
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSystematic investigation of ABO3 perovskite synthesis to generalise a La and Rh co-doping strategy for visible light photocatalysis†
Thomas D.
Small
 a,
Mahmoud Adel
Hamza
ab,
Yideng
Shen
a,
Cameron J.
Shearer
*a and
Gregory F.
Metha
*a
a,
Mahmoud Adel
Hamza
ab,
Yideng
Shen
a,
Cameron J.
Shearer
*a and
Gregory F.
Metha
*a
aDepartment of Chemistry, University of Adelaide, Adelaide, SA 5005, Australia. E-mail: cameron.shearer@adelaide.edu.au; greg.metha@adelaide.edu.au
bChemistry Department, Faculty of Science, Ain Shams University, Abbassia, Cairo, 11566, Egypt
First published on 28th May 2025
Abstract
Photocatalysts present a promising method of producing cheap and green hydrogen. However, most highly-efficient photocatalysts are active to only UV light, which is only 4% of the power in sunlight. Consequently, commercial photocatalysis can only be achieved by developing efficient materials active to visible light (47% of sunlight). Co-doping La and Rh into the perovskite oxide, SrTiO3 is known to create visible light photocatalysts as part of a Z-scheme system. Here, we rationally apply this strategy to SrTiO3 and five other ABO3 perovskites. The A and B-site elements (Sr, Ba, Zr, and Ti) were systematically varied within periodic groups to fabricate six different perovskites: SrZrO3, SrZr0.5Ti0.5O3, SrTiO3, BaZrO3, BaZr0.5Ti0.5O3, and BaTiO3. The elemental composition of the resulting perovskite affected particle size, crystallinity, morphology, and bandgap. All six perovskites were then co-doped with La and Rh and in every case the doped materials demonstrated red-shifted bandgap. Further, four La,Rh co-doped perovskites demonstrated photocatalytic hydrogen production under visible light (405 and 455 nm). Consequently, the La, Rh co-doping strategy was successfully generalised from SrTiO3 to ABO3 perovskites. La,Rh:BaTiO3 exhibited the narrowest bandgap (2.58 eV) and the second highest photocatalytic activity. These properties establish La,Rh:BaTiO3 as a potential commercial visible-light active photocatalyst.
1 Introduction
Preventing global temperature from rising 1.5 °C by 2050 is critical to continued human survival.1 This can only be delivered through an energy ecosystem of cost-competitive fossil fuel alternatives.1 Green hydrogen is a renewable, energy-dense chemical fuel that is a key part of this future ecosystem.2,3 However, green H2 is still prohibitively expensive4 rendering it currently unviable to either decarbonise the 2.5% of world CO2 emissions already due to hydrogen production or establish green hydrogen as an integral part of a renewable energy economy.4Photocatalysts are simple nanoparticulate semiconductors capable of producing green H2 by absorbing sunlight to convert water into H2 and O2.5 Compared to more mature green hydrogen technologies, e.g. photovoltaic-powered electrolysis, photocatalysts provide an inexpensive means of hydrogen production.6 However, state-of-the-art photocatalysts typically report sunlight to hydrogen efficiency (STH) of 1–2%,7–9 falling short of the 5–10% STH calculated by technoeconomic assessments for commercial implementation.6 Recently InGaN nanowires have demonstrated an STH over 9%.10 However, this was only achieved at concentrated solar conditions (33 suns and 70 °C). Further the nanowires were fabricated by molecular-beam epitaxy which could be expensive at scale. Regardless, the InGaN nanowires demonstrate that commercially viable STH values are achievable in photocatalysis.
High STH photocatalysts require narrow bandgap, high quantum efficiency, and redox stability.5 Photocatalysts which demonstrate high Apparent Quantum Yield (AQY) and redox stability for overall water-splitting at standard conditions have been reported (0.56–96%).7,11,12 However, such photocatalysts can only absorb UV light. UV-active photocatalysts are fundamentally unable to reach commercially viable STH as UV comprises only 4% of the power of sunlight.5 Consequently, much of the recent work in commercial photocatalytic water-splitting focuses on developing visible light active photocatalysts.13–15
The AQY of visible light active photocatalysts are smaller and typically require non-standard photocatalytic conditions such as reduced pressure, the addition of sacrificial reagents, or the photocatalyst itself quickly degrades.9,16–18 It is critical to develop visible light photocatalysts with the high AQY of UV photocatalysts at standard conditions to establish commercial photocatalysis.14,19
Perovskite materials are relatively simple, ABX3, rapidly maturing for use as highly efficient solar cells.20 Accordingly, ABO3 perovskite oxides demonstrate some of the highest reported photocatalytic STH16 and AQY.7,11 ABO3 perovskites also exhibit design flexibility. The A and B-site elements can be varied to tune the material properties and optimise photocatalytic water-splitting activity.21 Perovskites are also highly receptive to a wide variety of doping conditions.7,11,19,21–24
Co-doping La and Rh into SrTiO3 was recently demonstrated to confer visible light activity to the UV-only active base material without significantly compromising quantum efficiency.25 This is because filled Rh3+ 4d orbitals interact with O 2p orbitals, raising the valence band. Co-doped La cations, which can only occupy 3+ oxidation states, balance the Rh dopant charge. This suppresses the formation of Rh4+, which would introduce a filled mid-gap state from the unpaired 4d electron into the electronic band structure.25 La,Rh:SrTiO3 has demonstrated 30% AQY at 420 nm as part of a Z-scheme photocatalyst.25 Applied to oxides, which are typically redox stable, La,Rh co-doping is a potentially powerful technique to fabricate photocatalysts which meet the two major concerns of visible light activity and high quantum efficiency. However, this method has so far been demonstrated for only a single, UV-active perovskite, SrTiO3.
