
Peptide-based nanomaterials and their diverse applications
Tarak Nath
Das†
a,
Aparna
Ramesh†
bc,
Arghya
Ghosh†
a,
Sourav
Moyra†
bc,
Tapas Kumar
Maji
 *ad and
Goutam
Ghosh
*bc
*ad and
Goutam
Ghosh
*bc
aMolecular Materials Laboratory, New Chemistry Unit (NCU), Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR), Jakkur, Bangalore 560064, India. E-mail: tmaji@jncasr.ac.in
bCentre for Nano and Soft Matter Sciences (CeNS), Shivanapura, Dasanapura Hobli, Bengaluru, 562162, India. E-mail: gghosh@cens.res.in
cAcademy of Scientific and Innovation Research (AcSIR), Ghaziabad, 201002, India
dMolecular Materials Laboratory, Chemistry and Physics of Materials Unit (CPMU), International Centre for Materials Science (ICMS), School of Advanced Materials (SAMat), Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR), Jakkur, Bangalore 560064, India
First published on 19th November 2024
Abstract
The supramolecular self-assembly of peptides offers a promising avenue for both materials science and biological applications. Peptides have garnered significant attention in molecular self-assembly, forming diverse nanostructures with α-helix, β-sheet, and random coil conformations. These self-assembly processes are primarily driven by the amphiphilic nature of peptides and stabilized by non-covalent interactions, leading to complex nanoarchitectures responsive to environmental stimuli. While extensively studied in biomedical applications, including drug delivery and tissue engineering, their potential applications in the fields of piezoresponsive materials, conducting materials, catalysis and energy harvesting remain underexplored. This review comprehensively elucidates the diverse material characteristics and applications of self-assembled peptides. We discuss the multi-stimuli-responsiveness of peptide self-assemblies and their roles as energy harvesters, catalysts, liquid crystalline materials, glass materials and contributors to electrical conductivity. Additionally, we address the challenges and present future perspectives associated with peptide nanomaterials. This review aims to provide insights into the versatile applications of peptide self-assemblies while concisely summarizing their well-established biomedical roles that have previously been extensively reviewed by various research groups, including our group.
 Tarak Nath Das | Tarak Nath Das obtained his BSc in Chemistry from Ramakrishna Mission Vivekananda Centenary College, Rahara, Kolkata, in 2019. He completed his MS in Chemical Sciences from the New Chemistry Unit, Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR), Bangalore, in 2022. He is currently pursuing his PhD under the guidance of Prof. Tapas Kumar Maji at JNCASR, Bangalore, India. His research work mainly focuses on multifarious properties and applications of hybrid gel materials. |
 Aparna Ramesh | Aparna Ramesh obtained her BSc in Chemistry from Govt Victoria College Palakkad, University of Calicut, Kerala, India, in 2020. She completed her MSc in Chemistry from the Central University of Punjab, India, in 2022. She is currently pursuing her PhD under the guidance of Dr Goutam Ghosh at the Centre for Nano and Soft Matter Sciences (CeNS), Bangalore, India. Her research work mainly focuses on multicomponent peptide self-assembly and their numerous applications. |
 Arghya Ghosh | Arghya Ghosh obtained his BSc in Chemistry from Ramakrishna Mission Vidyamandira, West Bengal, India, in 2020. He completed his MS in Chemical Sciences from the New Chemistry Unit, Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR), Bangalore, in 2023. He is currently pursuing his PhD under the guidance of Prof. Tapas Kumar Maji at JNCASR, Bangalore, India. His research work mainly focuses on soft organic inorganic hybrid materials for energy storage and conversion applications. |
 Sourav Moyra | Sourav Moyra obtained his BSc in Chemistry from Ramakrishna Mission Vivekananda Centenary College, Rahara, Kolkata, India, in 2019. He completed his MSc in Chemistry from the University of Hyderabad, India in 2021. He is currently pursuing his PhD under the guidance of Dr Goutam Ghosh at the Centre for Nano and Soft Matter Sciences (CeNS), Bangalore, India. His research work mainly focuses on controlling the self-assembly behavior of aromatic peptides and their material properties. |
 Tapas Kumar Maji | Tapas Kumar Maji obtained his PhD in 2002 from the Indian Association for the Cultivation of Science (IACS), Kolkata. After a postdoctoral stint at Kyoto University, Japan, he joined Jadavpur University as a lecturer. Then, he moved to the Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR), Bangalore, where he is currently a professor in the Chemistry and Physics of Materials Unit. His research focuses on the design and synthesis of bulk and nanoscale metal–organic frameworks (MOFs), organic porous polymers and metal–organic hybrid ‘soft’ materials for energy-storage and conversion, photo-physical analyses and photocatalysis. Prof. Maji has published 257 peer-reviewed publications. |
 Goutam Ghosh | Goutam Ghosh received his PhD in 2014 from the University of Calcutta, Kolkata, India. After completing his PhD, he joined the Indian Association for the Cultivation of Science (IACS), Kolkata, as a postdoctoral researcher, where he worked on controlled supramolecular polymerization of π-systems and peptides. Later, he moved to Westfälische Wilhelms Universität (WWU) Münster, Germany, as a postdoctoral research fellow. Presently, he is an Assistant Professor (Ramanujan Faculty) at the Centre for Nano and Soft Matter Sciences (CeNS), Bangalore, India. His current research interest focuses on controlled supramolecular polymerization of peptides, amphiphilic molecules and polymers and their numerous applications in fields ranging from materials to biomedicine. |
1. Introduction
Self-assembly is a ubiquitous process in nature, involving the formation of ordered arrangements of small molecules without the guidance of any external influence.1–3 Molecular self-assemblies are primarily held together by non-covalent interactions, including H-bonding, π-interaction, van der Waals interactions, solvophobic effects, and halogen bonding.4 The combined effect of these non-covalent forces contributes to both the structural and chemical stability of the system. In recent years, adaptive molecular self-assemblies have attracted attention, with a focus on their reversibility and stimuli-responsiveness.5–10Given the rapid advancement of organic nanotechnology, the need to fine-tune physical and chemical properties has become essential. In this context, bottom-up stepwise self-assembly approaches have proven to be superior in manipulating nanoarchitectures.11–14 The self-assembly process guides several polypeptide chains to pack in a unique manner, forming higher-order nanostructures.15 Apart from the classical single-step self-assembly approach, recent reports indicate a non-classical multistep self-assembly approach of peptides through the liquid–liquid phase separation method.16 Improvements in understanding the thermodynamics and kinetics of the self-assembly process, along with complex phase structures, have led to the development of various advanced materials. Proteins and peptides emerge as captivating choices for the self-assembly process among other building blocks, primarily owing to their biological compatibility. Particularly, the self-assembly of peptides has consistently been a focal point of intensive research, showcasing significant potential applications in various fields such as biomedicine, bio-imaging, energy harvesting, catalysis, sensors, and actuators with simple and cost-effective synthesis methods.17,18 Peptides exhibit a variety of secondary structures, which further evolve into various nanostructures, such as nanofibers, nanotubes, nanotapes, 2D nanosheets, micelles, and vesicles.19–23 Naturally occurring peptides, such as amyloids, peptide hormones, collagen peptides, elastin-derived peptides, and fibrin, are essential for their specific functions in biological processes owing to their unique secondary and tertiary arrangements. Hence, achieving hierarchical packing arrangements is crucial for unlocking the full potential of supramolecular higher-order architectures across various application fields.24 However, designing and fabricating sophisticated functional systems with precise long-range molecular alignment under spatiotemporal control remain significant challenges. Hence, adjusting the self-assembly process is essential to manipulate various secondary and nanostructures of peptides, serving as a prerequisite for understanding and harnessing their material and biological functions.14,25–29 The swift progress in controlled supramolecular polymerization has empowered the control of spatial and temporal organization within nanostructures.30 Currently, the integrated approach to modulate peptide self-assemblies using diverse supramolecular techniques is of paramount importance, unlocking its potential for practical applications in everyday life. Furthermore, the stimuli-responsiveness of peptides endows them with the ability to adjust their structure under different experimental conditions, facilitating the creation of adaptable nanoarchitectures with diverse dimensions.31 Studies indicate that the biological responses associated with peptide self-assemblies are influenced, either directly or indirectly, by factors such as pH, temperature, concentration, ionic strength, redox conditions, and exposure to light.32–39 Conversely, there has been a notable lack of focus on modifying the material properties of the self-assembled peptide nanostructure via the adjustment of experimental parameters and exposure to various stimuli. The diverse array of amino acids, which vary in charge, size, hydrophobicity, and polarity, governs the physicochemical characteristics of peptides and provides a versatile foundation for the precise design and control of nanomaterials.40
Peptide self-assemblies are extensively documented in the field of biomedical applications because of their notable biocompatibility and biodegradability.41 Nevertheless, this category of systems holds significant potential in various fields of materials sciences. In particular, chromopeptides are well known for their crucial role in biomimetic catalysis, artificial photosynthesis, bio-imaging and diagnosis.42 In this review, we present an overview of the various applications of peptides such as piezoresponse, catalytic applications, biomedical applications, conductivity and various distinct states of peptide self-assemblies. Certain peptides exhibit piezoresponse, making them valuable for sensors and energy devices. Additionally, these systems can act as catalysts in various processes. Their ability to form liquid crystals also enables the development of advanced materials for optoelectronics and display technologies. A significant area of research on peptide assemblies is in biomedicine, where they are being explored as therapeutic agents, for targeted drug delivery and diagnostic imaging. Some peptides also demonstrate electrical conductivity, which could be applied in flexible electronics. Due to their structural versatility, peptides are beneficial across numerous fields, from medical science to materials engineering, and are expected to gain even greater significance as research uncovers new facets of their functionality. This will pave the way for novel therapeutic approaches, biotechnological uses, and advancements across a range of scientific domains. Along with the versatile properties and potential applications of peptide-based self-assembled systems, two distinct self-assembled states such as liquid crystals and peptide glasses, an emerging class of materials have been discussed comprehensively. Peptide glass is a new class of system which mimics the glassy disordered state of material with high biological significance.43 The diverse applications of these systems have not been extensively explored or collectively reviewed, as most research studies have focused on the biomedical applications of self-assembled peptides, which have already been thoroughly reviewed by multiple research groups, including ours.28,29,31,44–47
2. Peptide self-assemblies
Peptides, consisting of amino acids sequentially connected by covalent bonds, undergo various intermolecular non-covalent interactions to form self-assembled nanostructures. Moreover, peptides are well known for folding locally into specific patterns within a single peptide chain, primarily through hydrogen bonding (H-bonding), which gives rise to their secondary structure, whereas the tertiary structure of peptides refers to the overall 3D conformation of the peptide chain, which are stabilized via various non-covalent interactions. Peptide self-assembly involves multiple small peptide units coming together through non-covalent forces to form a larger, well-ordered nanostructure. These self-assembled arrangements exhibit various shapes, sizes, and surface areas attributed to different morphologies. The distinct physical, chemical, and biological characteristics of self-assembled peptides are predominantly influenced by the diverse conformations found in both secondary and tertiary structures.48 Small synthetic peptides self-assemble into various nanostructures following the classical nucleation growth mechanism, which involves a single-step nucleus formation process from a supersaturated peptide medium.16 However, this classical principle fails to explain the liquid-to-solid phase transition process.16,49 A thorough theoretical and experimental investigation indicates a non-classical model, where a meta-stable phase-mediated two-step nucleation process is involved prior to the peptide self-assembly. Recent reports have suggested a process called liquid–liquid phase separation (LLPS), where a peptide solution separates into a solvent-rich liquid phase and a solute-rich liquid droplet at the initial time frame. These liquid intermediates serve as precursors for nucleation, followed by the formation of nanostructures with the achievement of a thermodynamically stable state.50 These liquid droplets were beneficial not only for the nucleation event but also served as targets for adjusting the self-assembly pathway.51 Hence, to maintain control over the entire assembly process, it is essential to carefully balance attractive and repulsive forces. The highly flexible nature of peptides and their capability to form specific secondary structures present a novel framework for designing nanomaterials with adjustable structural characteristics. In order to create supramolecular structures, various peptide-based building blocks including cyclic peptides, amphiphilic peptides, co-polypeptides, surfactant-like oligopeptides, dendritic peptides, and aromatic dipeptides have been developed.2.1. Noncovalent interactions
The complete self-assembly evolution process involves various types of non-covalent interactions such as H-bonding, π-interactions, hydrophobic effects, van der Waals forces, and electrostatic interactions. These weak interactions have a significant impact on the self-assembly process of peptides, resulting in the formation of diverse nanostructures (Scheme 1). In most cases, the peptide self-assembly is driven by a combination of various non-covalent interactions.52 Cooperativity in these systems primarily stems from: (i) many-body interactions, (ii) secondary interactions, (iii) chelate effects, and (iv) conformational changes.53 Among these, many-body interactions play a major role in cooperativity, contributing either positive or negative feedback to the overall assembly process. Cooperativity via multivalent interactions has been suggested as a key physiological mechanism for regulating cellular functions.54,55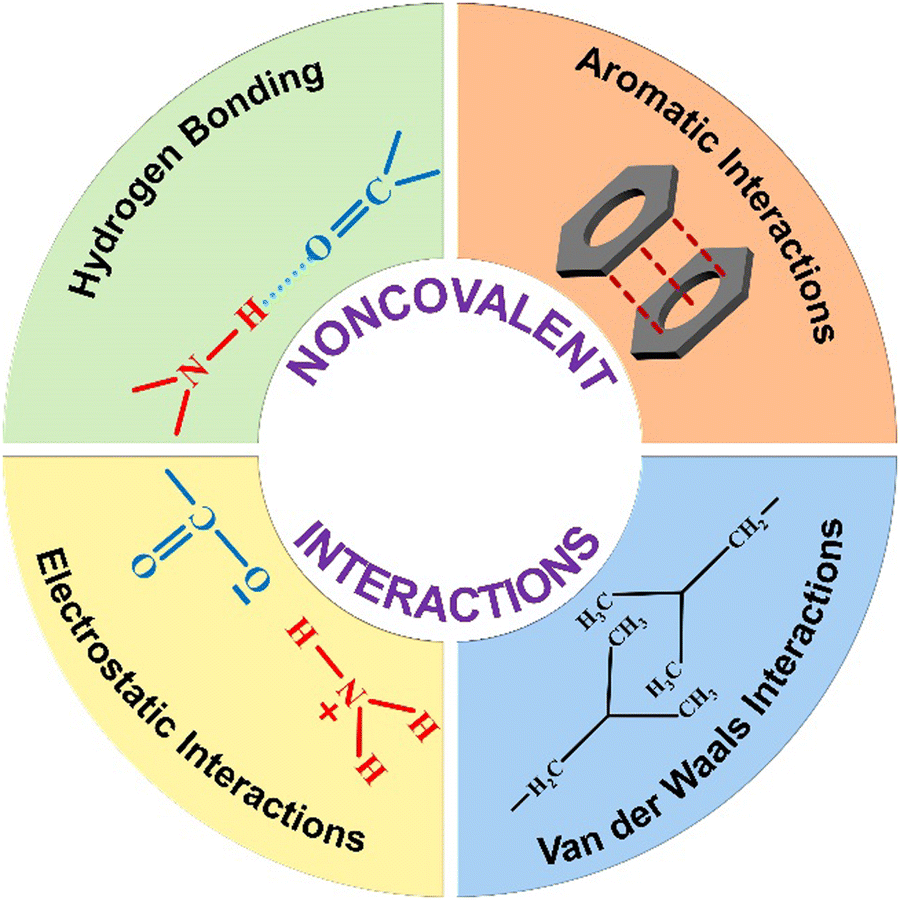 | ||
| Scheme 1 Schematic showing various non-covalent interactions present in the peptide self-assembly. | ||
H-bonding is recognized as the predominant force among all non-covalent interactions in the self-assembly of peptides. The essential role of H-bonding also comes into play during the stabilization of secondary structures of peptides or protein folding. The abundance of amide bonds and other functional groups such as amine and carboxyl in the peptide system makes peptides well-suited for directional H-bonding. As a result, a diverse range of nanostructures emerge, each possessing unique features. It was believed that water-assisted H-bonding serves as a driving force for the formation of protein fibrils (amyloid), considered as a significant factor in Alzheimer's disease and other neurodegenerative disorders.56–58 Topological control over the nanostructure and their growth mechanism highly depends on the H-bonding interactions. The formation of a 1D nanotubular structure of a cyclic peptide59–61 and its transformation into a 2D nanosheet was investigated by Montenegro and colleagues.62 The diverse nanostructures observed in the well-studied peptide diphenylalanine (FF) can be attributed to the precise adjustment of H-bonding interactions under different conditions.63 The H-bonding within the self-assembled nanostructure was found to play a crucial role in biomedical applications, catalytic processes, and the overall structural stability of the system.64–66
The self-assembly of peptides linked with an aromatic π-core is significantly driven by the strong force of π-stacking interaction. Peptide chains adorned with amino acids related to the π-system, such as phenylalanine, tyrosine, and tryptophan, or externally introduced with chromophores such as pyrene, anthracene, naphthalene diimide, and perylene diimide exhibit exceptional regulation over nanostructures due to interactions involving π–π stacking.67–72 We have illustrated how adjusting the effective area of a π-core for stacking can influence the self-assembly process of peptides.47,73 The overall self-assembly mechanism of a dipeptide (FF) is guided by the presence of a pyrene or naphthalene motif at the N-terminal, and this effect has been observed in the alteration of their secondary structure (Fig. 1). The larger π-surface of pyrene facilitates strong interactions, resulting in the arrangement of peptides in a parallel fashion. In contrast, the weaker interactions between naphthalene units lead to an antiparallel arrangement of peptides during aggregation. The impact of π–π interactions also modifies the mechanistic pathway of the self-assembly process, shifting from an isodesmic mechanism in the case of naphthalene-FF to a cooperative mechanism in the case of pyrene-FF. Pyrene-FF exhibited a secondary structure rich in β-sheets, while no distinctive secondary structure was observed in the case of naphthalene-FF. This distinction is primarily attributed to the influence of different π-stacking interactions. These π surfaces act as a hydrophobic interface in water-based assemblies, in addition to aiding in the assembly process via π-stacking interactions. In the case of pyrene-FF, a probable parallel packing was expected due to their strong π–π interactions, whereas a weak π-interaction of the naphthalene core lead to a change in the complete supramolecular polymerization pathway (Fig. 1). The directional expansion of the assembly is also supported by these strong π–π stacking interactions.73,74 The significance of π–π interactions in π-chromophore-conjugated peptide self-assemblies and their influence on tuning the assembly pathway have been recently documented by Ulrich and colleagues.69 Not only the π-stacking, but also the π-interaction involves cation–π, and CH–π interactions. These weak forces often play a crucial role in the overall structural stabilization of protein and ligand binding.75,76
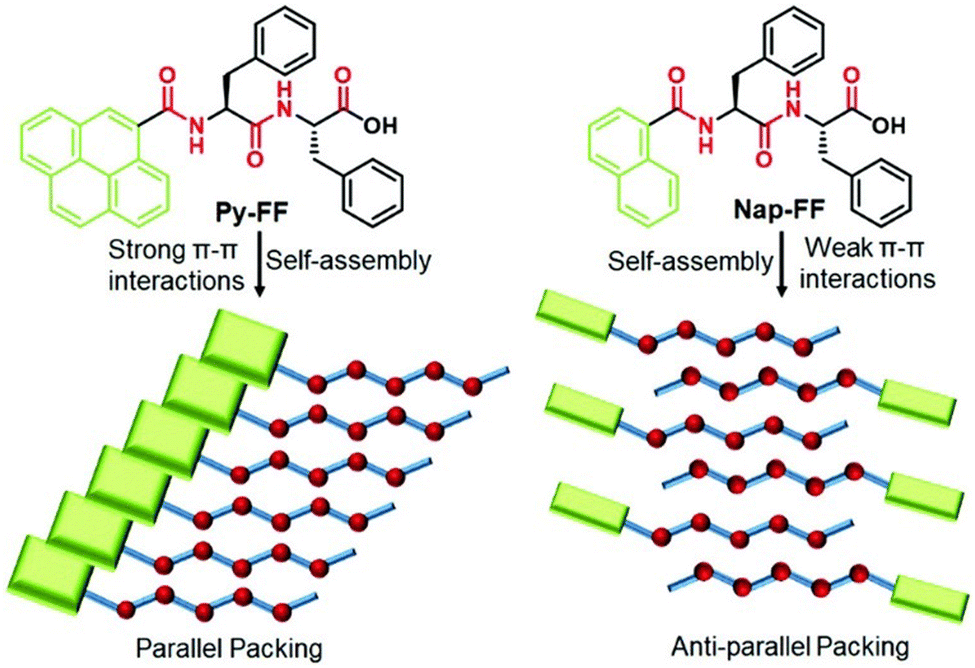 | ||
| Fig. 1 Chemical structure of two different peptides with the same amino acid sequence and different π-conjugated chromophoric moieties, and the schematic of the packing of the peptides during the self-assembly process. The peptide with a stronger π-interaction with larger π-surface (pyrene) directs a cooperative mechanism and the peptide with a weaker π-interaction (anthracene) leads to an isodesmic pathway. Adapted with permission from ref. 47. Copyright 2021 Royal Society of Chemistry. | ||
The peptide self-assembly frequently entails electrostatic interactions, which are considerably more robust than the typical H-bonding or π–π interactions. These coulombic forces arise from either ion pairs or the zwitterionic nature of the functional groups. By altering the system's environmental parameters such as pH and ionic strength, the interactions can be fine-tuned. This non-covalent interaction is well known to drive the self-assembly process in a layer-by-layer fashion.77 The importance of such kind of interaction towards the self-assembly process of charged peptides is well documented.78,79 Another class of non-covalent forces that contribute in stabilizing the peptide self-assembly are van der Waals interactions. The primary driving force behind the assembly in the amphiphilic system is the hydrophobic contact between the alkyl chain and the π-surface. Apart from π-interactions, aromatic motifs also contribute to the peptide assembly via the hydrophobic effect. The hydrophobic effect with aromatic groups is typically non-directional in nature, differing from the more organized nature of π–π interactions. Assemblies predominantly driven by these forces are often micellar in structure, minimizing surface contact with water. The amphiphilic system organizes itself in a manner where the hydrophobic functional chain is concealed within the structure, while the hydrophilic group is positioned externally, contributing to the stabilization of the entire system. Gçrbitz has reviewed the role of the hydrophobic effect of the dipeptide system on the architecture of the supramolecular self-assembled system. These hydrophobic dipeptide building units can construct microporous super structures for various applications in different fields such as storage and selective adsorption of gas.80 These types of interactions are also recognized for their role in securing the assembly against proteolytic susceptibility and enzymatic degradation.81 The role of a co-solvent with water is known to significantly impact the nanostructure and properties of the system.82,83 The solvent gradient in a mixed solvent system of water and acetone is known to induce the self-assembly of a peptide into a hollow nanocapsule.84 In another study, the impact of a second solvent alongside water was investigated on nanostructural transitions. Varying the amount of ethanol in water led to the peptide adopting different morphologies. At lower ethanol concentrations, the peptide formed short helical ribbons, while at higher percentages, it developed into longer twisted ribbons. These small-chain alcohol molecules are known to insert into the hydrophobic pockets of the assembly, altering the nanostructure's curvature by weakening the interactions.85–87 Apart from the aforementioned interactions, another significant non-covalent force that plays a crucial role in the peptide assembly is the van der Waals interaction. This force emerges from the partial and asymmetric distribution of electron density or dipole between two closely spaced molecules. Despite the weaker nature (∼5 kJ mol−1) of this force, it makes a substantial and widespread contribution to peptide self-assemblies.56 Yet, in very few instances, van der Waals interactions emerge as the dominant mechanism governing the self-assembly.88 The alkyl chain present in the peptide sequence mainly takes part in van der Waals interaction. The position and the number of alkyl chains control the overall stability of the assembled system.89 With the aid of molecular dynamics simulations, Schatz and colleagues elucidated how these interactions stabilize cylindrical fibers within the peptide assembly.90
2.2. Secondary and nano-structures
The unique folds in the structure of a peptide, giving rise to its secondary structure, stem primarily from the intramolecular H-bonding interactions among the integrated amino acid residues (Scheme 2).20 Furthermore, the hydrophobic effect drives the system toward its thermodynamic state, as hydrophobic functional groups in an aqueous solution self-organize toward the interior of the assembly, releasing free water molecules.91 The α-helix and β-sheet are the most abundant secondary structures found under the physiological condition. The nonordered state is referred to as random coil or irregular structure. Understanding this secondary structure of peptide assembly is important to realize its potential in various fields.92,93 NMR spectroscopy, X-ray crystallography, FTIR spectroscopy and circular dichroism are the most frequent techniques to analyse the secondary structure of the peptide.94 Minor structural alterations in a peptide system can induce the transformation in secondary structures, shifting from an α-helix to a β-sheet, and consequently, altering the nanostructure from a helical ribbon to a core–shell worm-like structure.74,95 Variations in secondary structures under diverse environmental conditions can improve the cell permeability, making peptides more effective for transporting genetic materials.96 Peptide secondary structures can also be maintained under co-assembly conditions by altering the system's overall stoichiometry.97 Guest-induced secondary structural regulation of a dipeptide leads to a change in photophysical properties. It has also been reported that the co-assembly of Zn-phthalocyanine and Fmoc-FF can tune the short-range secondary structure in helical supramolecular nanotubes, resulting in circularly polarized luminescence (CPL).98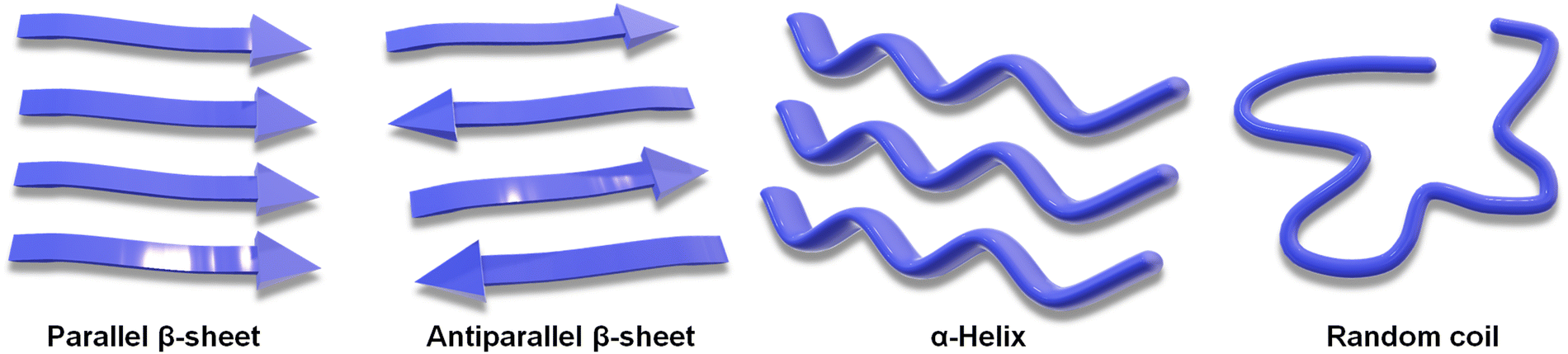 | ||
| Scheme 2 Schematic showing various secondary structures of a peptide. | ||
Non-covalent interactions not only stabilize the secondary structure of peptides but also facilitate intermolecular interactions, allowing peptides to self-assemble into various nanostructures. Common self-assembled nanostructures include vesicles, micelles, nanofibers, nanoribbons, and nanosheets (Scheme 3). Each of these comes with its set of advantages and disadvantages depending on their specific applications, and tuning non-covalent interactions allows modification of these nanostructures.19,91,99–101 Vesicular and micellar structures are commonly employed for the delivery of drugs or genetic materials, leveraging the hydrophobic pocket to facilitate efficient cargo loading.102,103 Vesicular nanostructures can be transformed from a fibrillar network upon dilution, which showed a higher cellular uptake.104 Nanofibers and nanotubes exemplify the anisotropic growth of the assembly, often resulting in gelation.105–107 Gel scaffolds are valuable in biomedical applications and piezoresponsive materials, owing to their directional array of polar functional groups throughout the peptide chain.108,109 Furthermore, nanoribbons and nanosheets are advantageous in catalysis due to their high surface area and active site exposure.110–112 The potential of these nanostructures for loading and functionality is practically infinite.113 Due to the wide versatility and adjustability of peptide self-assemblies, peptides are a potential material for driving innovation across various scientific and technological realms. Lastly, a clear difference between secondary structures and nanostructures can be pointed out in such a way that: Secondary structures are local conformations within the peptide chain, stabilized primarily by H-bonding at the molecular level, which determine the peptide's basic functional properties and folding. In the case of peptide nanostructures, these are larger assemblies formed from multiple peptides that organize into specific shapes. These nanostructures also contribute to the material's overall macroscopic properties and potential applications. It is noteworthy that, while there is no direct correlation between the nano- and secondary structure formation of self-assembled peptides, both play a pivotal role in controlling the material properties.
