
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Eco-friendly anaerobic oxidation of aryl diazo esters with heterocyclic N-oxide under ball milling: synthesis of 1,2-dicarbonyl systems†
Souvik
Guha
 a,
Subhabrata
Sen
*b and
Ludovic
Gremaud
*a
a,
Subhabrata
Sen
*b and
Ludovic
Gremaud
*a
aSchool of Engineering and Architecture of Fribourg, Department of Chemistry, Institute of Chemical Technology, HES-SO University of Applied Sciences and Arts Western Switzerland, Boulevard de Pérolles 80, 1700 Fribourg, Switzerland. E-mail: Ludovic.gremaud@hefr.ch
bDepartment of Chemistry, School of Natural Sciences, Shiv Nadar Institution of Eminence Deemed to be University, Chithera, Uttar Pradesh 201314, India. E-mail: subhabrata.sen@snu.edu.in
First published on 25th October 2024
Abstract
Herein we report anaerobic oxidation of metal carbenoids generated from aryl diazo esters under ball milling with heterocyclic N-oxide in the presence of catalytic copper(I) to afford 1,2-dicarbonyls in excellent yield. Efficiently progressing across a diverse spectrum of substrates, the reaction demonstrates exceptional tolerance to a variety of functional groups, under mild reaction conditions, at low catalyst loading and minimum volume of solvent as a Liquid-Assisted Grinding (LAG) auxiliary thereby demonstrating a practical strategy to generate these molecules.
Introduction
The 1,2-dicarbonyl motif is found extensively in natural products1–5 and contemporary pharmaceuticals.6–10 Hence it plays a crucial role in modern day drug discovery. Notable examples include licoagrodione, isolated from Glycyrrhiza glabra, which demonstrates antimicrobial activity, Tanshinone IIA, isolated from Salvia miltiorrhiza Bunge, which serves as a transcription factor inhibitor and Mansonone C, extracted from Mansonia altissima, which exhibits antifungal activity against P. parasitica which is a plant-damaging oomycete capable of causing enormous economic damage both within crops, as well as natural ecosystems (Fig. 1A).2–4 The ability of the dicarbonyl motif to bind to proteins in the body enhances their bioavailability. Accordingly, several clinically used drugs have been developed, such as the anticancer drug biricodar and the anti-HCV drug boceprevir (Fig. 1B).8,9 Furthermore, dicarbonyl-containing compounds frequently serve as valuable synthetic intermediates and precursors in organic synthesis and materials science. Aromatic substituted quinoxalines, synthesized from dicarbonyl-containing compounds, have broad applications as photoinitiators and fluorescence-based sensors.11–14 | ||
| Fig. 1 Significant molecules containing dicarbonyl moieties. (A) Natural products rich in dicarbonyl functionality. (B) Active pharmaceutical ingredients incorporating dicarbonyl motifs. | ||