This study begins with synthesising, and then La,Rh co-doping, the well-studied SrTiO3. The A and B-site elements were then systematically modified with same-group elements for the A and B-sites respectively. In total, six base perovskites were synthesised: SrZrO3, SrZr0.5Ti0.5O3, SrTiO3, BaZrO3, BaZr0.5Ti0.5O3, and BaTiO3. The six perovskites were then co-doped with La and Rh following the methods developed for SrTiO3. This resulted in six doped perovskites: La,Rh:SrZrO3, La,Rh:SrZr0.5Ti0.5O3, La,Rh:SrTiO3, La,Rh:BaZrO3, La,Rh:BaZr0.5Ti0.5O3, and La,Rh:BaTiO3. The six pristine and six La,Rh co-doped materials were tested for sacrificial photocatalytic water-splitting under visible light (405 and 455 nm). Four of the La,Rh co-doped perovskites demonstrated H2 production under visible light. Overall, this work generalises La,Rh co-doping as a strategy for obtaining active visible light photocatalysts.
2 Experimental
2.1 Materials and reagents
All materials were used as purchased unless noted in the experimental. SrCO3 powder (≥99.9%, Sigma-Aldrich). BaCO3 powder (≥99%, Sigma-Aldrich). TiO2 powder (≥99.5%, Aeroxide P25, Sigma-Aldrich). ZrO2 powder (≥99%, Sigma-Aldrich). NaCl (≥99%, Sigma-Aldrich). KCl (≥99%, Chem-supply). Rh2O3.5H2O (99.99%, 59% Rh, Premion, Alfa Aesar). La2O3 (99.99%, Sigma-Aldrich) and K2PtCl6 (≥99.9%, Sigma-Aldrich).2.2 Perovskite synthesis
The overall synthesis scheme for La,Rh:ABO3 perovskites is shown in Fig. 1. | ||
| Fig. 1 Overall synthesis scheme for ABO3 perovskites and subsequent La,Rh co-doping. | ||
Four ABO3 perovskites, SrZrO3, BaZrO3, SrTiO3, and BaTiO3, were synthesised in a solid-state reaction adapted from Wang et al.16 The A-site reagent, SrCO3 or BaCO3 (preheated in air at 573 K for 1 h), and the B-site reagent, TiO2 or ZrO2, were ground together with ethanol (3 × 1 mL) in a mortar and pestle for 20 min. The A/B element ratio was 1.05. The mixture was heated in an alumina crucible to 1423 K (5 K min−1 to 523 K then 10 K min−1 to 1423 K) and held at that temperature for 10 h before cooling naturally to room temperature.
Two mixed B-site perovskites, SrZr0.5Ti0.5O3, and BaZr0.5Ti0.5O3, were synthesised in a molten salt flux adapted from O'Donnell et al.26 The A-site reagent, SrCO3 or BaCO3, in 10% mole excess, was mixed with equimolar TiO2 and ZrO2 and ground in a mortar and pestle with ethanol (2 × 1 mL) for 10 min. Subsequently an equimolar mixture of NaCl and KCl with a salt-to-product molar ratio of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was added to the reagents and the mixture was ground for a further 10 min. The mixture was heated in an alumina crucible to 1473 K (5 K min−1 to 523 K then 10 K min−1 to 1423 K) and held at that temperature for 24 h before cooling naturally to room temperature. Excess salt was removed by washing the product with Milli-Q water and the product was collected by vacuum filtration. Repeated washings were performed until aq. AgNO3 (0.5 M) did not precipitate when added to the waste filtrate.
:1 was added to the reagents and the mixture was ground for a further 10 min. The mixture was heated in an alumina crucible to 1473 K (5 K min−1 to 523 K then 10 K min−1 to 1423 K) and held at that temperature for 24 h before cooling naturally to room temperature. Excess salt was removed by washing the product with Milli-Q water and the product was collected by vacuum filtration. Repeated washings were performed until aq. AgNO3 (0.5 M) did not precipitate when added to the waste filtrate.
2.3 Co-doping synthesis
All six perovskites were co-doped with La and Rh in a solid-state synthesis adapted from Wang et al.16 The perovskite was ground with La2O3 (pre-treated by heating in air at 1432 K for 10 h) and Rh2O3·xH2O (dehydrated in air at 973 K for 2 h) and ethanol (3 × 1 mL) in a mortar and pestle for 20 min. The mixture was then heated in an alumina crucible to 1473 K (5 K min−1 to 523 K then 10 K min−1 to 1473 K) and held at that temperature for 6 h before cooling to room temperature (50 K min−1). The target dopant levels were: La/(La + A) = 4 mol% and Rh/(Rh + B) = 4 mol%. The naming convention in this article follows: SrZrO3 is sometimes truncated to SZO. SrZr0.5Ti0.5O3 to SZT, SrTiO3 to STO, BaZrO3 to BZO. BaZr0.5Ti0.5O3 to BZT, BaTiO3 to BTO. Their co-doped counterparts follow suit (e.g. La,Rh:SZO).2.4 Characterisation
2.5 Photocatalytic hydrogen production
Photocatalytic hydrogen evolution was performed in a sealed batch reactor fitted with a quartz lid (1.7 cm2) for overhead-irradiation. In each reaction, 10 mg of sample was immersed in a 1:1 (by volume) mixture of distilled water and methanol and sonicated to homogeneous suspension. Air was evacuated from the reactor and replaced with Ar (1 atm). Each photocatalyst was surface-decorated with 1 wt% Pt nanoparticles by in situ photoreduction of aq. K2PtCl6 (80.03 μL, 3.11 mg mL−1) for 1 h. Photoreduction was performed with a UV LED (365 nm, 83 mW cm−2 HongKong UVET Co., UH-82F+L12). The stirring suspension was then irradiated with 405 and 455 nm LEDs. The evolved gases were sampled at 0 h, 1.5 h, and 3 h by gas chromatography (Agilent Technologies Micro GC 990, thermal conductivity detector, Ar carrier gas, molecular sieve 5 Å column).
The AQY measurements were performed in the same sealed batch reactor using a wider range of LED wavelengths. Before photocatalysis, air was evacuated from the reactor and replaced with Ar (1 atm). The stirring suspension was then irradiated with one LED for up to 50 min. During the reaction, Ar flowed through the reaction cell at 1 mL min−1. The evolved gases were sampled every 2 min, starting at 0 min, and analysed by gas chromatography (Agilent Technologies Micro GC 990, thermal conductivity detector, Ar carrier gas, molecular sieve 5 Å column). In this system with constant flow of carrier gas, the evolved hydrogen is mixed with the carrier gas and its relative % is determined from calibrated data. This value is converted to a hydrogen production rate.
