
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDendritic cell activation by iron oxide nanoparticles depends on the extracellular environment†
Mason
Song
a,
Robert
Ivkov
bcde and
Preethi
Korangath
 *bc
*bc
aDepartment of Biomedical Engineering, Whiting School of Engineering, Johns Hopkins University, Baltimore 21218, USA
bDepartment of Radiation Oncology and Molecular Radiation Sciences, School of Medicine, Johns Hopkins University, 1550 Orleans Street, Cancer Research Building – II, Rm 416, Baltimore, MD 21231, USA. E-mail: kpreeth1@jhmi.edu
cDepartment of Oncology, Sydney Kimmel Comprehensive Cancer Center, School of Medicine, Johns Hopkins University, Baltimore, MD 21231, USA
dDepartment of Mechanical Engineering, Whiting School of Engineering, Johns Hopkins University, Baltimore, 21218, USA
eDepartment of Materials Science and Engineering, Whiting School of Engineering, Johns Hopkins University, Baltimore, 21218, USA
First published on 12th November 2024
Abstract
Nanoparticles can exert immune modulating effects in a host depending on composition, mode of administration, and type of disease. Although the specific mechanisms of nanoparticle-induced immune responses remain unclear, their uptake by macrophages and other phagocytic innate immune cells is considered to be a key event. Our objective here was to ascertain if nanoparticle-mediated activation of dendritic cells (DCs) occurs in vitro or in vivo when exposed to hydroxyethyl starch-coated iron oxide nanoparticles. For the present studies, our choice of nanoparticles, animal model, and experimental design is motivated by our previously published observations that systemic exposure can induce antitumor adaptive immune responses in mouse models of metastatic breast cancer. Here, we began by assessing the potential toxicity of systemic exposure to commercially available starch-coated Bionized Nanoferrite® nanoparticles (BP) by measuring body weight, complete blood count, and enzyme parameters in healthy FVB/NJ mice after repeated BP dosing. We observed no evidence of toxicity at doses up to 25 mg Fe per mouse, five-fold higher than those used in subsequent in vivo experiments. We then measured the expression of surface maturation markers (CD86, MHC II) in DCs incubated with BP in vitro. Although DCs cultured with BP revealed high levels of nanoparticle uptake, neither JAWSII dendritic cells nor bone marrow derived dendritic cells (BMDCs) showed significant changes in marker expression to indicate stimulation of maturation and effector function. To assess whether BP interactions in vivo produced different effects, we analyzed CD80, CD86, and MHC II expression of DCs recovered from the livers, spleens, bone marrows, and lymph nodes of mice injected once with BP (5 mg Fe). Interestingly, only DCs in spleens and bone marrow cells responded to BP exposure. DCs recovered from other organs showed no evidence of increased activation. These findings highlight complex interactions between living systems and nanoparticles, and their potential to mediate context-specific and selective activation of innate immune cells. Our study also emphasizes that results obtained from in vitro experiments must be interpreted with caution, as they may not faithfully represent responses in living systems.
Introduction
Since the 1930s, iron-containing colloids, or nanoparticles are used as parenteral iron replacement therapy to treat iron deficiency anemia (IDA).1–4 Other clinical, and biomedical research applications with iron/iron oxide nanoparticles include magnetic hyperthermia cancer therapy,1 magnetic resonance (MR) contrast for diagnostic imaging1,2,5 and cell tracking.2,6,7 Most recently, magnetic iron-containing nanoparticles are being explored as magnetic particle imaging (MPI) tracers for diagnostic imaging and integrated theranostic applications.6–8 Despite the history, it remains unclear if, or how specific iron-nanoparticle compositions might interact with individual immune cell lineages to affect host immune responses. A growing body of evidence shows that these interactions can initiate complex disease-specific immune responses in the host through macrophages to suppress cancer progression.9–14Systemic exposure to hydroxyethyl starch-coated Bionized Nanoferrite nanoparticles (BP) can stimulate antitumor type I interferon (IFN) responses by activating toll-like receptor (TLR) that include signaling via toll/interleukin-1 receptor domain-containing adaptor inducing IFN-β (TRIF) pathway(s) leading to inhibition of tumor growth, metastasis, and disease progression in mouse models of metastatic breast cancer.15,16 When systemically delivered, BP nanoparticles ingested by phagocytic immune cells – monocytes, macrophages, dendritic cells and NK cells, was followed by recruitment of anti-tumor CD8+ T cells to tumor microenvironment in a time dependent manner in an orthotopic, syngeneic mouse model of breast cancer.15 Another study showed activation of TLR/TRIF pathway with downstream activation of IRF3, leading to activation of type 1 IFN anti-tumor immune response that suppressed metastases in both transgenic and syngeneic mouse models of metastatic breast cancer.16 The host immune profile was altered with increased numbers of activated T cells, NK T cells, and dendritic cells (DCs) in tumors, spleens and livers of tumor bearing mice after BP treatment.16 These results show that uptake of BP nanoparticles by innate immune cells can stimulate host-driven innate to adaptive anti-tumor immune signaling to suppress disease.
Adaptive anti-tumor immune responses require T cell activation, which implies cross presentation of antigens by innate immune cells. Although macrophages exhibit some antigen presenting function to activate antitumor T cells, their capacity to do so is limited compared to that of DCs.17,18 Indeed, there is less evidence directly linking specific macrophage phenotypes with increased CD8+ T cell activity in tumors than there is for specific DCs.19 Innate to adaptive immune signaling, e.g. via TRIF activation, stimulates maturation of DCs20 and their cross presentation of antigens.21 Immature DCs, frequently found in peripheral tissues, internalize pathogens or antigens through nonselective actin-mediated phagocytosis.22 Fragmented antigenic information is then presented on their surface, which is coupled with major histocompatibility complex (MHC) molecules.23 Activation and maturation of DCs typically begins when stimulated by danger signals known as pathogen-associated molecular patterns (PAMPs). DCs recognize PAMPs through pattern recognition receptors (PRRs), such as toll-like receptors (TLRs).24,25 Thus, DC maturation and activation is context-specific, depending on tissue type and host health.
Upon stimulation of DCs, the concentration of intracellular Ca2+ increases to activate transcription factors such as nuclear factor of activated T cells (NFAT) or nuclear factor-κB (NF-κB), leading to expression of high levels of surface markers (MHCII, CD80, CD86, and CD83), and homing receptor (CCR7).26,27 As part of their overall response, DCs secrete immunostimulatory cytokines such as IL-12 and TNF-α to activate other immune cell phenotypes in a concerted response. Subsequently, mature DCs migrate to lymph nodes in response to other chemokines (such as CCL-19 and CCL-21) secreted from the lymph nodes where T cells recognize antigen fragments presented by MHC molecules on the DC surface through T cell antigen receptors (TCRs). Simultaneously, T cells interact with DC costimulatory molecules CD80/CD86 via CD28. Naive T cells then differentiate into specialized cytotoxic T cells or helper T cells and exit the lymph nodes to combat pathogens or infected cells.22,28
Given the central role played by DCs in generating adaptive immune responses, we hypothesized that the observed anti-tumor adaptive immune responses generated by systemic exposure to BP nanoparticles occurs via DC activation in various immune rich organs of mice. To rule out the alternate hypothesis that the observed effects were caused by nanoparticle-induced immunotoxicity, or generalized toxicity, we assessed effects of high BP dose in FVB/NJ mice. We also assessed DC responses to BP exposure in vitro using the immortalized JAWS II dendritic cell line and cultured bone marrow derived dendritic cells extracted from FVB/NJ mice. Finally, we analyzed dendritic cells isolated from bone marrows, spleens, livers and lymph nodes of normal FVBN/J mice 24 hours after intravenous BP administration.
