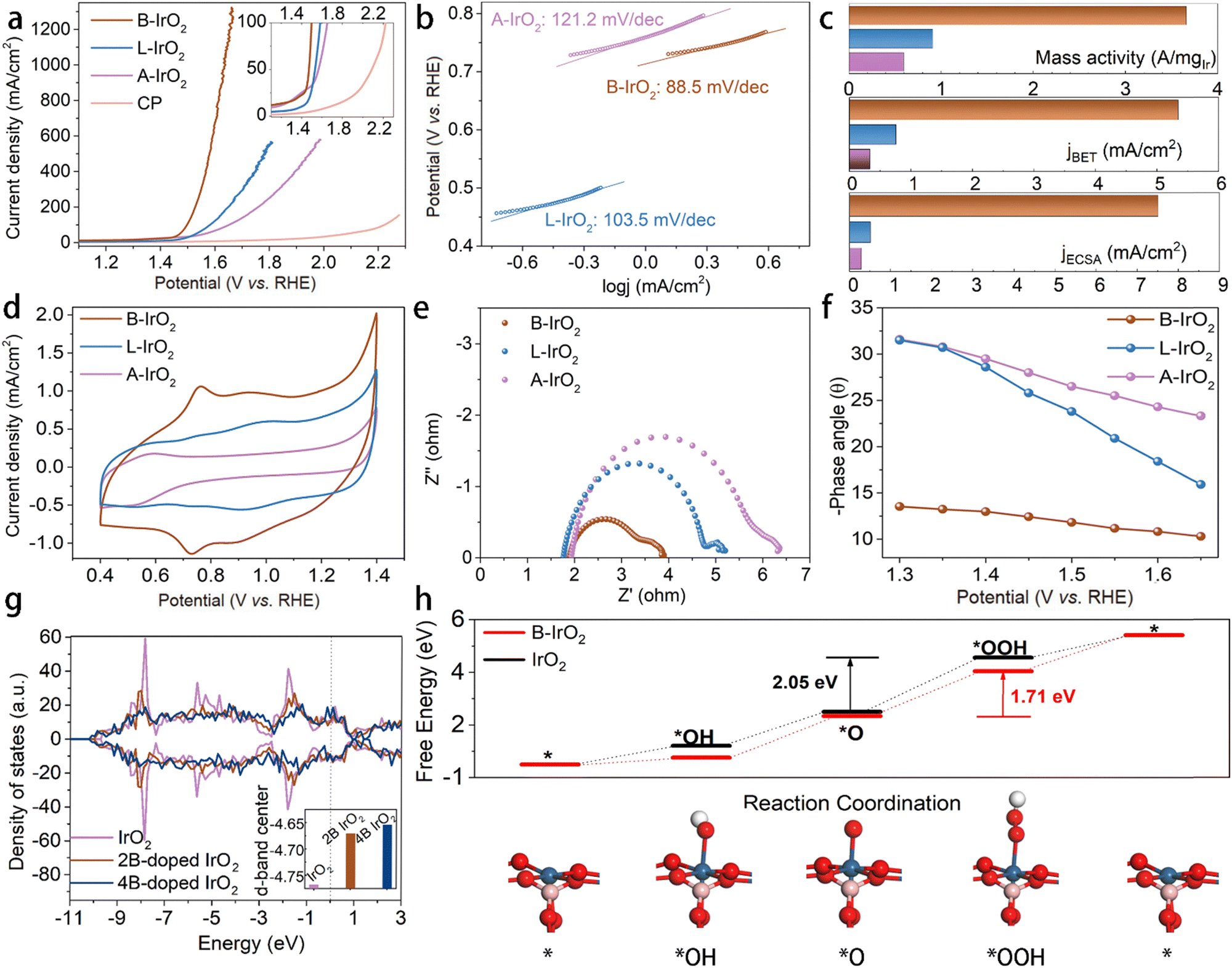 | ||
| Fig. 4 Electrochemical OER performance. (a) Polarization curves of A-IrO2, L-IrO2, and B-IrO2 in 0.5 M H2SO4 at a scanning rate of 5 mV s−1. (b) Tafel plots derived from the polarization curves in (a). (c) Comparison of mass activity and specific activities based on the BET and ECSA areas of A-IrO2, L-IrO2, and B-IrO2 at a potential of 1.55 VRHE. (d) CV curves for measuring the active Ir sites. (e) Nyquist plots of samples recorded at the 1.5 VRHE. (f) Phase peak angles of samples recorded at different potentials. (g) The calculated Ir 4f and O 2p states of IrO2 and B-IrO2. (h) Gibbs free energy diagram of the OER on B-IrO2 and IrO2, and the key parts of the optimized structural configurations for the OER over B-IrO2. Red, blue, pink, and grey spheres represent oxygen, iridium, boron, and hydrogen atoms, respectively. | ||
To delve into the intrinsic activity levels, we calculate the current densities at a 320 mV overpotential (corresponding to 1.55 VRHE) based on various benchmarks, as illustrated in Fig. 4c. The mass activity (MA) and specific activity values of B-IrO2 surpass those of L-IrO2 and A-IrO2. The MA is determined by normalizing the current with the Ir loading. At 1.55 VRHE, the mass activity of B-IrO2 stands at 3656.3 A gIr−1, outperforming L-IrO2 and A-IrO2 by factors of 4.02 and 6.18, respectively. This MA value closely aligns with that of nanoribbon layered IrO2 (∼22.53 μm in length) at 1.5 VRHE (2354.5 A gIr−1)24 and boron-doped amorphous IrOx at 1.53 VRHE (2779 A gIr−1).30 Compared with other Ir-based catalysts listed in Table S3 (ESI†), our B-IrO2 catalyst with a lower loading exhibits higher mass activity, but microgram (μg) loading is impractically low for real-world applications. By depositing B-IrO2 with a loading of 0.08 mgIr cm−2, a promising and reliable catalyst is developed for practical electrolyzer applications. The impact of the surface area aspect on OER performance is also scrutinized by normalizing the BET surface area and the electrochemically active surface area (ECSA, Fig. S9, ESI†). Despite that B-IrO2 possesses the lowest BET and ECSA surface area due to the needle-like morphology, the specific activities normalized by BET/ECSA of B-IrO2 are estimated to be 7.05/14.8 and 15.84/25.99 times greater than those of L-IrO2 and A-IrO2, respectively. All of the results demonstrate that B-IrO2 possesses exceptional intrinsic activity, superior to L-IrO2 and A-IrO2. The active-site-normalized activity is another key descriptor for intrinsic activity. We then measure the electrochemically active Ir sites (NactiveIrsite) through cyclic voltammetry (CV) between Ir3+ and Ir4+,30 revealing that B-IrO2 exhibits the highest NactiveIrsite among these samples, as illustrated in Fig. 4d. The argument for reducing the Ir-loading concerns the limited worldwide production of iridium metal to enable the deployment of electrolysis plants at the scale of GW per year (GW/a). According to the Department of Energy (DOE)'s estimates, achieving a target of less than 0.5 mg cm−2 of Ir appears feasible for reaching a mature state of development within the next decade, with advancements projected to reach up to 20 GW/a. To promote annual capacity additions of 100 GW/a which would position PEMWEs as the primary hydrogen-producing technology for renewable energy storage, Ir loading should be kept below 0.1 mg cm−2, as shown in Fig. S10 (ESI†).