LEDs used: 365 nm (31.46 mW cm−2 HongKong UVET Co., UH-82F+L12). 405 nm (mW cm−2 HongKong UVET Co., UH-82F+L12). 430 nm (28.9 mW cm−2 Thorlabs, Inc). 455 nm (27.3 mW cm−2 Thorlabs, Inc). 470 nm (20.8 mW cm−2 Thorlabs, Inc). 505 nm (25.6 mW cm−2 Thorlabs, Inc). The powers were selected to keep a constant photon flux of 6.25 × 1019γ s−1 cm−2 across all LEDs used.
3 Results and discussion
3.1 Characterisation
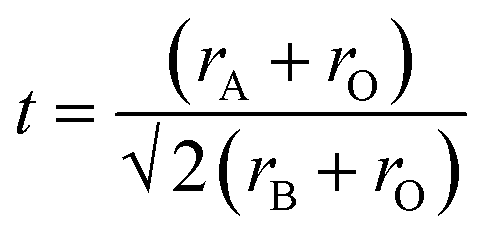 | (1) |
The effect of co-doped materials on t is not reported in previous results focused on doping SrTiO3.22,25,30 Consequently, a modified equation (t′) for the La,Rh:ABO3 perovskites were calculated by incorporating the La ionic radii (rLa) and Rh ionic radii (rRh) into the tolerance equation. The radii of each cation (dopant or base) in t′ were weighted by stoichiometry. Following previous reports, it was assumed La occupies the A-site and Rh occupies the B-site.22,31 The modified tolerance equation is:
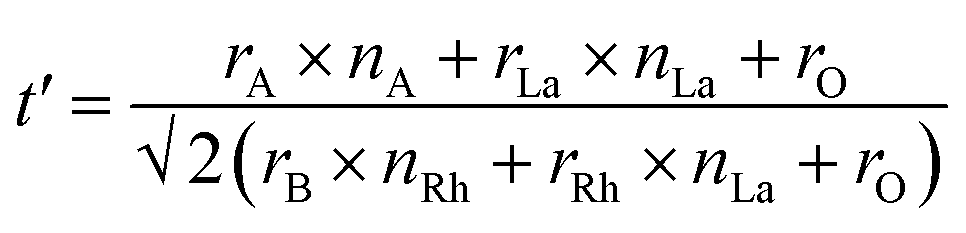 | (2) |
A-site elements have a coordination number (CN) of 12 and the B-site elements have a CN of 6 in a perovskite. A CN of 6 was used for the oxygen anions.28,29 The resulting ionic radii are Sr2+: 1.44 Å, Ba2+: 1.61 Å, Ti4+: 0.605 Å, Zr4+: 0.72 Å, La3+: 1.36 Å and Rh3+ 0.665 Å.32 The tolerance factors for the base perovskites are summarised in Table 1.
| Photocatalyst | t (a.u.) | t′ (a.u.) |
|---|---|---|
| SZO | 0.95 | 0.95 |
| BZO | 1.00 | 1.00 |
| SZT | 0.97 | 1.00 |
| BZT | 1.03 | 1.06 |
| STO | 1.00 | 1.00 |
| BTO | 1.06 | 1.06 |
The calculated t for all 12 materials are close to 1, ranging from 0.95 to 1.06. This indicates all the photocatalysts have, or are close to, ideal cubic perovskite bulk crystallinity. STO and BZO have ideal values of 1. SZT and SZO have t < 1 and likely incur slight orthorhombic distortion. Whereas BZT and BTO with t > 1 have slight hexagonal distortion.
Co-doping La and Rh at 4 mol% does not significantly alter t. This results from two factors. (1) The similarity in ionic radii of the dopants to their respective A or B-site elements. (2) The small amount of dopants used (4 mol%). These calculations suggest that co-doping will not significantly alter the crystal structure of the base materials.
 | ||
| Fig. 2 X-ray diffraction patterns of base ABO3 and La,Rh:ABO3 perovskites. | ||
 | ||
| Fig. 3 (a) Normalised Tauc plot of La,Rh:ABO3 perovskites. The inset contains Tauc plots for the base perovskites. (b) The Tauc of La,Rh co-doped and undoped BTO are plotted to demonstrate the visible light absorption conferred by La,Rh co-doping. | ||
| Photocatalyst | t (a.u.) | Crystal structure | Bandgap (eV nm−1) | Mode particle size (nm) | Particle morphology | 405 nm H2 prod. (nmol h−1) | 455 nm H2 prod. (nmol h−1) |
|---|---|---|---|---|---|---|---|
| SZO | 0.95 | Orthorhombic | 4.23/293 | 291 | Irregular | 0.00 | 0.00 |
| BZO | 1.00 | Cubic | 4.07/305 | 339 | Irregular | 0.00 | 0.00 |
| SZT | 0.97 | Orthorhombic | 3.32/374 | 335 | Cubic | 0.00 | 0.00 |
| BZT | 1.03 | Hexagonal | 3.40/365 | 382 | Cubic | 0.00 | 0.00 |
| STO | 1.00 | Cubic | 3.22/385 | 253 | Cubic | 95.0 | 0.00 |
| BTO | 1.06 | Hexagonal | 3.09/401 | 360 | Cubic | 79.3 | 0.00 |
| Photocatalyst | t (a.u.) | Crystal structure | Bandgap (eV nm−1) | Mode particle size (nm) | Size change (%) | Particle morphology | 405 nm H2 prod. (nmol h−1) | 455 nm H2 prod. (nmol h−1) |
|---|---|---|---|---|---|---|---|---|
| La,Rh:SZO | 0.95 | Orthorhombic | 3.40/365 | 347 | +17.6 | Irregular | 0.00 | 0.00 |
| La,Rh:BZO | 1.00 | Cubic | 3.32/374 | 420 | +21.3 | Irregular | 0.00 | 0.00 |
| La,Rh:SZT | 1.00 | Orthorhombic | 2.94/422 | 377 | +11.8 | Cubic | 66.6 | 0.00 |
| La,Rh:BZT | 1.06 | Hexagonal | 2.95/420 | 386 | +1.04 | Cubic | 16.5 | 0.00 |
| La,Rh:STO | 1.00 | Cubic | 2.67/460 | 350 | +32.2 | Cubic | 289.3 | 109.3 |
| La,Rh:BTO | 1.06 | Hexagonal | 2.58/480 | 451 | +22.4 | Cubic | 195.4 | 42.2 |
These findings are exemplified by La,Rh:BTO and SZO. La,Rh:BTO comprises the heaviest A-site element and lightest B-site element and demonstrates the narrowest bandgap of 2.58 eV. SZO is undoped and comprises the lightest A-site element and heaviest B-site element; this results in the widest bandgap of 4.23 eV.
