
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRhodium nanospheres for ultraviolet and visible plasmonics†
David
Muñeton Arboleda
 ab,
Vito
Coviello
a,
Arianna
Palumbo
a,
Roberto
Pilot
ac and
Vincenzo
Amendola
*a
ab,
Vito
Coviello
a,
Arianna
Palumbo
a,
Roberto
Pilot
ac and
Vincenzo
Amendola
*a
aDepartment of Chemical Sciences, University of Padova, Padova, Italy. E-mail: vincenzo.amendola@unipd.it
bCentro de Investigaciones Ópticas CIOp (CONICET-CIC-UNLP) and Facultad de Ingeniería UNLP, La Plata, Argentina
cConsorzio INSTM, UdR Padova, Padova, Italy
First published on 4th November 2024
Abstract
The development and understanding of alternative plasmonic materials are crucial steps for leveraging new plasmonic technologies. Although gold and silver nanostructures have been intensively studied, the promising plasmonic, chemical and physical attributes of rhodium remain poorly investigated. Here, we report the synthesis and plasmonic response of spherical Rh nanoparticles (NPs) with sizes in the 20–40 nm range. Due to the high cohesive energy of this metal, synthesis and experimental investigations of Rh nanospheres in this size range have not been reported; yet, it becomes possible here using a green and one-step laser ablation in liquid method. The localized surface plasmon (LSP) of Rh NPs falls in the ultraviolet spectral range (195–255 nm), but the absorption tail in the visible region increases significantly upon clustering of the nanospheres. The surface binding ability of Rh NPs towards thiolated molecules is equivalent to that of Au and Ag NPs, while their chemical and physical stability at high temperatures and in the presence of strong acids such as aqua regia is superior to those of Au and Ag NPs. The plasmonic features are well described by classical electrodynamics, and the results are comparable to Au and Ag NPs in terms of extinction cross-section and local field enhancement, although blue shifted. This allowed, for instance, their use as an optical nanosensor for the detection of ions of toxic metals in aqueous solution and for the surface enhanced Raman scattering of various compounds under blue light excitation. This study explores the prospects of Rh NPs in the realms of UV and visible plasmonics, while also envisaging a multitude of opportunities for other underexplored applications related to plasmon-enhanced catalysis and chiroplasmonics.
New conceptsPlasmonics has penetrated fields ranging from energy conversion to catalysis, biomedical analysis, nanomedicine, light management, and quantum phenomena. However, gold and silver nanostructures do not support plasmons in the ultraviolet (UV) region, while other UV-plasmonic compounds are not chemically stable and cannot be coated with functional molecules. Rh, on the other hand, has favorable characteristics for UV plasmonics, but its high cohesive energy leads to anisotropic nanocrystals with red-shifted plasmon resonance when their size exceeds 7–10 nm, which is the critical threshold for optimal performance. To address this issue, spherical Rh nanoparticles were synthesized by laser ablation in liquid in the range of 20–40 nm. Furthermore, these Rh nanospheres can be coated with ligands and their exploitation in the optical detection of toxic ions is demonstrated in the visible as well as in the UV range, which remained inaccessible to other plasmonic sensors of this type described so far. The spherical shape of Rh nanoparticles also facilitated benchmarking against classical electrodynamic models for plasmonic behavior. Finally, Rh nanoparticles offer an advantage over gold, silver and other materials because they are more resistant to harsh environments and high temperatures, while providing a comparable plasmonic response. Overall, Rh nanospheres have the required set of features to leverage plasmonic technologies in the underexploited UV region. |
Introduction
Localized surface plasmons (LSPs) are collective excitations of conduction electrons driven by the interaction of metallic nanostructures with electromagnetic radiation.1–4 To date, LSPs have been exploited for a wide number of applications spanning several technological and scientific branches, such as optical sensing,5 anticounterfeiting,6 ultradense data storage,7 theranostics,8 and photocatalysis,9 and various emerging fields including, for instance, quantum phenomena10 and chiro-selective photochemistry.11 The benchmark plasmonic materials are Au and Ag, which exhibit LSP resonances in the visible and near infrared spectral range, depending on the specific morphology of the nanostructure.1–3,12 Cu has also been studied for plasmonics but its applications are limited by the lower chemical and physical stability and by the presence of interband transitions in the visible range, which significantly affect the plasmonic performances.12,13 Indeed, these two features mostly determine the exploitability of any plasmonic material, which is true also for Au and Ag since they have interband transitions in the UV region, where they cannot support any LSP.3,12–14 Ultraviolet plasmonics is interesting because of its implications in advanced optical phenomena,15–17 plasmonic photocatalysis,18 thermal activation of catalytic processes,19 photodetection20 and sensing.21 UV photons can generate energetic hot-carriers in plasmonic nanostructures, unlocking catalytic processes with high energy barriers.15–17 Besides, provided that photodegradation is avoided, the Raman spectroscopy of organic compounds and biomolecules is boosted, because of the inverse dependence of the Raman scattering cross-section on the excitation frequency and the approaching to electronic transitions in molecules without resonances in the visible range.3,15–17 The combination of UV radiation and local field enhancement in plasmonic materials is also an emerging strategy for enhancing natural optical activity.11In order to expand the spectral range of plasmonic applications to the UV, a few materials with promising optical properties have been identified, such as Al, Mg, Bi, Ga and Rh.3,16,22,23 Other metals have plasmons in the UV region, such as Pd, Pt and some non-noble transition metals.3,16,22,23 However, their plasmonic properties are poor due to a small real component of the dielectric function compared to the imaginary component, either because of the presence of interband transitions in the UV range or because of their low electrical conductivity.3,12,13,24 Besides, non-noble metals undergo oxidation without the formation of a native shell of passivating oxide.
Unfortunately, Al, Mg and Bi are not chemically stable in air and in water,23,25–27 requiring further stabilization with thick passivating shells, while Ga is liquid at room temperature and may also undergo oxidation in the long term without appropriate protection.28 This is the main drawback of other plasmonic materials alternative to Au and Ag, such as nitrides.27
Rh is an exception, because it is renowned for its resistance to air and aqueous environments, without the formation of a native oxide coating, and for its high melting temperature (2236 K in the bulk).13,29 The chemical stability of Rh is a great advantage for its widespread use as a catalyst for hydrogenation, hydroformylation, NOx reduction, CO oxidation and hydrogen evolution.30–32 To date, Rh has been used in particular for surface enhanced Raman scattering (SERS),33–36 plasmon-enhanced fluorescence,37 UV photodetection38 and plasmonic photocatalysis,31,39 and even better functions are expected for Rh alloys.40,41 The main drawback of Rh, to date, is the limited earth abundance (0.0002 ppm) and the consequently high cost,32,39 which however is not the main problem for most plasmonic applications requiring only tiny amounts of metal (below milligrams), for which the prevailing cost sources are the synthesis, processing and durability of the device and the related sustainability and environmental impact.42
Several Rh nanostructures have been proposed for UV plasmonics, such as decahedra,29 nanocubes,31,34,35,43 nanomultipods,43 nanotetrahedra,39,43 nanoplates,34 and nanotriangles.44 The shape of plasmonic NPs impacts their optical and chemical properties, hence the controlled synthesis of distinct morphologies is important.29,44,45 The sphere is the simplest and most frequent shape of plasmonic NPs, and it is associated to the largest blue shift of the LSP resonance among all other geometries with equivalent volume and composition.1,2 Thus, Rh nanosperes exhibit plasmonic properties at shorter wavelengths than that of the other Rh nanostructures mentioned above. However, Rh nanospheres that have been reported to date are below the 5–7 nm size threshold.33,45 In this size range, some faceting already appeared and the plasmon properties are limited by the small dimension.44 Obtaining spherical Rh NPs with size >5–7 nm is still a challenge due to the exceptionally high surface energy and cohesive energy of Rh in its natural face centered cubic (FCC) structure, which exceed those of the other noble metals such as Pt, Pd, Au and Ag.29,44–46 For instance, spherical Pt NPs, spherical Pd NPs, but cubical Rh NPs were obtained under the same experimental wet chemistry conditions.44 This may explain the absence of literature reports about nanospheres with sizes above 10 nm obtained using chemical methods.
Another critical aspect of Rh NPs described in the literature is that they are obtained either on substrates or as a colloid with the use of capping agents and other stabilizing compounds.47 In the latter case, chemicals absorb UV light, overlapping with the LSP of rhodium nanostructures,29,33,43,44 and they hamper further functionalization with the desired organic molecules or the adsorption of analytes for SERS. Nonetheless, a considerable part of the success of “standard” Au and Ag plasmonic nanoparticles is connected to the easy surface chemistry based on the formation of metal–sulfur chemical bonds with any molecule exposing a thiol group.1,2 This is crucial for the stability of the colloid of NPs, also in complex environments at high ionic strength.48–50 Besides, surface functionalization is key to endowing plasmonic NPs with chemical selectivity, which is the basis for a series of analytical methods based on the change in the linear and nonlinear absorption spectra, as well as for analyte-selective surface enhanced spectroscopies.1,2 Hence, surface availability would be a key empowering feature for plasmonic Rh NPs.