Results and discussion
Systemic exposure to high dose of BP induced modest and transient changes in blood parameters with no evidence of toxicity in mice
Generalized toxicity arising from cell death can activate immune cells in an uncontrolled manner with subsequent and immediate release of sequestered iron by macrophages, culminating in widespread toxicity, cellular damage, and death.29,30 To rule out generalized toxicity caused by exposure to BP as a mechanism inducing the observed immune modulating effects, we exposed heathy mice to multiple i.v. doses of BP. Five normal female FVB/NJ mice (average body weight 23.0 ± 0.6 g) each received weekly injections of 5 mg Fe of BP nanoparticles into their tail vein for five consecutive weeks to a total of dose of 25 mg Fe. The dose of 5 mg Fe was determined from our previous studies and this amount is analogous to that of a human dose for FDA approved colloidal iron formulations.16 All mice were euthanized 24 hours after receiving the final dose to collect blood and other organs. We observed no weight loss or any toxicity-related changes in appearance including hunched posture, piloerection, and unkempt hair coat in mice during this period (Fig. 1A). In addition, we observed no behavioral changes such as decreased activity or lethargy to indicate adverse reaction(s) or declining health. Complete blood counts (Fig. 1B), liver function (Fig. 1C) and kidney function (Fig. 1D) analysis showed measurable differences between control and BP cohorts, but all changes were modest and results remained within normal ranges reported for this strain of mouse.31,32 Histopathological analysis of all vital organs collected from mice showed no changes (ESI Fig. 1A–H†). These data indicate that systemic exposure to BP following i.v. injection, at these doses, was well tolerated by this strain of mice. BP nanoparticles are endotoxin free16 and hence any possibility of endotoxin induced immune activation is not expected. From the results, we concluded that any changes observed in immune cell phenotypes could be attributed to direct effects of BP acting on immune cells, rather than immune response(s) to widespread cell death or generalized toxicity.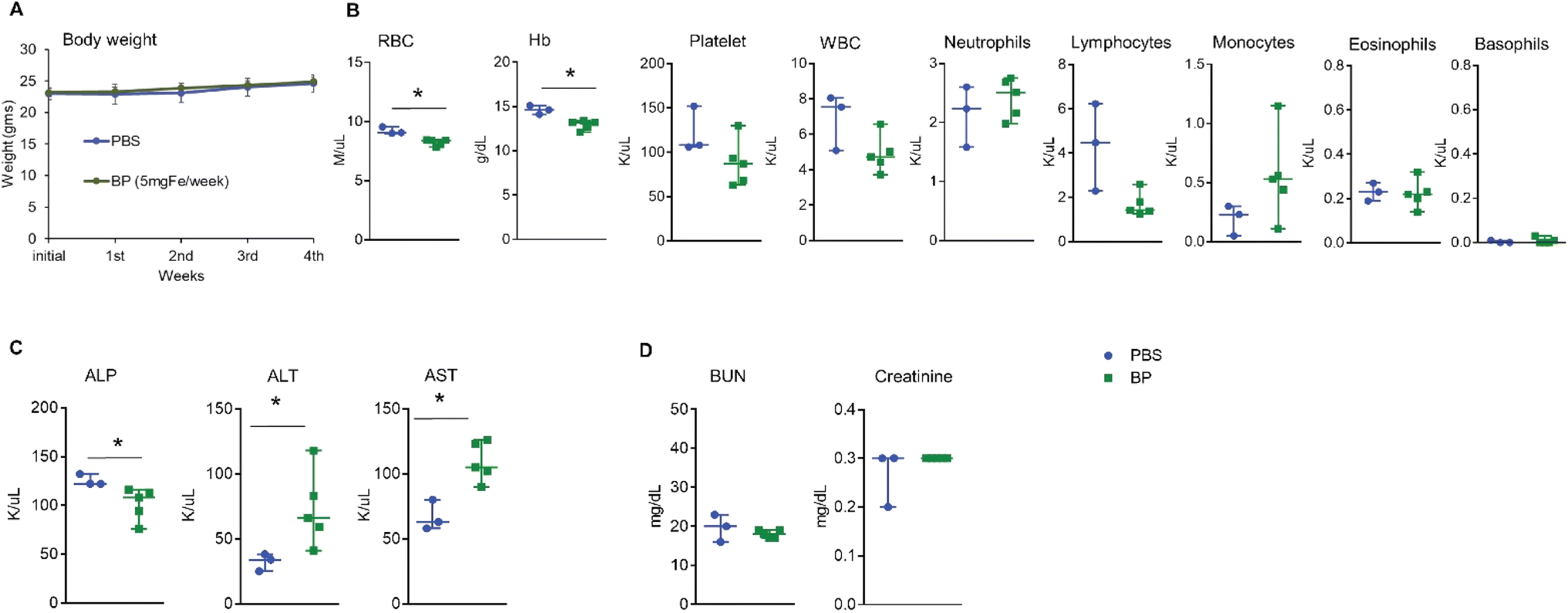 | ||
| Fig. 1 BP nanoparticles showed modest changes with no evidence of toxicity after repeated injection. (A) No change in body weight was observed between groups (n = 3 for PBS and n = 5 for BP). (B) Complete blood analysis showed a slight decrease in RBC, Hb, platelet, WBC, lymphocyte and a slight increase in neutrophils and monocytes. (C) Similarly, there were variations in liver enzyme parameters between control and BP treated group but they were within the normal range as reported before. (D) No changes in kidney function parameters were observed. All data points are shown in the figures with the median and range. | ||
Dendritic cells internalize BP in vitro but their activation depends upon specific conditions
Next, we tested whether DC activation occurs upon exposure to BP and requires direct uptake of the nanoparticles. We determined whether DCs would ingest BP in vitro by incubating JAWS II DCs with varying concentrations of BP for 24 hours. JAWS II DCs internalized BP depending on BP concentration in media as indicated by intracellular iron concentration, a measure of ingested BP (ESI Fig. 2A†). Prussian blue staining also showed internalized BP in JAWS II cells as dark blue punctate regions (ESI Fig. 2B). We measured no difference in live/dead cell numbers after incubating cells with BP, ruling out direct cytotoxicity as a factor affecting in vitro outcomes (data not shown). JAWS II cells were then assessed for changes in surface expression of CD86 and MHC II after 24 hour incubation with BP. We observed no differences in either CD86 or MHC II expression with 0.05 mg per mL or 0.25 mg per mL BP exposure (ESI Fig. 2C†). Since GM-CSF in JAWS II culture media itself is a pro-inflammatory/regulatory cytokine used to stimulate cells,33 we cultured JAWS II cells in the absence of