The Rh,La co-doping strategy successfully red-shifted the absorption of all 6 photocatalysts (Fig. 3a). This absorption was extended into visible light for La,Rh:SZT, La,Rh:BZT, La,Rh:STO and La,Rh:BTO. The perovskites from widest to narrowest bandgap are La,Rh:SZO, La,Rh:BZO, La,Rh:SZT, La,Rh:BZT, La,Rh:STO, La,Rh:BTO. The undoped perovskites follow an identical trend. La,Rh:STO and La,Rh:BTO demonstrated the greatest amount of visible light absorption and consequently have the greatest potential for visible light photocatalysis.
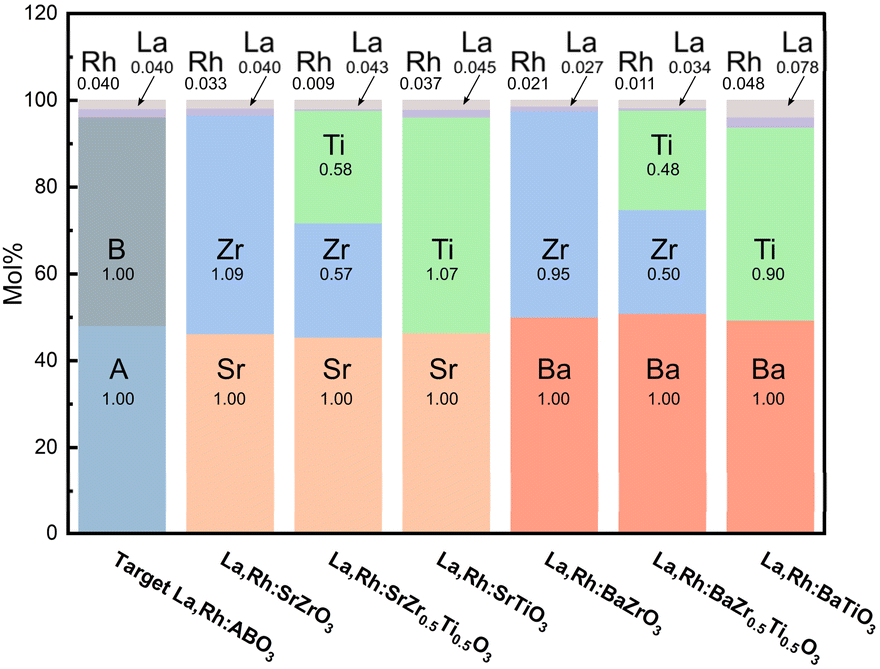 | ||
| Fig. 4 ICP-MS derived mol% of elements comprising the La,Rh:ABO3 perovskites. The corresponding stoichiometry is labelled in black (normalised to the A element, Sr or Ba). | ||
The expected stoichiometries for the co-doped materials is La0.04,Rh0.04:A0.96B0.96O3 (and La0.04,Rh0.04:A0.96Ti0.48Zr0.48O3 for the mixed oxides). Doping had little appreciable effect on the base element stoichiometry (Table S1†). This corroborates the PXRD, suggesting that particle crystallinity, morphology, and lattice structure are not detrimentally affected by La,Rh co-doping.
ICP-MS data confirms the presence of La and Rh in all 6 co-doped samples, roughly in accordance with the expected stoichiometry (Fig. 4). This substantiates the cause of the red-shifted DRS spectra of the doped perovskites. The combined ICP and DRS data confirm Rh and La are correctly incorporated into the atomic lattice of the base material, providing improved visible light absorption.
Generally, both titanates demonstrated dopant content closest to the expected stoichiometry. They were followed by the zirconates which were followed by the mixed-oxides. No systematic variation of La or Rh content was observed when comparing the strontium perovskites to the barium perovskites.
The Rh content in SZO, SZT, BZT, and BZT was lower (0.009–0.0021) than the expected stoichiometry (0.04). On the other hand, the two titanite samples had Rh in near-expected stoichiometry. This may indicate Ti is a better host element for Rh than Zr. That said, the Rh content in SZT and BZT was noticeably lower than SZO and BZO. This suggests mixed B-sites may suppress dopant incorporation. The La content in the mixed-oxides did not see a similar drop.
In all six samples the La content was higher than the corresponding Rh content. In general, the La content was closer to the expected stoichiometry than Rh. This could indicate La incorporation is more favourable than Rh incorporation. This could stem from the smaller ionic radius of La3+ than either of the A-site elements whereas Rh4+ is smaller than Zr4+ but larger than Ti3+. However, it should then follow that the Rh content of the zirconates would be higher than the Rh content of the titanates. In fact, the opposite is true. Alternatively, Rh may be less digestible in aqua regia which could result in lower % observed. Three samples, SZT, STO, and BTO, demonstrated slightly excessive La stoichiometries of 0.043, 0.045, and 0.078. This may mean the generally higher La concentrations stem from ICP-MS matrix effects.
 | ||
| Fig. 5 SEM images of La,Rh co-doped perovskites. (a) La,Rh:SrZrO3. (b) La,Rh:SrZrTiO3. (c) La,Rh:SrTiO3. (d) La,Rh:SrZrO3. (e) La,Rh:SrZrTiO3. (f) La,Rh:SrTiO3. | ||
The SEM images were used to calculate the size distribution of each sample. The size distribution was fit to a log-normal distribution and the mode particle size of the fit was used to compare between samples. Both zirconates have the narrowest size distributions (Fig. S2†). BZT and BTO samples have a broader range of particle sizes than their Sr counterparts.
