
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle-molecule force spectroscopy shows that side chain interactions govern the mechanochemical response of polypeptide α-helices and prevent the formation of β-sheets†
Marie
Asano
ab,
Damien
Sluysmans
 a,
Nicolas
Willet
a,
Colin
Bonduelle
b,
Sébastien
Lecommandoux
*b and
Anne-Sophie
Duwez
*a
a,
Nicolas
Willet
a,
Colin
Bonduelle
b,
Sébastien
Lecommandoux
*b and
Anne-Sophie
Duwez
*a
aMolecular Systems Research Unit, University of Liège, B6a Sart-Tilman, 4000 Liège, Belgium. E-mail: asduwez@uliege.be
bLaboratoire de Chimie des Polymères Organiques, Univ. Bordeaux, CNRS, Bordeaux INP, LCPO, UMR 5629, 16 Avenue Pey Berland, Pessac, F-33600, France. E-mail: Sebastien.Lecommandoux@enscbp.fr
First published on 26th August 2024
Abstract
Secondary α-helix and β-sheet structures are key scaffolds around which the rest of the residues condense during protein folding. Despite their key role in numerous processes to maintain life, little is known about their properties under force. Their stability under mechanical stress, as constantly experienced in the turbulent environment of cells, is however essential. Here, we designed and synthesized two pH-responsive polypeptides, poly(L-glutamic acid) and poly(L-lysine), for single-molecule mechanochemistry experiments using AFM to probe the mechanical unfolding of α-helix and β-sheet secondary motifs. The force experiments, supported by simulations, reveal a superior mechanical stability of the poly(L-lysine) α-helix, which we attribute to hydrophobic interactions of the alkyl side chains. Most importantly, our results show that these interactions play a key role in inhibiting the formation of a metastable β-sheet-like structure when the polypeptide is subjected to mechanical deformations, which might have important implications in the mechanism behind polyQ diseases.
Introduction
Proteins govern the most essential and fundamental processes needed to maintain life. It is thus not surprising that a wide range of debilitating illnesses result from errors in their structures. The misfolding of proteins and subsequent aggregation thereof, are thought to be the unifying cause and pathological mechanism behind a variety of neurodegenerative diseases.1 It has been shown that the folding of proteins occurs by the rapid folding of local secondary structures stabilized through interactions with key amino-acid residues, which forms a scaffold around which the rest of the residues condenses.2,3 The scaffold consists of the initial rapidly formed local secondary α-helix and β-sheet structures.2–4 Although these structures are of crucial importance, little is known about their mechanical stability. This is especially relevant if we consider that a combination of α-helices and β-sheets is found in nearly every native protein and that they are constantly submitted to mechanical forces and deformations in vivo.5AFM-based single-molecule force spectroscopy is a powerful method to probe molecular-level processes and mechanical forces with sub-nanometer resolution in (bio)molecules.6–12 However, no systematic studies on α-helices or β-sheets have been conducted so far. A reason for this is that these structures are embedded in the protein and it is relatively difficult to isolate the mechanical signature of the unfolding of single α-helices or β-sheets structures. Here, we synthesized