
Edge-doped substituents as an emerging atomic-level strategy for enhancing M–N4–C single-atom catalysts in electrocatalysis of the ORR, OER, and HER†
Liang
Xie
,
Wei
Zhou
 *,
Zhibin
Qu
,
Yuming
Huang
,
Longhao
Li
,
Chaowei
Yang
,
Junfeng
Li
,
Xiaoxiao
Meng
,
Fei
Sun
,
Jihui
Gao
and
Guangbo
Zhao
*,
Zhibin
Qu
,
Yuming
Huang
,
Longhao
Li
,
Chaowei
Yang
,
Junfeng
Li
,
Xiaoxiao
Meng
,
Fei
Sun
,
Jihui
Gao
and
Guangbo
Zhao
School of Energy Science and Engineering, Harbin Institute of Technology, Harbin, Heilongjiang 150001, P. R. China. E-mail: hitzhouw@hit.edu.cn
First published on 6th November 2024
Abstract
M–N4–C single-atom catalysts (MN4) have gained attention for their efficient use at the atomic level and adjustable properties in electrocatalytic reactions like the ORR, OER, and HER. Yet, understanding MN4's activity origin and enhancing its performance remains challenging. Edge-doped substituents profoundly affect MN4's activity, explored in this study by investigating their interaction with MN4 metal centers in ORR/OER/HER catalysis (Sub@MN4, Sub = B, N, O, S, CH3, NO2, NH2, OCH3, SO4; M = Fe, Co, Ni, Cu). The results show overpotential variations (0 V to 1.82 V) based on Sub and metal centers. S and SO4 groups optimize FeN4 for peak ORR activity (overpotential at 0.48 V) and reduce OER overpotentials for NiN4 (0.48 V and 0.44 V). N significantly reduces FeN4's HER overpotential (0.09 V). Correlation analysis highlights the metal center's key role, with ΔG*H and ΔG*OOH showing mutual predictability (R2 = 0.92). Eg proves a reliable predictor for Sub@CoN4 (ΔG*OOH/ΔG*H, R2 = 0.96 and 0.72). Machine learning with the KNN model aids catalyst performance prediction (R2 = 0.955 and 0.943 for ΔG*OOH/ΔG*H), emphasizing M–O/M–H and the d band center as crucial factors. This study elucidates edge-doped substituents' pivotal role in MN4 activity modulation, offering insights for electrocatalyst design and optimization.
New conceptsOur study introduces a novel concept in the realm of M–N4–C single-atom catalysts by systematically examining the influence of edge-doped substituents on the electrocatalytic performance of MN4 in the ORR, OER, and HER. The innovation here lies in our comprehensive exploration of the interactions between various substituents (B, N, O, S, CH3, NO2, NH2, OCH3, and SO4) and diverse metal centers (Fe, Co, Ni, and Cu). We demonstrate that strategic edge modification can drastically enhance catalytic activity, revealing unprecedented tunability in MN4 catalysts. This represents a significant deviation from the traditional focus on metal centers alone, highlighting the critical role of substituents. Our work provides new insights into nanoscience and nanotechnology by correlating the substituent electronic properties, metal center characteristics, and catalytic activity. We identify key determinants such as M–O/M–H bond characteristics and the d band center for efficiency. Furthermore, our application of machine learning algorithms, particularly the KNN model, to predict catalyst performance based on these factors marks a conceptual leap in catalyst design, surpassing conventional trial-and-error methods. This study not only underscores the importance of edge-doped substituents but also establishes a predictive framework for designing high-efficiency electrocatalysts, advancing the field towards more sustainable energy technologies. |
1. Introduction
The growing demand for clean and cost-effective energy conversion and storage has led to a search for efficient devices such as fuel cells, metal–air batteries, and water electrolysis systems.1–4 Key reactions in these processes include oxygen reduction (ORR), oxygen evolution (OER), and hydrogen evolution (HER). To enable widespread application of these technologies, it is crucial to develop low-cost, high-performance catalysts5,6 that can significantly lower the kinetic barriers of these reactions. However, many current catalysts still rely heavily on precious metals. Among various catalysts explored so far, such as precious metal alloys (e.g., Pt–Hg),7–10 inexpensive metals (iron, cobalt, nickel, etc.),11,12 and heteroatom-doped carbon materials,13–15 carbon-based catalysts with isolated transition metals fixed by nitrogen groups (MN4) have garnered increasing attention for their atomic-level utilization efficiency and adjustable properties.16–19Understanding the origin of true activity in MN4 catalysts and fine-tuning their performance present challenges for the practical application of MN4 catalysts.20,21 It is now recognized that the active sites in MN4 materials extend beyond the M–N4 structure (metal center and first coordination shell).22 Factors such as axial coordination,23–25 the second coordination shell,26,27 and edge/defect sites28,29 can also influence catalytic activity to varying degrees. Notably, during the synthesis of MN4 materials, heteroatoms from the carbon precursor exist not only in coordination with metal ions but also at the edges. These edge heteroatoms significantly impact the electronic structure of MN4 catalysts and, thus, their activity.30–33 For instance, S located at the edge of FeN4 greatly enhances the ORR activity. Mun et al. demonstrated that using SO2 to extract electrons from the Fe center could optimize intermediates' adsorption capacity, thus improving FeN4 performance.34 Subsequent research on S-doped FeN4 materials showed high ORR activity and increased stability.30 Moreover, the presence of O at the edge of CoN4 leads to the electrocatalytic synthesis of H2O2 instead of H2O. Oxygen-rich CoN4 catalysts showed enhanced reaction activity from the metal Co center and improved selectivity for H2O2 formation from oxygen.35 Recently, edge-enriched CoN4 materials with oxygen at the edge were found to enhance H2O2 synthesis.33 Additionally, N or O coordination at the edge of FeN4 contributes to improved ORR performance.31,36 Understanding the role of edge heteroatoms in MN4 catalysts and conducting extensive research with various metal centers, substituents, and catalyzed reactions (e.g., the ORR, OER and HER) are essential for practical applications of MN4 catalysts in different scenarios.
Most understanding of the impact of edge-doped substituents on MN4 centers currently relies on experimental “point-to-point” studies. However, achieving directed synthesis through theoretical guidance can lead to the cost-effective discovery of efficient catalysts.37,38 One widely accepted design criterion is the electron-withdrawing/donating strategy proposed by Mun et al. to regulate FeN4 catalysts.34 They use charge transfer numbers as descriptors to qualitatively assess the performance of Sub@FeN4, which has been widely adopted. Nevertheless, this approach only provides qualitative insights into Sub's influence on FeN4 performance, making it challenging to quantitatively compare Subs with similar properties. Additionally, the applicability of this strategy to other transition metals (e.g., Co, Ni, Cu) requires further confirmation. Recently, predictive approaches based on machine learning (ML) algorithms have gained attention. By training ML models with multiple descriptors, including charge, it becomes possible to predict the catalytic activity of catalysts, offering the potential for high-performance, cost-effective screening of Sub@MN4 systems.39–41
In this study, we extensively investigated the Sub@MN4 system (Sub = B, N, O, S, CH3, NO2, NH2, OCH3, SO4; M = Fe, Co, Ni, Cu) using density functional theory (DFT). The findings demonstrate that changing the edge-doped substituents and the type of metal centers significantly impact the ORR/OER/HER activity. Correlation analysis of substituent/metal electronegativity, M–N/M–O/M–H bond length, metal d-band center/d-orbital electron numbers, Fundamental Gap (Eg), and *OOH/*H adsorption free energies (ΔG*OOH/ΔG*H) reveals the dominant role of the metal center, with a strong mutual predictability between ΔG*H and ΔG*OOH (R2 = 0.92). For Sub@CoN4, Eg can predict ΔG*OOH/ΔG*H effectively (with R2 values of 0.96 and 0.72, respectively). Machine learning (ML) analysis shows that the KNN model effectively predicts catalyst performance (with R2 values of 0.955 and 0.943 for ΔG*OOH/ΔG*H), where M–O/M–H and d band center descriptors are primary factors characterizing ΔG*OOH/ΔG*H. Overall, this work provides insight into the complex regulatory mechanisms of edge-doped substituents on MN4 activity, offering guidance for understanding the true origin of MN4 catalyst activity and optimizing and designing efficient MN4 electrocatalysts.
2. Model construction and calculation method
All calculations were conducted using density functional theory (DFT) in the Gaussian 09 code.42 Geometry relaxation and frequency analysis were performed at the wb97xd/def2svp level.43,44 For the influence of the aqueous solution, the SMD implicit solvent model was applied.45 The formula for calculating the free energy (G) of the system is: G = Gvac + ΔGsol, where Gvac is the free energy under vacuum condition, and ΔGsol represents the influence of solvent environment on the free energy. Although the effect of hydrogen bonds on water molecules in explicit solvation has been found, here we focus more on the properties of Sub@MN4 systems in implicit solvation, which is sufficient for description. In addition, the focus of this work is on the influence of Sub on the electronic structure of MN4 and its adsorption performance. As for the solvation effect caused by the presence of Sub, it is more complicated and is not within the focus of this work.Fig. 1 depicts the schematic diagrams of edge-doped MN4 structures constructed in this study, surrounded by a graphene carbon framework containing 36 carbon atoms with edges saturated by hydrogen atoms.19,46,47 In the study we are preparing for submission (Fig. S39, ESI†), we found that the size of the carbon material affects the properties of the single atoms, but the size selection for different MN4 catalysts remains a mystery. Therefore, we comprehensively consider the computing power and the main concern of this work (the influence of edge substituents), and choose 36C@MN4 as the model for this paper, which is used in many studies.19,46,47 The model maintains 1500 fs through AIMD at 298.15 K, and still has good structure-preserving properties (Fig. S1, ESI†). The investigated metal centers include transition metals Fe, Co, Ni, and Cu, while the edge-doped substituents comprise B, N, O, S, CH3, NO2, NH2, OCH3, and SO4. Detailed optimized structures are presented in Fig. S2–S6 (ESI†).