After La,Rh co-doping, all of the samples demonstrated a greater mode particle size, growing by 50–100 nm (Fig. 6). La,Rh:BZT was an exception, increasing in mode particle size by only 10 nm. This indicates that co-doping La,Rh could lead to increased particle sizes. However, La and Rh were doped at temperatures 50 K higher than the synthesis temperature of the base perovskites and for 6 more hours. Additional time spent at elevated temperature could also increase particle size.
 | ||
| Fig. 6 Mode perovskite particle size comparison. (a) Undoped perovskites (b) La,Rh co-doped perovskites. | ||
Photocatalysts with Ba at the A-site were larger than their Sr counterparts in all cases (Fig. 6). The larger atomic radius of Ba (268 pm) compared to Sr (249 pm) is a potential cause. The synthetic conditions were adapted from conditions optimised for SrTiO3. BaTiO3 and BaCO3 also have lower melting points than SrTiO3 and SrCO3. This may indicate smaller BaTiO3 could be obtained from lower synthesis temperatures. Zr and Ti have near identical atomic radii (186 and 187 pm respectively). Consequently, the AZrO3 perovskites tended to similar sizes of their ATiO3 counterparts.
For the pristine perovskites, both mixed B-site perovskites exhibited a particle size increase. Given the atomic radius similarity of Ti and Zr, the size increase is attributed to the longer synthesis time (24 h vs. 10 h) needed to fabricate the mixed-oxide perovskites.
The co-doped photocatalysts reverse this trend. The mixed B-site perovskites are smaller compared to their AZO and ATO counterparts. That said, the AZT perovskites also demonstrate the smallest post-doping particle size increase (Table 3, vide infra). The smaller La,Rh:AZT size change is also attributed to the longer base material syntheses time.
 | ||
| Fig. 7 (a) High-angle annular dark-field (HAADF) TEM image of a representative La,Rh:SrTiO3 particle. (b) EDS map of the particle. (c) La,Rh:SrTiO3 atomic lattice of the region outlined in the red box in (a). (d) Atomic-resolution image of the red dotted-box in (c), depicting the cubic structure of the La,Rh-concentrated exterior of the particle. The A-site elements (Sr and La) are identified with blue circles and the B-site elements (Ti and Rh) are identified with red circles. The A-site elements both have higher atomic numbers than the corresponding B-site elements and therefore appear brighter in a HAADF image. (e) EDS line profile of net signal intensity for Ti and Rh across the arrow in (b). The white shaded region around the arrow depicts the integration area. A moving average (data window = 5) was applied to smooth the data. (f) Normalised atomic fraction EDS line profile. A moving average (data window = 5) was applied to smooth the data. | ||
Maintaining the perovskite crystallinity of the host material in the dopant-rich surface is critical to suppressing chemical or crystalline defects.25,38 Further, perovskite morphology enables facet-specific electronics which improve charge-transfer efficiency.7,34 The atomic lattice for STO and La,Rh:STO maintained cubic perovskite crystallinity. The shell lattice shows no discernible difference from the core lattice (Fig. 7c). Further, the typical cubic crystal structure of SrTiO3 is identifiable in the dopant-rich shell (Fig. 7d). This demonstrates that La and Rh are incorporated into the ATiO3 perovskite without appreciable disruption to the crystal lattice.
Two-step solid-state doping methods typically produce core–shell particles comprising a pristine core and the dopant-rich shell.19,22,24,39 Doping occurs in the second step by diffusion of the dopants into the base material's structure. The photocatalytic efficacy of core–shell nanoparticles is debated. Computational modelling has found such structures to be either advantageous or deleterious.19,40 More research is needed to determine the photocatalytic efficacy of particles with core–shell morphology.
3.2 Photocatalysis
Photocatalytic hydrogen evolution for all 12 samples was performed using 405 and 455 nm LEDs under sacrificial conditions (1:1 CH3OH:H2O, Fig. 8). Pt nanoparticles were photodeposited in situ during photocatalysis at 1 wt%. Pt nanoparticles have proven efficacy in enabling catalytic hydrogen production.41 Visible wavelengths were selected to confirm co-doping La and Rh conferred visible light activity.
 | ||
| Fig. 8 Photocatalytic H2 evolution of various perovskites from a 1:1 H2O and CH3OH mix using 405 (a) and 455 nm light (b). The shaded areas depict standard error. Conditions: 7 mg photocatalyst, 405 (228.0 mW cm−2) or 455 (88.4 mW cm−2) nm LED. | ||
As expected, all six undoped perovskites were photocatalytically inactive at 455 nm (Fig. 8b). Whereas four La,Rh co-doped materials (La,Rh:SrZr0.5Ti0.5O3, La,Rh:BaZr0.5Ti0.5O3, La,Rh:SrTiO3 and La,Rh:BaTiO3) achieved visible light hydrogen production at 405 nm. The co-doped titanates, La,Rh:SrTiO3 and La,Rh:BaTiO3 also demonstrated H2 evolution at 455 nm. On the other hand, La,Rh:SrZrO3 and La,RhBaZrO3 did not produce H2 at any wavelength. These results are all consistent with the DRS-determined bandgaps for each photocatalyst (Table 3, vide infra).
Photocatalysis was performed for up to 3 h with close to linear production which indicates the potential long-term activity of the materials. ABO3 and La,Rh:SrTiO3 perovskites have demonstrated long-term activity in the literature (up to 1000 + h), which provides further support to the long-term viability of the materials.8,42 However further testing of the photocatalysts described here is necessary to confirm similar longevity.
Two undoped materials, SrTiO3 and BaTiO3, unexpectedly evolved small amounts hydrogen at 405 nm (Table 2). The titanates have the narrowest bandgaps of the undoped photocatalysts and the LEDs emit in a Gaussian distribution, peak: 405 nm FWHM: 10.8 nm (Fig. S6†). Consequently, some UV photons present in the 405 nm LED could be absorbed and therefore explain the activity at 405 nm.
