
Understanding the effects of adduct functionalization on C60 nanocages for the hydrogen evolution reaction†
Joy
Spears
a,
Mina
Shawky Adly
b,
Edison
Castro
c,
Alain R.
Puente Santiago
 *d,
Luis
Echegoyen
c,
Tianwei
He
*e,
Christopher J.
Dares
d and
Mohamed
Noufal
*a
*d,
Luis
Echegoyen
c,
Tianwei
He
*e,
Christopher J.
Dares
d and
Mohamed
Noufal
*a
aDepartment of Chemical Engineering, Hampton University, Hampton, VA 23668, USA. E-mail: mohamed.noufal@hamptonu.edu
bDeparment of Chemistry, Faculty of Science, Mansoura University, Al-Mansoura 35516, Egypt
cDepartment of Chemistry, University of Texas at El Paso, 500 West University Avenue, El Paso, Texas 79968, USA
dFlorida International University (FIU), Department of Chemistry and Biochemistry, Miami, FL, USA. E-mail: alpuente@fiu.edu
eNational Center for International Research on Photoelectric and Energy Materials, Yunnan Key Laboratory for Micro/Nano Materials& Technology, School of Materials and Energy, Institute of International Rivers and Eco-Security, Yunnan University, Kunming 650091, China. E-mail: he.tianwei@ynu.edu.cn
First published on 22nd January 2025
Abstract
In this work, we use experimental and theoretical techniques to study the origin of the boosted hydrogen evolution reaction (HER) catalytic activity of two pyridyl-pyrrolidine functionalized C60 fullerenes. Notably, the mono-(pyridyl-pyrrolidine) penta-adduct of C60 has exhibited a remarkable HER catalytic activity as a metal-free catalyst, delivering an overpotential (η10) of 75 mV vs. RHE and a very low onset potential of −45 mV vs. RHE. This work addresses fundamental questions about how functionalization on C60 changes the electron density on fullerene cages for high-performance HER electrocatalysis.
New conceptsIn this work, we thoroughly investigated the electrocatalytic performance of two pyridyl-pyrrolidine functionalized C60 fullerenes for the hydrogen evolution reaction (HER) using advanced experimental and theoretical tools. These functionalized fullerenes showed an exceptional electron density relocation at the C60-ligand interfaces, which significantly impacts the electronic structure of the carbon atoms in the nearest regions, thus inducing a new type of highly active electrocatalytic site for the molecular generation of hydrogen. Notably, the mono-(pyridyl-pyrrolidine) penta-adduct of C60 has exhibited a remarkable HER catalytic activity as a metal-free catalyst, delivering an overpotential (η10) of 75 mV vs. RHE and a very low onset potential of −45 mV vs. RHE. Additionally, we identified the key factors that influence the intermolecular electron transfer at the C60-addend interface and their impact on the HER. This work introduces new chemistry concepts to understand the origin of the electrocatalytic activity of metal-free 0D-low-dimensional nanomaterials with curved surfaces. |
The HER must achieve technological maturity, relying on widely available carbon-based materials. Pt-group metal (PGM) catalysts constitute a significant portion of the design and cost for proton-exchange membrane fuel cells (PEMFCs) and electrolyzers.1–3 Over the past decade, substantial efforts have been made to develop PGM-free cathodes for HER, especially with the recent promising developments in metal–N–C-based and metal single-atom alternatives to PGMs.4 Most recent reports have focused on studies of metal-containing carbides and Ni-based materials for HER catalysis, which exhibit only moderate activities and require greater stability and reproducibility.4–6 Low-dimensional carbons, such as 1D-carbon nanotubes and 2D-graphene, have been used as metal-free catalysts for the electrochemical generation of molecular hydrogen because of their large surface area, excellent chemical stability, and high conductivity.7,8 Despite these advances, employing metal-free catalysts for HER and their applications remains challenging due to their large molecular footprint, poor durability, and limited electrochemical stability.9,10 With the latest progress in electrocatalysts, properties such as high electrochemical stability and high conductivity, are drawing researchers to use molecular catalysts for energy conversion applications.11 0D-based fullerene architectures have recently emerged as a unique alternative to reduce protons to hydrogen molecules for fuel cell applications.12–16 The distinctive chemical and physical characteristics of fullerenes and their derivatives primarily arise from their electron-accepting abilities and high charge transport capabilities in three dimensions. However, the role of interfacial chemistry at the C60-ligand heterointerface for hydrogen evolution reactions has not been completely unveiled.