Over the past few decades, an increasing number of reports have highlighted various organic transformations achieved through mechanochemical ball milling. These approaches are characterized by their eco-friendliness, as they are either solvent-free or involve microliter volumes of solvents, utilized as Liquid-Assisted Grinding (LAG) auxiliaries represented by η (μL mg−1), typically ranging between 0 and 1.15–23 Mechanochemical activation is a key driver methodology for a new chemical pathway allowing expansion of the process windows and chemistry space by having access to more selective and efficient syntheses without bulk dissolution of reactants.24 In this context, the International Union of Pure and Applied Chemistry (IUPAC) has recognized mechanochemistry as one of the ten innovations with the potential to make our planet more sustainable or an event to carry out transformative changes in the chemical world.25a Besides these high level sustainable considerations and aims, the mechanochemical approach offers notable advantages, including shorter reaction times, lower reaction temperatures, and improved “green” metrics compared to solution-based processes.25b
In the contemporary research, diazo compounds have been extensively explored as carbene/carbenoid precursors, utilizing thermal, photochemical, or metal-catalyzed processes to generate singlet and triplet carbenes or metal carbenoids.26a These processes have led to various essential organic transformations, yielding diverse value-added organic compounds.26b,c Since oxygenation of organic molecules holds significant importance in organic transformations, the oxidative denitrogenation of α-diazoesters, for the production of α-ketoesters is an interesting strategy which was explored by the research groups led by Chang, Stoltz, and Doyle.27 They have reported the oxygen-transfer process on the diazo esters under either transition-metal or metal-free reaction conditions.28 Traditional oxidation protocols of diazo esters often involved the use of dimethyldioxirane (DMDO) (Scheme 1a).29 Since DMDO was generated from oxone and acetone, a significant drawback of this strategy was limited functional group tolerance, and safety concerns for large-scale production due to the potent oxidizing ability of the oxidants. In another example, Hu et al. reported a di-rhodium acetate catalyzed oxidation of aryl diazoacetates with toluene, resulting in the formation of corresponding aryl α-ketoesters (Scheme 1b).30 It is noteworthy that this process required a stoichiometric amount of the dehydrogenation reagent diethyl azodicarboxylate (DEAD). Stoltz and co-workers reported that dimethyl sulfoxide (DMSO) could also act as an oxidant for the denitrogenation of aryl α-diazoesters. This process is well-tolerated only for substrates with electron-donating functionality (Scheme 1c).27b
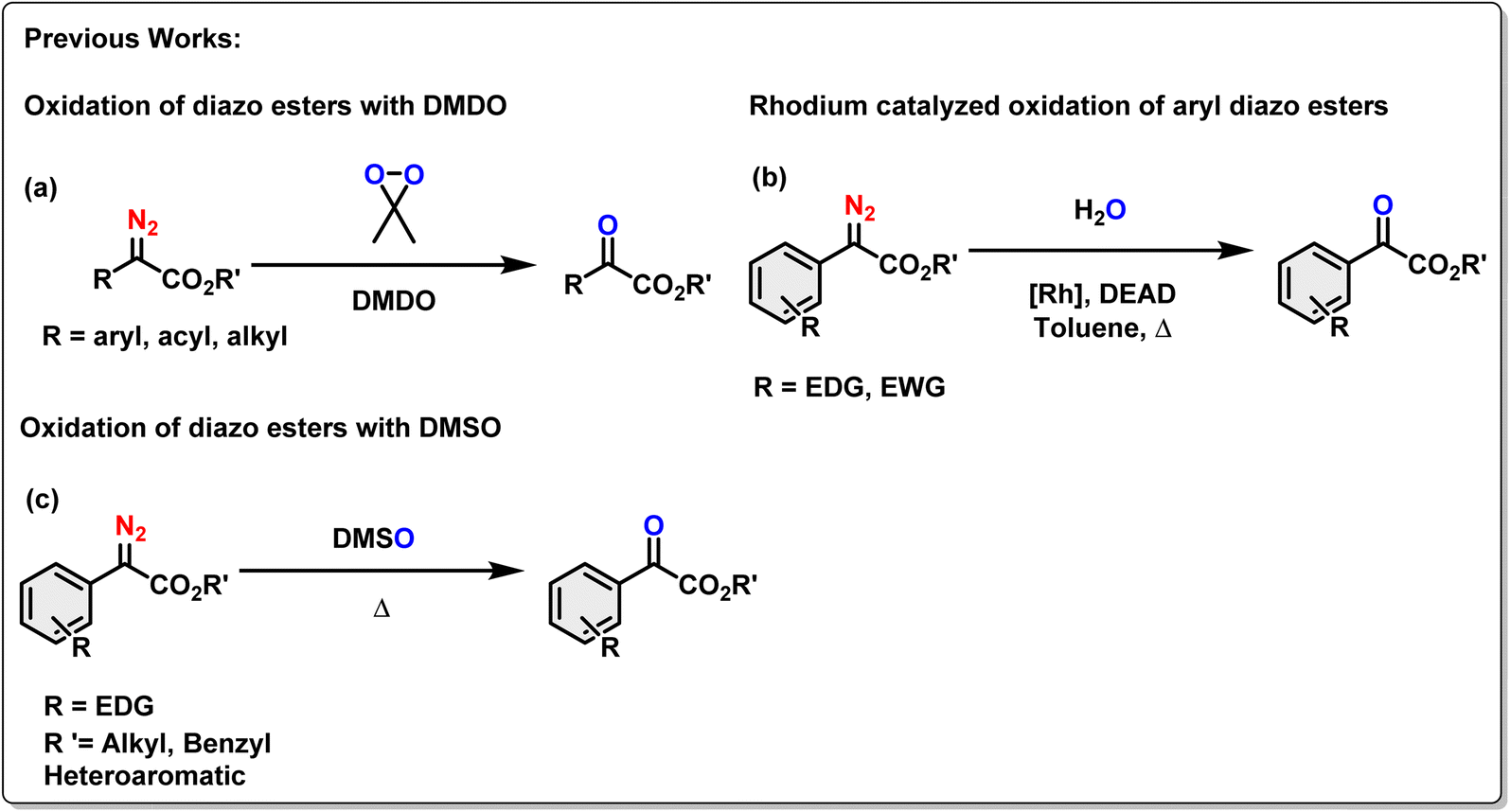 | ||
| Scheme 1 Literature precedence of the oxygen transfer process with diazo esters. | ||
Over the decades, transition-metal-catalyzed carbene generation has gained prime attention from the synthetic community due to its high efficiency and selectivity.31 As an abundant transition metal copper is considered as an ideal metal carbenoid precursor. Herein, we present a highly efficient catalytic approach for the anaerobic oxidation of α-diazoesters to α-ketoesters using heterocyclic N-oxides, with copper iodide as a catalyst (Scheme 2). The reaction happened in a RETSCH mixer mill (MM 400). Given its compatibility with diverse functional groups, we anticipate that this method will find extensive applications in the derivatization of 1,2-dicarbonyl systems.