polymer-polypeptides for optimal interfacing with an AFM force spectroscopy set-up to investigate their stability under external stress. The use of synthetic polymer-polypeptides allows us to tune and isolate desirable structures and, as a result, gain insight into more complex biological systems by observing an isolated property in a controlled environment. Indeed, polymer-polypeptides have been successfully used previously as model systems to decipher natural protein secondary structure at a time when their chemical conformations were not fully understood.13 We evidence here for the first time the key role of side chain interactions when the polypeptides are exposed to mechanical deformations, as constantly experienced in the turbulent environment of cells.
Results and discussion
Polypeptide design and synthesis
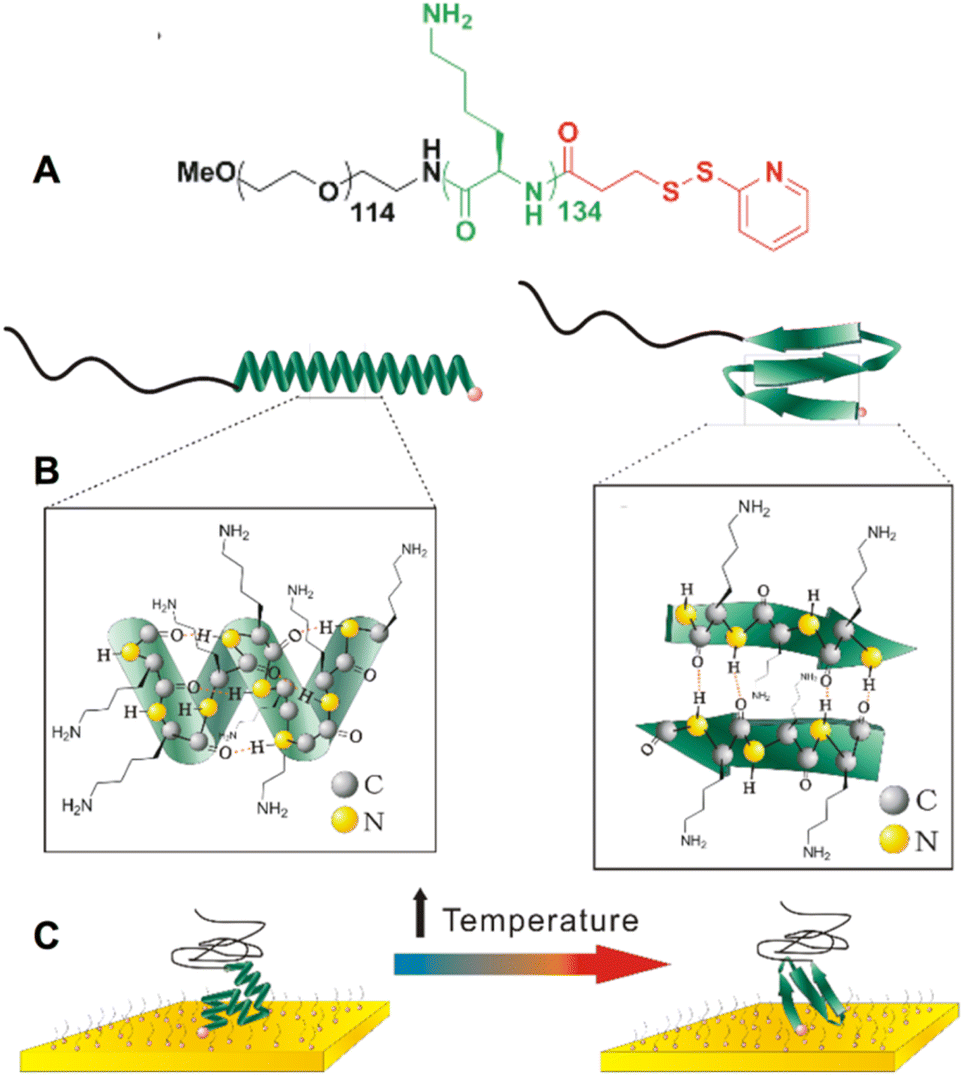
Two stimuli-responsive bioinspired polymers, poly(L-lysine) (PLys) and poly(L-glutamic acid) (PGA) were designed and synthesized for use in single-molecule force spectroscopy experiments (Fig. 1). Both molecules were prepared by the ring opening polymerization of amino-acid N-carboxyanhydrides (NCA) and subsequent coupling with N-succinimidyl 3-(2-pyridyldithio)-propionate (see synthesis and characterization details in ESI†). The design of the molecules consists of a PEG tether by which one end of the molecule can physisorb onto the AFM tip for pulling, and a 2-dipyridyl disulfide moiety for grafting the other end onto a gold surface through Au–S bonding (Fig. 1). We previously showed that this approach, based on the physisorption of caught PEG tethers onto the tip, provides a sufficiently strong and stable attachment of the molecule onto the tip to obtain reliable information on the system grafted on the surface.14–19 As a result of the different side chains (Fig. 1), the α-helices form under opposite pH conditions, pH > 11 for PLys and pH < 4 for PGA.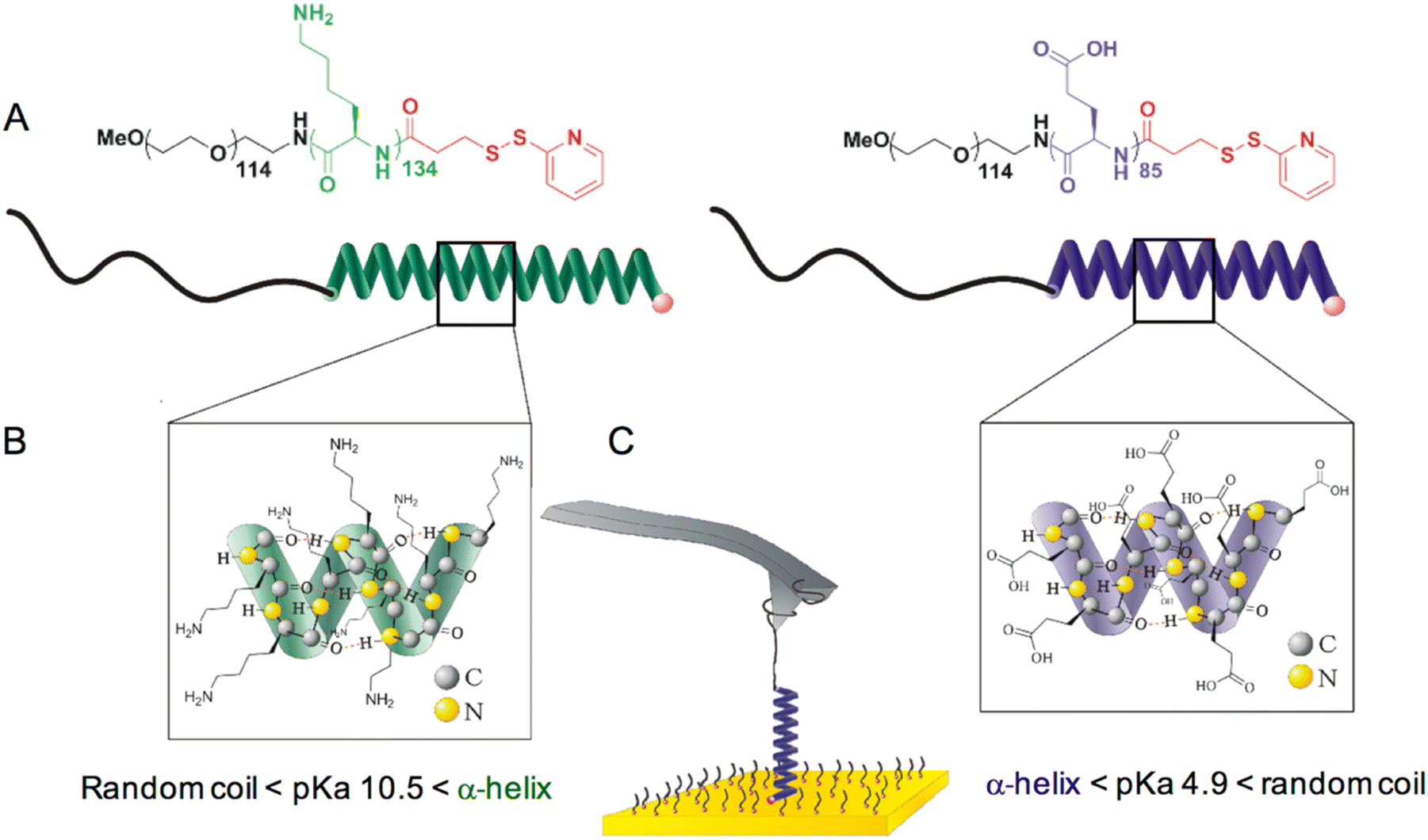 | ||
| Fig. 1 Chemical structures and schematic representations of the synthetic helices and AFM pulling experiment. (A) Chemical structure of poly(ethylene glycol)114-b-poly(L-lysine)134-(2-pyridyl disulfide) (left) and poly(ethylene glycol)114-b-poly(L-glutamic acid)85-(2-pyridyl disulphide) (right), as well as their schematic representation in a α-helix conformation. (B) Detailed representation of side chains and hydrogen bonding within the poly-L-lysine (left) and poly(L-glutamic acid) (right) α-helices. (C) Schematic representation of a molecule grafted onto a gold surface with the PEG chain caught by the AFM tip. The molecule is surrounded by PEG6-SH also grafted onto the surface as a passivating agent. | ||
Immediately prior to performing single-molecule force spectroscopy experiments, the gold substrates were dipped in a diluted solution of the molecules and a passivating agent PEG6-SH was added, to enable single molecules to be individually distributed and immobilized onto the cleaned surface (see Methods section). This approach, combined with the small size of the molecules, also prevents the unspecific adsorption of the polymer chains onto the surface and gives rise to very clean force curves without unspecific peaks.14,16,18–21
Poly(L-lysine) mechanical unfolding