 | ||
| Scheme 3 Schematic showing various nanostructures of the peptide. | ||
2.3. Factors affecting the peptide self-assembly
The secondary and associated nano-structure of the peptide assembly are significantly influenced by various stimuli such as pH, light, temperature, redox and environmental conditions of the systems.33,114 Peptides are frequently decorated with pH-responsive functional units such as free amines or acids, in their side chains and at the C-/N-terminus, making them susceptible to pH stimuli. The alterations in H-bonding and salt bridge interactions occur as a result of the protonation and deprotonation processes linked with pH fluctuations. In this regard, we demonstrated that by incorporating Asp and Lys as pH-responsive units, the secondary structure and self-assembly of an octapeptide (FALDLAKD) can be modified at three different pH levels (Fig. 2).45 At a physiological pH of 7.4, this peptide formed a hydrogel characterized by a fibrous nanostructure and a rich β-sheet structure. This β-sheet structure was observed to be disrupted under acidic conditions (pH = 5.5) or basic conditions (pH = 9 or 12), as confirmed by CD spectroscopy. The transformation of the self-assembled hydrogel was verified through TEM analysis, revealing a shift from its original nanofibrous structure to an irregular nanostructure when exposed to acidic or basic pH conditions. It has been hypothesized that alterations in pH levels, either lower or higher, result in the protonation or deprotonation of functional groups, causing an increase in electrostatic repulsion within the entire system. In a different peptide system (CLRVRGEV), we observed the breakdown of the β-sheet secondary structure as the pH transitioned from 7.4 to 5.5 (Fig. 2).46 The fractal-like nanostructure of the peptide underwent a conversion to a spherical morphology when transitioning to a lower pH. The arginine (Arg) unit present in the system becomes protonated at pH 5.5, leading to electrostatic repulsion and the destabilization of the self-assembled structure. Tanaka and co-workers demonstrated that another peptide consisting of cyclic α,α-disubstituted α-amino acid (dAA) undergoes a transformation from a cyclic acetal to an acyclic dAA at low pH, and the corresponding secondary structure also transformed from the α-helix to a random coil.115 In a separate study, we demonstrated how pH influences the secondary structure and the corresponding morphology. Under physiological pH conditions, the octapeptide was found to adopt a β-sheet structure, accompanied by the formation of a nanofibrillar architecture (Fig. 3). The secondary structure was shown to be stable at pH 13, but morphological modifications to fractals were seen as a result of dewetting processes and increased fibrillar overlap. Conversely, at a lower pH of 5.5, the assembly was disrupted, as indicated by the absence of circular dichroism (CD) signal and the presence of short nanorod structures.44 The self-assembly process of peptides, primarily guided by diverse noncovalent interactions, can be regulated by changing the temperature. Elevated temperatures cause the destabilization of various interactions by the supplied energy, occasionally leading to the degradation of the self-assembled state. Nevertheless, a noteworthy increase in hydrophobic interaction sometimes results in the formation of higher-order aggregates. A diphenylalanine (FF) peptide was found to exist in monomeric state at an elevated temperature of 90 °C; however, lowering the temperature leads to the formation of nanowire-like structures in an acetonitrile/water mix solvent.116 Elastin-like polypeptides (ELPs) remain as monomers at room temperature, but as the temperature increases, the heat energy triggers the assembly process due to hydrophobic interaction.117 In a polar solvent, the hydrophobic activity initiates the formation of the micellar nanostructure. Hamley and co-workers have shown that an amphiphilic peptide C16-KKFFVLK gets self-assembled into a nanotube and helical ribbon under ambient conditions. Remarkably, at a transition temperature of 55 °C, the nanostructure transforms into twisted nanotapes. Moreover, it undergoes a reversible transition back to its original structure upon cooling.38 We have demonstrated that the self-assembly of an octa peptide is also temperature-responsive (Fig. 3). A two-fold increase was observed in the CD signal, indicating the transition to a strong, well-defined β-sheet structure within the system. Significant nanostructural changes were observed, transforming from nanofibers to 2D nanosheets due to increased hydrophobic interactions, accompanied by robust π–π stacking and H-bonding.44,118,119 The effects of solvent polarity and H-bonding capability also affect the peptide self-assembly process. Ueda and colleagues have illustrated the influence of co-solvents (EtOH and ACN in H2O) in the self-assembly of an amphiphilic peptide {PSar30-(L-Leu-Aib)6(S30L12)}.87 A morphological transformation was observed from twisted nanoribbons to nanotubes in the EtOH/H2O mixture. These twisted nanoribbons were the kinetic trapped state, which act as primary precursors and evolved into the nanotube considered to be the thermodynamic state by a rapid rolling-up process. It was believed that ethanol (EtOH) molecules formed the H-bonded networks with water molecules, removing water from the hydrophobic hydration shell. This process strengthens the interactions between the peptide units. Das and colleagues demonstrated a morphological change in a perylene diimide-conjugated peptide (PDI-peptide) by altering the solvent polarity.120 In non-polar solvents such as tetrahydrofuran or chloroform, the peptide assembled into a nanofibrillar structure. Conversely, in polar solvents such as acetone, acetonitrile, methanol, and hexafluoro-2-propanol, it adopted a spherical structure. Interestingly, the PDI-peptide conjugate maintained a right-handed helical structure in both nanostructures, irrespective of the solvent polarity.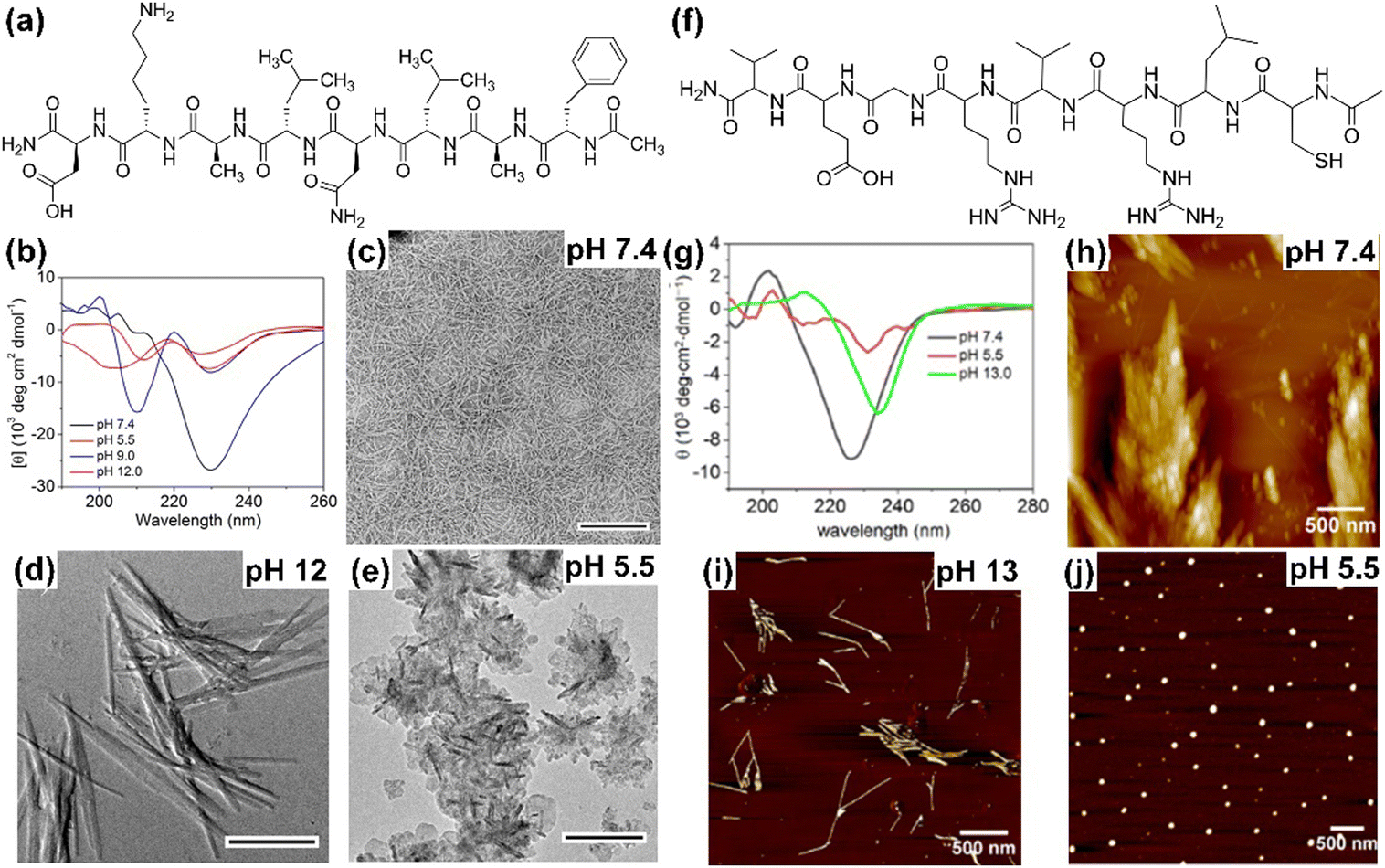 | ||
| Fig. 2 (a) Molecular structure of the pH-responsive octapeptide and (b) corresponding pH-responsive CD spectra demonstrating the disruption of the β-sheet secondary structure as pH shifts towards acidic or basic conditions. (c) HR-TEM image of the peptide at pH 7.4 showing entangled nanofibrillar morphology (scale bar = 500 nm). The HR-TEM image indicates the changes in the self-assembled nanostructure when moved to (d) basic (scale bar = 1 μm) and (e) acidic pH (scale bar = 200 nm). Adapted with permission from ref. 45, Copyright 2019 American Chemical Society. (f) Molecular structure of another pH-responsive peptide. (g) CD spectra of the pH-responsive peptide showing β-sheet formation at neutral pH (7.4), which is disrupted at acidic pH (5.5), while a more twisted β-sheet structure forms at basic pH (13), as indicated by the red-shifted CD spectrum. Corresponding different nanostructure formations at different pH conditions: (h) pH = 7.4, (i) pH = 13, and (j) pH = 5.5. Adapted with permission from ref. 46, Copyright 2020 Beilstein Institute for the Advancement of Chemical Sciences. | ||
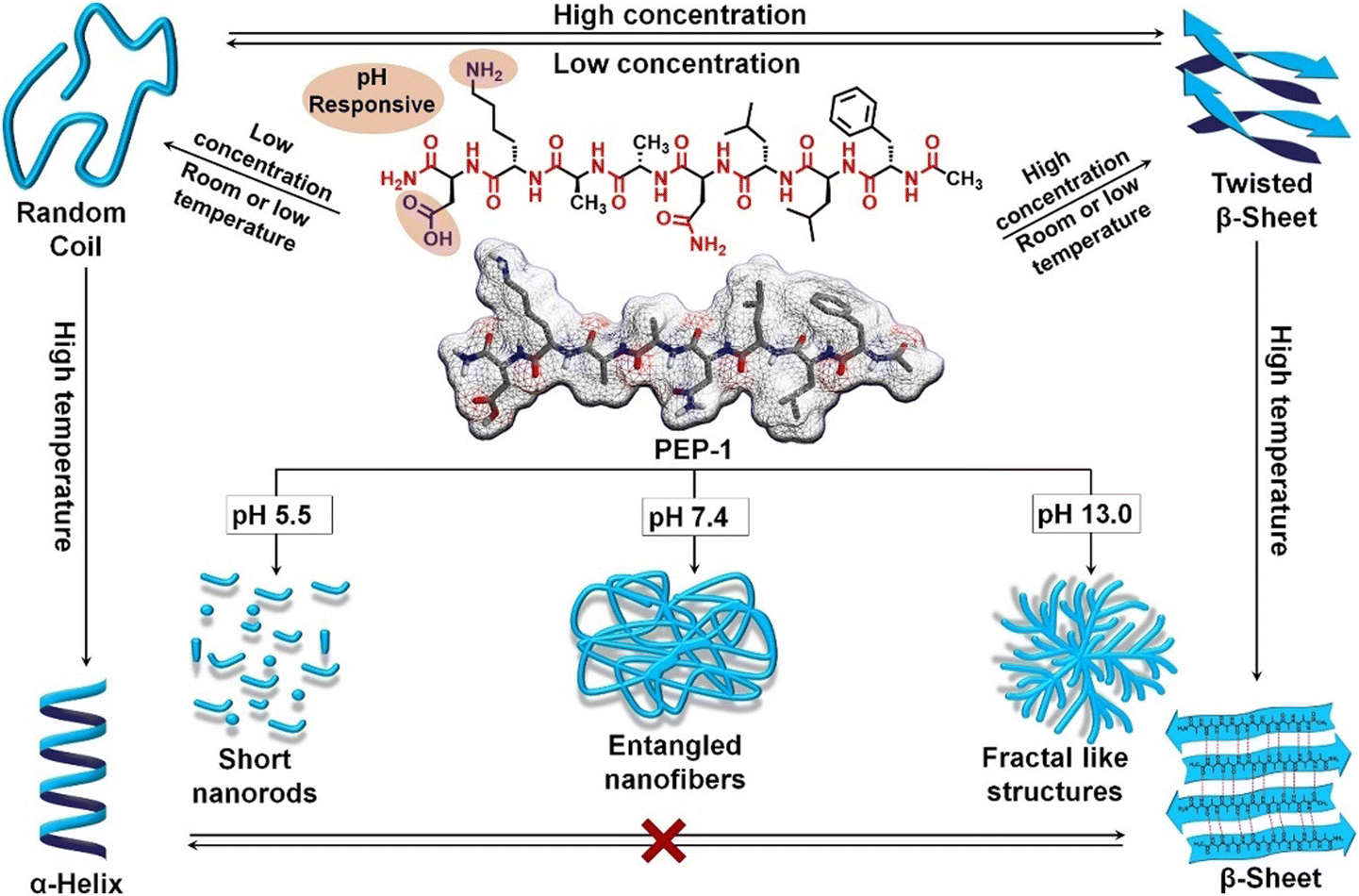 | ||
| Fig. 3 Molecular structure of an octapeptide, along with a schematic illustrating the controlled transformation of both secondary and nanostructures in response to various stimuli. Adapted with permission from ref. 44. Copyright 2022 Wiley-VCH. | ||
Change in the redox states can also serve as a key parameter in mediating the assembly of peptides. Shi and co-workers showed a gel-to-sol transition by the help of redox process in a thiol-rich peptide.121 Upon oxidation, the formation of the di-sulphide bond leads to an amphiphilic β-hairpin conformation and subsequently assembled into a rigid hydrogel. A gel-to-sol transition was found upon reduction due to the breaking of the disulphide bond. Light is recognized as a crucial stimulus capable of influencing assembly behaviour and serving as a fuel in the chemistry of peptide-based systems.122 Parquette and his team explored the impact of light on a tetrapeptide adorned with a light-responsive unit, spiropyran.123 Upon exposure to visible light, this peptide undergoes a transformation leading to transient self-assembly and eventual gelation in the TFA/H2O mix solvent. However, upon removal of the light source, the peptide reversibly comes back to its monomeric state. Visible light causes the formation of metastable spiropyran state and undergoes assembly into nanofibers, which got thermally relaxed into merocyanine state in the absence of light.
The peptide concentration significantly influences the packing in the self-assembled state. In a recent study, we have shown concentration-dependent adaptation of the secondary structure of a pH-responsive octapeptide (with the sequence FLLDAAKD).44 At a concentration of 0.5 mM (pH = 7.4), the peptide exhibited an enrichment of β-sheet structure (Fig. 3). In sharp contrast, the secondary structure changed to a random coil when the peptide was diluted to 0.05 mM, as evident from the CD and FTIR spectroscopic studies. The alteration in concentration not only impacts the secondary structure but also influences the nanostructure of the system. The nanofibrillar morphology gets transformed into nanoparticles at low peptide concentrations. Increased peptide concentration allows for closer contact, fostering stronger intermolecular interactions and resulting in a β-sheet structure. Conversely, intramolecular forces primarily drive the formation of a random coil structure at lower concentrations.44,46
The presence of noncovalent interactions in the system makes the peptides promising candidates for investigating the reversibility of their assembly. The change in pH has been shown to reversibly influence the nanostructure, transitioning it from nanorods to nanoparticles.124 In addition to enabling interchangeable nanostructures, pH can also affect the assembly and disassembly of the peptide network.125 It has also been known that certain peptides can switch between the nematic gel phase and isotropic fluid state through pH.126 Stupp and colleagues have studied the reversible self-assembly of peptide-DNA superstructures. Both theoretical and experimental findings indicated a large-scale spatial arrangement of the small molecular units, driven by strong noncovalent forces.127
There are several other factors such as the presence of metal ions, the ionic strength and the presence of enzymes, which play a significant role in altering the secondary or nanostructure of the peptide self-assembly.48,91
3. Applications of peptide-based self-assembled nanomaterials
Peptide-based self-assembled systems are a crucial category of materials with applications in chemistry, biology, and material sciences.25,128 In contrast to individual peptides, the advantages of self-assembled peptides lie in their highly organized and precisely controlled supramolecular structures, characterized by excellent thermal and mechanical stability.128,129 In addition to natural peptides, hybrid synthesized peptides with diverse chemical functionalities exhibit a wide range of applications in semiconductor devices, piezo-responsiveness, and optoelectronics.128,129 Every peptide system, with its unique aggregation method, gives rise to a variety of tunable secondary and corresponding nanostructures, and has a crucial impact on specific application fields. A detailed discussion of the distinctive features and excellent biocompatibility of these self-assembled peptide-based systems, along with their applications in various fields, can be found in the subsequent section of this review.3.1. Peptides as energy harvesters (piezoresponsive materials)
Out of all renewable energy harvesting processes, piezoelectric energy harvesting stands out as particularly convenient for directly converting mechanical energy into electrical output (Scheme 4).130 Piezoelectric materials constitute a distinct category of substances that can transform mechanical stress into electrical output, and conversely, this holds true as well. These materials have broad applications in piezotronics, sensors, actuators, and transformers, serving as an energy harvesting system.26,128,131,132 There are several benchmark piezoelectric materials with non-centrosymmetric space groups, which include lead zirconate titanate, zinc oxide, barium titanate, and molybdenum disulfide. The generation of piezoelectricity in this inorganic system stems from rearranging ions within a dielectric material lacking inversion symmetry in its crystal structure.133 However, their metal-associated toxicity restricts applications in biological fields. Furthermore, their processing frequently demands high-temperature techniques and solid-state synthesis methods with constrained control.134–139 A well-recognized subset within this domain includes biomaterials such as proteins (specifically collagen) and viruses. The presence of numerous H-bonds aligned in a specific direction results in a macroscopic dipole that can interact with external electric fields or which can be influenced by applied forces, leading to the generation of a piezoelectric.140 The obstacles associated with natural piezoelectric materials, particularly their low piezoelectric constants, have prompted significant strides in the progress of synthetic biocompatible piezoelectric materials. Within a synthetic system, peptides emerge as the most analogous component, offering the potential for realizing piezoelectric responses through the alignment of H-bonding dipoles in a specific direction.141–145 The application of mechanical stress induces polarization in the organic system as a result of the reorientation of these dipoles, leading to a piezoelectric response.146 Arranging the peptide in a specific order holds great promise for achieving piezoelectric response by ensuring the proper orientation of the functional groups. Within this framework, diphenylalanine (FF) has been extensively researched. Yang and co-workers successfully surmounted the significant challenge posed by random polarization in the self-assembled state.147 They demonstrated a seeded self-assembly technique under an applied electric field to form microrods of FF from a water solution at an elevated temperature of 55 °C (Fig. 4a and b). FF crystallizes in a chiral P61 space group, featuring directional polarization aligned with the applied electric field. The alignment of electrical polarization depends on whether the electric field is applied parallel or antiparallel to the substrate surface normal. The piezo force microscopy (PFM) technique was utilized to probe the polarization phase at the two opposing surfaces of the microrods, determining whether it is inward or outward. The even distribution of surface charge at the tip of the microrod was additionally validated by a Scanning Kelvin Probe Microscopy (SKPM) study. A significantly higher piezoelectric coefficient d33 = 17.9 pm V−1 was found in this system. The substantial rise in the piezoelectric coefficient and dipole strength was attributed to the electric field-induced stretching of the peptide backbone during the self-assembly process. They have also demonstrated the real-time application of this microrod towards power generation. The nanogenerator showed the open-circuit voltage (Voc) and short-circuit current (Isc) of 1.4 V and 39.2 nA, respectively under an applied force of 60 N (Fig. 4c–e).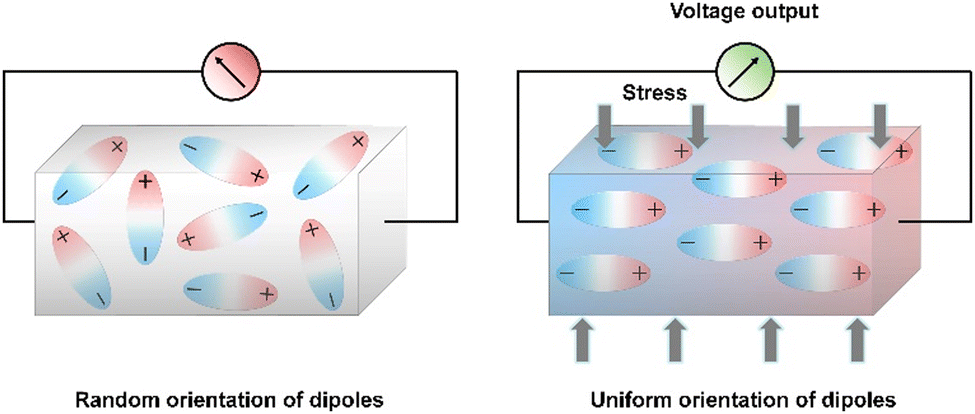 | ||
| Scheme 4 Schematic representation of the positive piezoelectric effect. Left: Random dipole orientation under ambient conditions. Right: Dipoles are aligned in an orientated fashion under stress that resulted in charge separation at the two ends of the material. | ||
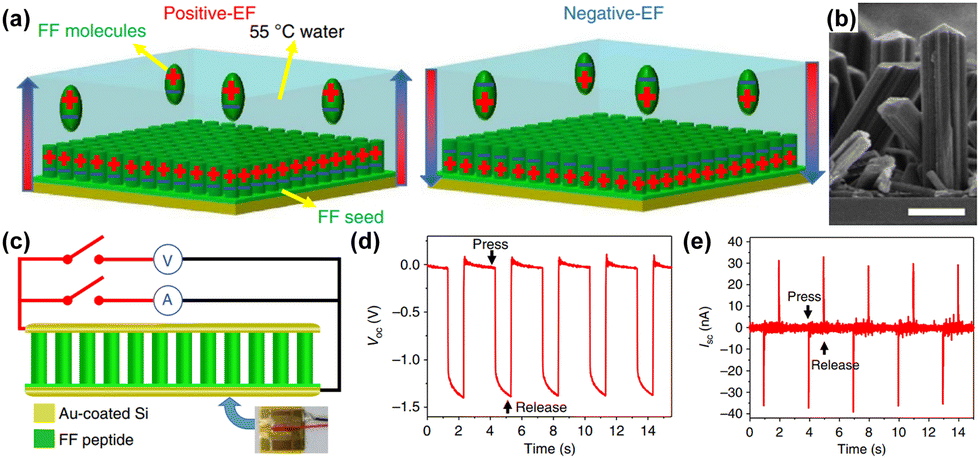 | ||
| Fig. 4 (a) Schematic showing the control growth of FF dipeptides under a controlled polarization direction. (b) Microscopic cross-sectional view of arrays of the FF dipeptide showing unidirectional nanostructure formation of the FF dipeptide. (c) Schematic of an FF dipeptide-based generator. (d) Open circuit voltage and (e) short circuit current obtained as output from the microrod peptide-based generator. Adapted with permission from ref. 147. Copyright 2016 Springer Nature. | ||
Numerous investigations into this specific system, involving various self-assembly processes and molecular orientations within the crystal, revealed a wide spectrum of piezoelectric coefficients, ranging from 5 to 30 pm V−1.147–150 Kholkin and co-workers have shown a temperature control piezo response modulation on FF nanotubes having an effective d33 value of 30 pm V−1. The robust polarization resulting from the oriented H-bonding among the FF monomers deteriorated as the temperature increased, leading to an irreversible phase transition in the system. As a result of that, PFM responses were also found to decrease gradually with the rise in temperature.149 The same group has also reported a high piezoelectric coefficient d15 of 80 ± 15 pm V−1.151 The substantial piezoelectric response was attributed to the pronounced dipole moments of six FF rings that were uniformly oriented in the same direction.149 Piezoelectric responses were also found in modified peptide based on FF. Rodriguez and co-workers have synthesized fluorenylmethyloxycarbonyl diphenylalanine (Fmoc–FF) nanofibrils. The piezoelectric response was attributed to the specific alignment of H-bonds within the anti-parallel β-sheet structure. The non-centrosymmetric nature of the β-sheet assembly was the fundamental reason behind the directional arrangements of the H-bonded dipolar amide bonds. Chemical modification, coupled with the presence of a strong antiparallel beta-sheet structure, resulted in a higher piezoelectric response compared to the pristine FF system. A shear piezoelectric constant (d15) of 33.7 pm V−1 was found in these nanofibrils.152
Gazit and colleagues have taken another stride towards emulating the properties of collagen, a widely recognized biocompatible piezoelectric system.140 They have modified the diphenylalanine by introducing hydroxyproline (Hyp), and for comparison, they conducted a control study using proline (Fig. 5a). This particular amino acid is present in collagen and is known to play a significant role in piezoelectric responses. The single-crystal X-ray diffraction study was used to ascertain the supramolecular arrangement and intermolecular interactions of these two tripeptides. A minor polar modification such as the addition of a hydroxy group in this case leads to a notable alteration in the overall arrangement of the crystals. The aromatic zipper-like structure formed via the π–π interaction of the phenyl group of Hyp-Phe-Phe has been demonstrated to reduce the crystal symmetry and shear stiffness, while simultaneously increasing the overall crystal dipole (Fig. 5b). A nearly two-fold enhancement in the effective vertical piezoelectric coefficient (d33) was detected in the case of Hyp-Phe-Phe compared to Pro-Phe-Phe. Furthermore, they achieved a remarkable shear piezoelectric coefficient (d34) of 16.12 ± 2.3 pm V−1, surpassing the benchmark inorganic material LiNbO3 (13 pm V−1). Molecular dynamics (MD) simulation study provides additional evidence of a robust H-bonding interaction with a well-balanced electrostatic interaction in Hyp-Phe-Phe. Finally, both tripeptides were utilized in the construction of nanogenerators, demonstrating a significantly higher open circuit potential and short circuit current compared to any other peptide-based system. The presence of the hydroxyl group has a notable impact in this real-time application. Pro-Phe-Phe generated a short circuit current (Isc) of 52 nA when subjected to a force of 55 N, while Hyp-Phe-Phe produced a short circuit current (Isc) of 39.3 nA under a force of 23 N. An overall output current comparison of three different systems is given in Fig. 5c. The piezoelectric response of the peptide is not limited in its crystal state. It has also been observed in its self- assembled state, where proper alignment of functional planarization of the azobenzene core resulted in its conversion into a photodormant state. The introduction of β-cyclodextrin led to the attainment of a chemodormant state via host–guest interactions. The photodormant state transformed into nanofibers upon exposure to visible light. Additionally, the introduction of a stronger binding agent to β-cyclodextrin and adamantane in the host–guest system resulted in a similar transformation, akin to the previous process. Internal alterations in the peptide arrangements during these transitions brought about changes in their macroscopic properties (Fig. 6a).153 The highest piezoelectric coefficient of this system was found to be d33 = 81 pm V−1, in case of the thermodynamic twisted nanofibrous state. The kinetic product (nanofibers) also showed a d33 value of 7 pm V−1, whereas the off-pathway dormant nanoparticle was piezo-silent (Fig. 6b–g). Remarkably, the nanofibers resulting from the transformation of the dormant state under applied stimuli exhibited piezoelectric activity. The study demonstrated that the pivotal directional alignment of the polar amide bond across the nanostructure led to an on–off piezoelectric response. The nanofibers showed a piezo-electric energy conversion owing to the unidirectional array of the amide functional group. In contrast, the random orientation of the H-bonded amide groups within the nanoparticle resulted in a piezo-off state. Furthermore, they constructed a nanogenerator using the thermodynamic peptide assembly, which exhibited a promising voltage response of 8 mV (peak to peak) upon finger tapping.
 | ||
| Fig. 5 (a) Chemical structure of tripeptides Pro-Phe-Phe and Hyp-Phe-Phe. (b) Crystal structure depicting the helical packing of Hyp-Phe-Phe with H-bonding stabilization and an aromatic zipper-like structure. (c) A comparison output current of three different peptides showing the highest obtained value for the helical Hyp-Phe-Phe system, indicating the impact of the hydroxyl group on polarization and corresponding piezoresponse. Adapted with permission from ref. 140. Copyright 2021 Springer Nature. | ||
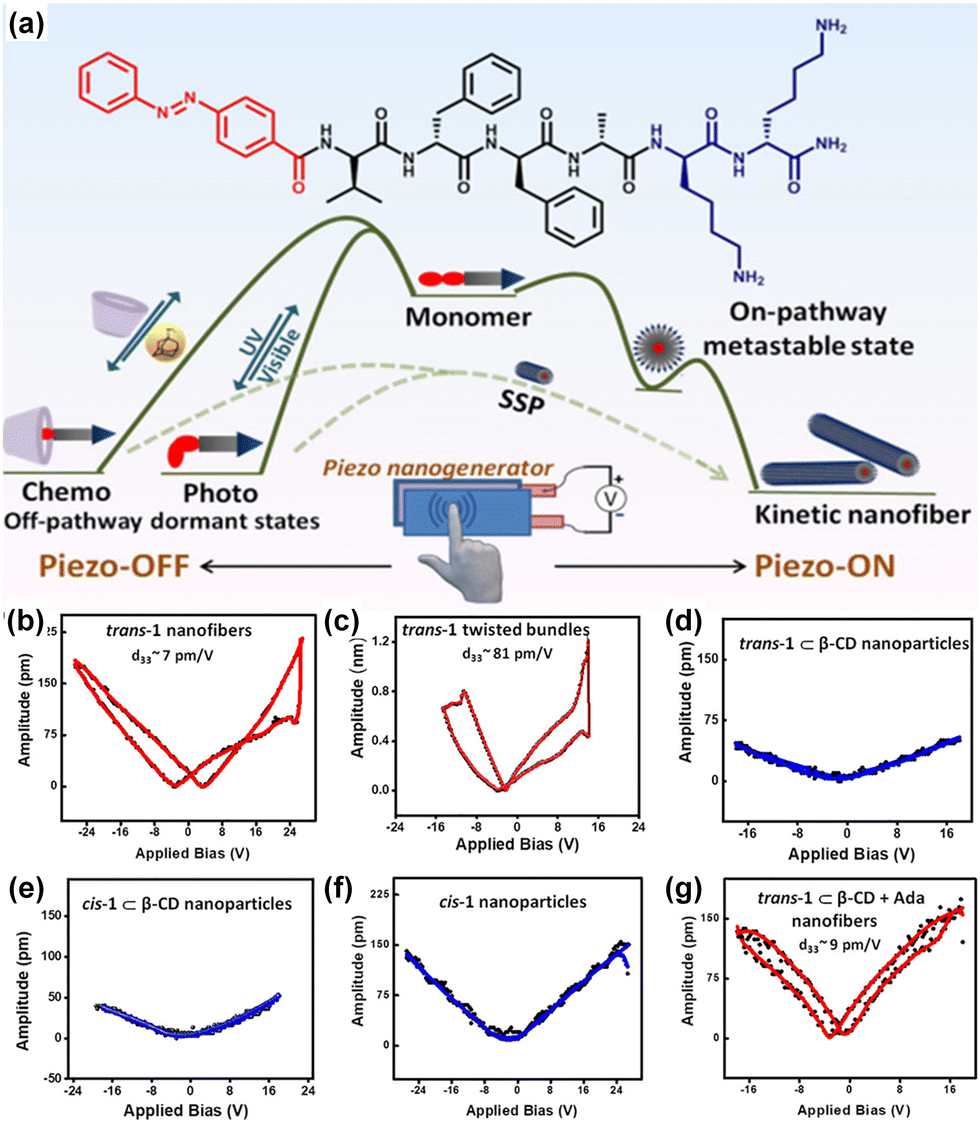 | ||
| Fig. 6 (a) Graphical representation of the peptide chemical structure and its pathway complexity in supramolecular polymerization under different control methods. (b)–(g) PFM amplitude loops for distinct aggregated states, highlighting the highest piezo-response observed in thermodynamically controlled twisted nanofibers, also indicating effective orientation of the dipoles in the fibrillar arrangement. Adapted with permission from ref. 153. Copyright 2022 American Chemical Society. | ||
A dipeptide based on phenylalanine, N-protected by tri-3,4,5-(n-dodecyloxy)benzoic acid, and C-protected by an aminomethyl pyrene unit has been recently reported by Bhattacharya et al.154 This specific versatile system resulted in the formation of an organogel in toluene, exhibiting a nanofibrillar network structure. The dipeptide followed an isodesmic polymerization pathway, as revealed by the temperature-dependent absorption study. In the cooling process, it adopted a nanofibrous structure at 30 °C, observed to exhibit circularly polarized luminescence (CPL) with a glum value of 3.0 (± 0.2) × 10−3. The piezoelectric response represents a kinetically stable state. Subsequent cooling to 10 °C led to transformation into a thermodynamically stable 2D nanosheet (Fig. 7a). These nanofibrous aggregates were assessed by performing a PFM analysis for both the kinetic and thermodynamic states (Fig. 7b). In the kinetic state, a piezoelectric coefficient (d33) of 64.1 pm V−1 was observed, in contrast to the thermodynamic state that displayed a notably higher value of 75.9 pm V−1 (Fig. 7c). The heightened piezoresponse was attributed to the enhanced alignment of the dipole moment and the one-directional characteristic of the H-bond, strengthened by the chiral arrangement of the π-stacked pyrene unit.
