
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembled rosette nanotubes from tetra guanine-cytosine modules†
Usha D.
Hemraz
 *abc,
Takeshi
Yamazaki
ab,
Mounir
El-Bakkari
ab,
Jae-Young
Cho
b and
Hicham
Fenniri
*abd
*abc,
Takeshi
Yamazaki
ab,
Mounir
El-Bakkari
ab,
Jae-Young
Cho
b and
Hicham
Fenniri
*abd
aDepartment of Chemistry, University of Alberta, 11227 Saskatchewan Drive, Edmonton, Alberta T6G 2G2, Canada
bNanotechnology Research Centre, National Research Council of Canada, 11421 Saskatchewan Drive, Edmonton, Alberta T6G 2M9, Canada. E-mail: Usha.Hemraz@nrc-cnrc.gc.ca
cHuman Health Therapeutics, National Research Council Canada, 6100 Royalmount Avenue, Montreal, Quebec H4P 2R2, Canada
dUniversity Mohammed VI Polytechnic, Lot 660, Hay Moulay Rachid, 43150 Benguerir, Morocco. E-mail: hicham.fenniri@um6p.ma
First published on 27th November 2024
Abstract
Self-assembly of small molecules into supramolecular architectures is a sustainable alternative to new advanced material design. Herein, the design and synthesis of a self-assembling system containing four covalently linked hybrid guanine and cytosine (G∧C) units that were connected through bifunctional amines are reported. These tetra G∧C motifs were characterized and self-assembled in water and methanol to produce discrete nanostructures. Each module has 24 sites for intermolecular hydrogen bonding and it is proposed that in solution the four G∧C units per molecule align into a linear stack which in turn self-assembles into a hexameric super-helix held together by 72 intermolecular hydrogen bonds. Stacking of these nano-helices led to the formation of quad rosette nanotubes.
Introduction
Nature relies on a number of large and complex architectures to perform sophisticated processes, which demand the co-ordination of both non-directional and directional noncovalent interactions.1–3 Achieving the level of complexity and molecular recognition ability as natural systems entail in-depth understanding of the forces and interactions that propel such organization. Still, a variety of artificial self-assembled structures have emerged.4–10 The majority of self-assembled supramolecular systems have been based on the built-in hydrogen bonding complementarity and directionality of the monomers, hydrophobic effects and aromatic stacking interactions.11–16Among such self-assembled systems, rosette nanotubes (RNTs) are biologically inspired nanomaterials, formed from the hierarchical self-assembly of a hybrid guanine-cytosine motif (G∧C) featuring self-complementary hydrogen-bonding arrays.17–20 In solution, the G∧C base forms six-membered rosettes, which then stack into a tubular architecture. The length and the diameter of RNTs can be readily tuned21–23 and their applications in tissue engineering,24–30 cargo delivery,31–33 simulation34,35 and material science,36–38 was made possible due to their synthetic accessibility and chemical tunability. Our group has worked on the design of two main families of RNTs: (1) a single G∧C base with six sites for intermolecular hydrogen bonding in a spatial arrangement that forces the modules to form a hexameric super-macrocycle, which are maintained by 18 H-bonds (Fig. 1A),16,17 and (2) two covalently-linked G∧C bases in a stacked syn arrangement that led into a six membered twin rosette system maintained by 36 H-bonds (Fig. 1B).22 Though both the single and twin G∧C systems have built-in ability to engage in intermolecular hydrogen-bonding, the twin G∧C variant is more stable and yield RNTs at a faster rate due to lower functional group density, reduced net charge, enhanced stability arising from preorganization, increased amphiphilic character, greater number of H-bonds per module, higher number of H-bonds per rosette (36 H-bonds instead of 18), less steric congestion and lower electrostatic repulsion.22 The design of the twin RNTs is so robust that it can withstand higher temperatures, broad pH range, and sterically demanding functional groups.23,25,26,39 To test the limits of this design, we synthesized a library of peptide-functionalized RNTs varying in length from 1 to 15 lysine residues, and investigated their self-assembling properties.23,33 While these modules formed RNTs, a decrease in stability was noted as the peptide moiety was lengthened. The modules bearing peptides of 10 or more amino acids required alkaline conditions to reduce the electrostatic repulsion and form RNTs. This observation prompted a quest to design a self-assembling core containing more than two G∧C units. We presumed that they would in turn be maintained by a higher number of intermolecular hydrogen bonds than the 36 H-bonds for the twin G∧C system. The new system could potentially withstand a higher steric bulk and self-assemble at physiological conditions for any prospective biological applications. Herein, we report the design and synthesis of a self-assembling system containing four covalently linked G∧C moieties that were connected using bifunctional amines. The presence of ample hydrogen bonding sites led to the self-assembly of the newly synthesized tetra G∧C molecules to produce nanotubes. This new self-assembling tetra G∧C system could be useful in the design of nanomaterials that could sustain higher steric bulk.
