
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrasmall Bi/Bi2O3 nanoparticles as biocompatible and efficient CT imaging contrast agents†
Le T. T.
Tam
 ab,
Duong T.
Ngoc
c,
Nguyen T. N.
Linh
d,
Le T.
Tam
e,
Nguyen V.
Dong
f,
Nguyen T.
Yen
b,
Nguyen T.
Suong
e,
Ngo T.
Dung
*b and
Le T.
Lu
ab
ab,
Duong T.
Ngoc
c,
Nguyen T. N.
Linh
d,
Le T.
Tam
e,
Nguyen V.
Dong
f,
Nguyen T.
Yen
b,
Nguyen T.
Suong
e,
Ngo T.
Dung
*b and
Le T.
Lu
ab
aGraduate University of Science and Technology, Vietnam Academy of Science and Technology, 18 Hoang Quoc Viet, Hanoi, Vietnam
bInstitute of Materials Science, Vietnam Academy of Science and Technology, 18 Hoang Quoc Viet, Hanoi, Vietnam. E-mail: ngodunght@gmail.com
cHanoi National University of Education, 136 Xuan Thuy, Cau Giay, Hanoi, Vietnam
dThai Nguyen University of Sciences, Tan Thinh Ward, Thai Nguyen City 25000, Thai Nguyen, Vietnam
eVinh University, 182 Le Duan, Vinh City, Vietnam
fHoan My Vinh Hospital, 99 Pham Đinh Toai, Vinh City, Vietnam
First published on 28th May 2025
Abstract
Computed tomography (CT) imaging is a widely used diagnostic tool, but conventional iodine-based contrast agents suffer from limitations such as short circulation time and potential nephrotoxicity. In this study, we present a simple one-pot thermal decomposition method for synthesizing ultrasmall Bi/Bi2O3 nanoparticles (NPs) using a commercial Bi(NO3)3 precursor and a surfactant mixture of oleic acid and oleylamine. Oleylamine acts as a reducing agent, facilitating the conversion of Bi3+ to metallic Bi, while the Bi2O3 oxide layer is controlled by adjusting the oleic acid-to-oleylamine ratio. To enhance biocompatibility and aqueous dispersibility, the NPs are further modified with polyacrylic acid (PAA), resulting in Bi/Bi2O3@PAA NPs with an ultra-small size, high Bi content, and stability. X-ray attenuation measurements reveal that Bi/Bi2O3@PAA NPs exhibit superior contrast enhancement compared to the traditional iodine-based contrast agents, with increasing efficacy at higher tube voltages. Given their facile synthesis, excellent biocompatibility, and outstanding imaging performance, Bi/Bi2O3@PAA NPs hold significant promise as next-generation CT contrast agents for clinical applications.
Introduction
Computed tomography (CT) imaging is an indispensable tool in modern diagnostic medicine, providing high spatial resolution and detailed visualization of internal structures.1,2 However, the inherent low contrast of soft tissues in CT scans necessitates the use of contrast agents to create distinct images of tissues with similar contrast. Currently, iodinated compounds such as iohexol, ioversol or iopamidol are the most commonly used contrast agents due to their strong X-ray attenuation and water solubility.3,4 Despite their effectiveness, these agents have significant limitations, including short circulation times, nonspecific distribution, the necessity of high doses, hypersensitivity reactions, and potential nephrotoxicity, especially in patients with pre-existing kidney conditions.5–8 These challenges drive the ongoing search for safer and more effective alternatives to traditional iodine-based agents.Recent advancements in nanotechnology have enabled the development of CT contrast agents based on metal NPs such as gold (Au), bismuth (Bi), and gadolinium (Gd), offering an alternative to traditional iodine-based agents.4,8–12 These NPs exhibit superior X-ray absorption due to their high atomic numbers.2 Among them, bismuth (Bi), the heaviest non-radioactive metal, has emerged as a particularly promising candidate due to its excellent X-ray attenuation (Z = 83) and ability to provide higher contrast than iodine and other high-Z materials in CT imaging. This is attributed to its high K-edge value (90.5 keV) and large X-ray attenuation coefficient (5.74 cm2 g−1 at 100 keV).13 Additionally, bismuth is the most cost-effective among heavy metal and is widely recognized for its biosafety.14–16 A series of bismuth-containing medications have been extensively used for treating gastrointestinal disorders, hypertension, infections, cancers, and inflammation, further supporting its potential as a high-performance CT contrast agents (CAs).
Various Bi-based nanomaterials have been explored as next-generation CT CAs, including small-molecule chelates such as Bi-DTPA,17–19 Bi-DOTA20 as well as NPs like metallic Bi,2,21 bismuth oxide (Bi2O3),22 and bismuth sulfide (Bi2S3).23–26 Among these, metallic Bi NPs are particularly attractive as CT contrast agents due to their high bismuth atom density, which enables strong X-ray attenuation within a relatively small volume. For example, Li et al. developed PEGylated metallic Bi nanocrystals (Bi-PEG NCs) using a chemical reduction method.11 These Bi-PEG NCs exhibited excellent CT contrast enhancement, good biocompatibility and safety. Similarly, Yang et al. synthesised Bi NPs using Bi(III) acetate as a precursor in a mixed solution of oleic acid and 1-dodecanethiol.27 The resulting Bi NPs were coated with 1,2-dilauroyl-sn-glycero-3-phosphocholine (Bi@DLPC NPs) through ultrasonic treatment, achieving a uniform size of 47 ± 3 nm. The CT imaging properties of Bi@DLPC NPs were concentration-dependent, with signal intensity and HU values significantly increasing as the concentration rose from 0.313 to 5 mg mL−1. Tarighatnia et al. synthesized Bi NPs via thermal decomposition and subsequently functionalized them with targeting biomolecules such as MUC16, leading to enhanced X-ray attenuation through specific targeting effects.28,29 Compared to gold NPs (Au NPs), which share similar synthesis versatility and tunability in size and morphology, Bi NPs offer a more cost-effective alternative, being approximately 2000 times less expensive.30,31
Despite these advantages, Bi NPs face several challenges, including poor stability, large particle sizes, labor-intensive postsynthetic surface modifications, and limited targeting properties.11,27 For instance, bare Bi NPs tend to undergo rapid oxidation and aggregation in biological environments, limiting their practical application.11 Meanwhile, Bi2O3 and Bi2S3 NPs have gained increasing attention due to their high stability, adjustable cytotoxicity, and cost-effective synthesis, making them promising candidates for CT diagnostic applications.32 However, a key limitation of these compounds is their lower bismuth atom concentration per particle due to the presence of oxygen or sulfur. Furthermore, Bi2S3 suffers from instability in aqueous media, where hydrolysis can lead to the release of toxic hydrogen sulfide gas, posing safety concerns.