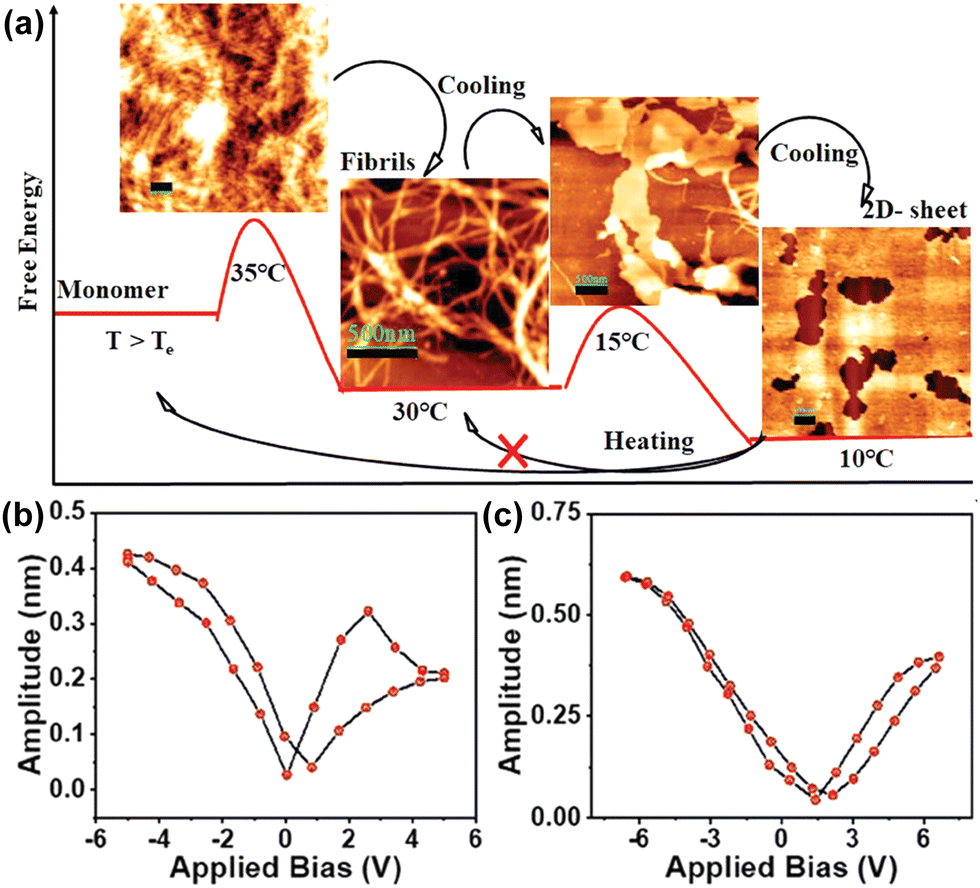 | ||
| Fig. 7 (a) Qualitative energy diagram and corresponding AFM images indicating the formation of different self-assembled states of the peptide at different temperatures. PFM amplitude loop of (b) the fibrillar nanostructure and (c) 2D-sheet. Adapted with permission from ref. 154. Copyright 2023 Wiley-VCH. | ||
Very recently, we have demonstrated a multi-responsive π-chromophore conjugated peptide self-assembly towards chiroptical switching and controlled piezo-response behaviour. The amphiphilic tetrapeptide appended with an NDI chromophore gets self-assembled into kinetically controlled short nanorods or nanoparticles depending upon the solvent environment, depicting the kinetic state of the system (Fig. 8a–c). This kinetic state further evolves into a stable thermodynamic state with crosslinked nanofibrillar architectures through processes such as increased temperature, denaturation, and co-solvent assistance. It was also observed that the CD signal corresponding to the peptide assembly reversed as it transitions from the kinetic to the thermodynamic state, indicating a change in the peptide's helical sense during packing. As a result of the nanostructural transition from nanoparticles to long-range nanofibers, a significant change in the long-range ordering of the amide H-bonds was anticipated. This led to a transition from the piezo off-state to the piezo on-state, as evidenced by the butterfly loop opening in the thermodynamic state (Fig. 8d–g). This study demonstrated the importance of packing of the peptide units and corresponding nanostructure formation towards modulating the piezo-responsiveness of a particular system.155
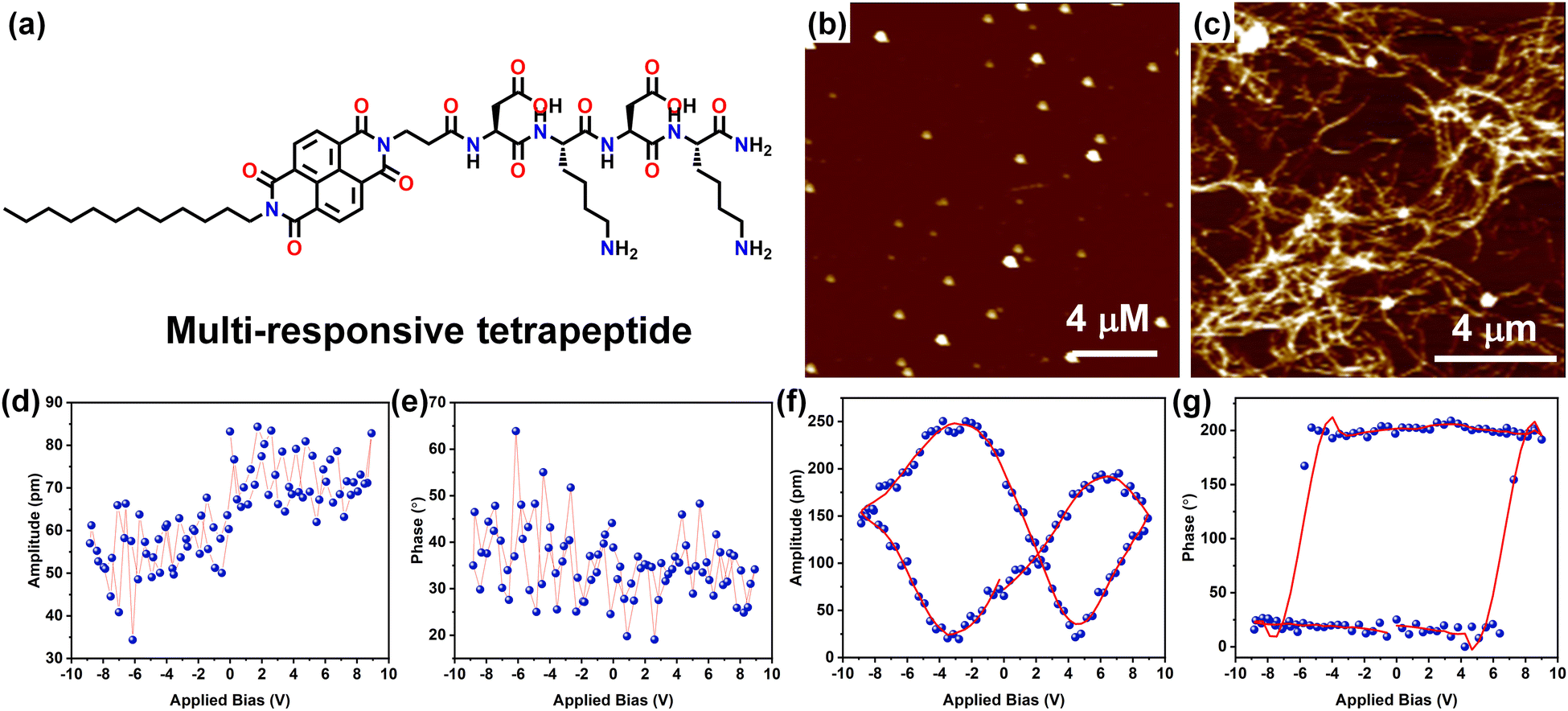 | ||
| Fig. 8 (a) Chemical structure of the multi-responsive NDI π-chromophore-appended tetrapeptide. AFM images of (b) the kinetic state (KS) display a nanoparticle morphology, which evolves into the (c) thermodynamic state (TS) characterized by cross-linked nanofibrillar architectures. PFM (d) and (e) amplitude-voltage butterfly loop (off state) and phase-voltage hysteresis loop (off state) obtained for the KS respectively. PFM (f) and (g) amplitude-voltage butterfly loop (off state) and phase-voltage hysteresis loop (off state) obtained for the TS respectively. Adapted with permission from ref. 155. Copyright 2024 Royal Society of Chemistry. | ||
The advantages of peptide-based piezoelectric systems are biocompatibility and biodegradability. Using sensor-based devices for monitoring the human body is a crucial component that contributes to improved healthcare. In this regard, piezoelectric nanomaterials are important, as they can efficiently diagnose diseases.154 Therefore, the rational design of converting mechanical energy into electrical power serves as a valuable energy source for various microelectronic applications. Researchers have explored the use of these biocompatible piezoelectric materials for potential applications in implantable medical electronics (IMEs), including cardiac pacemakers, active pressure sensors, and devices designed for the direct stimulation of tissues and living cells. Organic piezoelectric materials using peptides possess advantageous features such as biocompatibility, affordability, lightweight construction, and flexibility. Nevertheless, there are various challenges associated with the real-time application of such systems. Detailed mechanistic studies of these biomaterials are essential. Molecular dynamics (MD) simulations can contribute significantly in this regard by aiding in the comprehension of peptide packing and the generation of piezoresponse in these materials. Exploring the conductivity and semiconducting characteristics of such systems is essential for advancing modern devices across diverse fields.
3.2. Catalytic activity of peptides
Catalysis is a fundamental chemical process in which a reaction is accelerated by the addition of a mediator that is not consumed in the reaction. Catalysts can be classified based on their phases as either homogeneous (in the same phase as the reactants) or heterogeneous (in a different phase). It is also integral to numerous industrial processes such as petroleum refining, pharmaceutical manufacturing, food production, and environmental applications.156–159 Among the various bioactive molecules that show catalytic activity, peptide is one of the promising candidates because of its various advantages. The benefits associated with peptides, include a wide range of biological activities, high specificity and low side effect when developing new drugs, ease of synthesis via different routes, and structural diversity for customized applications. Their ability to mimic enzymes and perform specific reactions efficiently and selectively holds significant promise for future applications and research.160–162 Depending on the various functional moieties associated with synthetic peptide units, they can serve as catalysts for a range of reactions such as hydrolysis, coupling reactions and redox reactions.Das et al. investigated a short amyloid peptide capable of binding with a small molecular coenzyme, which can then polymerize and form catalytically active nonequilibrium amyloid microphases.163 It was demonstrated that a short amyloid-based hepta peptide (Im-KLVFFAE-NH2) with an imidazole moiety for hydrolytic competence and an amine group for forming a dynamic covalent imine bond interact through dynamic covalent linkage with a negatively charged low-molecular-weight ester ((2-(4-formylphenoxy)-2-oxoethanesulfonate) (AS) to form nonequilibrium amyloid polymers (Fig. 9). By degrading the ester and thereby giving a negative feedback to their assembly process, the polymers exhibit covalent catalysis, which leads to depolymerization. Through mixing them in various molar ratios (40% v/v, DMSO/HEPES buffer, 100 mM, pH 7), the ester's capacity to promote Im-K's self-assembly was examined. Interestingly, the mixture of Im-K (4 mM) and AS (8 mM) in a molar ratio of 1/2 quickly became viscous and eventually produced gel after 10 minutes. After 360 minutes of stability, the gel gradually weakened and underwent a gel-to-sol transition. The nonequilibrium self-assembled state shows selective hydrolysis of two charged substrates, p-nitrophenyl esters of positively charged betaine (PS) and negatively charged sulfoacetic acid (NS). These findings showed that dynamic catalytic competency switching was present on the surface of the non-equilibrium assemblies. The self-assembly of nucleic acid and peptide was investigated by Ding et al.164 The assembly with hemin as a cofactor forms catalytic nanoparticles that mimic enzymes and have customizable activities. Using the Autodock program, they demonstrated how the 32 His-containing peptide molecule (H32) can approach the folded, guanine-rich DNAzyme-I (GGGTAGGGCGGGTTGGG; DzI) from multiple angles, proving the shape complementarity between the two species. van der Waals force was found to be the driving force behind the DzI/H32 self-assembly, which was realised through molecular dynamics (MD) simulation studies. The complexation of hemin with H32 and DzI was successfully demonstrated by the elemental mapping and scanning transmission electron microscopy (STEM) imaging. To comprehend it further, the organization of the assembly was investigated by a UV-vis spectroscopic study. Compared to self-assembled systems containing only nucleic acids or peptides, the self-assembled nanoparticles incorporating hemin as a cofactor exhibited significantly higher peroxidase-mimicking activity in the oxidation of various reducing substrates. The catalytic efficiency was found to be similar to that of myoglobin, and peroxide's turnover number approached that of natural peroxidase. The hybrid nanoparticles’ functionality was largely attributed to the roles of hemin (a His-rich peptide), which facilitated catalysis by providing activation of the functional groups, and the guanine-rich DNA aids in hemin coordination (Fig. 10).
 | ||
| Fig. 9 Diagram illustrating the chemical structures of the peptide sequences utilized and the nonequilibrium polymerization of short amyloid peptides fueled by a negatively charged activated ester. The short peptides use a dynamic non-covalent linkage to interact with the negatively charged low-molecular-weight esters. β-Strands are represented by arrows. Adapted with permission from ref. 163. Copyright 2022 American Chemical Society. | ||
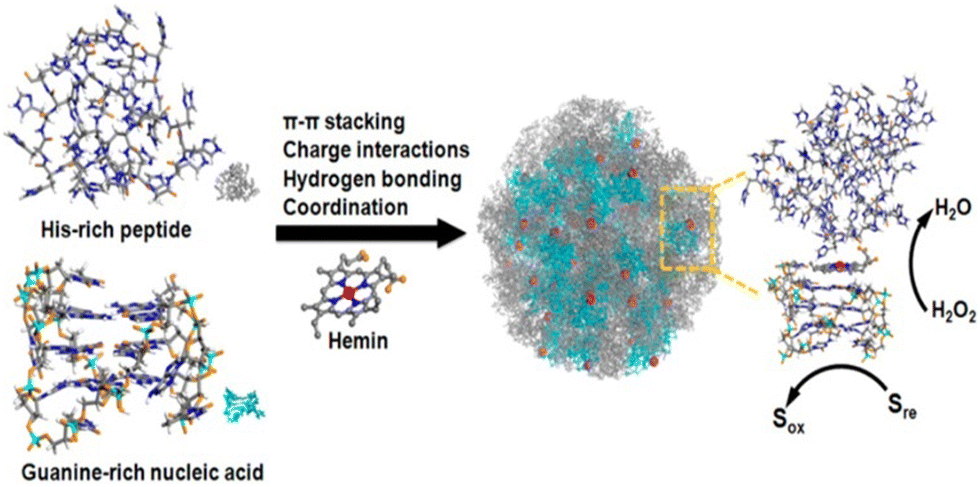 | ||
| Fig. 10 Schematic shows the self-assembly of the hybrid system consisting of the His-rich peptide and Guanine-rich nucleic acid into a ligand environment for hemin (cofactor). The coordination, catalytic characteristics and activation mechanism of hemin are shown. Adapted with permission from ref. 164. Copyright 2017 American Chemical Society. | ||
Metal composites, particularly those of gold and silver, have recently been created using peptides and proteins that serve as reducing and stabilizing agents.165 The size, composition, arrangement, and self-organization of gold nanoparticles determine their potential uses.166,167 Biocompatible and physicochemically advantageous self-assembling biomolecules such as oligonucleotides, proteins, and peptides have gained a lot of interest in the development of metallic composite nanoparticles.168 Peptides possess the capacity to adjust the physicochemical characteristics of metals and manage their size during the complexation process. In addition to serving as stabilizers and reducers for gold nanoparticles, peptide building blocks can also be used as a template to synthesize gold nanoparticles within a composite material.169
Hauser and coworkers presented a simple method for fabricating gold peptide nanoparticle (GPNP) hybrid composites using the UV-assisted self-assembly of the peptide AcIVFK-NH2 and gold salt, eliminating the need for extra capping or reducing agents through a photochemical reduction technique. Through this, they proposed an environment friendly, biologically sound, straightforward, and mechanistically understandable method for the catalytic reduction of pollutants instead of employing inorganic reagents in the synthesis of gold nanoparticles. The peptide's photoionization activity resulted from the aromatic residue's exposure to UV light, which allows the reduction of gold ions. The peptide simultaneously functioned as a capping, stabilizing, and reducing agent. Gold nanoparticles dispersed on peptide nanofibers had an average size of 5.16 nm. Additionally, it has been observed that gold-peptide composites can rapidly reduce the small-molecule pollutant p-nitrophenol to p-aminophenol (Fig. 11); the catalytic reaction rate constant was 0.057 min−1 at a 50-fold diluted sample of 2 mg mL−1 and 0.72 mM gold concentration in the composites.170 Nevertheless, since the conversion of the organic pollutant took place in two minutes when they used peptide–gold nanocomposites without dilution, the rate of reaction depended on their concentration. These peptide-metal hybrid composites, synthesized by an eco-friendly method, will pave the way for new advancements in environmental applications and biocatalysis.
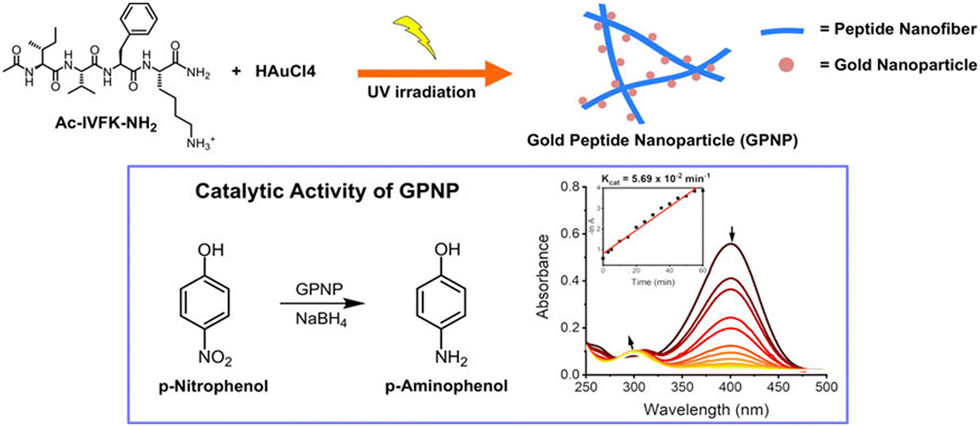 | ||
| Fig. 11 Chemical structure of the tetramer peptide and the UV-assisted fabrication of GPNP composites (254 nm). The catalytic activity of GPNPs for the reduction of the small-molecule pollutant p-nitrophenol to the least toxic compound p-aminophenol, and the formation of p-aminophenol was detected simultaneously at around 400 nm, which indicates the catalytic activity of GPNP composites. Adapted with permission from ref. 170. Copyright 2022 American Chemical Society. | ||
The solid-phase peptide method with microwave assistance was used to synthesize cyclic-dihistidine (cyclo-HH). This was employed as a component in the construction of a hydrolytically active nano-superstructure based on the supramolecular assembly. Cyclo-HH was combined with ZnI2 in a regulated hydrothermal environment to facilitate self-assembly, which led to the formation of nanostructures (Fig. 12). X-ray crystallography analysis and all-atom MD simulations were used to study the self-assembly mechanism of cyclo-HH and zinc iodide (cyclo-HH-ZnI2). With remarkable stability and recyclable nature, the cyclo-HH-ZnI2 nanowires demonstrated hydrolytic activity. The hydrolytic activity of the cyclo-HH-ZnI2 nanowires was investigated using p-nitrophenyl acetate hydrolysis as a probe reaction. By tracking the time-dependent absorbance changes of the reaction product, p-nitrophenol, at 405 nm, the hydrolysis activity of cyclo-HH-ZnI2 nanowires toward p-nitrophenyl acetate was examined. The cyclo-HH-ZnI2 nanowire catalyst was found to be capable of ester hydrolysis, as evidenced by the colorless p-nitrophenyl acetate solution turning yellow to signify the formation of the reaction product, p-nitrophenol. This was an interesting finding about the capabilities of cyclo-HH-ZnI2 nanowires. Moreover, a clear increase in the reaction rate was observed with the increase in cyclo-HH-ZnI2 nanowire concentrations. Excellent catalytic activity was demonstrated by Cyclo-HH-ZnI2 nanowires towards two additional ester substrates: 4-nitrophenyl butyrate and 4-nitrophenylvalerate.171 Beyond providing a more reliable and effective substitute for commercial and scholarly enzymatic catalysis, the creation of an enzyme-inspired peptide supramolecular catalyst is anticipated to provide a deeper understanding of the native enzyme's genesis.
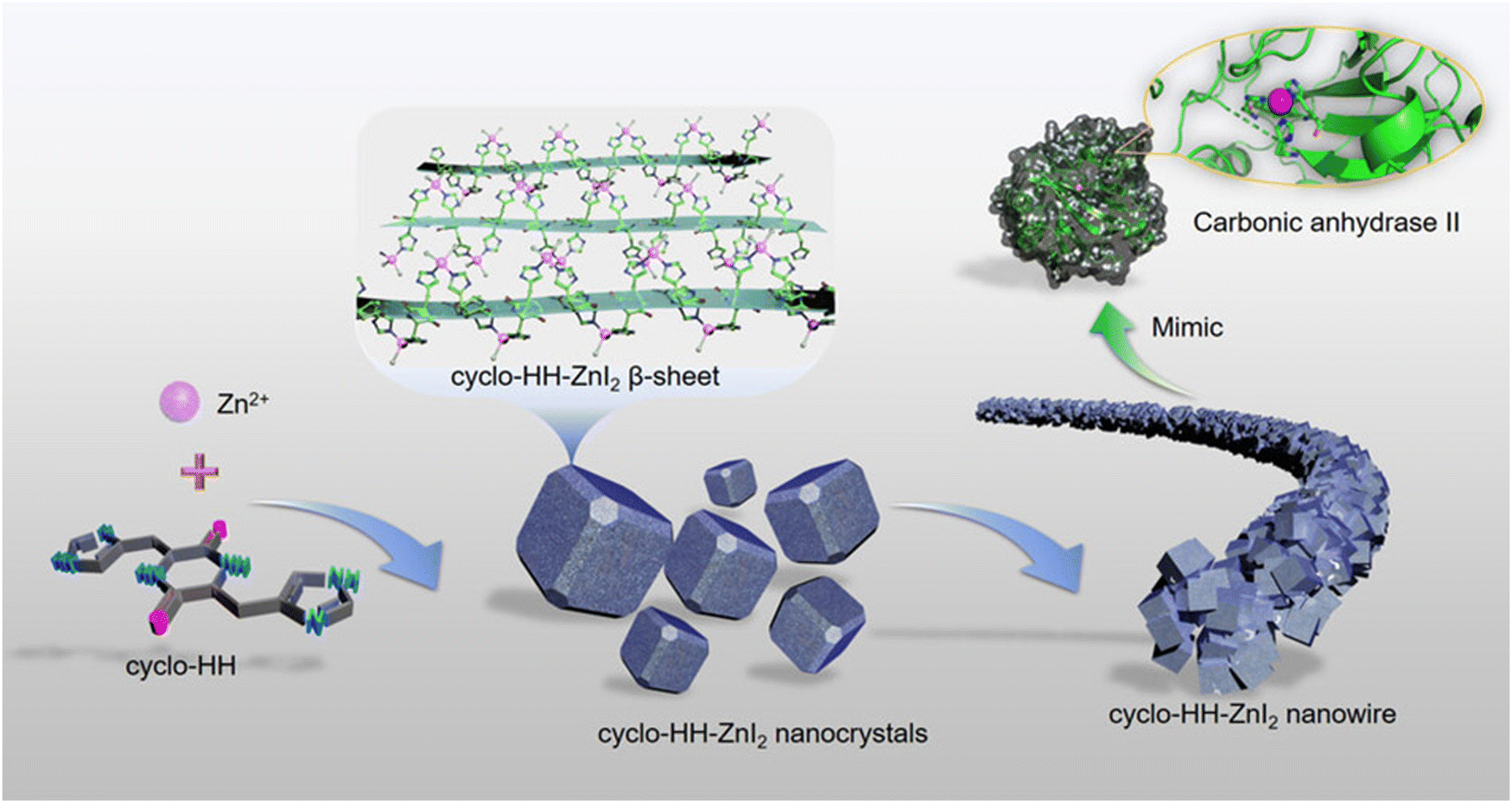 | ||
| Fig. 12 Design of a nano-superstructure based on the supramolecular assembly of a dihistidine containing a cyclic peptide and zinc iodide, which mimic the molecular structure of carbonic anhydrase II's active site. Adapted with permission from ref. 171. Copyright 2021 Wiley-VCH GmbH, Weinheim. | ||
A supramolecular catalytic system by the self-assembly of small peptides on the surface of gold monolayer-protected clusters (Au MPCs) was synthesized by Prins et al.172 They demonstrated how histidine-containing peptides H1–H3 self-assemble on AuMPC1's surface, initiating their esterolytic activity and producing nanostructures that accelerate the reaction rate 100 folds (Fig. 13). Since the peptides were completely inactive as catalysts in the absence of Au MPCs, self-assembly was a crucial prerequisite for catalysis to take place. Significantly, adjusting the catalytic activity is greatly aided by fluorophores used to study the binding of peptides to Au MPCs1 through fluorescence titrations. The p-nitrophenyl ester of N-carboxy benzyl phenylalanine (Cbz-Phe-ONP) was trans-esterified to assess the catalytic activity of the developed systems. The peptides H1–H3 at their respective surface saturation concentrations and a solution of Au MPC in 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2O/CH3CN buffered at pH 7.0 at 37 °C were combined with the substrate (10 μM) to perform kinetic experiments. They found that the cleavage of Cbz-Phe-ONP was accelerated by at least two orders of magnitude in all cases of the complexes H1–3/Au MPC. Furthermore, a 20-fold increase in the hydrolysis rate was seen upon the straightforward addition of Au MPC to Cbz-Phe-ONP.
:1 H2O/CH3CN buffered at pH 7.0 at 37 °C were combined with the substrate (10 μM) to perform kinetic experiments. They found that the cleavage of Cbz-Phe-ONP was accelerated by at least two orders of magnitude in all cases of the complexes H1–3/Au MPC. Furthermore, a 20-fold increase in the hydrolysis rate was seen upon the straightforward addition of Au MPC to Cbz-Phe-ONP.