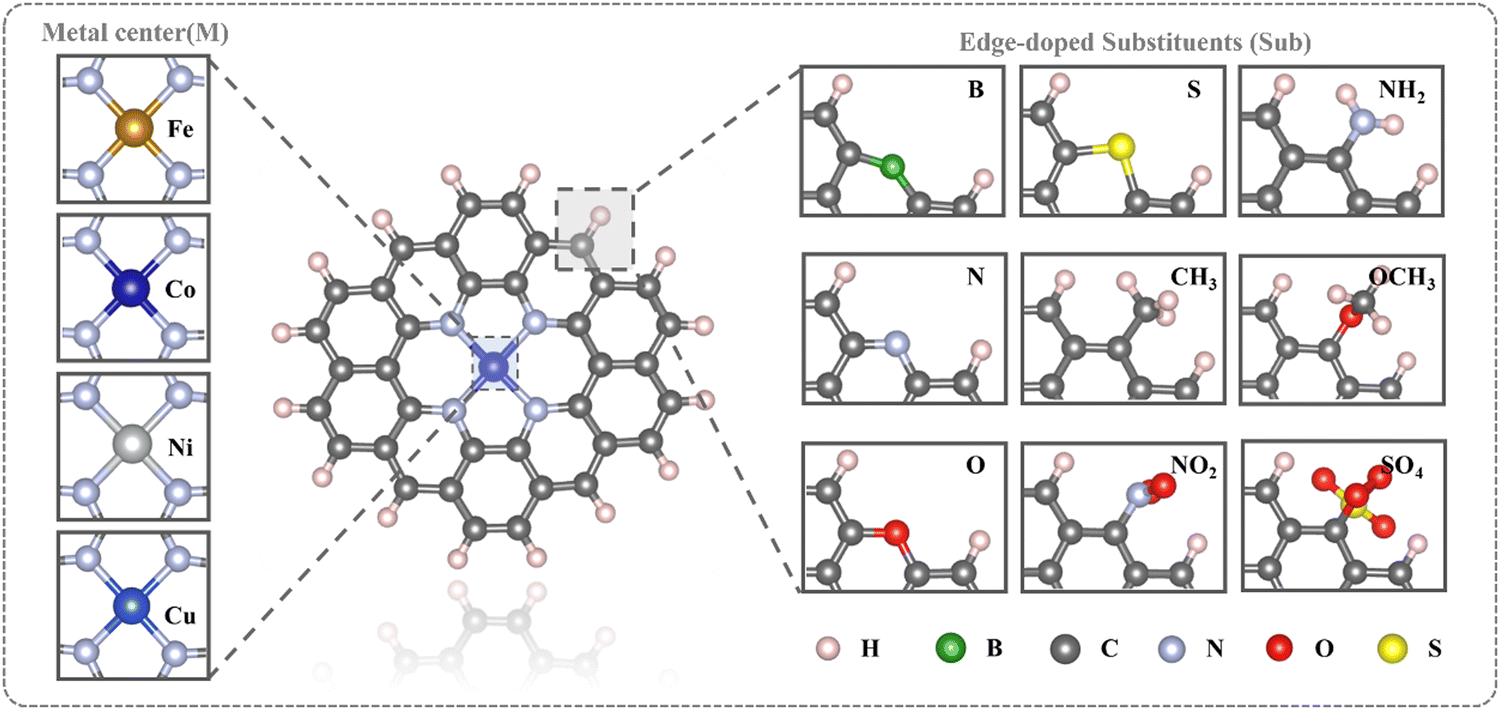 | ||
| Fig. 1 Schematic representation of the Sub@MN4 structures (Sub = B, N, O, S, CH3, NO2, NH2, OCH3, SO4; M = Fe, Co, Ni, Cu). | ||
2.1 Calculation of Gibbs free energy for the ORR/OER process
The ORR and OER are two reversible reactions, as shown in the following equations. Under acidic conditions, the reaction from left to right represents the ORR, which produces H2O, while the reaction from right to left represents the OER, which generates O2. Here, we do not consider new mechanisms, such as those occupied by *OH,48 but rather focus on the intrinsic properties of Sub@MN4 under traditional mechanisms.| 4H+ + O2 + 4e− ↔ 2H2O | (1) |
The entire ORR process consists of four steps, each of which involves proton-coupled electron transfer reactions, as previously reported:49,50
| * + O2 + H+ + e− → *OOH | (2) |
| *OOH + H+ + e− → *O + H2O | (3) |
| *O + H+ + e− → *OH | (4) |
| *OH + H+ + e− → * + H2O | (5) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) ln
ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 × pH. The calculation of the Gibbs free energy change for each step is as follows:
10 × pH. The calculation of the Gibbs free energy change for each step is as follows:| ΔG3 = ΔG*OOH − 4.92 eV + eU + kbTln10 × pH | (6) |
| ΔG4 = ΔG*O − ΔG*OOH + eU + kbTln10 × pH | (7) |
| ΔG5 = ΔG*OH − ΔG*O + eU + kbTln10 × pH | (8) |
| ΔG6 = −ΔG*OH + eU + kbTln10 × pH | (9) |
The values of ΔG*OOH, ΔG*O, and ΔG*OH are calculated through the following reactions (2H2O + * → *OOH + 3/2H2, H2O + * → *O + H2, H2O + * → *OH + 1/2H2):
| ΔG*OOH = G*OOH − G* − (2GH2O − 3/2GH2) | (10) |
| ΔG*O = G*O − G* − (GH2O − GH2) | (11) |
| ΔG*OH = G*OH − G* − (GH2O − 1/2GH2) | (12) |
Among these four steps, the rate-determining step is defined as the one with the largest Gibbs free energy change. Therefore, the overpotential of the ORR can be calculated using the following equation:
| ηORR = max{ΔG3, ΔG4, ΔG5, ΔG6}/e + 1.23 | (13) |
As the OER is the reversible reaction of the ORR, the overpotential can be calculated using the following equation:
| ηOER = −min{ΔG3, ΔG4, ΔG5, ΔG6}/e − 1.23 | (14) |
It is worth noting that the HER, OER, and ORR are reactions that occur at different electrode potentials, and thus, they do not compete selectively with each other. Specifically, in the water splitting system, when a positive potential exceeding 1.23 V is applied, the OER reaction takes place at the anode, whereas when a negative potential below 0 V is applied, the HER reaction occurs at the cathode. In the case of metal–air batteries, the ORR will occur when the applied positive potential is between 0 to 1.23 V, while the OER will occur when the applied positive potential exceeds 1.23 V.
2.2 Calculation of Gibbs free energy for the HER process
The HER reaction energy can be described by the following equation52 (here, we are not focusing on some recent new mechanisms for the HER, such as *H occupying,53 but rather on the performance of MN4 on traditional mechanisms):| 2H+ + 2e− → H2 | (15) |
This process is divided into the following two steps:
| *+ H+ + e− → *H | (16) |
| *H + H+ + e− → *+ H2 | (17) |
In the equations provided, * represents the active site of the catalyst, and *H is the adsorbed intermediate of the HER process. The CHE model is used to equate the chemical potential of gaseous hydrogen under standard conditions to the chemical potential of the proton–electron pair.51 The influence of electrode potential U on the electron e is expressed through −eU, and the effect of pH is calculated using kbTln10 × pH. The calculation of the Gibbs free energy change for each step is as follows:51
| ΔG16 = ΔG*H + eU +kbTln10 × pH | (18) |
| ΔG17 = −ΔG*H+ eU +kbTln10 × pH | (19) |
The overpotential of the entire reaction is:
| HHER = max{ΔG16, ΔG17}/e | (20) |
2.3 Calculation of electronic structure
The analysis of charge and d-band center was conducted using Multiwfn.54 The charge was calculated using the atomic dipole moment correction Hirshfeld (ADCH) method,55 and the numerical value of the d-band center of the metal M in the MN4 catalyst was obtained through the following formula:56,57 d band center = Ec,M – EF. Here, Ec,M refers to the energy of the d-band position center of the metal M, which is calculated using this formula: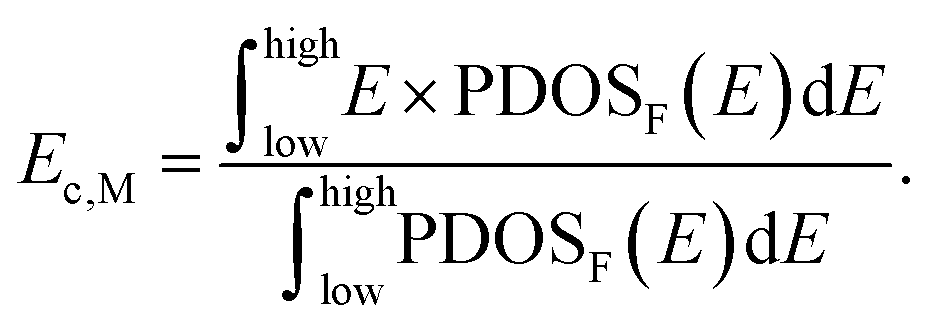 EF refers to the Fermi energy level, and for the isolated system model constructed in this study, we adopt the value of the highest occupied molecular orbital (HOMO) energy level.
EF refers to the Fermi energy level, and for the isolated system model constructed in this study, we adopt the value of the highest occupied molecular orbital (HOMO) energy level.
The fundamental gap (Eg) was proposed to describe the electron supply capacity of the Sub@MN4 system.58,59 It is obtained by subtracting the vertical electron affinity (VEA) from the vertical ionization potential (VIP): Eg = VIP – VEA = (E(N − 1) − E(N)) + (E(N + 1) − E(N)) = E(N − 1) + E(N + 1) − 2*E(N). Here, E(N − 1), E(N + 1), and E(N) represent the electronic energies of the Sub@MN4 model with N − 1 electrons, N + 1 electrons, and N electrons, respectively.