Here we report the preparation and study of Rh nanospheres with a size of 20–45 nm, which exhibit a well-defined and intense LSP in the UV region and flexibility upon surface functionalization with thiolated ligands. The spherical Rh NPs were obtained using a laser ablation in liquid synthetic approach, which provides several benefits such as the use of a bulk target instead of more expensive chemical precursors and the operation at room temperature and ambient pressure.22,40,51,52 Most importantly, no surface coating agents or chemical contaminants are present on the surface of Rh NPs, which is left ready for functionalization with the desired molecules after or during the synthesis. The plasmonic properties of Rh NPs are described using classical electrodynamics and indicate good resistance to acid environments (aqua regia) and high temperature because of the excellent chemical stability of rhodium.
The set of properties of the Rh NPs allowed, for the first time, their exploitation as an optical sensor for the detection of toxic ions in aqueous solution and confirmed their potential for the collection of SERS spectra of various analytes upon excitation with blue light.
Overall, these results demonstrate the useful properties of Rh nanospheres for UV and visible plasmonics, opening the way for a multitude of opportunities for other underexplored applications in a broad range of the electromagnetic spectrum, including advanced plasmonic-related phenomena such as plasmon-enhanced catalysis and chiroplasmonics.
Results
The synthesis protocol follows what was previously reported for Au and Ag nanospheres53,54 and consists of laser ablation with 1064 nm laser pulses (6 ns) of a bulk rhodium plate in distilled water (Fig. 1A). Resorting to bulk pure metal is an advantage in the case of expensive elements like Rh because it avoids extra costs due to the preparation, purification and management of metal precursors,44,51,55 which in some cases also pose concerns for safety and formation of pollutant waste.32 Another advantage of the LAL is the straightforward automation and compatibility with remote control from a PC or a smartphone,54 as well as the scalability to gram-scale synthesis without compromising any of the typical features of the lab-scale procedure.56 Compared to other scalable chemical methods for the production of spherical UV plasmonic NPs, which may require several days,57 LAL is much faster. The choice of NIR laser pulses is crucial to avoid overlap with the plasmon absorption of the NPs, which induces photofragmentation exacerbating the polydispersity of the products. NIR light is also preferred to limit the formation of strongly oxidising species by decomposition of water molecules,25 which react even with noble metals such as Rh forming its oxide.32,43,58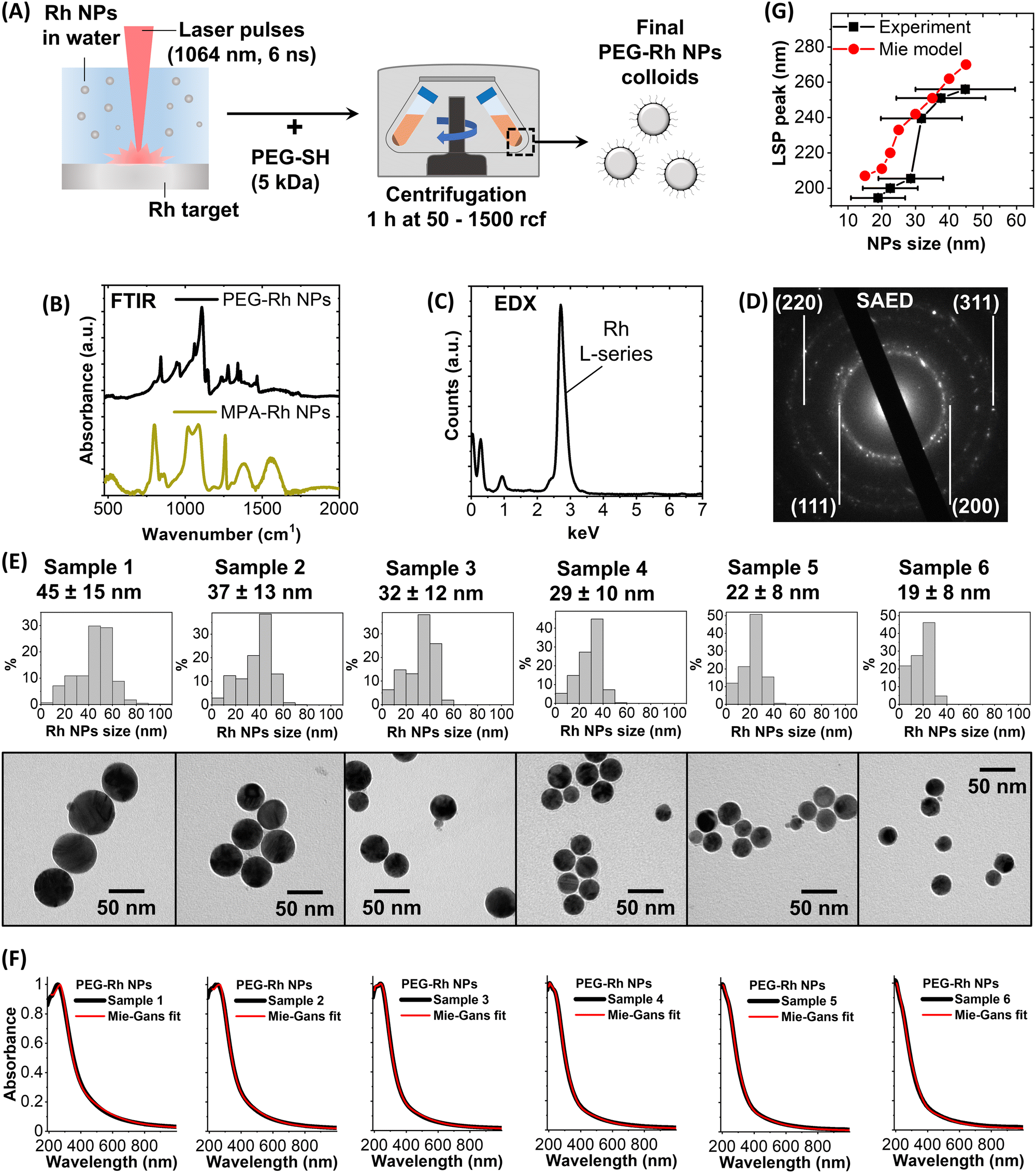 | ||
| Fig. 1 (A) Sketch of the LAL synthesis followed by the SBS protocol for the size selection of the six Rh NP samples. (B) FTIR spectra of PEG-Rh NPs and MPA-Rh showing the distinctive bands of, respectively, PEG and MPA. (C) and (D) TEM-EDX spectrum (C) and TEM-SAED pattern (D) of a group of PEG-Rh NPs. (E) TEM images and size histograms of the six PEG-Rh NP samples. (F) UV-visible spectra (190–1000 nm) of the six PEG-Rh samples (black lines) and fittings with the Mie-Gans model (red lines). (G) Plot of the LSP peak position versus the NP size for the six PEG-Rh NP samples (black squares) and from the Mie model simulations for Rh nanospheres in water. | ||
The absence of coating agents and other chemicals permits the functionalization of Rh NPs with the desired ligands and their separation from the synthesis solution by centrifugation or dialysis. Thiolated polyethylene glycol (PEG-SH) was added to the colloid after the synthesis, to ensure stability in an aqueous solution. The successful coating of Rh NPs with PEG was assessed by Fourier-transform infrared (FTIR) spectroscopy of the particles collected after dialysis to remove the unbound polymer (Fig. 1B). The FTIR spectrum of the dried PEG-Rh NP pellet shows the vibrational fingerprint of the polyethylene glycol backbone, such as the intense C–O–C stretching at 1100 cm−1 and the bands in the 800–1500 cm−1 region, including the vibrations of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–H bonds.49 Besides, to demonstrate that the Rh NPs can be coated also with other thiolated compounds, mercaptopropionic acid (MPA) was used, and this will be of interest for the applications described in the following section. Again, the successful coating of the dialysed and pelleted MPA-Rh NPs was assessed by FTIR (Fig. 1B) by identifying several distinctive bands such as those of the CO of the carboxylic group (1550 cm−1), the CH2 wagging mode (1380 cm−1), C–O stretching (1260 cm−1) and the O–CO symmetric stretching bands (1020 and 1080 cm−1).59
O and C–H bonds.49 Besides, to demonstrate that the Rh NPs can be coated also with other thiolated compounds, mercaptopropionic acid (MPA) was used, and this will be of interest for the applications described in the following section. Again, the successful coating of the dialysed and pelleted MPA-Rh NPs was assessed by FTIR (Fig. 1B) by identifying several distinctive bands such as those of the CO of the carboxylic group (1550 cm−1), the CH2 wagging mode (1380 cm−1), C–O stretching (1260 cm−1) and the O–CO symmetric stretching bands (1020 and 1080 cm−1).59
Energy dispersive X-ray (EDX) spectroscopy (Fig. 1C) and selected area electron diffraction (SAED, Fig. 1B) were performed on the PEG-Rh NPs within a transmission electron microscope (TEM) to identify the spectral signature of rhodium (L-series in the EDX spectrum) and the reflections from the (111), (200), (220) and (311) lattice planes, expected for the FCC rhodium structure.