GM-CSF once they were thawed. JAWS II cells cultured without GM-CSF (JAWS II-GM) still internalized large quantities of BP to a similar extent as those cultured with GM-CSF (Fig. 2A and B). We then assessed gene expression changes for JAWS II cells treated with 0.5 mg per mL BP grown without GM-CSF because this group had comparable intracellular iron levels to JAWS II cells grown with GM-CSF incubated with 0.25 mg per mL BP (∼8.0 pg Fe per cell). JAWS II without GM-CSF cells exposed to 0.5 mg per mL BP for 24 hours significantly upregulated expression of iNOS, with a five-fold increase in expression following exposure to BP; however, we measured only a slight increase in TNF-α (Fig. 2C). Despite the relatively high uptake of BP and increased expression of iNOS, we measured minimal but significantly higher expression of CD86 and MHC II on the surface of JAWS II-GM cells (Fig. 2D).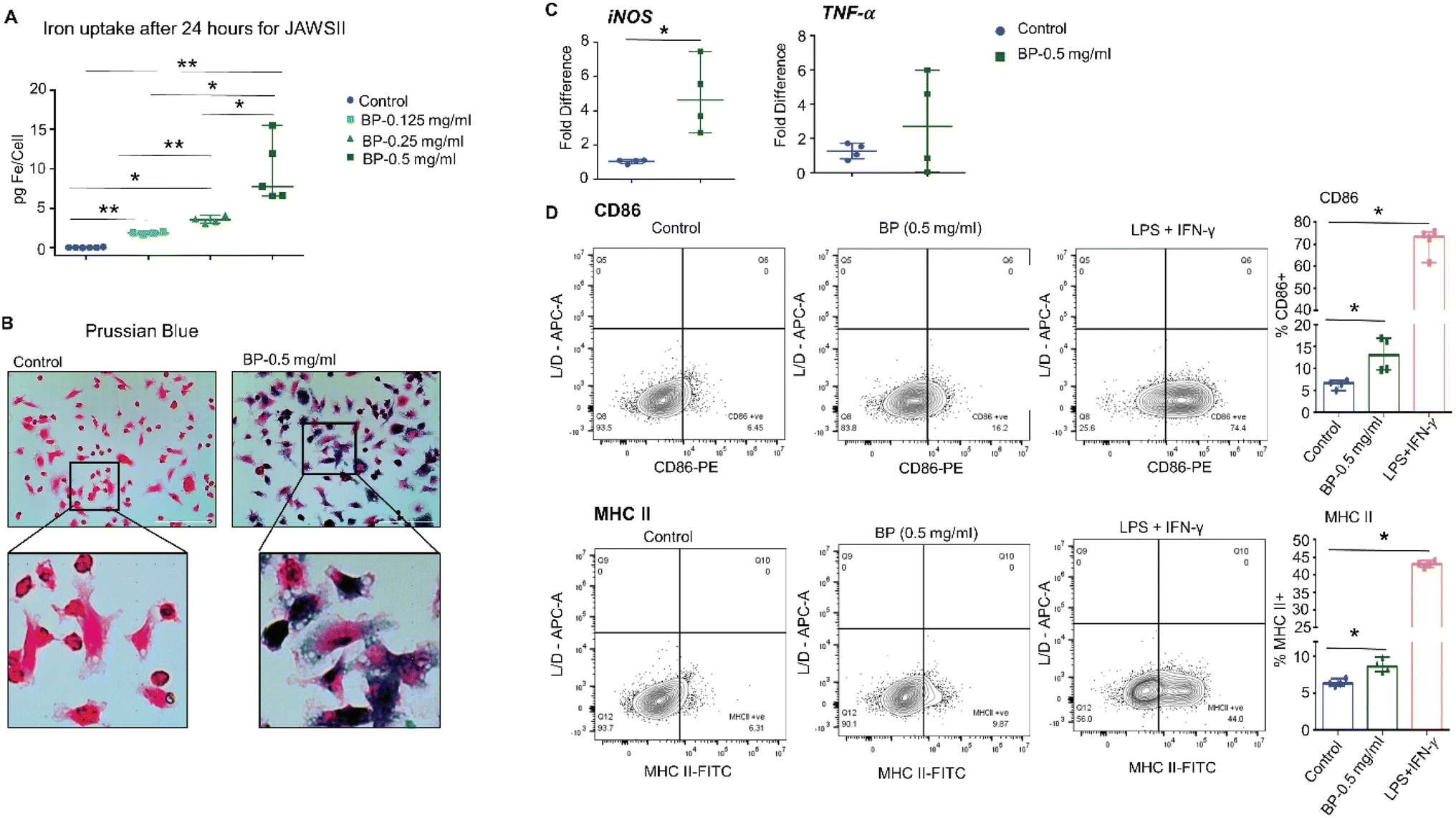 | ||
| Fig. 2 JAWS II dendritic cells grown without GM-CSF internalize BP in a dose dependent manner and activates proinflammatory gene and surface markers of activation. JAWS II dendritic cells grown without GM-CSF were incubated with BP in vitro for 24 hours. (A) JAWS II dendritic cells showed dose dependent internalization of BP nanoparticles. (B) Prussian blue staining revealed the nanoparticles inside the cells as seen by the dark blue color in the cells. (C) After 24 hours of incubation with 0.5 mg per mL BP, significant upregulation of proinflammatory gene iNOS with only a slight increase in TNF-α gene was noted. (D) Interestingly, JAWS II dendritic cells grown without GM-CSF significantly upregulated both CD86 and MHC II when incubated with 0.5 mg per mL BP. LPS + IFN-γ treatment was used as positive control. All data points from three to six independent experiments are shown in the figures with the median and range. *p ≤ 0.05, **p ≤ 0.01 – Mann Whitney test. | ||
In vitro, BMDCs harvested from mice displayed negligible evidence of activation after exposure to BP
To test whether the observed results with JAWS II cells were specific to those observed with the immortalized cell line, we assessed responses of BMDCs isolated from FVB/NJ mice. After harvest, cultured BMDCs were treated in vitro with BP for 24 hours. As with JAWS II cells, the BMDCs internalized BP, though to a lesser degree when exposed to BP at the same concentration of 0.5 mg per mL (Fig. 3A). Prussian blue staining also confirmed BP localization within cells (Fig. 3B). Despite internalizing BP, the BMDCs also showed minimal changes in expression of CD86 or MHC II on their membranes indicating little or no activation of effector immune function (Fig. 3C). These findings suggest that while BP may be ingested by DCs and may induce pro-inflammatory changes in DCs in culture, they do not directly induce or activate DC effector functions in this context.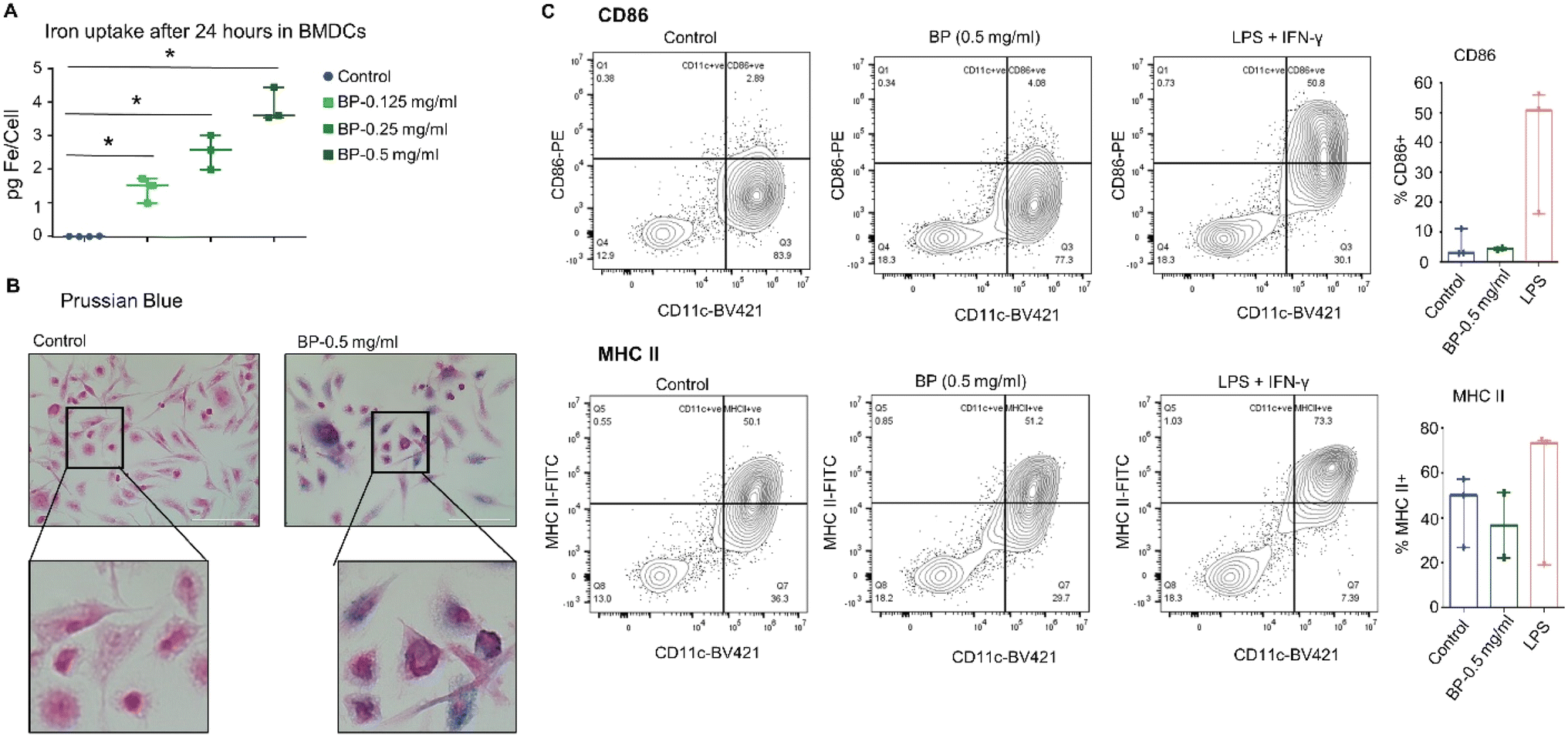 | ||
| Fig. 3 BMDCs internalize BP nanoparticles in a dose dependent manner but does not induce surface markers. BMDCs were incubated with different concentrations of BP in vitro for 24 hours. After 24 hours, intracellular iron content was quantified by ferene assay and visualized by Prussian blue. (A) BMDCs show maximum uptake when incubated at 0.5 mg per mL of BP, although at a relatively lower quantity compared to JAWS II cells. (B) Prussian blue staining confirms this observation as seen by the dark blue color in the cells. (C) Flow cytometry performed on BMDCs incubated with 0.5 mg per mL BP for 24 hours showed negligible effect on expression of CD86 and MHC II. LPS treatment was used as a positive control. All data points from three independent experiments are shown in the figures with the median and range. *p ≤ 0.05 – Mann Whitney test. | ||
Considering that DCs can be activated either directly or indirectly,34,35 we hypothesized that direct addition of BP to cells in in vitro culture lacked context provided by a living system. To test this, we queried DCs extracted from normal FVB/NJ mice after they were injected with BP.