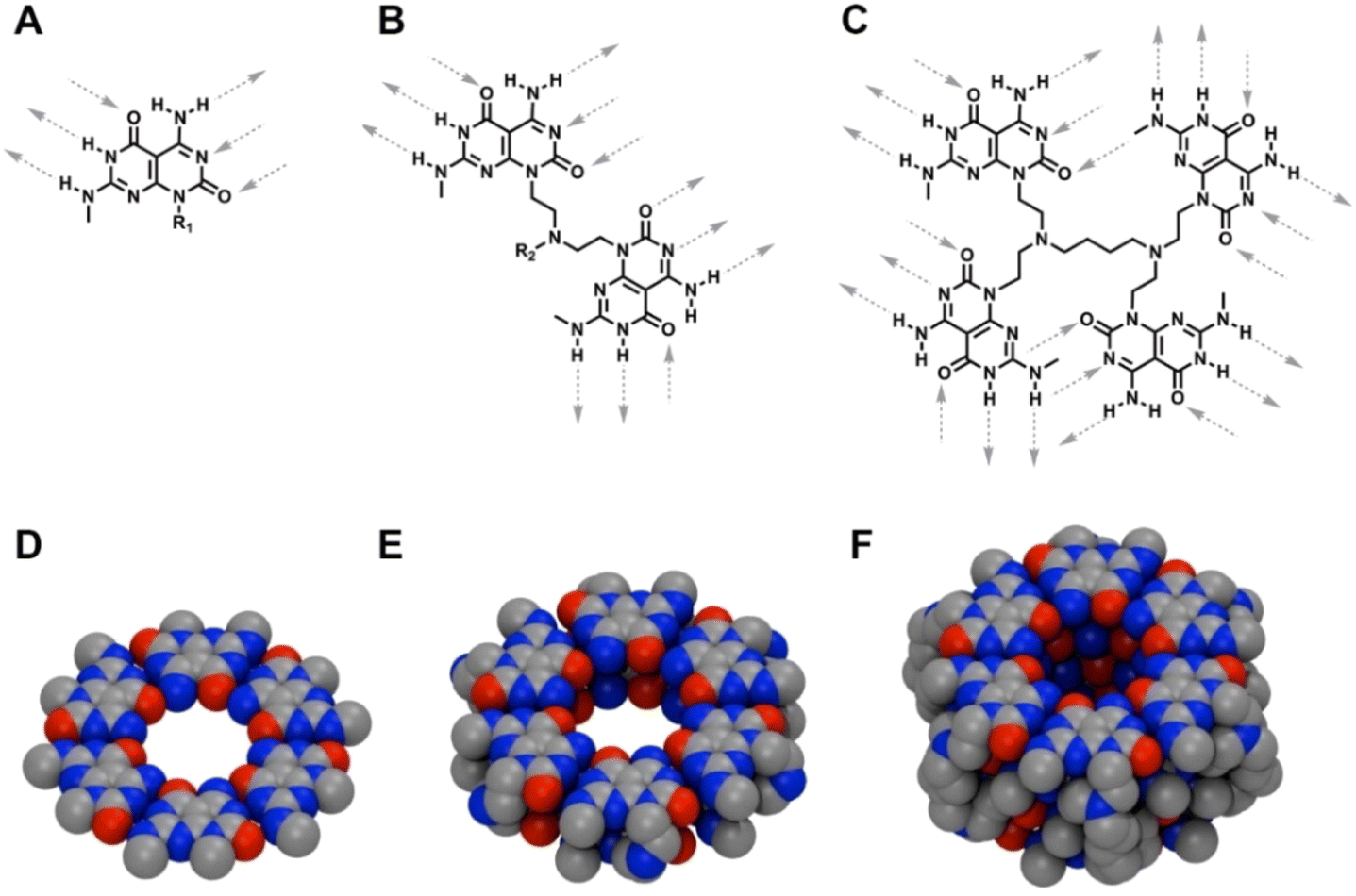 | ||
| Fig. 1 Structures of: single G∧C rosette with 3 donor and 3 acceptor sites for hydrogen bonding (A); twin G∧C rosette with 6 donor and 6 acceptor sites for hydrogen bonding (B) and tetra G∧C rosette with 12 donor and 12 acceptor sites for hydrogen bonding (C). Top view of: single G∧C rosette (D), twin G∧C rosette (E) and tetra G∧C rosette (F), held by 18, 36 and 72 hydrogen bonds, respectively and obtained through simulation. | ||
Results and discussion
The tetra-G∧C module 4a was synthesized in two steps starting from G∧C aldehyde 2 (Scheme 1). The latter, which was synthesized starting from barbituric acid accordingly to a previously reported procedure,19,20 was coupled with the commercially available 1,4 diaminobutane 1a through a reductive amination reaction to yield the benzyl and Boc-protected tetra-G∧C 3a. Removal of the protecting groups under acidic conditions afforded 4a as a hydrochloride salt in quantitative yield. A similar procedure was applied for the synthesis of monomers 4b and 4c using oligoethyleneoxide diamines 1b and 