In order to investigate the effect of Sub on MN4, we consider the Sub as a whole and calculate their average electronegativity (χSub). The formula for calculating by harmonic average method is as follows:
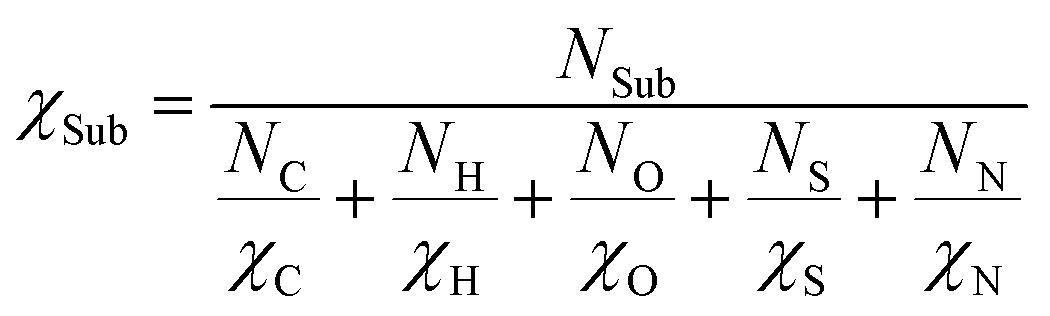 | (21) |
3. Results and analysis
3.1 Influence of edge-doping on the ORR/OER/HER performance of the MN4 structure
This study investigated the regulatory effect of edge-doping coordination on the MN4 structure by constructing 36 configurations. Four common transition metals (Fe, Co, Ni, Cu) were selected to form the MN4 configurations, and nine types of edge-doping groups, including four heteroatoms (B, N, O, S) and five substituents (CH3, NO2, NH2, OCH3, SO4), were doped at the same edge position to create different Sub@MN4 models.First, we evaluated the impact of different Sub on the ORR/OER performance of MN4. Both the ORR and OER involve three intermediate species (*OOH, *O, *OH) and four elementary steps.49 A strong linear correlation between ΔG*OOH and ΔG*OH (Fig. 2a) resulted in a minimum overpotential of 0.63 V at the top of the volcano plot (Fig. S8, ESI†). For the ORR, the free energy evolution of various transition metal systems is shown in Fig. 2b and Fig. S7 (ESI†). FeN4 exhibits a strong adsorption ability, with most Sub@FeN4 models limited by the last step (*OH desorption) and located on the left side of the volcano plot (Fig. S8e, ESI†). Three groups (O, S, SO4) further weaken FeN4's *OH adsorption, reducing overpotentials to 0.53 V, 0.56 V, and 0.56 V, respectively, thereby decreasing the overpotential of FeN4 (1.71 V) by 1.15 V (Fig. 2c). The presence of the S/O group (O, S, SO4) shifts FeN4 to the top of the volcano plot, consistent with the results of Mun et al. and Maouchue et al. regarding S regulation30,34 and Ni et al.'s understanding of O regulation.36,60 In comparison to Sub@FeN4, the Sub@CoN4 system exhibits weaker *OH adsorption (Fig. S8, ESI†), and the B, CH3, N and NH2 groups further regulate CoN4's intermediate adsorption, reducing overpotentials to 0.48 V, 0.63 V, 0.66 V and 0.58 V, respectively. Additionally, the NO2 group enhances CoN4's *OH adsorption, lowering the overpotential to 1.43 V (Fig. 2c). Compared to Sub@FeN4 and Sub@CoN4, the Sub@NiN4 system demonstrates weaker oxygen binding ability, making the first step (*OOH adsorption) the rate-determining step, located on the right side of the volcano plot (Fig. S8, ESI†). Among these groups, NH2 exhibits a better regulation ability, reducing NiN4's overpotential from 1.56 V to 0.91 V. As for the Sub@CuN4 system, the differentiation in group regulation is more evident. The O group significantly decreases the CuN4's overpotential to 0.63 V, while other groups (such as SO4) raise the overpotential of CuN4 to 1.44 V. Regarding the Sub@MN4 catalyzing the OER, certain similarities to the ORR exist, and the overpotentials are summarized in Fig. 2c and compared with the ORR.
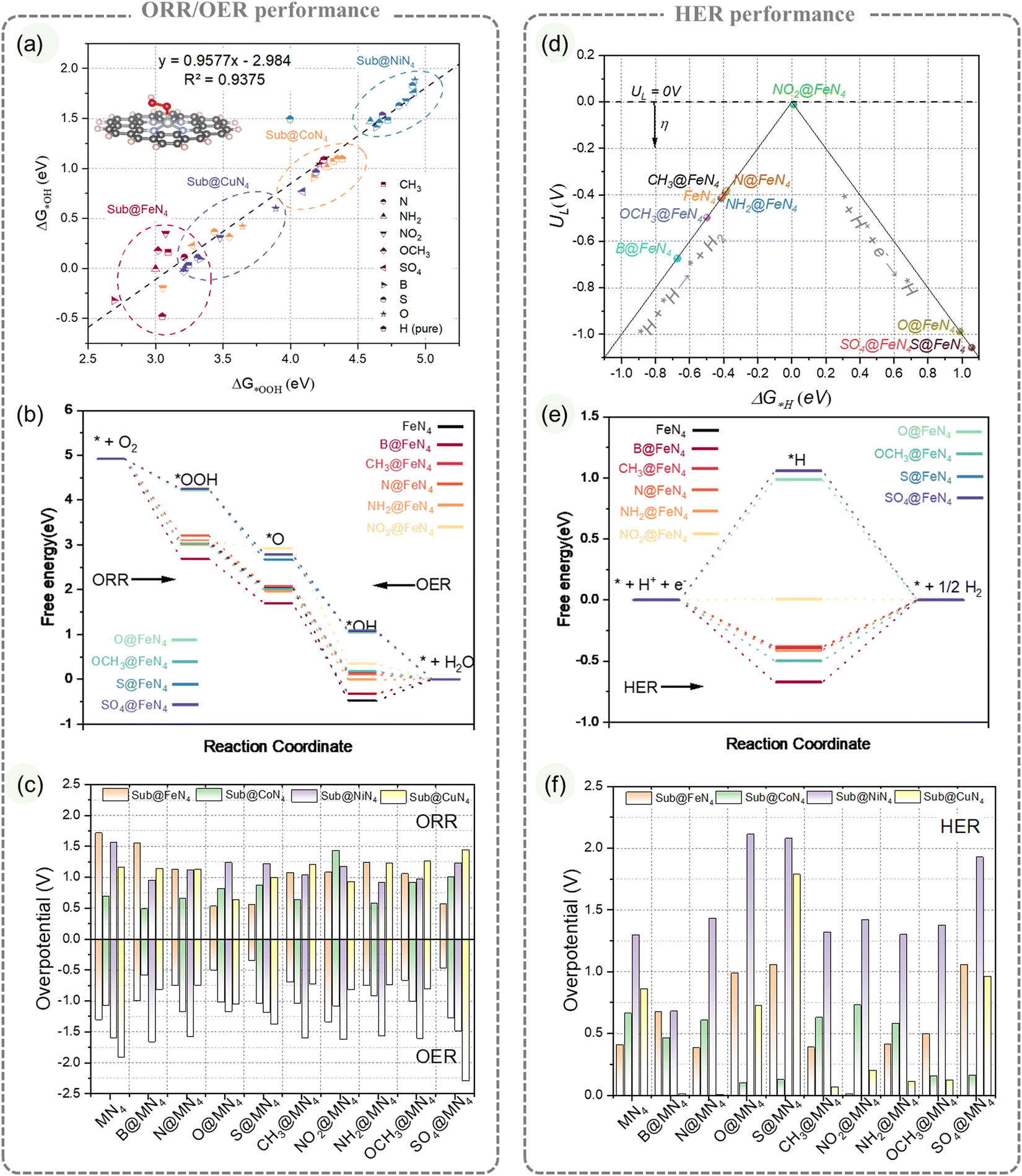 | ||
| Fig. 2 Influence of edge-doping on the ORR/OER/HER performance of the MN4 structure. (a) Linear correlation between the adsorption free energies of reaction intermediates *OOH and *OH on Sub@FeN4 catalysts, (b) free energy profiles and (c) overpotentials for OER/ORR pathways on Sub@MN4, (d) Activity volcano plot, (e) free energy profiles, and (f) overpotentials for HER pathways on Sub@MN4. | ||
The impact of different Sub on the HER performance of MN4 is depicted in Fig. 2d–f. Both Sub@CoN4 and Sub@CuN4 systems show promising HER potentials, with overpotentials ranging from 0.10 V to 0.73 V and from 0.01 V to 1.79 V, respectively. However, the Sub@NiN4 system exhibits an overpotential of at least 0.68 V. For FeN4, NO2 weakens ΔG*H from −0.41 eV to 0.01 eV, while N enhances ΔG*H to −0.67 eV (Fig. 2d and e). It is worth noting that S/O functional groups (O, S, SO4) also weaken ΔG*H but cross the top of the volcano plot to the right side (Fig. S9e, ESI†), resulting in no significant increase in overpotential (0.98–1.05 V). In the Sub@CoN4 system, different N functional groups have varying effects on CoN4: N and NH2 have little influence on ΔG*H, only changing it from 0.66 eV to 0.61 eV and 0.59 eV, while NO2 enhances CoN4's ΔG*H to −0.73 eV (Fig. S9b and f, ESI†). These differences indicate that even with similar elements in the substituents, the introduction of other elements can lead to significant variations. In the Sub@NiN4 system, most substituents primarily weaken its ΔG*H (similar to the weakening of oxygen adsorption energy). O, S, and SO4 weaken its ΔG*H to above 1.93 eV (Fig. S9c and g, ESI†). In contrast, in the Sub@CuN4 system, the substituents primarily enhance ΔG*H, with B and CH3 showing the most significant enhancement, crossing the reducing ΔG*H to 0.01 eV and −0.07 eV, respectively (Fig. S9d and h, ESI†). The summarized overpotentials for the HER are presented in Fig. 2f.
The analysis of the impact of edge substituent on the ORR/OER/HER performance of MN4 structures reveals diverse and sometimes contrasting effects on different metals. Additionally, the same edge substituent may yield varying outcomes on different metals, indicating the potential for edge substituent regulation varies. These observations imply complex interactions between edge substituents and metal centers. Understanding the underlying regulatory mechanisms and providing quantitative descriptions will aid in the design and optimization of Sub@MN4-based catalysts.