Furthermore, the influence of synthesis parameters on the OER performance is evaluated (Fig. S11, ESI†). As shown in Fig. S11a (ESI†), the electrocatalytic OER performance of the B-IrO2 electrode is varied with the mass of boric acid. The activity peaks at a B/Ir atomic ratio of 8 (corresponding to the B8-IrO2 sample), showing a decline with both higher and lower doping amounts of boric acid (MHB3O3). The activity of different B-IrO2 electrodes is estimated by analyzing the electrochemically active Ir sites based on CV measurements (Fig. S12, ESI†). The optimal doping of boron, achieved with a B-to-Ir precursor molar ratio of 8, results in the B-IrO2 (i.e., B8) sample bearing the largest number of electrochemically active Ir sites. This suggests that this specific amount of boric acid facilitates the exposure of more active Ir sites. However, when the B doping is further increased by raising the B-to-Ir precursor molar ratio to 11, the number of electrochemically active Ir sites of the resulting B11 sample is decreased. This reduction is likely due to the structure perturbation caused by excessive B doping. By fixing the B/Ir ratio at 8, it is found that 1 M cysteamine·HCl is the optimal amount to achieve peak performance, as shown in Fig. S11b (ESI†). The impact of calcination parameters on the OER performance is also investigated. Various calcination temperatures ranging from 350 to 650 °C and holding times from 1 min to 4 hours are tested, with the results outlined in Fig. S11c and d (ESI†). The most favorable performance is observed in the case of calcination at 550 °C for 30 min. Additionally, the OER performance of B-IrO2 was assessed by loading different amounts of Ir (ranging from 0.01 to 0.08 mgIr cm−2). As depicted in Fig. S11e (ESI†), a loading of 0.08 mgIr cm−2 is deemed cost-effective, as evidenced by the corresponding reduction in overpotentials with increasing Ir loading.
The exceptional catalytic performance of B-IrO2 may also be attributed to the increasing anisotropy. With high crystallinity, the elongated nanoneedles intertwined, thus promoting fast ionic conductivity and enhanced electrical conductivity. To investigate the impact of shape on electron transfer, the electrical conductivity of B-IrO2, L-IrO2, and A-IrO2 was directly measured using a collinear four-point probe method. The electrical conductivity is measured to be 1.715 S cm−1 for B-IrO2 on CP, greater than 1.629 and 1.215 S cm−1 for L-IrO2 and A-IrO2 on CP, respectively. To gain deeper insights into the kinetics, we employed an in situ technology, electrochemical impedance spectroscopy (EIS), to observe the OER process. A series of impedance spectra recorded at different potentials in the Nyquist and Bode format are presented in Fig. S13 (ESI†). Fig. S13a–c (ESI†) illustrates a decrease in the semicircle as the potential increases, indicating a favorable OER process. Fig. 4e compares the Nyquist plots of A-IrO2, L-IrO2, and B-IrO2 at a fixed overpotential of 1.5 VRHE, revealing that B-IrO2 exhibits significantly lower charge transfer resistance (Rct), implying a faster catalytic process. The Bose phase plots in Fig. S13d–f (ESI†) display the phase angle as a function of frequency at various potentials. The decreased phase peak angle of B-IrO2 (Fig. 4f) suggests that the introduction of boron into IrO2 can enhance the charge transfer at the electrolyte–catalyst interface, thereby expediting the OER kinetics. The reduced phase peak angle of B-IrO2 under a high overpotential indicates excellent conductivity, likely stemming from their unique morphological features.
Based on the EIS results, we fitted with an appropriate equivalent circuit model,66,67 which contains three resistors (RΩ, R1, and Rct) along with two constant phase elements (Q1 and Qct). RΩ refers to the uncompensated electrolyte resistance. The Q1 in parallel with R1 models the interfacial transport impedance. The polarization resistance Rct is considered to be the total charge transfer resistance for the overall OER rate. The fitting data obtained from the equivalent circuit model (Fig. S14, ESI†) are plotted as a function of applied potential in Fig. S13g and h (ESI†). This analysis provides a detailed insight into the individual contributions to the overall faradaic impedance. As the applied potential increases, the diminished value of Rct with increasing potential indicates enhanced electron transfer kinetics.