Systemic exposure of BP triggers activation of dendritic cells in vivo
We injected BP (5 mg Fe per mouse) i.v. into normal wild type female FVB/NJ mice (n = 4 per group) to assess changes in DCs induced by nanoparticle exposure 24 hours after injection. Having previously noted tissue-specific differences in immune cell responses to systemic BP exposure,16 we analyzed DCs harvested from individual immune organs separately. We measured significant differences in activation of DCs residing in spleens and bone marrows. Splenic DCs, identified as CD11c+ cells, significantly upregulated surface expression of CD86 (median: 26.4% cells positive, IQR: 25.7–27.8%) compared to control (median: 9.55%, IQR: 7.8–9.9%) (Fig. 4A and B). Similarly, bone marrow-derived DCs upregulated surface expression of CD86 (median: 34.3%, IQR: 24.9–41.7%) compared to control (median: 2.7%, IQR: 1.4–4.8%), and surface expression of MHC II (median: 48.9%, IQR: 44.0–54.2%) compared to control (median: 24.8%, IQR: 20.8–28.4%) (Fig. 4C and D). On the other hand, we measured negligible changes in expression of CD80 and population counts of DCs in all tissues (ESI Fig. 3 and 4†). Surface expression of CD86 and MHC II markers remained unchanged in DCs from livers and lymph nodes (ESI Fig. 5†). | ||
| Fig. 4 Splenic and bone marrow dendritic cells upregulate activation markers after 24 hours of BP injection. PBS or BP (5 mg per mouse) were injected through the tail vein of female FVB/NJ mice (n = 4/group). After 24 hours, spleen and bone marrow were harvested for analyzing MHC II and CD86 expression on CD11c+ dendritic cells. (A) Representative flow cytometry dot plot of the spleen cells after treatment. (B) Statistically significant upregulation of CD86 on CD11c+ve dendritic cells was seen after BP treatment whereas there was no change in MHC II expression. (C) Representative flow cytometry dot plot of the bone marrow cells after treatment. (D) Both CD86 and MHC II were significantly upregulated after BP treatment in bone marrow cells. All data points are shown in the figures with the median and range. *p ≤ 0.05 – Mann Whitney test. | ||
The variation in DC activation may be attributed to differences in role of tissue-resident DC subset populations in each organ.36 Hepatic DCs are responsible in maintaining peripheral tolerance in a healthy context,37 which suggests BPs at this dose are insufficient to induce liver-resident DCs. On the other hand, the spleen, bone marrow, and lymph node play a crucial role in host immune regulation.38–40 Indeed, DC subsets in these organs process antigens and induce an adaptive immune response when exposed to infectious agents. Bone marrows house conventional type 2 dendritic cells (cDC2).41 cDC2s process and present antigens to CD4 T cells.42 Similarly, CD8+ DCs are among the subsets found in the spleen and lymph nodes, and these DCs are crucial for anti-viral immunity and inducing T cell responses.37 However, DC activation in lymph nodes, or lymph node involvement is often associated with a specific and developed disease.
Thus, the lack of evidence showing DC activation in lymph nodes (and potentially livers) found here may be attributed to the specific (non-diseased) biological context, and possibly time after exposure. Our chosen time point was 24 h after i.v. injection of BP in healthy FVB/NJ mice. The mice used here lacked disease-associated dysfunction, e.g. cell death, release of antigens, danger signals, etc. to stimulate robust immune responses; therefore, the chosen time may be shorter than that required to develop a full systemic host adaptive immune response(s). By comparison, when transgenic (HuHER2) FVB/NJ mice bearing HER2+ tumors were injected with BP at the same dose used here, substantial changes were noted in gene expression among multiple immune cell lineages extracted from tumor-draining lymph nodes seven days after injection.16 We thus conclude that the healthy mice used in the current study lacked appropriate biological (disease) context to generate DC activation in lymph nodes. Nevertheless, our measured upregulation of activation surface markers in DCs harvested from spleens and bone marrows 24 hours after i.v. injection of BP into healthy mice reinforces the prominent role of these tissues/organs in early response(s) to exposure by pathogens or other agents.
Our results also suggest an indirect mechanism of DC activation.43,44 Functional properties of DCs are influenced by their extracellular environment.45 Nanoparticles in the bloodstream will adsorb serum proteins, such as opsonins, complement proteins, immunoglobulins, and dysopsonins.46,47 These proteins can contribute to the nanoparticle biological identity, which consequently modulates or dictates nanoparticle immune recognition or its immunogenicity.47,48 We also expect that coronal composition within an individual could influence nanoparticle retention in tissues and thus which tissue resident cells will react and ingest the nanoparticles. Therefore, differences in the coronal composition may explain some of the observed discrepancies between in vivo and in vitro experiments conducted here as the latter lack much of the biological complexity of a living system.
Results presented here support an interpretation that BP-mediated activation of DC effector function in mice depends on the microenvironment in which the DCs reside. Various secretory factors produced by other cell types often dictate functional changes of innate immune cells. Böttcher et al., shown that cDC1 accumulation in mouse tumors often depends on chemoattractants produced by co-resident NK cells.49 DC maturation can be directly influenced by their interaction with NK cells.50 In this study, we did not consider the relative influence of indirect, i.e. cell-mediated DC activation that may be stimulated by other (innate) immune cell phenotypes, e.g. macrophages or NK cells, after they internalize BP. Nanoparticle composition, specifically varied coatings (e.g. dextran, polyethylene glycol, etc.) may also influence DC activation by altering biodistribution and/or DC uptake. We noted higher variations in certain markers in organs like lymph nodes. Inherent variability of immune cells, especially in the more rare populations such as DCs (which typically comprise only about 1% of all myeloid cells; which comprise a very small population of all cells, ∼1%), is a known challenge in the immunology field. Within the stated limitations, our results here demonstrate that DC activation can be stimulated by exposure to BP nanoparticles, but that the activation is context specific. Thus, DC activation by BP may require additional and specific interactions with other immune subtypes, as arising from disease, to receive appropriate signaling cues to fully engage their effector function(s).51 Ascertaining which of these mechanisms predominates, and in what specific disease context(s), requires further study with a focus on the potential crosstalk among immune cell types.