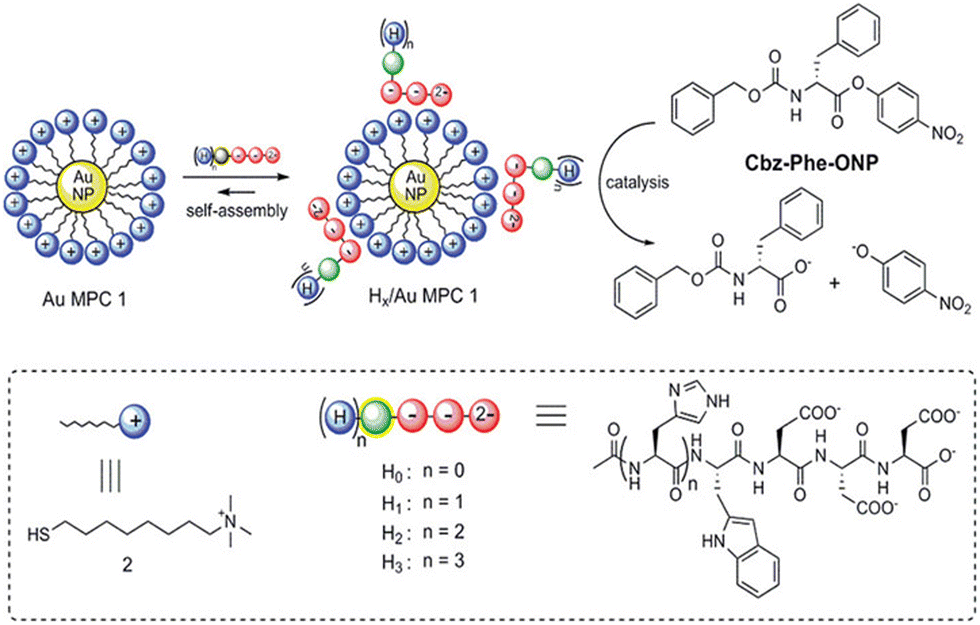 | ||
| Fig. 13 Self-assembly of peptides H0–H3 on the surface of an Au MPC, followed by the formation of Hx/Au MPC complexes that (x = 1–3) that can catalyze the transesterification of the substrate Cbz-Phe-ON. Adapted with permission from ref. 172. Copyright 2012 American Chemical Society. | ||
Knecht et al. presented a novel idea for the development of multifunctional nanoparticle ligands that produce single nanoparticle cascade catalysts through the use of biomimetic techniques. In order to create a multistep catalytic nanoparticle system, biomolecules were designed by incorporating Au nanoparticle binding domain (AuBP1) and a catalytic domain (CPN3), resulting in either AuBP1–CPN3 or CPN3–AuBP1, two distinct peptide ligands (Table 1). The catalytic CPN3 domain was located at the C-terminus in the first example, and the Au-binding AuBP1 domain was located at the N-terminus. The two domains were inverted for the latter sequence. A 4-glycine spacer was added in between the two distinct domains in both instances. Following the synthesis of the two chimeric peptides, QCM was used to determine both their affinity for Au and that of the catalytic CPN3 peptide. After confirming the chimeric peptides’ ability to form nanoparticles, their catalytic reactivity was examined. Using only the peptides, the hydrolysis of 4-nitrophenyl acetate to 4-nitrophenol, the first reaction of the tandem process, was investigated. The reactivity of the peptide ligands for the first step of the tandem reaction was clarified, and then the catalytic activity of the underlying Au core for the second step of the reduction of 4-nitrophenol to 4-aminophenol was investigated (Fig. 14). After the hydrolysis of 4-nitrophenyl acetate and 4-nitrophenol reduction was confirmed, the tandem reactivity of AuBP1–CPN3-capped Au nanoparticles was investigated. No further investigation was conducted into the reactivity of the CPN3–AuBP1-based system for the tandem reaction, as it showed minimal reactivity for 4-nitrophenyl acetate hydrolysis.173 Bedford et al. employed eco-friendly methods to synthesize peptide-promoted catalytic Au nanoparticles (PEPCANs) and used a combined experimental and computational approach to study the sequence-dependent structural relationships of Au NPs. Their findings indicated that the catalytic activity varies based on the peptide's interaction with the Au surface, surface disorder, and the amount of exposed Au, influencing the reduction of 4-nitrophenol.174
| Name | Sequence | ΔG (kJ mol−1) | Diameter (nm) |
|---|---|---|---|
| CPN3 | DLRSCTACAVNA | −28.7 ± 0.9 | 4.1 ± 0.8 |
| AuBP1 | WAGAKRLVLRRE | −40.7 ± 2.1 | 3.1 ± 0.7 |
| AuBP1–CPN3 | WAGAKRLRREGGGG-DLRSCTACAVNA | −28.6 ± 1.0 | 2.7 ± 0.7 |
| CPN3–AuBP1 | DLRSCTACAVNAGGGG-WAGAKRLVLRRE | −27.4 ± 1.6 | 2.8 ± 0.8 |
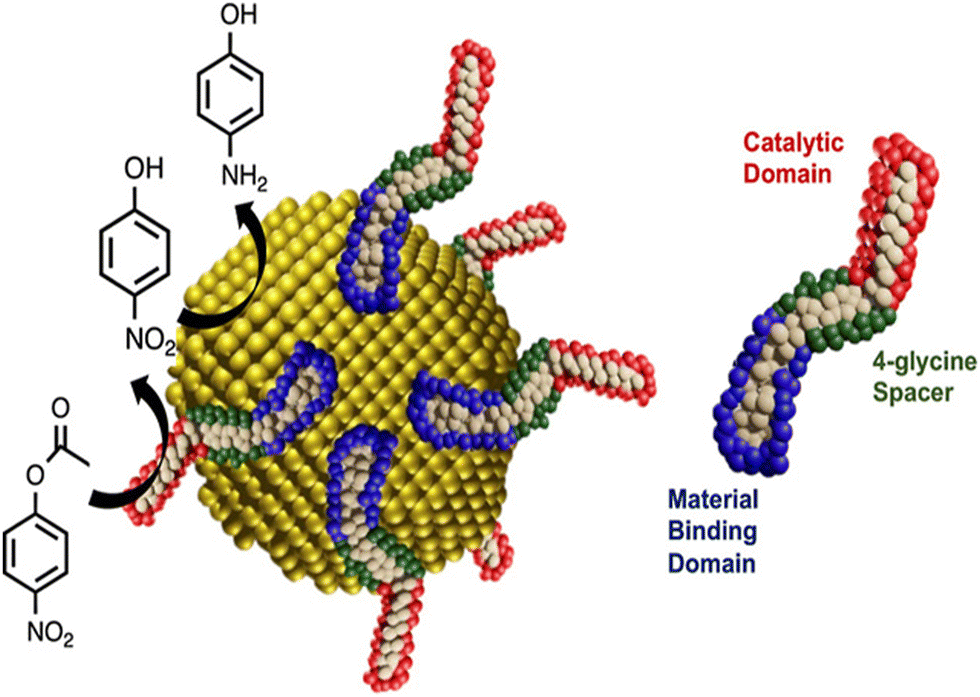 | ||
| Fig. 14 Scheme showing the Tandem catalysis in bio-interfaces. The multistep reaction is driven by the binding of catalytic peptides to Au nanoparticles. Right side is the diagram of multicomponent peptide. The peptide interface stimulated the hydrolysis of 4-nitrophenyl acetate to produce 4-nitrophenol. The multistep activity was then demonstrated at the Au surface by converting this intermediate to 4-aminophenol. Adapted with permission from ref. 173. Copyright 2023 American Chemical Society. | ||
Banerjee and Das et al. developed a cladding technique using covalent organic frameworks (COFs). It has also been shown that these co-assemblies could function as catalysts in organic solvents. The process produced a uniform dispersion of peptide nanotubes within the COF backbone, and a potent noncovalent interaction kept the peptides from leaking out of the COF. Fmoc-based solid-phase synthesis was utilized to create peptide-amphiphiles (C10FFVK and C10FFVR) functionalized with decanoic acid. Homogeneous nanotubular structures were produced by the C10FFVK and C10FFVR peptide-amphiphiles self-assembling in 40% acetonitrile/water (0.1%TFA) for 30 days. The peptide-amphiphile nanotubes C10FFVK demonstrated enzyme-like characteristics and effectively catalyzed the cleavage of C–C bonds in a buffer medium with a pH of 7.5, using interfacial crystallization (IC). They have also synthesized TpAzo–C10FFVK and TpDPP–C10FFVK nanotubular structures via COF cladding (Fig. 15). The armoured peptide nanotube's high stability and ease of separation allowed for up to ten cycles of C–C bond cleavage catalysis.175
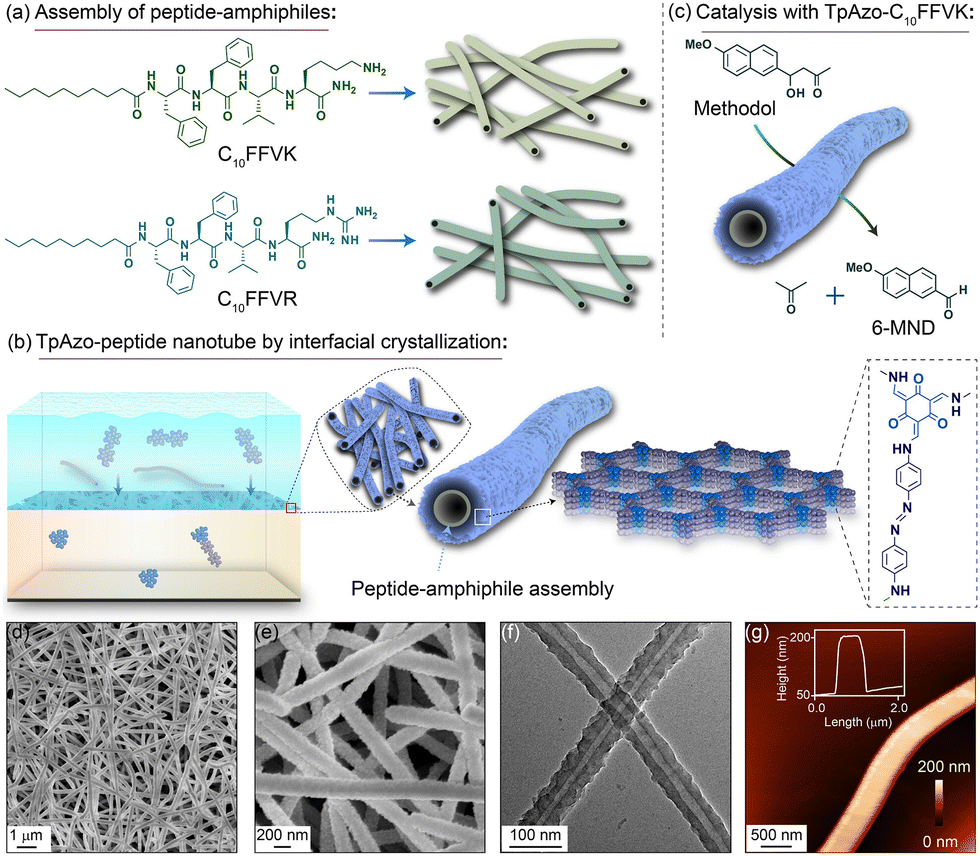 | ||
| Fig. 15 Diagrammatic representation of the process: (a) self-assembly of C10FFVK and C10FFVR peptides into nanotubes, (b) synthesis of TpAzo-peptide nanotubes via interfacial crystallization, and (c) catalysis with TpAzo–C10FFVK nanotubes. (d) and (e) SEM images, (f) TEM image, and (g) AFM image of TpAzo–C10FFVK nanotubes. Adapted with permission from ref. 175. Copyright 2023 American Chemical Society. | ||
Dai et al. demonstrated how azobenzene-terminated peptides (Azo-GFGH) could self-assemble on the surface of β-cyclodextrin-capped gold nanoparticles (AuNP@CDs) to produce a cascade catalyst (AuNP@CDs-Azo-GFGH) with dual-catalytic sites. The peptide assembly disintegrated when exposed to UV radiation because of the photoisomerization of the azobenzene molecule. By using AuNP@CDs as a catalyst and NaBH4 as a reducing agent, the hydrolysis product 4-nitrophenol can be reduced to 4-aminophenol (Fig. 16). The catalyst combines hydrogenation and esterase activities in a single pot, a process known as photo-switchable cascade catalysis.176
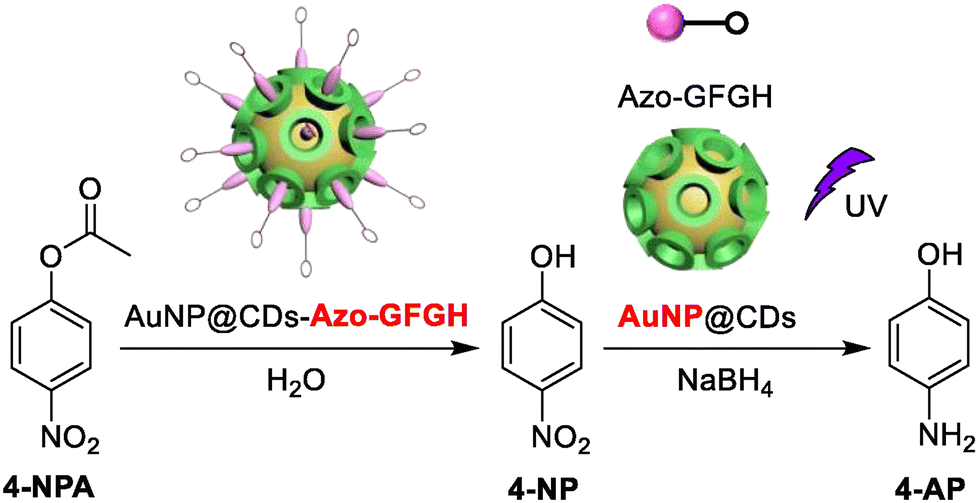 | ||
| Fig. 16 Schematic showing the hydrolysis reaction of 4-nitrophenyl acetate followed by the reduction of 4-nitrophenol. Adapted with permission from ref. 176. Copyright 2021 Elsevier Inc. | ||
Koksch et al. presented the first example of a Pep-Au-MPC catalyst, called Au@E1H8. It combined the esterase and hydrogenation activities to catalyze two different chemical reactions in a controlled manner in the same pot. With an excess NaBH4, the Au-NP surface can act as catalyst under the same reaction conditions (Fig. 17). A crucial property that nature uses to control the catalytic activities of enzymes is the self-assembled peptide monolayer's ability to mimic the hydrogenation catalyst for the subsequent reduction of a nitro-containing substrate. The peptide–gold nanoparticle conjugate that was created offers a proof of concept for the formation of peptide-noble metal catalysts that can effectively carry out two different reactions in succession in a single pot.177 The location adjusted the catalytic activity of peptide-Au-MPCs. Three conjugates of peptide and gold nanoparticles, which differ only in the catalytic unit location, were examined. It was discovered that the substrate's hydrophobicity and the area where catalysis occurs are related to the rates of catalyzed ester hydrolysis.177 Over the past decade, peptide catalysts have emerged as versatile alternatives to enzymes and small synthetic catalysts for a wide range of reactions, with their modular nature and ease of synthesis allowing for simple structural and functional modifications that enable fine-tuning of catalytic properties.178
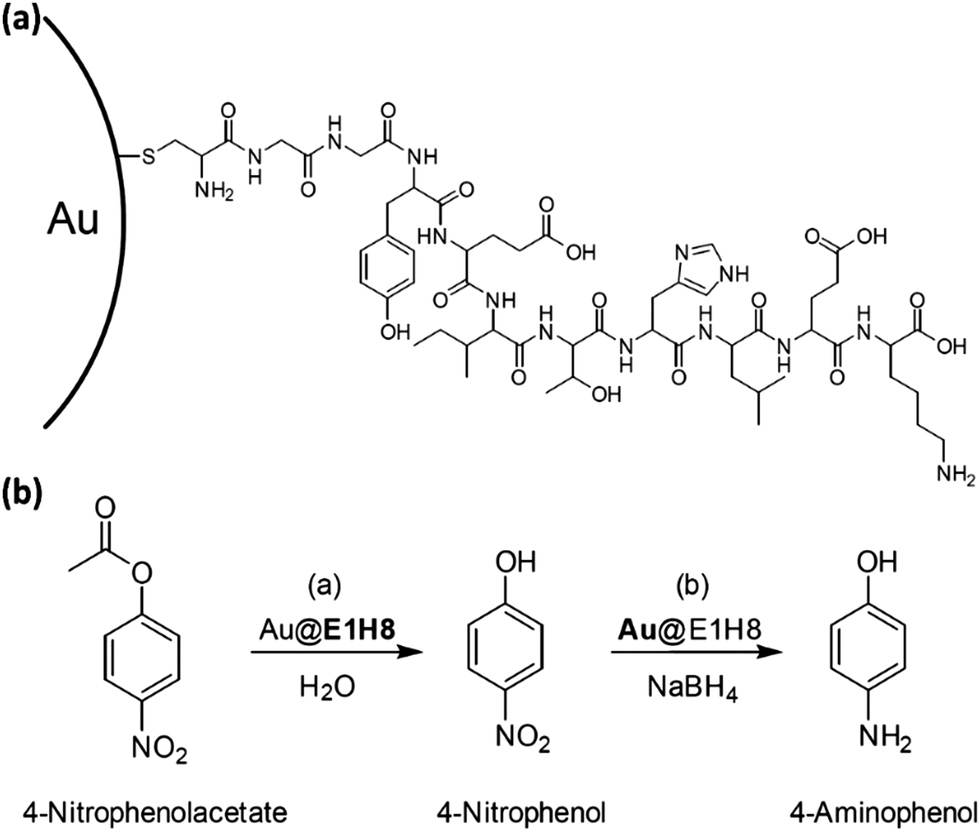 | ||
| Fig. 17 (a) Diagram illustrating the conjugation of the E1H8-peptide chain to the Au-NP surface. (b) Diagram showing the hydrolysis reaction of 4-nitrophenolacetate to 4-nitrophenol, followed by the reduction to 4-aminophenol using NaBH4 as a reducing agent and Au@E1H8 as a catalyst. Adapted with permission from ref. 177. Copyright 2018 Wiley-VCH GmbH, Weinheim. | ||
Recently, Shen and coworkers have synthesised molecules of the form HS-Cn+1-His-Phe (n = 4, 7, 11, 15, and 17) with the SH group and His-Phe dipeptide in the terminal positions, which were connected by hydrophobic alkyl chains of different lengths. This dipeptide molecule helped to catalyze the hydrolysis reactions of various carboxylic esters, namely DNPB, p-NPB, DNPA and p-NPA. Interestingly, multivalent nanofibers were produced due to the self-assembly of this dipeptide molecule in aqueous solutions, which was fueled by the H-bonding and hydrophobic interactions between two His-Phe dipeptides. The catalytic activity was measured using the Michalis–Menten kinetics and it was found to increase with the increase in the chain length between SH group and His-Phe dipeptide.179
Peptides have the ability to mimic the enzyme system, enabling them to facilitate various catalytic processes.180 Numerous artificial enzymes based on peptides can exhibit catalytic activities akin to that of natural enzymes such as hydrolases, peroxidases, laccases, and aldolases. Aldolases are enzymes that have a key function in metabolic processes and which are capable of catalyzing the synthesis or cleavage of carbon–carbon (C–C) bonds to produce various organic molecules. In this regard, Wennemers and coworkers showed that a self-assembled amphiphilic tripeptide (H-D-Pro-Pro-Glu-NHC12H25) mediated asymmetric conjugated addition reactions of aldehydes to nitroolefins in water with high stereoselectivity, as opposed to the relatively low stereoselectivity of its parent tripeptide (H-D-Pro-Pro-Glu-NH2).181 An important breakthrough was made by Das and coworkers when they created an amyloid-like nanotube that could catalyze the forward and retro-aldol reactions. The amphiphile peptide C10-FFVK was utilized to form the nanotube with exposed Lys residues, demonstrating clear catalytic activity in the direction of the methodol to 6-methoxy-2-naphthaldehyde hydrophobic interactions. This was the first known artificial retro-aldol reaction. The high-density array of proximal Lys and Tyr residues on the surface of the amyloid-like nanotube could be the result of Fmoc-Tyr molecules adhering to it through an enzyme made of peptides that was able to catalyze a cascade reaction involving the cleavage and formation of C–C bonds.182 One common family of copper-dependent oxidases is laccases, which oxidize various organic substrates by using oxygen as an electron acceptor. Laccases are therefore frequently used to break down organic pollutants and have long been regarded as green catalysts. In natural laccases, the coordination of copper ions with His and Cys is predominant. He and coworkers used self-assembled Cys-His dipeptide and copper chloride as a precursor to hydrothermally synthesise an artificial enzyme that mimics laccase. The chromogenic reaction involving 2,4-dichlorophenol and 4-aminoantipyrine was employed to assess the catalytic capability of the nanocatalyst. This artificial enzyme demonstrated a higher catalytic activity than that of laccase.183 Using a similar synthetic method, they developed another artificial enzyme mimicking laccase, this time using Cys-Asp as the dipeptide precursor instead of Cys-His, and the Cys-Asp-based enzyme demonstrated superior catalytic activity to the Cys-His-based one.184
Peroxidases are enzymes that catalyze oxidation reactions between peroxides and reducing substrates. Yan and coworkers synthesized peroxidase-mimicking nanozymes by co-assembling Fmoc-His and hemin, mimicking the heme-His coordination found in the active site of horseradish peroxidase (HRP). The enzyme's peak catalytic performance (FH/hemin = 4:1) was comparable to that of HRP.185 He and coworkers used ferrocene (Fc) as a prosthetic group in place of heme to create an artificial enzyme based on a peptide that mimics peroxidase. By self-assembling ferrocene-modified tripeptide (ferrocenyl-Phe-Phe-X), where X stood for Phe, Asp, His, or Arg, the artificial enzyme was created.186 According to research by Korendovych and coworkers, the self-assembled peptide Ac-VHVHVQV-NH2 exhibited peroxidase-like activity without needing a cofactor. They postulated that the catalytic activity could be attributed to the substrate's altered redox potential following adsorption onto the self-assembled peptides.187
Stupp and coworkers created a peptide amphiphile by combining a short peptide sequence (including two His residues) with a palmitoyl group. The peptide amphiphile formed ordered nanofibers with a high aspect ratio. This catalyst showed significant esterase-like activity in the hydrolysis of 2,4-dinitrophenyl acetate (DNPA).188 Das et al. reported that the Im-KLVFFAL-NH2 peptide (Im-KL) formed an amyloid nanotube. The hydrolysis of ester substrates was facilitated by the self-assembled nanotube via the establishment of reversible covalent bonds. The imidazole group and terminally adjacent Lys residue in the intricately constructed peptide sequence were visible on the nanotube surface during self-assembly. The imidazole group hydrolyzed the substrate's ester bond, while the Lys residue helped in anchoring the substrate by forming a Schiff imine. When the Lys residue was swapped out for Arg, ornithine, or Glu, the self-assembled nanotube's esterase-like catalytic activities dramatically decreased.189 Pal and coworkers created two different peptide amphiphiles (Fmoc-VFFAHH and Cou-VFFAHH) by integrating the amyloid-forming peptide fragment (VFFA). The Fmoc- and Cou-groups at the N-terminal were employed as stimuli-response units, and the His residues at the C-terminal acted as active sites for catalyzing the hydrolysis reactions. When exposed to external stimuli such as heat, light, or chemical cues, the two peptide amphiphiles demonstrated pathway-driven self-assembly behaviours that resulted in various nanostructures and adaptable esterase-like catalytic activities directed toward p-nitrophenyl acetate hydrolysis.190 The aforementioned group additionally exhibited seed-promoted elongation of homochiral nanofibers from enantiomeric peptide amphiphiles (C10-L/D-VFFAKK) and chirality-driven self-sorting of the VFFA fragment. This opened up the possibility of creating supramolecular active sites based on peptides that might have high catalytic stereo-selectivity and chiral configuration.191
3.3. Biomedical applications
Self-assembled peptides offer a versatile platform for various biomedical applications due to their unique properties such as tunable nanostructures, biocompatibility, and the ability to mimic the extracellular matrix. In this section, we describe some of the key biomedical applications192–194 of self-assembled peptides, though detailed biomedical applications of peptides have been reviewed in recent years.17,32,195–197 These self-assembled nanomaterials provide a versatile and customizable platform for addressing various challenges in tissue engineering and regenerative medicine. Their biomimetic nature (mimicking the extracellular matrix) and the ability to fine-tune their properties make them promising candidates for developing advanced solutions to promote cell adhesion, proliferation, tissue repair and regeneration. Ongoing research continues to explore innovative applications and refine the design of peptide-based nanomaterials for enhanced clinical outcomes.198,199 Peptide-based nanostructures have attracted considerable attention in the field of drug delivery due to their various challenges associated with traditional drug delivery, offering improved targeting, reduced side effects, and increased therapeutic effectiveness. Ongoing research aims to optimize the biomedical advantages.These nanostructures can be engineered to encapsulate therapeutic agents, protect them from degradation, and enable controlled release.21,200–209 Peptide-based drug delivery systems leverage the unique properties of peptides to enhance the delivery of therapeutic agents. These systems show promise in addressing and expanding the applications of peptide-based drug delivery in various medical contexts.17,208,210,211 Along with other stimuli, pH plays a crucial role in drug delivery and tissue engineering within self-assembled peptide-based systems. The pH is particularly significant since it changes abruptly in the cells affected by various diseases. The drug molecules are encapsulated within the self-assembled system, which can subsequently disassemble to release the drug based on the pH of the affected cell. This approach consistently serves as a targeted drug delivery agent to diseased cells. It has been reported that the presence of certain acidic and basic amino acids enhances the self-assembly process of the system, which can then disassemble at a specific pH to release the drug at the target site. In this regard Lei et al. demonstrated a lignin-H nanoparticle, which encapsulated the drug 10-hydroxycampothecin (HCPT).212 Their study demonstrated that nanoparticles of size 30–40 nm could penetrate the cell membrane and release the drug in the acidic tumor microenvironment, owing to the presence of imidazole groups in the molecule. Ghosh et al. also demonstrated that an octapeptide, a double mutant of Galectin-1, could form a hydrogel that exhibited pH-responsive behavior in different environments (Fig. 18).45 This discovery showed that the peptide could encapsulate dyes at a physiological pH of 7.4, while get disassembled at pH 5.5 under acidic conditions. They demonstrated the biocompatibility of the peptide and its potential as a targeted drug delivery agent for cancer cells. The presence of free acidic and amine groups in the peptide facilitated disassembly at pH 5.5, aiding in drug release. In a recent review, Nisbet et al. discussed the potential of self-assembled peptide-based hydrogels as effective drug delivery systems for transporting drugs to cancer cells.213 They have demonstrated that chemotherapeutic agents such as Docetaxel, Doxorubicin, Doxorubicin, Erlotinib, Oxaliplatin, Paclitaxel, 5-Fluorouracil, and 10-Hydroxycamptothecin can be delivered to targeted cancer cells using specific peptide sequences.
 | ||
| Fig. 18 Molecular structure of the peptide and hydrogel formed by the peptide with nanofibrillar morphology. The peptide can encapsulate the dye after the formation of self-assemblied nanofibers at pH 7.4 and release it at pH 5.5, thus acting as a targeted drug delivery agent. Adapted with permission from ref. 45. Copyright 2019 American Chemical Society. | ||
Peptide-based nanomaterials represent a cutting-edge approach in cancer therapy, leveraging the unique properties of peptides to design advanced platforms for diagnosis and treatment. These nanomaterials, typically self-assembled peptides or peptide-functionalized nanoparticles, exhibit several advantages and can be explored in treating cancers in numerous ways213–220 as follows: (a) targeted drug delivery: peptide-based nanomaterials can be engineered to selectively target cancer cells by incorporating specific peptide ligands. This targeted approach improves drug delivery precision, reducing off-target effects and enhancing therapeutic efficacy. (b) Photothermal therapy: some self-assembled peptides can absorb near-infrared light, enabling photothermal therapy for cancer treatment by inducing localized hyperthermia and cell death. (c) Nanocarriers for chemotherapy: peptide-based nanomaterials serve as efficient carriers for chemotherapeutic drugs.221–223 These nanocarriers protect the drugs from degradation, improve their solubility, and enable controlled release at the tumor site. (d) pH-responsive systems: some peptide-based nanomaterials are designed to respond to the acidic tumor micro-environment.36,214,224 This pH responsiveness can be exploited for triggered drug release, enhancing therapeutic efficacy.213,216–220,225–231 The microtubule, one of the three cytoskeleton types found in eukaryotes, is vital to the cell cycle. For the treatment of cancer, microtubule-targeting medicines remain the most reliable class of antineoplastic medications. Ghosh et al. designed an antimitotic peptide, which has depolymerizing effects on the microtubule. The peptide generated β-sheet structures upon self-assembly. AFM studies revealed the formation of a homogeneous vesicle structure when 400 μM of the peptide was incubated at 37 °C. Moreover, at a 400 μM concentration, the peptide showed considerable cytotoxicity on tumorigenic Michigan Cancer Foundation-7 (MCF7) cell lines while remaining non-cytotoxic towards Michigan Cancer Foundation-10 A (MCF10 A), which corresponds to a non-cancerous epithelial cell line, according to in vitro experiments conducted on two distinct cell lines. Through various in vitro analyses, including turbidity assay and 4′,6-diamidino-2-phenylindole (DAPI) fluorescence quenching, they concluded that the peptide interferes with the tubulin–tubulin interaction between important microtubule subunits, hence disrupting the microtubule assembly. The drug loading and release efficiency of the peptide vesicles were examined using a fluorescently tagged Docetaxel. The analysis of intracellular microtubule structures using immunocytochemistry and cell viability studies showed that the peptide vesicles can hold both Docetaxel and RAD001, which significantly lowers the IC50 (half-maximal inhibitory concentration) value when compared to treating Docetaxel and RAD001 separately to MCF7 cell lines.232 Das et al. demonstrated that two peptides (S1 and S2) with opposite charges may self-assemble when a Phe-Leu dipeptide unit was fused with either the negatively charged amino acid L-Glu (S1) or the positively charged amino acid L-Arg (S2). S1 and S2 self-assemble to form distinct supramolecular nanostructures (Fig. 19a and b). These self-assembled structures are non-toxic and proteolytically stable. Additionally, the chemotherapeutic drug (Dox) can be encapsulated by these self-assembled superstructures made from S1 and S2, efficiently transporting it into malignant cells while preserving its therapeutic efficacy. Dox-encapsulated S1 and S2 (Dox-S1 and Dox-S2) were identical to the unencapsulated medication in their capacity to successfully stop the growth and induce cell death in cancer cells (Fig. 19c).233
 | ||
| Fig. 19 (a) Molecular structures of S1 and S2, (b) and (c) FESEM images showing the morphology of S1 and S2 assemblies in 100% aqueous conditions. (d) A comparative analysis of their drug release capabilities. Adapted with permission from ref. 233. Copyright 2022 Wiley-VCH. | ||
Neurodegenerative diseases are a group of disorders characterized by the progressive degeneration of the structure and function of the nervous system, particularly the neurons. These conditions are often chronic, debilitating, and can lead to severe impairment of cognitive, motor, and/or sensory functions. Some of the most common neurodegenerative diseases are Alzheimer's disease (AD) and Parkinson's disease (PD) where AD is a progressive and irreversible neurodegenerative disorder that primarily affects the brain, leading to a decline in cognitive function, memory loss, and changes in behavior. It is the most common cause of dementia, accounting for a significant portion of dementia cases in the elderly population.234,235 PD is also a progressive neurodegenerative disorder that primarily affects movement. Parkinson's disease is marked by the degeneration of dopaminergic neurons in the substantia nigra, leading to a dopamine deficiency that disrupts motor function regulation.236–238 Multiple ways have been employed for using peptide-based nanomaterials to treat AD and PD, which include (a) amyloid beta (Aβ) aggregation inhibition; (b) drug delivery across the blood–brain barrier (BBB); (c) targeted drug delivery to neurological cells; (d) use of neuroprotective nanomaterials; (e) β-amyloid clearance; (f) dopamine replacement and delivery; (g) and the inhibition of α-synuclein aggregation.236,239–245 Self-assembled peptide-based nanomaterials have demonstrated promising antibacterial activities and are being explored as potential candidates for combating bacterial infections. The unique properties of these nanomaterials, such as their tunable structures and biomimetic nature, make them attractive for developing innovative antibacterial strategies. Self-assembled peptide-based nanomaterials demonstrate antibacterial activities via several mechanisms: (a) membrane disruption techniques where self-assembled peptide-based nanostructures can interact with bacterial membranes. These interactions may lead to membrane disruption, causing leakage of intracellular contents and bacterial cell death. The amphiphilic nature of some self-assembled peptides facilitates their interaction with bacterial membranes; (b) selective targeting techniques where peptide sequences can be designed to selectively target bacterial cells while sparing host cells.246,247 This selectivity enhances the specificity of antibacterial action, minimizing potential side effects.23,44,247–250
In a recent review, Xiong et al. highlighted self-assembled peptides as delivery agents and the potential of various antimicrobial drugs such as Pexiganan, Iseganan, D2A21, and Omiganan.23 The secondary structure of self-assembled peptides plays a crucial role in their antimicrobial and hemolytic activities. Typically, charged peptides with free acid and amine groups enable self-assembled peptides to interact with bacterial membranes and kill the bacteria. In particular, the α-helix and β-sheet structures formed by these peptides contribute to these processes. Antimicrobial peptides (AMPs) capable of conformational changes in response to specific infectious microenvironments can enhance selectivity while minimizing toxicity. However, significant progress is still required to advance these conformation-switching AMPs for clinical use. To date, the exact mechanism by which radially amphiphilic (RA) AMPs interact with and disrupt bacterial cell membranes remains unclear and requires further investigation. Alongside, numerous studies and reviews have explored the connection between helical structures and antibacterial activities.