3.2 Effects of edge substituent coordination on MN4 structure
The varying degrees of influence of edge substituent on the catalytic performance of the metal center are closely related to the structural variations in MN4. The metal center charge and metal–nitrogen (M–N) bond lengths serve as direct indicators of the edge substituent effects on the MN4 structure.61 To address this, we measured the bond lengths of the four M–N bonds in Sub@MN4 and analyzed their average as an indicator. Additionally, the metal center ADCH charges were examined to understand the charge transfer between the edge substituent groups and the metal center.55 In the following sections, we will delve into how these physical structures vary with different edge substituent groups.The influence of the edge substituent on the average M–N bond length in the MN4 structure is illustrated in Fig. 3a and b. The type of metal center M significantly impacts the M–N bond length. In Sub@FeN4, the Fe–N bond length ranges from 1.88 to 1.93 Å, in Sub@CoN4, the Co–N bond length ranges from 1.86 to 1.90 Å, in Sub@NiN4, the Ni–N bond length ranges from 1.86 to 1.89 Å, and in Sub@CuN4, the Cu–N bond length ranges from 1.92 to 2.00 Å. Generally, the order of M–N bond length from largest to smallest in Sub@MN4 is: Sub@CuN4 > Sub@FeN4 > Sub@CoN4 > Sub@NiN4. However, there are two edge substituents with some differences: for O-doping, the order is Sub@CuN4 > Sub@FeN4 > Sub@NiN4 > Sub@CoN4; for NH2-doping, the order is Sub@CuN4 > Sub@CoN4 > Sub@FeN4 > Sub@NiN4. This indicates that these two edge substituents have specific characteristics concerning the metal center. In Sub@FeN4, B elongates the Fe–N bond, while CH3 and NO2 have minimal effects, and other groups shorten the bond length. In Sub@CoN4, except for N and O which shorten the Co–N bond, other groups elongate it. In Sub@NiN4, except for B, S, and SO4 which elongate the Ni–N bond, other groups have little influence. In the Sub@CuN4 system, all other Sub groups, except for the N group, elongate the Cu–N bond, especially S, which can increase the Cu–N bond length from 1.93 Å to 1.99 Å.
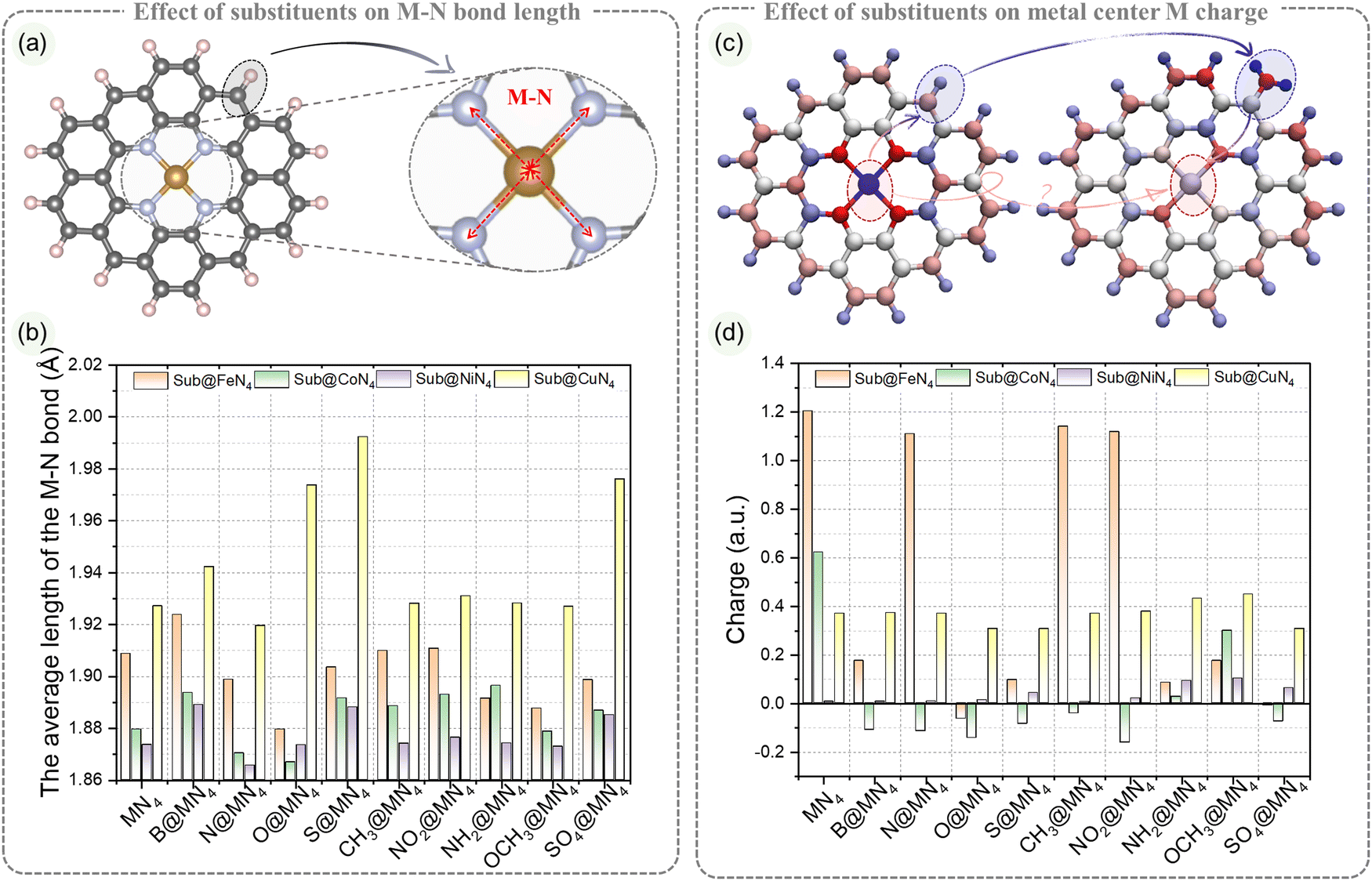 | ||
| Fig. 3 Effects of edge substituent coordination on the MN4 structure. (a) Schematic representation of M–N bonds in Sub@MN4. (b) Average M–N bond lengths in Sub@MN4. (c) Schematic representation of the influence of substituent groups on the charge of the metal center in Sub@MN4. (d) Charge of the metal center in Sub@MN4. | ||
The influence of edge substituent on the charge of the metal center M in the MN4 structure is demonstrated in Fig. 3c and d. Sub effectively regulates the charge of the metal center M in FeN4 and CoN4 over a wide range, within −0.06 to 1.20 a.u. and −0.16 to 0.62 a.u., respectively. In the Sub@FeN4 system, N, CH3, and NO2 have a weak effect on the Fe center, with only a slight change from 1.2 a.u. to 1.11 a.u., while other groups can significantly lower the charge by at least 1.02 a.u. On the other hand, in the Sub@CoN4 system, except for OCH3, which reduces the Co charge from 0.62 a.u. to a higher 0.30 a.u., other groups can transfer the Co charge to negative values. In contrast, the ranges of the metal center M charge in Sub@NiN4 and Sub@CuN4 are concentrated in 0.01–0.10 a.u. and 0.31–0.45 a.u., respectively, and the changes in charge due to the substituent are within ±0.08 a.u. These indicate different potentials for charge regulation in different metal centers. To measure the changes in electronic transfer during the adsorption of H/OOH in Sub@MN4, we also calculated and compared the charges before and after H/OOH adsorption, as shown in Fig. S8–S20 (ESI†). Notably, for the Sub@FeN4 and Sub@CoN4 systems, the metal center M after H/OOH adsorption remains within a relatively small range, contrasting with the larger range of charges before adsorption. For Sub@FeN4, except for S/SO4@FeN4 with charges of 0.04 and 0.19 a.u. after adsorbing *H, the remaining Sub@FeN4 have Fe charges within the range of 0.32–0.43 a.u. after *H adsorption. In Sub@CoN4, the charges of Co after *H adsorption for S/SO4@CoN4 are 0.11 and 0.20 a.u., while the others are between 0.23 and 0.26 a.u. A similar trend is observed for *OOH adsorption. In Sub@NiN4, the charges before and after adsorption follow the trend Sub@NiN4 < Sub@NiN4–H < Sub@NiN4–OOH, with relatively larger fluctuations in charge compared to the previous cases. For Sub@CuN4, in the unadsorbed configurations, the charge of the metal center is at least above 0.3 a.u., while after *H adsorption, the Cu charge remains around 0.2 a.u. (except for O/S@CuN4, which is around 0.3 a.u.), and after *OOH adsorption, the Cu charge remains around 0.3 a.u. (except for O/S@CuN4, which is above 0.4 a.u.).
The analysis above reveals that even when the Subs are positioned at least 4.68 Å away from the metal center M in the MN4 structure, they exert a substantial influence on the coordination of the metal M with surrounding nitrogen atoms and the charge of the metal center M. Consequently, this results in interactions between the metal M and the reaction intermediates (*OOH, *H) in the ORR/OER/HER, including charge transfer, leading to varying degrees of catalytic performance. These qualitative insights provide a clear understanding of the underlying mechanisms. However, for the development of MN4 catalysts, it is crucial to quantitatively evaluate and guide the prediction of the performance and synthesis of Sub@MN4 catalysts, which has significant importance.