In this work, we report an activation approach involving the regio- and stereoselective synthesis of hybrid fullerene C60 penta (P-C60) and hexa-adduct (H-C60).17 These functionalized fullerenes showed an exceptional electron density relocation at the C60-ligand interfaces, which significantly impacts the electronic structure of the carbon atoms in the nearest regions, thus inducing a new type of highly active electrocatalytic sites for the molecular generation of hydrogen. Hexa and penta adduct of C60 are prepared as shown in the scheme presented in Fig. 1 and Supporting information 1 (ESI†).
 | ||
| Fig. 1 Synthesis of penta and hexa-adducts of C60: hexa-adduct (C2h-symmetric trans-1-(bis-pyrrolidine)-tetra-malonate hexa-adducts of C60) and penta-adduct-C60 (mono-pyrrolidine penta-adduct of C60) were synthesized through the reaction of C60-TM with methyl glycine ester, using diacetoxyiodobenzene (DIB) and sodium carbonate decahydrate as reagents in ortho-dichlorobenzene (o-DCB), under sonication at room temperature. | ||
The 13C NMR spectrum (Fig. S1–S3, ESI†) of H and P-C60 exhibited resonances analogous featuring carbonyl group signals at δ = 163.8 and 163.4 ppm, seventeen signals between 156.1 and 122.6 ppm corresponding to twelve sp2 fullerene carbon atoms and five 2-pyridyl groups, pyrrolidine addend signals at δ = 70.6 and 76.5 ppm, methylene group signals at δ = 62.7 and 62.1 ppm, and methyl group signals at δ = 14.1 ppm. Furthermore, five additional signals at δ = 72.7, 70.9, 66.2, 45.4, and 44.1 ppm confirmed the presence of the cis–anti–cis isomer.18 The crystal data of the as-separated structures of penta and hexa adduct are presented in (Fig. S4 and Supporting information 2, ESI†). The exclusive formation of cis–anti–cis hexa-adduct isomers was primarily ascribed to the interactions between the pendant groups in the malonate and the pyrrolidine rings. These interactions, crucial in the gas phase (van der Waals forces), are diminished when the solvent (o-DCB) is present.
The hydrogen evolution characteristics of functionalized fullerenes were investigated through linear sweep voltammetry (LSV) in 0.5 M H2SO4 (Experimental details in Supporting information 3, ESI†). Catalytic activities for hydrogen evolution are depicted in Fig. 2A, showcasing pristine C60, P-C60, and H-C60, alongside the benchmark Pt/C. The onset potentials and current densities for hexa and penta functionalized fullerenes (−90 mV and −45 mV vs. RHE) significantly exceed those of pristine C60, suggesting that organic functionalization decreases the uphill thermodynamic barrier towards the hydrogen adsorption in the fullerene cage.17 Notably, the catalytic activity of P-C60 surpasses that of the H-C60, reflecting the importance of the group, as well as the degree of functionalization and symmetry.17 Remarkably, P-C60, with an onset potential of −45 mV vs. RHE, an overpotential of 75 mV to reach 10 mA cm−2 and a high current density of 160 mA cm−2 at −0.6 V vs. RHE, outperforms the HER activity of the state-of-the-art metal-free carbon nanostructure electrocatalysts (Tables S1 and Supporting information 4, ESI†). The superior catalytic activity of the P-C60 can be associated with the unique distribution of ester-pyrrolidine motif in the carbon cage, which activates the nearest carbon atoms at the C60-ligand interfaces, thus creating new highly active centers, enhancing the adsorption of catalytic intermediates, and improving the catalytic rates for the HER. Tafel plots provide insights into the HER mechanisms of the functionalized C60 surfaces, with the penta-C60 adduct displaying a Tafel slope of 48 mV dec−1 (Fig. 2B). It is worth pointing out that the Tafel slope of the functionalized C60 molecules is by far lower than that of pristine C60, thus implying a more efficient HER pathway, possibly the Volmer–Heyrovsky mechanism, and an improved kinetic efficiency upon the fullerene functionalization.