1c, respectively. Unlike the tetra-G∧C module 4a where the linker consisted of a four-carbon aliphatic chain, the tetra-G∧C modules 4b and 4c were made of ether linkages of varied length.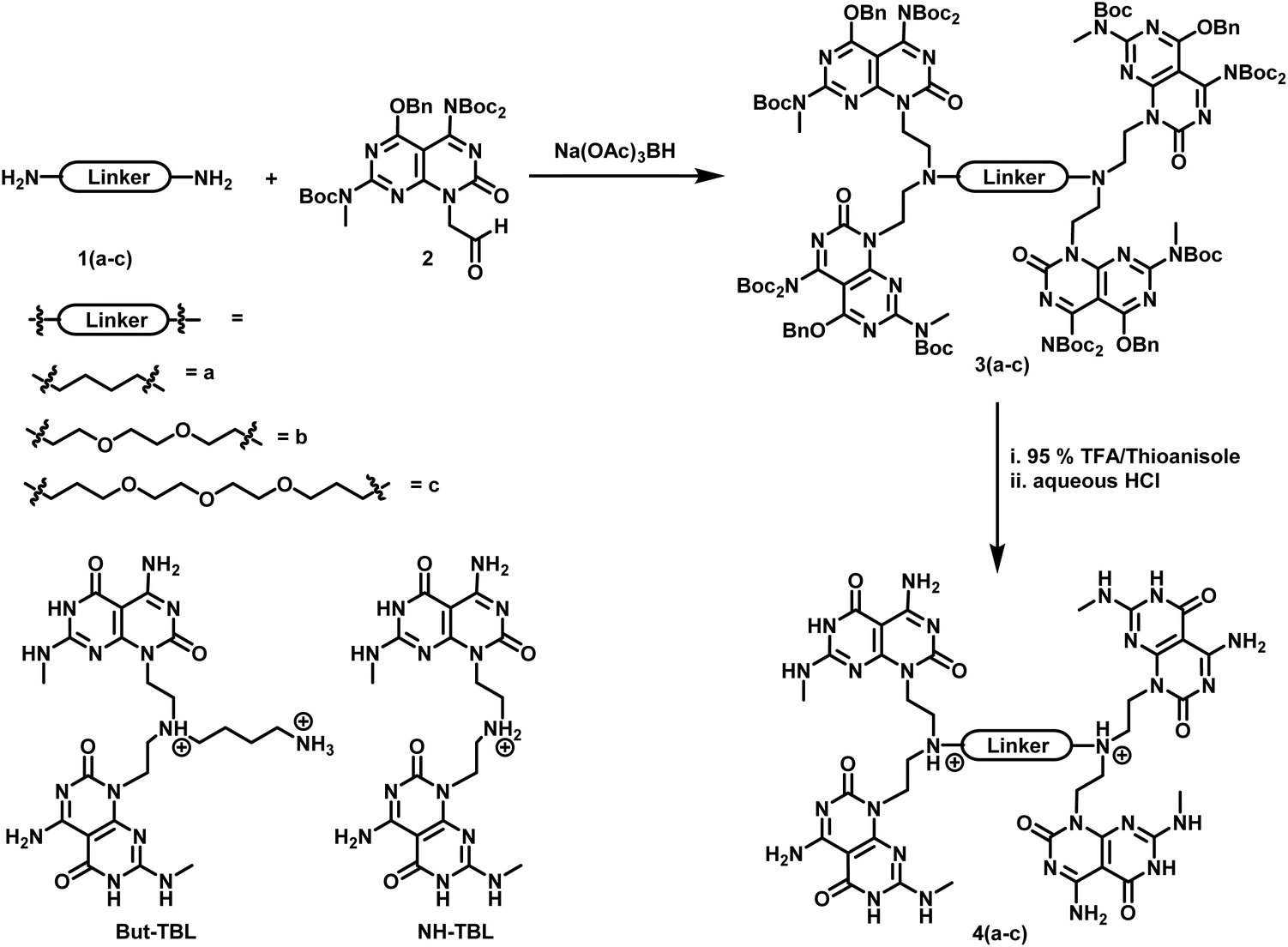 | ||
| Scheme 1 Synthetic route for the formation of tetra G∧C modules 4(a–c) and structures of twin G∧C modules But-TBL and NH-TBL. | ||
By design, the G∧C motif has built-in hydrogen bonding sites that allows the system to self-assemble. We have previously shown the design and self-assembly of the single and twin G∧C building blocks. Unlike the single (Fig. 1A) and twin (Fig. 1B) G∧C motifs that have six and twelve sites available for intermolecular hydrogen bonding per molecule, the newly synthesized tetra-G∧C module possesses twenty-four intermolecular hydrogen-bonding sites (Fig. 1C). To maximize the use of all twenty-four intermolecular hydrogen-bonding sites per module, the tetra-G∧C building blocks could self-organize into linear stacks, which would in turn self-assemble into a hexameric super-helix held together by 72 intermolecular hydrogen bonds (Fig. 1F).