Conclusion
Here we document evidence showing that in healthy FVB/NJ mice, BP can activate dendritic cells in an organ-specific manner. We found no evidence of acute toxicity in mice at the doses tested. This is important as generalized toxicity can produce immune responses to confound interpretation.52–54 We also found little evidence of DC activation following exposure to BP in cell culture. Within the limits of the present study, we conclude that systemic exposure to BP can stimulate immune responses; however, the degree of stimulation to exert effector function depends on the extracellular environment. Taken together, these data suggest that BP can stimulate DC activation, but that DCs require additional (co-) stimulatory cues to become fully activated. Elucidating mechanistic details of BP induced DC stimulation requires further investigation.Experimental
Nanoparticle
Hydroxyethyl starch-coated Bionized Nanoferrite® (BP) nanoparticles (Micromod Partikeltechnologie, GmbH, Rostock, Germany) were purchased as suspensions in sterile water (20 mg Fe per mL) and were used as received from the manufacturer for all experiments. We have previously characterized the nanoparticles for physical and magnetic properties, chemical composition, and endotoxin content.16,55–57 They comprise a multi-crystallite core, ∼50 nm diameter containing a mixture of Fe3O4, γ-Fe2O3 and Fe(OH)2 parallelepiped-shaped grains.55,56 When coated with hydroxyethyl starch, z-average hydrodynamic diameter measured by dynamic light scattering is ∼100 nm with ∼0.7 polydispersity index and zeta potential of ∼0 mV to −5 mV (Table S1†). Endotoxin testing by us and manufacturer revealed no contamination within detection limits.16 Hydrodynamic diameter, surface charge, and concentration data for each BP batch used in the present study were provided by the manufacturer (Table S1†).Animal study
All animal studies were performed in strict accordance with the NIH guidelines for the care and use of laboratory animals (Guide for the Care and Use of Laboratory Animals, 8th edition, National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals. Washington (DC): National Academies Press (US); 2011). The animal experiment protocol used in this study was approved by the Institutional Animal Care and Use Committees (IACUC) of the Johns Hopkins University and the animals were treated according to the policies and guidelines of the Johns Hopkins University Animal Care and Use Committee, Baltimore, Maryland. Female FVB/NJ mice having varied ages were purchased (Jackson Laboratory, Bar Harbor, ME) and were maintained on 12 h day/night cycle and fed ad libitum. For in vivo toxicity studies, eight mice (8 weeks old) were used. For in vivo immune cell analysis eight mice (10–12 weeks old) were used, and seven mice (7–16 weeks old) were used for in vitro BMDC culture.In vivo toxicity analysis
Mice were given weekly injections of BP (n = 5) (5 mg Fe per mouse, for up to five weeks or 25 mg Fe total dose) or PBS (n = 3) intravenously (i.v.) for up to 5 weeks to assess potential for toxicity. Body weight was measured weekly, and the animals were monitored for indications of toxicity such as lethargy and ruffled hair. For the study, animals were euthanized 24 h after the final dose. Whole blood was collected by heart puncture and submitted to the JHU core facility for complete blood count (CBC analysis). Serum was separated to analyze alanine transaminase (ALT), alkaline phosphatase (ALP), aspartate transferase (AST), blood urea nitrogen (BUN), and creatine levels. We also harvested and fixed the lungs, kidney, intestine, heart, bone marrow, spleen, lymph node, and liver in 10% formalin for 48 hours and paraffin embedded. 5 μm thick slices were cut on to charged slides to perform hematoxylin and eosin (H&E) staining through Johns Hopkins core facility. Images were viewed and taken with Aperio ImageScope software (version 12.4.0.5043).JAWS II cell culture
JAWS II dendritic cell line was purchased from the American Type Culture Collection (ATCC, CRL-11904). JAWS II cells were cultured under 5% CO2 at 37 °C with complete media consisting of 80% alpha Modified Essential Medium (α MEM) (Sigma Aldrich, St Louis, MO), 20% FBS (HyClone, South Logan, Utah), 4 mM L-glutamine (Gibco, Grand Island, NY), and 1 mM sodium pyruvate (Sigma Aldrich, UK). JAWS II were cultured either with or without 5 ng per mL murine granulocyte macrophage colony-stimulating factor (GM-CSF) (PeproTech, Cranbury, NJ) to account for its pro-inflammatory properties. JAWS II cells (P5) were used for all experiments after growing them for 48 hours. All in vitro experiments were performed in triplicate and repeated at least 4 times or more independently unless mentioned otherwise in the methods below. Flow cytometry analysis showed comparable expression of CD86 and MHC II protein expression on JAWS II cells grown with or without GM-CSF (Table S2†).BMDC culture
Bone marrow derived dendritic cells (BMDCs) culture media and specifications were adapted and modified from prior literature.58 For this, healthy female FVB/NJ mouse (n = 7) (Jackson Laboratory, Bar harbor, ME) aged 7–16 weeks were sacrificed to harvest bone marrow cells (day 0). Femurs and tibias were collected from each mouse and flushed into a conical tube with RPMI (Sigma Aldrich, St. Louis, MO). The conical tube was centrifuged at 200 g for 5 minutes at 4 °C, with cell suspension aspirated. To remove non-cellular debris, the cell pellet was then resuspended with fresh RPMI and passed through a 100 μm cell strainer (Corning Inc., Corning, NY). The cells were then centrifuged under the same conditions. ACK lysis buffer (1 mL) (Quality Biological, Gaithersburg, MD) was added to remove red blood cells for 5 minutes at room temperature. Incomplete dendritic cell media (4 mL) containing 90% Iscove's Modified Dulbecco's Medium (Thermo Fisher Scientific, Waltham, MA), 10% HI-FBS (Corning, Woodland, CA), 2 mM L-glutamine (Gibco, Grand Island, NY), 100 IU per mL pen. (Gibco, Grand Island, NY), 100 mg per mL strep. (Gibco, Grand Island, NY), and 50 μM 2-mercaptoethanol (Sigma Aldrich, St Louis, MO) were added to stop the lysis. The cell suspension was centrifuged one final time at the same conditions, counted, and seeded into complete media containing 20 ng per mL GM-CSF (PeproTech, Cranbury, NJ) and 20 ng per mL IL-4 (Miltenyi Biotec, Auburn, CA). Bone marrow cells were cultured in 100 mm culture dishes at a density of 0.25 × 106 cells per mL in complete media for a total volume of 12 mL. The cells were split on day 3. On day 5, 75% of the old media was replaced with new media. The cells were collected on day 7 for subsequent experiments. Experiments using BMDCs were done in triplicate (technical replicates) and repeated three times independently.Quantitative real time polymerase chain reaction (qRT-PCR)