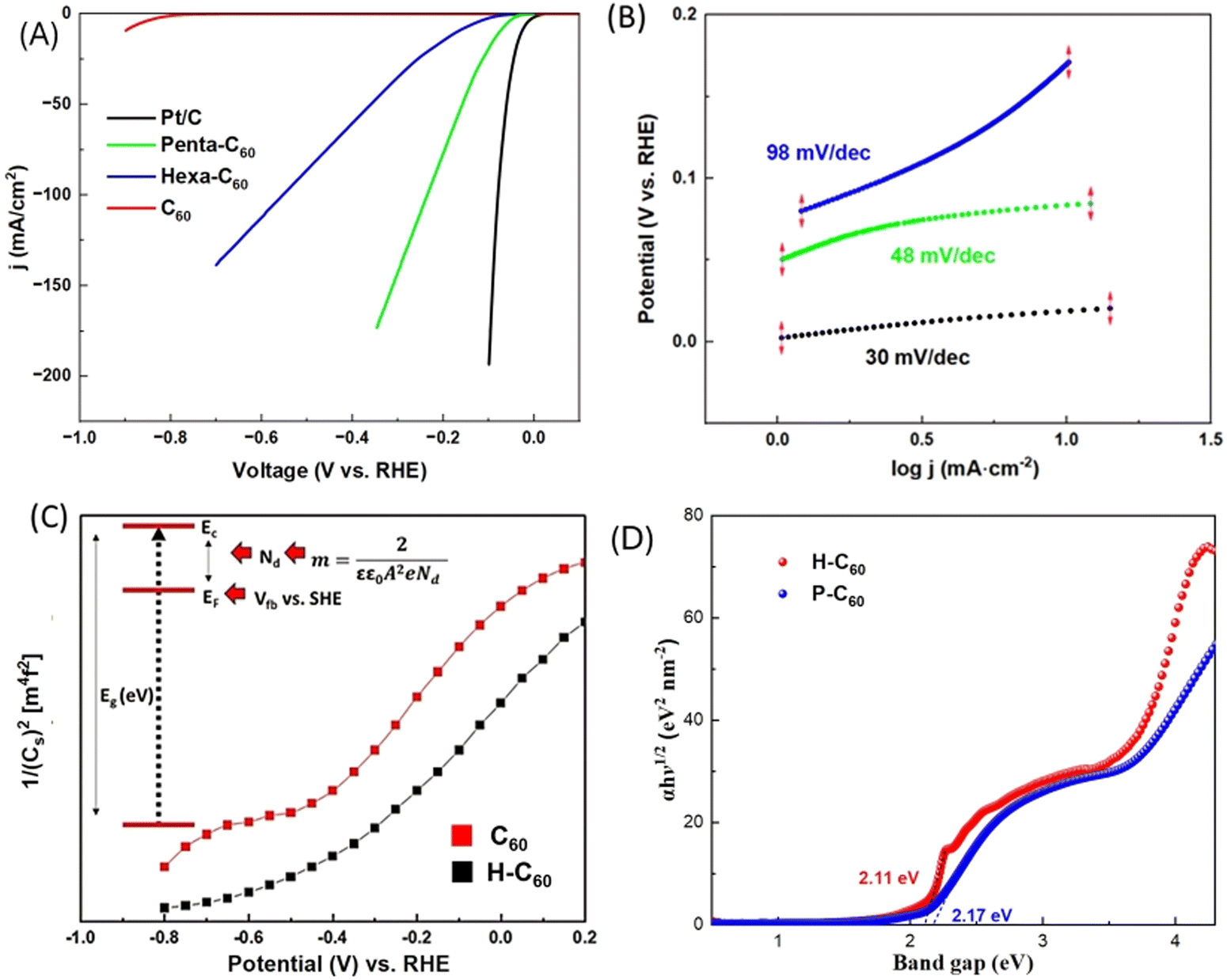 | ||
| Fig. 2 (A) Hydrogen evolution linear sweep voltammetry profile of pristine C60, hexa adduct of C60, H-C60 and penta adduct P-C60 the benchmark Pt/C. (B) Tafel slope of the optimum penta adduct C60 and Pt/C; (C) Mott–Schottky analysis of C60 and H-C60. (D) DRS profile of the hexa (red) and penta (blue) adduct of C60. | ||
Furthermore, the P-C60 demonstrates remarkable electrochemical stability in acidic conditions, maintaining 97% of the initial current density after 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000s, as evidenced in Fig. S5 and Supporting information 4 (ESI†). Mott–Schottky (M–S) and diffuse reflectance spectroscopy (bandgap) analyses were conducted to assess the charge carrier densities (Nd) for the HER of the electrochemical materials (Fig. 2C and D).
000s, as evidenced in Fig. S5 and Supporting information 4 (ESI†). Mott–Schottky (M–S) and diffuse reflectance spectroscopy (bandgap) analyses were conducted to assess the charge carrier densities (Nd) for the HER of the electrochemical materials (Fig. 2C and D).
The well-established inverse relationship between the M–S and Nd suggests that a decrease in slope corresponds to an increase in Nd, thereby enhancing the charge carrier densities and consequently, the electron transfer kinetics. The P-C60 exhibited a lower slope than non-functionalized C60 fullerenes, suggesting that functionalization improves the charge carriers, significantly boosting the charge transfer at the M–S interfaces. Furthermore, the flat band potential shifted from −0.6 V for pristine C60 to −0.4 V for the P-C60, suggesting a higher