In this study, we propose a simple and effective synthesis method for ultrasmall Bi/Bi2O3 NPs via a one-pot thermal decomposition of a commercial Bi(NO3)3 precursor in the presence of a surfactant mixture containing oleic acid and oleylamine. In this process, oleylamine acts as a reducing agent to efficiently convert Bi3+ to metallic Bi, while the formation of the Bi2O3 oxide layer is controlled by adjusting the oleic acid-to-oleylamine ratio. Further surface modification of Bi@Bi2O3 NPs can be easily achieved with polyacrylic acid (PAA) to make the sample hydrophilic and biocompatible. The obtained Bi/Bi2O3@PAA NPs exhibited an ultra-small size (3 nm), outstanding water solubility, high Bi content and biocompatibility. The X-ray attenuation ability of Bi/Bi2O3@PAA NPs is more than 2-fold higher than that of some iodine-based clinical contrast agents, and the superiority becomes increasingly pronounced with increasing tube voltage.
Compared to previously reported methods, the proposed strategy offers several distinct advantages for the large-scale production of high-performance CT contrast agents: (i) a simple and cost-effective synthesis utilizing commercially available reagents, eliminating the need of the specially designed precursors or multi-step procedures; (ii) the synthesis of ultrasmall, monodisperse NPs with tunable structure and composition by adjusting OA/OLA ratio; and (iii) straightforward surface modification with ligands such as PAA, resulting in enhanced aqueous stability and biocompatibility. These advantages underscore the high potential of Bi/Bi2O3@PAA NPs as promising next-generation CT contrast agents.
Experimental
Chemicals
All used chemicals are at least analytical grade without further purification. Bismuth nitrate pentahydrate (99.99%), dibenzyl ether (90%), 1-octadecene (90%), oleic acid (OA) (99%), oleylamine (OLA) (70%), N,N-dimethylformamide (DMF), nitrosonium tetrafluoroborate (NOBF4, 95%), polyacrylic acid (PAA, Mw = 1800), ethanol (≥96%), n-hexane (99%) and chloroform (≥99%) were purchased from Sigma-Aldrich.Synthesis of Bi/Bi2O3 NPs
Ultrasmall Bi/Bi2O3 NPs were synthesized via a thermal decomposition method. Initially, 10 mmol of Bi(NO3)3·5H2O was dissolved in a 100 mL three-neck round-bottom flask containing a surfactant mixture of OA and OLA in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio. 40 mL solvent mixture of octadecene and dibenzyl ether in a 1:1 volume ratio was then added to the flask. The reaction mixture was stirred at 600 rpm and heated to 100 °C for 30 minutes under a nitrogen atmosphere to remove air and prevent subsequent oxidation of the NPs. The temperature was subsequently increased to 200 °C and maintained for 1 hour. Following this step, the mixture was further heated to 300 °C and held for an additional hour. After the reaction, the solution was allowed to cool naturally to room temperature. The synthesised NPs were collected, washed 2–3 times with a mixture of n-hexane/ethanol (1:1), precipitated by centrifugation, and then dispersed in n-hexane.
:1 molar ratio. 40 mL solvent mixture of octadecene and dibenzyl ether in a 1:1 volume ratio was then added to the flask. The reaction mixture was stirred at 600 rpm and heated to 100 °C for 30 minutes under a nitrogen atmosphere to remove air and prevent subsequent oxidation of the NPs. The temperature was subsequently increased to 200 °C and maintained for 1 hour. Following this step, the mixture was further heated to 300 °C and held for an additional hour. After the reaction, the solution was allowed to cool naturally to room temperature. The synthesised NPs were collected, washed 2–3 times with a mixture of n-hexane/ethanol (1:1), precipitated by centrifugation, and then dispersed in n-hexane.
Surface modification of Bi/Bi2O3 NPs with PAA
Bi/Bi2O3 NPs were surface modified with PAA through ligand exchange according to the following procedure: firstly, 10 mL of a Bi/Bi2O3 NPs solution was sonicated for 3–5 minutes to ensure uniform dispersion. Subsequently, 10 mL of a chloroform/DMF solution containing 0.01 M NOBF4 was added to the NP solution at room temperature. The mixture was sonicated until a precipitate formed, typically within 5 minutes. The NPs were purified by washing multiple times with a toluene/hexane mixture (1:1 volume ratio) followed by centrifugation at 12000 rpm for 8–10 minutes.
The purified NPs were then redispersed in 10 mL of an aqueous solution containing PAA. This mixture was continuously stirred at room temperature for 8 hours to facilitate surface modification. After the reaction, the product was washed thoroughly with distilled water until the solution reached a neutral pH (pH = 7). The resulting Bi/Bi2O3@PAA NPs are good dispersible in water, making them suitable for further applications.