Relative gene expression changes were assessed using qRT-PCR in JAWS II cells. Tumor necrosis factor-α (TNF-α) and inducible nitric oxide synthase (iNOS) expression were quantified for JAWS II cells. These genes were chosen for their relevance in immune cell effector function. TNF-α is a proinflammatory cytokine that is a maturation marker in dendritic cells,59,60 whereas iNOS expression regulates the immune function in dendritic cells.61 Ribosomal RNA 36B4 was used as the housekeeping gene for JAWS II. Briefly, RNA was isolated by TRIzol method (Ambion, Austin, TX). Cell pellets after incubation were dissolved in 1 mL of TRIzol followed by the addition of chloroform for phase separation. The cells were then pelleted by centrifugation. Supernatant containing RNA was collected into a new tube and precipitated out with isopropanol. The samples were centrifuged and washed twice with 75% EtOH/DEPC H2O. After the final wash, all the supernatants were discarded, and the RNA pellet was dissolved with RNase free water. Concentration was quantified using the NanoDrop spectrophotometer (Fisher Scientific, Hampton, NH). RNA (2 μg) from each sample was treated with DNase I for 15 minutes at room temperature. EDTA was added to the sample to stop enzyme activity and heated at 65 °C for 10 minutes in the PCR machine (Applied Biosystems, Waltham, MA). cDNA was then synthesized by adding reverse transcriptase, 5× reaction, and nuclease free water as per manufacturer's instruction. cDNA was synthesized in a PCR machine. After cDNA synthesis, cDNA, primers, and SYBR Green Supermix were added to a 96 well plate. qRT-PCR was performed with the Bio-Rad CFX opus 96 (Bio-Rad Laboratories, Hercules, CA), and the relative expression of each gene was calculated using the ΔΔCT method and shown as fold change relative to controls grown in absence of any immune stimulants.Primer sequences used were:
| iNOS – forward: GAGACAGGGAAGTCTGAAGCAC |
| iNOS – reverse: CCAGCAGTAGTTGCTCCTCTTC |
| TNF-α – forward: GGATCTCAAAGACAACCAAC |
| TNF-α – reverse: ACAGAGCAATGACTCCAAAG |
| 36B4 – forward: AGATTCGGGATATGCTGTTGGC |
| 36B4 – reverse: TCGGGTCCTAGACCAGTGTTC |
Iron uptake quantification
Ferene-s assay was used to quantify the intracellular nanoparticle content, as previously described.62 Briefly, one million cells were suspended in 1 mL of complete media in 4 mL polystyrene tubes. Aliquots of the stock BP were added to the cell suspensions to nanoparticle concentrations of 0.125 mg mL−1, 0.25 mg mL−1, or 0.5 mg mL−1. After 24 hours of incubation at 37 °C and 5% CO2, samples were processed to measure iron content at a wavelength of 595 nm.Iron uptake visualization
Prussian blue staining was used to visualize intracellular iron oxide nanoparticles. Briefly, cells were seeded onto a four chamber well slide (Thermo Fisher Scientific, Rochester, NY) after 24 hour incubation with BP. Cells were incubated for another 48 hours to adhere onto the well surface and fixed using formalin. Fixed cells were incubated with a working solution of hydrochloric acid and potassium ferrocyanide for 20 minutes to detect the presence of ferric ions. Afterwards, each well was counterstained with nuclear fast red (Abcam, Cambridge, UK) and imaged with the 473 M Evos microscope at 40× magnification (Thermo Fisher Scientific, Waltham, MA).In vitro surface marker characterization
The surface markers of interest are CD80, CD86, and MHC II because they are needed for antigen presentation and represents mature and activated dendritic cells.63,64 For BMDCs, CD11c was also used to determine the successful differentiation into dendritic cells.65 JAWS II cells and BMDCs (0.5 × 106 cells) were incubated with nanoparticles in 6-well plates (Corning Inc, Corning, NY) for 24 h at 37 °C, 5% CO2. As a positive control, JAWS II cells incubated with lipopolysaccharide (LPS) (1 μg mL−1) and IFN-γ (10 ng mL−1), while BMDCs were incubated with 500 ng per mL LPS. After incubation, cells were centrifuged, washed, and suspended with FACS buffer consisting of live/dead fixable far red (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000) (Molecular Probes, Eugene, OR), antibodies CD80–PerCP-Cy5.5 (1:20) (Biolegend, San Diego, CA), CD86–PE (1:80) (Biolegend, San Diego, CA), and MHC II–FITC (1:200) (Biolegend, San Diego, CA) in the dark, on ice, for 30 minutes. BMDCs were also stained with CD11c–BV421 (1:40) (Biolegend, San Diego, CA). Afterwards, all tubes were washed with FACs buffer 2 times before analyzing on the flow cytometer (Beckman Coulter, Brea, CA). Flow cytometry experiments were repeated three times for JAWS II cells grown with GM-CSF, four times for JAWS II cells grown without GM-CSF, and three times for BMDCs. Subsequent analysis was performed using FlowJo software (version 10.8).
:1000) (Molecular Probes, Eugene, OR), antibodies CD80–PerCP-Cy5.5 (1:20) (Biolegend, San Diego, CA), CD86–PE (1:80) (Biolegend, San Diego, CA), and MHC II–FITC (1:200) (Biolegend, San Diego, CA) in the dark, on ice, for 30 minutes. BMDCs were also stained with CD11c–BV421 (1:40) (Biolegend, San Diego, CA). Afterwards, all tubes were washed with FACs buffer 2 times before analyzing on the flow cytometer (Beckman Coulter, Brea, CA). Flow cytometry experiments were repeated three times for JAWS II cells grown with GM-CSF, four times for JAWS II cells grown without GM-CSF, and three times for BMDCs. Subsequent analysis was performed using FlowJo software (version 10.8).
In vivo immune cell analysis
Female FVB/NJ mice aged 10–12 weeks were assigned to either the control or the BP-treated group. PBS or 5 mg of BP (170 μL) were injected through the tail vein for control and treated groups, respectively. The mice were sacrificed 24 hours after, and the liver, spleen, lymph node, and bone marrow were harvested for further analysis because we have previously seen BP accumulate in these immune cell rich organs.16,66 The methods were adapted from prior publication.67 Liver was digested and Percoll extraction was performed as described below. All other organs were simply mashed and filtered through 70 μm strainers before ACK lysing and proceeded as given below. The liver samples were minced and transferred into 50 mL conical tubes containing 25 mL digestion media consisting of 0.1% collagenase type 4 (Worthington Biochemical Corporation, Lakewood, NJ), 0.005% hyaluronidase (MP Biomedicals LLC, Solon, OH), and 10% HI-FBS in DMEM (Life Technologies, Carlsbad, CA). Tubes were incubated at 37 °C in a rotor for 30 minutes. All samples were mashed through 70 μm strainers and washed with DMEM w/ 10% HI-FBS and centrifuged at 1400 rpm for 10 minutes at 4 °C. After discarding the media, cell pellet was resuspended with DMEM with 10% HI-FBS. Next, all samples are centrifuged at 1400 rpm for 10 minutes at 4 °C. After decanting the supernatants, 4 mL of ACK lysis buffer (Quality Biological, Gaithersburg, MD) was added to each sample for 5 minutes. DMEM (6 mL) with 10% HI-FBS was added to quench the reaction. The samples were then centrifuged at 1400 rpm for 10 minutes. The liver cell pellets were resuspended in freshly made 5 mL of 40% Percoll (in 1× PBS), and underlayed with 5 mL of 80% Percoll (in 1× PBS). Both Percoll solutions were prepared from 1× Percoll (Cytiva, Marlborough, MA) which was diluted with 10× PBS in 9:1 ratio. The samples were then centrifuged at 3200 rpm for 25 minutes. Once complete, the middle cell layer was collected, mixed with 20 mL of DMEM with 10% HI-FBS, and centrifuged at 1400 rpm for 10 minutes and total number of cells were counted using the Cellometer (Nexcelom Bioscience, Lawrence, MA).