To elucidate the impact of B doping on the electronic structure and OER activity of the IrO2 nanoneedle network, we explored the variation of the d-band centers of IrO2 catalysts with B doping based on theoretical calculations, aiming to compare the binding energy between the catalysts and intermediates. Analysis of the density of states of IrO2 (Fig. 4g) reveals that the 5d-band center of Ir ions is lower at −4.768 eV compared with the counterparts with B doping exhibiting a shift of the d-band center of Ir ions towards the Fermi level. The replacements with two and four B atoms in IrO2 lattices result in the d-band center of Ir ions at −4.671 eV and −4.654 eV, respectively. This positive shift in the d-band center of Ir ions establishes a balance between the adsorption and desorption of oxygen intermediates, enabling the self-adjustment of the four-electron OER processes to achieve a harmonized free energy profile with reduced overpotential. We further conducted DFT calculations to investigate the atomic-scale mechanism of the OER, as illustrated in Fig. 4h and Fig. S15, S16 (ESI†). The results indicate that the rate-determining step (RDS) for both B-IrO2 and IrO2 samples is the conversion of the *O species to the *OOH intermediate. The RDS energy barrier for B-IrO2 is calculated to be 1.71 eV, which is lower than that for IrO2 (2.05 eV). This suggests that the B doping facilitates the protonation of *O into *OOH, thereby enhancing the OER performance. Those microscopic perspectives suggest that boron doping promotes the release of oxygen from the surface-active sites of IrO2, thereby enhancing the OER catalytic activity.
In addition to the activity, the stability of catalysts is also a crucial indicator for practical applications. The stability of the present electrocatalyst under high current density conditions is further assessed by chronopotentiometry (CP) methodology. As illustrated in Fig. 5a, B-IrO2 demonstrates remarkable OER stability of sustaining 10 mA cm−2 for a minimum of 150 h in challenging acidic environments. Specifically, merely a 60 mV increase in overpotential can be detected after 150 h of continuous durability testing. In contrast, under the same conditions, L-IrO2 and A-IrO2 show contrasting behaviors. While A-IrO2 experiences rapid degradation, L-IrO2 demonstrates significant deterioration within 50 h. Following the stability assessment, we delve into the XPS spectra of iridium (Fig. S17, ESI†). A moderate decrease can be noted in the peak area ratio of IrIII from 56.91% to 46.88% after the long-term durability test, indicating the decent stability of the active Ir oxides. The enhanced stability of the composite catalysts can be primarily attributed to the B-induced strengthening of the Ir bonds, which help resist electrochemical dissolution.
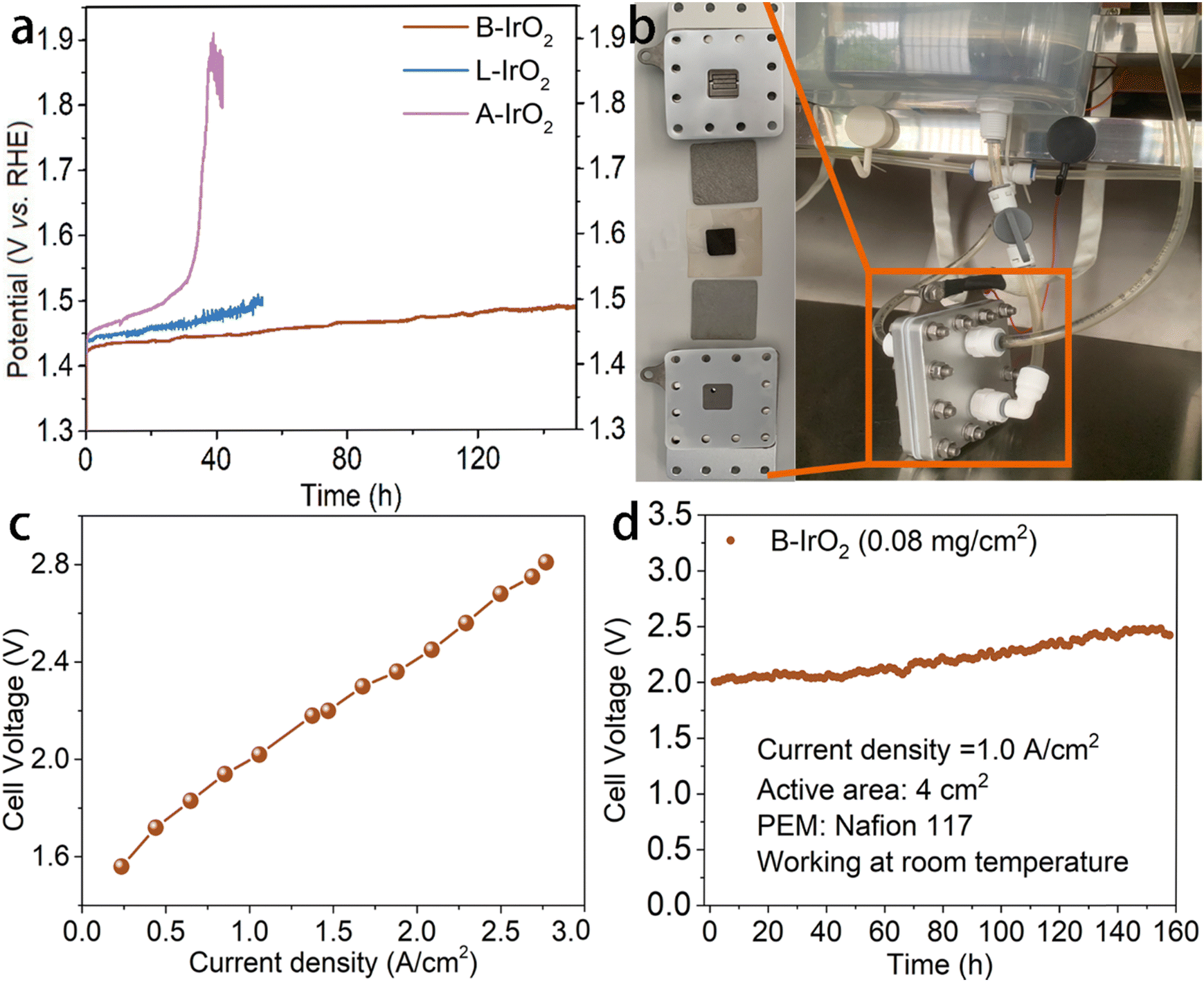 | ||