Next, the self-assembly of tetra-G∧C modules were investigated. The self-assembly of 4a was carried out in methanol and water, whereby discrete structures were observed in both solvents (Fig. 2A and B). The nanostructures seemed to form larger aggregates in water (Fig. 2B, S1 and S3†) compared to methanol (Fig. 2A and S2†). Since the SEM images of 4a in water showed a larger diameter than what is usually observed for a RNT, RNTs self-assembled from 4a and a twin G∧C module (But-TBL, Scheme 1) separately were imaged on the same TEM grid (Fig. 2C) to have a better visual comparison of dimensions. The twin G∧C module But-TBL self-assembles into a hexameric macrocycle held together by 36 hydrogen bonds, with a theoretical diameter of 3.4 nm.31 The electron microscopy data displayed nanostructures of two distinct dimensions that were obtained through measurements, with a larger diameter of 7.3 ± 0.3 nm corresponding to 4a and a smaller diameter of 3.3 ± 0.2 nm resulting from But-TBL.25,31 However, a careful analysis of the TEM images revealed that the larger nanostructures were in fact made up of two or three thinner tube-like structures (Fig. 2D and S3†), with an average diameter of a single tube of 3.2 ± 0.1 nm in both solvents. The same difference in the theoretical diameters of the hexameric macrocycles from 4a (3.1 nm) and But-TBL (3.4 nm) is attributed to the butyl ammonium side chain in the latter.
 | ||
| Fig. 2 SEM images of 4a module in methanol (A), in water (B), co-deposited with But-TBL in water (C), TEM image of 4a in water (D), and an inset to show individual RNTs (E). Scale bar = 100 nm. | ||
The simplest way that the tetra-G∧C system 4a could yield a diameter of about 3.2 nm was to mimic the self-assembly of the twin G∧C system. As mentioned earlier, self-assembly in a twin G∧C system involves the syn intramolecular stacking of the two G∧C units, followed by the formation of a six membered macrocycle held by 36 H-bonds. The four G∧C units in module 4a can be considered as a pair of twin G∧C stacks tethered by a linker. Due to the favourable π–π interactions between the G∧C units and intramolecular hydrogen bonding of the ammonium ion and carbonyl oxygen of the cytosine ring, we presumed that the four G∧C units could stack into a 4-fold pile. This hypothesis was verified computationally (Fig. 3). Starting from a quad-rosette ring consisting of six motifs with a syn–syn–syn conformation (SSS) (Fig. 3B), the geometry of the ring was optimized, following previous modeling studies for single and twin G∧C systems. Upon optimization, it was found that the G∧C units aligned themselves in a displaced stacking fashion, with a zig-zag type arrangement of the methyl groups (Fig. 3C and D). This could have been triggered by the relatively short length of the linker in 4a that could exert a certain degree of strain on this strand due to the limited flexibility of the tether. We believe that alignment in a displaced fashion could allow the G∧C units to offset this strain. The outer diameter of this quad-rosette was calculated to be 3.1 nm. With this arrangement, there was an optimal use of the hydrogen-bond donor and acceptor sites, where each molecule was involved in 24 H-bonds, and a total of 72 intermolecular H-bonds per quad-rosette. A similar quad-rosette ring was constructed with a syn–anti–syn (SAS) arrangement of the G∧C moieties to give a rosette ring with an outer diameter of 3.1 nm (Fig. 3E and F).