Approximately 1–2 million cells were seeded into 96 well U-bottom plates. After decanting and washing each well with 200 μL of PBS with 5% HI-FBS, cells were stained with 100 μL of live/dead fixable far red (1:1000) (Molecular Probes, Eugene, OR) in the dark for 30 minutes at room temperature. Afterwards, the samples were blocked with 100 μL of anti-CD16/32 (Biolegend, San Diego, CA) for 10 minutes in the dark on ice. For dendritic cells, samples were then stained with a 50 μL of single stain or master mix containing CD80–PerCP-Cy5.5 (1:20) (Biolegend, San Diego, CA), CD86–PE (1:80) (Biolegend, San Diego, CA), MHC II–FITC (1:200) (Biolegend, San Diego, CA), and CD11c–BV421 (1:40) (Biolegend, San Diego, CA) for 30 minutes in the dark at room temperature. Once the staining is done, the cell samples are centrifuged and washed 3 times before proceeding with flow cytometry (CytoFLEX, Beckman Coulter, Brea, CA). Compensation beads (Invitrogen, Waltham, MA) were used for single stain antibodies. Data were analyzed with FlowJo version 10.8 (Becton, Dickinson and Company, Franklin Lakes, NJ).
Statistical analysis
For all statistical analysis, we used Graphpad Prism 6 (Insight Partners, New York City, NY). For tests of significance, non-parametric Mann Whitney tests were performed. For this study, a p value of less than or equal to 0.05 was deemed to be statistically significant.Data availability
All raw data including excel files, .fcs files and original image files are accessible through https://doi.org/10.7281/T1/AOTRD6, Johns Hopkins Research Data Repository, V1.Author contributions
Investigation, data collection, and data curation: M. S., P. K.; writing – original draft preparation: M. S., P. K.; writing – review & editing: M. S., R. I., P. K.; study design, conceptualization, supervision, project administration and funding acquisition: R. I., P. K.Conflicts of interest
R. I. is an inventor listed on several nanoparticle patents. All patents are assigned to either Johns Hopkins University or Aduro Biosciences, Inc. All other authors report no other competing interests.Acknowledgements
This work was funded by Jayne Koskinas Ted Giovanis (JKTG) Foundation for Health and Policy. Animal and other core facilities were supported in part by the NIH/NCI grant P30 CA006973. R. I. and P. K. received additional funding from NIH/NCI grant R01 CA257557. The content is solely the responsibility of the authors and does not necessarily represent the official views of Johns Hopkins University, JKTG Foundation, or the National Institutes of Health.References
- A. K. Hauser, M. I. Mitov, E. F. Daley, R. C. McGarry, K. W. Anderson and J. Z. Hilt, Biomaterials, 2016, 105, 127–135 CrossRef CAS PubMed.
- A. Khurana, H. Nejadnik, F. Chapelin, O. Lenkov, R. Gawande, S. Lee, S. N. Gupta, N. Aflakian, N. Derugin, S. Messing, G. Lin, T. F. Lue, L. Pisani and H. E. Daldrup-Link, Nanomedicine, 2013, 8, 1969–1983 CrossRef CAS PubMed.
- R. Provenzano, B. Schiller, M. Rao, D. Coyne, L. Brenner and B. J. G. Pereira, Clin. J. Am. Soc. Nephrol., 2009, 4, 386–393 CrossRef CAS.
- B. S. Spinowitz, A. T. Kausz, J. Baptista, S. D. Noble, R. Sothinathan, M. V. Bernardo, L. Brenner and B. J. G. Pereira, J. Am. Soc. Nephrol., 2008, 19, 1599 CrossRef CAS PubMed.
- T. Matsushita, Y. Kusakabe, H. Fujii, K. Murase, Y. Yamazaki and K. Murase, Magn. Reson. Imag., 2011, 29, 173–178 CrossRef CAS PubMed.
- G. Song, M. Chen, Y. Zhang, L. Cui, H. Qu, X. Zheng, M. Wintermark, Z. Liu and J. Rao, Nano Lett., 2018, 18, 182–189 CrossRef CAS PubMed.
- B. Zheng, T. Vazin, P. W. Goodwill, A. Conway, A. Verma, E. U. Saritas, D. Schaffer and S. M. Conolly, Sci. Rep., 2015, 5, 14055 CrossRef PubMed.
- C. Li, Nat. Mater., 2014, 13, 110–115 CrossRef CAS PubMed.
- H. H. Gustafson, D. Holt-Casper, D. W. Grainger and H. Ghandehari, Nano Today, 2015, 10, 487–510 CrossRef CAS.
- C. M. Thielmann, M. Costa da Silva, T. Muley, M. Meister, E. Herpel and M. U. Muckenthaler, Sci. Rep., 2019, 9, 11326 CrossRef PubMed.
- S. Zanganeh, G. Hutter, R. Spitler, O. Lenkov, M. Mahmoudi, A. Shaw, J. S. Pajarinen, H. Nejadnik, S. Goodman, M. Moseley, L. M. Coussens and H. E. Daldrup-Link, Nat. Nanotechnol., 2016, 11, 986–994 CrossRef CAS.
- W. Zhang, S. Cao, S. Liang, C. H. Tan, B. Luo, X. Xu and P. E. Saw, Front. Bioeng. Biotechnol., 2020, 8, 537 CrossRef PubMed.
- E. Cendrowicz, Z. Sas, E. Bremer and T. P. Rygiel, Cancers, 2021, 13, 1946 CrossRef CAS.
- M. Bied, W. W. Ho, F. Ginhoux and C. Blériot, Cell. Mol. Immunol., 2023, 20, 983–992 CrossRef CAS.
- P. Korangath, J. D. Barnett, A. Sharma, E. T. Henderson, J. Stewart, S.-H. Yu, S. K. Kandala, C.-T. Yang, J. S. Caserto, M. Hedayati, T. D. Armstrong, E. Jaffee, C. Gruettner, X. C. Zhou, W. Fu, C. Hu, S. Sukumar, B. W. Simons and R. Ivkov, Sci. Adv., 2020, 6, eaay1601 CrossRef CAS.
- P. Korangath, L. Jin, C.-T. Yang, S. Healy, X. Guo, S. Ke, C. Grüttner, C. Hu, K. Gabrielson, J. Foote, R. Clarke and R. Ivkov, ACS Nano, 2024, 18(15), 10509–10526 CrossRef CAS.
- V. Yao, C. Platell and J. C. Hall, ANZ J. Surg., 2002, 72, 501–506 CrossRef PubMed.
- J. M. H. de Jong, D. H. Schuurhuis, A. Ioan-Facsinay, M. M. Welling, M. G. M. Camps, E. I. H. van der Voort, T. W. J. Huizinga, F. Ossendorp, J. S. Verbeek and R. E. M. Toes, Immunology, 2006, 119, 499–506 CrossRef CAS PubMed.
- E. M. Muntjewerff, L. D. Meesters and G. van den Bogaart, Front. Immunol., 2020, 11, 1276 CrossRef CAS PubMed.
- H. Weighardt, G. Jusek, J. Mages, R. Lang, K. Hoebe, B. Beutler and B. Holzmann, Eur. J. Immunol., 2004, 34, 558–564 CrossRef CAS.
- S. Burgdorf, C. Schölz, A. Kautz, R. Tampé and C. Kurts, Nat. Immunol., 2008, 9, 558–566 CrossRef CAS.
- J. A. Villadangos and P. Schnorrer, Nat. Rev. Immunol., 2007, 7, 543–555 CrossRef CAS PubMed.
- J. Banchereau and R. M. Steinman, Nature, 1998, 392, 245–252 CrossRef CAS PubMed.
- N. W. Palm and R. Medzhitov, Immunol. Rev., 2009, 227, 221–233 CrossRef CAS.
- K. Takeda, T. Kaisho and S. Akira, Annu. Rev. Immunol., 2003, 21, 335–376 CrossRef CAS PubMed.
- M. Vukcevic, F. Zorzato, G. Spagnoli and S. Treves, J. Biol. Chem., 2010, 285, 16003–16011 CrossRef CAS.