Gene delivery using self-assembled peptides is a promising area for non-viral gene therapy due to the customizable and biocompatible nature of peptides. Peptides can be engineered to create nanoparticles that enclose DNA, allowing for secure and efficient delivery to target cells. The process begins with the electrostatic interaction between the positively charged peptides and the negatively charged DNA, leading to the formation of nanoparticles (Fig. 20).251 These nanoparticles not only protect the DNA from degradation but also facilitate its internalization into cells. Peptides offer several advantages in gene delivery, including their ability to self-assemble into well-ordered structures under specific conditions, forming stable carriers for genetic materials.128,252 One of the key challenges in gene therapy is overcoming biological barriers such as endosomal escape and enabling the successful release of DNA into the cell's cytoplasm. Peptides can be engineered to respond to environmental cues such as pH changes, facilitating DNA release from the nanoparticle through endosomal disruption mechanisms such as the proton sponge effect. For instance, peptides with histidine residues that have a pKa close to the endosomal pH can cause membrane destabilization, facilitating the release of DNA. Combining peptides with other materials such as lipids or synthetic chemistry can improve the efficiency of DNA transfection. Additionally, innovative DNA designs such as synthetic or modified plasmids can be combined with peptide carriers to extend gene expression and enhance therapeutic results. Overall, self-assembled peptide-based gene delivery systems provide a flexible, efficient, and biocompatible platform that could revolutionize non-viral gene therapies. Their programmability, stability, and ability to form nanoparticles make them a highly promising alternative to viral delivery methods. Drugs containing nucleic acids have also been known to treat viral infections, cancers, and AIDS.253–256
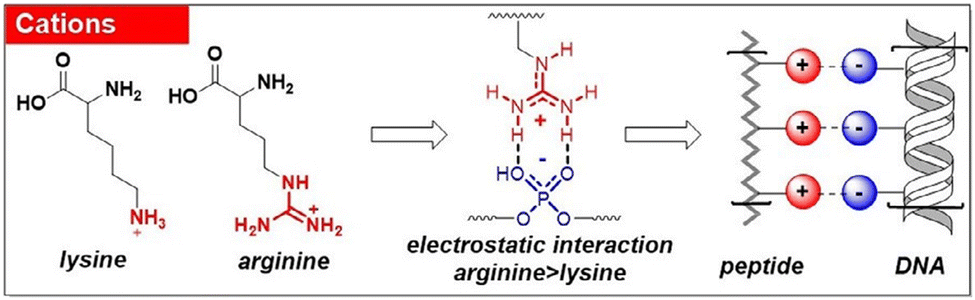 | ||
| Fig. 20 Molecular structure of cationic peptides, which contains positively charged regions, helps them bind to the negatively charged phosphate groups of DNA via electrostatic interactions. This interaction plays a key role in supporting the self-assembly of peptides for gene therapy. Adapted with permission from ref. 251. Copyright 2022 Elsevier Inc. | ||
Zhang et al. developed multifunctional dipeptide nanoparticles (DNPs) using the dipeptide Trp-Phe, which self-assembled through chelation with zinc ions, an essential metal in cellular processes. To improve targeting and therapeutic efficacy, they combined the chemotherapy drug Clofarabine (Clolar) with the AS1411 aptamer (which targets the role in cell division; Fig. 21). Additionally, Doxorubicin (DOX), a chemotherapy drug, was incorporated via π–π stacking interactions, resulting in composite nanoparticles DNPs/Clolar/AS1411/HA/RNA/DOX. This design aimed to provide a combination therapy, using both chemotherapy and gene silencing to enhance the antitumor effects. The multifunctional nanoparticle system demonstrated successful MCF-7 cells and an influenza hemagglutinin peptide (HA) to promote endosomal escape. The DNPs were further functionalized by modifying the carboxyl groups to attach siRNA, targeting Thymidine Kinase 1 (TK1), which plays a crucial endosomal escape and significant antitumor activity in vitro and in vivo, showcasing its potential as an advanced targeted cancer therapy.257 Self-assembled peptide-based nanomaterials have gained considerable interest in the field of bioimaging due to their unique properties, and the ability to encapsulate or conjugate imaging agents. These nanomaterials offer versatile platforms for designing contrast agents, probes, and imaging systems for various biomedical applications.32,41,258 Self-assembled peptide-based nanomaterials are utilized in bioimaging through various techniques including nuclear imaging (PET and SPECT), targeted imaging, responsive imaging, and multimodal imaging.32,52,258–261 Wang et al. have demonstrated a peptide-based probe in the near-infrared range that can be utilized for tumor imaging. The molecule is designed with a peptide backbone and a cyanine probe that includes hydrophilic motifs, a tailoring motif, a self-assembly motif, and a cyanine dye. They synthesized four different molecules and demonstrated that molecule 1 was responsive to fibroblast activation protein-α (FAP-α), forming nanofibers in situ on cancer-associated fibroblasts (CAFs). A 5.5-fold signal enhancement was observed after 48 hours of applying the material to the cells (Fig. 22). The molecule was capable of detecting tumors as small as 2 nm in diameter. The molecule is designed with a peptide backbone and a cyanine probe that includes hydrophilic motifs, a tailoring motif, a self-assembly motif, and a cyanine dye. When the molecule was attached to CAFs, it broke down into two components, as confirmed by HPLC. This breaking facilitated the molecule's reassembly into a β-sheet secondary structure with fibrillar morphology. As a result of this, the properties enhanced 5.5 times more than those of normal cells.262 Another type of peptide-based hydrogel has been identified for biosensing applications, capable of detecting superoxide anions released by cells in 3D culture in response to drug treatments. For instance, King et al. have shown an octapeptide linked to a DNA recognition sequence for detecting hybridizing DNA via fluorescence. For detecting superoxide anions from cells in 3D culture, Lian and colleagues developed a Fmoc dipeptide hydrogel with embedded HeLa cells, HRP, and superoxide dismutase (SOD). This setup provided accurate and sensitive detection of superoxide anions under physiological conditions.263
 | ||
| Fig. 21 Chemical structure of the dipeptide Trp-Phe and schematic representation of its self-assembly into nanoparticles in the presence of Zn2+ ions. The assembled dipeptide nanoparticles (DNPs) are utilized for targeted cancer therapy and gene therapy applications, incorporating chemotherapy drugs, aptamers, and gene-silencing molecules for enhanced therapeutic efficacy. Adapted with permission from ref. 257. Copyright 2021 Elsevier Inc. | ||
 | ||
| Fig. 22 (a) Schematic diagram illustrating molecular recognition and tailoring, along with their self-assembly, which leads to the formation of a β-structure and fibrillar network, enhancing tumor imaging sensitivity. (b) and (c) Molecular structure of molecule-1 and its various motifs that contributed to its self-assembly. Adapted with permission from ref. 262. Copyright 2019 Wiley-VCH. | ||
In addition, peptide-based nanomaterials have been used for the purposes of vaccination, and treatment of asthma, cardiovascular diseases (CVDs), and hypertension, and particular bioactive collagen peptides have been exploited to enhance muscular strength and improve body composition.258–260,264
3.4. Peptide as a conducting material
The presence of diverse directionally oriented functional groups in the peptide self-assembly is recognized for enhancing electrical transport within that specific configuration. It has been found that the peptides can participate in electronic conduction in two different ways: “Short-range electron transfer (ET)”, where electron exchange takes place via an oxidation–reduction pathway between the ionically active electrolyte and the peptide. The screening of electrons in the solution leads to a modification in electrical potential throughout this process. Furthermore, short-range electron transport (ETp) occurs by the electron transport through the peptide without any electrolyte or electrolytic participation.265–268 The mechanism behind the electrical conductance is not very clear and often poorly understood. There are several mechanisms for short-range electron conduction where electron tunnelling occurs through a single-step electron transfer process between the donor and acceptor molecules. Such kind of electron transport rate depends on the distance between the donor and acceptor molecules. Temperature has been observed to play a crucial role in influencing this tunnelling process. To understand solid-state electron transfer across single peptides in the ETp process, a number of multistep electron transport models such as hopping, super-exchange, and flickering resonance have been developed.269The band gap order in the majority of peptides was typically calculated to fall within the range from semiconducting (≤4 eV) to insulating (>4 eV) (Scheme 5). The presence of π-electron-containing residues was discovered to have a substantial influence on the electron transport process within the peptide nanostructure. The presence of π–π stacking in these aromatic rings may lead to a reduction in the band gap (Eg) compared to peptides without aromatic residues. This π-stacking plays a crucial role in regulating the semiconductor or metallic characteristics exhibited by peptides, while electron transport facilitates the organic supramolecular polymeric peptide to function as a conducting material. The electrical transport phenomenon throughout the assembly is also influenced by both the secondary and nanostructure of the material. As in the secondary structure, the molecular dipole generated by the peptide self-assembly plays a major role in the dipole orientation from the N-terminus to the C-terminus. The impact of dipole moment, induced by the helical structure, increases by 5.0 D per residue for alpha helices. In contrast, for the beta strand structure, the dipole moment was elevated by 0.25 D per residue.270
 | ||
| Scheme 5 (a) Schematic of the band diagram of typical metal, semiconductor and Insulator. (b) General schematic representation of a device fabricated with self-assembled peptide systems for various applications. | ||
In this regard, Amdursky and co-workers have shown the effects of the π-electronic surface of the peptide residue in electrical conduction of the overall system. A small change of the π-residue from phenylalanine (in FF) to tryptophan (in FW) showed a significant increment in the transport process. Moving from FF to FW of the nanotube, the band gap was found to decrease from 4.48 eV to 3.04 eV. The conductivity measurement was performed by conductive probe atomic force microscopy, revealing that FW was five times more efficient than FF.271 The electrical conductivity in the solid peptide assembly was attributed to the ETp process in these nanorods. Meanwhile, the arrangement of these dipeptides played a crucial role in controlling the overall electronic structure and associated properties of the system. The system with FW with a larger π-surface exhibited a lower tunnelling barrier and band gap, making it more efficient in the electrical transport process than FF. The tryptophan unit is well known to impart a strong effect on the conductivity of the peptide system. A separate study conducted by Cahen and his co-workers demonstrated that monolayers of homopeptides containing tryptophan exhibited the most efficient electron transport, followed by lysine, glutamic acid, and alanine.272 Subsequent experimental and theoretical investigation explained the steady drop in conductivity, which was attributed to the secondary structure of the peptide and the residue charge of the constituent amino acid. DFT calculations revealed that the HOMO–LUMO energy barriers for helical peptides were lower than those for small homopeptides, an observation consistent with the experimental findings. The aggregation nature of small molecules plays a crucial role in the formation of organic supramolecular polymeric systems. This process aids in the creation of a polymer-like nanofibrous network, facilitating the conductivity of electrons and rendering the system semiconducting in nature.273 Peptide-based nanowires demonstrate impressive inherent conductivity attributed to electron delocalization facilitated by closely aligned aromatic amino acids. Additionally, the conductivity is influenced by a hopping/charge transfer mechanism, contributing to the understanding of self-assembled peptides as conductive materials.
Ing et al. demonstrated that only lower concentrations of peptides (relating to lower assembly and branching) can increase the conductance, due to decreasing fibril–fibril contact resistance. As peptide concentrations are reduced, the chance of more assembly and branching among peptides will also be reduced due to the decrease in fibril–fibril contact resistance.274 The conductivity of dried hydrogel films surprisingly decreased as the peptide concentration increased. They compared the values obtained from electrical impedance spectroscopy (EIS), and the conductivity of the hydrogels were also measured. They also found concentration-dependent conductance variance, as reported earlier.274 Nanofibers formed by self-assembly are more conductive in nature than the normal amino acids or proteins. This is due to easier electron transport that occurs over a long distance and nanofibers formed by more π-stacking, extended conjugation or redox centres than the normal amino acid.275 Laycock et al. showcased two peptides, with one incorporating phenylalanine as an aromatic residue and the other being replaced with leucine. They employed two different peptide sequences and compared the conductivity within the peptide gel (Fig. 23). Although both peptides were capable of forming fibrillar networks via a self-assembly process in water, the conductivity was observed to be higher for the phenyl group than the leucine moiety. The electrochemical transport test revealed that the fibres exhibited both ohmic electronic transport and a metallic-like temperature dependency of conductance in an aqueous buffer. A strong π-interaction between the phenyl residue makes the system efficient in electron transport process. These findings suggest that conductivity is likely dependent on the supramolecular structure present in the highly organized fiber shape at low peptide concentrations, indicating that the peptide itself may not be intrinsically conductive. As the concentration increases, the fibre length and density increase, making the system more complicated and less conductive in nature. The ohmic conduction observed in these two peptide nanofibers does not rely on redox hopping, π-stacking, or other thermally activated mechanisms for facilitating charge transport over micrometer-scale distances. Instead, it is insensitive to environmental changes and shows dependence on pH and ionic strength also. This study indicated that the peptide nanofiber formed due to supramolecular interaction with the help of aromatic amino acid side chains by overlapping their π-surface showed electron transportation through the micrometer long-range nano-assembly without any redox-active metal components.
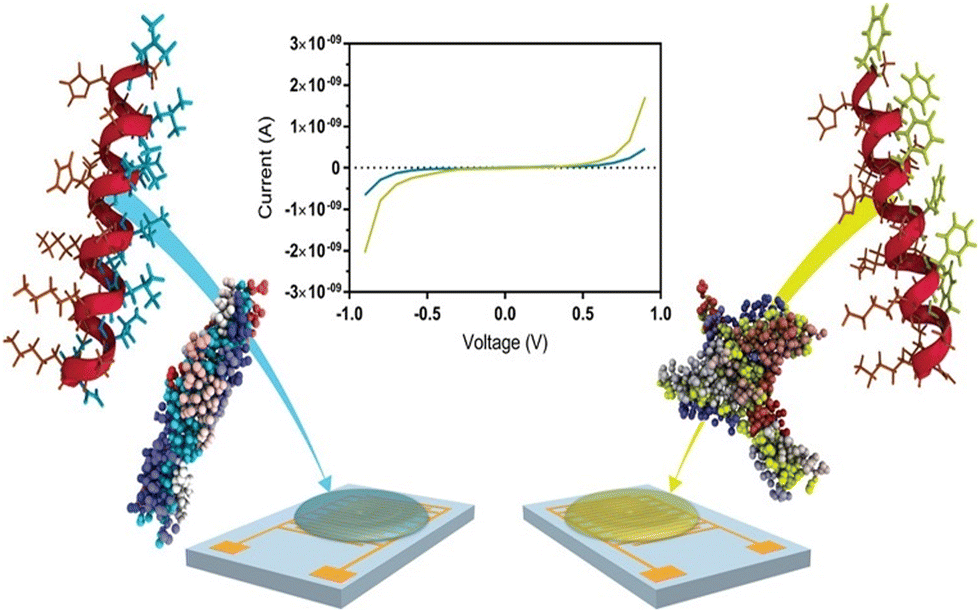 | ||
| Fig. 23 Comparison of the conductivity studies between the phenyl alanine and lysin motif and schematic of the conductivity comparison in the gel state. It is clearly mentioned that the lysin motif gave better results than those of the phenyl group owing to the free NH2 group that aids in giving more conductivity. Adapted with permission from ref. 275. Copyright 2019 American Chemical Society. | ||
In a recent study, Banerjee and colleagues synthesized a symmetric NDI (naphthalene diimide) molecule with terminal valine and histidine amino acids. This molecule displayed the monomeric state in DMSO (dimethyl sulfoxide) and transitioned to an aggregated state in the DMSO/buffer mix solvent system, forming a J-type aggregate (Fig. 24).273 The higher order molecular aggregation leads to gelation with a clear aggregation-induced emission property. The TEM image confirmed the presence of a 1D fibrillar network in the gel state, supporting the supramolecular polymerization via π–π stacking. They discovered that the nanofibrous network created by the molecule was sufficient to establish a channel for electron transport through a hopping mechanism, a phenomenon that is not possible in a non-aggregated or non-dispersed state. When the voltage gradient was applied, the movement of electrons was detected by measuring the current of the dry gel state. However, zero conductivity was observed in the dispersion state. The overall electrical conductivity arose from the synergistic effect of ionic conductivity contributed by Na+ ions in the phosphate buffer system and the conductivity originating from the one-dimensional orientation of the π-core. The molecule showed a conductance of 3.5 μS cm−1 due to the presence of conductive nanofibers in the aggregated state in the gel state, which remained intact after drying the gel. In the nanofibrillar aggregated state, the closely packed NDI core ultimately helped in lowering down the energy gap between HOMO and LUMO in this π-conjugated system, as reflected in the band gap measurements and conductivity. The photo-induced current was measured to account for the presence of photo-responsive NDI moieties in the system, revealing an Ion/Ioff ratio of 5.58. In temperature-dependent experiments, it was observed that the resistance of the dried gel decreased, indicative of the semiconducting properties of the material. Interestingly, the NDI-based peptide in the dried gel state exhibits no conductivity at a lower voltage bias; however, at a higher voltage bias, there was a sudden increase in conductivity. The diode-like nature of the I–V curve was ascribed to the movement of electrons at a lower voltage bias, while at a higher voltage bias, conductivity resulted from the contributions of both ions and electrons present in the system.
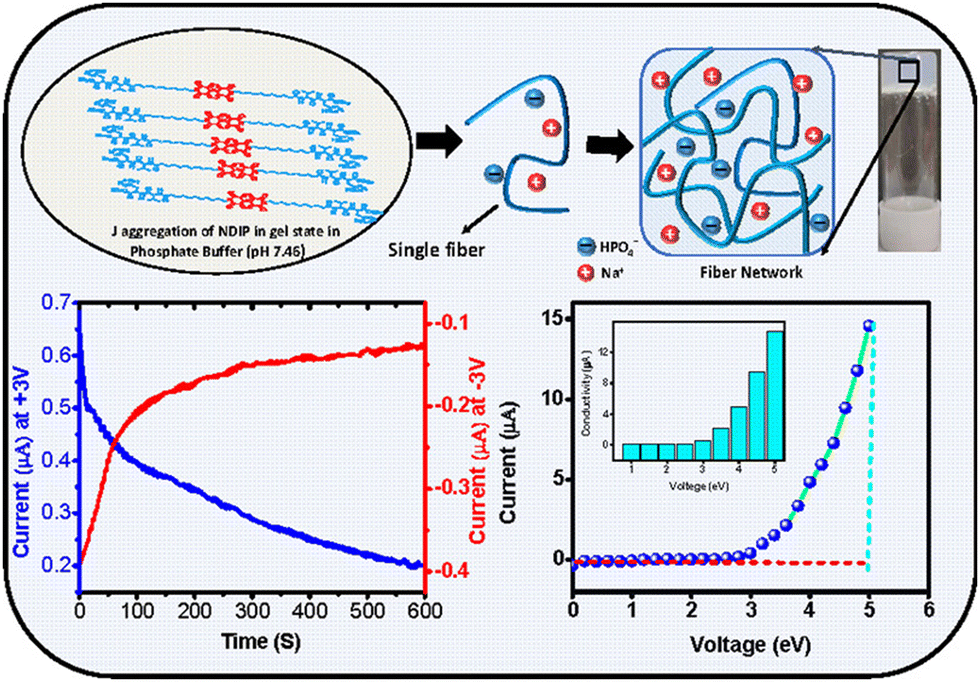 | ||
| Fig. 24 Schematic representation of the NDI-based peptide in a buffer medium (up). Current vs. time curve (down left) and current vs. voltage plot with statistical diagram in the inset (down right). The ionic conductivity is increased after the aggregation occurs, confirming the role of the single fibre to show more conductivity than others. Adapted with permission from ref. 273. Copyright 2024 American Chemical Society. | ||
In another example Das et al. reported the design, synthesis, and self-assembly of a peptide-quinquethiophene-based π-gel, which demonstrated the electrochromic property. The molecular design suggested a bolaamphiphile-type structure, featuring a redox-active photochromic π-core and peptide motifs at the ends, facilitating self-assembly. It formed a J-type aggregate through π–π stacking and H-bonding interactions in an aqueous medium, and the SEM imaging confirmed the fibrillar network structure formed by the molecular self-assembly (Fig. 25a). According to the earlier report, the molecule formed with oligothiophenes was found to exhibit effective charge carrier mobility by forming various nanostructures.276 The redox-active behavior of the system was confirmed through cyclic voltammogram (CV) experiments, where in the gel state, an oxidation peak at 1.21 V and a reduction potential peak at 0.58 V were observed. Further, the electrochromic property was studied by preparing a thin film of the gel on the ITO-coated glass surface. The yellow colour of the gel was found to disappear upon oxidation at 1.21 V, which reversibly returned to its original colour upon reduction at 0.6 V (Fig. 25b and c).
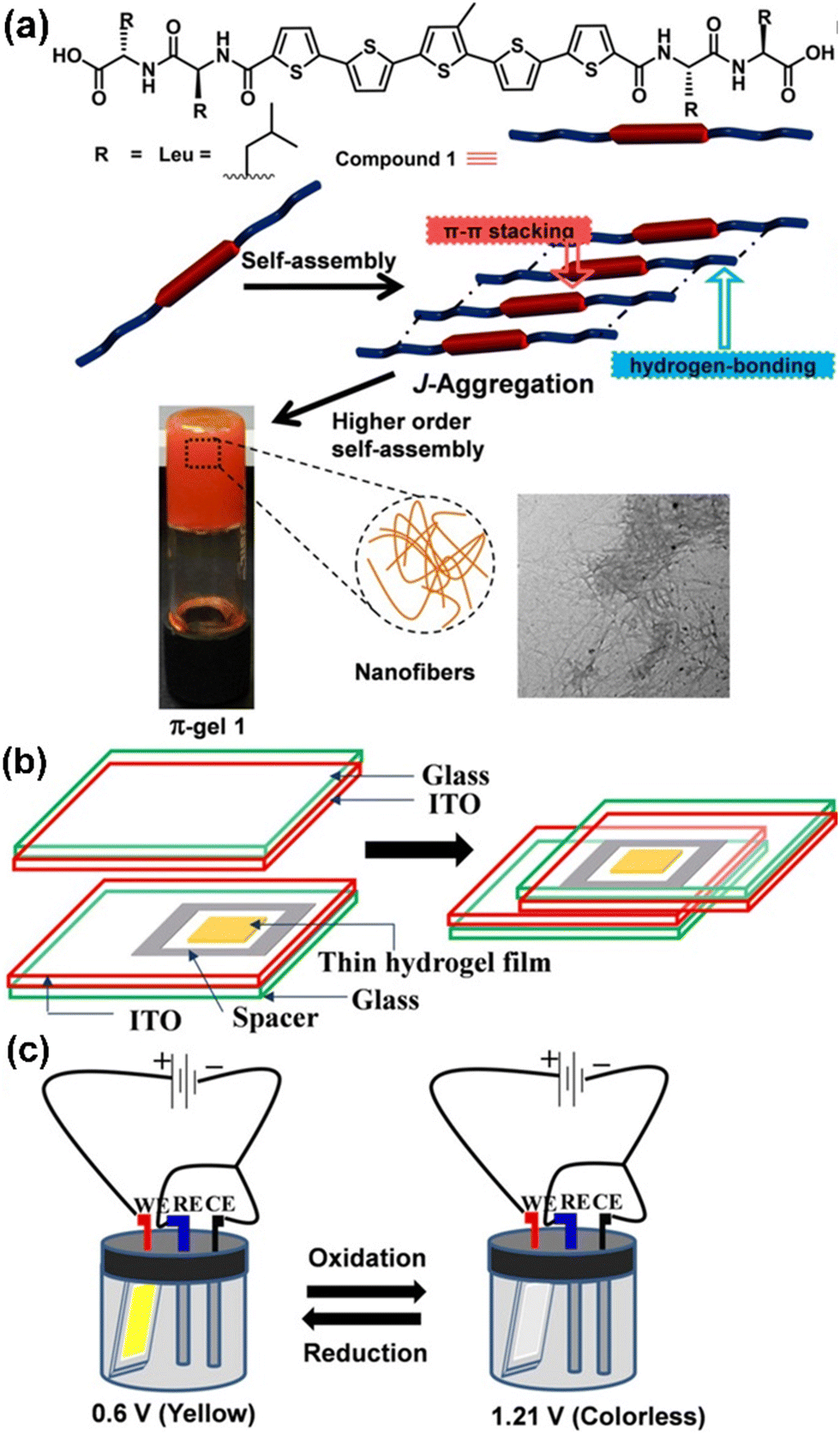 | ||
| Fig. 25 (a) Molecular structure of compound 1 and schematic representation of the gelation mechanism. The self-assembly process leads to the formation of nanofibrils in water and, consequently, gels. (b) Schematic of the thin-film device and (c) corresponding its electrochromic property upon oxidation and reduction. Fibrillar network formed upon aggregation helps the molecule to show conductivity. Adapted with permission from ref. 276. Copyright 2018 Wiley-VCH. | ||
Effective excitonic separation upon light irradiation is an exciting field of research that has found significant applications in areas such as photocatalysis, photoresponsivity, and photoconductivity. The donor–acceptor co-assembly is a well-recognized technique for effectively separating the electrons and holes produced following photoexcitation, resulting in extended lifetimes for both. In this regard, Martin et al. have reported the self-assembly and co-assembly of positively charged perylene di-imide-based system (p-type acceptor PDI) and negatively charged π-extended peptide-conjugated tetrathiafulvalene (n-type donor TTF).277 Both π-chromophoric units underwent self-assembly individually in water with H-type aggregation, leading to the formation of nanostructures with a fibrillar morphology. Moreover, when these individual nano-assemblies were coupled, they formed co-assemblies in water via dipolar interactions between the charged surfaces of the discrete nanofibers. The establishment of a highly organized nonfibrous co-assembly with alternating n and p characteristics was validated through UV-vis, CD spectroscopic studies and XRD analysis. The charge carrier mobility was examined by flash-photolysis time-resolved microwave conductivity measurement. Upon photoexcitation with a 355 nm laser pulse, the co-assembly showed a significantly higher conductivity than that of the individual assembly. The co-assembly showed effective photo charge carrier generation along with a longer lifetime of >3 ms. This study indicated facile charge carrier generation at the n/p heterojunction and effective transportation through the 1D π-stacked assembly (Fig. 26). They also discovered that variations in the terminal groups of PDI did not result in significant changes in the lifetime values, indicating that the value depends primarily on the π-site rather than the terminal long chain.
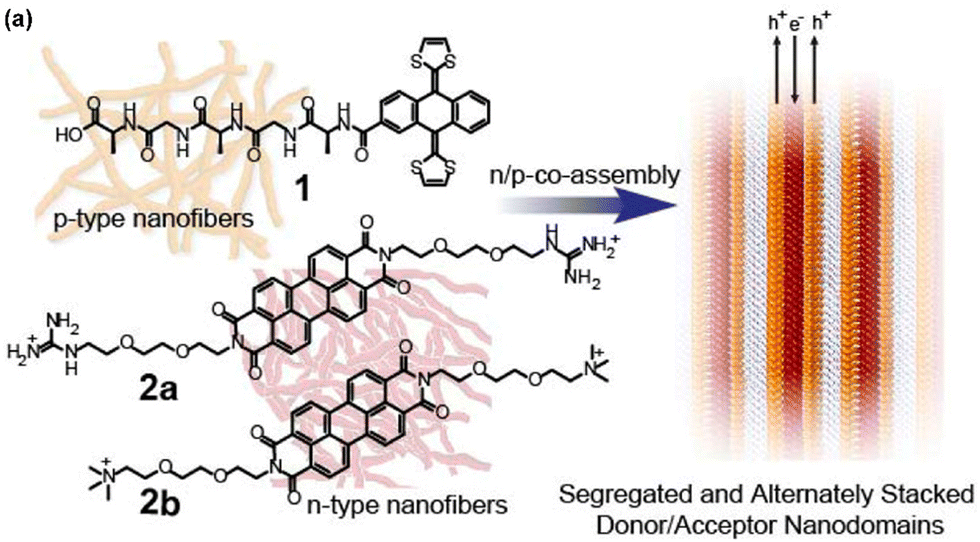 | ||
| Fig. 26 (a) Molecular structure with the schematic of nanofibrillar aggregated (left). Schematic representation of n/p co-assembly for efficient photoexcited charge carrier mobility. It is shown that higher conductivity is found for the co-assembled system compare to the individual system. This confirms the role of making the proper channel between the p-type and n-type material. Adapted with permission from ref. 277. Copyright 2015 American Chemical Society. | ||
In summarizing various examples of peptide motifs demonstrating conductivity, it can be concluded that to achieve conductivity in the peptide-based assembly, the molecular design should incorporate aromatic rings for π–π stacking and better conductance. Opting for a 1D fibrillar network in an aggregated state is consistently favoured due to its anisotropic nature, which imparts directionality. Nevertheless, achieving an optimal crosslinking between the fibers is essential to enhance conductivity, as indicated by the concentration-dependent analysis. These peptide nano-assembly systems exhibit semiconducting properties as well, with temperature-dependent conducting properties. The significance of the D/A approach is also important as shown in the TTF and PDI-based co-assembly. The enhancement of photoconductivity occurs through the coupling of n- and p-type semiconductors, leading to increased charge carrier separation and mobility. With the easy modulation of the sequence elements, these peptide nanofibers offer a viable experimental platform for additional research into the structure–property correlations of conductive amino acid materials. Their well-defined surface chemistry and biocompatibility make them suitable for optimal, naturally inspired bioelectronic interface materials. The use of these peptide building blocks lies in their ability to form via highly reversible non-covalent interactions, making them valuable and versatile components in the expanding toolkit of synthetic biology.