The typical force-extension profile for PLys in α-helix conformation at pH 12 shows a plateau at 34 ± 4 pN (Fig. 2). The general shape of the curves can be interpreted as a sequence of four events. Due to the axial force that is applied by the cantilever displacement, the molecule aligns towards the direction of the displacement (I). A characteristic plateau is then observed and attributed to the unravelling of the α-helix. The appearance of a plateau is consistent with the sequential breaking of identical intra-molecular interactions in series, i.e. both the hydrogen bonds in the main chain and the interactions between the side chains (II). As the cantilever displacement continues, the main peak is observed which resembles that of a random coil extension as the PEG tether and peptide segment are further extended (III). Finally, the molecule detaches from the AFM tip as the applied force exceeds the force of interaction between the molecule and tip (IV). The appearance of a constant force plateau supports the turn-by-turn unravelling of the helix predicted theoretically.22,23 The length of the obtained plateau, 27 ± 4 nm, is in good agreement with the theoretical length difference between the folded and the stretched α-helix of the molecule calculated from the degree of polymerization obtained by NMR (≈27 nm) (details in ESI, Fig. S13†).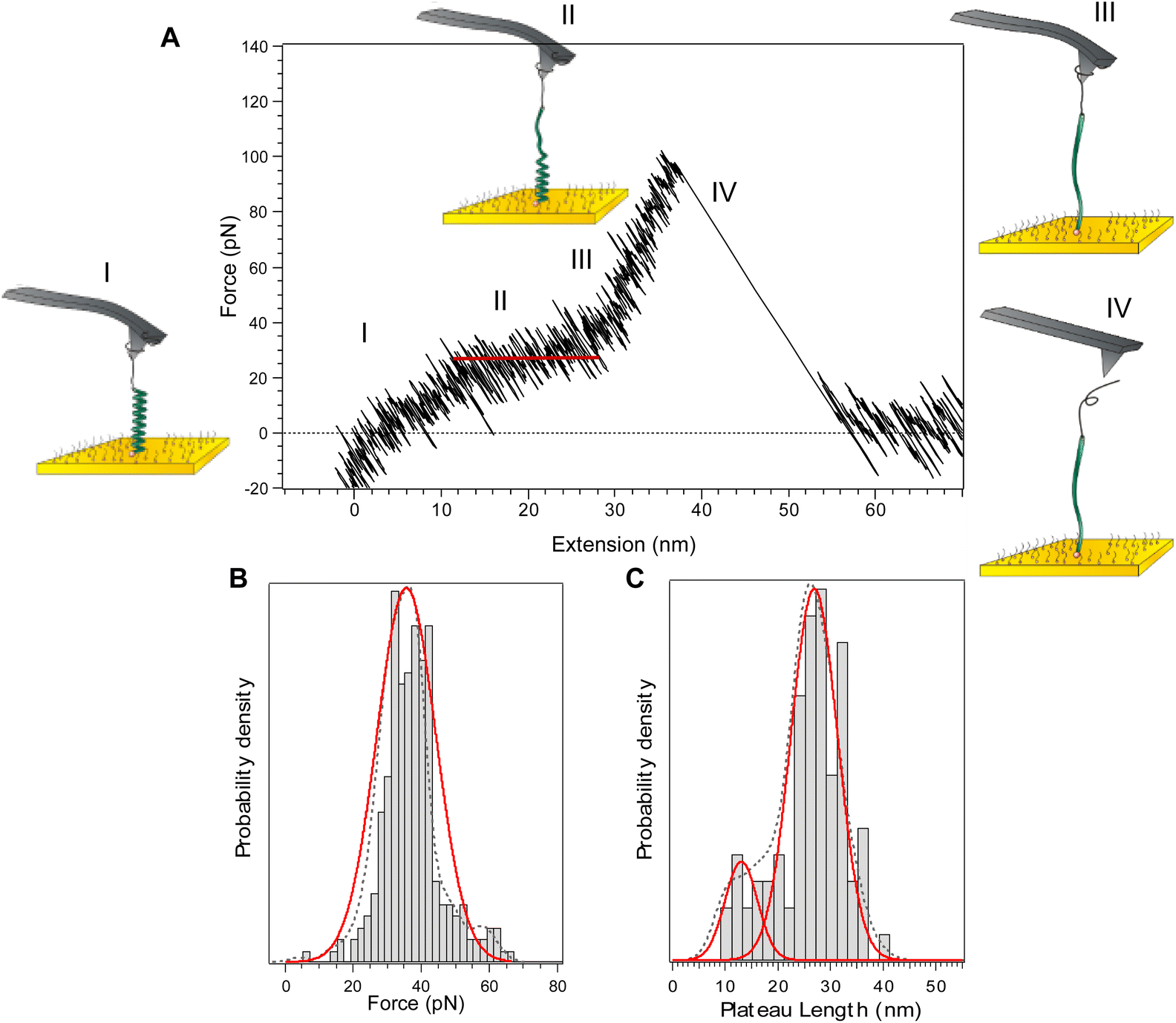 | ||
| Fig. 2 Force-extension curve for poly(L-lysine) in a α-helix conformation (pH 12) and corresponding histograms of plateau force and length. (A) A typical force-extension profile showing an unwinding plateau. Roman numerals and the corresponding diagrams represent an interpretation of the events during the force-extension cycle. (B) Histogram, probability density function (dotted line), and Gaussian distribution (red line) of the plateau forces. The most probable force is 34 ± 4 pN (±s.d., n = 253). (C) Histogram, probability density function (dotted line), and Gaussian distributions (red lines) of plateau lengths. The most probable lengths appear at 27 ± 4 nm (84%) and 13 ± 3 nm (16%) (±s.d., n = 253). | ||
Literature data report that polypeptides fold into a higher order structure with the α-helix segments separated by denatured residues in between. The most stable and thermodynamically favoured length of a single α-helix was shown to be below 20 amino acids.16,24 While this suggests that we should detect the mechanical unfolding of several helices of 20 residues, only one plateau is observed in the force curves and not a series of consecutive plateaus. We have shown recently16 that when the cantilever displacement begins to align the whole molecule backbone in the pulling direction, the higher order structure is lost as the separated α-helix segments align on top of each other. It allows the denatured residues between the helical segments to refold, resulting in a single, uniform α-helix strand.16,25–29 The presence of a second population of plateau length at 13 ± 3 nm (Fig. 2C) is reminiscent of the fact that separated α-helical segments do not always fold back perfectly resulting in a single uniform helix. Consequently, sometimes only a shorter, isolated helical segment was extended resulting in a shorter plateau length.16
As a control experiment, we measured the force-extension profile of PLys at pH 7, at which the amino groups are protonated, which prevents the formation of the helical structure. The profile (Fig. S15†) describes the behaviour of an ideal, random coil polymer without plateau.