| Fig. 5 (a) Chronopotentiometric measurements of A-IrO2, L-IrO2, and B-IrO2 at 10 mA cm−2, with carbon paper used as the catalyst support. (b) Photograph showing the PEMWE structure. (c) Polarization j–V curves (measurement conditions: room temperature, 0.08 mgIr cm−2 on a Ti felt for the anode, 1.0 mgPt cm−2 of 40% Pt/C on a carbon paper for the cathode, Nafion 117 membrane, and 4 cm2 electrodes). (d) Static durability test of a PEMWE single cell equipped with the B-IrO2 catalyst through galvanostatic operation at 1.0 A cm−2 for 160 h. | ||
In summary, boron doping alters the crystal lattice and facilitates anisotropic growth. The unique 1D anisotropic structure not only demonstrates improved conductivity and reduced charge transfer resistance but also exhibits increased percentages of active IrIII species and hydroxyl groups (OOH), aligning with the estimated number of electrochemically active sites (NactiveIrsite). DFT calculations reveal that B doping moderately increases the d-band center energy level and significantly lowers the free energy barrier for the conversion of *O to *OOH, thereby improving the intrinsic activity.
Subsequently, we assemble the PEM electrolyzer to validate the catalyst's performance in a practical device setting. Electrodes are prepared by spraying the B-IrO2 anode and commercial Pt/C cathode onto a Ti-felt substrate and carbon paper, respectively (each with a surface area of 4 cm2). The membrane electrode assembly (MEA) is crafted by hot-pressing a Nafion membrane between the B-IrO2 anode and the commercial Pt/C cathode. The visual representation of the PEM electrolyzer setup is captured (Fig. 5b), while the single-cell test results of the PEM electrolyzer are shown in Fig. 5c. Each data point on the polarization curves is acquired through galvanostatic measurements. The j–V curves in Fig. 5c reveal that the PEM electrolyzer with B-IrO2 requires voltages of 1.611, 1.998, and 2.679 V to achieve current densities of 300, 1000, and 2500 mA cm−2, respectively. The distinct structure of B-IrO2 on the Ti-felt substrate is presented in Fig. S18 (ESI†), showing sufficient porosity and open spaces that facilitate the rapid release of oxygen bubbles. Moreover, this structure promotes electrocatalyst–electrolyte interactions by enhancing the accessibility of electrolyte molecules to the deeper regions of the electrode/catalyst interface.
After the PEMWE test, boron is confirmed to be retained in the catalyst through ICP-OES analysis, which indicates a B/Ir molar ratio of 0.34:1. This finding reinforces the stability and reliability of boron doping in the IrO2 nanoneedle network. The stability of the PEMWE cell is evaluated under a current density of 1.0 A cm−2 (Fig. 5d) and 2.8 A cm−2 (Fig. S19, ESI†). The home-assembled PEMWE delivers long-term stability for at least 160 h at 1.0 A cm−2, during which the cell voltage increases from 2.0 V to 2.4 V. However, the performance gradually deteriorates within 35 h at a current density of 2.8 A cm−2. While B-IrO2 exhibits excellent OER performance in acidic solutions, it demonstrates greater stability in a 1 M KOH alkaline solution albeit with a somewhat reduced activity, as shown in Fig. S20 (ESI†).