 | ||
| Scheme 2 Mechanochemical anaerobic oxidation of diazo esters. | ||
Results and discussion
Initially, in a bid to functionalize quinoline/pyridines through C–H activation pathways, we explored the conversion of methyl 2-diazo-2-phenylacetate (2a) by reacting it with pyridine N-oxide (1a) under Cu-catalyzed and ball milling conditions. We had anticipated that this would yield C2-arylated quinoline/pyridines (3a′).32 Surprisingly, the α-ketoester (3a) was obtained as the major product with no trace of 3a′ (Scheme 3).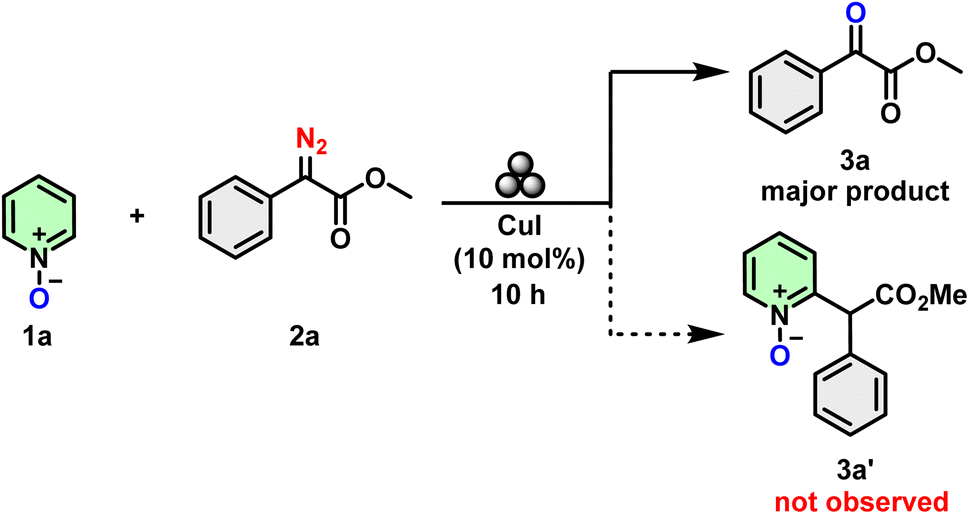 | ||
| Scheme 3 Observation during the Minisci-type functional group addition reaction. | ||
Considering these preliminary results, the synthesis of α-ketoester 3a was optimized using Cu salt, pyridine N-oxide 1a (1 eq.) and methyl 2-diazo-2-phenylacetate 2a (2 eq.). When 1a and 2a underwent a reaction at 30 Hz for 2 hours through mechanochemical ball milling in a Retsch MM 400 mixer mill, utilizing solid-state grinding in a stainless steel (ss) jar (5 mL) with three ss balls of 5 mm diameter, the desired product 3a was obtained in a yield of 38% (Table 1, entry 1). Employing the same conditions with 2 equivalents of 2a and extending the reaction time show a slight improvement in the yield (Table 1, entry 2). In the following reaction (with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio of 1a and 2a), using Cu(OTf)2 as a Cu catalyst decreases the reaction yield (Table 1, entry 3). Subsequently, the inclusion of Al2O3 and aluminum foil as grinding auxiliaries, along with an extension of the reaction time up to 5 hours, did not yield any improvement in the reaction (Table 1, entries 4 and 5). However, the use of 4 Å molecular sieves slightly improved the results, likely due to their high surface area and porous structure (Table 1, entry 6).