Poly(L-glutamic acid) mechanical unfolding
For PGA in a α-helical conformation at pH 3, a plateau is observed at 19 ± 2 pN (Fig. S16†), in only 20% of the cases. The vast majority of the observed profiles (80% of the cases) show a characteristic small peak or inflection at 18 ± 4 pN (indicated in Fig. 3A by a red arrow, see Fig. 3B for histogram). The appearance of this small peak is in very good agreement with the simulated curves (see below Fig. 5B). It means that in this case, only a partially helical molecule was extended. The extension of this partially helical PGA is thus highly biased towards the extension of the denatured segments22 resulting in a force-extension profile resembling that of a random coil (Fig. 3). The inflection at 18 ± 4 pN indicates the unravelling of the isolated helical segments. This inflection and the plateau (19 ± 2 pN, Fig. S16†), appear at a very similar force, providing further evidence that the unravelling of the helical segments begins at this force. The lower force of unfolding and the lower occurrence of plateau for PGA indicate that PGA forms less stable helices than PLys, in agreement with circular dichroism experiments (see ESI†), which show a small helix content of about 30% for PGA, as compared to 70% for PLys. As a control experiment, we measured the force-extension profile of PGA at pH 7, at which the carboxylic acid groups are deprotonated, which prevents the formation of the helical structure. The profile (Fig. S15†) describes the behaviour of an ideal, random coil polymer. | ||
| Fig. 3 Force-extension curve for poly(L-glutamic acid) in a α-helix conformation (pH 3) and corresponding histogram of force. (A) Typical force profile showing the inflection. The red line is a WLC fit (lp = 0.4 nm, Lc = 38 nm) used as a guide to the eye to facilitate the observation of a deviation from the typical random-coil behaviour. Roman numerals and the corresponding diagrams represent an interpretation of the events during the force-extension cycle. (B) Histogram, probability density function (dotted line), and Gaussian distribution (red line) of the inflection forces. Most probable force: 18 ± 4 pN (±s.d., n = 232). | ||
Stabilization by side chains interactions
We suggest that the higher hydrophobicity of the lysine side chains helps to stabilize the PLys helix, which leads to a higher unfolding force (≈30 pN) and the occurrence of long plateaus characteristic of stable helices. The lysine is more hydrophobic due its longer aliphatic chain, and the presence of an amine group, as compared to a shorter aliphatic chain and a carboxyl moiety for the glutamic acid side chain. To test our hypothesis on the stabilizing hydrophobic interactions between the side chains, force spectroscopy experiments for PLys at pH 12 were conducted in a higher salt concentration (50 mM NaCl). It has been shown that even a small increase in the salt concentration from 10 mM to 50 mM screens the weak van der Waals interactions between the hydrophobic lysine side chains.30 In addition, the increased concentration of sodium ions increases the solubility of the side chains by interacting with the amine groups. Despite this higher solubility effect, the helix forming ability is not affected by this salt concentration. Indeed, it has been demonstrated previously that micelles and vesicles of synthetic polypeptides were shown to be pH responsive up to 1 M NaCl due to conformational change.31,32 From this, we infer that at 50 mM NaCl, only the hydrophobic interactions between the lysine side chains are affected and not the main hydrogen bonding of the α-helix. Moreover, it is also known that ions increase the strength of H-bonding in water.33 Therefore, in the event that the mechanical stability would exclusively be attributable to the H-bonding, the force to unfold the helix would be even higher because ions would increase the H-bonding strength and thus the mechanical stability of the polypeptide. The obtained curves (Fig. 4) resemble the force extension profiles obtained for PGA, evidencing a destabilization of the helix. The profiles were characterized by a reoccurring inflection at 17 ± 4 pN, which is similar to the inflection observed for the helix of glutamic acid at 18 ± 4 pN. The results demonstrate that the hydrophobic interactions between the side chains play a major role in the helix formation and stabilization. | ||
| Fig. 4 Force-extension curve for poly(L-lysine) in a α-helix conformation at 50 mM NaCl (pH 12) and corresponding histograms of force. (A) Typical force profile showing the inflection (red arrow). The red line is a WLC fit (lp = 0.4 nm, Lc = 43 nm) used as a guide to the eye to facilitate the observation of a deviation from the typical random-coil behaviour. Roman numerals and corresponding diagrams represent an interpretation of the events during the force-extension cycle. (B) Histogram, probability density function (dotted line), and Gaussian distribution (red line) of the inflection forces. Most probable force: 17 ± 4 pN (±s.d., n = 167). | ||
Unfolding simulations