Conclusions
We demonstrate a simple and scalable method for fabricating a porous network of B-IrO2 nanoneedles. With a low B-IrO2 loading of 0.08 mg cm−2, robust catalytic performance in acidic media is maintained. While cysteamine·HCl acts as a shaping agent to induce the nanoneedle structure, boric acid promotes the unidirectional growth of IrO2 nanoneedles during a modified Adams fusion process. The distinctive geometry of B-IrO2 catalysts not only enhances electronic conductivity but also elevates the concentration of active Ir oxides and hydroxyl groups, thereby improving the intrinsic activity. Consequently, B-IrO2 exhibits a superior mass activity of 3656.3 A g Ir−1 at an Ir loading of 0.08 mg cm−2. This enhanced catalytic performance is also attributed to a moderate increase in d-band center energy levels induced by boron doping, which harmonizes the adsorption of oxygen intermediates and, meanwhile, expedites desorption during the OER process. When employed as the anode catalyst, B-IrO2 demonstrates a substantial current density of up to 2.8 A cm−2 and maintains operation for at least 160 h at a current density of 1.0 A cm−2 in the PEMWE. Our findings provide a practical roadmap by employing a combined strategy of morphology tuning and boron doping to minimize iridium usage while delivering impressive electrocatalytic performance in scaling up PEMWE applications.Experimental
Raw materials
Iridium(III) chloride hydrate (H2IrCl6·6H2O; 98%), cysteamine hydrochloride (H2SCH2Ch2NH2·HCl; ≥98%), and Nafion perfluorinated resin solution (5 wt% in isopropyl alcohol) were purchased from Sigma-Aldrich. Sodium nitrate (NaNO3; 98%) was obtained from Sinopharm Chemical Reagent Co., Ltd. Deionized (DI) water with a resistivity of 18.5 MΩ cm used in the experiment was generated through an ultra-pure purification system. All the chemicals were utilized as received from manufacturers without further purification.Synthesis of B-IrO2
A modified Adams fusion method was employed to synthesize B-IrO2. In a typical B-IrO2 synthesis, 100 mg of H2IrCl6·6H2O, 1 g of NaNO3, 27 mg of cysteamine·HCl, and 89 mg of H3BO3 (with a H3BO3-to-H2IrCl6·6H2O molar ratio of 8) were dissolved in DI water. The solution was stirred at 80 °C for 1 h and then transferred to a convection oven at 80 °C to evaporate the water. The resulting dried powders were finely ground and then charged into an alumina crucible for calcination in a furnace. The calcination process was carried out at temperatures ranging from 350 to 650 °C for durations of 1 min to 4 h. Subsequently, the calcined samples were allowed to cool naturally to room temperature before being thoroughly washed with DI water. The resultant typical sample was called B-IrO2 or B8 throughout the article unless otherwise indicated. For comparison, we also prepared B-doped IrO2 with H3BO3-to-H2IrCl6·6H2O molar ratios of 3, 5, 11, and 13 other than 8, resulting in the samples designated as B3, B5, B11, and B13, respectively. For comparison, L-IrO2 was synthesized under the same conditions as those for B-IrO2 except for that H3BO3 was not added. Additionally, unshaped IrO2 nanoparticles (i.e., A-IrO2) were synthesized without H3BO3 or cysteamine·HCl under identical conditions.Electrochemical measurements
Half-cell electrochemical measurements were carried out using a potentiostat (CHI 760E, CH Instruments) with a Pt wire counter electrode and an Ag/AgCl (3 M NaCl) reference electrode. Catalyst inks were prepared by dispersing 24.5 mg of catalysts in a solution containing 3.74 mL of isopropanol (IPA), 1.24 mL of DI water, and 20 μL of a 5 wt% Nafion solution. The working electrode with an area of 0.5 cm2 was prepared by dropping the catalyst ink on the carbon paper. The electrolyte solution used was Ar-purged 0.5 M H2SO4, and the cell temperature was maintained at room temperature. Linear sweep voltammetry (LSV) was conducted from 1.1 VRHE to 2.3 VRHE at a scan rate of 5 mV s−1. All the LSV curves were plotted after iR compensation. Electrochemical impedance spectroscopy (EIS) was performed at 1.35–1.65 VRHE over a frequency range from 10−2 to 106 Hz. All potentials were calibrated with a reversible hydrogen electrode (RHE). Charge double layer capacitance (Cdl) was measured using cyclic voltammetry (CV) in the non-faradaic current regions. The electrochemical surface area (ECSA) was calculated by dividing the Cdl by the specific capacitance (CS, 0.03 mF cm−2). Chronopotentiometry at a current density of 10 mA cm−2 was conducted to assess the durability of the B-IrO2 catalysts.Single-cell fabrication and operation in a PEMWE
The membrane electrode assembly (MEA) was prepared using commercial 40 wt% Pt/C (Premetek) as the cathode, Nafion 117 as the proton exchange membrane, and the synthesized IrO2-based catalysts as the anode, with a geometric area of 4 cm2. Catalyst inks were prepared by dispersing the catalysts in a solution of IPA, water, and a 5% Nafion solution. The Pt/C ink was sprayed onto a carbon paper (SGL) for cathode preparation, and the IrO2 ink was sprayed onto a Ti-felt substrate for anode preparation. The Pt/C-coated carbon-paper cathode, IrO2-coated Ti-felt anode, and Nafion 117 membrane were hot-pressed to form the MEA. The Ti diffusion layer was pretreated in boiling oxalic acid before washing with DI water. The MEA and pretreated Ti diffusion layer were assembled into a single cell, with a Pt/C catalyst loading of 1.0 mgPt cm−2 for the cathode and an IrO2 loading of 0.08 mg cm−2 for the anode. Ti plates served as current collectors on both the anode and cathode. The electrolyzer cell was operated at room temperature, with DI water supplied to the anode from a water sink. Oxygen and hydrogen were released from the anode and cathode, respectively. Data points on the polarization curves were obtained through galvanostatic measurements at current densities of up to 2.8 A cm−2.Characterization