:2 ratio of 1a and 2a), using Cu(OTf)2 as a Cu catalyst decreases the reaction yield (Table 1, entry 3). Subsequently, the inclusion of Al2O3 and aluminum foil as grinding auxiliaries, along with an extension of the reaction time up to 5 hours, did not yield any improvement in the reaction (Table 1, entries 4 and 5). However, the use of 4 Å molecular sieves slightly improved the results, likely due to their high surface area and porous structure (Table 1, entry 6).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ratio1a:2a |
Reaction time [h] | Grinding auxiliary | Catalysta | Yieldb,c [%] |
| a 10 mol% catalyst loading. b Unless otherwise mentioned, all the reactions were carried out on a 0.52 mmol scale with respect to 1a (1 equiv.). c Yield based on the isolated amount after column chromatography. d Grinding auxiliary taken was 200 mg. | |||||
| 1 | 1:1 |
2 | Neat | CuI | 38 |
| 2 | 1:2 |
4 | Neat | CuI | 41 |
| 3 | 1:2 |
4 | Neat | Cu(OTf)2 | 32 |
| 4c | 1:2 |
5 | Al2O3 | CuI | 40 |
| 5 | 1:2 |
5 | Al foil | CuI | 37 |
| 6d | 1:2 |
5 | 4 Å MS | CuI | 47 |

With this moderate improvement in hand, we began to explore the reaction conditions further, particularly focusing on the choice of LAG (η = 0.5 μL mg−1) in a variety of solvents, such as tetrahydrofuran (THF), methanol (MeOH), ethyl acetate (EtOAc), acetonitrile (ACN), dichloromethane (DCM), chloroform (CHCl3), 1,2-dichloroethane (1,2-DCE). A variety of solvents were tested from weakly polar to highly polar as well as protic and aprotic solvents (Table 2, entries 1–7). In the first set of trials, no trend was observed except that chlorinated solvent allows better yield to be obtained (Table 2, entry 7) and both acetone and methanol are not efficient with no conversion (NR) observed (Table 2, entries 4 and 6). By increasing the reaction time from 5 to 7 hours in the presence of DCM as a LAG auxiliary the reaction was pushed but not significantly (Table 2, entry 7 vs. entry 8), and even poorer yield was obtained with CHCl3 (Table 2, entry 9). In an attempt to improve the reaction yield, we performed experiments using excess heterocyclic N-oxide as the oxidant. However, further increasing the amount of the oxidant did not enhance the yield. To our satisfaction, we noted that employing 1,2-dichloroethane as a LAG auxiliary, along with an extended reaction time of 7 hours, yielded the most favorable results (Table 2, entry 10).
|
|
|||
|---|---|---|---|
| Entrya | Reaction time [h] | Liquid assisted grinding | Yieldb [%] |
|
a Reaction scale: 0.52 mmol of 1a (1 equiv.), 1:2 ratio of 1a and 2a, 10 mol% catalyst loading, η = 0.5 μL mg−1.
b Yield based on the isolated amount after column chromatography.
|
|||
| 1 | 5 | EtOAc | 52 |
| 2 | 5 | THF | 41 |
| 3 | 5 | CH3CN | 56 |
| 4 | 5 | Acetone | NR |
| 5 | 5 | Toluene | 30 |
| 6 | 5 | MeOH | NR |
| 7 | 5 | DCM | 70 |
| 8 | 7 | DCM | 76 |
| 9 | 7 | CHCl3 | 72 |
| 10 | 7 | 1,2-DCE | 82 |

To investigate the catalyst's role, various Cu halides were introduced into the reaction (Table 3, entries 1 and 2). Among the diverse transition metals investigated as catalysts in metal-carbenoid chemistry, copper stands out as particularly appealing due to its status as an economical and abundant first-row transition metal.33 Several copper salts were tested, but none yielded improved results. Notably, with CuI, the reaction was observed to proceed significantly faster compared to other copper salts. This observation indicates that the selection of CuI was optimal for the mechanochemical oxygen transfer reaction. Decreasing the catalyst loading of CuI to 5 mol% or increasing the number of equivalents of 2a to 3 equiv. negatively distorts the yield (Table 3, entries 3 and 4). Subsequent attempts with a reduced number of balls (2) and larger ball sizes, such as 10 mm, which are known to contribute to more aggressive milling with higher impact energy and faster breakage of larger particles or conducting the reaction in zirconium (Zr) and polytétrafluoroéthylène (PTFE) jar-balls, did not lead to any further improvement in the yield of 3a (Table 3, entries 5–7).