Then, to study the optical properties of PEG-Rh NPs, a well-established selective sedimentation protocol was applied, working in distilled water, to isolate different size fractions.53,60 From the TEM images (Fig. 1E) we evaluated the spherical morphology, typical of noble metal particles obtained by LAL,52 of the six fractions of PEG-Rh NPs, which have the average sizes of 45 ± 15 nm (Sample 1), 37 ± 13 nm (Sample 2), 32 ± 12 nm (Sample 3), 29 ± 10 nm (Sample 4), 22 ± 8 nm (Sample 5) and 19 ± 8 nm (Sample 6). These are the only examples of spherical Rh NPs in these size ranges reported to date. Benefiting from the absence of other chemical compounds with absorption bands in the UV region, the optical properties of the aqueous solutions of PEG-Rh NPs in quartz cuvettes were measured by UV-visible spectroscopy in the 190–1000 nm spectral range (Fig. 1F). All samples have a plasmon band centered in the UV region with a tail extending to the visible region. The LSP maximum is at longer wavelengths for the biggest NPs (256 nm for Sample 1) and at shorter wavelengths for the smallest NPs (195 nm for Sample 6). The trend of the LSP position is in general agreement with the predictions of the Mie model for spherical Rh NPs in water (Fig. 1G). Therefore, we used a classical electrodynamics approach based on the Mie and Gans models2,61 to fit the experimental spectra, which provided remarkable agreement as shown by the red lines in Fig. 1F. Besides, the model succeeded in predicting the average size of the samples with an accuracy of 0–16% in all cases (45 nm, 43 nm, 34 nm, 29 nm, 24 nm and 21 nm for, respectively, samples 1, 2, 3, 4, 5 and 6) and without any prior calibration, which instead is required for Au NPs.2,61 This is the first example of the accurate replication of the experimental optical properties of Rh NPs using classical electrodynamics, indicating that the dielectric function reported in the literature and the theoretical models are exploitable for the prediction of plasmonic properties in rhodium nanostructures. For this purpose, the availability of NPs with a simple spherical geometry represented an advantage for benchmarking the modelling capabilities.
Despite Rh being known for its high melting temperature and resistance to strong acids, the plasmonic properties of Rh NPs have not been tested previously against these extreme conditions, also in comparison to Au and Ag NPs. In fact, the plasmon band of the PEG-coated Au and Ag NPs in water (Fig. 2A) disappeared completely (an LSP intensity change, ΔLSP, of −100%) already at a concentration of aqua regia of 2 v/v% for Ag and 10 v/v% for Au, which is the most noble of the two plasmonic metals. In contrast, the plasmon band of PEG-coated Rh NPs persisted, with a ΔLSP of −17 ± 2% at 2 v/v% and −35 ± 3% at 10 v/v%. This decay is also contributed by particle aggregation at low pH and by the oxidising conditions affecting both the polymeric coating and the low-coordinated surface Rh atoms in the nanostructures.43 The effect of aqua regia was investigated more at the single NP level by depositing PEG-Rh and PEG-Au NPs on a crystalline Si substrate and exposing it to an acid-saturated environment for 15′ (Fig. 2B). The scanning electron microscopy (SEM) images confirmed that the Au NPs are completely degraded by the interaction with the vapours of aqua regia, whereas Rh NPs maintained their initial morphology and size, although a coating appeared on the surface, which is compatible with the formation of a passivating oxide layer.32,43
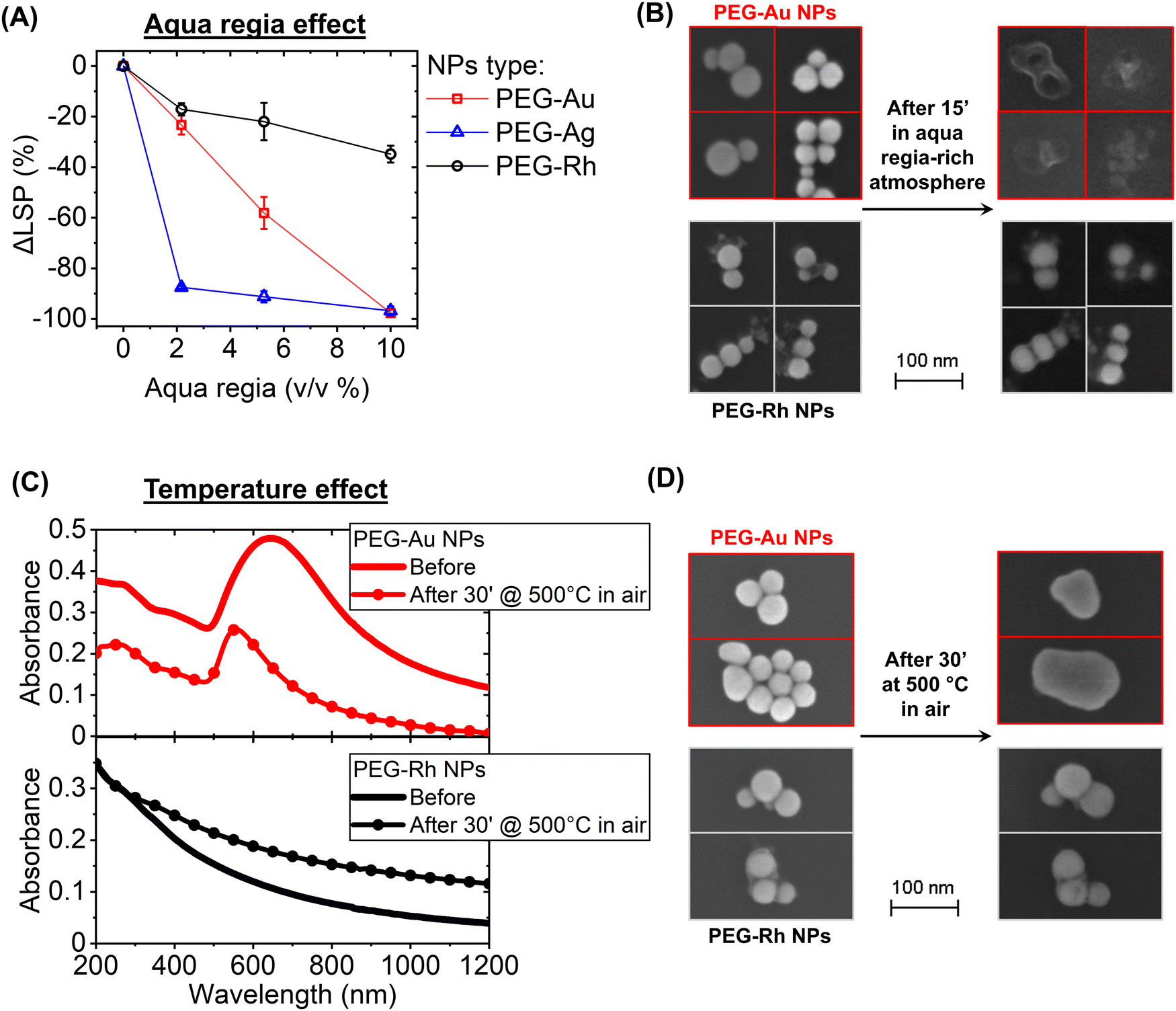 | ||
| Fig. 2 (A) Change in LSP intensity (ΔLSP), in water dispersions of PEG-coated Rh, Au and Ag NPs. The error bars are the average of two independent experiments. (B) SEM images of the same PEG-Rh and PEG-Au NPs on a crystalline Si substrate before and after exposure to a saturated aqua regia atmosphere for 15′. (C) Optical absorption spectra of PEG-Rh and PEG-Au NPs deposited on quartz before and after heating in air at 500 °C for 30′. (D) SEM images of the same PEG-Rh and PEG-Au NPs on a crystalline Si substrate before and after heating in air at 500 °C for 30′. | ||
The effect of temperature on the LSP and the morphology of Rh NPs, compared to that of Au NPs with equivalent size and coating, was also tested. The PEG-Rh and PEG-Au NPs were deposited on quartz and crystalline Si to record optical properties and morphology before and after heating in air at 500 °C for 30′. It is well known that the melting temperature and thermal stability of Au and Ag nanostructures are dramatically reduced at the nanoscale compared to the bulk.27 In fact, despite a bulk gold melting temperature of 1064 °C, the LSP of Au NPs undergoes blue shifting and shrinking at 500 °C, with a significant reduction in the scattering background. These changes are indicative of the transition from the aggregates of the pristine Au NPs on the quartz substrate to single submicrometric NPs due to melting and merging (Fig. 2C top). This is confirmed by the SEM images of the PEG-Au NPs on the Si substrate (Fig. 2D top). In contrast, the optical properties of Rh NPs do not change in the UV region, although a significant increase of the scattering background is observed after the heat treatment (Fig. 2C bottom). However, the SEM images indicated that the morphology of the Rh NPs remained unmodified, with no signs of melting or merging (Fig. 2D bottom). Since the PEG coating is oxidised at 500 °C in air, the change in the optical properties is, thus, compatible with the coarsening of the Rh NPs in the macroscopic deposits on the quartz substrate due to the removal of the organic shell.