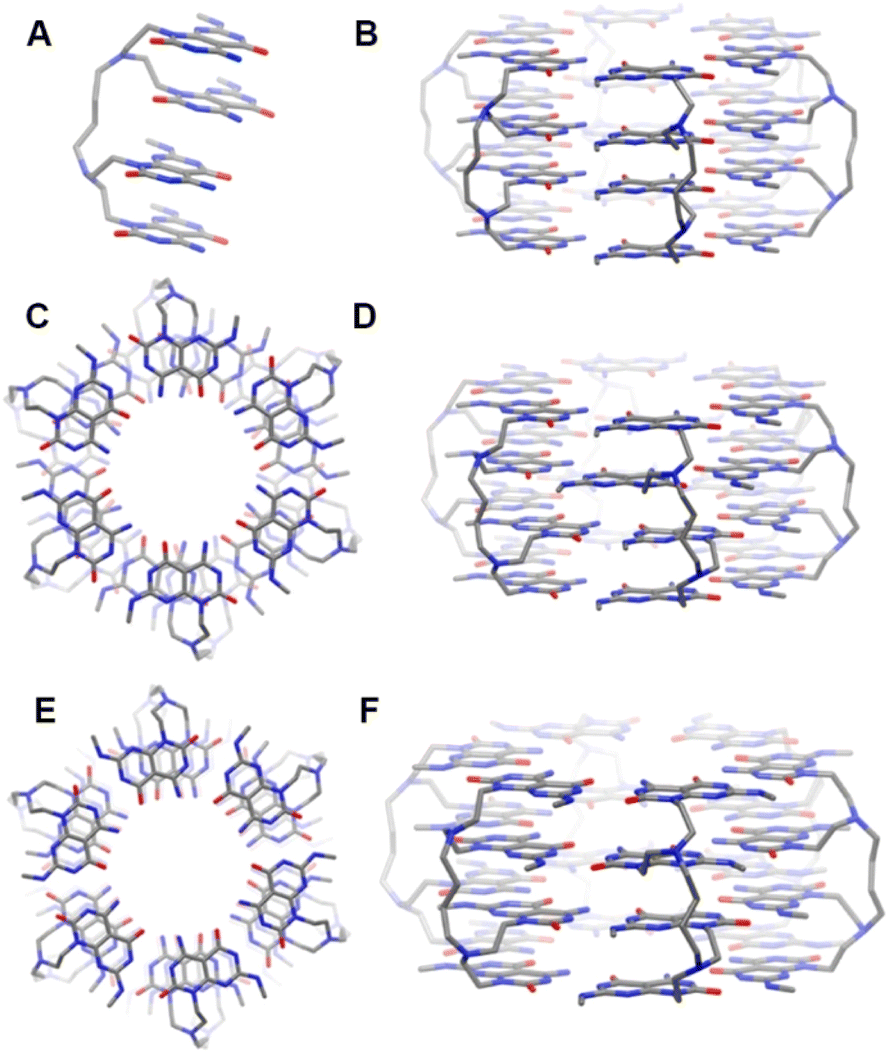 | ||
| Fig. 3 Side view of an all-syn quad-rosette from the 4a monomer (A) and side view of the unoptimized quad-rosette ring (B); top view (C) and side view (D) of the optimized quad-rosette ring with a SSS conformation, showing displaced stacks of G∧C units; and top view (E) and side view (F) of the optimized quad-rosette ring from 4a with a SAS conformation and displaced stacks of G∧C units. | ||
Experimentally, the tetra-G∧C module 4a could form nanotubes (Fig. 2) and the quad-rosettes from 4a of SSS and SAS configurations produced nanotubes with a calculated outer diameter of 3.1 nm (Fig. 4A, B, respectively and S6†). Next, the relative stabilities of the resulting nanotubes were calculated and compared to the twin RNT NH-TBL (resulting from coupling of ammonia with two molecules of a G∧C derivative, Scheme 1). To justify for the difference in the number of the G∧C units, each quad-rosette ring from 4a was accounted for two twin rosette rings. From the association free energy plot, it was found that NH-TBL and the two possible conformers of the 4a module had comparable stabilities (Fig. 4D). Although in this work a comparison of a twin-G∧C containing a bulky side chain was not carried out against the tetra-G∧C module with a similar pendant, it is speculated that the tetra-G∧C would have a higher stability when carrying an analogous bulk as compared to twin-G∧C due to the increase in the number of hydrogen bonding per rosette. Despite the limited flexibility of the hydrophobic butyl linker in the strand, negative association free energies of the SSS and SAS nanotubes in water suggested that the solvent plays an important role in the formation of such superstructures. The solvophobicity of the 4a module in water is greater than that in methanol. As such, the higher solvophobic interactions in water leads to the formation of RNTs' aggregates, which could explain the formation of RNTs' bundles in water and the isolated RNT in methanol.
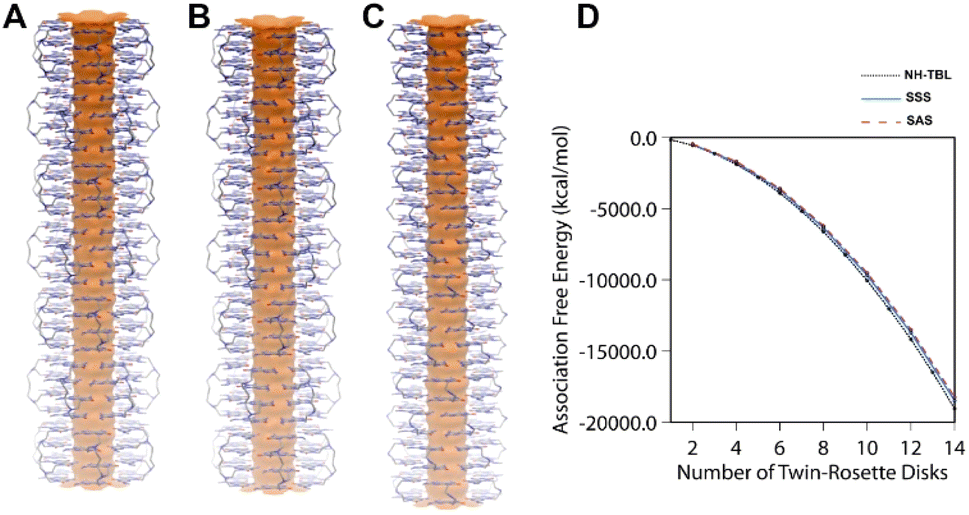 | ||
| Fig. 4 Nanotubes made up of 7 quad-rosettes from 4a with SSS (A) and SAS (B) arrangements of the G∧C units, and nanotube derived from NH-TBL (C), generated by the stacking of 14 twin-rosettes through simulation. Comparison of association free energies of nanotubes from 4a with SSS and SAS arrangements of the G∧C units with increasing number of rosette stacks (up to 7 stacks), and of NH-TBL (up to 14 stacks) (D). | ||
Apart from the SSS and SAS conformations for 4a using molecular modelling, two other supramolecular structures were explored (Fig. S7†). Firstly, a six-membered ring structure with a 5.1 nm diameter was studied, whereby only two of the four G∧C units were involved in intermolecular hydrogen bonding (Fig. S8†). While this arrangement did mimic the twin-RNT system, the free hydrogen-bond donor and acceptor sites of the two remaining G∧C units were free to engage in intermolecular interactions. In the second case, a four-membered ring structure with an outer diameter of 3.6 nm was obtained (Fig. S9†). Both conformations were unlikely since they cannot produce discrete architectures and do not have the correct geometry for hydrogen bonding and intermolecular π–π stacking of the G∧C units.