The morphology and structure of the as-prepared samples were characterized using a scanning electron microscope (Hitachi Regulus 8100) with an acceleration voltage of 200 kV and a high-angle annular dark field scanning transmission electron microscope (HAADF-STEM, FEI Tecnai F20). TEM samples were prepared by placing a drop of the liquid onto lacy carbon-coated copper grids and allowing for natural drying. The phase and crystalline structures of the products were characterized using an X-ray diffractometer (XRD, Bruker, AXS) with Cu Kα radiation. The elemental content was determined through inductively coupled plasma emission spectroscopy (ICP-OES, PerkinElmer 8300). X-ray photoelectron spectroscopy (XPS) was employed to characterize the surface composition and valence states of the samples, with binding energies calibrated using the C 1s signal at 284.8 eV as a reference. The BET surface areas of the prepared catalysts were measured using an ASAP2460 (Micromeritics). A collinear four-point probe method was adopted to measure the electrical conductivity of the prepared samples at room temperature. The probes were equally spaced with a spacing of 1 mm.DFT computation
DFT calculations were utilized to gain a deeper understanding of the relationship between the structures of electrocatalysts and their electrocatalytic activities. To elucidate the effect of B doping on the electronic properties of IrO2, we selected a 2 × 2 IrO2 bulk crystal structure containing 48 atoms. B atoms were randomly substituted for two or four Ir atoms in the IrO2 crystal to construct the calculation models of B-IrO2. Spin-polarized DFT calculations were performed using the Vienna ab initio simulation package (VASP), and the Perdew–Burke–Ernzerhof (PBE) form of the generalized gradient approximation (GGA) was employed as the exchange–correlation function. A 4 × 4 × 4 Monkhorst–Pack special K-point grid was used to sample the first Brillouin zone of all calculation models, with a plane-wave energy cutoff of 400 eV. The total energy was converged to 1.0 × 10−4 eV and the force on each atom was kept below 0.02 eV Å−1. The optimized lattice parameters of IrO2 closely matched experimental and previous calculation results, indicating that our calculation parameters and models were appropriate and reliable.43,44Author contributions
Fei Hu: writing – original draft, methodology, conceptualization, and funding acquisition. Peiyu Huang: investigation. Xu Feng: investigation. Changjian Zhou: investigation. Xinjuan Zeng: investigation. Congcong Liu: writing – review & editing. Guangjin Wang: writing – computational calculation. Xiaowei Yang: writing – review & editing. Huawen Hu: writing – review & editing and funding acquisition.Data availability
Data will be made available on request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22078059, 12102310), the Project of Educational Commission of Guangdong Province of China (2020ZDZX2050, 2021ZDJS104), the Key Project of Guangdong Basic and Applied Basic Research Foundation (2020B1515120081), the Project of Foshan Education Bureau (2021JNHB18), the National Natural Science Foundation of China (32072304), and the Guangdong Basic and Applied Basic Research Foundation (2023A1515140180).Notes and references
- J. Hao, K. Wu, C. Lyu, Y. Yang, H. Wu, J. Liu, N. Liu, W.-M. Lau and J. Zheng, Mater. Horiz., 2023, 10, 2312–2342 RSC.
- B. Yu, J.-H. Liu, S. Guo, G. Huang, S. Zhang, S. Chen, X. Li, Y. Wang and L.-P. Lv, Mater. Horiz., 2023, 10, 4589–4596 RSC.
- A. Muzammil, R. Haider, W. Wei, Y. Wan, M. Ishaq, M. Zahid, W. Yaseen and X. Yuan, Mater. Horiz., 2023, 10, 2764–2799 RSC.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- L. Li, P. Wang, Q. Shao and X. Huang, Chem. Soc. Rev., 2020, 49, 3072–3106 RSC.
- Z. Wei, J. Sun, Y. Li, A. K. Datye and Y. Wang, Chem. Soc. Rev., 2012, 41, 7994 RSC.
- B. Zhang, X. Zheng, O. Voznyy, R. Comin, M. Bajdich, M. García-Melchor, L. Han, J. Xu, M. Liu, L. Zheng, F. P. García de Arquer, C. T. Dinh, F. Fan, M. Yuan, E. Yassitepe, N. Chen, T. Regier, P. Liu, Y. Li, P. De Luna, A. Janmohamed, H. L. Xin, H. Yang, A. Vojvodic and E. H. Sargent, Science, 2016, 352, 333–337 CrossRef CAS.
- Y. Shi, Z. Lu, L. Guo, Z. Wang, C. Guo, H. Tan and C. Yan, Int. J. Hydrogen Energy, 2018, 43, 9133–9143 CrossRef CAS.
- Z. Cheng, B. Huang, Y. Pi, L. Li, Q. Shao and X. Huang, Natl. Sci. Rev., 2020, 7, 1340–1348 CrossRef CAS.
- L. An, C. Wei, M. Lu, H. Liu, Y. Chen, G. G. Scherer, A. C. Fisher, P. Xi, Z. J. Xu and C. H. Yan, Adv. Mater., 2021, 33, 2006328 CrossRef CAS.
- Q. Li, J. Li, J. Xu, N. Zhang, Y. Li, L. Liu, D. Pan, Z. Wang and F. L. Deepak, ACS Appl. Energy Mater., 2020, 3, 3736–3744 CrossRef CAS.
- D. Jin, H. Yoo, Y. Lee, C. Lee and M. H. Kim, ACS Appl. Energy Mater., 2022, 5, 3810–3820 CrossRef CAS.
- G. Gao, Z. Sun, X. Chen, G. Zhu, B. Sun, Y. Yamauchi and S. Liu, Appl. Catal., B, 2024, 343, 123584 CrossRef CAS.
- U. Babic, M. Suermann, F. N. Büchi, L. Gubler and T. J. Schmidt, J. Electrochem. Soc., 2017, 164, F387–F399 CrossRef CAS.
- J. E. Park, S. Kim, O.-H. Kim, C.-Y. Ahn, M.-J. Kim, S. Y. Kang, T. I. Jeon, J.-G. Shim, D. W. Lee, J. H. Lee, Y.-H. Cho and Y.-E. Sung, Nano Energy, 2019, 58, 158–166 CrossRef CAS.
- T. Reier, Z. Pawolek, S. Cherevko, M. Bruns, T. Jones, D. Teschner, S. Selve, A. Bergmann, H. N. Nong, R. Schlögl, K. J. J. Mayrhofer and P. Strasser, J. Am. Chem. Soc., 2015, 137, 13031–13040 CrossRef CAS PubMed.