A great advantage of plasmonic NPs is the possibility of adding chemical functionalities that allow the specific recognition of analytes through the change of their colloidal state from isolated to aggregated particles, which implies a modification of the optical properties due to the LSP hybridization between particles.1,2,5,50 However, there are no examples in the literature that describe this phenomenon and its application with Rh NPs. Hence, the response of a colloid of MPA-Rh NPs was tested in the presence of a series of toxic metal ions in aqueous solutions buffered at pH 9. This pH is required for the dissociation of the proton from the carboxylic acid of MPA, providing the surface of the MPA-Rh NPs with a high density of negative charges, which is necessary for colloidal stability in the absence of metal ions and for the coordination of metal ions and consequent NP aggregation when the analyte is present in solution (Fig. 3A).62,63 For instance, after increasing the concentration of Cd(II) in the MPA-Rh NP solution, a clear modification of the optical extinction spectrum was observed, that is, the decrease of the absorbance in the 230–400 nm range (the buffer absorbs at wavelengths < 225 nm) and its increase beyond 400 nm (arrows in Fig. 3B). This behaviour is typical of plasmonic optical nanosensors due to the aggregation of NPs promoted by the analyte,2,50,62,63 as demonstrated by the TEM images of the MPA-Rh NPs before and after the addition of Cd(II) at 159 μM concentration (Fig. 3C). The responsivity of the optical metal-ion sensor based on MPA-Rh NPs was also tested with other toxic cations (Pb(II), Co(II), Hg(II) and Mn(II), Fig. 3D), and it was found to be higher for lead, intermediate for cadmium, cobalt and manganese and lower for mercury but always with a sensitivity in the range of a few units of μM and with a saturation beyond 200 μM under our experimental conditions (0.03 mg mL−1 Rh NP concentration). Note that 550 nm was identified as a convenient wavelength to track the response of the optical nanosensor, although other wavelengths can be selected because of the response of the Rh NPs in a wide spectral range from 230 nm to the NIR, which has no precedents for plasmonic particles. For instance, optical sensors based on the UV plasmon band of these Rh NPs are relevant in environments containing species that absorb in the visible range, for which Au and Ag NPs are not exploitable.
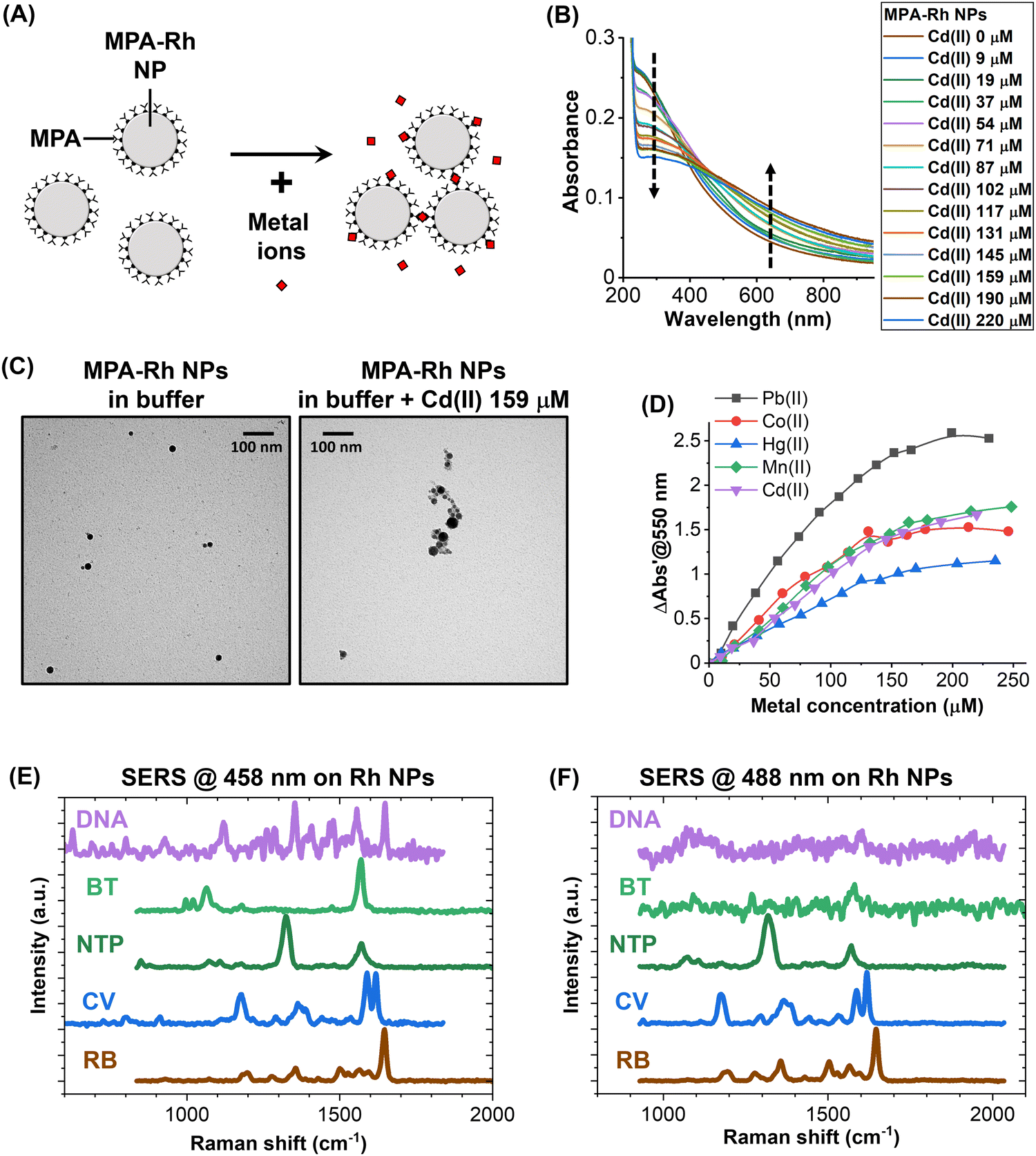 | ||
| Fig. 3 (A) Sketch of the MPA-Rh NP optical sensor based on the coordination of metal ions by the carboxylic groups of MPA. (B) Evolution of the optical spectra of MPA-Rh NPs while increasing the concentration of Cd(II). (C) TEM images of the MPA-Rh NPs before and after the addition of Cd(II). (D) Response of the optical sensor for different metal ions. (E) and (F) SERS spectra of bare Rh NPs excited at 458 nm (E) and 488 nm (F). | ||
UV plasmonic nanomaterials, including Rh nanostructures, have previously been studied as substrates for SERS at various wavelengths in the UV and visible ranges.33–35 In this case, uncoated Rh NPs were collected from the liquid after the LAL synthesis and tested for SERS with 458 and 488 nm laser excitation (Fig. 3E and F) in the presence of various analytes such as rhodamine B (RB), crystal violet (CV), nitro-thiophenol (NTP), benzenethiol (BT) and DNA. The Raman fingerprints of the five compounds, deposited at millimolar concentration on the Rh NPs (for details see Methods), were recorded in all cases with excitation at 458 nm (Fig. 3E). With 488 nm laser excitation, only the signal of RB, CV, NTP and a weak signal from BT were detected. At both excitation wavelengths, the signals were detected only after optimization of the exposure time and laser intensity to prevent the rapid degradation of the analytes. This effect has been observed systematically in the literature on Rh NPs excited with blue and UV light and has been attributed to the photochemical effects of photons in this spectral range combined with the high catalytic activity of rhodium.34,35 In our case, the degradation of the analytes is favoured because the surface of Rh NPs obtained by LAL is free of capping agents or other chemical compounds.
Discussion
In this study, rhodium nanospheres with sizes in the 20–45 nm range were obtained by LAL synthesis in water (Fig. 4A). Previous reports on the synthesis of Rh NPs with equivalent size by chemical routes only reported anisotropic morphologies, which in some cases were also affected by oxidation or partial reshaping under real conditions with the alteration of their optical and chemical properties.31,43 With chemical synthesis, faceting and formation of elongated shapes were already observed for sizes below 5–7 nm (Fig. 4B),44 because Rh is known for its surface energy higher than those of all the other plasmonic noble metals.29,44–46 In LAL with nanosecond pulses, a thin metal plume is detached from the bulk Rh target after absorption of beam energy, which expands towards the underlying solution while also transforming into liquid metal droplets through a process of spinodal decomposition (Fig. 4A).64 In this way, Rh hot vapours and liquid metal droplets are ejected into the liquid solution, where they will be cooled down to room temperature on the timescale of hundreds of milliseconds.51,52,64 Hence, the formation of LAL generated Rh NPs is orders of magnitude faster than in chemical reduction, allowing