The most active samples were La,Rh:SrTiO3 and La,Rh:BaTiO3. These materials have the narrowest bandgaps and Ti as the B-site element. Further, both materials outperform their pristine counterparts at 405 and 455 nm. La,Rh:BaTiO3, which demonstrated the greatest visible light absorption, and the 2nd highest photocatalytic activity was then compared against La,Rh:SrTiO3 over a range of LED wavelengths from 365–505 nm (Fig. 9). AQY was obtained at each wavelength and plotted with the DRS data, as shown in Fig. 9.
 | ||
| Fig. 9 Kubelka–Munk transform of UV-Vis diffuse reflectance and wavelength dependence on apparent quantum yield (AQY) of La,Rh:SrTiO3 and La,Rh:BaTiO3 from 365–505 nm. 7 mg photocatalyst, varying LED wavelength, constant photon flux (6.25 × 1019γ s−1 cm−2). | ||
Both La,Rh co-doped materials demonstrated visible light activity, evolving H2 up to 505 nm which is well into the visible region of sunlight. La,Rh:BaTiO3 shows lower activity at shorter wavelengths, but as the wavelength of light extends into the visible region it begins to outperform La,Rh:SrTiO3. This is in line with the absorption profiles obtained in Fig. 3. However, the AQY also drops as the light absorption falls which is also in line with UV-Vis absorption data. This indicates synthetic modifications which increase the visible light absorption, bringing it in line with the UV light absorption, could yield better catalysts. Fig. S7† contains the gas chromatographs of the H2 peaks for photocatalysis of La,Rh:SrTO3 at 455, 470 and 505 nm and confirms that the H2 peak is readily distinguishable from the baseline trace and dark peak at lower AQY. The bandgaps of La,Rh:SrTiO3 and La,Rh:BaTiO3 were determined to be 2.67 eV (460 nm) and 2.58 eV (480 nm) respectively. This could indicate absorbance and activity at 505 nm is due to Rh4+ defect states rather than Rh3+ valence band states and the former are correlated to lower activity.43 However, the relatively broad spectral output of the 505 nm LED depicted in Fig. S6† means some photons will have sufficient energy to excite the Rh3+ modified bandgap, particularly for La,Rh:BaTiO3, which could also explain the activities observed. The AQY obtained here for La,Rh:SrTiO3 is lower than the AQY reported for the same material by Wang et al. (30%).16 However, the latter material was used as a component in a Z-scheme system which used conductive gold mediator and was measured at reduced pressure.
Given the better light absorption profile and similar photocatalytic rates of La,Rh:BaTiO3 compared with La,Rh:SrTiO3, La,Rh:BaTiO3 is a promising alternative candidate for hydrogen evolution in a Z-scheme photocatalyst. La,Rh:BaTiO3 was fabricated using synthetic conditions optimised for La,Rh:SrTiO3. Tailoring the synthetic conditions to La,Rh:BaTiO3 could further improve the efficiency of La,Rh:BaTiO3.
4 Discussion
6 base perovskites were synthesised and the effect of systematically varying both the A and B sites was investigated. For the A-site element, moving down group 2 on the periodic table from Sr to Ba was found to narrow the bandgap. The opposite was true for the B-site element. Moving up the Group 4 elements to Zr from Ti was found to narrow the bandgap. Consequently, BaTiO3 had the narrowest bandgap of the six perovskites tested. Ti-containing perovskites were found to achieve greater photocatalytic hydrogen production. This activity tracks with bandgap, Goldschmidt tolerance factor (t), and particle size. Only STO and BTO have bandgaps able to absorb 405 nm light. STO are smaller particles and attains an ideal t of 1 (Table 2).The 6 perovskites were then co-doped with La and Rh, characterised, and tested for photocatalytic hydrogen evolution (Table 3). Bandgap narrowing from La,Rh co-doping was previously only established for SrTiO3. The combined DRS and ICP-MS, results of La,Rh:STO and 5 other La,Rh:ABO3 perovskites successfully generalise this bandgap narrowing strategy to ABO3 perovskites. PXRD, SEM and TEM measurements show La,Rh co-doping doesn't significantly alter bulk crystallinity, particle morphology or particle size. TEM elemental mapping for La,Rh:SrTiO3 revealed the doped particles comprise base material cores surrounded by dopant rich shells. Visible light (405 and 455 nm) photocatalytic hydrogen evolution was demonstrated for four perovskites, La,Rh:SrZr0.5Ti0.5O3, La,Rh:BaZr0.5Ti0.5O3, La,Rh:SrTiO3 and La,Rh:BaTiO3.
The four active materials at 405 nm demonstrated H2 activity rates in order of La,Rh:BZT < La,Rh:SZT < La,Rh:BTO < La,Rh:STO. Particle morphology was similar across all four samples. Particle size, likewise, does not explain the relative activities; for example, La,Rh:BTO has the largest particles but reaches the second highest production rate. Ba appears to be a less efficient A-site element than Sr. Of the four active co-doped materials, perovskites with A-site Sr have close to ideal t. Their Ba equivalents demonstrated slight distortion. The H2 activities at 455 nm reflect the latter point. As the BaBO3 perovskites were uniformly larger than their SrBO3 counterparts, this may indicate photocatalytic improvements could be achieved with reduced particle size of BaBO3 perovskites.
Finally, the novel material La,Rh:BTO exhibits the most favourable bandgap for operation under solar radiation, as exemplified by its improved performance at 470 and 505 nm (Fig. 9). Further, Ti at the B-site was found to be more favourable for charge separation in comparison with a mixed B-site analogue. The synthetic conditions used were based on the synthesis and doping of SrTiO3. Synthetic conditions tuned to BaTiO3 may improve quantum efficiency and enable the material to make full use of its bandgap.
5 Conclusion
Six base perovskites were synthesised: SrZrO3, SrZr0.5Ti0.5O3, SrTiO3, BaZrO3, BaZr0.5Ti0.5O3, and BaTiO3. A and B-site variation was found to modify particle size, morphology, crystallinity, and the bandgap. The perovskite bandgap was determined to be tuneable by up to 108 nm through A and B-site variation.The six perovskites were then co-doped with La and Rh following the methods developed for SrTiO3. This resulted in six doped perovskites: La,Rh:SrZrO3, La,Rh:SrZr0.5Ti0.5O3, La,Rh:SrTiO3, La,Rh:BaZrO3, La,Rh:BaZr0.5Ti0.5O3, and La,Rh:BaTiO3. The bandgap of all six perovskites was successfully narrowed – redshifting the bandgap by ca. 60–80 nm. Additionally, co-doping was found to increase particle size slightly and maintain base particle morphology and crystallinity.