|
|
||||
|---|---|---|---|---|
| Entrya | Jar-ball (# – size) | Catalyst | Catalyst loading [mol%] | Yieldb [%] |
|
a Unless otherwise mentioned, all the reactions were carried out on reaction scale: 0.52 mmol of 1a (1 equiv.), 1:2 ratio of 1a and 2a, 10 mol% catalyst loading, η = 0.5 μL mg−1.
b Yield based on the isolated amount after column chromatography.
c 3 equiv. 2a.
|
||||
| 1 | SS (3–5 mm) | CuCl | 10 | 58 |
| 2 | SS (3–5 mm) | CuBr | 10 | 62 |
| 3 | SS (3–5 mm) | CuI | 5 | 66 |
| 4c | SS (3–5 mm) | CuI | 10 | 77 |
| 5 | SS (2–10 mm) | CuI | 10 | 80 |
| 6 | Zr (3–5 mm) | CuI | 10 | 70 |
| 7 | Teflon (3–5 mm) | CuI | 10 | 72 |

Next, we explored the reagent scope of the heterocyclic N-oxides. The deoxygenation reaction proceeded smoothly regardless of the electronic variation of the heterocyclic N-oxides (1b–1c, Scheme 4). However, in the case of non-aromatic heterocyclic N-oxides, the yield of 3a was lower, and the reaction proceeded very slowly. This could be due to the steric hindrance around the N-oxide (1d).
 | ||
| Scheme 4 Screening of heterocyclic N-oxides. | ||
With the optimized conditions in hand, we explored the scope of this anaerobic oxidation reaction using a series of α-diazoesters (Scheme 5). It is noteworthy that the reaction exhibited tolerance towards these diversely substituted substrates in 66 to 86% yield. Observations revealed that neutral or electron-rich aryl diazoesters, such as 2a, 2d, 2e, 2l (see the ESI†) exhibited a slightly better reaction preference, with yields ranging from 82 to 86%, compared to their electron-poor counterparts, namely 2b, c, 2f–i (see the ESI†). In general, α-diazoesters with benzyl and heteroaromatic substituents at R′ yielded α-ketoesters (3ne–3qh and 3ad–3nd) in moderate to high yields.
 | ||
| Scheme 5 Substrate scope of the outlined procedure. | ||
Additionally, under standard reaction conditions, the reaction of ethyl diazoacetate, methyl diazoacetate and aryl diazoacetate related to the isatin moiety did not reach completion, resulting in poor conversion to the end product (2ca–2cd, ESI†). The scope of various N-oxide compounds was subsequently investigated. As expected, pyridine N-oxide, halogenated pyridine N-oxide, and quinoline N-oxide were deoxygenated without difficulty (1a–c). To our delight, the desired reduction of N-Me morpholine N-oxide also proceeded under standard conditions (1d) to give 3a in significant amounts (Scheme 4).
To showcase the practical applicability of this methodology, we conducted large-scale experiments involving model substrates 1a and 2a. The reaction was carried out with 2 stainless-steel balls (∅ = 10 mm, mtot = 2.71 g) in a stainless-steel ball milling jar (10 mL). Employing 1,2-DCE as a LAG auxiliary (η = 0.5 μL mg−1) at 30 Hz for 10 hours successfully completed the reaction (Scheme 6).
 | ||
| Scheme 6 Large scale anaerobic oxidation of α-diazoesters. | ||
As mentioned in the Introduction, α-ketoamides play a crucial role in drug discovery, contributing to both biological activities and synthetic transformations. The efficient transfer of a 1,2-dicarbonyl fragment to secondary α-ketoamides has been achieved with good yields. The generation of α-ketoamides was performed using the crude reaction mixture of product 3a, with the addition of morpholine or piperidine, resulting in the production of α-ketoamides in an 83% and 79% yield respectively (Scheme 7).