In addition to the pulling experiments, we utilized a helix-coil transition model AGADIR34 coupled with a random-coil polypeptide elastic model proposed by Torabi and Schatz (see ESI†).35 For the simulations, the N-terminal was left protected to approximate our molecule which was grafted on to a gold surface by a 2-pyridyl disulfide group. The C-terminal was left unprotected approximating the high solubility of the PEG tether in water. An average value of the cantilever spring constant of 9.5 pN nm−1 was inserted, which was obtained by the thermal noise method prior to each experiment. The model accurately predicts the shape of the force-extension curves obtained for the two different helices (Fig. 5). For the PLys helix, it predicts the occurrence of a constant force plateau. The force of the plateau obtained in the simulation is in the 30 pN range, which is in agreement with the force plateau we were able to experimentally show at 34 ± 4 pN. For PGA, the model predicts the random coil nature evidenced in the experimental force-extension profiles. The inflection observed experimentally at 18 ± 4 pN is also evident in the simulated profile at approximately 15 pN.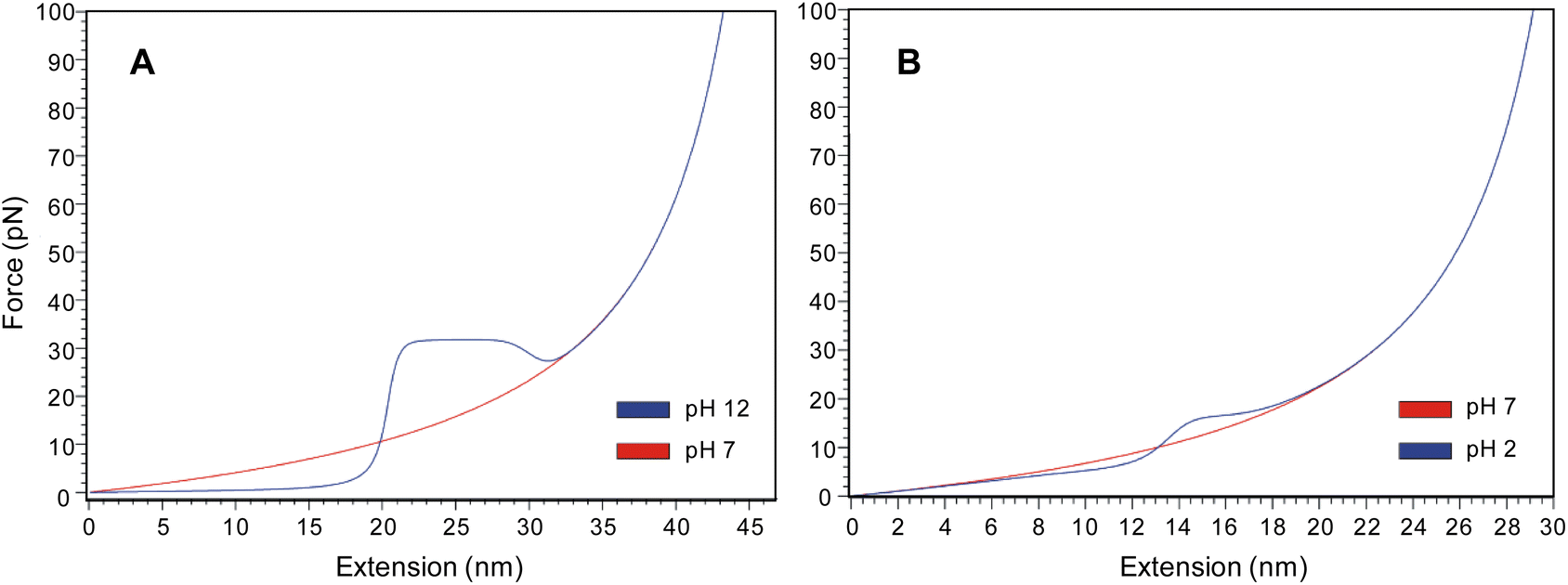 | ||
| Fig. 5 Force extension profiles obtained using the helix-coil transition model AGADIR proposed by Torabi and Schatz for poly(L-lysine) and poly(L-glutamic acid) in both random coil and α-helix conformations. (A) Force extension profiles for poly(L-lysine) at pH 12 (blue line, α-helix) and pH 7 (red line, random coil). (B) Force extension profiles for poly(L-glutamic acid) at pH 2 (blue line, α-helix) and pH 7 (red line, random coil). | ||
β-Sheet formation under mechanical load
In 60% of the PGA force-extension curves that displayed the first inflection at 18 ± 4 pN, a second inflection at a higher force of 60 ± 9 pN (cyan arrow in Fig. 6) can be identified. The second inflection is observed only in force-extension profiles where the first inflection is observed, indicating that the presence of the second inflection is dependent on the mechanical unfolding of a pre-existent α-helix secondary structure. It is well known that a transition from a α-helix to a β-sheet structure can occur in response to pH, temperature, solvent composition, DNA binding, but also mechanical deformation.36 However, α–β transitions by mechanical deformation have only been reported for protein coiled-coil structures.36–38 Nevertheless, a theoretical study by Ding et al.39 has suggested the presence of a metastable β-hairpin intermediate state, and most importantly, they indicated that although the potential energy of the β-hairpin state is indeed higher than the α-helix state, the entropy of the β-hairpin intermediate is significantly higher due to less constraints imposed by the hydrogen bonding. We postulate that the formation of a metastable β-hairpin-like interaction occurs during the unfolding of the α-helix structure as a result of the β-hairpin structure possessing a higher entropy, and thus occupying an energy minimum in conditions where the entropic forces dominate. As the force applied to the partially helical PGA becomes increasingly larger, the helix structure starts to destabilize further as the residues within undergo increasing spontaneous fluctuations between their random coil and helix states. As enough force is applied to break the hydrogen bonding of the helix, the β-hairpin structure is expected to form starting from the available denatured segments. In literature39 the α-helix to β-hairpin transition was found to proceed via a random coil structure, so it would make sense if such a structure were to form in a transitional state when enough mechanical force has been applied to pull the helix out of its energy minimum, yet not enough force has been applied to pull the molecule completely taunt, allowing backbone entropic forces to dominate. When such an intermediate force is applied, it allows enough conformational freedom for a few hydrogen bonding sites to come into close enough contact to form the β-hairpin interaction.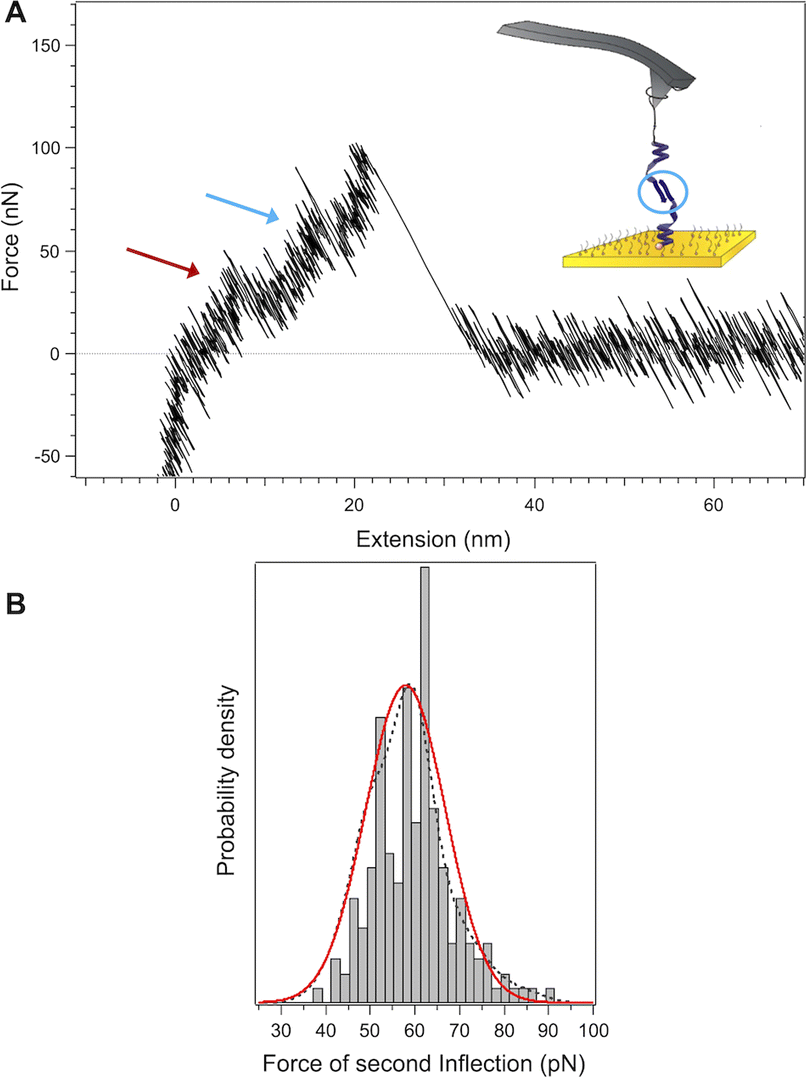 | ||
| Fig. 6 Force-extension curve for poly(L-glutamic acid) in a α-helix conformation (pH 3) where a second inflection at a higher force was observed and corresponding histogram of force. (A) Typical force profile showing the first inflection (red arrow) and second inflection (cyan arrow). (B) Histogram, probability density function (dotted line), and Gaussian distribution (red line) of the second inflection force. Most probable force: 60 ± 9 pN (±s.d., n = 177). | ||
We propose here that the formation of a metastable β-hairpin-like interaction is a property unique to poor helix formers, such as poly(L-glutamic acid), that do not possess side chains that can effectively form helix-stabilizing hydrophobic interactions. Simulations performed on a peptide with increasingly hydrophobic side chains have shown that the increasing hydrophobicity of side chains stabilize the helix structure, and prevent the formation of the metastable β-hairpin.39