the preservation of the spherical morphology of the original droplets. Instead, the metal vapours can either nucleate and grow into smaller Rh NPs or react with the oxidising compounds generated by the decomposition of water molecules at the interface with the hot metal plume ejected from the target.25,32,58 The latter pathway leads to the formation of rhodium oxide, as verified in a previous study about LAL of bulk Rh in water.32 Here, Rh oxide was removed through the dialysis and the SBS of the colloids. Hence, from the perspective of the recovery of precious metals and for the global efforts towards the development of sustainable processes, Rh oxide can be collected without the need for solvent evaporation, contrary to what happens for the unreacted molecular Rh precursors that persist in chemical synthesis.29,44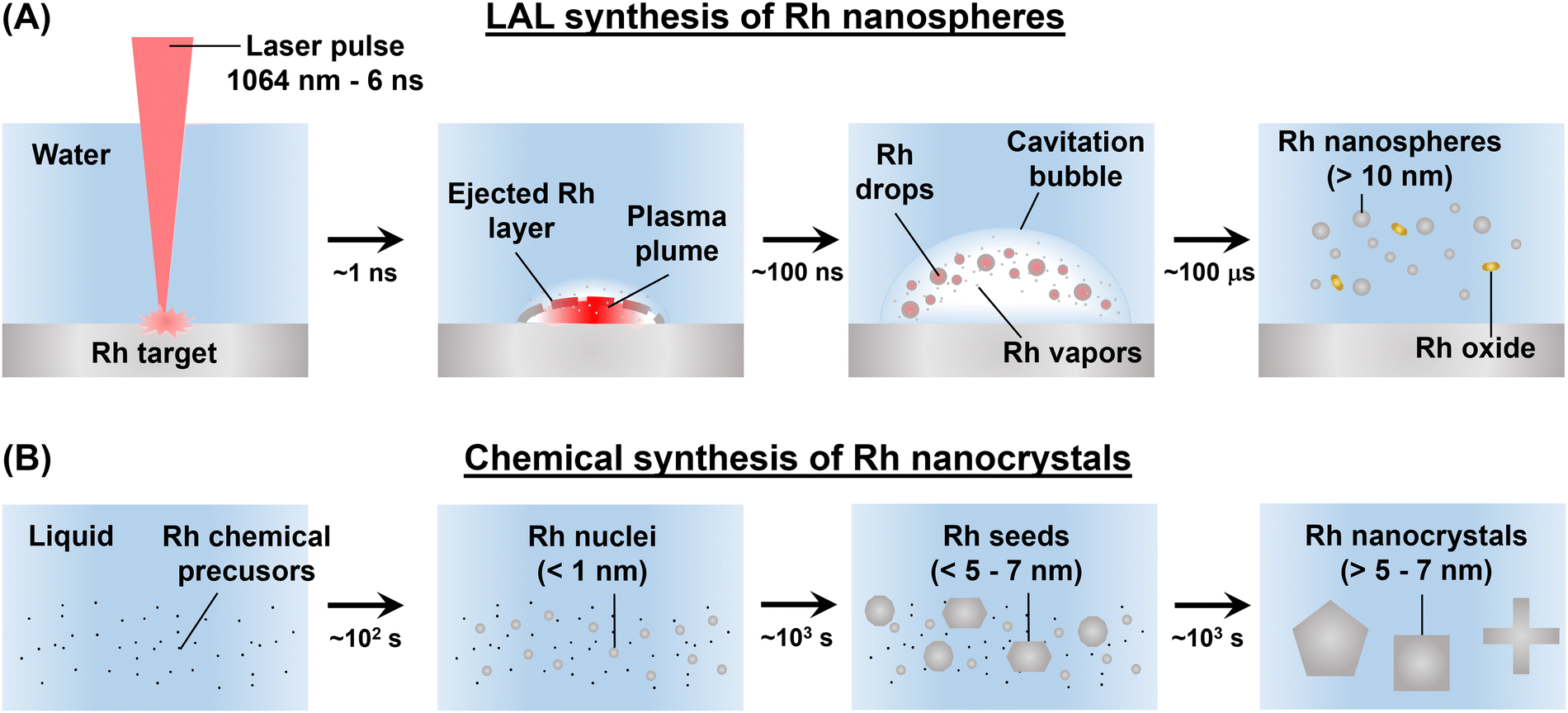 | ||
| Fig. 4 Sketch for the comparison of Rh NP synthesis by LAL (A) and chemical methods (B). (A) In LAL, a layer of the bulk metal target is ejected into the solution (pure water) where it undergoes a spinodal decomposition into liquid droplets and some Rh vapours. This material initially cools down inside a cavitation bubble formed in the liquid solution underlying the ablated metal target, before reaching the thermal equilibrium with the liquid phase in less than 100 ms. Rh vapours and clusters can form Rh oxide compounds, while the metal droplets form the final Rh nanospheres. (B) In the chemical synthesis, starting from a Rh precursor in the presence of various compounds (reducing agents, stabilizers, and templating agents), subnanometric nuclei form on a typical time scale of 1–100 s, and subsequently coalesce in seeds and grow into the final nanocrystals on a time scale of thousands of seconds. | ||
The extinction spectra of the six samples of Rh NPs were successfully fitted with the classical electrodynamics model, using the experimental dielectric function of Rh from the literature (see Methods). The model was also able to identify the average size of each sample, without any prior calibration or other external input. There are no other examples in the literature that indicate such a good agreement, meaning that the plasmonic properties of Rh nanospheres can be predicted and modelled in the same way as it is done for Ag and Au NPs. This offers the opportunity to use the Mie model for a direct comparison of the extinction, scattering and absorption cross-sections of 10 nm and 50 nm nanospheres of the three metals (Rh, Ag, and Au) in water (Fig. 5A and B). One can observe that the extinction cross-section of Rh NPs is comparable to that of Ag and Au NPs of the same size and shape, although the LSP of each metal is in different spectral regions. This was not obvious from the plasmonic quality factor (QLSP), which for a nanosphere in the low loss limit and in the quasistatic regime can be approximated as minus the ratio of the real (e′) to the imaginary (e′′) components of the dielectric function.3,12,13 The QLSP in the visible range is much higher for Ag and Au than that for Rh, but Rh prevails in the UV range, where interband transitions in Ag and Au are located (Fig. 5C). One of the requisites for appreciable plasmonic behaviours is the high density of conduction electrons and the high conductivity,27 which at the nanoscale are larger in Rh compared to Ag and Au,65,66 leading to a high real dielectric function in the UV-visible range. However, the Fermi level of Rh overlaps with the energy of d-levels, which are partly empty,67 leading to intraband and interband transitions and high imaginary component of the dielectric function in the visible range,16,67 with a negative impact on the plasmonic quality factor.12,68
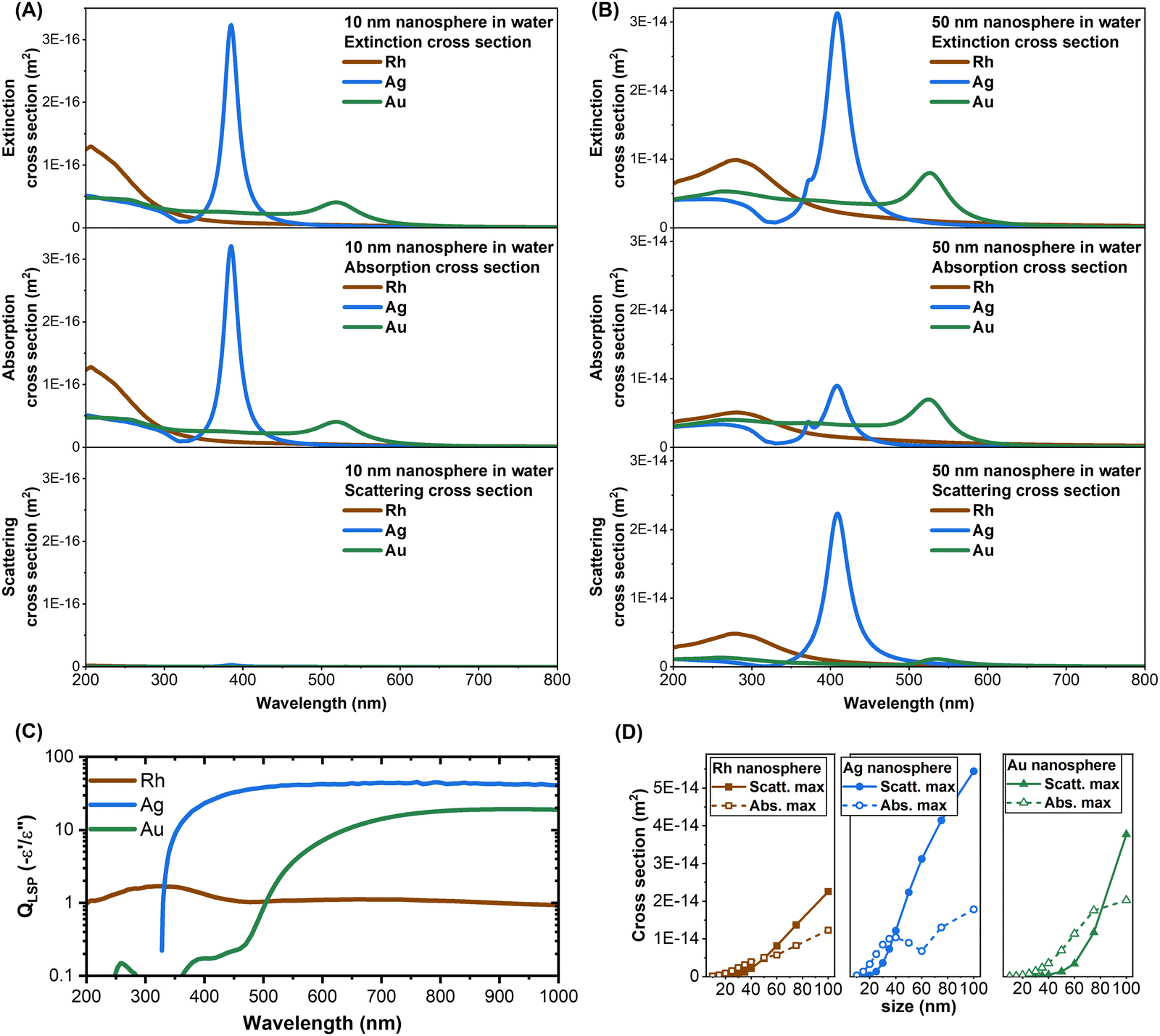 | ||
| Fig. 5 (A) and (B) Comparison of the extinction, absorption and scattering cross-sections of Rh, Ag and Au NPs in water with a diameter of 10 nm (A) and 50 nm (B). (C) Plasmonic quality factor for spherical NPs in the quasistatic regime obtained from the dielectric functions reported in the literature. (D) Plot of the scattering (full circles) and absorption (hollow circles) cross-section maxima in Rh, Ag and Au nanospheres in water versus the particle size. The cross-sections in (A), (B) and (D) are calculated using the Mie model. | ||
Another feature emerging from the comparison of the cross-sections in Fig. 5A and B is that, when going from the 10 nm to the 50 nm NPs, the LSP of the Rh nanosphere red shifts more (73 nm) than that for the Ag (24 nm) and Au (7 nm) equivalents, because of the retardation effects occurring at the shorter wavelength of UV light.69 The combination of retardation effects and the electronic properties of Rh results in the domination of the extrinsic size effect on the LSP bandwidth, while the intrinsic size effect is negligible for nanospheres bigger than 10 nm. This explains the excellent fitting with the MG model without any calibration of Kreibig's A parameter.2,61 Besides, the scattering and absorption contributions to the extinction cross-section of the 50 nm Rh NPs are comparable, as in Ag, while in Au the absorption prevails. This is best shown by the plot of the calculated maximum of the scattering and absorption cross-sections in nanospheres versus their size (Fig. 5D), showing a crossover at 50 nm for Rh, 38 nm for Ag and 80 nm for Au. Like Ag, this makes Rh interesting for near field and nanolensing phenomena even at small NP sizes.