Characterisation
Morphology of Bi/Bi2O3 NPs was studied by transmission electron microscopy (TEM, JEM1010-JEOL). The phase structure and composition of the NPs were determined by X-ray diffraction analysis. UV-vis (Jasco V-670) spectrometer was employed to record absorption spectra of the samples. The distribution of the elements within the Bi/Bi2O3 samples were determined by the Energy Dispersive X-ray spectroscopy (EDX) analysis on the Hitachi S-4800 FESEM equipped. X-ray photoelectron spectroscopy measurements (XPS, Thermo Fisher) were carried out using monochromatic Al Kα radiation (hν = 1486.6 eV). FT-IR spectroscopy (Nicolet 6700) and TGA (Nezsch, Germany) analyses were used to characterise the surface structure of Bi/Bi2O3 NPs. Dynamic light scattering (DLS) and zeta potential measurements were used to investigate the distribution and stability of Bi/Bi2O3 NPs in aqueous medium. ICP-MS was obtained on an Agilent Technologies, Japan.Cell viability measurement
The cytotoxicity of Bi/Bi2O3 NPs was evaluated using the methyl thiazolyl tetrazolium (MTT) assay with Vero cells (kidney cells from the African green monkey) as a model. First, Vero cells (1.5 × 105) were seeded in a 96-well plate and incubated at 37 °C with 5% CO2 for 24 hours in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. The medium was then replaced with fresh medium containing Bi/Bi2O3 NPs at concentrations of 12.5, 25, 50, 100, and 200 μg mL−1, followed by incubation for 72 hours. Cell viability was assessed by adding 20 μL of MTT solution to each well and incubating for 4 hours at 37 °C. The resulting formazan crystals were dissolved in dimethyl sulfoxide (DMSO, Sigma-Aldrich), and optical density was measured at 540 nm using a Tecan Spark microplate reader (Männedorf, Switzerland).CT phantom image measurements
The CT imaging was performed using 128-Somatom perspective CT scanner (Siemens, Germany) with the following imaging parameters: source voltage 80–130 kV, 80 mAs, slice thickness 0.75 mm, field of view DFOV x–y 290 × 320 mm2, matrix size 532 × 532. Samples, dispersed in water with different metal concentrations (agarose 2%, 2, 4, 6, 9 and 12 mM), were placed in 2 mL wells and positioned in a custom-made scanning support. X-ray attenuation intensity was assessed by loading digital CT images into a standard display program. For each sample, a uniform circular region of interest (ROI) with an area of approximately 0.3 cm2 was consistently selected at the center of the well to minimize variability from edge effects or positioning differences. Contrast enhancement was quantified in Hounsfield units (HU) for each sample at different metal concentrations. The measurements (ROIs in cm2) were obtained using eFilm workstations (Merge Healthcare, Chicago, IL, USA) and used for image reconstruction. Prior to scanning, quality control procedures were performed on the CT system to ensure accurate and reproducible measurements.Results and discussion
In this study, we developed a straightforward strategy to synthesise ultra-small Bi/Bi2O3 NPs through the thermal decomposition of the commercial precursor Bi(NO3)3 in the presence of oleic acid (OA) and oleylamine (OLA) as surfactants. Notably, OLA also served as a reducing agent, efficiently converting Bi3+ to metallic Bi. Meanwhile, the surface oxidation level of Bi NPs was controlled by adjusting the OA/OLA ratio during the synthesis process. The surface of the Bi/Bi2O3 NPs was subsequently modified with the polyacrylic acid (PAA) using a ligand exchange method. The synthesis and surface modification process of Bi/Bi2O3 NPs are illustrated in Scheme 1. This scheme also includes photographs of the NP solutions before and after ligand exchange, visually indicating their distinct physicochemical properties. The dark brown solution in the upper non-polar phase (n-hexane) represents the as-synthesized hydrophobic Bi/Bi2O3 NPs, stabilized by OA/OLA ligands. In contrast, the milky white solution in the lower aqueous phase corresponds to the PAA-coated NPs, suggesting successful ligand exchange and enhanced colloidal stability in water. This clear visual distinction provides qualitative evidence of successful surface modification and indicates the effectiveness of the strategy in improving NP dispersibility and chemical compatibility for potential biomedical applications.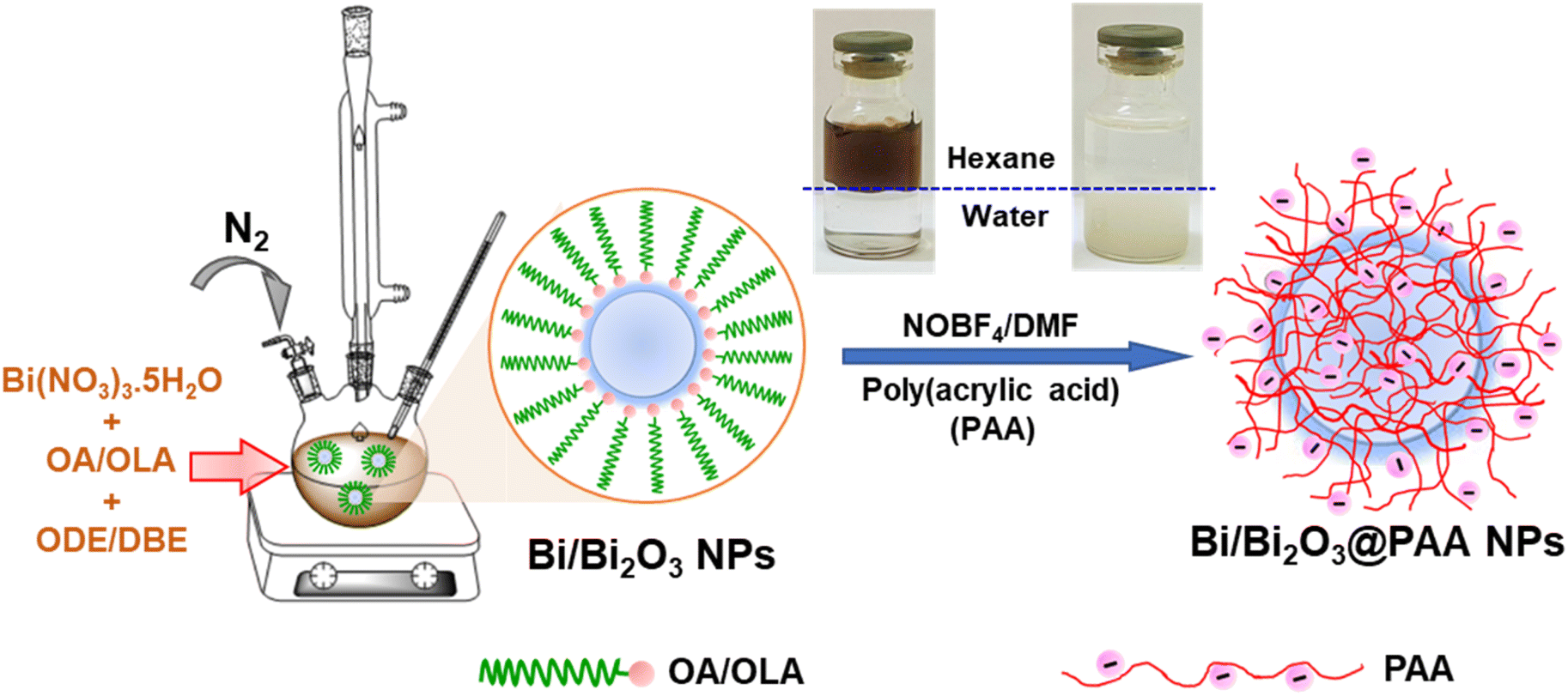 | ||
| Scheme 1 Schematic illustration of one-pot synthesis of Bi/Bi2O3 NPs as a high-performance CT contrast agent. | ||
Fig. 1a and b display TEM image and size distribution of as-synthesised Bi/Bi2O3 NPs. The TEM image revealed that the synthesised Bi/Bi2O3 NPs are monodisperse and spherical with a small size of ∼3 nm. Additionally, a high-resolution TEM image and a fast Fourier transform (FFT) analysis of a single nanoparticle (Fig. 1c) confirm the presence of a distinct lattice fringe measured at 2.25 Å, corresponding to the (110) diffraction planes of the rhombohedral structure of bismuth.