3.5. Peptide-based liquid crystals and their potential applications
Soft functional materials offer remarkable opportunities for the development of cutting-edge devices due to their dynamic properties.278 Among various soft materials such as polymers, gels, colloids, biomaterials, and liquid crystals (LCs), LCs are functional organic molecules, distinguished by order and mobility. The LC is an intermediate state between crystalline solid and isotropic liquid states which is a fourth (actual) state of matter (Scheme 6).278 There are two main types of LCs: lyotropic and thermotropic LCs. With the addition of solvents, lyotropic LCs developed a long-range order and are impacted by amphiphilic molecules, additives,279,280 solvent polarities,281,282 and external influences such as temperature, pressure, light, and magnetic field.283 Thermotropic LCs are anisotropic liquids that have both liquid and crystal phases. A number of distinct liquid crystalline phases can only be realised with the impact of temperature. The thermotropic LCs are the main components of many electronic gadgets including cell phones and digital notebooks. While the functionality and biocompatibility of peptide-based LCs hold great promise, their use as drug carriers, tissue scaffolds, biosensors, or bioimaging agents, and for soft robotics and wearable technologies, are still in the infancy stage.284–286 Peptide nanotubes are a particular topic of interest, because they allow for multiple chemical modifications and the application of biological systems.81 Gazit and coworkers reported the potential of very short nanotubular aromatic peptides (hexapeptides and shorter) to form well-ordered amyloid fibrils.287–289 All amyloid fibrils possess a comparable and structurally unrelated set of proteins. They have shown that the dipeptides can form self-assembled nanotubes in an aqueous medium to cast silver nanowires using them as nanoscale molds.81 Inspired by their work, Kim and co-workers have discovered the liquid crystalline peptides, where the peptide generated rigid nanowires that are dispersed individually in an organic solvent (CS2), a unique material for nanofabrication.290 The stiffness and high aspect ratio of the nanowires led to the formation of a colloidal liquid crystalline phase throughout a wide concentration range. The ability to quickly fabricate a morphology that is well-aligned across a large region was made possible by the liquid crystallinity in an extremely volatile organic solvent. An isotropic-to-liquid crystalline phase transition was indicated by the dispersion, which contained 0.5–7% weight percent of nanowires, spontaneously separating into isotropic and birefringent phases. The polydispersity of nanowires is the source of such a wide-phase transition range. The entire dispersion exhibited optical birefringence at concentrations between 7 and 10 wt%, with no phase separation. The birefringent dispersion exhibited mechanical elasticity above the high concentration of 10 wt%, which is characteristic of nematic gel or nematic glass. They have demonstrated the hierarchical structure of liquid crystalline peptide nanowires, which are made up of nanoscale liquid crystalline ordering and highly precise peptide assembly, offering a quick and effective route towards unique nanoarchitecture (Fig. 27). Liquid crystalline peptide nanowires have the potential to be used in nanopatterning and as reinforcing materials for nanocomposites.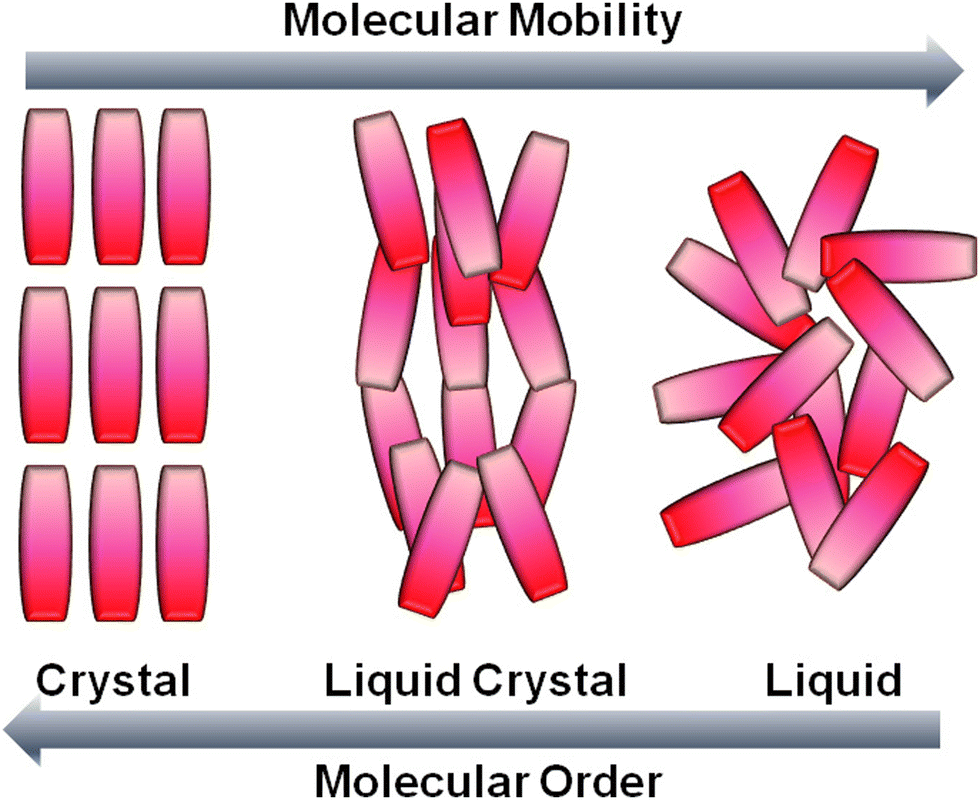 | ||
| Scheme 6 Orientation of molecules in Crystal, LC, and liquid state where LC lies between crystalline solid and isotropic liquid states. | ||
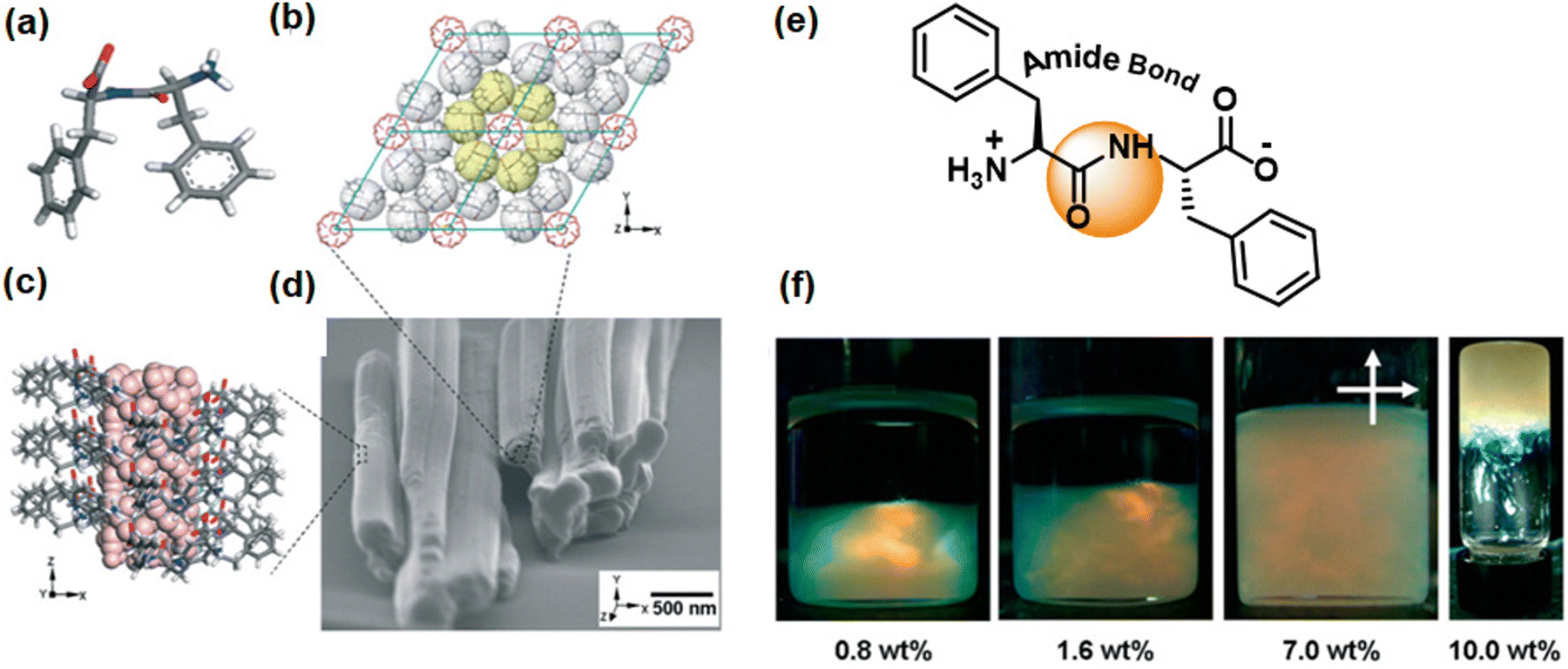 | ||
| Fig. 27 (a) Dipeptide molecule in its molecular form, where red, blue, grey, and white represented oxygen and nitrogen, carbon and hydrogen atoms, respectively. (b) Dipeptide molecules are arranged in a hexagonal pattern around a centre water molecule. Two types of intermolecular H-bonding (FF–FF and FF–water) hold dipeptide molecules in place, and each nanotube's outside diameter was around 2.4 nm. Each nanowire, which is a collection of helical nanotubes, was created by hydrophobic phenyl rings in the dipeptide molecule. (c) 3D depiction showing dipeptide self-assembly into a helical architecture via multilayer H-bonding. (e) SEM image of nanowires. The average diameter of the nanowires was 346 ± 103.9 nm, the length was 8.6 ± 4.4 nm, and the aspect ratio was 25.1 ± 11.1. (e) Zwitterionic form of L-Phenylalanine–L-Phenylalanine dipeptide which has the ability to undergo non-covalent interactions, including π—π interactions (FF–FF) and H-bonding (FF–H2O). (f) Nanowire dispersions in glass vials held between crossed polarizers. The black isotropic phase is separated from the yellow birefringent nematic liquid crystalline phase. Adapted with permission from ref. 290. Copyright 2007 Wiley-VCH. | ||
Mezzenga et al. reported the discovery of cholesteric phases in amyloids using β-lactoglobulin fibrils shortened by shear pressures, which exhibit unparalleled structural complexity.291 Abbott and Gellman et al. reported a variety of well-designed β-polypeptides which form liquid crystals, and the phase behaviour could be adjusted by varying the peptide sequence and modifications.292–294 These peptide LCs have biocompatibility and structural variety, making them ideal for guiding cell orientation or building highly ordered materials.295 He and coworkers investigated the lyotropic liquid-crystalline behaviour of a set of minimalistic peptides using small-angle X-ray scattering and molecular dynamics simulations (Fig. 28).296 Their findings imply that the amino acid sequence controls spontaneous crystallization in the networks of self-assembled nanofilaments as well as the short-range interactions between the peptides. For every peptide, a colloidal liquid-crystalline phase including a rigid FF section and a repulsive terminal was observed. The peptides formed high-aspect ratio nanofilaments by self-assembly, measuring ∼3.3 nm in diameter for Fmoc-diphenylalanine (Fmoc-FF) and ∼4.2 nm for Fmoc-Phe-Phe-Ser (Fmoc-FFS), Fmoc-Phe-Phe-Asp (Fmoc-FFD), and Fmoc-Phe-Phe-Glu (Fmoc-FFE), confirmed by TEM images (Fig. 29a–d). The biocompatibility and structural variety of peptide LCs make them attractive as potential templates for highly ordered material manufacturing or functioning as stereoselective sensor probes. Liquid crystals are a popular choice for biomolecule detection due to their high resolution, sensitivity, and cost-effectiveness.297,298 It may be possible to convert chemical binding events into amplified optical signals that are visible to the naked eye due to the orientational order and optical anisotropy of the liquid-crystalline molecules. They observed that the condensation of long-range ordered peptide nanofilaments by multivalent cations is extremely stereoselective, probably due to the intrinsic chirality of the self-assembled peptide nanofilaments (Fig. 29e). Over the past few decades, extensive research has been focused on developing a peptide-based technique for creating nanostructured inorganic materials.299,300 Hartgerink and coworkers observed that hollow silica nanotubes were designed utilizing peptide nanofiber templates that self-assembled from ultrashort peptides.301 He and coworkers demonstrated that at ambient temperature and neutral pH, liquid-crystalline peptide nanofilaments might be used as templates for the biomimetic production of well-ordered mesoporous silica nanofibers (Fig. 29f). The findings offer a new perspective on the synthesis of peptide LCs, which will increase the applicability of peptide-based materials for various uses such as chemical sensing probes and templates for the design of other functional nanomaterials.
In the past few decades, it has been observed that the molecular self-assembly process can yield a supramolecular hydrogel.302,303 According to recent studies, the nanofibers can produce anisotropic nanoscale crystals and higher-ordered assemblies, which could be aligned by external forces to display anisotropic characteristics on a macroscopic scale.304–306 Wang and co-workers have shown that in aqueous solutions (pH 7.4), spontaneously aligned nanofiber bundles and biocompatible liquid crystalline hydrogels can be prepared by regulating the intermolecular interaction of two oppositely charged small molecules (e.g., tetrapeptide, MW < 700 Da) without hydrophobic modification. The authors demonstrated a rational design of a tetrapeptide without N-terminal modification and chemical conjugation that used intermolecular interactions to induce the formation of nanofiber bundles in a two-component system that is not accessible by a single component. Self-assembled complementary charged tetrapeptides exhibit geometrical anisotropy, leading to the formation of a liquid crystalline microdomain in hydrogels. It appeared that a lyotropic liquid crystal would form based on the concentration-dependent formation of liquid crystalline hydrogels. According to the results, complementary heterochiral pairings of peptides show a tendency toward aggregation and fast gelation when the hydrogel is formed. Because the guanidyl group can connect to the carboxyl group in different orientations, the mechanical properties of the hydrogel are improved when lysine (K) in the tetrapeptide is changed to arginine (R). In vitro cytotoxicity tests revealed that the peptide mixture has negligible toxicity on cell growth (human bone osteosarcoma Saos-2 cells and marrow stromalHS-5 cells), suggesting the biocompatibility of the hydrogel.307
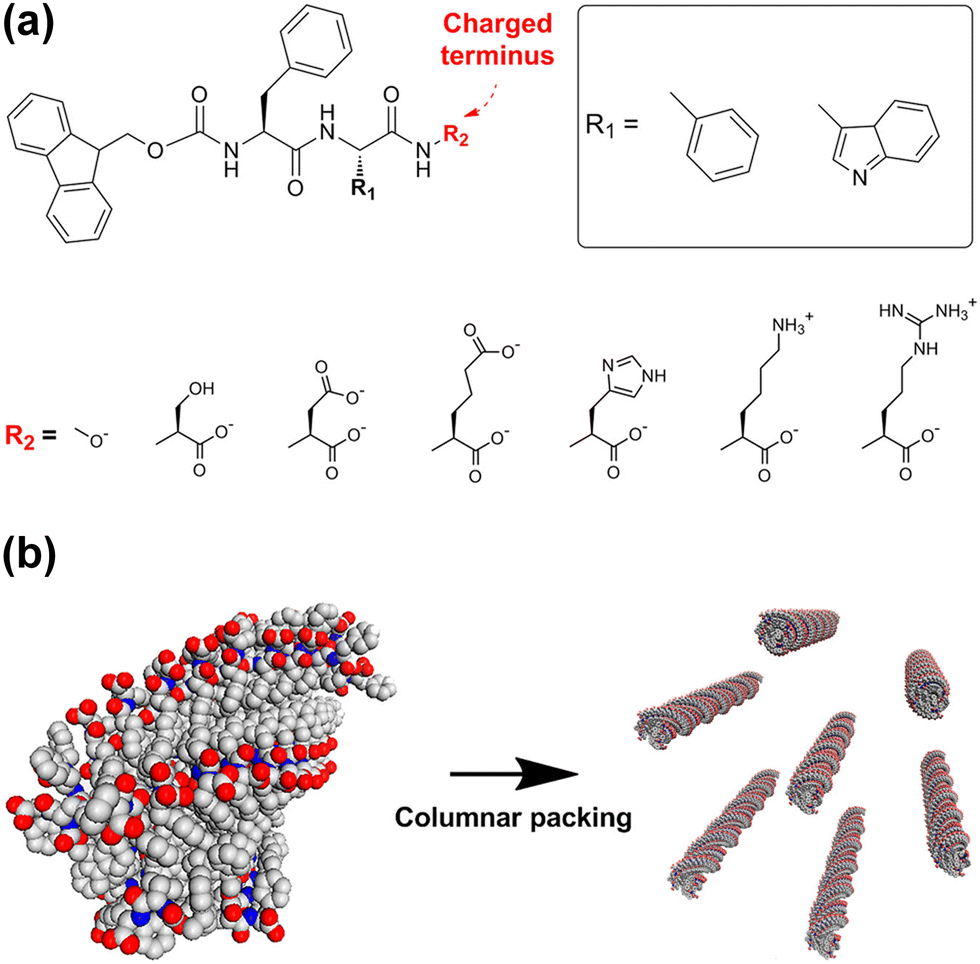 | ||
| Fig. 28 (a) Amphiphilic peptide molecular structure. (b) Illustration of the highly charged hexagonal liquid-crystalline phase with a large d spacing during the hierarchical chiral self-assembly of the peptides. Adapted with permission from ref. 296. Copyright 2018 American Chemical Society. | ||
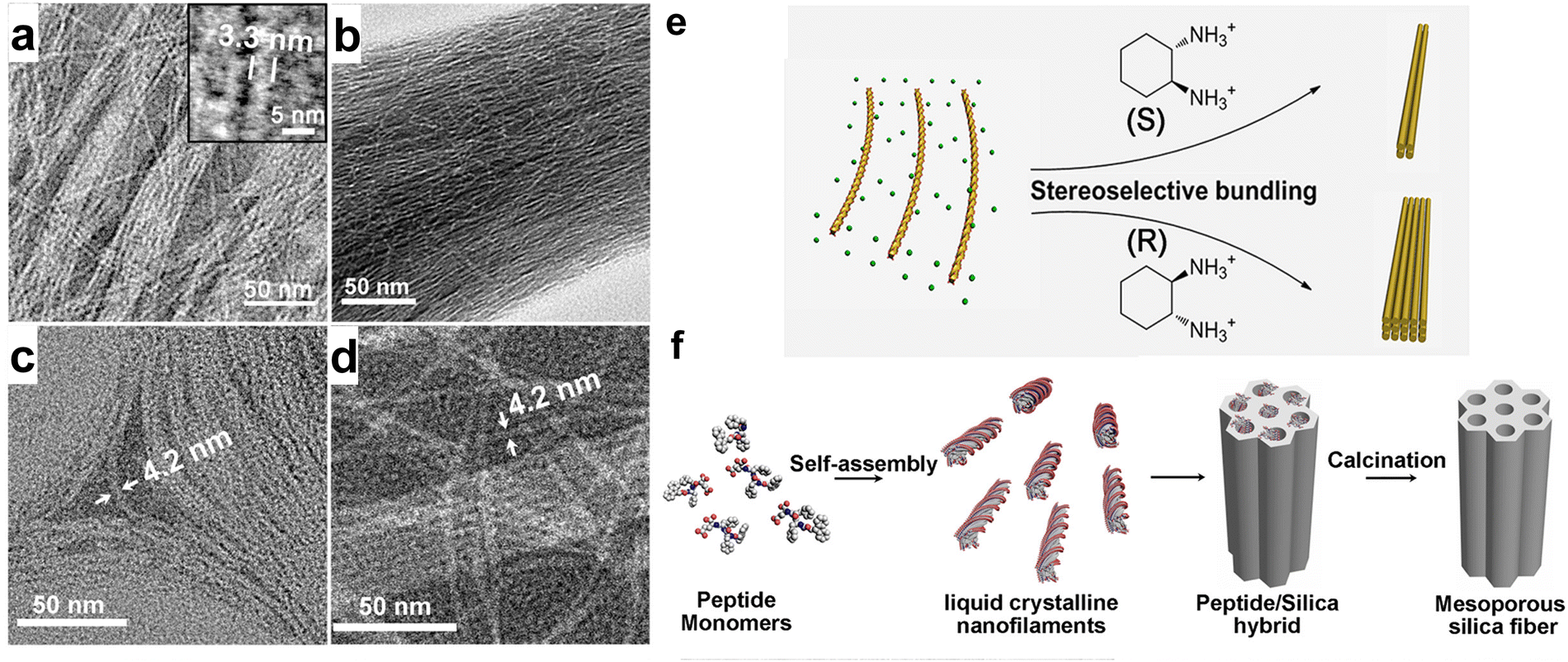 | ||
| Fig. 29 (a)–(d) TEM images of the self-assembled peptide nanofilaments; Fmoc-FF, Fmoc-FFS, Fmoc-FFD, and Fmoc-FFE, respectively with a diameter of ∼3.3 nm for Fmoc-FF and ∼4.2 nm for Fmoc-FFS, Fmoc-FFD, and Fmoc-FF. (e) Diagram illustrating the use of a chiral dipeptide in the stereoselective condensation of peptide nanofilaments [(1S,2S)-(−)-1,2-diaminocyclohexane and (1R,2R)-(−)-1,2-diaminocyclohexane]. (f) Diagram represents the synthesis of ordered cavities in mesoporous silica using peptide templates at neutral pH and room temperature. Adapted with permission from ref. 296. Copyright 2018 American Chemical Society. | ||
Over the few decades, supramolecular chemistry has gained great interest to develop peptide-based liquid crystals from discrete molecular motifs to complex molecular architectures.308 Liquid crystals based on peptides will be potential candidates for various biomedical applications such as biomaterials and drug delivery systems.285,309–311 Future research may focus on designing peptide-based liquid crystals that can encapsulate and release drugs in a controlled manner, as well as developing materials for tissue engineering and regenerative medicine. Liquid crystals based on peptides with distinctive optical characteristics may be investigated for application in optoelectronic devices such as digital displays and sensors.312–315 Further exploration of the self-assembly properties of peptide-based LCs could lead to the development of nanoscale structures in a device biomineralization strategy and biological phase-separated systems.316–318 It may become more of an emphasis to use ecologically responsible and sustainable processes for the synthesis and manufacturing of peptide-based LCs. This might entail producing these materials through the use of renewable or biobased resources.
3.6. Peptides as glass materials
Silicate glass is a widely utilized material, composed of silicon dioxide. It is known for its excellent optical clarity, chemical durability, and thermal resistance. It has a wide range of applications including windows, bottles, and various containers. However, these glasses are brittle and are prone to shattering, making them less durable in high-impact situations. Additionally, its production has a significant environmental impact which can result in long-term environmental consequences for the green life. Interestingly, these glasses serve as the foundation for bioglass and biomolecular glass, which is modified with additional oxides to enhance its bioactivity and ability to bond with biological tissues for medical applications.Bioglass is mainly used in bone repair, tissue engineering, and wound healing, while biomolecular glass is employed in 3D printing, additive manufacturing, and mold casting, offering minimal environmental impact. It has been found that when amino acids and peptides are chemically modified at the ends with hydrophobic groups, their heat stability improves significantly, making them suitable for glass processing. Recently, Yan and coworkers have synthesized biomolecular glass from Ac-modified amino acid (Ac-F) and a Cbz-modified tripeptide [Cbz-(D)-FF-glycine (Cbz-FFG)]. The respective compounds were melted to form supercooled liquid and quenched to form biomolecular glass (Fig. 30). These glasses were found to have exceptional optical properties, mechanical properties, processability, and flexibility. The glasses were found worthy for three-dimensional-printed additive manufacturing and mold casting. Most importantly, the glasses demonstrate biocompatibility, biodegradability, and biorecyclability that surpass that of the plastic materials and noncovalent glass (MIBNG) using a simple amino acid derivative, Fmoc-Leu-OH. This MIBNG has ceramic-like properties including hardness, flexibility, and wear resistance, which makes it superior to normal biomolecular glasses.43,319 Pal and his group discovered commercial glasses currently in use. Moreover, the same group has reported the development of metal ion-coordinated biomolecular pathway-driven self-assembled peptide nanostructures, which can be used as a template to yield bioactive glass (Fig. 31). Solid-phase synthesis was used to form the peptide amphiphile by attaching the hydrophobic moiety of Fmoc at the N-terminus and two hydrophilic lysine units at the C-terminus of a short peptide sequence VFFA. The peptide self-assembled into kinetically controlled nanofibers and thermodynamically stable twisted helical bundles. Compared to nanofibers, twisted helical bundles increased the percentage of nonbridging oxygen (NBO) sites on the surface of the bioglass, which improved the mineralization efficacy of hydroxyapatite. When the bioglass composites were incubated in a simulated body fluid, they exhibited a tunable bioactive response that was topologically and structurally controlled to cause the deposition of carbonate hydroxyapatite, octacalcium phosphate, and multiphasic calcium phosphate.320 Rijt and coworkers developed a system for immobilizing vascular endothelial growth factor (VEGF) using nanosized bioactive glass nanoparticles (nBGs) embellished with a 12-mer peptide sequence that originates from the transmembrane glycoprotein prominin-1 (PR1P: DRVQRQTTTVVA43), which exhibits high specificity in binding pro-angiogenic growth factor VEGF165. This is a viable approach to address the issue of inadequate neovascularization in the regeneration of massive bone defects.321 Peptide glass represents a groundbreaking material with immense potential for future applications. Its unique properties make it ideal for biomedical applications. Additionally, peptide glass can be engineered to display tailored optical and electronic properties, along with a bioactive response that stimulates multiphasic deposition, opening new pathways in flexible electronics and photonics.
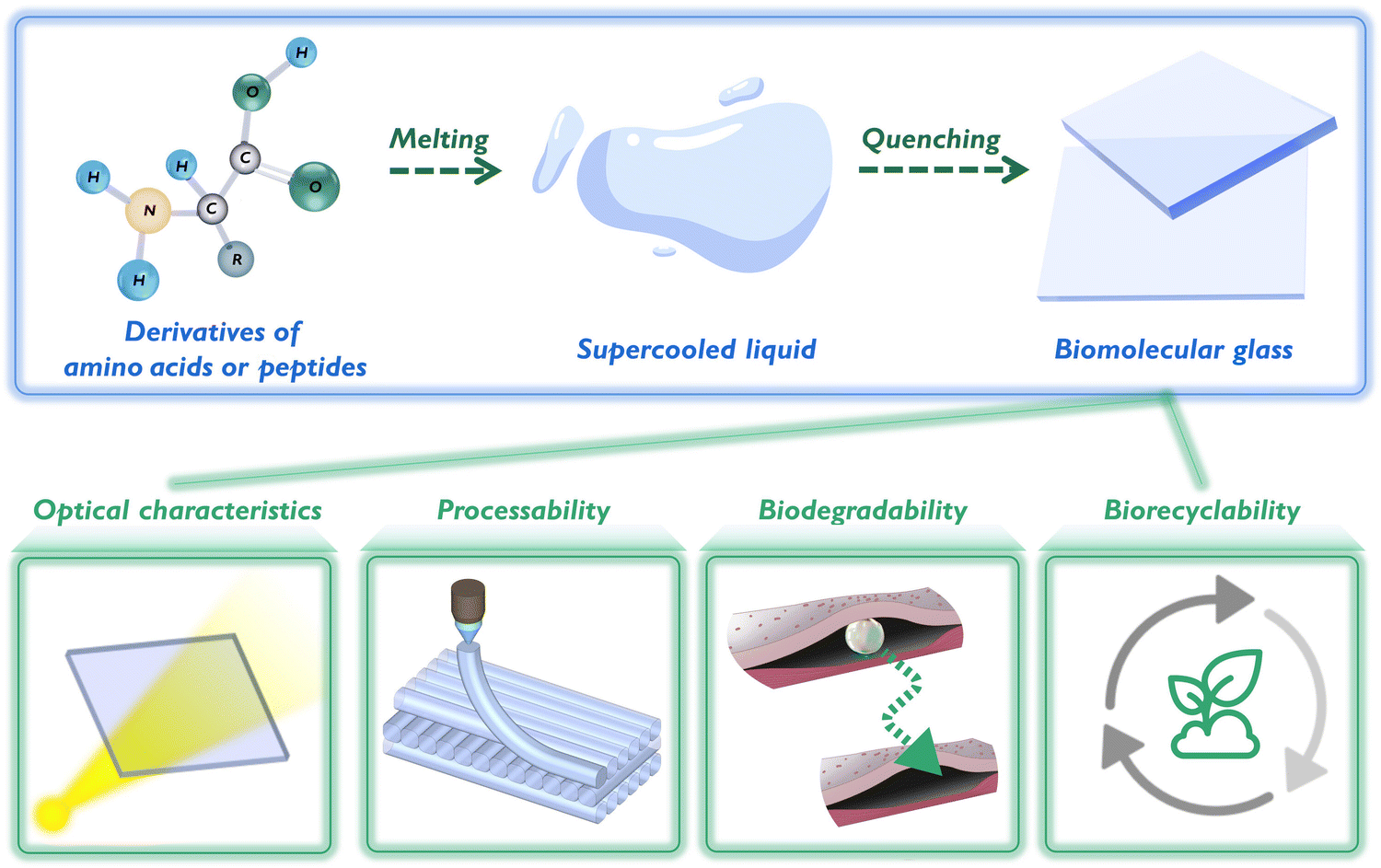 | ||
| Fig. 30 Schematic illustration of the preparation of the peptide glass through high-temperature melting, followed by a quenching process. This category of the peptide has excellent optical characteristics, high processability, and good biodegradability. Adapted with permission from ref. 43. Copyright 2023 American Association for the Advancement of Science. | ||
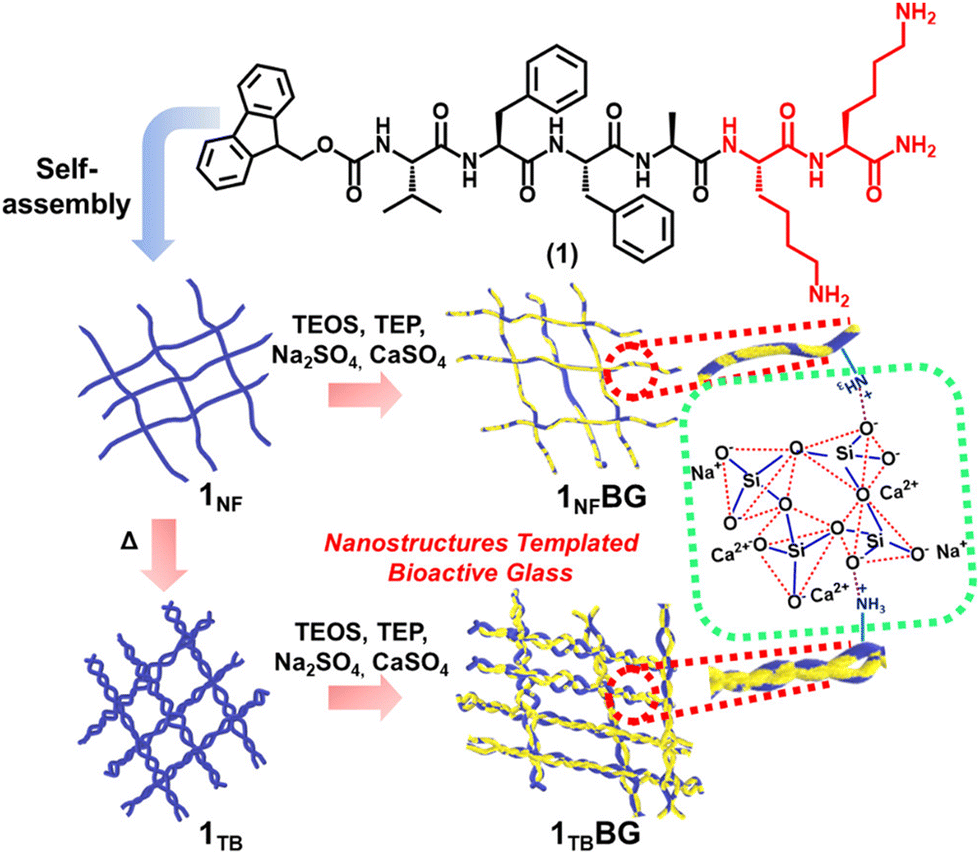 | ||
| Fig. 31 Molecular Structures of the peptide amphiphile Fmoc-VFFAKK and its pathway-driven self-assembly to form different nanostructures, kinetically controlled nanofibers (1NF) and thermodynamically stable twisted helical bundles (1TB), which act as a template for the synthesis of bioglass composites 1NFBG and 1TBBG. As precursors to the Si, P, Na, and Ca contents of bioactive glass, tetraethyl orthosilicate (TEOS), triethylphosphate (TEP), sodium sulfate, and calcium sulfate were utilized. Adapted with permission from ref. 320. Copyright 2021 American Chemical Society. | ||
4. Conclusions and future outlook
In this review, we demonstrated how the various non-covalent interactions play crucial roles in creating secondary and nanostructures of peptides during the self-assembly. We have discussed peptides as piezoelectric energy harvesters by modulating the structural building blocks or functionalities of peptides. There were several contexts in which the catalytic activity of peptide-based self-assembly was investigated, such as enzymatic catalysis and specific chemical reactions. The field of biomedical research including drug delivery, tissue engineering, regenerative medicine, cancer therapy, neurodegenerative disease, and antimicrobial activity has attracted a lot of attention towards peptides and peptide-based nanostructures in recent years because of their distinctive properties.19,322 Peptide-based liquid crystals and biomolecular glass have also been investigated for their considerable potential in various applications, particularly in the fields of nanoarchitectonics and biomaterials.The future outlook for the application of peptide-based nanomaterials across various fields is highly promising. Here is a comprehensive overview of potential developments: peptide-based nanomaterials offer exciting prospects for piezoelectric power generation due to their tunable properties and biocompatibility. Future advancements may focus on the development of peptide-based nanomaterials capable of efficiently converting mechanical energy from vibrations or movements into electrical energy. The integration of peptide nanomaterials with emerging nanogenerators, such as triboelectric or thermoelectric devices, could lead to the fabrication of self-powered wearable electronics, sensors, and biomedical implants. Peptide-based nanomaterials hold significant promise for catalytic processes, offering high specificity and tunable reactivity. Future developments may focus on designing multifunctional peptide nanomaterials capable of targeted drug delivery, controlled release, and theragnostic applications. Peptide-based nanomaterials could also find applications in regenerative medicine, biosensing, and personalized medicine, offering innovative solutions for disease diagnosis, treatment, and monitoring. Future research may explore the use of peptide nanomaterials as additives or templates to manipulate the phase behavior, alignment, and responsiveness of liquid crystals in display technologies, optical devices, and biosensors. Tailoring peptide sequences and structures could enable precise control over liquid crystal properties, leading to the development of advanced materials with enhanced functionality and performance. Peptide-based nanomaterials exhibit intriguing electrical properties, including conductivity, semiconductivity, and tunable electronic structures. Future research may explore the use of peptide nanomaterials in electronic and optoelectronic devices, such as transistors, sensors, and photodetectors. Designing peptide-based nanomaterials with hierarchical structures and ordered assemblies could enhance their electrical conductivity and charge transport properties, enabling the development of high-performance nanoelectronics and flexible electronic devices.