The module 4a self-assembled readily in water. However, given that built-in linker was hydrophobic, aggregation was observed in water. For any prospective biological applications, we believe that it is important for the monomers to self-assemble into well-dispersed nanostructures under physiological conditions. Therefore, analogues (4b and 4c) of the 4a motif were designed to test the effect of longer and more hydrophilic linkers on self-assembly and whether the aggregation observed was only a consequence of the hydrophobic nature of the linker. The ether linkages incorporated in the system design of 4b and 4c provided a boost in the hydrophilicity of the monomers. The presence of the oxygen atoms in the linker resulted into well-dispersed single tubes in water, as observed from the microscopy images (Fig. 5A, B, S4 and S5†). The measured diameters obtained by TEM were 3.4 ± 0.2 nm and 3.4 ± 0.1 nm respectively. This data was in agreement with TEM measurement observed for the 4a building block and is therefore in line with the proposed formation mechanism of the self-assembled structures. The present observation also provides evidence that RNTs consisting of the 4a motif form bundles based on the solvophobic (hydrophobic) interactions in water, where the tetra-G∧C system had preferential parallel formation of nanotubes.
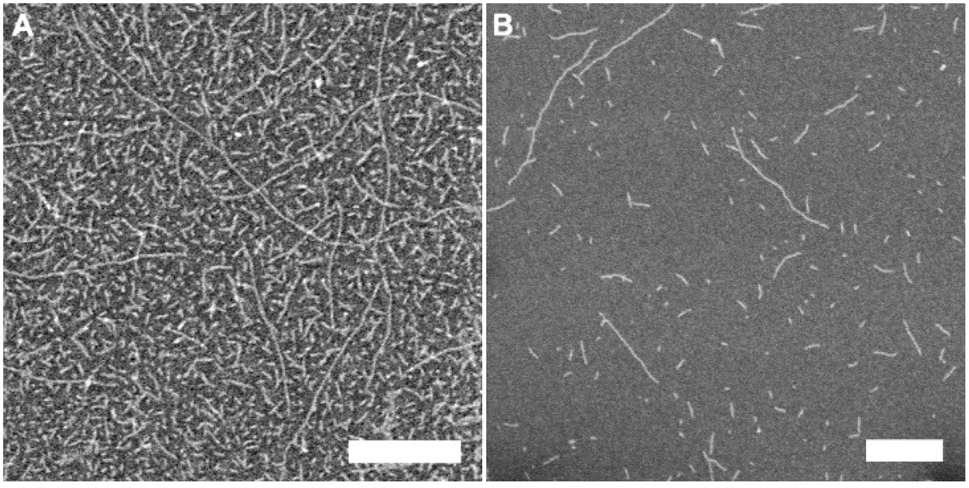 | ||
| Fig. 5 SEM images of 4b (A) and 4c (B) modules in water. Scale bar = 100 nm. | ||
Experimental section
General methods
Unless stated otherwise, all reactions were performed under N2 atmosphere. Reagent grade solvents (DCM, 1,2 DCE) were purified using the solvent purification system and all other commercial reagents and solvents were used as purchased. The G∧C aldehyde 2 was synthesized according to a previously reported procedure. Reactions were monitored by TLC using silica-coated TLC plated and were visualized under UV light. The NMR data is presented as follows: chemical shift, multiplicity, coupling constant, integration. The following abbreviations were used for the multiplicities: s = singlet, d = doublet, t = triplet m = multiplet, bs = broad singlet. Residual 1H shifts in CDCl3 (7.24 ppm), d1-TFA (11.50 ppm), d6-DMSO (2.50 ppm) and CD2Cl2 (5.30 ppm) were used as internal references. Similarly, CDCl3 (77.23 ppm), d1-TFA (164.2 ppm), d6-DMSO (39.51 ppm) and CD2Cl2 (53.8 ppm) were used as internal references for 13C NMR. The 1H NMR and 13C NMR spectra were calibrated using 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt (TMS = 0.0) as the internal reference.Synthetic procedures
1H NMR (600 MHz, CD2Cl2) δ 7.46–7.30 (m, 20H), 5.57 (s, 8H), 4.35 (t, J = 7.2 Hz, 8H), 3.52–3.48 (m, 8H), 3.47 (s, 12H), 2.95–2.90 (m, 12H), 1.54 (s, 36H), 1.31 (s, 72H); 13C NMR (150 MHz, CD2Cl2) δ 171.2, 166.1, 161.8, 161.6, 160.7, 155.9, 153.1, 149.8, 135.8, 129.0, 128.98, 128.95, 93.2, 83.9, 83.2, 70.8, 70.4, 70.3, 60.7, 54.1, 52.0, 41.9, 35.4, 28.4, 28.1; HRMS (ESI+) calcd for (C130H176N26Na2O34)0.5 [M/2 + Na]+ 1345.6313; found 1345.6306.