 | ||
| Fig. 1 (a) TEM image, (b) size distribution of Bi/Bi2O3 NPs, (c) HRTEM image of Bi/Bi2O3 NPs with an inset FFT image and (d) Rietveld refinement profile of XRD data of as prepared Bi/Bi2O3 NPs. The black line correspond to the experimental data and the red line is for the Rietveld refinement fit. The lower curve is the difference between the observed and calculated at each step. | ||
To further evaluate the phase composition and crystallinity of the Bi/Bi2O3 NPs, X-ray diffraction (XRD) analysis was performed (Fig. 1d). The diffraction peaks align well with standard patterns for rhombohedral metallic bismuth (PDF 01-085-1329) and tetragonal β-Bi2O3 (PDF 01-074-1374), confirming the formation of a Bi/Bi2O3 hybrid structure. The presence of Bi2O3 suggests partial surface oxidation of bismuth during synthesis, while the absence of impurity peaks indicating the high purity of the synthesised NPs. The sharp and well-defined diffraction peaks further demonstrate the crystalline nature of the sample, which is consistent with previously reported XRD data for ultra-small Bi NPs.21,33
The extent of oxidation of Bi NPs was found to be significantly influenced by the OA/OLA ratio, as evidenced by the XRD and TEM analyses (Fig. S1 and S2†). In OA-rich condition (OA/OLA = 3/1), the elevated concentration of OA promoted significant oxidation of bismuth, leading to the predominant formation of Bi2O3 NPs with an average size of 3.3 ± 0.5 nm. In contrast, in OLA-rich liquids (OA/OLA = 1/3), OLA served as the dominant reducing agent, effectively suppressing oxidation and promoting the formation of metallic Bi NPs with the size of 2.9 ± 0.4 nm. At an intermediate OA/OLA ratio of 2/1, a controlled oxidation process was achieved, leading to the formation of Bi/Bi2O3 hybrid NPs while maintaining a small and uniform particle size (3.0 ± 0.4 nm). These findings demonstrate the pivotal role of the OA/OLA ratio in tuning the oxidation state of the synthesised NPs.
To quantitatively determine the phase composition, Rietveld refinement was performed using the FullProf software (pseudo-Voigt function). The analysis revealed that the sample prepared at OA/OLA ratio of 2:1 consists of 75.1% metallic Bi and 24.9% Bi2O3, indicating that this ratio achieves a well-balanced composition between Bi and Bi2O3. This precise control over the oxidation state is essential for tuning the physicochemical properties of the NPs, thereby enhancing their potential in biomedical applications such as CT imaging.
To further characterise the synthesised Bi/Bi2O3 NPs, UV-vis spectroscopy was also conducted. The optical transmission spectra of the Bi/Bi2O3 NPs solution, recorded in wavelength range of 200 and 800 nm, exhibited a distinct absorbance peak at 273 nm, as shown in Fig. 2a. This peak is characteristic of Bi2O3 NPs and is consistent with previous reports,34 thereby confirming the presence of the Bi2O3 phase within the hybrid structure.
 | ||
| Fig. 2 (a) UV-vis absorption spectrum and (b) the optical band gap energy (Eg) of the Bi/Bi2O3 NPs. | ||
The optical band gap energy (Eg) of Bi/Bi2O3 NPs is calculated using the Tauc equation:35,36
| (αhν) = A(hν − Eg)n |
For Bi/Bi2O3 NPs, a value of n = 2 was used in the Tauc equation, indicating a direct allowed transition. By extrapolating the linear portion of the Tauc plot to the x-axis, the optical band gap energy was determined to be 3.88 eV (Fig. 2b). This value is good agreement with previously reported values for Bi2O3, which range from 2 to 3.96 eV.35–38 The observed increase in band gap energy compared to bulk material is likely due to the quantum confinement effect, which becomes significant as the particle size decreases to the nanoscale.35
EDX analysis (Fig. 3) confirms the presence of Bi, O, and C elements, where C originating from surfactant molecules on the NPs surface. EDX elemental mapping (Fig. 2c and d) shows a dominant Bi signal alongside a relatively weak O signal, supporting the formation of the Bi/Bi2O3 structure, consistent with the XRD results. Furthermore, the elemental mapping demonstrates the homogeneous distribution of Bi and O throughout the sample.
 | ||
| Fig. 3 (a) SEM image, (b) EDX spectrum and (c and d) elemental mapping of the Bi/Bi2O3 NPs. | ||
Further insights into the surface chemical composition of the Bi/Bi2O3 NPs were obtained via XPS analysis, as shown in Fig. 4. The wide-scan XPS survey spectrum (Fig. 4a) confirmed the presence of Bi, O and C elements on the NPs surface. The high resolution C 1s spectrum (Fig. 4b) was deconvoluted into three peaks at binding energies of 284.7 eV, 285.1 eV and 288 eV, corresponding to C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–O and O–CO bonds, respectively.
C, C–O and O–CO bonds, respectively.
 | ||
| Fig. 4 XPS spectra of Bi/Bi2O3 NPs: (a) scan survey; high-resolution XPS spectra of (b) C 1s, (c) Bi 4f and (d) O 1s. | ||
The Bi 4f spectrum (Fig. 4c) exhibits two distinct doublets. The peak at 158.6 eV and 163.9 eV are assigned to metallic Bi (Bi0), while the peaks at 159.1 eV and 164.4 eV are associated with Bi3+ species, originating from Bi2O3 phase. The coexistence of Bi0 and Bi3+ signals provide evidence of partial surface oxidation, consistent with previous reports.29,39,40 These results further suggest that Bi2O3 is effectively anchored to the surface shell of the Bi NPs, forming a core–shell Bi/Bi2O3 hybrid nanostructure.