Finally, all 12 materials were tested for sacrificial photocatalytic water-splitting under visible light (405 and 455 nm). Of the six doped materials, the two doped titanates, La,Rh:SrTiO3 and La,Rh:BaTiO3, achieved the best performance. This trend was followed for the six base counterparts. Further, 4 La,Rh-doped perovskites demonstrated H2 production under visible light. Overall, this work generalises La,Rh co-doping as a strategy for obtaining active visible light perovskite oxides.
The high performance of BaTiO3 and La,Rh:BaTiO3 is noteworthy, especially at longer wavelengths. The two photocatalysts are redshifted, close-analogues of the better studied SrTiO3 and La,Rh:SrTiO3. Numerous modifications to SrTiO3, such as synthesis under flux-conditions and co-catalyst optimisation over the past 20 years have culminated in a material which achieves a 96% AQY for UV-light. Given the similarity of BaTiO3 to SrTiO3 similar AQY improvements are possible and in conjunction with the narrower bandgap of BaTiO3 and La,Rh:BaTiO3 could lead to a material with a higher theoretical STH. Detailed investigations of La,Rh:BaTiO3 such as optimised syntheses could lead to new perovskites which outperform SrTiO3 for visible light water-splitting.
Data availability
All data is provided in the ESI .xlsx file.†Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
This work was supported by funding from the Future Fuels CRC. TDS acknowledges the Future Fuels CRC for awarding a postgraduate scholarship. The authors acknowledge the facilities of Adelaide Microscopy (University of Adelaide). CJS acknowledges funding support from The Australian Research Council (ARC) for a Future Fellowship (FT190100854).References
- IEA, Net Zero by 2050, IEA, 2021, https://www.iea.org/reports/net-zero-by-2050 Search PubMed.
- BP, BP - Energy Outlook 2020, 2020, https://www.bp.com/content/dam/bp/business-sites/en/global/corporate/pdfs/energy-economics/energy-outlook/bp-energy-outlook-2020.pdf Search PubMed.
- DCCEEW 2024, National Hydrogen Strategy 2024, Department of Climate Change, Energy, the Environment and Water, Canberra, CC BY 4.0., http://www.dcceew.gov.au/sites/default/files/documents/national-hydrogen-strategy-2024.pdf Search PubMed.
- IEA, Global Energy Review 2025, https://www.iea.org/reports/global-energy-review-2025 Search PubMed.
- Q. Wang and K. Domen, Particulate Photocatalysts for Light-Driven Water Splitting: Mechanisms, Challenges, and Design Strategies, Chem. Rev., 2020, 120, 919–985, DOI:10.1021/acs.chemrev.9b00201.
- B. A. Pinaud, et al., Technical and economic feasibility of centralized facilities for solar hydrogen production via photocatalysis and photoelectrochemistry, Energy Environ. Sci., 2013, 6, 1983–2002, 10.1039/c3ee40831k.
- T. Takata, et al., Photocatalytic water splitting with a quantum efficiency of almost unity, Nature, 2020, 581, 411–414, DOI:10.1038/s41586-020-2278-9.
- Q. Wang, et al., Particulate Photocatalyst Sheets Based on Carbon Conductor Layer for Efficient Z-Scheme Pure-Water Splitting at Ambient Pressure, J. Am. Chem. Soc., 2017, 139, 1675–1683, DOI:10.1021/jacs.6b12164.
- J. Liu, et al., Metal-free efficient photocatalyst for stable visible water splitting via a two-electron pathway, Science, 2015, 347, 970–974, DOI:10.1126/science.aaa3145.
- P. Zhou, et al., Solar-to-hydrogen efficiency of more than 9% in photocatalytic water splitting, Nature, 2023, 613, 66–70, DOI:10.1038/s41586-022-05399-1.
- H. Kato, K. Asakura and A. Kudo, Highly Efficient Water Splitting into H2 and O2 over Lanthanum-Doped NaTaO3 Photocatalysts with High Crystallinity and Surface Nanostructure, J. Am. Chem. Soc., 2003, 125, 3082–3089, DOI:10.1021/ja027751g.
- Y. Sakata, et al., Remarkable improvement of the photocatalytic activity of Ga2O3 towards the overall splitting of H2O, ChemSusChem, 2011, 4, 181–184, DOI:10.1002/cssc.201000258.
- H. Shi, et al., Visible Light Photoanode Material for Photoelectrochemical Water Splitting: A Review of Bismuth Vanadate, Energy Fuels, 2022, 36, 11404–11427, DOI:10.1021/acs.energyfuels.2c00994.
- J. Xiao, T. Hisatomi and K. Domen, Narrow-Band-Gap Particulate Photocatalysts for One-Step-Excitation Overall Water Splitting, Acc. Chem. Res., 2023, 56, 878–888, DOI:10.1021/acs.accounts.3c00011.
- K. Rajeshwar, P. A. Maggard and S. O'Donnell, Search of the “Perfect” Inorganic Semiconductor/Liquid Interface for Solar Water Splitting, Electrochem. Soc. Interface, 2021, 30, 47–51, DOI:10.1149/2.f07211if.
- Q. Wang, et al., Scalable water splitting on particulate photocatalyst sheets with a solar-to-hydrogen energy conversion efficiency exceeding 1%, Nat. Mater., 2016, 15, 611–615, DOI:10.1038/nmat4589.
- Q. Wang, et al., Oxysulfide photocatalyst for visible-light-driven overall water splitting, Nat. Mater., 2019, 18, 827–832, DOI:10.1038/s41563-019-0399-z.
- K. Chen, et al., Overall Water Splitting by a SrTaO(2)N-Based Photocatalyst Decorated with an Ir-Promoted Ru-Based Cocatalyst, J. Am. Chem. Soc., 2023, 145, 3839–3843, DOI:10.1021/jacs.2c11025.
- S. O'Donnell, et al., Prediction and Kinetic Stabilization of Sn(II)-Perovskite Oxide Nanoshells, Chem. Mater., 2022, 34, 8054–8064, DOI:10.1021/acs.chemmater.2c02192.
- Y. Wang, et al., Direct Solar Hydrogen Generation at 20% Efficiency Using Low-Cost Materials, Adv. Energy Mater., 2021, 11, 2101053, DOI:10.1002/aenm.202101053.