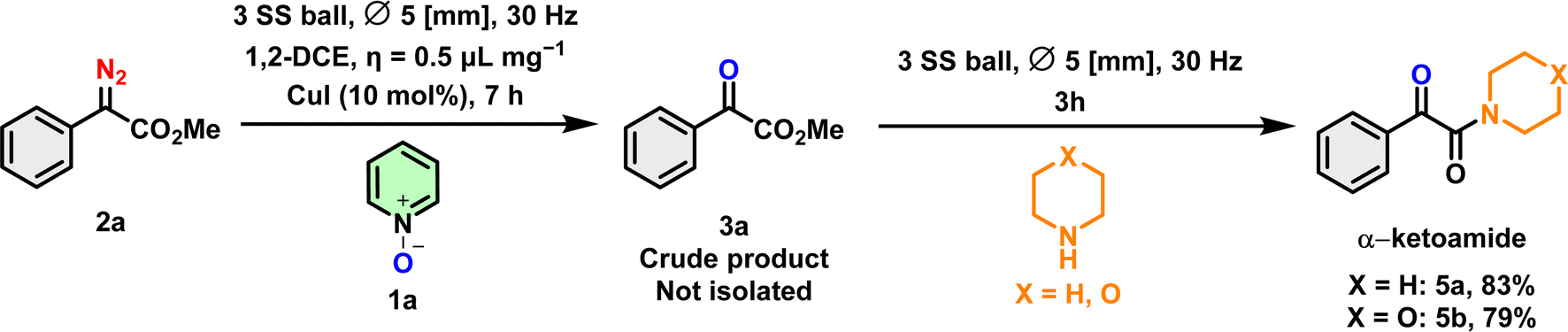 | ||
| Scheme 7 Post-synthetic transformation to α-ketoamide. | ||
To highlight the practical utility of our strategy, we had conducted syntheses of pharmaceutically relevant precursor molecule 8 for natural products. The anticancer drug indibulin and the natural product polyandrocarpamide C7 could be prepared using the following methodology (Scheme 8).
 | ||
| Scheme 8 Application of the methodology to the synthesis of active pharmaceutical ingredients. | ||
To gain insight into the mechanism, control reactions were conducted. When 1a was subjected to reaction with 2a without the use of a Cu catalyst, the TLC of the reaction showed several unwanted spots, with a significant amount of the starting material remaining with less than 10% of 3a formation (Scheme 9A). This observation suggests the importance of metal catalysts in the reaction. Additionally, the use of radical quenchers such as TEMPO and BHT separately could not inhibit product formation. Compound 3a was obtained as the major product in the reaction, yielding 78% (Scheme 9B). Based on the control experiments and drawing from precedent literature, a plausible oxygenation pathway is illustrated in Scheme 9C. It is initially postulated that diazoacetate reacts with copper species to form copper-carbenoid (complex I) accompanied by the evolution of nitrogen gas.34 The subsequent attack at the electrophilic carbene carbon by the oxygen atom of pyridine N-oxide generates an oxonium ylide (complex II). It is proposed that the decomposition of complex II occurs in the final step, potentially through two pathways. In one scenario, the reductive elimination of Cu results in the formation of an azabicyclo ring (intermediate A as observed in HRMS), regenerating Cu(I) for the next catalytic cycle. Simultaneously, it undergoes fragmentation to generate the final product (Scheme 9C). In an alternative pathway, the elimination of Cu and the heterocycle leads to the formation of α-ketoester and the deoxygenated product.
 | ||
| Scheme 9 (A) & (B) Control experiments; (C) proposed mechanistic pathway. | ||
Ultimately, green metrics were computed to evaluate the environmental impact, efficacy, and sustainability of our mechanochemical protocol with the solution process.35 The analysis of the data presented in Fig. 2 revealed an E-factor nearly sixty times lower for our mechanochemically promoted oxygen transfer reactions with heterocyclic N-oxide in the synthesis of α-ketoester (3da) However, the atom economy was comparable, largely due to the lack of optimization in downstream operations. The primary goal was to produce a broad range of compounds efficiently. This indicates its advantage over heterogeneous solvent-based strategies for the same synthesis. However, the eco-scale values, which gauge the greener aspect of a process based on factors such as yield, chemical cost, safety, technical setup, reaction conditions (i.e. temperature and time), and reaction work-up/purification methods, were found to be comparable. Beyond the numerical results of the green metrics, it is essential to highlight that our procedure eliminates the need for additional heating, preventing potential degradation of heat-labile compounds. This methodology also avoids the use of large amounts of solvents, contributing to its environmental benefits.