In Fig. 4d, the high resolution O 1s spectrum was deconvoluted into two peaks located at 530.7 eV and 531.8 eV. These binding energies are attributed to the lattice oxygen in bismuth oxide and the surface-adsorbed oxygen species, respectively.41
The as-synthesised Bi/Bi2O3 NPs exhibited hydrophobicity due to the presence of OA and OLA on their surface. To enable biomedical application, a surface modification via ligand exchange was performed to render the NPs hydrophilic. The surface modification process was monitored using FT-IR spectroscopy, as shown in Fig. 5a. The FTIR spectrum of OA/OLA-coated Bi/Bi2O3 NPs displayed strong symmetric and asymmetric C–H stretching vibrations at 2850 cm−1 and 2920 cm−1, respectively, characteristic of the alkyl chain of OA and OLA. A broad absorption band at 3435 cm−1 corresponds to the vibration of O–H. Additional peaks observed at 1463 cm−1, 1542 cm−1 and 1635 cm−1, are attributed to the –CH2– bending, and the symmetric and asymmetric stretching vibration of the carboxylate (COO−) group, respectively, indicating the coordination of OA and OM molecules to the NPs surface, possibly as OA/OLA complexes.42–44
 | ||
| Fig. 5 (a) FTIR spectra and (b) thermogravimetric analysis (TGA) plots of Bi/Bi2O3 NPs before surface modification (Bi/Bi2O3@OA/OLA) and after modification with PAA. | ||
Upon treatment with NOBF4, the FTIR spectrum of BF4−-coated Bi/Bi2O3 NPs, reveals the disappearance of these characteristic bands, alongside the appearance of new peaks at 1084 cm−1 and 1643 cm−1, which are assigned to BF4− anions and CO stretching vibrations from DMF solvent molecules, respectively. These spectral changes confirm the successful removal of OA/OLA ligands and their replacement with BF4− anions.
Following PAA modification, the FTIR spectrum of PAA-coated Bi/Bi2O3 NPs exhibited a peak at 1712 cm−1, corresponding to the CO stretching vibration of free carboxylic acid groups (COOH). A strong band at 1553 cm−1 is attributed to the C–O stretching of carboxylate ions, indicating partial deprotonation of the PAA at neutral pH, where both protonated and deprotonated carboxyl groups coexist.45 Additionally, weak peaks observed at 550 cm−1 and 462 cm−1 correspond to Bi–O stretching vibrations, further confirming the presence of Bi2O3 in the NP structure.46,47
To further confirm the successful coating of PAA onto Bi/Bi2O3 NPs, thermogravimetric analysis (TGA) was performed. The TGA curves of the NPs before and after PAA modification are presented in Fig. 5b. For OA/OM-coated Bi/Bi2O3 NPs, a substantial weight loss is observed between 200 and 550 °C, which is attributed to the decomposition of OA and OM molecules. After PAA modification, the onset of decomposition shifts to a higher temperature (260 °C), suggesting stronger binding interactions of PAA molecules to the particle surface, consistent with the FTIR results.
The colloidal stability of Bi/Bi2O3@PAA NPs was evaluated using DLS analysis, and the results are presented in Fig. 6. As shown in Fig. 6a, the NPs exhibit a narrow hydrodynamic size distribution with an average hydrodynamic diameter of 36.8 nm, indicating uniform dispersion of Bi/Bi2O3@PAA NPs. The zeta potential measurement (Fig. 6b) reveals a surface charge of −38.8 mV, which is attributed to the presence of negative charged carboxyl groups from PAA coating. This strong electrostatic repulsion contributes to enhanced colloidal stability by effectively preventing NP aggregation.
 | ||
| Fig. 6 (a) Hydrodynamic diameter, (b) zeta potential, (c) DLS profiles of Bi/Bi2O3@PAA NPs at different pH values and (d) long-term colloidal stability of the NPs over time, with corresponding solution photographs (inset). | ||
Further investigation of size distribution under varying pH conditions reveals that the NPs remain stable across a wide pH range (2 to 12), while a highly acidic environment (pH = 1) induces a significant change in particle size, suggesting pH-dependent surface interactions (Fig. 6c). After two months of storage, both the hydrodynamic size and zeta potential remain nearly unchanged, confirming the long-term colloidal stability of the NPs (Fig. 6d).
The colloidal stability of the Bi/Bi2O3@PAA NPs is further supported by the visual appearance of their dispersions at different concentrations. At high concentration, the solution exhibits a milky white appearance. Notably, no visible aggregation or sedimentation was observed after two months of storage, indicating excellent long-term colloidal stability under ambient conditions. Upon dilution to a concentration of 5 mM, the solution becomes optically transparent, consistent with the expected behavior of ultrasmall NPs, as reported in the literature. These findings suggest the effectiveness of PAA surface modification in enhancing the colloidal stability of Bi/Bi2O3 NPs, supporting their potential for imaging diagnostic applications.
In vitro toxicity assessment
The biocompatibility of Bi/Bi2O3@PAA NPs was evaluated through MTT assay, measuring the viability of Vero cells after 72 hours of incubation with varying NP concentrations (Fig. 7). The results demonstrated that cell viability remained above 72% even at concentrations as high as 200 μg mL−1, indicating low cytotoxicity and suitable biocompatibility of the NPs for potential in vivo CT applications. | ||
| Fig. 7 Cellular cytotoxicity in Vero cells after 72 h incubation. | ||
Performance on CT imaging in vitro
The CT imaging capability of Bi/Bi2O3@PAA NPs was evaluated in vitro using CT phantom imaging. Solutions with varying Bi concentrations (2, 4, 6, 9 and 12 mM) were imaged under tube voltages ranging from 80 to 130 kV. As shown in Fig. 8, Bi/Bi2O3@PAA NPs exhibited progressively enhanced brightness with increasing Bi concentrations, indicating a concentration-dependent enhancement in X-ray attenuation. | ||
| Fig. 8 (a) In vitro CT images and (b) corresponding CT signals curves of Bi/Bi2O3@PAA with different concentration under different tube ranging from 80 to 130 kV. | ||
This effect arises from the increasing density of high atomic number atoms (Bi3+) in the medium, as CT contrast is directly correlated with the concentration of contrast-generating atoms within a given volume. Quantitative analysis revealed a linear relationship between the Hounsfield unit (HU) values and Bi concentration, with a calculated slope of 14.32 ± 1.14 HU mM−1 at 80 kV. This value exceeds that of several iodine-based clinical contrast agents and previously reported Bi-based agents, as summarized in Table 1.