Hence, specific information on the local field enhancement was obtained from numerical calculations with the discrete dipole approximation. The calculations considered the ideal case of a dimer of Rh nanospheres separated by a gap of 2 nm and excited at wavelengths from UV to NIR, which are typical of commercial spectrometers (Fig. 6A). The plots of Fig. 6A report the bidimensional map of the SERS enhancement factor (GSERS) when the light is polarized along the dimer axis, i.e. when the electromagnetic hot spots are generated in the gap between the NPs, considering nanospheres with radii of 20, 30 and 40 nm. GSERS at each point surrounding the NP can be evaluated with good approximation as the 4th power of the ratio between the local and the incident electric field.1,2,70 As expected, the maximum GSERS is achieved in the gap of the dimer, even for the shortest wavelengths and the 40 nm NPs, where retardation effects are more important.1,2 The maximum GSERS at 350 nm and 458 nm are on the order of 107 and 106 for the 30 and 40 nm NPs, which are values comparable to those predicted at longer wavelengths (532 and 633 nm) in Ag and Au NPs with equivalent morphology.49,71,72 This is in agreement with previous numerical analysis based on the rhodium dielectric function.3,13,16,73 The maximum GSERS in the dimers is reported versus the excitation wavelength in Fig. 6B, showing that the best performance is expected for 350 nm excitation, while the enhancement is much lower at 258 nm and 633–758 nm. This trend is qualitatively in agreement with the experimental evidence of better SERS signals with excitation at 458 nm than at 488 nm (Fig. 3E and F). As for the reference plasmonic materials (Ag and Au)71 and also for Rh, there is a clear dependence of the local field enhancement on the size of the NPs, with 40 nm NPs performing better than the 30 and 20 nm ones, despite the spectral trend of the GSERS remaining the same for the three sizes. Due to retardation effects, which red-shift the dipolar LSP in larger nanospheres,1,2 the GSERS at 258 nm for 20 and 30 nm NPs are close to that for 40 nm NPs. At longer wavelengths, the calculations indicate that the presence of NPs with sizes below 30 nm in a polydisperse sample of Rh NPs is not beneficial for all those phenomena relying on the enhancement of the local electric field. However, small NPs are suitable for thermoplasmonic phenomena and, in the case of a catalytic metal such as Rh, for photodegradation of organic compounds.2,3,8 Regarding the possibility of improving the size control of Rh NPs generated by LAL, different methods have been proposed in the literature,51,52,74 which can be summarised as follows: addition of stabilizing ligands during LAL or laser fragmentation of LAL-generated Rh NPs allows reducing the average size and relative standard deviation down to the range of a few nanometers; laser melting in liquid of LAL-generated Rh NPs allows increasing the average size and relative standard deviation up to the submicron range. In addition, the combination of the SBS protocol with different LAL conditions in terms of pulse wavelength, duration, repetition rate and liquid environment has also been reported as a suitable approach to access laser-generated NPs in a variety of size ranges as required for each specific application.51–53,60,71,74
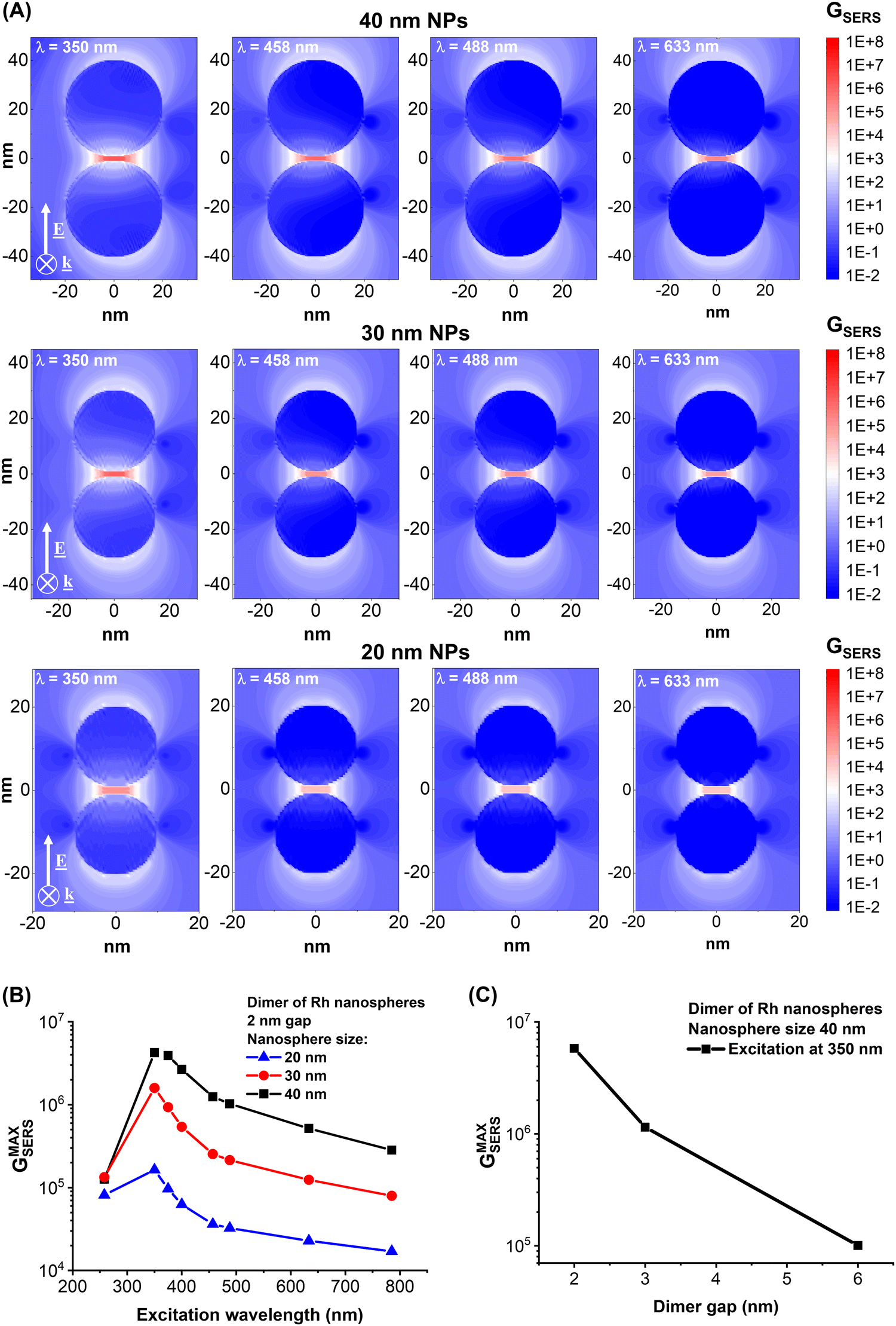 | ||
| Fig. 6 (A) Bidimensional maps of the GSERS in the equatorial plane of Rh NP dimers with a gap of 2 nm and sizes of 20, 30 and 40 nm excited at 350, 458, 488 and 633 nm in a medium with refractive index n = 1.334. (B) Plot of the GSERS maximum versus the excitation wavelength for the three dimer sizes considered. (C) The plot of the GSERS maximum versus the gap size for an excitation wavelength of 350 nm and dimers with a size of 40 nm. | ||
The increase of the gap of the dimer also leads to the typical exponential decay of GSERS (Fig. 6C). Considering this result and the rapid photodegradation of analytes reported in the literature,34,35 and confirmed in this study, it can be concluded that maximum local field performances can be achieved by coating the Rh NPs with a subnanometric shell of an inert material to benefit from local field effects while minimizing analyte decomposition, which remains an open challenge for Rh nanostructures.