3.3 Exploring the origin of activity in Sub@MN4
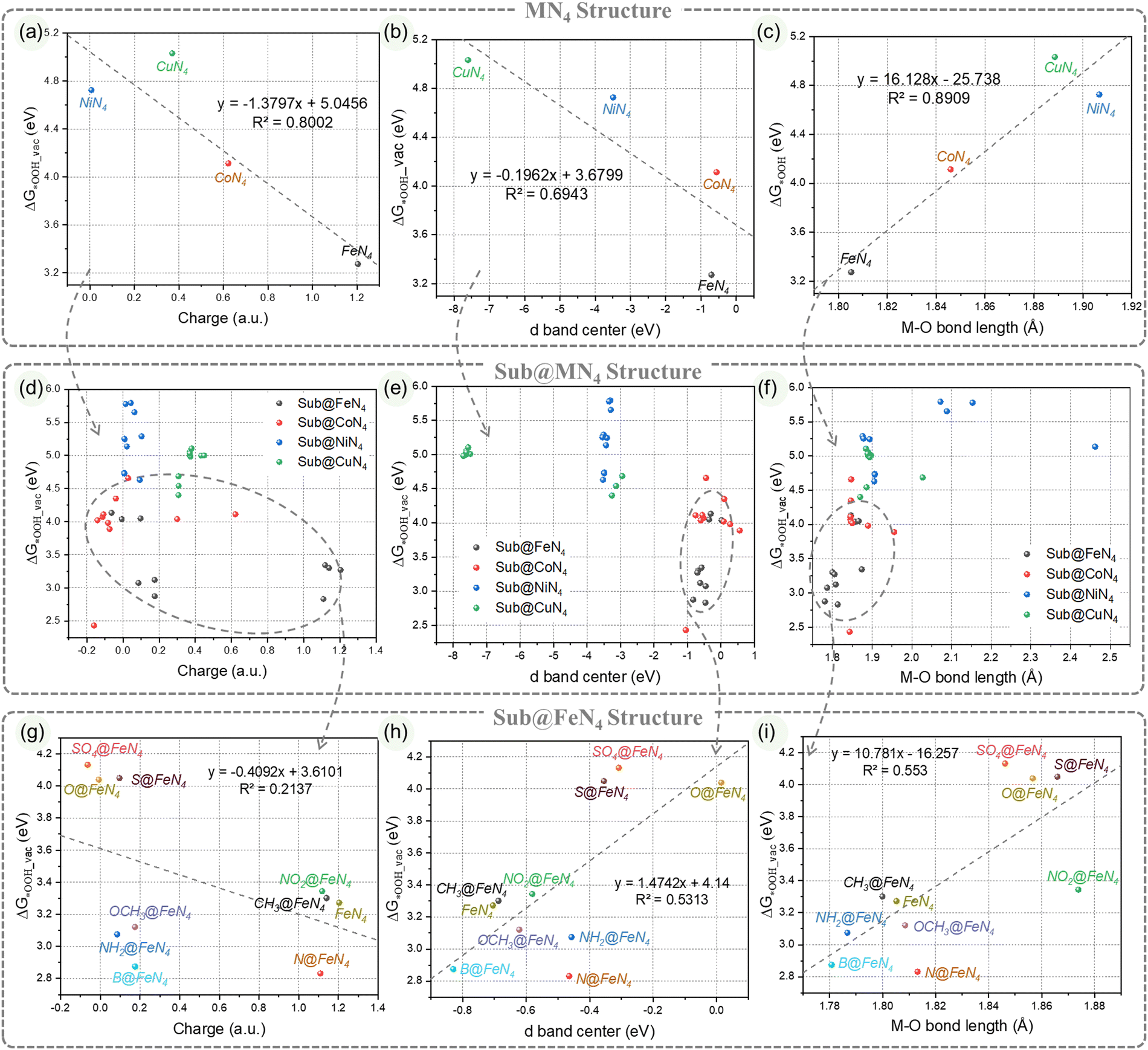 | ||
| Fig. 4 Correlations of ΔG*OOH_vac with (a) metal center charge, (b) d-band center, and (c) M–O bond length in pristine MN4 structures; correlations of ΔG*OOH_vac with (d) metal center charge, (e) d-band center, and (f) M–O bond length in Sub@MN4 structures; correlations of ΔG*OOH_vac with (g) metal center charge, (h) d-band center, and (i) M–O bond length in Sub@FeN4 structures. | ||
We further categorized the data according to different metals and extracted the information (Fig. 4g–i). The structure–performance relationship of Sub@FeN4 is presented in Fig. 4g–i. When attempting to fit the data points of Sub@FeN4 with straight lines, it was found that the correlation was not ideal (R2 ranging from 0.21 to 0.55). This implies that there is some correlation between them, but it is difficult to quantitatively describe it with a simple formula. Therefore, we are exploring the possibility of predicting and guiding the performance of this system based on a comprehensive understanding of the catalytic origin of Sub@MN4, thereby facilitating the synthesis of potentially efficient catalysts.
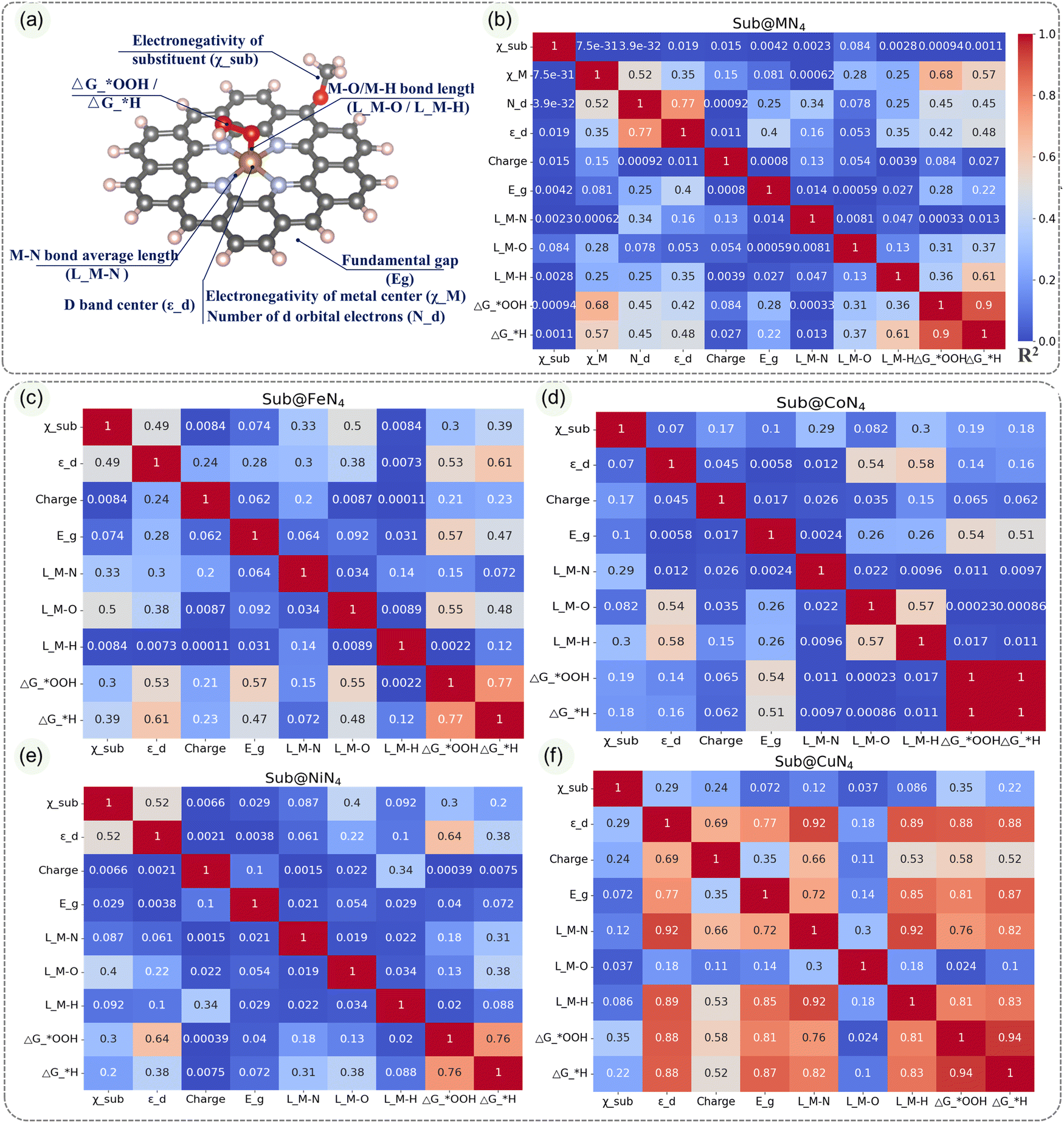 | ||
| Fig. 5 (a) Schematic representation of the descriptors analyzed in Sub@MN4. Correlation analysis of descriptors among (b) Sub@MN4, (c) Sub@FeN4, (d) Sub@CoN4, (e) Sub@NiN4, and (f) Sub@CuN4 structures, as well as their correlation with the adsorption free energies of intermediates under vacuum conditions. | ||
In order to further understand these influencing factors, the correlations among various indicators in Sub@MN4, as shown in Fig. 5a, are presented in the heatmap of Fig. 5b. It is noticeable that there is an almost linear relationship between the ΔG*OOH and ΔG*H (correlation coefficient R2 = 0.9), indicating that they can be mutually predicted. Another significant correlation is observed between the d-band center and the number of d electrons (correlation coefficient R2 = 0.77), which is consistent with our general understanding. The M–O/M–H bond lengths also show certain correlations with the catalytic performance (R2 = 0.31, 0.61).61 It is worth noting that in Sub@MN4, the contribution of the edge-doping group to the ΔG*OOH/ΔG*H seems negligible compared to the contribution of the metal center (R2 = 0.001, 0.001), which is contrary to our subconscious perception. Therefore, we further analyzed the correlations between various factors within different metals (Sub@FeN4, Sub@CoN4, Sub@NiN4, Sub@CuN4) as shown in Fig. 5c–f. In these more detailed heatmaps, we discovered strong correlations that were not evident in Fig. 5b, which may be attributed to the specific characteristics of different metals. In the Sub@FeN4 system, there are strong correlations between the d-band center, fundamental gap, M–O bond lengths and the ΔG*OOH/ΔG*H (R2 = 0.53/0.61, 0.57/0.47, 0.55/0.48).62,67 Particularly in the Sub@CoN4 system, the Eg can be used to predict the ΔG*OOH/ΔG*H effectively (R2 = 0.54, 0.51).46 For the Sub@CuN4 system, there are strong correlations between the d-band center, fundamental gap, M–N/M–H bond lengths and the ΔG*OOH/ΔG*H (R2 = 0.88/0.88, 0.81/0.87, 0.76/0.82, 0.81/0.83).
:1, and the effectiveness of the models is evaluated using R2 and RMSE metrics. Among all the tested models (Fig. S30–S37, ESI†), the KNN model (Fig. 6b and c) demonstrates excellent predictive performance.41 For ΔG*OOH prediction, the R2 is 0.955, and RMSE is 0.102; for ΔG*H prediction, the R2 is 0.943, and RMSE is 0.177. The GBR model closely follows, with an R2 of 0.723 and RMSE of 0.267 for ΔG*OOH prediction, and an R2 of 0.995 and RMSE of 0.066 for ΔG*H prediction. Other models show less satisfactory predictive abilities.