As a control experiment to confirm that the inflection at 60 pN is indeed due to a β-sheet structure, we probed PLys in its β-sheet conformation. Poly(L-lysine) can be considered as an ideal model polypeptide to study conformational transitions between random coil, α-helix and β-sheet structures, as it is capable of exhibiting all three secondary structures by varying pH and temperature.40,41 The α–β transition of the molecule was followed by circular dichroism where it is confirmed that the lysine segment of the molecule undergoes a α–β transition for pH ≥ 11 and temperatures of 50 °C (see ESI Fig. S12†). We performed pulling experiments after a heat treatment of the functionalized surface at pH 12 (Fig. 7). While the force-extension curves for PLys in the α-helical conformation show a plateau (Fig. 2), an inflection at an average force of 65 pN appears instead of this plateau when the molecules were exposed to elevated temperatures, confirming our interpretation that the appearance of the second inflection for PGA was due to the formation of a β-sheet conformation (Fig. 8).
 | ||
| Fig. 7 (A) Chemical structure of poly(ethylene glycol)114-b-poly(L-lysine)134-(2-pyridyl disulfide), as well as its schematic representation in α-helical and anti-parallel β-sheet conformation. (B) Detailed representation of side chains and hydrogen bonding within poly(L-lysine). (C) Schematic representation of the α–β transition that occurs after the surface functionalized with poly(L-lysine) is exposed to elevated temperatures needed to induce the transition. | ||
 | ||
| Fig. 8 Example of a force-extension profile of poly(L-lysine) presumed to be in the β-form after being exposed to high temperatures (50° during 20 minutes, pH 12), with an observed inflection at an average force of 65 pN. | ||
Conclusion
In summary, our results indicate that the mechanical behaviour of a α-helix under tension is strongly governed by the interactions of the side chains. The higher hydrophobicity of the lysine side chains helps to stabilize the helix, which leads to a higher unfolding force and the occurrence of long plateaus characteristic of stable helices. Theoretical studies had suggested previously that a stronger hydrophobicity of the side chains support a longer α-helix with greater stability.24 A α-helix behaves as a multistate system consisting of numerous helical and random coil segments that form at different locations along the chain and a typical helix fluctuates between these interchanging states at different locations along the entirety of its length.35,42 The hydrophobicity of the lysine residue increases the free energy barrier between these two states. It therefore inhibits somewhat these fluctuations, allowing the denatured segments between the separated helical segments to reform more easily when a small axial force is applied. On the contrary, the glutamic acid side chains, which are not as hydrophobic, cannot inhibit the spontaneous helix-coil fluctuations as successfully, leading to a lower free energy barrier between these two states. Consequently, it is more difficult for the PGA helix to refold into a complete α-helix when a small force is applied. Even when a plateau is evident, it occurs less frequently (20% of the cases) and at a lower force (19 pN) than that of the PLys helix (100% of the cases and a force of 34 pN). As a result of the instability of its helix, PGA is also able to form metastable β-hairpin-like interactions during the unfolding event of the helix.Although the pivotal role that the side chain interactions play in stabilizing the α-helix has been appreciated for some time,43–45 it is to our knowledge the first time that their major role on the mechanical behaviour is demonstrated. It is also the first time that their role in preventing the formation of a metastable β-sheet-like structure when the polypeptide is exposed to mechanical deformations is evidenced. This might have important implications in the mechanism behind polyQ diseases. Although speculative, due to the structural similarities between polyglutamic acid and polyglutamine, the mechanical properties and instability of our PGA system might mirror the mechanism behind polyglutamine (polyQ) neurodegenerative diseases where an elongation of a polyQ tract causes the misfolding of proteins and inevitably the presence of mutant protein aggregates. The nine different polyQ diseases share some common pathological features such as the longer polyglutamine tract. Perhaps this points to a more generic, structural mechanism, where the native secondary structure formed from long strands of polyglutamine are inherently structurally unstable and can easily form metastable β-sheet like interactions upon some mechanical extension, which forms the precursor to polyQ protein aggregates leading to the onset of the disease.46
Methods
Immobilization of the polymer-polypeptides onto Au/Si surfaces
2 cm by 2 cm Au/Si surfaces (Sigma-Aldrich) were cleaned by using a TL1 cleaning solution (H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NH3:H2O2, 5:1:1, v/v/v) at 70 °C for 15 minutes. The cleaned surfaces were carefully rinsed with ultra-pure water (milliQ) before drying under nitrogen flow. The functionalization solution was prepared by either dissolving 0.1 mg ml−1 of PEG114-b-PGA85-(2-pyridyl disulphide) or 0.1 mg ml−1 of PEG114-b-PLys134-(2-pyridyl disulphide), with a short PEG6-SH oligomer (Polypure) with a molar ratio of (80:20, PEG-SH:copolymer) in 100 mM KH2PO4 (Sigma Aldrich) at pH 7.0 adjusted with NaOH or HCl (Sigma Aldrich), respectively. After functionalization, the surfaces were rinsed repeatedly with ultra-pure water (milliQ) and used immediately for single-molecule force spectroscopy experiments.