 | ||
| Fig. 2 Green metrics comparison for the preparation of 3 dicarbonyl motifs. | ||
Conclusion
In conclusion, we have devised a convenient, environmentally friendly, highly atom-economical, and cost-effective methodology for displacing dinitrogen from α-diazocarbonyls through copper-catalyzed anaerobic oxidation using mechanochemical mixing-milling in a RETSCH mixer mill (MM 400) at ambient temperature, with η = 0.5 μL mg−1 1,2-DCE serving as a LAG auxiliary. The reaction proved remarkably mild, efficient, and chemoselective, accommodating a wide range of functional groups. With its compatibility with readily available aryl diazoacetates, we anticipate that this oxidation method will emerge as a valuable route for the synthesis of α-ketoesters.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and/or its ESI.†Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors would like to thank the State Secretariat for Education, Research and Innovation (SERI) of the Swiss Confederation to have received a research grant commissioned by ZHAW: “Leading House South Asia and Iran, Zurich University of Applied Sciences”. L. G. would like to thank the Chemistry Department, as well as the ChemTech Institute, of University of Applied Sciences Western Switzerland, Faculty of Engineering and Architecture, for the technical, operational as well as financial support. S. S. is thankful to Shiv Nadar Institution of Eminence, deemed to be University for funding and facility.References
- J. Staunton and B. Wilkinson, Chem. Rev., 1997, 97, 2611–2630 CrossRef.
- W. Li, Y. Asada and T. Yoshikawa, Planta Med., 1998, 64, 746–747 CrossRef CAS.
- S. I. Jang, et al. , Planta Med., 2003, 69, 1057–1059 CrossRef CAS.
- G. B. M. Bettòlo, C. G. Casinovi and C. A. Galeffi, Tetrahedron Lett., 1965, 52, 4857–4864 CrossRef.
- C. C. Shen, et al. , J. Nat. Prod., 2006, 69, 1237–1240 CrossRef CAS.
- H. H. Otto and T. Schirmeister, Chem. Rev., 1997, 97, 133–172 CrossRef CAS PubMed.
- M. Knaack, et al. , Eur. J. Org Chem., 2001, 2001, 3843–3847 CrossRef.
- M. D. Armistead, W. M. Harding, J. O. Saunders and S. J. Boger, US Pat., US 5723459, 1998 Search PubMed.
- B. R. Bacon, et al. , N. Engl. J. Med., 2011, 364, 1207–1217 CrossRef.
- T. F. Hartley, et al. , J. Allergy Clin. Immunol., 1985, 75, 501–507 CrossRef.
- T. D. Ashton, K. A. Jolliffe and F. M. Pfeffer, Chem. Soc. Rev., 2015, 44, 4547–4595 RSC.
- L. Wang, et al. , Angew. Chem., Int. Ed., 2013, 52, 1768–1772 CrossRef PubMed.
- C. T. Chen, et al. , J. Am. Chem. Soc., 2006, 128, 10992–10993 CrossRef PubMed.
- J. L. Sessler, D. G. Cho and V. Lynch, J. Am. Chem. Soc., 2006, 128, 16518–16519 CrossRef PubMed.
- For reviews: (a) S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. Harris, G. Hyett, W. Jones and A. Krebs, Chem. Soc. Rev., 2012, 41, 413–447 RSC; (b) X. Liu, Y. Li, L. Zeng, X. Li, N. Chen, S. Bai, H. He, Q. Wang and C. Zhang, Adv. Mater., 2022, 34, 2108327 CrossRef; (c) R. T. O'Neill and R. Boulatov, Nat. Rev. Chem, 2021, 5, 148–167 CrossRef; (d) J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080–3094 RSC; (e) J. L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed; (f) X. L. Wu, J. J. Xia and G. W. Wang, Org. Biomol. Chem., 2008, 6, 548–553 RSC; (g) G. W. Wang and X. L. Wu, Adv. Synth. Catal., 2007, 349, 1977–1982 CrossRef CAS; (h) D. Virieux, F. Delogu, A. Porcheddu, F. García and E. Colacino, J. Org. Chem., 2021, 86, 13885–13894 CrossRef CAS; (i) G. W. Wang, Chin. J. Chem., 2021, 39, 1797–1803 CrossRef CAS; (j) Y. Gao, C. Feng, T. Seo, K. Kubota and H. Ito, Chem. Sci., 2022, 13, 430–438 RSC; (k) A. C. Jones, W. I. Nicholson, J. A. Leitch and D. L. Browne, Org. Lett., 2021, 23, 6337–6341 CrossRef CAS; (l) L. Li, C. Niu and G. W. Wang, Chin. J. Chem., 2022, 40, 2539–2545 CrossRef CAS; (m) R. K. Fang, Z. C. Yin, J. S. Chen and G. W. Wang, Green Chem. Lett. Rev., 2022, 15, 519–528 CrossRef CAS.
- L. Takacs, Chem. Soc. Rev., 2013, 42, 7649–7659 RSC.
- G. W. Wang, Chem. Soc. Rev., 2013, 42, 7668–7700 RSC.
- V. Šepelák, A. Düvel, M. Wilkening, K. D. Becker and P. Heitjans, Chem. Soc. Rev., 2013, 42, 7507–7520 RSC.