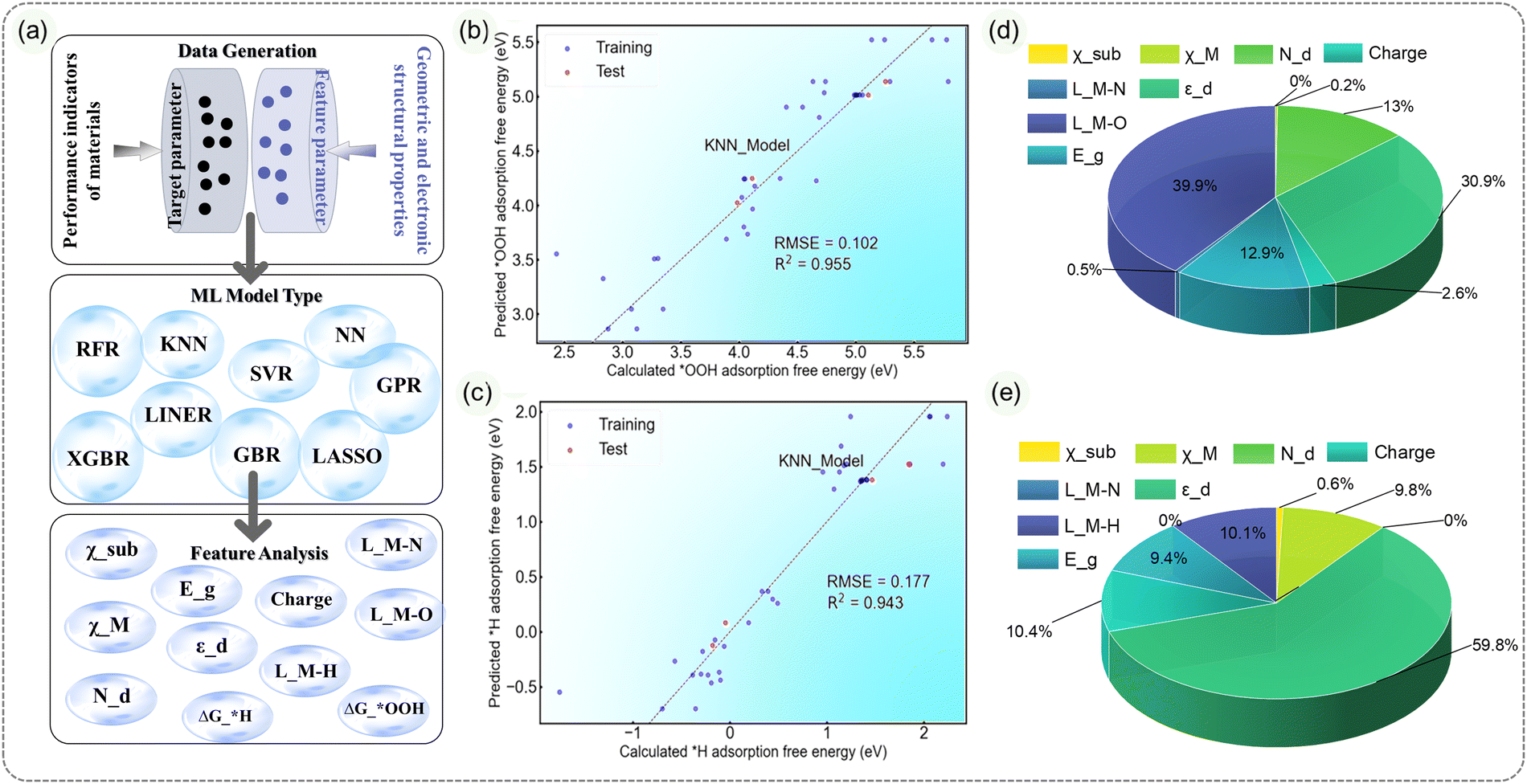 | ||
| Fig. 6 (a) A schematic representation of the machine learning (ML) process, divided into three parts: data generation, ML model type selection, and feature importance analysis. (b) Comparison of ΔG*OOH and (c) ΔG*H calculated from the KNN model with DFT calculations. (d) Importance analysis of various features for ΔG*OOH and (e) ΔG*H. | ||
During the feature importance analysis based on the GBR model, we find that, for ΔG*OOH prediction, the M–O bond length, d band center, Nd and Eg are the primary contributing factors, accounting for 39.9%, 30.9%, 12.9% and 13.0%, respectively. On the other hand, for ΔG*H prediction, the d band center is the dominant factor, accounting for 59.8% of importance. Charge, M–H bond length, metal center M electronegativity and Eg follow with importance values of 10.4%, 10.1%, 9.8% and 9.4%, respectively. It is worth noting that even if the M–O/M–H bond length is removed as a feature parameter, KNN and GBR still have good predictability, and the d band center still plays the most important role among all feature parameters (Fig. S38, ESI†), which also means that the ML models of these two have good predictive performance.
In conclusion, we have selected the K-Nearest Neighbors (KNN) model as the ML model for predicting the Sub@MN4 system. The feature importance analysis highlights the d band center and the significance of local M–O/M–H bond lengths. Additionally, in comparison to substituents, the metal center M plays a more crucial role in determining the ORR/OER/HER performance of the material. These analyses can serve as a reference for further investigations into more realistic and complex MN4 catalysts. Furthermore, they can offer a more efficient, rapid, and cost-effective approach for predicting and guiding the performance and synthesis of Sub@MN4 catalysts.
For practical applications, the ultimate goal of improving MN4 catalysts is to enable sustained operation at high current densities.69–71 Although this paper focuses on the theoretical understanding of atomically regulated MN4 catalysts, the proposed Sub@MN4 as a potential catalyst exhibits an overpotential as low as 0.09 V for the HER. This also suggests that combining our findings on the interactions between substituents and the M–N4 framework could lead to the development of M–N4 catalysts capable of high current density electrocatalysis. To this end, we believe that experimental verification of computational predictions under high current density conditions should be pursued, as well as the exploration of new materials and designs that can further enhance catalytic performance.
For the synthesis during the experimental process, approaches such as “modification of substituents based on the MN4 structure” or “selection of carbon precursors with substituents during the synthesis process” can be employed. The identification of specific structures is facilitated by the combination of characterization techniques such as X-ray photoelectron spectroscopy (XPS) and nuclear magnetic resonance (NMR).
4. Conclusion
In this study, we systematically investigated the effects of edge substituent and its interaction with the MN4 metal center on the catalytic performance of ORR/OER/HER reactions (Sub@MN4, Sub = B, N, O, S, CH3, NO2, NH2, OCH3, SO4; M = Fe, Co, Ni, Cu). The results demonstrated that edge coordination can influence the metal center charge, metal–nitrogen (M–N) bond lengths, and other geometric/electronic structures, thereby regulating their ORR/OER/HER activities. Notably, certain substituents, such as S, O, and SO4, can significantly reduce the overpotential of FeN4 by more than 1.15 V, while others like NO2 can increase the overpotential of CuN4 by more than 0.73 V. Additionally, for the OER, B, N, CH3, NO2, OCH3 and NH2 can reduce the overpotential of NiN4 by more than 1.0 V, while SO4 can increase the overpotential of NiN4 by 0.32 V. Furthermore, for the HER, O, S, and SO4 can increase the overpotential of NiN4 to at least 1.93 V.To understand the regulatory mechanisms, we conducted correlation analysis among edge substituents/metal electronegativity, M–N/M–O/M–H bond lengths, metal d-band center/d-orbital electrons, fundamental gap (Eg), and ΔG*OOH/ΔG*H. The results indicated that edge substituents remotely influence the electronic structure of the single-atom metal center and also affect the overall electronic structure of Sub@MN4, thereby impacting its adsorption capacity for key intermediates like *OOH/*H.
Moreover, to further describe and predict Sub@MN4, we employed machine learning analysis and found that the K-Nearest Neighbors (KNN) model can effectively guide the prediction of catalyst performance (with R2 values of 0.955 and 0.943 for predicting ΔG*OOH_vac/ΔG*H_vac, respectively). The d band center and M–O/M–H bond length descriptors were identified as the main factors characterizing the adsorption energies.
In conclusion, our research reveals the regulatory role of edge substituent on the MN4 catalyst's activity, providing guidance for the design and further optimization of efficient electrocatalysts. These findings contribute to a comprehensive understanding of the catalytic origin of Sub@MN4 systems, offering valuable insights for the development of high-performance electrocatalysts.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was financially supported by the Natural Science Foundation of Heilongjiang Province (No. YQ2022E027), the Natural Science Foundation of China (No. 52106237) and the Fundamental Research Funds for the Central Universities (No. FRFCU5710051422).References
- K. Jiao, J. Xuan, Q. Du, Z. Bao, B. Xie, B. Wang, Y. Zhao, L. Fan, H. Wang, Z. Hou, S. Huo, N. P. Brandon, Y. Yin and M. D. Guiver, Designing the next generation of proton-exchange membrane fuel cells, Nature, 2021, 595, 361–369, DOI:10.1038/s41586-021-03482-7.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Design principles for oxygen-reduction activity on perovskite oxide catalysts for fuel cells and metal-air batteries, Nat. Chem., 2011, 3, 546–550, DOI:10.1038/nchem.1069.
- S. C. Perry, D. Pangotra, L. Vieira, L. I. Csepei, V. Sieber, L. Wang, C. Ponce de León and F. C. Walsh, Electrochemical synthesis of hydrogen peroxide from water and oxygen, Nat. Rev. Chem., 2019, 3, 442–458, DOI:10.1038/s41570-019-0110-6.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Design of electrocatalysts for oxygen- and hydrogen-involving energy conversion reactions, Chem. Soc. Rev., 2015, 44, 2060–2086, 10.1039/c4cs00470a.
- Z. W. She, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Combining theory and experiment in electrocatalysis: Insights into materials design, Science, 2017, 355, 146, DOI:10.1126/science.aad4998.
- I. Roger, M. A. Shipman and M. D. Symes, Earth-abundant catalysts for electrochemical and photoelectrochemical water splitting, Nat. Rev. Chem., 2017, 1, 0003, DOI:10.1038/s41570-016-0003.
- X. Wang, S. Il Choi, L. T. Roling, M. Luo, C. Ma, L. Zhang, M. Chi, J. Liu, Z. Xie, J. A. Herron, M. Mavrikakis and Y. Xia, Palladium-platinum core-shell icosahedra with substantially enhanced activity and durability towards oxygen reduction, Nat. Commun., 2015, 6, 1–8, DOI:10.1038/ncomms8594.