Anal. calcd for C42H56N26O10(CF3CO2H)5.15(H2O)0.25(C4H10O), C, 38.62; H, 4.12; N, 20.80; found C, 38.85, H, 4.09, N, 20.67.
For the self-assembly study, an aliquot of the TFA salt of 4b was converted to the HCl salt. Compound 4b (TFA salt) (0.035 g, 0.020 mmol) was dissolved in 1 M hydrochloric acid (15 mL), followed by removal of water under reduced pressure. This process was repeated two more times before the solid was dried under vacuum for 72 h to give the HCl salt of 4b as a white solid (C42H56N26O10 + 8 HCl + 2H2O + 0.5 Et2O, 87%). MALDI-TOF MS (THAP, positive mode) calcd for C42H57N26O10 [M + H]+ 1085.4745; found 1085.4730; anal. calcd for C42H56N26O10(HCl)8(H2O)2(C4H10O)0.5, C, 36.45; H, 5.08; N, 25.12; found C, 36.44, H, 5.24, N, 24.91.
1H NMR (600 MHz, d1-TFA) δ 4.78–4.62 (m, 8H), 3.90–3.71 (m, 24H), 3.14 (d, J = 7.8 Hz, 12H), 2.18 (s, 4H); 13C NMR (150 MHz, D2O/d1-TFA) δ 163.39, 161.49, 157.45, 157.26, 150.63, 83.92, 71.05, 70.83, 69.03, 53.87, 52.49, 38.23, 28.77, 24.38; MALDI-TOF MS (THAP, positive mode) calcd for C46H65N26O11 [M + H]+ 1157.5320; found 1157.5316; anal. calcd for C46H64N26O11(CF3CO2H)5.25(H2O)(C4H10O)0.25, C, 38.53; H, 4.15; N, 20.32; found C, 38.63, H, 4.17, N, 20.39.
For self-assembly, an aliquot of the TFA salt of 4c was converted to the HCl salt. Compound 4c (TFA salt) (0.036 g, 0.020 mmol) was dissolved in 1 M hydrochloric acid (15 mL), followed by removal of water under reduced pressure. This process was repeated two more times before the solid was dried under vacuum for 72 h to give the HCl salt of 4c as a white solid (C46H64N26O11 + 8 HCl + 2.5H2O + 0.6 Et2O, 62%). MALDI-TOF MS (HCCA, positive mode) calcd for C46H65N26O11 [M + H]+ 1157.5320; found 1157.5307; anal. calcd for C46H64N26O11(HCl)8(H2O)2.5(C4H10O)0.6; C, 37.79; H, 5.44; N, 23.67, found C, 37.81, H, 5.47, N, 23.53.
Conclusions
We have designed a tetra-G∧C system, which has the ability to self-assemble into nanotubes of about 3 nm in diameter, by adopting a 4-fold stack. The rosette formation relied mainly on the 72 intermolecular hydrogen bonding and stacking of the intramolecular G∧C units. The relative stabilities of the different conformers were evaluated computationally. Despite the limited flexibility of the hydrophobic butyl linker in the strand, the negative association free energies of different conformations in water suggested that the solvent plays an important role in the formation of such superstructures. We anticipate that this quad-RNT system will allow the attachment of functional groups with higher steric bulk on the periphery of the nanotubes compared to the single or twin G∧C system and would be useful towards the design of advanced functionalized RNTs for cargo-carrying capabilities under physiological conditions in targeted applications.Data availability
The data supporting this article are included as part of the ESI.†Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was funded by the National Research Council of Canada, a Federal Research and Development Organization of the Government of Canada and the University of Alberta.References
- A. Klug, Angew Chem. Int. Ed. Engl., 1983, 22, 565–582 CrossRef.
- N. C. Seeman, Trends Biotechnol., 1999, 17, 437–443 CrossRef CAS.
- T. L. Schlick, Z. Ding, E. W. Kovacs and M. B. Francis, J. Am. Chem. Soc., 2005, 127, 3718–3723 CrossRef CAS.
- D. Ajami, J. L. Hou, T. J. Dale, E. Barrett and J. Rebek Jr, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10430–10434 CrossRef CAS PubMed.
- A. M. Carmona-Ribeiro, Chem. Soc. Rev., 2001, 30, 241–247 RSC.
- C. Schmuck, Angew Chem. Int. Ed. Engl., 2007, 46, 5830–5833 CrossRef CAS PubMed.
- C. F. van Nostrum, Adv. Mater., 1996, 8, 1027–1030 CrossRef CAS.
- N. P. Dharmarajan, D. Vidyasagar, J. H. Yang, S. N. Talapaneni, J. Lee, K. Ramadass, G. Singh, M. Fawaz, P. Kumar and A. Vinu, Adv. Mater., 2024, 36, e2306895 CrossRef PubMed.
- F. B. Ilhami, Y. S. Birhan and C. C. Cheng, ACS Biomater. Sci. Eng., 2024, 10, 234–254 CrossRef CAS PubMed.
- S. Huang, H. Yu and Q. Li, Adv. Sci., 2021, 8, 2002132 CrossRef.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS.
- J.-M. Lehn, Angew Chem. Int. Ed. Engl., 1990, 29, 1304–1319 CrossRef.