- N. R. Rightmire and T. P. Hanusa, Dalton Trans., 2016, 45, 2352–2362 RSC.
- T. Friščić, Chem. Soc. Rev., 2012, 41, 3493–3510 RSC.
- A. Delori, T. Friščić and W. Jones, CrystEngComm, 2012, 14, 2350–2362 RSC.
- A. L. Garay, A. Pichon and S. L. James, Chem. Soc. Rev., 2007, 36, 846–855 RSC.
- P. Baláž, M. Achimovičová, M. Baláž, P. Billik, Z. Cherkezova-Zheleva, J. M. Criado, F. Delogu, E. Dutková, E. Gaffet, F. J. Gotor and R. Kumar, Chem. Soc. Rev., 2013, 42, 7571–7637 RSC.
- J. L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed.
- (a) S. Ni, M. Hribersek, S. K. Baddigam, F. J. Ingner, A. Orthaber, P. J. Gates and L. T. Pilarski, Angew. Chem., Int. Ed., 2021, 60, 6660–6666 CrossRef CAS; (b) R. Geib, E. Colacino and L. Gremaud, ChemSusChem, 2024, e202301921 CrossRef CAS.
- (a) Y. He, Z. Huang, K. Wu, J. Ma, Y. G. Zhou and Z. Yu, Chem. Soc. Rev., 2022, 51, 2759–2852 RSC; (b) T. Curtius, Ber. Dtsch. Chem. Ges., 1883, 16, 2230–2231 CrossRef; (c) Y. Y. Ren, S. F. Zhu and Q. L. Zhou, Org. Biomol. Chem., 2018, 16, 3087–3094 RSC.
- (a) J. Jeong, D. Lee and S. Chang, Chem. Commun., 2015, 51, 7035–7038 RSC; (b) N. R. O'Connor, P. Bolgar and B. M. Stoltz, Tetrahedron Lett., 2016, 57, 849 CrossRef; (c) Y. Yu, Q. Sha, H. Cui, K. S. Chandler and M. P. Doyle, Org. Lett., 2018, 20, 776–779 CrossRef CAS.
- (a) L. Melone and C. Punta, Beilstein J. Org. Chem., 2013, 9, 1296–1310 CrossRef CAS; (b) C. Bian, A. K. Singh, L. Niu, H. Yi and A. Lei, Asian J. Org. Chem., 2017, 6, 386–396 CrossRef CAS; (c) A. Petrosyan, R. Hauptmann and J. Pospech, Eur. J. Org Chem., 2018, 2018, 5237–5252 CrossRef CAS; (d) B. S. Anju, R. Bhattacharjee, S. Gupta, S. Ahammad, A. Datta and S. Kundu, Chem. Commun., 2020, 56, 12166–12169 RSC.
- (a) T. Ye and M. A. Mckervey, Chem. Rev., 1994, 94, 1091–1160 CrossRef; (b) M. Ma, C. Li, L. Peng, F. Xie, X. Zhang and J. Wang, Tetrahedron Lett., 2005, 46, 3927–3929 CrossRef; (c) P. Truong, C. S. Shanahan and M. P. Doyle, Org. Lett., 2012, 14, 3608–3611 CrossRef; (d) M. B. Rubin and R. Gleiter, Chem. Rev., 2000, 100, 1121–1164 CrossRef.
- Z. Guo, H. Huang, Q. Fu and W. Hu, Synlett, 2006, 15, 2486–2488 Search PubMed.
- For reviews: (a) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides, Wiley, New York, 1998, p. 652 Search PubMed; (b) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981–10080 CrossRef.
- A. K. Jha and N. Jain, Chem. Commun., 2016, 52, 1831–1834 RSC.
- (a) M. P. Doyle, M. A. McKervey and T. Ye, Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides, Wiley, New York, 1998, p. 652, ISBN: 978-0-471-13556-2 Search PubMed; (b) X. Zhao, Y. Zhang and J. Wang, Chem. Commun., 2012, 48, 10162–10173 RSC.
- C. D. Lu, H. Liu, Z. Y. Chen, W.-H. Hu and A. Q. Mi, Chem. Commun., 2005, 20, 2624–2626 RSC.
- C. Xu, Y. Wang and L. Bai, J. Org. Chem., 2020, 85, 12579–12584 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional references cited within the ESI. See DOI: https://doi.org/10.1039/d4mr00097h |
| This journal is © The Royal Society of Chemistry 2025 |