electron density near the Fermi level in the case of the P-C60. Additionally, XPS experiments were carried out before and after a chronoamperometric experiment with the F-C60 (Fig. S6 and Supporting information 5, ESI†). Ex situ XPS after the chronoamperometry experiment shows a slight shift in the N 1s band most likely due to the electron transfer processes in the C60-addend interface (Fig. S6b, ESI†). The faradaic efficiency (FE) of P-C60 catalysts was also determined by dividing the measured H2 produced by the expected amount based on the charge passed during controlled-potential electrolysis measurements. For longer 24 hour experiments, the FE was found to be 89%, a promising value for a metal-free HER catalyst.19 (see Table S2 and Supporting information 5, ESI†).
To unlock the effects of the ligand functionalization strategy in the HER electrocatalytic properties of C60 fullerenes and gain an in-depth mechanistic understanding, we employed density functional theory (DFT) calculations. We first calculated the electronic properties of H-C60 and P-C60 adducts. The density of states of the H-C60 and P-C60 molecules was studied using different link groups (Fig. 3A and D). For the H-C60 adduct, the group-1 has a higher contribution to the C60 compared to the group-2 (Fig. 3A). The density of states near the Fermi level was mainly contributed by the 2p orbitals of C and N atoms (Fig. 3G). Moreover, the 2p orbital of O atoms from the addends is hybridized with the 2p orbital of C atoms in the cage, indicating that the addends exhibit a strong interaction with the C60 cage. The P-C60 adduct shows the same trend (Fig. 3D and H).