While peptide-based nanostructures offer numerous advantages for various applications, they also face several challenges that need to be addressed for their widespread adoption. Here are some key challenges associated with self-assembled peptide-based nanostructures such as controlled assembly, scalability, toxicity, functionalization and modification, structural stability in biological environments, regulatory approval and commercialization, and proteolytic cleavage of the peptide-based drug delivery. Addressing these challenges will require interdisciplinary efforts involving peptide chemistry, materials science, biotechnology, and biomedical engineering. Continued research and innovation in peptide design, self-assembly strategies, characterization techniques, and application-specific optimization are essential for realizing the full potential of peptide-based nanostructures across diverse fields. A more detailed theoretical understanding of peptide self-assemblies is crucial for investigating the packing dynamics of the peptide assembly process. To model the aggregation of peptide conjugates, cutting-edge simulation techniques can be used. However, this requires large-scale modelling to capture extended self-assembled structures from multiatomic molecules, similar to modelling of smaller peptides. Artificial intelligence (AI) has significant potential for navigating the huge search space of amino acid combinations and presenting a subset of the most promising options. These techniques can screen and find functional peptides and assemblies, potentially leading to new functional molecules in the field of material research. Proteolysis resistant self-assembled peptides can be fabricated as an efficient non-cytotoxic drug delivery vehicle.323 The peptide co-assembly produces materials whose emergent properties are drastically altered when compared to the self-assembled materials of the individual components. There is a need and opportunity to investigate and understand these multi-component materials. Peptide-based nanomaterials are potential components in environmental applications such as removing contaminants and poisons from wastewater through filtration or adsorption. Functionalized peptides, leading to the formation of heterostructures such as peptide metal–organic cages (MOCs), peptide metal–organic frameworks (MOFs), peptide covalent organic frameworks (COFs), and peptide coordination polymer gels (CPGs), hold potential for addressing the global energy crisis through applications in energy storage, conversion, and solar cells. Overall, the future integration of peptide-based nanomaterials across these diverse fields holds tremendous promise for addressing global challenges in healthcare, energy, and technology, driving innovation and creating new opportunities for scientific exploration and technological advancement.
Author contributions
GG conceptualized the topic of the review. All the authors contributed equally in surveying literature and writing the manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. N. D. and A. G. acknowledge to JNCASR for the fellowship, A. R. and S. M. acknowledge to DST Inspire (DST/INSPIRE/03/2023/002028) and CeNS for the fellowship respectively. T. K. M. thankfully acknowledges SERB, Dept. of Science and Technology (DST), Govt. of India, for financial support. SAMat, ICMS, Sheikh Saqr senior fellowship (T. K. M.) are also gratefully acknowledged, G. G. thanks the Ramanujan Fellowship (file no. RJF/2022/000002), SERB, Government of India and CeNS (Project no.: CeNSitize-3-GG) for the funding support.References
- J.-M. Lehn, Makromol. Chem., Macromol. Symp., 1993, 69, 1–17 CrossRef CAS.
- B. E. Dial and K. D. Shimizu, Appl. Supramol. Chem., 2012, 301–320 CAS.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed.
- E. Krieg, M. M. C. Bastings, P. Besenius and B. Rybtchinski, Chem. Rev., 2016, 116, 2414–2477 CrossRef CAS.
- T. F. A. De Greef and E. W. Meijer, Nature, 2008, 453, 171–173 CrossRef CAS.
- X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042–6065 RSC.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS.
- L. Yang, X. Tan, Z. Wang and X. Zhang, Chem. Rev., 2015, 115, 7196–7239 CrossRef CAS.
- R. Dong, Y. Zhou, X. Huang, X. Zhu, Y. Lu and J. Shen, Adv. Mater., 2015, 27, 498–526 CrossRef CAS.
- S. L. Li, T. Xiao, C. Lin and L. Wang, Chem. Soc. Rev., 2012, 41, 5950–5968 RSC.
- R. Orbach, I. Mironi-Harpaz, L. Adler-Abramovich, E. Mossou, E. P. Mitchell, V. T. Forsyth, E. Gazit and D. Seliktar, Langmuir, 2012, 28, 2015–2022 CrossRef CAS PubMed.
- V. L. Sedman, X. Chen, S. Allen, C. J. Roberts, V. V. Korolkov and S. J. B. Tendler, J. Microsc., 2013, 249, 165–172 CrossRef CAS PubMed.
- S. Yuran, Y. Razvag and M. Reches, ACS Nano, 2012, 6, 9559–9566 CrossRef CAS PubMed.
- L. Adler-Abramovich and E. Gazit, Chem. Soc. Rev., 2014, 43, 6881–6893 RSC.
- J. Zhu, N. Avakyan, A. Kakkis, A. M. Hoffnagle, K. Han, Y. Li, Z. Zhang, T. S. Choi, Y. Na, C. J. Yu and F. A. Tezcan, Chem. Rev., 2021, 121, 13701–13796 CrossRef CAS.
- C. Yuan, Q. Li, R. Xing, J. Li and X. Yan, Chem, 2023, 9, 2425–2445 CAS.
- S. La Manna, C. Di Natale, V. Onesto and D. Marasco, Int. J. Mol. Sci., 2021, 22, 12662 CrossRef CAS PubMed.
- K. Tao, H. Wu, L. Adler-Abramovich, J. Zhang, X. Fan, Y. Wang, Y. Zhang, S. A. M. Tofail, D. Mei, J. Li and E. Gazit, Prog. Mater. Sci., 2024, 142, 101240 CrossRef CAS.
- N. Habibi, N. Kamaly, A. Memic and H. Shafiee, Nano Today, 2016, 11, 41–60 CrossRef CAS.
- R. V. Ulijn and A. M. Smith, Chem. Soc. Rev., 2008, 37, 664–675 RSC.
- T. Fan, X. Yu, B. Shen and L. Sun, J. Nanomater., 2017, 2017, 562474, DOI:10.1155/2017/4562474.
- I. W. Hamley, Angew. Chem., Int. Ed., 2007, 46, 8128–8147 CrossRef CAS.
- Y. Liang, X. Zhang, Y. Yuan, Y. Bao and M. Xiong, Biomater. Sci., 2020, 8, 6858–6866 RSC.
- C. Yuan, W. Ji, R. Xing, J. Li, E. Gazit and X. Yan, Nat. Rev. Chem., 2019, 3, 567–588 CrossRef CAS PubMed.
- A. Dasgupta and D. Das, Langmuir, 2019, 35, 10704–10724 CrossRef CAS PubMed.
- S. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS PubMed.
- A. Lakshmanan, S. Zhang and C. A. E. Hauser, Trends Biotechnol., 2012, 30, 155–165 CrossRef CAS.
- U. Ghosh and G. Ghosh, Acta Sci. Pharm. Sci., 2022, 6, 01–02 Search PubMed.
- G. Ghosh, Acta Sci. Pharm. Sci., 2021, 5, 5423 Search PubMed.
- G. Ghosh, P. Dey and S. Ghosh, Chem. Commun., 2020, 56, 6757–6769 RSC.
- G. Ghosh, Giant, 2023, 14, 100160 CrossRef CAS.
- U. Ghosh, N. Kumar and G. Ghosh, Pharm. Appl. Supramol., 2023, 273–299 Search PubMed.
- Y. Zhou, Q. Li, Y. Wu, X. Li, Y. Zhou, Z. Wang, H. Liang, F. Ding, S. Hong, N. F. Steinmetz and H. Cai, ACS Nano, 2023, 17, 8004–8025 CrossRef CAS PubMed.
- D. Gupta, V. Gupta, D. Nath, C. Miglani, D. Mandal and A. Pal, ACS Appl. Mater. Interfaces, 2023, 15, 25110–25121 CrossRef CAS PubMed.
- Y. Liu, Y. Yang, C. Wang and X. Zhao, Nanoscale, 2013, 5, 6413–6421 RSC.
- Z. Li, Y. Zhu and J. B. Matson, ACS Appl. Bio Mater., 2022, 5, 4635–4651 CrossRef CAS.
- T. J. Moyer, J. A. Finbloom, F. Chen, D. J. Toft, V. L. Cryns and S. I. Stupp, J. Am. Chem. Soc., 2014, 136, 14746–14752 CrossRef CAS PubMed.
- I. W. Hamley, A. Dehsorkhi, V. Castelletto, S. Furzeland, D. Atkins, J. Seitsonen and J. Ruokolainen, Soft Matter, 2013, 9, 9290–9293 CAS.
- Y. M. Abul-Haija, G. G. Scott, J. K. Sahoo, T. Tuttle and R. V. Ulijn, Chem. Commun., 2017, 53, 9562–9565 CAS.
- D. Mandal, A. Nasrolahi Shirazi and K. Parang, Org. Biomol. Chem., 2014, 12, 3544–3561 RSC.
- G. Bin Qi, Y. J. Gao, L. Wang and H. Wang, Adv. Mater., 2018, 30, 1703444 Search PubMed.
- R. Chang, L. Zhao, R. Xing, J. Li and X. Yan, Chem. Soc. Rev., 2023, 52, 2688–2712 RSC.
- R. Xing, C. Yuan, W. Fan, X. Ren and X. Yan, Sci. Adv., 2023, 9, 1–12 Search PubMed.
- G. Ghosh, R. Barman, A. Mukherjee, U. Ghosh, S. Ghosh and G. Fernández, Angew. Chem., Int. Ed., 2022, 61, e202113403 CrossRef CAS.
- G. Ghosh, R. Barman, J. Sarkar and S. Ghosh, J. Phys. Chem. B, 2019, 123, 5909–5915 CrossRef CAS.
- G. Ghosh and G. Fernández, Beilstein J. Org. Chem., 2020, 16, 2017–2025 CrossRef PubMed.
- G. Ghosh, K. K. Kartha and G. Fernández, Chem. Commun., 2021, 57, 1603–1606 RSC.
- S. Lee, T. H. T. Trinh, M. Yoo, J. Shin, H. Lee, J. Kim, E. Hwang, Y. B. Lim and C. Ryou, Int. J. Mol. Sci., 2019, 20, 5850 CrossRef CAS PubMed.
- R. Chang, C. Yuan, P. Zhou, R. Xing and X. Yan, Acc. Chem. Res., 2024, 57, 289–301 CrossRef CAS PubMed.
- P. Zhou, R. Xing, Q. Li, J. Li, C. Yuan and X. Yan, Matter, 2023, 6, 1945–1963 CrossRef CAS.
- C. Yuan, R. Xing, J. Cui, W. Fan, J. Li and X. Yan, CCS Chem., 2023, 6, 255–265 CrossRef.
- Z. Du, B. Fan, Q. Dai, L. Wang, J. Guo, Z. Ye, N. Cui, J. Chen, K. Tan, R. Li and W. Tang, Giant, 2022, 9, 100082 CrossRef CAS.
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2016, 116, 2775–2825 CrossRef CAS.
- Y. Li, Y. Wang, G. Huang and J. Gao, Chem. Rev., 2018, 118, 5359–5391 CrossRef CAS PubMed.
- C. Dalal and N. R. Jana, J. Phys. Chem. B, 2017, 121, 2942–2951 CrossRef CAS.
- J. Wang, K. Liu, R. Xing and X. Yan, Chem. Soc. Rev., 2016, 45, 5589–5604 RSC.
- M. G. Krone, L. Hua, P. Soto, R. Zhou, B. J. Berne and J. E. Shea, J. Am. Chem. Soc., 2008, 130, 11066–11072 CrossRef.
- T. P. Knowles, A. W. Fitzpatrick, S. Meehan, H. R. Mott, M. Vendruscolo, C. M. Dobson and M. E. Welland, Science, 2007, 318, 1900–1903 CrossRef CAS PubMed.
- R. J. Brea, C. Reiriz and J. R. Granja, Chem. Soc. Rev., 2010, 39, 1448–1456 RSC.
- Y. Zhao, L. J. Leman, D. J. Search, R. A. Garcia, D. A. Gordon, B. E. Maryanoff and M. R. Ghadiri, ACS Cent. Sci., 2017, 3, 639–646 CrossRef CAS PubMed.
- J. D. Hartgerink, J. R. Granja, R. A. Milligan and M. R. Ghadiri, J. Am. Chem. Soc., 1996, 118, 43–50 CrossRef CAS.
- I. Insua and J. Montenegro, J. Am. Chem. Soc., 2020, 142, 300–307 CrossRef CAS PubMed.
- X. Yan, P. Zhu and J. Li, Chem. Soc. Rev., 2010, 39, 1877–1890 RSC.
- J. Y. Rho, H. Cox, E. D. H. Mansfield, S. H. Ellacott, R. Peltier, J. C. Brendel, M. Hartlieb, T. A. Waigh and S. Perrier, Nat. Commun., 2019, 10, 4708 CrossRef CAS PubMed.
- Y. Yang, X. Wang, X. Wu, S. Guo, H. Yang, J. Lu and H. Dong, J. Am. Chem. Soc., 2024, 146, 13488–13498 CrossRef CAS.
- Y. Liu, L. Wang, L. Zhao, Y. Zhang, Z. T. Li, Z. T. Li, F. Huang and F. Huang, Chem. Soc. Rev., 2024, 53, 1592–1623 RSC.
- P. Dowari, S. Roy, S. Das, S. Chowdhuri, R. Kushwaha, B. K. Das, A. Ukil and D. Das, Chem. - Asian J., 2022, 17, e202200550 CrossRef CAS PubMed.
- B. K. Das, B. Pramanik, S. Chowdhuri, O. A. Scherman and D. Das, Chem. Commun., 2020, 56, 3393–3396 RSC.
- M. Coste, E. Suárez-Picado and S. Ulrich, Chem. Sci., 2022, 13, 909–933 RSC.
- J. K. Wychowaniec, R. Patel, J. Leach, R. Mathomes, V. Chhabria, Y. Patil-Sen, A. Hidalgo-Bastida, R. T. Forbes, J. M. Hayes and M. A. Elsawy, Biomacromolecules, 2020, 21, 2670–2680 CrossRef CAS.
- I. W. Hamley, ACS Appl. Bio Mater., 2023, 6, 384–409 CrossRef CAS.
- K. I. Min, S. W. Lee, E. H. Lee, Y. S. Lee, H. Yi and D. P. Kim, Adv. Funct. Mater., 2018, 28, 1–8 Search PubMed.
- P. Zhu, X. Yan, Y. Su, Y. Yang and J. Li, Chem. - Eur. J., 2010, 16, 3176–3183 CrossRef CAS PubMed.
- J. Li, X. Du, S. Hashim, A. Shy and B. Xu, J. Am. Chem. Soc., 2017, 139, 71–74 CrossRef CAS PubMed.
- J. Shao, B. P. Kuiper, A. M. W. H. Thunnissen, R. H. Cool, L. Zhou, C. Huang, B. W. Dijkstra and J. Broos, J. Am. Chem. Soc., 2022, 144, 13815–13822 CrossRef CAS PubMed.
- K. Carter-Fenk, M. Liu, L. Pujal, M. Loipersberger, M. Tsanai, R. M. Vernon, J. D. Forman-Kay, M. Head-Gordon, F. Heidar-Zadeh and T. Head-Gordon, J. Am. Chem. Soc., 2023, 145, 24836–24851 CAS.
- J. J. Richardson, M. Björnmalm and F. Caruso, Science, 2015, 348, 6233 CrossRef.
- T. H. Rehm and C. Schmuck, Chem. Soc. Rev., 2010, 39, 3597–3611 RSC.
- C. F. J. Faul and M. Antonietti, Adv. Mater., 2003, 15, 673–683 CrossRef CAS.
- C. H. Görbitz, Chem. - Eur. J., 2007, 13, 1022–1031 CrossRef PubMed.
- M. Reches and E. Gazit, Science, 2003, 300, 625–627 CrossRef CAS.
- A. Del Giudice, A. Rüter, N. V. Pavel, L. Galantini and U. Olsson, Langmuir, 2020, 36, 8451–8460 CrossRef CAS PubMed.
- S. Kashyap, V. K. Pal, S. Mohanty and S. Roy, ChemBioChem, 2024, 25, e202300835 CrossRef CAS PubMed.
- H. Li, X. Qian, H. Mohanram, X. Han, H. Qi, G. Zou, F. Yuan, A. Miserez, T. Liu, Q. Yang, H. Gao and J. Yu, Nat. Nanotechnol., 2024, 19, 1141–1149 CrossRef CAS PubMed.
- M. A. J. Gillissen, M. M. E. Koenigs, J. J. H. Spiering, J. A. J. M. Vekemans, A. R. A. Palmans, I. K. Voets and E. W. Meijer, J. Am. Chem. Soc., 2014, 136, 336–343 CrossRef CAS PubMed.
- P. A. Korevaar, C. J. Newcomb, E. W. Meijer and S. I. Stupp, J. Am. Chem. Soc., 2014, 136, 8540–8543 CrossRef CAS.
- A. Nandakumar, Y. Ito and M. Ueda, J. Am. Chem. Soc., 2020, 142, 20994–21003 CrossRef CAS PubMed.
- K. Tahara, S. Lei, J. Adisoejoso, S. De Feyter and Y. Tobe, Chem. Commun., 2010, 46, 8507–8525 RSC.
- Y. Hu, K. Miao, L. Xu, B. Zha, X. Miao and W. Deng, RSC Adv., 2017, 7, 32391–32398 RSC.
- O. S. Lee, S. I. Stupp and G. C. Schatz, J. Am. Chem. Soc., 2011, 133, 3677–3683 CrossRef CAS PubMed.
- M. E. Pitz, A. M. Nukovic, M. A. Elpers and A. A. Alexander-Bryant, Macromol. Biosci., 2022, 22, 1–16 CrossRef.
- A. S. De Toma, S. Salamekh, A. Ramamoorthy and M. H. Lim, Chem. Soc. Rev., 2012, 41, 608–621 RSC.
- X. Qiao, L. Qu, Y. Guo and T. Hoshino, J. Phys. Chem. B, 2021, 125, 11374–11385 CrossRef CAS PubMed.
- M. Amorín, L. Castedo and J. R. Granja, J. Am. Chem. Soc., 2003, 125, 2844–2845 CrossRef.
- D. Missirlis, A. Chworos, C. J. Fu, H. A. Khant, D. V. Krogstad and M. Tirrell, Langmuir, 2011, 27, 6163–6170 CrossRef CAS PubMed.
- H. Yamashita, T. Kato, M. Oba, T. Misawa, T. Hattori, N. Ohoka, M. Tanaka, M. Naito, M. Kurihara and Y. Demizu, Sci. Rep., 2016, 6, 2–9 CrossRef.
- W. Ji, C. Yuan, P. Chakraborty, S. Gilead, X. Yan and E. Gazit, Commun. Chem., 2019, 2, 65 CrossRef.
- X. Wang, L. Zhao, C. Wang, X. Feng, Q. Ma, G. Yang, T. Wang, X. Yan and J. Jiang, Small, 2022, 18, 1–9 Search PubMed.
- E. De Santis and M. G. Ryadnov, Chem. Soc. Rev., 2015, 44, 8288–8300 RSC.
- A. Levin, T. A. Hakala, L. Schnaider, G. J. L. Bernardes, E. Gazit and T. P. J. Knowles, Nat. Rev. Chem., 2020, 4, 615–634 CrossRef CAS.
- Q. Song, Z. Cheng, M. Kariuki, S. C. L. Hall, S. K. Hill, J. Y. Rho and S. Perrier, Chem. Rev., 2021, 121, 13936–13995 CrossRef CAS PubMed.
- L. Liu, K. Xu, H. Wang, P. K. Jeremy Tan, W. Fan, S. S. Venkatraman, L. Li and Y. Y. Yang, Nat. Nanotechnol., 2009, 4, 457–463 CrossRef CAS.
- R. Coppage, J. M. Slocik, B. D. Briggs, A. I. Frenkel, R. R. Naik and M. R. Knecht, ACS Nano, 2012, 6, 1625–1636 CrossRef CAS PubMed.
- X. Yan, Q. He, K. Wang, L. Duan, Y. Cui and J. Li, Angew. Chem., Int. Ed., 2007, 46, 2431–2434 CrossRef CAS PubMed.
- B. Mondal, D. Bairagi, N. Nandi, B. Hansda, K. S. Das, C. J. C. Edwards-Gayle, V. Castelletto, I. W. Hamley and A. Banerjee, Langmuir, 2020, 36, 12942–12953 CrossRef CAS PubMed.
- J. M. Carter, Y. Qian, J. C. Foster and J. B. Matson, Chem. Commun., 2015, 51, 13131–13134 RSC.
- A. Baral, S. Basak, K. Basu, A. Dehsorkhi, I. W. Hamley and A. Banerjee, Soft Matter, 2015, 11, 4944–4951 RSC.
- S. Zhang, F. Gelain and X. Zhao, Semin. Cancer Biol., 2005, 15, 413–420 CrossRef.
- P. Zhang, A. G. Cheetham, Y. A. Lin and H. Cui, ACS Nano, 2013, 7, 5965–5977 CrossRef CAS PubMed.
- I. W. Hamley, Biomacromolecules, 2021, 22, 1835–1855 CrossRef CAS.
- S. Song, J. Wang, N. Song, H. Di, D. Liu and Z. Yu, Nanoscale, 2020, 12, 2422–2433 RSC.
- C. Ghosh, S. Menon, S. Bal, S. Goswami, J. Mondal and D. Das, Nano Lett., 2023, 23, 5828–5835 CrossRef CAS.
- J. E. Aguilar-Toalá, A. Hernández-Mendoza, A. F. González-Córdova, B. Vallejo-Cordoba and A. M. Liceaga, Peptides, 2019, 122, 170170 CrossRef PubMed.
- Y. Xiang, C. Liu, S. Ma, X. Wang, L. Zhu and C. Bao, Adv. Funct. Mater., 2023, 33, 1–10 Search PubMed.
- K. Furukawa, M. Oba, K. Toyama, G. O. Opiyo, Y. Demizu, M. Kurihara, M. Doi and M. Tanaka, Org. Biomol. Chem., 2017, 15, 6302–6305 RSC.
- R. Huang, Y. Wang, W. Qi, R. Su and Z. He, Nanoscale Res. Lett., 2014, 9, 1–9 CrossRef CAS.
- W. Hassouneh, K. Fischer, S. R. MacEwan, R. Branscheid, C. L. Fu, R. Liu, M. Schmidt and A. Chilkoti, Biomacromolecules, 2012, 13, 1598–1605 CrossRef CAS.
- B. Kemper, L. Zengerling, D. Spitzer, R. Otter, T. Bauer and P. Besenius, J. Am. Chem. Soc., 2018, 140, 534–537 CrossRef CAS PubMed.
- A. Singh, J. P. Joseph, D. Gupta, I. Sarkar and A. Pal, Chem. Commun., 2018, 54, 10730–10733 RSC.
- G. Echue, I. Hamley, G. C. Lloyd Jones and C. F. J. Faul, Langmuir, 2016, 32, 9023–9032 CrossRef CAS PubMed.
- H. Dong, M. Wang, S. Fan, C. Wu, C. Zhang, X. Wu, B. Xue, Y. Cao, J. Deng, D. Yuan and J. Shi, Angew. Chem., Int. Ed., 2022, 61, e202212829 CrossRef CAS PubMed.
- A. Mukherjee and G. Ghosh, Nanoscale, 2024, 2169–2184 RSC.
- M. Liu, C. N. Creemer, T. J. Reardon and J. R. Parquette, Chem. Commun., 2021, 57, 13776–13779 RSC.
- M. G. Ciulla, F. Fontana, R. Lorenzi, A. Marchini, L. Campone, E. Sadeghi, A. Paleari, S. Sattin and F. Gelain, Mater. Chem. Front., 2023, 7, 3680–3692 RSC.
- H. M. Dao, S. Parajuli, E. Urena-Benavides and S. Jo, Colloid Interface Sci. Commun., 2020, 39, 100325 CrossRef CAS.
- A. Aggeli, M. Bell, L. M. Carrick, C. W. G. Fishwick, R. Harding, P. J. Mawer, S. E. Radford, A. E. Strong and N. Boden, J. Am. Chem. Soc., 2003, 125, 9619–9628 CrossRef CAS PubMed.
- R. Freeman, M. Han, Z. Álvarez, J. A. Lewis, J. R. Wester, N. Stephanopoulos, M. T. McClendon, C. Lynsky, J. M. Godbe, H. Sangji, E. Luijten and S. I. Stupp, Science, 2018, 362, 808–813 CrossRef CAS PubMed.
- T. Li, X. M. Lu, M. R. Zhang, K. Hu and Z. Li, Bioact. Mater., 2022, 11, 268–282 CAS.
- A. Boruah and A. Roy, Biomater. Sci., 2022, 10, 4694–4723 RSC.
- N. Sezer and M. Koç, Nano Energy, 2021, 80, 105567 CrossRef CAS.
- S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150–8177 RSC.
- C. Chen, K. Liu, J. Li and X. Yan, Adv. Colloid Interface Sci., 2015, 225, 177–193 CrossRef CAS.
- C. Li and G. J. Weng, J. Appl. Mech. Trans., 2002, 69, 481–488 CrossRef.
- D. J. Singh, M. Ghita, M. Fornari and S. V. Halilov, Ferroelectrics, 2006, 338, 73–79 CrossRef CAS.
- Z. Fan, K. Sun and J. Wang, J. Mater. Chem. A, 2015, 3, 18809–18828 RSC.
- A. Datta, P. E. Sanchez-Jimenez, R. A. R. Al Orabi, Y. Calahorra, C. Ou, S. L. Sahonta, M. Fornari and S. Kar-Narayan, Adv. Funct. Mater., 2017, 27, 1701169 CrossRef.
- S. Barman, A. Pal, A. Mukherjee, S. Paul, A. Datta and S. Ghosh, Chem. – Eur. J., 2024, 30, e202303120 CrossRef CAS PubMed.
- J. Wang, D. Gao and P. S. Lee, Adv. Mater., 2021, 33, 1–20 Search PubMed.
- L. W. Martin and A. M. Rappe, Nat. Rev. Mater., 2016, 2, 1–15 Search PubMed.
- S. Bera, S. Guerin, H. Yuan, J. O’Donnell, N. P. Reynolds, O. Maraba, W. Ji, L. J. W. Shimon, P. A. Cazade, S. A. M. Tofail, D. Thompson, R. Yang and E. Gazit, Nat. Commun., 2021, 12, 2634 CrossRef CAS.
- S. Almohammed, A. Finlay, D. Duleba, S. Cosgrave, R. Johnson, B. J. Rodriguez and J. H. Rice, ACS Mater. Lett., 2024, 6, 1863–1869 CrossRef CAS.
- T. Kurita, T. Terabayashi, S. Kimura, K. Numata and H. Uji, Biomacromolecules, 2021, 22, 2815–2821 CrossRef CAS PubMed.
- Y. Kim, H. Park, Y. Kim, C. Lee, H. Park and J. H. Lee, ACS Appl. Mater. Interfaces, 2022, 14, 38778–38785 CrossRef CAS.
- G. Finkelstein-Zuta, R. Shitrit, S. Gilead, S. Rencus-Lazar and E. Gazit, Isr. J. Chem., 2022, 62, 1–7 CrossRef.
- H. Park, Y. Kim, Y. Kim, C. Lee, H. Park, H. Joo, J. Hun Lee and J. H. Lee, Appl. Surf. Sci., 2023, 618, 156588 CrossRef CAS.
- A. Kholkin, N. Amdursky, I. Bdikin, E. Gazit and G. Rosenman, ACS Nano, 2010, 4, 610–614 CrossRef CAS.
- V. Nguyen, R. Zhu, K. Jenkins and R. Yang, Nat. Commun., 2016, 7, 1–6 Search PubMed.
- H. Yuan, P. Han, K. Tao, S. Liu, E. Gazit and R. Yang, Research, 2019, 2019, 9025939 CrossRef CAS.
- A. Heredia, I. Bdikin, S. Kopyl, E. Mishina, S. Semin, A. Sigov, K. German, V. Bystrov, J. Gracio and A. L. Kholkin, J. Phys. D: Appl. Phys., 2010, 43, 462001 CrossRef.
- I. Bdikin, V. Bystrov, I. Delgadillo, J. Gracio, S. Kopyl, M. Wojtas, E. Mishina, A. Sigov and A. L. Kholkin, J. Appl. Phys., 2012, 111, 074104 CrossRef.