| Contrast agent | Morphology, size (nm) | CT value (HU mM−1) | Tube voltage (kVp) | References |
|---|---|---|---|---|
| Bi/Bi 2 O 3 @PAA NPs | Spherical, 3.0 | 14.32 | 80 | This work |
| 14.04 | 110 | |||
| 13.42 | 130 | |||
| Bi2O3@PAA NPs | Spherical, 2.3 | 11.7 | 70 | 4 |
| Bi2O3@dextran NPs | Spherical, 3.4 | 6.945 | 80 | 22 |
| 5.837 | 120 | |||
| Bi2S3@polyvinyl pyrrolidone (PVP) | Nanosheet, (10–50) × (3–4) | 9.7 | 50 | 48 |
| Bi@poly(DL-lactic-co-glycolic acid) | Spherical, 38 | 6.6 | 80 | 49 |
| 5.3 | 120 | |||
| Bi@1,2-propanediol and glucose | Faceted, 74 | 5.9 | 80 | 50 |
| Bi@Oligosaccharide | Spherical, 22 | 8.5 | 80 | 51 |
| 6.4 | 120 | |||
| Bi@PVP nanodot | Spherical, 2.7 | 7.064 | 120 | 52 |
| Bi-SR-PEG NPs | Spherical, 40 | 6.1 | 120 | 53 |
| Bi-DTPA chelate | NA | 7.3 | 120 | 54 |
| Ultravist | NA | 4.4 | 70 | 4 |
| Iobitridol | NA | 3.850 | 120 | 52 |
| Iohexol | NA | 4.88 | 120 | 53 |
While Table 1 shows that several Bi-based agents with larger particle sizes exhibited lower CT numbers than those obtained in our study, the work by Tarighatnia et al.,28 which reported Bi NPs with an average size of 32.17 nm, demonstrated higher CT numbers under similar concentration conditions. In our study, the effect of particle size on CT contrast was not systematically investigated and thus was not a primary focus of the discussion. Nonetheless, the observed discrepancies suggest that nanoparticle size may influence CT attenuation.
To date, the literature has reported conflicting findings regarding the relationship between NP size and CT contrast. For instance, Xu et al.55 and Khademi et al.56 found that smaller Au NPs (<60 nm) exhibited higher X-ray attenuation than larger ones, possibly due to improved dispersion and higher surface-to-volume ratios. In contrast, Dou et al. observed that larger Au NPs produced greater contrast, possibly due to size-dependent variations in the spatial distribution of Bi atoms that influence the frequency of secondary atomic interactions.57 Meanwhile, Ross et al. reported size-independent attenuation for Au NPs up to 76 nm.58 Similarly, Dong et al. concluded that no statistically significant difference in CT contrast was observed across Au NPs of various sizes (4–152 nm) in phantom imaging using different CT systems, as the mass concentration of attenuating material remained constant regardless of particle size.59 These inconsistencies highlight that CT contrast is not determined solely by NP size, but rather by a combination of physicochemical factors. Parameters such as surface coating, hydration state, dispersion level, and colloidal stability can all influence effective electron density and, consequently, X-ray attenuation. For instance, smaller particles may form overly fine dispersions or acquire thick hydration layers, reducing their X-ray blocking efficiency.
To better elucidate the influence of Bi NP size on CT contrast, we plan to conduct controlled experiments in future studies that isolate particle size while keeping other variables constant.
Additionally, the effect of tube voltage on CT attenuation was evaluated across all tested Bi concentrations. The results showed a slight decrease in CT numbers with increasing tube voltage, which is a common trend attributed to the diminished contribution of the photoelectric effect at higher photon energies. Nevertheless, the CT attenuation values of Bi/Bi2O3@PAA NPs remained relatively stable across the voltage range, which can be attributed to the high K-edge energy of bismuth (90.5 keV). These findings suggest that Bi/Bi2O3@PAA NPs offer superior and voltage-independent CT imaging performance compared to conventional iodine-based contrast agent.
Our study demonstrates the potential of the synthesized ultrasmall Bi/Bi2O3 NPs as promising CT contrast agents, supported by favorable physicochemical characteristics and strong in vitro imaging performance. However, there are several limitations. For example, in vivo studies were not conducted, so biodistribution, clearance, and safety profiles remain unclear. Cellular uptake was not assessed, limiting insights into nanoparticle–cell interactions. Moreover, although the NPs showed good CT contrast, the effect of particle size on CT attenuation was not systematically explored. Future studies addressing these limitations are necessary to further validate and optimize their biomedical application.
Conclusions
In summary, we successfully synthesised ultrasmall Bi/Bi2O3 NPs using straightforward one-pot thermal decomposition method, yielding a core–shell structure. Surface modification with polyacrylic acid (PAA) further improved the aqueous dispersibility and biocompatibility of the NPs, enabling their suitability for biomedicine. The resulting Bi/Bi2O3@PAA NPs exhibited excellent X-ray attenuation performance, with Hounsfield unit (HU) values surpassing those of the conventional iodine-based contrast agents. Owing to their ultra-small size, high Bi content, superior stability, and low cytotoxicity, Bi/Bi2O3@PAA NPs present a promising alternative to traditional iodine-based CT contrast agents. Their facile synthesis and outstanding imaging properties highlight their potential for future clinical translation in diagnostic imaging.Data availability
The data supporting this article have been included in the main text and as part of the ESI.†Author contributions
Le T. T. Tam conceptualized the study, designed the methodology and drafted the manuscript. Duong T. Ngoc, Nguyen T. Yen and Nguyen T. Suong conducted the experiments and collected the associated data. Nguyen T. N. Linh carried out the toxicity tests and collected the associated data, while Le T. Tam and Nguyen V. Dong performed the CT measurements. Ngo T. Dung and Le T. Lu acquired funding and supervised the overall project. All authors reviewed and approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the Development program in the Field of Physics for the 2021–2025 period, through the Ministry of Science and Technology of Vietnam (Grant Number: ĐTĐL.CN.16/23). Le Thi Thanh Tam was funded by the Master, PhD Scholarship Programme of Vingroup Innovation Foundation (code: VINIF.2023.TS.104).References
- C. H. McCollough, S. A. Leng, L. F. Yu and J. G. Fletcher, Radiology, 2015, 276, 637–653 CrossRef PubMed.
- H. Bi, F. He, Y. Dong, D. Yang, Y. Dai, L. Xu, R. Lv, S. Gai, P. Yang and J. Lin, Chem. Mater., 2018, 30, 3301–3307 CrossRef CAS.
- H. Lusic and M. W. Grinstaff, Chem. Rev., 2013, 113, 1641–1666 CrossRef CAS PubMed.