 | ||
| Fig. 3 The calculated density of states and charge density difference maps of different ligands for H-C60 (A)–(C) and P-C60 (D)–(F); the calculated density of states for different elements contribution of H-C60 (G) and P-C60 (H); (I) the calculated Gibbs free energies for different C sites of H-C60 and penta-C60, the pristine C60 was included for comparison (the iso-surface value is 0.0015 e Å−3). | ||
We further calculated the charge distribution of the H-C60 and P-C60 to uncover the charge change of the C atoms at the C60-addend interfaces. The calculated Bader analysis shows that there are significant electron transfers between the ligands and C60 atoms. Depending on the distance from the functional group, the C atoms of C60 will gain or lose different electrons. For the P-C60, the first nearest C atoms that are directly connected to the functional group will gain 0.02–0.12 electrons from the functional groups. The second nearest C atoms may gain or lose electrons according to their distance from the other functional groups. For H-C60, the first nearest C atoms that are directly connected to the functional group lose 0.08 electrons while the second nearest C atoms all gain 0.01–0.3 electrons. The C atoms of pristine C60 are not active for HER due to the weak interaction with the H.20
After the functionalization, the electronic properties of C atoms that are near the addends were drastically affected. There are two different types of addends for the H-C60 and P-C60 adducts (pyridine and ethyl ester groups, respectively). We have calculated the charge density difference maps of H-C60 and P-C60 for each type of addend, respectively. There is a charge transfer process from the ligands to the C atoms of the C60 cages (Fig. 3B, C and E, F). The calculated Bader analysis shows that the ligands release electrons and the C atoms near the ligands gain electrons, thus promoting an intramolecular electron transfer phenomenon. Moreover, the charge distribution of C atoms around the functional groups on the C60 cage can also influence the H absorption strength during the electrocatalytic reaction. To verify this prediction, the Gibbs free energy values of the absorbed H on different C sites were calculated. As expected, the ideal HER catalyst requires a value of ΔGH* close to zero. According to the distance between the C atoms of C60 and the ligands, two types of catalytic sites, including the first and second nearest C atoms, were investigated (Fig. S7, S8 and Supporting information 6, ESI†). The results show that the first nearest C atoms were the most active sites for HER with Gibbs free energies of −0.28 and 0.27 eV for H-C60 and P-C60, respectively. As the distance between the C atoms and ligands increases, the Gibbs free energy of the absorbed H becomes more positive (0.42 and 0.37 eV for H-C60 and P-C60, which is close to the 0.44 eV of pristine C60), suggesting a substantial change in the HER efficiency (Fig. 3I).
In summary, we have disclosed the principal factors that control the intermolecular electron transfer at the C60-addend interface and their implication for the molecular generation of hydrogen. The C60-addend interface can finely tailor the nearest carbon atoms' electronic structure and decrease the H adsorption's thermodynamic barrier, thus promoting the overall catalytic activity. We have also proven the importance of the pyrrolidine addends, being more active in the ester groups when compared with those from the pyridine. This work offers a unique approach to the rational design of functionalized fullerene-based molecular catalysts for efficient electrochemical hydrogen evolution.
Data availability
The authors confirm that the data supporting the findings of this work is available within the article and its ESI.† The raw data is also accessible from the corresponding author upon reasonable request.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
MN acknowledges the financial support provided by the Partnership for Research and Education in Materials (PREM) by the U. S. National Science Foundation (NSF) (NSF PREM: award # 1827820). MN acknowledges the financial support from the new investigator award (Virginia Space Center and NASA). CD acknowledges the financial support provided by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Heavy Element Chemistry program under Award Number DE-SC0023050.References
- X. Cui, M. Wu, X. Liu, B. He, Y. Zhu, Y. Jiang and Y. Yang, Chem. Soc. Rev., 2024, 53, 1447–1494 RSC.
- D. Zhang, Z. Wang, F. Liu, P. Yi, L. Peng, Y. Chen, L. Wei and H. Li, J. Am. Chem. Soc., 2024, 146, 3210–3219 CrossRef CAS PubMed.
- A. R. Puente Santiago, M. F. Sanad, A. Moreno-Vicente, M. A. Ahsan, M. R. Cerón, Y.-R. Yao, S. T. Sreenivasan, A. Rodriguez-Fortea, J. M. Poblet and L. Echegoyen, J. Am. Chem. Soc., 2021, 143, 6037–6042 CrossRef CAS PubMed.