- S. Vasilev, P. Zelenovskiy, D. Vasileva, A. Nuraeva, V. Y. Shur and A. L. Kholkin, J. Phys. Chem. Solids, 2016, 93, 68–72 CrossRef CAS.
- K. Ryan, J. Beirne, G. Redmond, J. I. Kilpatrick, J. Guyonnet, N. V. Buchete, A. L. Kholkin and B. J. Rodriguez, ACS Appl. Mater. Interfaces, 2015, 7, 12702–12707 CrossRef CAS.
- D. Gupta, A. Bhatt, V. Gupta, C. Miglani, J. P. Joseph, J. Ralhan, D. Mandal, M. E. Ali and A. Pal, Chem. Mater., 2022, 34, 4456–4470 CrossRef CAS.
- Umesh, S. Bera and S. Bhattacharya, Small, 2023, 2308104, 1–10 Search PubMed.
- A. Ramesh, T. N. Das, T. K. Maji and G. Ghosh, Chem. Sci., 2024, 16355–16366 RSC.
- J. James and B. K. Simpson, Crit. Rev. Food Sci. Nutr., 1996, 36, 437–463 CrossRef CAS PubMed.
- J. N. Armor, Catal. Today, 2011, 163, 3–9 CrossRef CAS.
- M. Hao, M. Qiu, H. Yang, B. Hu and X. Wang, Sci. Total Environ., 2021, 760, 143333 CrossRef CAS.
- R. Bellabarba, P. Johnston, S. Moss, C. Sievers, B. Subramaniam, C. Tway, Z. Wang and H. Zhu, ACS Catal., 2023, 13, 7917–7928 CrossRef CAS.
- Y. Lou, B. Zhang, X. Ye and Z. G. Wang, Mater. Today NANO, 2023, 21, 100302 CrossRef CAS.
- J. S. Möhler, M. Pickl, T. Reiter, S. Simić, J. W. Rackl, W. Kroutil and H. Wennemers, Angew. Chem., Int. Ed., 2024, 63, e202319457 CrossRef.
- T. Schnitzer, M. Schnurr, A. F. Zahrt, N. Sakhaee, S. E. Denmark and H. Wennemers, ACS Cent. Sci., 2024, 10, 367–373 CrossRef CAS.
- S. Goswami, A. Reja, S. Pal, A. Singh and D. Das, J. Am. Chem. Soc., 2022, 144, 19248–19252 CrossRef CAS.
- Q. Liu, H. Wang, X. Shi, Z. G. Wang and B. Ding, ACS Nano, 2017, 11, 7251–7258 CrossRef CAS PubMed.
- M. Abbas, A. Atiq, R. Xing and X. Yan, J. Mater. Chem. B, 2021, 9, 4444–4458 RSC.
- A. I. Abdelrahman, A. M. Mohammad, T. Okajima and T. Ohsaka, J. Phys. Chem. B, 2006, 110, 2798–2803 CrossRef CAS.
- Y. Qian, W. Wang, Z. Wang, X. Jia, Q. Han, I. Rostami, Y. Wang and Z. Hu, ACS Appl. Mater. Interfaces, 2018, 10, 7871–7881 CrossRef CAS.
- M. J. Kogan, I. Olmedo, L. Hosta, A. R. Guerrero, L. J. Cruz and F. Albericio, Nanomedicine, 2007, 2, 287–306 CrossRef CAS PubMed.
- Y. Dou, H. Xu and J. Hao, Soft Matter, 2013, 9, 5572–5580 RSC.
- M. Abbas, H. H. Susapto and C. A. E. Hauser, ACS Omega, 2022, 7, 2082–2090 CrossRef CAS.
- Y. Chen, Y. Yang, A. A. Orr, P. Makam, B. Redko, E. Haimov, Y. Wang, L. J. W. Shimon, S. Rencus-Lazar, M. Ju, P. Tamamis, H. Dong and E. Gazit, Angew. Chem., Int. Ed., 2021, 60, 17164–17170 CrossRef CAS.
- D. Zaramella, P. Scrimin and L. J. Prins, J. Am. Chem. Soc., 2012, 134, 8396–8399 CrossRef CAS PubMed.
- Y. Perdomo, J. M. Slocik, D. M. Phillips and M. R. Knecht, J. Am. Chem. Soc., 2023, 145, 16650–16657 CrossRef CAS.
- N. M. Bedford, Z. E. Hughes, Z. Tang, Y. Li, B. D. Briggs, Y. Ren, M. T. Swihart, V. G. Petkov, R. R. Naik, M. R. Knecht and T. R. Walsh, J. Am. Chem. Soc., 2016, 138, 540–548 CrossRef CAS PubMed.
- A. Kumar Mahato, S. Pal, K. Dey, A. Reja, S. Paul, A. Shelke, T. G. Ajithkumar, D. Das and R. Banerjee, J. Am. Chem. Soc., 2023, 145, 12793–12801 CrossRef CAS.
- X. Tan, Y. Xu, S. Lin, G. Dai, X. Zhang, F. Xia and Y. Dai, J. Catal., 2021, 402, 125–129 CrossRef CAS.
- D. J. Mikolajczak and B. Koksch, ChemCatChem, 2018, 10, 4324–4328 CrossRef CAS.
- H. Wennemers, Chem. Commun., 2011, 47, 12036–12041 RSC.
- N. Liu, S. B. Li, Y. Z. Zheng, S. Y. Xu and J. S. Shen, ACS Omega, 2023, 8, 2491–2500 CrossRef CAS.
- Q. Liu, A. Kuzuya and Z. G. Wang, iScience, 2023, 26, 105831 CrossRef CAS PubMed.
- J. Duschmalé, S. Kohrt and H. Wennemers, Chem. Commun., 2014, 50, 8109–8112 RSC.
- A. Reja, S. P. Afrose and D. Das, Angew. Chem., Int. Ed., 2020, 59, 4329–4334 CrossRef CAS.
- J. Wang, R. Huang, W. Qi, R. Su, B. P. Binks and Z. He, Appl. Catal., B, 2019, 254, 452–462 CrossRef CAS.
- X. Xu, J. Wang, R. Huang, W. Qi, R. Su and Z. He, Catal. Sci. Technol., 2021, 11, 3402–3410 RSC.
- R. Geng, R. Chang, Q. Zou, G. Shen, T. Jiao and X. Yan, Small, 2021, 17, 1–6 Search PubMed.
- Y. Feng, Y. Wang, J. Zhang, M. Wang, W. Qi, R. Su and Z. He, Adv. Mater. Interfaces, 2019, 6, 1–9 CAS.
- O. Zozulia, L. R. Marshall, I. Kim, E. M. Kohn and I. V. Korendovych, Chem. - Eur. J., 2021, 27, 5388–5392 CrossRef CAS PubMed.
- M. O. Guler and S. I. Stupp, J. Am. Chem. Soc., 2007, 129, 12082–12083 CrossRef CAS PubMed.
- B. Sarkhel, A. Chatterjee and D. Das, J. Am. Chem. Soc., 2020, 142, 4098–4103 CrossRef CAS.
- A. Singh, J. P. Joseph, D. Gupta, C. Miglani, N. A. Mavlankar and A. Pal, Nanoscale, 2021, 13, 13401–13409 RSC.
- D. Gupta, R. Sasmal, A. Singh, J. P. Joseph, C. Miglani, S. S. Agasti and A. Pal, Nanoscale, 2020, 12, 18692–18700 RSC.
- P. Katyal, M. Meleties and J. K. Montclare, ACS Biomater. Sci. Eng., 2019, 5, 4132–4147 CrossRef CAS PubMed.
- A. Lampel and M. Reches, Isr. J. Chem., 2022, 62, 1–2 CrossRef.
- S. K. H. Shah, P. Chauhan, S. Karmakar, T. Mehmood, J. P. Reddy and P. Prabhakaran, in Design, Principle and Application of Self-Assembled Nanobiomaterials in Biology and Medicine, ed. A. Pandya, R. S. Bhosale and V. B. T.-D. Singh, Principle and Application of Self-Assembled Nanobiomaterials in Biology and Medicine, Elsevier, 2022, pp. 173–192 Search PubMed.
- J. Chen and X. Zou, Bioact. Mater., 2019, 4, 120–131 Search PubMed.
- S. Eskandari, T. Guerin, I. Toth and R. J. Stephenson, Adv. Drug Delivery Rev., 2017, 110–111, 169–187 CrossRef CAS PubMed.
- U. Ghosh and G. Ghosh, Supramolecular Self-Assembled Peptide-Based Nanostructures and Their Applications in Biomedicine, in Pharmaceutical Applications of Supramolecules, Springer, Cham., 2023, pp. 241–271 Search PubMed.
- J. Banerjee, E. Radvar and H. S. Azevedo, Self-assembling peptides and their application in tissue engineering and regenerative medicine, Elsevier Ltd, 2018 Search PubMed.
- S. Koutsopoulos, J. Biomed. Mater. Res., Part A, 2016, 104, 1002–1016 CrossRef CAS PubMed.
- X. Y. Guo, L. Yi, J. Yang, H. W. An, Z. X. Yang and H. Wang, Chem. Commun., 2024, 36, 2009–2021 RSC.
- R. C. Guo, X. H. Zhang, P. S. Fan, B. L. Song, Z. X. Li, Z. Y. Duan, Z. Y. Qiao and H. Wang, Angew. Chem., Int. Ed., 2021, 60, 25128–25134 CrossRef CAS.
- M. Di Wang, D. Y. Hou, G. T. Lv, R. X. Li, X. J. Hu, Z. J. Wang, N. Y. Zhang, L. Yi, W. H. Xu and H. Wang, Biomaterials, 2021, 278, 121139 CrossRef PubMed.
- M. Shahriari, M. Zahiri, K. Abnous, S. M. Taghdisi, M. Ramezani and M. Alibolandi, J. Controlled Release, 2019, 308, 172–189 CrossRef CAS PubMed.
- F. Araste, K. Abnous, M. Hashemi, S. M. Taghdisi, M. Ramezani and M. Alibolandi, J. Controlled Release, 2018, 292, 141–162 CrossRef CAS PubMed.
- Y. S. Zhu, K. Tang and J. Lv, Trends Pharmacol. Sci., 2021, 42, 857–869 CrossRef CAS PubMed.
- Y. Wang, A. G. Cheetham, G. Angacian, H. Su, L. Xie and H. Cui, Adv. Drug Delivery Rev., 2017, 110–111, 112–126 CrossRef CAS.
- E. I. Vrettos, G. Mező and A. G. Tzakos, Beilstein J. Org. Chem., 2018, 14, 930–954 CrossRef CAS.
- J. Yang, H. W. An and H. Wang, ACS Appl. Bio Mater., 2021, 4, 24–46 CrossRef CAS PubMed.
- M. Di Wang, D. Y. Hou, G. T. Lv, R. X. Li, X. J. Hu, Z. J. Wang, N. Y. Zhang, L. Yi, W. H. Xu and H. Wang, Biomaterials, 2021, 278, 121139 CrossRef PubMed.
- Z. Song, X. Chen, X. You, K. Huang, A. Dhinakar, Z. Gu and J. Wu, Biomater. Sci., 2017, 5, 2369–2380 RSC.
- S. Yang, M. Wang, T. Wang, M. Sun, H. Huang, X. Shi, S. Duan, Y. Wu, J. Zhu and F. Liu, Mater. Today Bio, 2023, 20, 100644 CrossRef CAS PubMed.
- J. Zhao, D. Zheng, Y. Tao, Y. Li, L. Wang, J. Liu, J. He and J. Lei, Biochem. Eng. J., 2020, 156, 107526 CrossRef CAS.
- S. J. Franks, K. Firipis, R. Ferreira, K. M. Hannan, R. J. Williams, R. D. Hannan and D. R. Nisbet, Mater. Horiz., 2020, 7, 1996–2010 RSC.
- V. B. Kumar, B. Ozguney, A. Vlachou, Y. Chen, E. Gazit and P. Tamamis, J. Phys. Chem. B, 2023, 127, 1857–1871 CrossRef CAS.
- Z. Li, Y. Zhu and J. B. Matson, ACS Appl. Bio Mater., 2022, 5, 4635–4651 CrossRef CAS.
- D. Bairagi, P. Biswas, K. Basu, S. Hazra, D. Hermida-Merino, D. K. Sinha, I. W. Hamley and A. Banerjee, ACS Appl. Bio Mater., 2019, 2, 5235–5244 CrossRef CAS PubMed.
- R. Curvello, V. S. Raghuwanshi and G. Garnier, Adv. Colloid Interface Sci., 2019, 267, 47–61 CrossRef CAS.
- M. Sedighi, F. Rahimi, M. A. Shahbazi, A. H. Rezayan, H. Kettiger, T. Einfalt, J. Huwyler and D. Witzigmann, ACS Appl. Bio Mater., 2020, 3, 239–251 CrossRef CAS PubMed.
- D. H. Zhao, J. Yang, R. X. Xia, M. H. Yao, R. M. Jin, Y. Di Zhao and B. Liu, Chem. Commun., 2018, 54, 527–530 RSC.
- S. Chibh, A. Kour, N. Yadav, P. Kumar, P. Yadav, V. S. Chauhan and J. J. Panda, ACS Omega, 2020, 5, 3365–3375 CrossRef CAS.
- R. Chang, Q. Zou, L. Zhao, Y. Liu, R. Xing and X. Yan, Adv. Mater., 2022, 34, 1–9 Search PubMed.
- J. Han, H. Li, L. Zhao, G. Kim, Y. Chen, X. Yan and J. Yoon, Chem. Sci., 2022, 13, 7814–7820 RSC.
- Z. Li, S. Li, Y. Guo, C. Yuan, X. Yan and K. S. Schanze, ACS Nano, 2021, 15, 4979–4988 CrossRef CAS.
- S. Cazzamalli, B. Ziffels, F. Widmayer, P. Murer, G. Pellegrini, F. Pretto, S. Wulhfard and D. Neri, Clin. Cancer Res., 2018, 24, 3656–3667 CrossRef CAS PubMed.
- Y. Cai, C. Zheng, F. Xiong, W. Ran, Y. Zhai, H. H. Zhu, H. Wang, Y. Li and P. Zhang, Adv. Healthcare Mater., 2021, 10, 1–23 Search PubMed.
- F. Raza, Y. Zhu, L. Chen, X. You, J. Zhang, A. Khan, M. W. Khan, M. Hasnat, H. Zafar, J. Wu and L. Ge, Biomater. Sci., 2019, 7, 2023–2036 RSC.
- L. Kennedy, J. K. Sandhu, M. E. Harper and M. Cuperlovic-culf, Biomolecules, 2020, 10, 1–27 CrossRef.
- S. Koutsopoulos, J. Biomed. Mater. Res., Part A, 2016, 104, 1002–1016 CrossRef CAS.
- R. Bazak, M. Houri, S. El Achy, W. Hussein and T. Refaat, Mol. Clin. Oncol., 2014, 2, 904–908 CrossRef PubMed.
- M. Sedighi, N. Shrestha, Z. Mahmoudi, Z. Khademi, A. Ghasempour, H. Dehghan, S. F. Talebi, M. Toolabi, V. Préat, B. Chen, X. Guo and M. A. Shahbazi, Polymers, 2023, 15, 1160 CrossRef CAS PubMed.
- K. Han, Z. Ma and H. Han, J. Mater. Chem. B, 2017, 6, 25–38 RSC.
- N. Mukherjee, R. Roy, S. Ghosh and S. Ghosh, Isr. J. Chem., 2022, 62, e202200019 CrossRef CAS.
- S. Sivagnanam, K. Das, V. Sivakadatcham, T. Mahata, M. Basak, I. Pan, A. Stewart, B. Maity and P. Das, Isr. J. Chem., 2022, 62, e202200001 CrossRef CAS.
- Y. Yu, X. Jiang, S. Gong, L. Feng, Y. Zhong and Z. Pang, Nanoscale, 2014, 6, 3250–3258 RSC.
- C. Ryou, J. Microbiol. Biotechnol., 2007, 17, 1059 CAS.
- Y. Zheng, L. Zhang, J. Xie and L. Shi, Front. Aging Neurosci., 2021, 13, 636726 Search PubMed.
- L. B. Vong, Y. Sato, P. Chonpathompikunlert, S. Tanasawet, P. Hutamekalin and Y. Nagasaki, Acta Biomater., 2020, 109, 220–228 CrossRef CAS.
- O. T. Korkmaz and N. Tunçel, Curr. Pharm. Des., 2019, 24, 4693–4701 CrossRef PubMed.
- M. H. Baig, K. Ahmad, G. Rabbani and I. Choi, Front. Aging Neurosci., 2018, 10, 1–6 CrossRef PubMed.
- C. Li, S. Grajales, S. Shuang, C. Dong and M. Nair, J. Anal. Test., 2017, 1, 15 CrossRef.
- G. Eskici and M. Gur, PLoS One, 2013, 8 Search PubMed.
- S. Jagota and J. Rajadas, Med. Chem. Res., 2013, 22, 3991–4000 CrossRef CAS.
- J. Liu, W. Wang, Q. Zhang, S. Zhang and Z. Yuan, Biomacromolecules, 2014, 15, 931–939 CrossRef CAS.
- R. J. Chalifour, R. W. McLaughlin, L. Lavoie, C. Morissette, N. Tremblay, M. Boulé, P. Sarazin, D. Stéa, D. Lacombe, P. Tremblay and F. Gervais, J. Biol. Chem., 2003, 278, 34874–34881 CrossRef CAS.
- C. W. Wei, Y. Peng, L. Zhang, Q. Huang, M. Cheng, Y. N. Liu and J. Li, Bioorganic Med. Chem. Lett., 2011, 21, 5818–5821 CrossRef CAS.
- M. S. Zharkova, D. S. Orlov, O. Y. Golubeva, O. B. Chakchir, I. E. Eliseev, T. M. Grinchuk and O. V. Shamova, Front. Cell. Infect. Microbiol., 2019, 9, 128 CrossRef CAS PubMed.
- A. Gupta, S. Mumtaz, C. H. Li, I. Hussain and V. M. Rotello, Chem. Soc. Rev., 2019, 48, 415–427 RSC.
- X. Tian, F. Sun, X. R. Zhou, S. Z. Luo and L. Chen, J. Pept. Sci., 2015, 21, 530–539 CrossRef CAS.
- T. Rounds and S. K. Straus, Int. J. Mol. Sci., 2020, 21, 1–15 Search PubMed.
- A. F. Chu-Kung, R. Nguyen, K. N. Bozzelli and M. Tirrell, J. Colloid Interface Sci., 2010, 345, 160–167 CrossRef CAS.
- R. Hadianamrei and X. Zhao, J. Controlled Release, 2022, 343, 600–619 CrossRef CAS PubMed.
- M. Urello, W. H. Hsu and R. J. Christie, Acta Biomater., 2020, 117, 40–59 CrossRef CAS.
- D. Bumcrot, M. Manoharan, V. Koteliansky and D. W. Y. Sah, Nat. Chem. Biol., 2006, 2, 711–719 CrossRef CAS PubMed.
- F. Mingozzi and K. A. High, Nat. Rev. Genet., 2011, 12, 341–355 CrossRef CAS PubMed.
- L. Crombez, G. Aldrian-Herrada, K. Konate, Q. N. Nguyen, G. K. McMaster, R. Brasseur, F. Heitz and G. Divita, Mol. Ther., 2009, 17, 95–103 CrossRef CAS.
- W. Li, D. Wang, X. Shi, J. Li, Y. Ma, Y. Wang, T. Li, J. Zhang, R. Zhao, Z. Yu, F. Yin and Z. Li, Mater. Horiz., 2018, 5, 745–752 RSC.
- Y. Guo, Y. Hu, X. Zheng, X. Cao, Q. Li, Z. Wei, Z. Zhu and S. Zhang, Talanta, 2021, 221, 121572 CrossRef CAS.
- X. Liu, X. Sun and G. Liang, Biomater. Sci., 2021, 9, 315–327 RSC.
- X. Ma and Y. Zhao, Chem. Rev., 2015, 115, 7794–7839 CrossRef CAS PubMed.
- Q. Jiang, X. Liu, G. Liang and X. Sun, Nanoscale, 2021, 13, 15142–15150 RSC.
- Y. Deng, W. Zhan and G. Liang, Adv. Healthcare Mater., 2021, 10, 1–16 Search PubMed.
- X. X. Zhao, L. L. Li, Y. Zhao, H. W. An, Q. Cai, J. Y. Lang, X. X. Han, B. Peng, Y. Fei, H. Liu, H. Qin, G. Nie and H. Wang, Angew. Chem., Int. Ed., 2019, 58, 15287–15294 CrossRef CAS.
- M. Lian, L. Xu, X. Zhu, X. Chen, W. Yang and T. Wang, Anal. Chem., 2017, 89, 12843–12849 CrossRef CAS PubMed.
- Y. Deng, W. Zhan and G. Liang, Adv. Healthcare Mater., 2021, 10, 2001211 CrossRef CAS PubMed.
- K. Tao, P. Makam, R. Aizen and E. Gazit, Science, 2017, 358, eaam9756 CrossRef PubMed.
- O. Silberbush, M. Engel, I. Sivron, S. Roy and N. Ashkenasy, J. Phys. Chem. B, 2019, 123, 9882–9888 CrossRef CAS PubMed.
- A. Fortunato, R. C. Hensel, S. Casalini and M. Mba, Molecules, 2023, 28, 1–11 CrossRef.
- N. Forlano, R. Bucci, A. Contini, M. Venanzi, E. Placidi, M. L. Gelmi, R. Lettieri and E. Gatto, Nanomaterials, 2023, 13, 333 CrossRef CAS.
- N. L. Ing, M. Y. El-Naggar and A. I. Hochbaum, J. Phys. Chem. B, 2018, 122, 10403–10423 CrossRef CAS PubMed.
- Y. Gyo, K. Shin, M. D. Newton and S. S. Isied, J. Am. Chem. Soc., 2003, 125, 3722–3732 CrossRef PubMed.
- N. Amdursky, Phys. Chem. Chem. Phys., 2013, 15, 13479–13482 RSC.
- L. Sepunaru, S. Refaely-Abramson, R. Lovrinčić, Y. Gavrilov, P. Agrawal, Y. Levy, L. Kronik, I. Pecht, M. Sheves and D. Cahen, J. Am. Chem. Soc., 2015, 137, 9617–9626 CrossRef CAS PubMed.
- N. Hazra, S. Hazra, K. Gayen, P. Ghosh, A. Palui and A. Banerjee, ACS Appl. Polym. Mater., 2024, 6, 859–869 CrossRef CAS.
- N. L. Ing, R. K. Spencer, S. H. Luong, H. D. Nguyen and A. I. Hochbaum, ACS Nano, 2018, 12, 2652–2661 CrossRef CAS.
- R. C. G. Creasey, A. B. Mostert, A. Solemanifar, T. A. H. Nguyen, B. Virdis, S. Freguia and B. Laycock, ACS Omega, 2019, 4, 1748–1756 CrossRef CAS.
- M. Konda, S. Maiti, R. G. Jadhav and A. K. Das, Chem. - Asian J., 2018, 13, 204–209 CrossRef CAS.
- J. López-Andarias, M. J. Rodriguez, C. Atienza, J. L. López, T. Mikie, S. Casado, S. Seki, J. L. Carrascosa and N. Martín, J. Am. Chem. Soc., 2015, 137, 893–897 Search PubMed.
- H. K. Bisoyi and Q. Li, Chem. Rev., 2022, 122, 4887–4926 Search PubMed.
- X. Hu, M. Liao, H. Gong, L. Zhang, H. Cox, T. A. Waigh and J. R. Lu, Curr. Opin. Colloid Interface Sci., 2020, 45, 1–13 CrossRef CAS.
- M. H. Shah and A. Paradkar, Eur. J. Pharm. Biopharm., 2007, 67, 166–174 CrossRef CAS PubMed.
- P. Wadsten-Hindrichsen, J. Bender, J. Unga and S. Engström, J. Colloid Interface Sci., 2007, 315, 701–713 CrossRef CAS PubMed.
- J. Barauskas and T. Landh, Langmuir, 2003, 19, 9562–9565 CrossRef CAS.
- Y. Huang and S. Gui, RSC Adv., 2018, 8, 6978–6987 RSC.
- S. J. Woltman, G. D. Jay and G. P. Crawford, Nat. Mater., 2007, 6, 929–938 CrossRef CAS.
- K. Iwabata, U. Sugai, Y. Seki, H. Furue and K. Sakaguchi, Molecules, 2013, 18, 4703–4717 CrossRef CAS.
- M. E. Prévôt, S. Ustunel and E. Hegmann, Materials, 2018, 11, 377 CrossRef PubMed.
- R. Azriel and E. Gazit, J. Biol. Chem., 2001, 276, 34156–34161 CrossRef CAS PubMed.
- Y. Mazor, S. Gilead, I. Benhar and E. Gazit, J. Mol. Biol., 2002, 322, 1013–1024 CrossRef CAS.
- M. Reches, Y. Porat and E. Gazit, J. Biol. Chem., 2002, 277, 35475–35480 CrossRef CAS PubMed.
- T. H. Han, J. Kim, J. S. Park, C. B. Park, H. Ihee and S. O. Kim, Adv. Mater., 2007, 19, 3924–3927 CrossRef CAS.
- G. Nyström, M. Arcari and R. Mezzenga, Nat. Nanotechnol., 2018, 13, 330–336 CrossRef.
- R. P. Cheng and W. F. DeGrado, J. Am. Chem. Soc., 2002, 124, 11564–11565 CrossRef CAS PubMed.
- P. Ji, X. Liu, J. Xu, X. Zhang, J. Guo, W. Chen and B. Zhao, Angew. Chem., Int. Ed., 2022, e202206111 CAS.
- W. C. Pomerantz, N. L. Abbott and S. H. Gellman, J. Am. Chem. Soc., 2006, 128, 8730–8731 Search PubMed.
- S. Zhang, M. A. Greenfield, A. Mata, L. C. Palmer, R. Bitton, J. R. Mantei, C. Aparicio, M. O. De La Cruz and S. I. Stupp, Nat. Mater., 2010, 9, 594–601 CrossRef CAS PubMed.
- Y. Wang, W. Qi, J. Wang, Q. Li, X. Yang, J. Zhang, X. Liu, R. Huang, M. Wang, R. Su and Z. He, Chem. Mater., 2018, 30, 7902–7911 Search PubMed.
- V. K. Gupta, J. J. Skaife, T. B. Dubrovsky and N. L. Abbott, Science, 1998, 279, 2077–2080 CrossRef CAS PubMed.
- S. Serizawa, K. Miyamichi, H. Nakatani, M. Suzuki, M. Saito, Y. Yoshihara and H. Sakano, Science, 2003, 302, 2088–2094 CrossRef CAS PubMed.
- B. Liu, Y. Cao, Z. Huang, Y. Duan and S. Che, Adv. Mater., 2015, 27, 479–497 CrossRef CAS PubMed.
- C. L. Chen and N. L. Rosi, Angew. Chem., Int. Ed., 2010, 49, 1924–1942 CrossRef CAS PubMed.
- V. M. Yuwono and J. D. Hartgerink, Langmuir, 2007, 23, 5033–5038 CrossRef CAS PubMed.
- L. Wang and Q. Li, Chem. Soc. Rev., 2018, 47, 1044–1097 RSC.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1217 CrossRef CAS.
- M. Wallace, A. Z. Cardoso, W. J. Frith, J. A. Iggo and D. J. Adams, Chem. - Eur. J., 2014, 20, 16484–16487 CrossRef CAS.
- K. Aoki, N. Tamaoki, A. Seki, K. Narazaki, D. Takahashi and K. Horitsugu, Langmuir, 2021, 37, 13160–13169 CrossRef CAS PubMed.
- I. Ziemecka, G. J. M. Koper, A. G. L. Olive and J. H. Van Esch, Soft Matter, 2013, 9, 1556–1561 RSC.
- B. Wu, S. Zhao, X. Yang, L. Zhou, Y. Ma, H. Zhang, W. Li and H. Wang, ACS Nano, 2022, 16, 4126–4138 CrossRef CAS PubMed.
- T. Z. Jia and T. P. Fraccia, Crystals, 2020, 10, 1–21 Search PubMed.
- Z. Zhang, X. Yang, Y. Zhao, F. Ye and L. Shang, Adv. Mater., 2023, 35, 1–29 Search PubMed.
- V. P. Chavda, S. Dawre, A. Pandya, L. K. Vora, D. H. Modh, V. Shah, D. J. Dave and V. Patravale, J. Controlled Release, 2022, 349, 533–549 CrossRef CAS PubMed.
- N. Yadav, M. K. Chauhan and V. S. Chauhan, Biomater. Sci., 2020, 8, 84–100 RSC.
- S. Setia, S. Sidiq, J. De, I. Pani and S. K. Pal, Liq. Cryst., 2016, 43, 2009–2050 CrossRef CAS.
- Y. Wang, D. Fang, T. Fu, M. U. Ali, Y. Shi, Y. He, Z. Hu, C. Yan, Z. Mei and H. Meng, Mater. Chem. Front., 2020, 4, 3546–3555 RSC.
- M. M. Abadla, H. A. Elsayed and A. Mehaney, Phys. Scr., 2020, 95, 085508 CrossRef CAS.
- M. Nakayama and T. Kato, Acc. Chem. Res., 2022, 55, 1796–1808 Search PubMed.
- G. Zhang, Q. Li, J. Li, M. Pan, Y. Wang, R. Su, W. Qi and W. Zhang, Langmuir, 2023, 39, 8779–8786 CrossRef CAS PubMed.
- T. P. Fraccia and T. Z. Jia, ACS Nano, 2020, 14, 15071–15082 CrossRef CAS.
- W. Q. Ding, H. Liu, S. Y. Qin, Y. Jiang, X. Lei and A. Q. Zhang, ACS Appl. Bio Mater., 2020, 3, 8989–8996 CrossRef CAS PubMed.
- S. Cao, W. Fan, R. Chang, C. Yuan and X. Yan, CCS Chem., 2024, 1–11 Search PubMed.
- N. Gupta, A. Singh, N. Dey, S. Chattopadhyay, J. P. Joseph, D. Gupta, M. Ganguli and A. Pal, Chem. Mater., 2021, 33, 589–599 CrossRef CAS.
- M. Schumacher, P. Habibović and S. Van Rijt, ACS Appl. Mater. Interfaces, 2022, 14, 4959–4968 CrossRef CAS PubMed.
- P. Zou, W. T. Chen, T. Sun, Y. Gao, L. L. Li and H. Wang, Biomater. Sci., 2020, 8, 4975–4996 RSC.
- P. Tiwari, R. Verma, A. Basu, R. M. Christman, A. K. Tiwari, D. Waikar and A. Dutt Konar, ChemistrySelect, 2017, 2, 6623–6631 Search PubMed.
Footnote |
| † These authors contributed equally to this manuscript |
| This journal is © The Royal Society of Chemistry 2025 |