- A. S. Altowyan, M. Shaban, K. Abdelkarem and A. M. El Sayed, Nanomaterials, 2022, 12, 3272 CrossRef CAS PubMed.
- G. Liu, J. Xu, Y. Wang, J. Jiang and X. Wang, Int. J. Hydrogen Energy, 2014, 39, 14531–14539 CrossRef CAS.
- Z. Liu, Y. Lu, Z. Cui and R. Qi, ACS Appl. Mater. Interfaces, 2023, 15, 10606–10620 CrossRef CAS.
- G. Jiang, H. Yu, Y. Li, D. Yao, J. Chi, S. Sun and Z. Shao, ACS Appl. Mater. Interfaces, 2021, 13, 15073–15082 CrossRef CAS.
- L. Li, P. Wang, Z. Cheng, Q. Shao and X. Huang, Nano Res., 2021, 15, 1087–1093 CrossRef.
- J. Lim, G. Kang, J. W. Lee, S. S. Jeon, H. Jeon, P. W. Kang and H. Lee, J. Power Sources, 2022, 524, 231069 CrossRef CAS.
- J. Lim, D. Park, S. S. Jeon, C. W. Roh, J. Choi, D. Yoon, M. Park, H. Jung and H. Lee, Adv. Funct. Mater., 2017, 28, 1704796 CrossRef.
- F. Liao, K. Yin, Y. Ji, W. Zhu, Z. Fan, Y. Li, J. Zhong, M. Shao, Z. Kang and Q. Shao, Nat. Commun., 2023, 14, 1248 CrossRef CAS PubMed.
- I. C. Man, H. Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- H. Tian, W. Zhu, Q. Shi, S. Ding, Z. Lyu, M. Xu, X. Pan, M. H. Engelhard, D. Dan and Y. Lin, J. Mater. Chem. A, 2022, 10, 11196–11204 RSC.
- M. Zhu, Q. Shao, Y. Qian and X. Huang, Nano Energy, 2019, 56, 330–337 CrossRef CAS.
- W. Lu, P. Yuan, F. Wei, K. Cheng, W. Li, Y. Zhou, W. Zheng and G. Zhang, Int. J. Electrochem. Sci., 2018, 13, 3235–3245 CrossRef CAS.
- N. Yan, R. J. Detz, N. Govindarajan, J. M. Koelewijn, B. Hua, P. Li, E. J. Meijer and J. N. H. Reek, J. Mater. Chem. A, 2019, 7, 23098–23104 RSC.
- Z. Cheng, Y. Pi, Q. Shao and X. Huang, Sci. China: Mater., 2021, 64, 2958–2966 CAS.
- J. Lim, S. Yang, C. Kim, C.-W. Roh, Y. Kwon, Y.-T. Kim and H. Lee, Chem. Commun., 2016, 52, 5641–5644 RSC.
- F. Hu, H. Wang, Y. Zhang, X. Shen, G. Zhang, Y. Pan, J. T. Miller, K. Wang, S. Zhu, X. Yang, C. Wang, X. Wu, Y. Xiong and Z. Peng, Small, 2019, 15, 1901020 CrossRef PubMed.
- R. Li, H. Wang, F. Hu, K. C. Chan, X. Liu, Z. Lu, J. Wang, Z. Li, L. Zeng, Y. Li, X. Wu and Y. Xiong, Nat. Commun., 2021, 12, 3540 CrossRef CAS.
- Y. Qiao, P. Yuan, C.-W. Pao, Y. Cheng, Z. Pu, Q. Xu, S. Mu and J. Zhang, Nano Energy, 2020, 75, 104881 CrossRef CAS.
- L. Zhang, J. Lu, S. Yin, L. Luo, S. Jing, A. Brouzgou, J. Chen, P. K. Shen and P. Tsiakaras, Appl. Catal., B, 2018, 230, 58–64 CrossRef CAS.
- X. Bai, X. Zhang, Y. Sun, M. Huang, J. Fan, S. Xu and H. Li, Angew. Chem., Int. Ed., 2023, 62, e202308704 CrossRef CAS.
- V. Voorhees and R. Adams, J. Am. Chem. Soc., 2002, 44, 1397–1405 CrossRef.
- G. Liu, J. Xu, Y. Wang and X. Wang, J. Mater. Chem. A, 2015, 3, 20791–20800 RSC.
- J. Chen, P. Cui, G. Zhao, K. Rui, M. Lao, Y. Chen, X. Zheng, Y. Jiang, H. Pan, S. X. Dou and W. Sun, Angew. Chem., Int. Ed., 2019, 58, 12540–12544 CrossRef CAS.
- Y. C. Cho, S. Lee, M. Ajmal, W.-K. Kim, C. R. Cho, S.-Y. Jeong, J. H. Park, S. E. Park, S. Park, H.-K. Pak and H. C. Kim, Cryst. Growth Des., 2010, 10, 2780–2784 CrossRef CAS.
- H. Bishara, S. Lee, T. Brink, M. Ghidelli and G. Dehm, ACS Nano, 2021, 15, 16607–16615 CrossRef CAS.
- S. D. Ghadge, O. I. Velikokhatnyi, M. K. Datta, P. M. Shanthi, S. Tan, K. Damodaran and P. N. Kumta, ACS Catal., 2019, 9, 2134–2157 CrossRef CAS.
- C. Koenigsmann and S. S. Wong, Energy Environ. Sci., 2011, 4, 1161–1176 RSC.
- Z. Cui and R. Qi, Appl. Surf. Sci., 2021, 554, 149591 CrossRef CAS.
- D. Von Dreifus, A. J. A. de Oliveira, A. V. do Rosario and E. C. Pereira, J. Supercond. Novel Magn., 2012, 26, 2319–2321 CrossRef.