- A. Ghazanfari, S. Marasini, X. Miao, J. A. Park, K. H. Jung, M. Y. Ahmad, H. Yue, S. L. Ho, S. Liu, Y. J. Jang, K. S. Chae, Y. Chang and G. H. Lee, Colloids Surf., A, 2019, 576(5), 73–81 CrossRef CAS.
- C. H. Hunt, R. P. Hartman and G. K. Hesley, Am. J. Roentgenol., 2009, 193, 1124–1127 CrossRef PubMed.
- K. Ai, Y. Liu, J. Liu, Q. Yuan, Y. He and L. Lu, Adv. Mater., 2011, 23, 4886–4891 CrossRef CAS PubMed.
- N. Gharehaghaji, B. Divband and F. Bakhtiari-Asl, BioNanoScience, 2020, 10, 909–916 Search PubMed.
- M. Yektamanesh, Y. Ayyami, M. Ghorbani, M. Dastgir, R. Malekzadeh and T. Mortezazadeh, Int. J. Pharm., 2024, 659, 124264 CrossRef CAS PubMed.
- C. Xu, Y. L. Wang, C. Y. Zhang, Y. W. Jia, Y. J. Luo and X. Y. Gao, Nanoscale, 2017, 9, 4620–4628 RSC.
- J. R. Ashton, K. D. Castle, Y. Qi, D. G. Kirsch, J. L. West and C. T. Badea, Theranostics, 2018, 8, 1782–1797 Search PubMed.
- Z. Li, J. Liu, Y. Hu, Z. Li, X. Fan, Y. Sun, F. Besenbacher, C. Chen and M. Yu, Biomaterials, 2018, 161, 279–291 CrossRef PubMed.
- R. Malekzadeh, M. Ghorbani, P. Faghani, B. B. Abdollahi, T. Mortezazadeh and B. Farhood, J. Radiat. Res. Appl. Sci., 2023, 16, 100490 CAS.
- B. M. Yeh, P. F. FitzGerald, P. M. Edic, J. W. Lambert, R. E. Colborn, M. E. Marino, P. M. Evans, J. C. Roberts, Z. J. Wang, M. J. Wong and P. J. Bonitatibus, Adv. Drug Delivery Rev., 2017, 113, 201–222 CrossRef CAS PubMed.
- D. M. Griffith, H. Y. Li, M. V. Werrett, P. C. Andrews and H. Z. Sun, Chem. Soc. Rev., 2021, 50, 12037–12069 Search PubMed.
- W. C. Huang, J. Zhu, M. K. Wang, L. P. Hu, Y. F. Tang, Y. Q. Shu, Z. J. Xie and H. Zhang, Adv. Funct. Mater., 2021, 31, 2007584 CrossRef CAS.
- H. Yu, H. Guo, Y. Wang, Y. Wang and L. Zhang, Wiley Interdiscip. Rev.:Nanomed. Nanobiotechnol., 2022, 14, e1801 CAS.
- W. Liao, P. Lei, J. Pan, C. Zhang, X. Suna, X. Zhang, C. Yu and S. K. Sun, Biomaterials, 2019, 203, 1 CrossRef CAS PubMed.
- J. Fu, J. Guo, A. Qin, X. Yu, Q. Zhang, X. Lei, Y. Huang, M. Chen, J. Li, Y. Zhang, J. Liu and Y. Dang, J. Nanobiotechnol., 2020, 18, 110 CrossRef CAS PubMed.
- G. Shu, L. Zhao, F. Li, Y. Jiang, X. Zhang, C. Yu, J. Pan and S. K. Sun, Biomaterials, 2024, 305, 122422 CrossRef CAS PubMed.
- G. D. Dai, Y. Zhang, X. M. Wang, X. Y. Wang, J. Jia, F. Jia, L. Yang and C. M. Yang, Front. Oncol., 2022, 12, 813955 CrossRef CAS PubMed.
- X. Yu, A. Li, C. Zhao, K. Yang, X. Chen and W. Li, ACS Nano, 2017, 11(4), 3990–4001 CrossRef CAS PubMed.
- G. Shu, C. Zhang, Y. Wen, J. Pan, X. Zhang and S. K. Sun, Biomaterials, 2024, 311, 122658 CrossRef CAS PubMed.
- J. M. Kinsella, R. E. Jimenez, P. P. Karmali, A. M. Rush, V. R. Kotamraju, N. C. Gianneschi, E. Ruoslahti, D. Stupack and M. J. Sailor, Angew. Chem., Int. Ed., 2011, 50, 12308–12311 CrossRef CAS PubMed.
- J. Chen, X. Q. Yang, Y. Z. Meng, M. Y. Qin, D. M. Yan, Y. Qian, G. Q. Xu, Y. Yu, Z. Y. Ma and Y. D. Zhao, Chem. Commun., 2013, 49, 11800–11802 RSC.
- Y. Wang, Y. Y. Wu, Y. J. Liu, J. Shen, L. Lv, L. B. Li, L. C. Yang, J. F. Zeng, Y. Y. Wang, L. S. W. Zhang, Z. Li, M. Y. Gao and Z. F. Chai, Adv. Funct. Mater., 2016, 26, 5335–5344 CrossRef CAS.
- Y. Lu, L. H. Li, Z. F. Lin, M. Li, X. M. Hu, Y. Zhang, M. Y. Peng, H. Xia and G. Han, Adv. Healthcare Mater., 2018, 7, 1800602 CrossRef PubMed.
- C. Yang, C. Guo, W. Guo, X. Zhao, S. Liu and X. Han, ACS Appl. Nano Mater., 2018, 1, 820–830 CrossRef CAS.
- A. Tarighatnia, M. H. Abdkarimi, N. D. Nader, T. Mehdipour, M. R. Fouladi, A. Aghanejad and H. Ghadiri, New J. Chem., 2021, 45, 18871 RSC.
- A. Tarighatnia, M. R. Fouladi, M. R. Tohidkia, G. Johal, N. D. Nader, A. Aghanejad and H. Ghadiri, J. Drug Delivery Sci. Technol., 2021, 66, 102895 CrossRef CAS.
- M. A. Shahbazi, L. Faghfouri, M. P. A. Ferreira, P. Figueiredo, H. Maleki, F. Sefat, J. Hirvonen and H. A. Santos, Chem. Soc. Rev., 2020, 49, 1253–1321 RSC.
- C. Gomez, G. Hallot and S. Laurent, Pharmaceutics, 2021, 13(11), 1793 CrossRef CAS PubMed.