- I. Mellman and R. M. Steinman, Cell, 2001, 106, 255–258 CrossRef CAS.
- K. Palucka and J. Banchereau, Nat. Rev. Cancer, 2012, 12, 265–277 CrossRef CAS PubMed.
- J. E. Cassat and E. P. Skaar, Cell Host Microbe, 2013, 13, 509–519 CrossRef CAS PubMed.
- A. Shander, M. D. Cappellini and L. T. Goodnough, Vox Sang., 2009, 97, 185–197 CrossRef CAS.
- MPD, Mouse strain: FVB/NJ, https://phenome.jax.org/strains/9, accessed April 10, 2024 Search PubMed.
- K. Schneck, M. Washington, D. Holder, K. Lodge and S. Motzel, Comp. Med., 2000, 50, 32–35 CAS.
- P. Bhattacharya, I. Budnick, M. Singh, M. Thiruppathi, K. Alharshawi, H. Elshabrawy, M. J. Holterman and B. S. Prabhakar, J. Interferon Cytokine Res., 2015, 35, 585–599 CrossRef CAS PubMed.
- C. M. Leopold Wager and F. L. Wormley, Mucosal Immunol., 2014, 7, 1023–1035 CrossRef CAS PubMed.
- J. Vega-Ramos and J. A. Villadangos, Mol. Immunol., 2013, 55, 175–178 CrossRef CAS.
- M. Merad and M. G. Manz, Blood, 2009, 113, 3418–3427 CrossRef CAS PubMed.
- R. Kushwah and J. Hu, Immunology, 2011, 133, 409–419 CrossRef CAS.
- E. Zhao, H. Xu, L. Wang, I. Kryczek, K. Wu, Y. Hu, G. Wang and W. Zou, Cell. Mol. Immunol., 2012, 9, 11–19 CrossRef CAS PubMed.
- V. Bronte and M. J. Pittet, Immunity, 2013, 39, 806–818 CrossRef CAS.
- S. Liao and T. P. Padera, Lymphatic Res. Biol., 2013, 11, 136–143 CrossRef CAS.
- J. Zhang, T. Supakorndej, J. R. Krambs, M. Rao, G. Abou-Ezzi, R. Y. Ye, S. Li, K. Trinkaus and D. C. Link, J. Clin. Invest., 2019, 129, 2920–2931 CrossRef.
- Y. Saito, S. Komori, T. Kotani, Y. Murata and T. Matozaki, Cancers, 2022, 14, 1976 CrossRef CAS PubMed.
- M. Bardou, J. Postat, C. Loaec, F. Lemaître, G. Ronteix, Z. Garcia and P. Bousso, EMBO J., 2021, 40, e107176 CrossRef CAS.
- M. P. Longhi, C. Trumpfheller, J. Idoyaga, M. Caskey, I. Matos, C. Kluger, A. M. Salazar, M. Colonna and R. M. Steinman, J. Exp. Med., 2009, 206, 1589–1602 CrossRef CAS.
- L. Minarrieta, L. N. Velasquez, T. Sparwasser and L. Berod, Curr. Opin. Biotechnol., 2021, 68, 202–212 CrossRef CAS PubMed.
- F. Chen, G. Wang, J. I. Griffin, B. Brenneman, N. K. Banda, V. M. Holers, D. S. Backos, L. Wu, S. M. Moghimi and D. Simberg, Nat. Nanotechnol., 2017, 12, 387–393 CrossRef CAS PubMed.
- S. Panico, S. Capolla, S. Bozzer, G. Toffoli, M. Dal Bo and P. Macor, Pharmaceutics, 2022, 14, 2605 CrossRef CAS PubMed.
- M. Debnath, J. I. Forster, A. Ramesh and A. Kulkarni, Bioconjugate Chem., 2023, 34, 1766–1779 CrossRef CAS PubMed.
- J. P. Böttcher, E. Bonavita, P. Chakravarty, H. Blees, M. Cabeza-Cabrerizo, S. Sammicheli, N. C. Rogers, E. Sahai, S. Zelenay and C. Reis e Sousa, Cell, 2018, 172, 1022–1037.e14 CrossRef.
- D. Piccioli, S. Sbrana, E. Melandri and N. M. Valiante, J. Exp. Med., 2002, 195, 335–341 CrossRef CAS PubMed.
- N. C. Fernandez, A. Lozier, C. Flament, P. Ricciardi-Castagnoli, D. Bellet, M. Suter, M. Perricaudet, T. Tursz, E. Maraskovsky and L. Zitvogel, Nat. Med., 1999, 5, 405–411 CrossRef CAS PubMed.
- M. A. Anwar, M. Shah, J. Kim and S. Choi, Med. Res. Rev., 2019, 39, 1053–1090 CrossRef CAS PubMed.
- C. Rolfo, E. Giovannetti, P. Martinez, S. McCue and A. Naing, npj Precis. Oncol., 2023, 7, 1–11 CrossRef.
- J. S. Weber, J. C. Yang, M. B. Atkins and M. L. Disis, J. Clin. Oncol., 2015, 33, 2092–2099 CrossRef CAS PubMed.
- C. L. Dennis, K. L. Krycka, J. A. Borchers, R. D. Desautels, J. van Lierop, N. F. Huls, A. J. Jackson, C. Gruettner and R. Ivkov, Adv. Funct. Mater., 2015, 25, 4300–4311 CrossRef CAS.
- C. L. Dennis, A. J. Jackson, J. A. Borchers, P. J. Hoopes, R. Strawbridge, A. R. Foreman, J. van Lierop, C. Grüttner and R. Ivkov, Nanotechnology, 2009, 20, 395103 CrossRef CAS PubMed.
- L. C. Liporagi Lopes, P. Korangath, S. R. Dos Santos, K. L. Gabrielson, R. Ivkov and A. Casadevall, Antimicrob. Agents Chemother., 2022, 66, e0239921 CrossRef PubMed.
- M. P. Matheu, D. Sen, M. D. Cahalan and I. Parker, J. Visualized Exp., 2008, 773 Search PubMed.
- D. Laha, R. Grant, P. Mishra and N. Nilubol, Front. Immunol., 2021, 12, 656908 CrossRef PubMed.
- N. J. Maney, G. Reynolds, A. Krippner-Heidenreich and C. M. U. Hilkens, J. Immunol., 2014, 193, 4914–4923 CrossRef CAS PubMed.
- Q. Xue, Y. Yan, R. Zhang and H. Xiong, Int. J. Mol. Sci., 2018, 19, 3805 CrossRef.
- M. Hedayati, B. Abubaker-Sharif, M. Khattab, A. Razavi, I. Mohammed, A. Nejad, M. Wabler, H. Zhou, J. Mihalic, C. Gruettner, T. DeWeese and R. Ivkov, Int. J. Hyperthermia, 2018, 34, 373–381 CrossRef CAS PubMed.
- A. M. Dudek, S. Martin, A. D. Garg and P. Agostinis, Front. Immunol., 2013, 4, 438 Search PubMed.
- J. C. Mbongue, H. A. Nieves, T. W. Torrez and W. H. R. Langridge, Front. Immunol., 2017, 8, 327 Search PubMed.
- H. Wang, D. Kwak, J. Fassett, X. Liu, W. Yao, X. Weng, X. Xu, Y. Xu, R. J. Bache, D. L. Mueller and Y. Chen, Basic Res. Cardiol., 2017, 112, 25 CrossRef PubMed.
- Y. Tang, X. Wang, J. Li, Y. Nie, G. Liao, Y. Yu and C. Li, ACS Nano, 2019, 13, 13015–13026 CrossRef CAS PubMed.
- P. Korangath and R. Ivkov, Bio-Protoc., 2020, 10, e3822 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4na00561a |
| This journal is © The Royal Society of Chemistry 2025 |