- D. Chen, R. Lu, R. Yu, H. Zhao, D. Wu, Y. Yao, K. Yu, J. Zhu, P. Ji, Z. Pu, Z. Kou, J. Yu, J. Wu and S. Mu, Nano-Micro Lett., 2023, 15, 168 CrossRef CAS.
- M. Zhou, X. Jiang, W. Kong, H. Li, F. Lu, X. Zhou and Y. Zhang, Nano-Micro Lett., 2023, 15, 166 CrossRef CAS PubMed.
- Y. Xia, R. Mokaya, G. S. Walker and Y. Zhu, Adv. Energy Mater., 2011, 1, 678–683 CrossRef CAS.
- X. Guo, X. Zhao, Z. He, T. Dong, R. Yang, Y. Hou, F. Wang and S. He, Energy Fuels, 2023, 37, 8284–8295 CrossRef CAS.
- V. Pfeifer, T. E. Jones, J. J. Velasco Vélez, C. Massué, M. T. Greiner, R. Arrigo, D. Teschner, F. Girgsdies, M. Scherzer, J. Allan, M. Hashagen, G. Weinberg, S. Piccinin, M. Hävecker, A. Knop-Gericke and R. Schlögl, Phys. Chem. Chem. Phys., 2016, 18, 2292–2296 RSC.
- V. Pfeifer, T. E. Jones, J. J. Velasco Vélez, C. Massué, R. Arrigo, D. Teschner, F. Girgsdies, M. Scherzer, M. T. Greiner, J. Allan, M. Hashagen, G. Weinberg, S. Piccinin, M. Hävecker, A. Knop-Gericke and R. Schlögl, Surf. Interface Anal., 2015, 48, 261–273 CrossRef.
- V. Pfeifer, T. E. Jones, S. Wrabetz, C. Massué, J. J. Velasco Vélez, R. Arrigo, M. Scherzer, S. Piccinin, M. Hävecker, A. Knop-Gericke and R. Schlögl, Chem. Sci., 2016, 7, 6791–6795 RSC.
- H. G. Sanchez Casalongue, M. L. Ng, S. Kaya, D. Friebel, H. Ogasawara and A. Nilsson, Angew. Chem., Int. Ed., 2014, 53, 7169–7172 CrossRef CAS PubMed.
- C. Spöri, P. Briois, H. N. Nong, T. Reier, A. Billard, S. Kühl, D. Teschner and P. Strasser, ACS Catal., 2019, 9, 6653–6663 CrossRef.
- L. Ding, K. Li, W. Wang, Z. Xie, S. Yu, H. Yu, D. A. Cullen, A. Keane, K. Ayers, C. B. Capuano, F. Liu, P.-X. Gao and F.-Y. Zhang, Nano-Micro Lett., 2024, 16, 203 CrossRef CAS.
- Y. Wang, R. Yang, Y. Ding, B. Zhang, H. Li, B. Bai, M. Li, Y. Cui, J. Xiao and Z.-S. Wu, Nat. Commun., 2023, 14, 1412 CrossRef CAS.
- J.-C. Dupin, D. Gonbeau, P. Vinatier and A. Levasseur, Phys. Chem. Chem. Phys., 2000, 2, 1319–1324 RSC.
- Z. Yu, J. Xu, Y. Li, B. Wei, N. Zhang, Y. Li, O. Bondarchuk, H. Miao, A. Araujo, Z. Wang, J. L. Faria, Y. Liu and L. Liu, J. Mater. Chem. A, 2020, 8, 24743–24751 RSC.
- T. Zhang, M.-Y. Wu, D.-Y. Yan, J. Mao, H. Liu, W.-B. Hu, X.-W. Du, T. Ling and S.-Z. Qiao, Nano Energy, 2018, 43, 103–109 CrossRef CAS.
- R. Ge, L. Li, J. Su, Y. Lin, Z. Tian and L. Chen, Adv. Energy Mater., 2019, 9, 1901313 CrossRef.
- Y. Cheng, J. Kang, P. Yan, J. Shen, Z. Chen, X. Zhu, Q. Tan, L. Shen, S. Wang and S. Wang, Appl. Catal., B, 2024, 341, 123325 CrossRef CAS.
- I. M. Nadeem, G. T. Harrison, A. Wilson, C. L. Pang, J. Zegenhagen and G. Thornton, J. Phys. Chem. B, 2017, 122, 834–839 CrossRef.
- N. G. Petrik and G. A. Kimmel, J. Phys. Chem. C, 2015, 119, 23059–23067 CrossRef CAS.
- X. Li, Z. Gu, J. Cheng, G. Zhang, F. Zheng, J. Huang, J. Xu, G. Wei and J. Zhang, ACS Appl. Nano Mater., 2024, 7, 12958–12969 CrossRef CAS.
- L. Li, G. Zhang, J. Xu, H. He, B. Wang, Z. Yang and S. Yang, Adv. Funct. Mater., 2023, 33, 2213304 CrossRef CAS.
- L. Ding, K. Li, Z. Xie, G. Yang, S. Yu, W. Wang, H. Yu, J. Baxter, H. M. Meyer, D. A. Cullen and F.-Y. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 20070–20080 CrossRef CAS PubMed.
- L. Ding, K. Li, Z. Xie, G. Yang, S. Yu, W. Wang, D. A. Cullen, H. Yu and F. Zhang, Electrochim. Acta, 2021, 395, 139199 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Figures and tables, including Fig. S1–S20 and Tables S1–S3. See DOI: https://doi.org/10.1039/d4mh01358a |
| This journal is © The Royal Society of Chemistry 2025 |