- L. Dong, P. Zhang, X. Liu, R. Deng, K. Du, J. Feng and H. Zhang, ACS Appl. Mater. Interfaces, 2019, 11(8), 7774–7781 CrossRef CAS PubMed.
- L. Jiao, Q. X. Li, J. J. Deng, N. Okosi, J. F. Xia and M. Su, Nanoscale, 2018, 10, 6751–6757 RSC.
- M. Mohammadi, A. Tavajjohi, A. Ziashahabi, N. Pournoori, S. Muhammadnejad, H. Delavari and R. Poursalehi, Nano-Micro Lett., 2019, 14, 239–244 CrossRef CAS.
- C. Li, K. P. O'Halloran, H. Ma and S. Shi, J. Phys. Chem. B, 2009, 113, 8043–8048 CrossRef CAS PubMed.
- X. Liu, H. Cao and J. Yin, Nano Res., 2011, 4(5), 470–482 CrossRef CAS.
- S. Dadashi, R. Poursalehi and H. Delavari, Mater. Res. Bull., 2018, 97, 421–427 CrossRef CAS.
- S. S. Shaker, R. A. Ismail and D. S. Ahmed, J. Inorg. Organomet. Polym. Mater., 2022, 32, 1381–1388 CrossRef CAS.
- Z. Feng, Y. Fu, Z. Yang, Y. He, C. Feng, B. Gao, P. Zhang, X. An, A. Abudula and G. Guan, J. Colloid Interface Sci., 2025, 678, 913–923 Search PubMed.
- Z. Yang, X. An, Z. Feng, Y. Ramli, C. Feng, P. Wang, J. Wang, S. Li, X. Hao, H. Lu, A. Abudula and G. Guan, Sep. Purif. Technol., 2025, 354, 129205 CrossRef CAS.
- S. Singh, R. K. Sahoo, N. M. Shinde, J. M. Yun, R. S. Mane, W. Chung and K. H. Kim, RSC Adv., 2019, 9, 32154–32164 RSC.
- L. J. Treadwell, T. J. Boyle, N. S. Bell, M. A. Rodriguez, B. R. Muntifering and K. Hattar, J. Mater. Sci., 2017, 52, 8268–8279 CrossRef CAS.
- M. T. Nguyen, K. Wongrujipairoj, H. Tsukamoto, S. Kheawhom, S. Mei, V. Aupama and T. Yonezawa, ACS Sustainable Chem. Eng., 2020, 8, 18167–18176 CrossRef CAS.
- N. T. T. Khue, L. T. T. Tam, N. T. Dung, L. T. Tam, N. X. Chung, N. T. N. Linh, N. D. Vinh, B. M. Quy and L. T. Lu, ChemistrySelect, 2022, 7(34), e202202062 CrossRef.
- T. Placido, L. Tognaccini, B. D. Howes, A. Montrone, V. Laquintana, R. Comparelli, M. L. Curri, G. Smulevich and A. Agostiano, ACS Omega, 2018, 3, 4959–4967 CrossRef CAS PubMed.
- M. Sarani, M. Roostaee, M. Adeli-Sardou, D. Kalantar-Neyestanaki, S. A. A. Mousavi, A. Amanizadeh, M. Barani and A. Amirbeigi, J. Trace Elem. Med. Biol., 2024, 81, 127325 Search PubMed.
- M. Prakash, H. P. Kavitha, S. Arulmurugan, J. P. Vennila, S. Abinaya, D. Lohita and R. Suresh, J. Sol-Gel Sci. Technol., 2024, 110, 807–818 CrossRef CAS.
- O. Rabin, J. M. Perez, J. Grimm, G. Wojtkiewicz and R. Weissleder, Nat. Mater., 2006, 5, 118–122 CrossRef CAS PubMed.
- E. R. Swy, A. S. Schwartz-Duval, D. D. Shuboni, M. T. Latourette, C. L. Mallet, M. Parys, D. P. Cormode and E. M. Shapiro, Nanoscale, 2014, 6, 13104–13112 RSC.
- A. L. Brown, P. C. Naha, V. Benavides-montes, H. I. Litt, A. M. Goforth and D. P. Cormode, Chem. Mater., 2014, 26, 2266–2274 CrossRef CAS PubMed.
- B. Wei, X. Zhang, C. Zhang, Y. Jiang, Y. Y. Fu, C. Yu, S. K. Sun and X. P. Yan, ACS Appl. Mater. Interfaces, 2016, 8, 12720–12726 CrossRef CAS PubMed.
- P. Lei, R. An, P. Zhang, S. Yao, S. Song, L. Dong, X. Xu, K. Du, J. Feng and H. Zhang, Adv. Funct. Mater., 2017, 1702018 CrossRef.
- N. Yu, Z. Wang, J. Zhang, Z. Liu, B. Zhu, J. Yu, M. Zhu, C. Peng and Z. Chen, Biomaterials, 2018, 161, 279–291 CrossRef CAS PubMed.
- J. Fu, J. Guo, A. Qin, X. Yu, Q. Zhang, X. Lei, Y. Huang, M. Chen, J. Li, Y. Zhang, J. Liu, Y. Dang, D. Wu, X. Zhao, Z. Lin, Y. Lin, S. Li and L. Zhang, J. Nanobiotechnol., 2020, 18, 110 CrossRef CAS PubMed.
- C. Xu, G. A. Tung and S. Sun, Chem. Mater., 2008, 20, 4167–4169 CrossRef CAS PubMed.
- S. Khademi, S. Sarkar, S. Kharrazi, S. M. Amini, A. Shakeri-Zadeh, M. R. Ay and H. Ghadiri, Phys. Med., 2018, 45, 127–133 CrossRef PubMed.
- Y. Dou, Y. Guo, X. Li, X. Li, S. Wang, L. Wang, G. Lv, X. Zhang, H. Wang, X. Gong and J. Chang, ACS Nano, 2016, 10, 2536–2548 CrossRef CAS PubMed.
- R. D. Ross, L. E. Cole, J. M. R. Tilley and R. K. Roeder, Chem. Mater., 2014, 26, 1187–1194 CrossRef CAS.
- Y. C. Dong, M. Hajfathalian, P. S. N. Maidment, J. C. Hsu, P. C. Naha, S. Si-Mohamed, M. Breuilly, J. Kim, P. Chhour, P. Douek, H. I. Litt and D. P. Cormode, Sci. Rep., 2019, 9, 14912 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5na00389j |
| This journal is © The Royal Society of Chemistry 2025 |