:NH3:H2O2, 5:1:1, v/v/v) at 70 °C for 15 minutes. The cleaned surfaces were carefully rinsed with ultra-pure water (milliQ) before drying under nitrogen flow. The functionalization solution was prepared by either dissolving 0.1 mg ml−1 of PEG114-b-PGA85-(2-pyridyl disulphide) or 0.1 mg ml−1 of PEG114-b-PLys134-(2-pyridyl disulphide), with a short PEG6-SH oligomer (Polypure) with a molar ratio of (80:20, PEG-SH:copolymer) in 100 mM KH2PO4 (Sigma Aldrich) at pH 7.0 adjusted with NaOH or HCl (Sigma Aldrich), respectively. After functionalization, the surfaces were rinsed repeatedly with ultra-pure water (milliQ) and used immediately for single-molecule force spectroscopy experiments.
Single-molecule force spectroscopy experiments
SMFS was performed on a PicoPlus 5500 (Agilent Technologies). A conventional fluid cell was used during experimentation. Force–distance curves were obtained by using gold coated silicon-nitride cantilevers (OBL series, Brucker) with a nominal spring constant of 0.06 N m−1. The spring constant of the cantilevers were determined by the thermal noise method after every experiment. Two different types of salt solutions were used depending on the copolymer. All salt solutions, bases and acids, were prepared with ultra-pure water, filtered and prepared fresh prior to every experiment. For measurements done with PEG114-b-PGA85-(2-pyridyl disulphide), 10 mM NaCl with 5 mM KH2PO4 was used. The pH of the solution was adjusted to either pH 7 (random coil) or pH 3 (helix) with the addition of NaOH or HCl. The pH was monitored throughout the experiment. For measurements done with PEG114-b-P(Lys)134-(2-pyridyl disulphide), 10 mM and 50 mM NaCl was used. The pH of the solution was adjusted to either pH 7 (random coil) or pH 12 (helix) with the addition of NaOH or HCl. The pH was monitored throughout the experiment. For the experiments on PEG114-b-poly(L-lysine)134-(2-pyridyl disulphide) in the β-form, the Au/Si surfaces grafted with PLys were heated in 20 mM NaCl solution at pH 12 at 50 °C for 60 minutes to induce the α–β transition before performing SMFS experiments.Statistical mechanical model developed by Schatz and Torabi
The model is freely available. Access was obtained by registering an account at http://www.nanohub.org/. For all the simulations, an ionic concentration of 10 mM was used. To mimic the architecture of our molecules as close as possible, the N terminal was assumed to be protected and the C terminal was left unprotected. A spring constant of 9.5 pN nm−1 was used, which is the average value of spring constants of the OBL cantilevers obtained during the SMFS experiments by thermal noise. Simulations were done at pH 7 and 12 for poly(lysine)134, and at pH 2 and 7 for poly(glutamic acid)85.Data availability
The data supporting this article have been included as part of the ESI.† The Statistical mechanical model developed by Schatz and Torabi is freely available. Access can be obtained by registering an account at http://www.nanohub.org/.Author contributions
ASD and SL designed the research. MA and CB synthesized and characterized the polymers. MA performed the AFM experiments. MA, NW and DS analyzed the data. The manuscript was written through contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the IDS FunMat program (ERASMUS MUNDUS agency EACEA, contract number 2010-0004/0001) for financial support. N. Guidolin and A.-L. Wirotius are thanked for their assistance with SEC and NMR analyses, respectively. D. S. is a Post-doctoral Researcher of the FRS-FNRS.References
- C. Soto, Nat. Rev. Neurosci., 2003, 4, 49–60 CrossRef PubMed.
- A. R. Dinner, A. Sali, L. J. Smith, C. M. Dobson and M. Karplus, Trends Biochem. Sci., 2000, 25, 331–339 CrossRef CAS.
- V. Daggett and A. Fersht, Nat. Rev. Mol. Cell Biol., 2003, 4, 497–502 CrossRef CAS PubMed.
- A. R. Fersht, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 1525–1529 CrossRef CAS.
- C. M. Dobson, Nature, 2003, 426, 884–890 CrossRef CAS.
- T. E. Fisher, P. E. Marszalek and J. M. Fernandez, Nat. Struct. Biol., 2000, 7, 719–724 CrossRef CAS PubMed.
- C. Bustamante, Y. R. Chemla, N. R Forde and D. Izhaky, Annu. Rev. Biochem., 2004, 73, 705–748 CrossRef CAS.
- K. C. Neuman and A. Nagy, Nat. Methods, 2008, 5, 491–505 CrossRef CAS.
- E. M. Puchner and H. E. Gaub, Curr. Opin. Struct. Biol., 2009, 19, 605–614 CrossRef.
- J. Liang and J. M. Fernandez, ACS Nano, 2009, 3, 1628–1645 CrossRef.
- Molecular Manipulation with Atomic Force Microscopy, ed. A.-S. Duwez and N. Willet, CRC Press, Boca Raton, 2012 Search PubMed.