Conclusions
In summary, this study contributed to expanding the toolbox of plasmonic materials with Rh nanospheres in a size range of 20–45 nm, where the optical properties are very appealing for applications under UV and visible light conditions. The spherical morphology was achieved through a laser-assisted synthesis, which avoids the spontaneous formation of anisotropic shapes typical of chemically synthesized Rh nanocrystals. The surface of the Rh NPs is available for conjugation with thiolated molecules, which is a key advantage of the reference plasmonic materials based on Ag and Au. The optical properties of the experimental samples of Rh NPs are modelled using classical electrodynamics. The calculations also indicated that the plasmonic behaviour of Rh nanospheres is comparable to that of Ag and Au NPs in terms of extinction cross-section and local field enhancement, although shifted to the near-UV spectral region, which is inaccessible to silver and gold nanostructures. As a proof of concept, SERS spectra were recorded for various analytes at 458 and 488 nm, and it was demonstrated that the functionalized Rh NPs can be used as an optical nanosensor for detection of toxic ions in aqueous solution. It is also shown that the chemical and physical stability at high temperatures and in the presence of strong acids (aqua regia) is superior to those of Au and Ag NPs. Besides, Rh nanospheres can be used in harsher chemical environments and at temperatures higher than those of Au and Ag equivalents. Overall, the plasmonic, chemical and physical attributes of these 20–45 nm Rh nanospheres highlight their importance in the realms of UV and visible plasmonics, while also envisaging a multitude of opportunities for other underexplored applications such as plasmon-enhanced catalysis and chiroplasmonics.Methods
Scanning electron microscopy (SEM) was performed with a Zeiss Sigma HD microscope, equipped with a Schottky FEG source, one detector for backscattered electrons and two detectors for secondary electrons (InLens and Everhart Thornley).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 vol/vol with a 10 mg mL−1 aqueous solution of poly(vinyl alcohol) (18 kDa, Sigma) and deposited on the TEM grid, to prevent clustering of the NPs while drying the liquid, as described in ref. 50. For the analysis of the spectra versus metal concentration, the difference in absorbance at 550 nm between the solution with the metal ions and the initial MPA-Rh NP solution (ΔAbs′@550 nm) was calculated after normalization at 255 nm of the experimental absorption spectra.
:10 vol/vol with a 10 mg mL−1 aqueous solution of poly(vinyl alcohol) (18 kDa, Sigma) and deposited on the TEM grid, to prevent clustering of the NPs while drying the liquid, as described in ref. 50. For the analysis of the spectra versus metal concentration, the difference in absorbance at 550 nm between the solution with the metal ions and the initial MPA-Rh NP solution (ΔAbs′@550 nm) was calculated after normalization at 255 nm of the experimental absorption spectra.
Uncoated Rh NPs were obtained by centrifugation of the LAL colloid (50 rcf at 1 h to discard the first sediment followed by 1000 rcf for 1 h twice to collect the sediment and wash it with water) and resuspension in distilled water at 0.1 mg mL−1 Rh concentration. For each analyte, 50 mL of the Rh NP solution was drop-cast on a microscope slide and dried at room temperature for 1 h, then 10 mL of the analyte solution was drop-cast on the deposit of Rh NPs and dried at room temperature for 30 minutes before analysis. The following analytes were used: rhodamine B chloride (RB, 6 mM in ethanol–water 1:1, Sigma), crystal violet chloride (CV, 6 mM in ethanol–water 1:1, Sigma), nitrothiophenol (NTP, 100 mM in ethanol–water 1:1, Sigma) benzenethiol (BT, 100 mM in ethanol–water 1:1, Sigma), and deoxyribonucleic acid sodium salt from herring testes (DNA, 500 mM base pairs in water, Sigma).
The GSERS bidimensional maps were calculated by the discrete dipole approximation (DDA) method, using the DDSCAT code and the related DDFIELD package,80 considering the equatorial plane of the dimers of Rh NPs, for an incident electric field linearly polarized along the dimer axis. The calculations were performed considering light excitation at 258, 350, 458, 488, 633, and 785 nm and that Rh NPs are embedded in a homogeneous medium of relative permittivity 1.77 to reproduce the influence of the substrate. The target consisted of a number of dipoles of 0.95 × 105 (20 nm NPs), 3.1 × 105 (30 nm NPs) and 7.4 × 105 (40 nm NPs), to ensure that the interdipole spacing was small compared to the NP size and incident wavelengths.2,80
Data availability
The data supporting the findings of this study are available from the corresponding author upon request. Source data are provided as ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Carlo Bravin and Matteo Moretto are acknowledged for helping with the optical sensor experiment. V. A. acknowledges the University of Padova P-DiSC project “DYNAMO”.References
- S. A. Maier, Plasmonics: fundamentals and applications, Springer, 2007 Search PubMed.
- V. Amendola, R. Pilot, M. Frasconi, O. M. Maragò and M. A. Iatì, J. Phys.: Condens. Matter, 2017, 29, 203002 CrossRef.
- Y. Gutiérrez, M. Losurdo, F. González, H. O. Everitt and F. Moreno, J. Phys. Chem. C, 2020, 124, 7386–7395 CrossRef.
- K. I. Saitow, J. Phys. Chem. C, 2024, 128, 5367–5393 CrossRef CAS.
- K. Kant, R. Beeram, Y. Cao, P. S. S. dos Santos, L. Gonzá lez-Cabaleiro, D. Garcí-Lojo, H. Guo, Y. Joung, S. Kothadiya, M. Lafuente, Y. Xiang Leong, Y. Liu, Y. Liu, S. Satya Bharati Moram, S. Mahasivam, S. Maniappan, D. Quesada-González, D. Raj, P. Weerathunge, X. Xia, Q. Yu, S. Abalde-Cela, R. A. Alvarez-Puebla, R. Bardhan, V. Bansal, J. Choo, L. C. C. Coelho, M. José, M. M. de Almeida, S. Gó mez-Grañ, M. Grzelczak, P. Herves, J. Kumar, T. Lohmueller, A. Merkoçi, J. Luis Montaño-Priede, X. Yi Ling, R. Mallada, J. Pérez-Juste, M. P. Pina, S. Singamaneni, V. Rao Soma, M. Sun, L. Tian, J. Wang, L. Polavarapu and I. Pastoriza Santos, Nanoscale Horiz., 10.1039/D4NH00226A.
- X. Duan and N. Liu, Acc. Chem. Res., 2019, 52, 1979–1989 CrossRef CAS PubMed.
- N. Zhou, X. Xu, A. T. Hammack, B. C. Stipe, K. Gao, W. Scholz and E. C. Gage, Nanophotonics, 2014, 3, 141–155 CAS.
- G. Baffou, F. Cichos and R. Quidant, Nat. Mater., 2020, 19, 946–958 CrossRef CAS PubMed.
- Z. Geng, Y. Yu and J. Liu, J. Phys. Chem. C, 2023, 127, 17723–17731 CrossRef CAS.
- N. S. Mueller, Y. Okamura, B. G. M. Vieira, S. Juergensen, H. Lange, E. B. Barros, F. Schulz and S. Reich, Nature, 2020, 583, 780–784 CrossRef CAS.
- T. R. Leite, L. Zschiedrich, O. Kizilkaya and K. M. McPeak, Nano Lett., 2022, 22, 7343–7350 CrossRef CAS PubMed.
- M. G. Blaber, M. D. Arnold and M. J. Ford, J. Phys.: Condens. Matter, 2010, 22, 143201 CrossRef CAS PubMed.
- B. Doiron, M. Mota, M. P. Wells, R. Bower, A. Mihai, Y. Li, L. F. Cohen, N. M. N. Alford, P. K. Petrov, R. F. Oulton and S. A. Maier, ACS Photonics, 2019, 6, 240–259 CrossRef CAS.
- J. M. Sanz, D. Ortiz, R. Alcaraz De La Osa, J. M. Saiz, F. González, A. S. Brown, M. Losurdo, H. O. Everitt and F. Moreno, J. Phys. Chem. C, 2013, 117, 19606–19615 CrossRef CAS.
- J. M. McMahon, G. C. Schatz and S. K. Gray, Phys. Chem. Chem. Phys., 2013, 15, 5415–5423 RSC.
- Y. Gutiérrez, R. A. de la Osa, D. Ortiz, J. M. Saiz, F. González and F. Moreno, Appl. Sci., 2018, 8, 64 CrossRef.
- M. B. Ross and G. C. Schatz, J. Phys. Chem. C, 2014, 118, 12506–12514 CrossRef CAS.
- D. F. Swearer, H. Robatjazi, J. M. P. Martirez, M. Zhang, L. Zhou, E. A. Carter, P. Nordlander and N. J. Halas, ACS Nano, 2019, 13, 8076–8086 CrossRef CAS.
- C. Wang, W. C. D. Yang, D. Raciti, A. Bruma, R. Marx, A. Agrawal and R. Sharma, Nat. Mater., 2021, 20, 346–352 CrossRef CAS.
- M. Yu, C. Yang, X. M. Li, T. Y. Lei, H. X. Sun, L. P. Dai, Y. Gu, X. Ning, T. Zhou, C. Wang, H. B. Zeng and J. Xiong, Nanoscale, 2017, 9, 8716–8722 RSC.
- J. Zheng, W. Yang, J. Wang, J. Zhu, L. Qian and Z. Yang, Nanoscale, 2019, 11, 4061–4066 RSC.
- A. G. Bezerra, P. Cavassin, T. N. Machado, T. D. Woiski, R. Caetano and W. H. Schreiner, J. Nanopart. Res., 2017, 19, 1–7 CrossRef CAS.
- V. Lomonosov, J. Yang, Y. Fan, S. Hofmann and E. Ringe, Nano Lett., 2024, 24, 7084–7090 CrossRef CAS.
- S. De Marchi, S. Núñez-Sánchez, G. Bodelón, J. Pérez-Juste and I. Pastoriza-Santos, Nanoscale, 2020, 12, 23424–23443 RSC.
- A. Barulin, J. B. Claude, S. Patra, A. Moreau, J. Lumeau and J. Wenger, J. Phys. Chem. Lett., 2019, 10, 5700–5707 CrossRef CAS PubMed.
- J. Asselin, E. R. Hopper and E. Ringe, Nanoscale, 2021, 13, 20649–20656 RSC.
- G. Albrecht, M. Ubl, S. Kaiser, H. Giessen and M. Hentschel, ACS Photonics, 2018, 5, 1058–1067 CrossRef CAS.