- T. Wang, A. Chutia, D. J. L. Brett, P. R. Shearing, G. He, G. Chai and I. P. Parkin, Palladium alloys used as electrocatalysts for the oxygen reduction reaction, Energy Environ. Sci., 2021, 14, 2639–2669, 10.1039/d0ee03915b.
- K. Jiang, D. Zhao, S. Guo, X. Zhang, X. Zhu, J. Guo, G. Lu and X. Huang, Efficient oxygen reduction catalysis by subnanometer Pt alloy nanowires, Sci. Adv., 2017, 3, 2–10, DOI:10.1126/sciadv.1601705.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Trends in electrocatalysis on extended and nanoscale Pt-bimetallic alloy surfaces, Nat. Mater., 2007, 6, 241–247, DOI:10.1038/nmat1840.
- C. Zhang, S. Yang, J. Wu, M. Liu, S. Yazdi, M. Ren, J. Sha, J. Zhong, K. Nie, A. S. Jalilov, Z. Li, H. Li, B. I. Yakobson, Q. Wu, E. Ringe, H. Xu, P. M. Ajayan and J. M. Tour, Electrochemical CO2 Reduction with Atomic Iron-Dispersed on Nitrogen-Doped Graphene, Adv. Energy Mater., 2018, 8, 1703487, DOI:10.1002/aenm.201703487.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, High-performance electrocatalysts for oxygen reduction derived from polyaniline, iron, and cobalt, Science, 1979, 332(2011), 443–447, DOI:10.1126/science.1200832.
- X. Liu and L. Dai, Carbon-based metal-free catalysts, Nat. Rev. Mater., 2016, 1, 16064, DOI:10.1038/natrevmats.2016.64.
- C. Hu and L. Dai, Carbon-Based Metal-Free Catalysts for Electrocatalysis beyond the ORR, Angew. Chem., Int. Ed., 2016, 55, 11736–11758, DOI:10.1002/anie.201509982.
- R. Ma, G. Lin, Y. Zhou, Q. Liu, T. Zhang, G. Shan, M. Yang and J. Wang, A review of oxygen reduction mechanisms for metal-free carbon-based electrocatalysts, NPJ Comput. Mater., 2019, 5, 78, DOI:10.1038/s41524-019-0210-3.
- J. Zhang, H. Yang and B. Liu, Coordination Engineering of Single-Atom Catalysts for the Oxygen Reduction Reaction: A Review, Adv. Energy Mater., 2021, 11, 2002473, DOI:10.1002/aenm.202002473.
- C. X. Zhao, B. Q. Li, J. N. Liu and Q. Zhang, Intrinsic Electrocatalytic Activity Regulation of M–N–C Single-Atom Catalysts for the Oxygen Reduction Reaction, Angew. Chem., Int. Ed., 2021, 60, 4448–4463, DOI:10.1002/anie.202003917.
- X. Guo, S. Lin, J. Gu, S. Zhang, Z. Chen and S. Huang, Simultaneously Achieving High Activity and Selectivity toward Two-Electron O2 Electroreduction: The Power of Single-Atom Catalysts, ACS Catal., 2019, 9, 11042–11054, DOI:10.1021/acscatal.9b02778.
- J. Gao, H. bin Yang, X. Huang, S. F. Hung, W. Cai, C. Jia, S. Miao, H. M. Chen, X. Yang, Y. Huang, T. Zhang and B. Liu, Enabling Direct H2O2 Production in Acidic Media through Rational Design of Transition Metal Single Atom Catalyst, Chem, 2020, 6, 658–674, DOI:10.1016/j.chempr.2019.12.008.
- Y. Wang, H. Su, Y. He, L. Li, S. Zhu, H. Shen, P. Xie, X. Fu, G. Zhou, C. Feng, D. Zhao, F. Xiao, X. Zhu, Y. Zeng, M. Shao, S. Chen, G. Wu, J. Zeng and C. Wang, Advanced Electrocatalysts with Single-Metal-Atom Active Sites, Chem. Rev., 2020, 120, 12217–12314, DOI:10.1021/acs.chemrev.0c00594.
- S. Chen, T. Luo, X. Li, K. Chen, J. Fu, K. Liu, C. Cai, Q. Wang, H. Li, Y. Chen, C. Ma, L. Zhu, Y. R. Lu, T. S. Chan, M. Zhu, E. Cortés and M. Liu, Identification of the Highly Active Co-N4Coordination Motif for Selective Oxygen Reduction to Hydrogen Peroxide, J. Am. Chem. Soc., 2022, 144, 14505–14516, DOI:10.1021/jacs.2c01194.
- Y. Sun, L. Silvioli, N. R. Sahraie, W. Ju, J. Li, A. Zitolo, S. Li, A. Bagger, L. Arnarson, X. Wang, T. Moeller, D. Bernsmeier, J. Rossmeisl, F. Jaouen and P. Strasser, Activity-Selectivity Trends in the Electrochemical Production of Hydrogen Peroxide over Single-Site Metal-Nitrogen-Carbon Catalysts, J. Am. Chem. Soc., 2019, 141, 12372–12381, DOI:10.1021/jacs.9b05576.
- L. Li, S. Huang, R. Cao, K. Yuan, C. Lu, B. Huang, X. Tang, T. Hu, X. Zhuang and Y. Chen, Optimizing Microenvironment of Asymmetric N,S-Coordinated Single-Atom Fe via Axial Fifth Coordination toward Efficient Oxygen Electroreduction, Small, 2022, 18, 2105387, DOI:10.1002/smll.202105387.
- X. Wang, Y. An, L. Liu, L. Fang, Y. Liu, J. Zhang, H. Qi, T. Heine, T. Li, A. Kuc, M. Yu and X. Feng, Atomically Dispersed Pentacoordinated-Zirconium Catalyst with Axial Oxygen Ligand for Oxygen Reduction Reaction, Angew. Chem., Int. Ed., 2022, 61, e202209746, DOI:10.1002/anie.202209746.
- J. Wang, M. Zheng, X. Zhao and W. Fan, Structure-Performance Descriptors and the Role of the Axial Oxygen Atom on M-N4-C Single-Atom Catalysts for Electrochemical CO2 Reduction, ACS Catal., 2022, 12, 5441–5454, DOI:10.1021/acscatal.2c00429.
- E. Jung, H. Shin, B. H. Lee, V. Efremov, S. Lee, H. S. Lee, J. Kim, W. Hooch Antink, S. Park, K. S. Lee, S. P. Cho, J. S. Yoo, Y. E. Sung and T. Hyeon, Atomic-level tuning of Co–N–C catalyst for high-performance electrochemical H2O2 production, Nat. Mater., 2020, 19, 436–442, DOI:10.1038/s41563-019-0571-5.
- Q. Zhang, X. Tan, N. M. Bedford, Z. Han, L. Thomsen, S. Smith, R. Amal and X. Lu, Direct insights into the role of epoxy groups on cobalt sites for acidic H2O2 production, Nat. Commun., 2020, 11, 4181, DOI:10.1038/s41467-020-17782-5.
- Q. Wang, Y. Tan, S. Tang, W. Liu, Y. Zhang, X. Xiong and Y. Lei, Edge-Hosted Mn-N4-C12Site Tunes Adsorption Energy for Ultralow-Temperature and High-Capacity Solid-State Zn-Air Battery, ACS Nano, 2023, 17, 9565–9574, DOI:10.1021/acsnano.3c02521.
- X. Wang, Y. Jia, X. Mao, D. Liu, W. He, J. Li, J. Liu, X. Yan, J. Chen, L. Song, A. Du and X. Yao, Edge-Rich Fe−N4 Active Sites in Defective Carbon for Oxygen Reduction Catalysis, Adv. Mater., 2020, 32, 2000966, DOI:10.1002/adma.202000966.
- C. Maouche, J. Yang, S. H. Al-Hilfi, X. Tao and Y. Zhou, Sulfur-Doped Fe-N-C Nanomaterials as Catalysts for the Oxygen Reduction Reaction in Acidic Medium, ACS Appl. Nano Mater., 2022, 5, 4397–4405, DOI:10.1021/acsanm.2c00501.
- Y. Lin, K. Liu, K. Chen, Y. Xu, H. Li, J. Hu, Y. R. Lu, T. S. Chan, X. Qiu, J. Fu and M. Liu, Tuning Charge Distribution of FeN4via External N for Enhanced Oxygen Reduction Reaction, ACS Catal., 2021, 11, 6304–6315, DOI:10.1021/acscatal.0c04966.
- Y. Ma, M. Zhang, J. Wu, Y. Zhao, X. Du, H. Huang, Y. Zhou, Y. Liu and Z. Kang, The Key Effect of Carboxyl Group and CuN2O2 Coordinate Structure for Cu, N Co-Doped Carbon Dots with Peroxidase-Like Property, Small, 2023, 19, 2300883, DOI:10.1002/smll.202300883.
- Y. Tian, M. Li, Z. Wu, Q. Sun, D. Yuan, B. Johannessen, L. Xu, Y. Wang, Y. Dou, H. Zhao and S. Zhang, Edge-hosted Atomic Co−N4 Sites on Hierarchical Porous Carbon for Highly Selective Two-electron Oxygen Reduction Reaction, Angew. Chem., Int. Ed., 2022, 61, e202213296, DOI:10.1002/anie.202213296.
- Y. Mun, S. Lee, K. Kim, S. Kim, S. Lee, J. W. Han and J. Lee, Versatile Strategy for Tuning ORR Activity of a Single Fe-N4 Site by Controlling Electron-Withdrawing/Donating Properties of a Carbon Plane, J. Am. Chem. Soc., 2019, 141, 6254–6262, DOI:10.1021/jacs.8b13543.
- B. Q. Li, C. X. Zhao, J. N. Liu and Q. Zhang, Electrosynthesis of Hydrogen Peroxide Synergistically Catalyzed by Atomic Co–N x –C Sites and Oxygen Functional Groups in Noble-Metal-Free Electrocatalysts, Adv. Mater., 2019, 1808173, 1–8, DOI:10.1002/adma.201808173.