- M. L. Hughes and L. Dougan, Rep. Prog. Phys., 2016, 79, 076601 CrossRef.
- (a) H. Schlaad, in Peptide Hybrid Polymers, ed. H.-A. Klok and H. Schlaad, Springer Berlin Heidelberg, 2006, vol. 202, pp. 53–73 Search PubMed; (b) C. Bonduelle, Polym. Chem., 2018, 9, 1517–1529 RSC.
- P. Lussis, T. Svaldo-Lanero, A. Bertocco, C.-A. Fustin, D. A. Leigh and A.-S. Duwez, Nat. Nanotechnol., 2011, 6, 553–557 CrossRef PubMed.
- A. Van Quaethem, P. Lussis, D. A. Leigh, A.-S. Duwez and C.-A. Fustin, Chem. Sci., 2014, 5, 1449–1452 RSC.
- D. Sluysmans, N. Willet, J. Thevenot, S. Lecommandoux and A.-S. Duwez, Nanoscale Horiz., 2020, 5, 671–678 RSC.
- D. Sluysmans, L. Zhang, X. Li, A. Garci, J. F. Stoddart and A.-S. Duwez, J. Am. Chem. Soc., 2020, 142, 21153–21159 CrossRef.
- D. Sluysmans, P. Lussis, C.-A. Fustin, A. Bertocco, D. A. Leigh and A.-S. Duwez, J. Am. Chem. Soc., 2021, 143, 2348–2352 CrossRef CAS PubMed.
- F. Devaux, X. Li, D. Sluysmans, V. Maurizot, E. Bakalis, F. Zerbetto, I. Huc and A.-S. Duwez, Chem, 2021, 7, 1333–1346 CAS.
- D. Sluysmans, S. Hubert, C. J. Bruns, Z. Zhu, J. F. Stoddart and A.-S. Duwez, Nat. Nanotechnol., 2018, 13, 209–213 CrossRef CAS PubMed.
- D. Sluysmans, F. Devaux, C. J. Bruns, J. F. Stoddart and A.-S. Duwez, Proc. Natl. Acad. Sci. U.S.A., 2018, 115, 9362–9366 CrossRef CAS.
- B. Chakrabarti and A. J. Levine, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2006, 74, 031903 CrossRef PubMed.
- R. Rohs, C. Etchebest and R. Lavery, Biophys. J., 1999, 76, 2760–2768 CrossRef CAS PubMed.
- Z. Qin, A. Fabre and M. J. Buehler, Eur. Phys. J. E, 2013, 36, 1–12 CrossRef.
- H. Wada and R. R. Netz, Europhys. Lett., 2007, 77, 68001 CrossRef.
- S. Zhang, L.-J. Qu, T. Suo, Z. Liu and D. Yan, J. Chem. Phys., 2017, 146, 174904 CrossRef.
- S. Sivaramakrishnan, B. J. Spink, A. Y. L. Sim, S. Doniach and J. A. Spudich, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13356–13361 CrossRef CAS.
- M. J. Williams and M. Bachmann, J. Chem. Phys., 2017, 147, 024902 CrossRef PubMed.
- F. C. Zegarra, G. N. Peralta, A. M. Coronado and Y. Q. Gao, Phys. Chem. Chem. Phys., 2009, 11, 4019–4024 RSC.
- H. Lodish, A. Berk, S. L. Zipursky, P. Matsudaira, D. Baltimore and J. Darnell, Molecular Cell Biology, W. H. Freeman, New York, 4th edn, 2000, p. 1084 Search PubMed.
- F. Chécot, A. Brûlet, J. Oberdisse, Y. Gnanou, O. Mondain-Monval and S. Lecommandoux, Langmuir, 2005, 21, 4308–4315 CrossRef.
- F. Chécot, S. Lecommandoux, Y. Gnanou and H.-A. Klok, Angew. Chem., Int. Ed., 2002, 41, 1339–1343 CrossRef.
- T. Urbič, Chem. Phys. Lett., 2014, 610–611, 159–162 CrossRef.
- E. Lacroix, A. R. Viguera and L. Serrano, J. Mol. Biol., 1998, 284, 173–191 CrossRef.
- K. Torabi and G. C. Schatz, Macromolecules, 2013, 46, 7947–7956 CrossRef.
- (a) Z. Qin and M. J. Buehler, Phys. Rev. Lett., 2010, 104, 198304 CrossRef PubMed; (b) M. Nguyen, J.-L. Stigliani, G. Pratviel and C. Bonduelle, Chem. Commun., 2017, 53, 7501–7504 RSC.
- L. Kreplak, J. Doucet, P. Dumas and F. Briki, Biophys. J., 2004, 87, 640–647 CrossRef CAS PubMed.
- L. Kreplak, H. Herrmann and U. Aebi, Biophys. J., 2008, 94, 2790–2799 CrossRef CAS.
- F. Ding, J. M. Borreguero, S. V. Buldyrey, H. E. Stanley and N. V. Dokholyan, Proteins, 2003, 53, 220–228 CrossRef CAS.
- W. Dzwolak, T. Muraki, M. Kato and Y. Taniguchi, Biopolymers, 2004, 73, 463–469 CrossRef CAS PubMed.
- J. J. Grigsby, H. W Blanch and J. M. Prausnitz, Biophys. Chem., 2002, 99, 107–116 CrossRef CAS PubMed.
- A. Chakrabartty and R. L. Baldwin, in Advances in Protein Chemistry, C. B. Anfinsen, F. M. Richards, J. T. Edsall and D. S Eisenberg, Academic Press, 1995, vol. 46, pp. 141–176 Search PubMed.
- S. Marqusee and R. L. Baldwin, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 8898–8902 CrossRef CAS.
- J. M. Scholtz and R. L. Baldwin, Annu. Rev. Biophys. Biomol. Struct., 1992, 21, 95–118 CrossRef CAS.
- J. M. Scholtz, H. Qian, V. H. Robbins and R. L. Baldwin, Biochemistry, 1993, 32, 9668–9676 CrossRef CAS PubMed.
- H.-C. Fan, L.-I. Ho, C.-S. Chi, S.-J. Chen, G.-S. Peng, T.-M. Chan, S.-Z. Lin and H.-J. Harn, Cell Transplant., 2014, 23, 441–453 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mr00068d |
| This journal is © The Royal Society of Chemistry 2025 |