- Z. Wang, et al., Advances in engineering perovskite oxides for photochemical and photoelectrochemical water splitting, Applied Physics Reviews, 2021, 8, 021320, DOI:10.1063/5.0039197.
- Q. Wang, T. Hisatomi, S. S. K. Ma, Y. Li and K. Domen, Core/Shell Structured La- and Rh-Codoped SrTiO3 as a Hydrogen Evolution Photocatalyst in Z-Scheme Overall Water Splitting under Visible Light Irradiation, Chem. Mater., 2014, 26, 4144–4150, DOI:10.1021/cm5011983.
- B. Modak and S. K. Ghosh, Enhancement of Visible Light Photocatalytic Activity of SrTiO3: A Hybrid Density Functional Study, J. Phys. Chem. C, 2015, 119, 23503–23514, DOI:10.1021/acs.jpcc.5b06667.
- A. S. Alotabi, et al., Reduction and Diffusion of Cr-Oxide Layers into P25, BaLa(4)Ti(4)O(15), and Al:SrTiO(3) Particles upon High-Temperature Annealing, ACS Appl. Mater. Interfaces, 2023, 15, 14990–15003, DOI:10.1021/acsami.3c00250.
- B. Moss, et al., Linking in situ charge accumulation to electronic structure in doped SrTiO3 reveals design principles for hydrogen-evolving photocatalysts, Nat. Mater., 2021, 20, 511–517, DOI:10.1038/s41563-020-00868-2.
- S. O'Donnell, et al., Pushing the Limits of Metastability in Semiconducting Perovskite Oxides for Visible-Light-Driven Water Oxidation, Chem. Mater., 2020, 32, 3054–3064, DOI:10.1021/acs.chemmater.0c00044.
- P. Makula, M. Pacia and W. Macyk, How To Correctly Determine the Band Gap Energy of Modified Semiconductor Photocatalysts Based on UV-Vis Spectra, J. Phys. Chem. Lett., 2018, 9, 6814–6817, DOI:10.1021/acs.jpclett.8b02892.
- H. Onishi, Sodium Tantalate Photocatalysts Doped with Metal Cations: Why Are They Active for Water Splitting?, ChemSusChem, 2019, 12, 1825–1834, DOI:10.1002/cssc.201802935.
- M. Pidburtnyi, B. Zanca, C. Coppex, S. Jimenez-Villegas and V. Thangadurai, A Review on Perovskite-Type LaFeO3 Based Electrodes for CO2 Reduction in Solid Oxide Electrolysis Cells: Current Understanding of Structure–Functional Property Relationships, Chem. Mater., 2021, 33, 4249–4268, DOI:10.1021/acs.chemmater.1c00771.
- Q. Jia, A. Iwase and A. Kudo, BiVO4–Ru/SrTiO3:Rh composite Z-scheme photocatalyst for solar water splitting, Chem. Sci., 2014, 5, 1513–1519, 10.1039/c3sc52810c.
- R. Asai, et al., A visible light responsive rhodium and antimony-codoped SrTiO3 powdered photocatalyst loaded with an IrO2 cocatalyst for solar water splitting, Chem. Commun., 2014, 50, 2543–2546, 10.1039/c3cc49279f.
- R. Shannon, Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides, Acta Crystallogr., Sect. A, 1976, 32, 751–767, DOI:10.1107/S0567739476001551.
- Y. Yang, X. Ning, S. Luo, F. Dong and L. Li, Electron Irradiation Effects of SrZrO3 Ceramic for Radioactive Strontium Immobilization, Procedia Environ. Sci., 2016, 31, 330–334, DOI:10.1016/j.proenv.2016.02.044.
- H. Kato, M. Kobayashi, M. Hara and M. Kakihana, Fabrication of SrTiO3 exposing characteristic facets using molten salt flux and improvement of photocatalytic activity for water splitting, Catal. Sci. Technol., 2013, 3, 1733–1738, 10.1039/c3cy00014a.
- S. S. Arbuj, R. R. Hawaldar, S. Varma, S. B. Waghmode and B. N. Wani, Synthesis and Characterization of ATiO3 (A = Ca, Sr and Ba) Perovskites and Their Photocatalytic Activity Under Solar Irradiation, Sci. Adv. Mater., 2012, 4, 568–572, DOI:10.1166/sam.2012.1320.
- T. Takata and K. Domen, Defect Engineering of Photocatalysts by Doping of Aliovalent Metal Cations for Efficient Water Splitting, J. Phys. Chem. C, 2009, 113, 19386–19388 CrossRef CAS.
- D. K. Bhat, H. Bantawal and U. S. Shenoy, Rhodium doping augments photocatalytic activity of barium titanate: effect of electronic structure engineering, Nanoscale Adv., 2020, 2, 5688–5698, 10.1039/d0na00702a.
- F. E. Osterloh, in Integrated Solar Fuel Generators Energy and Environment Series, 2018, vol. 214–280 Search PubMed.
- H. Sudrajat, et al., Water-Splitting Activity of La-Doped NaTaO3 Photocatalysts Sensitive to Spatial Distribution of Dopants, J. Phys. Chem. C, 2020, 124, 15285–15294, DOI:10.1021/acs.jpcc.0c03822.
- B. Zutter, et al., Single-Particle Measurements Reveal the Origin of Low Solar-to-Hydrogen Efficiency of Rh-Doped SrTiO3 Photocatalysts, ACS Nano, 2023, 17(10), 9405–9414 CrossRef CAS PubMed.
- C. Shearer, J. Alvino, M. Batmunkh and G. Metha, Pt Nanocluster Co-Catalysts for Photocatalytic Water Splitting, C, 2018, 4, 64, DOI:10.3390/c4040064.
- H. Lyu, et al., An Al-doped SrTiO3 photocatalyst maintaining sunlight-driven overall water splitting activity for over 1000 h of constant illumination, Chem. Sci., 2019, 10, 3196–3201, 10.1039/c8sc05757e.
- D. H. K. Murthy, et al., Revealing the role of the Rh valence state, La doping level and Ru cocatalyst in determining the H2 evolution efficiency in doped SrTiO3 photocatalysts, Sustainable Energy Fuels, 2019, 3, 208–218, 10.1039/c8se00487k.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5na00338e |
| This journal is © The Royal Society of Chemistry 2025 |