- W. Ni, Y. Gao, Y. Zhang, H. A. Younus, X. Guo, C. Ma, Y. Zhang, J. Duan, J. Zhang and S. Zhang, O-Doping Boosts the Electrochemical Oxygen Reduction Activity of a Single Fe Site in Hydrophilic Carbon with Deep Mesopores, ACS Appl. Mater. Interfaces, 2019, 1–25, DOI:10.1021/acsami.9b18510.
- X. Zhao, Z. H. Levell, S. Yu and Y. Liu, Atomistic Understanding of Two-dimensional Electrocatalysts from First Principles, Chem. Rev., 2022, 122, 10675–10709, DOI:10.1021/acs.chemrev.1c00981.
- H. Y. Zhuo, X. Zhang, J. X. Liang, Q. Yu, H. Xiao and J. Li, Theoretical Understandings of Graphene-based Metal Single-Atom Catalysts: Stability and Catalytic Performance, Chem. Rev., 2020, 120, 12315–12341, DOI:10.1021/acs.chemrev.0c00818.
- J. Liu, W. Luo, L. Wang, J. Zhang, X. Z. Fu and J. L. Luo, Toward Excellence of Electrocatalyst Design by Emerging Descriptor-Oriented Machine Learning, Adv. Funct. Mater., 2022, 32, 2110748, DOI:10.1002/adfm.202110748.
- N. Ma, Y. Zhang, Y. Wang, C. Huang, J. Zhao, B. Liang and J. Fan, Machine learning-assisted exploration of the intrinsic factors affecting the catalytic activity of ORR/OER bifunctional catalysts, Appl. Surf. Sci., 2023, 628, 157225, DOI:10.1016/j.apsusc.2023.157225.
- X. Liu, Y. Zhang, W. Wang, Y. Chen, W. Xiao, T. Liu, Z. Zhong, Z. Luo, Z. Ding and Z. Zhang, Transition Metal and N Doping on AlP Monolayers for Bifunctional Oxygen Electrocatalysts: Density Functional Theory Study Assisted by Machine Learning Description, ACS Appl. Mater. Interfaces, 2022, 14, 1249–1259, DOI:10.1021/acsami.1c22309.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- J. Da Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620, 10.1039/b810189b.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305, 10.1039/b508541a.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions, J. Phys. Chem. B, 2009, 113, 6378–6396, DOI:10.1021/jp810292n.
- L. Xie, W. Zhou, Z. Qu, Y. Ding, J. Gao, F. Sun and Y. Qin, Understanding the activity origin of oxygen-doped carbon materials in catalyzing the two-electron oxygen reduction reaction towards hydrogen peroxide generation, J. Colloid Interface Sci., 2022, 610, 934–943, DOI:10.1016/j.jcis.2021.11.144.
- Y. Huang, W. Zhou, L. Xie, J. Li, Y. He, S. Chen, X. Meng, J. Gao and Y. Qin, Edge and defect sites in porous activated coke enable highly efficient carbon-assisted water electrolysis for energy-saving hydrogen production, Renewable Energy, 2022, 195, 283–292, DOI:10.1016/j.renene.2022.06.037.
- Y. Wang, Y. J. Tang and K. Zhou, Self-Adjusting Activity Induced by Intrinsic Reaction Intermediate in Fe-N-C Single-Atom Catalysts, J. Am. Chem. Soc., 2019, 141, 14115–14119, DOI:10.1021/jacs.9b07712.
- X. Chen, H. Zhu, J. Zhu and H. Zhang, Indium-based bimetallic clusters anchored onto silicon-doped graphene as efficient multifunctional electrocatalysts for ORR, OER, and HER, Chem. Eng. J., 2023, 451, 138998, DOI:10.1016/j.cej.2022.138998.
- X. Chen, Y. Zhang, X. Zhao, H. Yu and H. Zhang, Effect of the Axial Halogen Ligand on the Oxygen Reduction Reaction Performance of Transition Metal–Nitrogen–Carbon Catalysts, J. Phys. Chem. C, 2023, 127, 14107–14116, DOI:10.1021/acs.jpcc.3c02628.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, Origin of the overpotential for oxygen reduction at a fuel-cell cathode, J. Phys. Chem. B, 2004, 108, 17886–17892, DOI:10.1021/jp047349j.
- X. Liu, Y. Jiao, Y. Zheng, K. Davey and S. Z. Qiao, A computational study on Pt and Ru dimers supported on graphene for the hydrogen evolution reaction: New insight into the alkaline mechanism, J. Mater. Chem. A, 2019, 7, 3648–3654, 10.1039/c8ta11626a.
- G. Di Liberto, L. A. Cipriano and G. Pacchioni, Role of Dihydride and Dihydrogen Complexes in Hydrogen Evolution Reaction on Single-Atom Catalysts, J. Am. Chem. Soc., 2021, 143, 20431–20441, DOI:10.1021/jacs.1c10470.
- T. Lu and F. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592, DOI:10.1002/jcc.22885.
- T. Lu and F. Chen, Atomic dipole moment corrected Hirshfeld population method, J. Theor. Comput. Chem., 2012, 11, 163–183, DOI:10.1142/S0219633612500113.
- J. K. Nørskov, F. Abild-Pedersen, F. Studt and T. Bligaard, Density functional theory in surface chemistry and catalysis, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 937–943, DOI:10.1073/pnas.1006652108.
- S. Bhattacharjee, U. V. Waghmare and S. C. Lee, An improved d-band model of the catalytic activity of magnetic transition metal surfaces, Sci. Rep., 2016, 6, 35916, DOI:10.1038/srep35916.
- J. L. Bredas, Mind the gap!, Mater. Horiz., 2014, 1, 17–19, 10.1039/c3mh00098b.
- J. Zirzlmeier, S. Schrettl, J. C. Brauer, E. Contal, L. Vannay, É. Brémond, E. Jahnke, D. M. Guldi, C. Corminboeuf, R. R. Tykwinski and H. Frauenrath, Optical gap and fundamental gap of oligoynes and carbyne, Nat. Commun., 2020, 11, 4797, DOI:10.1038/s41467-020-18496-4.
- M. Liu, Z. Liu, W. Chen, Z. Liu, Z. Li, X. Pi, Q. Du, X. Lai, Y. Xia and Y. Li, NiFe Nanoparticle-Encapsulated Ultrahigh-Oxygen-Doped Carbon Layers as Bifunctional Electrocatalysts for Rechargeable Zn-Air Batteries, Inorg. Chem., 2023, 62, 11199–11206, DOI:10.1021/acs.inorgchem.3c01375.
- K. Liu, J. Fu, Y. Lin, T. Luo, G. Ni, H. Li, Z. Lin and M. Liu, Insights into the activity of single-atom Fe-N-C catalysts for oxygen reduction reaction, Nat. Commun., 2022, 13, 2075, DOI:10.1038/s41467-022-29797-1.
- H. Xu, D. Cheng, D. Cao and X. C. Zeng, A universal principle for a rational design of single-atom electrocatalysts, Nat. Catal., 2018, 1, 339–348, DOI:10.1038/s41929-018-0063-z.
- P. Zhu, X. Xiong, X. Wang, C. Ye, J. Li, W. Sun, X. Sun, J. Jiang, Z. Zhuang, D. Wang and Y. Li, Regulating the FeN4Moiety by Constructing Fe-Mo Dual-Metal Atom Sites for Efficient Electrochemical Oxygen Reduction, Nano Lett., 2022, 22, 9507–9515, DOI:10.1021/acs.nanolett.2c03623.
- X. Li, J. Liu, Q. Cai, Z. Kan, S. Liu and J. Zhao, Engineering d-band center of iron single atom site through boron incorporation to trigger the efficient bifunctional oxygen electrocatalysis, J. Colloid Interface Sci., 2022, 628, 331–342, DOI:10.1016/j.jcis.2022.07.158.
- H. Sun, M. Wang, X. Du, Y. Jiao, S. Liu, T. Qian, Y. Yan, C. Liu, M. Liao, Q. Zhang, L. Meng, L. Gu, J. Xiong and C. Yan, Modulating the d-band center of boron doped single-atom sites to boost the oxygen reduction reaction, J. Mater. Chem. A, 2019, 7, 20952–20957, 10.1039/c9ta06949f.
- F. Zheng, Y. Ji, H. Dong, C. Liu, S. Chen and Y. Li, Edge Effect Promotes Graphene-Confining Single-Atom Co–N4 and Rh–N4 for Bifunctional Oxygen Electrocatalysis, J. Phys. Chem. C, 2022, 126, 30–39, DOI:10.1021/acs.jpcc.1c07691.
- C. Ren, S. Lu, Y. Wu, Y. Ouyang, Y. Zhang, Q. Li, C. Ling and J. Wang, A Universal Descriptor for Complicated Interfacial Effects on Electrochemical Reduction Reactions, J. Am. Chem. Soc., 2022, 144, 12874–12883, DOI:10.1021/jacs.2c04540.
- C. Jia, Q. Wang, J. Yang, K. Ye, X. Li, W. Zhong, H. Shen, E. Sharman, Y. Luo and J. Jiang, Toward Rational Design of Dual-Metal-Site Catalysts: Catalytic Descriptor Exploration, ACS Catal., 2022, 12, 3420–3429, DOI:10.1021/acscatal.1c06015.
- H. Liu, R. Xie, Y. Luo, Z. Cui, Q. Yu, Z. Gao, Z. Zhang, F. Yang, X. Kang, S. Ge, S. Li, X. Gao, G. Chai, L. Liu and B. Liu, Dual interfacial engineering of a Chevrel phase electrode material for stable hydrogen evolution at 2500 mA cm−2, Nat. Commun., 2022, 13, 6382, DOI:10.1038/s41467-022-34121-y.
- S. Hu, S. Ge, H. Liu, X. Kang, Q. Yu and B. Liu, Low-Dimensional Electrocatalysts for Acidic Oxygen Evolution: Intrinsic Activity, High Current Density Operation, and Long-Term Stability, Adv. Funct. Mater., 2022, 32, 2201726, DOI:10.1002/adfm.202201726.
- Y. Luo, L. Tang, U. Khan, Q. Yu, H. M. Cheng, X. Zou and B. Liu, Morphology and surface chemistry engineering toward pH-universal catalysts for hydrogen evolution at high current density, Nat. Commun., 2019, 10, 269, DOI:10.1038/s41467-018-07792-9.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nh00424h |
| This journal is © The Royal Society of Chemistry 2025 |