- J. Durst, A. Siebel, C. Simon, F. Hasché, J. Herranz and H. A. Gasteiger, Energy Environ. Sci., 2014, 7, 2255–2260 RSC.
- Y. Lu, L. Tang, P. Wang, M. He, C. Yang and Z. Li, ACS Catal., 2023, 13, 13804–13815 CrossRef CAS.
- Z. Jiang, W. Zhou, A. Hong, M. Guo, X. Luo and C. Yuan, ACS Energy Lett., 2019, 4, 2830–2835 CrossRef CAS.
- H. Huang, M. Yan, C. Yang, H. He, Q. Jiang, L. Yang, Z. Lu, Z. Sun, X. Xu, Y. Bando and Y. Yamauchi, Adv. Mater., 2019, 31, 1903415 CrossRef CAS.
- S. Pal, M. Sahoo, V. T. Veettil, K. K. Tadi, A. Ghosh, P. Satyam, R. K. Biroju, P. M. Ajayan, S. K. Nayak and T. N. Narayanan, ACS Catal., 2017, 7, 2676–2684 CrossRef CAS.
- M. Batabyal, S. Jaiswal, R. K. Jha and S. Kumar, J. Am. Chem. Soc., 2024, 146, 57–61 Search PubMed.
- A. A. Feidenhans’l, Y. N. Regmi, C. Wei, D. Xia, J. Kibsgaard and L. A. King, Chem. Rev., 2024, 124, 5617–5667 CrossRef PubMed.
- K. Muñoz-Becerra, F. J. Recio, R. Venegas and J. H. Zagal, Curr. Opin. Electrochem., 2023, 42, 101387 Search PubMed.
- A. R. Puente Santiago, T. He, O. Eraso, M. A. Ahsan, A. N. Nair, V. S. N. Chava, T. Zheng, S. Pilla, O. Fernandez-Delgado, A. Du, S. T. Sreenivasan and L. Echegoyen, J. Am. Chem. Soc., 2020, 142, 17923–17927 Search PubMed.
- L. Song, T. Li and S. Zhang, J. Phys. Chem. C, 2017, 121, 293–299 CrossRef CAS.
- A. C. Bevilacqua, M. H. Köhler and P. C. Piquini, J. Phys. Chem. C, 2019, 123, 20869–20876 Search PubMed.
- M. F. Sanad, H. M. Franklin, B. A. Ali, A. R. Puente Santiago, A. N. Nair, V. S. N. Chava, O. Fernandez-Delgado, N. K. Allam, S. Stevenson, S. T. Sreenivasan and L. Echegoyen, Angew. Chem., Int. Ed., 2022, 61, e202116727 CrossRef CAS PubMed.
- W. Yang, Q. Huang, Y. Yan, Y. Li, T. Xu, A. Yu, Y. Zhao, P. Peng, Y. Wang, L. Echegoyen and F.-F. Li, Angew. Chem., Int. Ed., 2024, e202414149 Search PubMed.
- E. Castro, K. Azmani, A. H. Garcia, A. Aghabali, S. Liu, A. J. Metta-Magana, M. M. Olmstead, A. Rodríguez-Fortea, J. M. Poblet and L. Echegoyen, Chem. – Eur. J., 2017, 23, 15937–15944 Search PubMed.
- E. Castro, K. Azmani, A. H. Garcia, A. Aghabali, S. Liu, A. J. Metta-Magana, M. M. Olmstead, A. Rodríguez-Fortea, J. M. Poblet and L. Echegoyen, Chem. – Eur. J., 2017, 23, 15841 Search PubMed.
- M. Cai, L. Xu, J. Guo, X. Yang, X. He and P. Hu, J. Mater. Chem. A, 2024, 12, 592–612 Search PubMed.
- T. He, G. Gao, L. Kou, G. Will and A. Du, J. Catal., 2017, 354, 231–235 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic information and 1H- and 13C-NMR spectra for P-C60 and H-C60. See DOI: https://doi.org/10.1039/d4nh00586d |
| This journal is © The Royal Society of Chemistry 2025 |