- F. J. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS PubMed.
- J. P. Mathias, J. A. Zerkowski, C. T. Seto, E. E. Simanek and G. M. Whitesides, J. Am. Chem. Soc., 1994, 116, 4316–4325 CrossRef CAS.
- S. Kabata Glowacki, K. Koszinowski, D. Hubner, H. Frauendorf, P. Vana and U. Diederichsen, Chemistry, 2020, 26, 12145–12149 CrossRef CAS PubMed.
- D. T. Bong, T. D. Clark, J. R. Granja and M. R. Ghadiri, Angew Chem. Int. Ed. Engl., 2001, 40, 988–1011 CrossRef CAS PubMed.
- A. Marsh, M. Silvestri and J.-M. Lehn, Chem. Commun., 1996, 1527–1528 RSC.
- M. Mascal, N. M. Hext, R. Warmuth, M. H. Moore and J. P. Turkenburg, Angew Chem. Int. Ed. Engl., 1996, 35, 2204–2206 CrossRef CAS.
- H. Fenniri, P. Mathivanan, K. L. Vidale, D. M. Sherman, K. Hallenga, K. V. Wood and J. G. Stowell, J. Am. Chem. Soc., 2001, 123, 3854–3855 CrossRef CAS PubMed.
- H. Fenniri, B.-L. Deng and A. E. Ribbe, J. Am. Chem. Soc., 2002, 124, 11064–11072 CrossRef CAS PubMed.
- G. Borzsonyi, A. Alsbaiee, R. L. Beingessner and H. Fenniri, J. Org. Chem., 2010, 75, 7233–7239 CrossRef CAS.
- J. G. Moralez, J. Raez, T. Yamazaki, R. K. Motkuri, A. Kovalenko and H. Fenniri, J. Am. Chem. Soc., 2005, 127, 8307–8309 CrossRef CAS PubMed.
- U. D. Hemraz, M. El-Bakkari, T. Yamazaki, J.-Y. Cho, R. L. Beingessner and H. Fenniri, Nanoscale, 2014, 6, 9421–9427 RSC.
- A. L. Chun, J. G. Moralez, H. Fenniri and T. J. Webster, Nanotechnology, 2004, 15, S234–S239 CrossRef CAS.
- L. Zhang, U. D. Hemraz, H. Fenniri and T. J. Webster, J. Biomed. Mater. Res., Part A, 2010, 95A, 550–563 CrossRef CAS.
- A. Childs, U. D. Hemraz, N. J. Castro, H. Fenniri and L. G. Zhang, Biomed. Mater., 2013, 8, 065003 CrossRef CAS PubMed.
- L. Sun, L. Zhang, U. D. Hemraz, H. Fenniri and T. J. Webster, Tissue Eng., 2012, 18, 1741–1750 CrossRef CAS.
- L. Sun, D. Li, U. D. Hemraz, H. Fenniri and T. J. Webster, J. Biomed. Mater. Res., Part A, 2013, 102A(10), 3446–3451 CrossRef PubMed.
- L. Zhang, F. Rakotondradany, A. J. Myles, H. Fenniri and T. J. Webster, Biomaterials, 2009, 30, 1309–1320 CrossRef CAS.
- X. Zhou, S. Tenaglio, T. Esworthy, S. Y. Hann, H. Cui, T. J. Webster, H. Fenniri and L. G. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 33219–33228 CrossRef CAS.
- J.-Y. Cho, P. Bhowmik, P. L. Polowick, S. G. Dodard, M. El-Bakkari, G. Nowak, H. Fenniri and U. D. Hemraz, ACS Omega, 2020, 5, 24422–24433 CrossRef CAS.
- T. Yamazaki and H. Fenniri, J. Mol. Liq., 2016, 217, 70–74 CrossRef CAS.
- U. Ho, M. El-Bakkari, A. Alshamsan, J. Y. Cho, T. Yamazaki, U. D. Hemraz and H. Fenniri, Biomater. Sci., 2023, 11, 7169–7178 RSC.
- H. N. M. Ali and A. A. Gonzales 3rd, Molecules, 2023, 28, 7853 CrossRef CAS PubMed.
- D. Roy and A. Kovalenko, Thermo, 2023, 3, 375–395 CrossRef.
- P. Tripathi, L. Shuai, H. Joshi, H. Yamazaki, W. H. Fowle, A. Aksimentiev, H. Fenniri and M. Wanunu, J. Am. Chem. Soc., 2020, 142, 1680–1685 CrossRef CAS PubMed.
- J. Raez, J. G. Moralez and H. Fenniri, J. Am. Chem. Soc., 2004, 126, 16298–16299 CrossRef CAS PubMed.
- B.-L. Deng, R. L. Beingessner, R. S. Johnson, N. K. Girdhar, C. Danumah, T. Yamazaki and H. Fenniri, Macromolecules, 2012, 45, 7157–7162 CrossRef CAS.
- C. Igci, O. Karaman, Y. Fan, A. A. Gonzales, H. Fenniri and G. Gunbas, Sci. Rep., 2018, 8, 15949 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4na00567h |
| This journal is © The Royal Society of Chemistry 2025 |