- P. C. Wu, T.-H. Kim, A. S. Brown, M. Losurdo, G. Bruno and H. O. Everitt, Appl. Phys. Lett., 2007, 90, 103119 CrossRef.
- S. R. Lee, M. Vara, Z. D. Hood, M. Zhao, K. D. Gilroy, M. Chi and Y. Xia, ChemNanoMat, 2018, 4, 66–70 CrossRef CAS.
- L. Luo, H. Li, Y. Peng, C. Feng and J. Zeng, ChemNanoMat, 2018, 4, 451–466 CrossRef CAS.
- X. Zhang, X. Li, D. Zhang, N. Q. Su, W. Yang, H. O. Everitt and J. Liu, Nat. Commun., 2017, 8, 1–9 CrossRef.
- G. A. Volpato, D. Muneton Arboleda, R. Brandiele, F. Carraro, G. B. Sartori, A. Cardelli, D. Badocco, P. Pastore, S. Agnoli, C. Durante, V. Amendola and A. Sartorel, Nanoscale Adv., 2019, 1, 4296–4300 RSC.
- S. Kundu, Y. Chen, W. Dai, L. Ma, A. M. Sinyukov and H. Liang, J. Mater. Chem. C, 2017, 5, 2577–2590 RSC.
- G. Kumar and R. K. Soni, J. Raman Spectrosc., 2022, 53, 1890–1903 CrossRef CAS.
- X. Zhang, P. Li, Á. Barreda, Y. Gutiérrez, F. González, F. Moreno, H. O. Everitt and J. Liu, Nanoscale Horiz., 2016, 1, 75–80 RSC.
- J. Piaskowski, G. Haberfehlner, T. Bartschmid, G. Kothleitner, M. Steinhart and G. R. Bourret, J. Mater. Chem. C, 2023, 11, 10872–10883 RSC.
- P. Roy, S. Zhu, J. B. Claude, J. Liu and J. Wenger, ACS Nano, 2023, 17, 22418–22429 CrossRef CAS PubMed.
- X. Hu, B. Chen, C. Huang, H. Qiu, N. Gao, Y. Wu, D. Cai, K. Huang, J. Kang and R. Zhang, Nanoscale, 2023, 15, 9684–9690 RSC.
- C. S. Kuo, C. R. Kao, W. J. Chen, M. Y. Lu, D. A. Cullen, B. T. Sneed, Y. C. Chuang, C. C. Yu and C. H. Kuo, Chem. Mater., 2018, 30, 4448–4458 CrossRef CAS.
- K. A. Kane and M. F. Bertino, Beilstein J. Nanotechnol., 2019, 10, 1958–1963 CrossRef CAS PubMed.
- M. Vanzan, R. M. Jones, S. Corni, R. D’Agosta and F. Baletto, ChemPhysChem, 2022, 23, e202200035 CrossRef CAS PubMed.
- D. Raabe, Chem. Rev., 2023, 123, 2436–2608 CrossRef CAS PubMed.
- T. Wakita and H. Yao, Chem. Phys. Lett., 2021, 779, 138866 CrossRef CAS.
- A. J. Biacchi and R. E. Schaak, ACS Nano, 2015, 9, 1707–1720 CrossRef CAS PubMed.
- L. Xu, D. Liu, D. Chen, H. Liu and J. Yang, Heliyon, 2019, 5, e01165 CrossRef PubMed.
- B. Jiang, C. Li, Ö. Dag, H. Abe, T. Takei, T. Imai, M. S. A. Hossain, M. T. Islam, K. Wood, J. Henzie and Y. Yamauchi, Nat. Commun., 2017, 8, 3–4 CrossRef PubMed.
- E. Demishkevich, A. Zyubin, A. Seteikin, I. Samusev, I. Park, C. K. Hwangbo, E. H. Choi and G. J. Lee, Materials, 2023, 16, 3342 CrossRef CAS PubMed.
- T. M. R. Wayman, V. Lomonosov and E. Ringe, J. Phys. Chem. C, 2024, 128, 4666–4676 CrossRef CAS.
- A. Guadagnini, S. Agnoli, D. Badocco, P. Pastore, D. Coral, M. B. Fernàndez van Raap, D. Forrer and V. Amendola, J. Colloid Interface Sci., 2021, 585, 267–275 CrossRef CAS.
- C. Bravin and V. Amendola, ACS Appl. Nano Mater., 2022, 5, 578–586 CrossRef CAS.
- V. Amendola, D. Amans, Y. Ishikawa, N. Koshizaki, S. Scirè, G. Compagnini, S. Reichenberger and S. Barcikowski, Chem. – Eur. J., 2020, 26, 9206–9242 CrossRef CAS PubMed.
- V. Amendola and M. Meneghetti, Phys. Chem. Chem. Phys., 2013, 15, 3027–3046 RSC.
- F. Bonaccorso, M. Zerbetto, A. C. C. Ferrari and V. Amendola, J. Phys. Chem. C, 2013, 117, 13217–13229 CrossRef CAS.
- S. Crivellaro, A. Guadagnini, D. M. D. M. Arboleda, D. Schinca and V. Amendola, Rev. Sci. Instrum., 2019, 90, 033902 CrossRef.
- S. Jendrzej, B. Gökce, M. Epple and S. Barcikowski, ChemPhysChem, 2017, 18, 1012–1019 CrossRef CAS PubMed.
- I. Y. Khairani, Gladys Mínguez-Vega, Carlos Doñate-Buendía and Bilal Gökce, Phys. Chem. Chem. Phys., 2023, 25, 19380–19408 RSC.
- M. Castilla, S. Schuermans, D. Gérard, J. Martin, T. Maurer, U. Hananel, G. Markovich, J. Plain and J. Proust, ACS Photonics, 2022, 9, 880–887 CrossRef CAS.
- E. Fazio, S. G. Leonardi, M. Santoro, N. Donato, G. Neri and F. Neri, Sens. Actuators, B, 2018, 262, 79–85 CrossRef CAS.
- T. Burks, M. Avila, F. Akhtar, M. Göthelid, P. C. Lansåker, M. S. Toprak, M. Muhammed and A. Uheida, J. Colloid Interface Sci., 2014, 425, 36–43 CrossRef CAS PubMed.
- V. Coviello, D. Badocco, P. Pastore, M. Fracchia, P. Ghigna, A. Martucci, D. Forrer and V. Amendola, Nat. Commun., 2024, 15, 1–13 Search PubMed.
- V. Amendola and M. Meneghetti, J. Phys. Chem. C, 2009, 113, 4277–4285 CrossRef CAS.
- C. C. Huang and H. T. Chang, Chem. Commun., 2007, 1215–1217 RSC.
- E. Priyadarshini and N. Pradhan, Sci. Rep., 2017, 7, 1–8 CrossRef CAS.
- C.-Y. Shih, M. V. Shugaev, C. Wu and L. V. Zhigilei, Phys. Chem. Chem. Phys., 2020, 22, 7077–7099 RSC.
- D. Gall, J. Appl. Phys., 2016, 119, 85101 CrossRef.
- A. Jog, T. Zhou and D. Gall, IEEE Trans. Electron Devices, 2021, 68, 257–263 CAS.
- G. S. Tripathi, N. E. Brener and J. Callaway, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 38, 10454 CrossRef CAS PubMed.
- D. T. L. Alexander, D. Forrer, E. Rossi, E. Lidorikis, S. Agnoli, G. D. Bernasconi, J. Butet, O. J. F. Martin and V. Amendola, Nano Lett., 2019, 19, 5754–5761 CrossRef CAS.
- H. Yu, P. Zhang, S. Lu, S. Yang, F. Peng, W. S. Chang and K. Liu, J. Phys. Chem. Lett., 2020, 11, 5836–5843 CrossRef CAS.
- E. C. Le Ru and P. G. Etchegoin, Chem. Phys. Lett., 2006, 423, 63–66 CrossRef CAS.
- V. Amendola and M. Meneghetti, Adv. Funct. Mater., 2012, 22, 353–360 CrossRef CAS.
- S. Scaramuzza, S. Polizzi and V. Amendola, Nanoscale Adv., 2019, 1, 2681–2689 RSC.
- A. Ahmadivand, R. Sinha, S. Kaya and N. Pala, Plasmonics, 2016, 11, 839–849 CrossRef CAS.
- D. Zhang, B. Gökce and S. Barcikowski, Chem. Rev., 2017, 117, 3990–4103 CrossRef CAS PubMed.
- E. D. Palik, Handbook of Optical Constants of Solids, Academic Press, 1985 Search PubMed.
- R. L. Olmon, B. Slovick, T. W. Johnson, D. Shelton, S.-H. Oh, G. D. Boreman and M. B. Raschke, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 235147 CrossRef.
- P. B. Johnson and R. W. Christy, Phys. Rev. B: Solid State, 1972, 6, 4370–4379 CrossRef CAS.
- G. M. Hale and M. R. Querry, Appl. Opt., 1973, 12, 555 CrossRef CAS PubMed.
- D. M. Arboleda, J. M. J. Santillán, L. J. Mendoza Herrera, D. Muraca, D. C. Schinca and L. B. Scaffardi, J. Phys. D: Appl. Phys., 2016, 49, 075302 CrossRef.
- B. T. Draine and P. J. Flatau, arXiv, 2013, 1305.6497 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nh00449c |
| This journal is © The Royal Society of Chemistry 2025 |