
Sustainable, precious-metal-free C–N cross coupling through photocatalysis†
Mohammad
Hassam
 *,
Samuelu
Mamidipalli
,
Akhila
Ailaveni
,
Pankajkumar
Singh
and
Shambabu Joseph
Maddirala
*,
Samuelu
Mamidipalli
,
Akhila
Ailaveni
,
Pankajkumar
Singh
and
Shambabu Joseph
Maddirala
Chemveda Life Sciences Pvt Ltd, Plot No. B-11/1, IDA, Uppal, Hyderabad 500039, Telangana, India. E-mail: mohammad.hassam@chemvedals.com
First published on 4th February 2025
Abstract
Photoredox catalysis has evolved as a sustainable method of constructing C–N bonds. Herein, we have reported a visible-light organic-photoredox-catalyzed method that enables C–N cross-coupling under mild and sustainable reaction conditions. This catalytic system is suitable for both aromatic and aliphatic amines, as well as electron-rich and -deficient aryl bromides, exhibits broad functional group tolerance, and provides good-to-excellent yields. The noteworthy aspects of milder reaction conditions at room temperature without the addition of any additional ligands make this procedure attractive.
Nitrogen-containing organic structure units constitute the basic building blocks of life, such as proteins, DNA, and RNA. Moreover, most natural products, pharmaceuticals and agrochemicals require a nitrogen-containing moiety for their particular activity (Fig. 1).1 Considering the wide-spread importance of arylated/aliphatic amines in pharmaceuticals, numerous metal-mediated methodologies have been developed during the past three decades for C–N bond formation. Major development has been observed in the Pd-catalysed C–N cross coupling of amines with aryl halides to form N-substituted anilines.2 Recently, aminative Suzuki–Miyaura coupling was reported, which involved nitrene insertion for C–N bond formation.3 In most cases, Pd-catalysed methods depend on sterically hindered, expensive ligands and elevated temperatures; these harsh conditions sometimes cause undesired outcomes, such as deprotection of the protecting group, hydrolysis, charring and the possibility of the formation of color impurities.4 In addition to these limitations, removal of trace Pd metal from active pharmaceutical ingredients is another challenge.5 The development of greener and more convenient ways to form C–N bonds has become one of the hottest research goals in synthetic chemistry to overcome these challenges.6
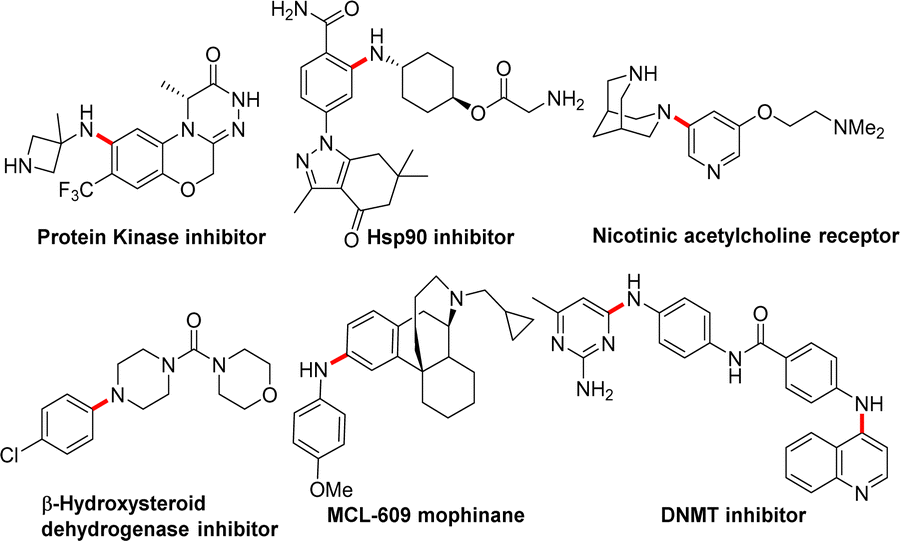 | ||
| Fig. 1 Involvement of Pd-mediated C–N cross-coupling in potential pharmaceutical compounds. | ||
One of the prime requirements in chemical processes is reducing or eliminating the use or formation of hazardous substances. Methods based on green chemistry principles7 are being embraced to meet this requirement. Photochemistry-based applications, such as photoredox catalysis, wherein redox reactions are accomplished in a catalytic manner, have become methodologies8 of choice for achieving challenging carbon–heteroatom bond formations under mild conditions,9 making this application a greener, more cost-effective and more-sustainable one. Photoredox catalysis, especially in the last decade, has become one of the trusted options to make new molecules by over-coming the limitations of the past, and impressive methodologies have been developed. Photoredox catalysis, in recent times, has also emerged as a highly efficient and mild strategy for late-stage diversification and access to synthetically challenging bioisosteres.10 Asymmetric photoredox catalysis that enables enantioselective versions of useful methods for chemical synthesis, is gaining momentum to harness the stereoselectivity that is possible in this platform.11 Efforts/explorations in appending flow chemistry to photoredox catalysis research are in progress for the adoption photoredox catalysis to achieve carbon–heteroatom couplings on a large scale.12
Metal-based photocatalysts, mainly the polypyridine complexes of Ru(II) and Ir(III), have been explored extensively as visible-light photocatalysts with good success. However, the environmental issues related to these applications of rare metals have led to the consideration of alternatives to these metal-based photocatalysts. Organic dyes have been found to be economical and effective visible-light photoredox organocatalysts (PROC), wherein the photogenerated radical ion of the dye can promote a chemical reaction. The low cost and ease of preparation of these dyes have made them attractive alternatives to transition metal complexes; in some cases, even better photocatalytic performances with respect to their metal counterparts have been observed.13
Furthermore, the synergistic action of photoredox catalysis with other catalytic processes has a profound impact on the reactivity profiles of many traditional synthetic routes.14 Numerous organic molecules, such as methylene blue, rose bengal, eosin Y, acridinium salts, and cyanoarenes, have been reported as organic photocatalysts for organic transformations.15 Application of organic photocatalysts such as phenoxazine, anthrazoline, and dihydrophenazine for C–N cross coupling between aromatic halides with aromatic/aliphatic amines has also been reported (Scheme 1).16
 | ||
| Scheme 1 Earlier photoredox approaches for C–N cross coupling. | ||
C–N bond formation under extremely mild conditions with high functional group tolerance is a need in the pharmaceutical industry. To facilitate a library project on anilines under photoredox conditions, we wanted to have a photoredox catalyst that could work well, preferably providing good yields, for a wider substrate scope with regards to both aryl halides and the primary and secondary amines (both cyclic and acyclic).
Herein, we report a general and attractive methodology for C–N cross-coupling using inexpensive, easily synthesizable 4DPAIPN as a photoredox catalyst. To realize the viability of the reaction, a model reaction employing 2-bromonaphthalene and p-toluidine was carried out using 450 nm blue light. Initially, we screened different organic dyes at room temperature for 12 h; dicyanoarene-based 4DPAIPN emerged as the most promising one for the coupling with an excellent yield of 82% (Table 1, entry 2). Other dicyanoarene-based organic dyes (4CzPIN, 3DPA2FBN, dicyanoanthracene) were found to be the next-most effective ones and gave the product in good yields (Table 1, entry 1, 3, 4). The remaining organic dyes were unable to generate any significant impact on product formation (Table 1, entry 5–8). Moreover, when the reaction was performed without 4DPAIPN, there was no coupling, which showed the significant role of DPAIPN in the C–N cross coupling (Table 1, entry 9). Encouraged by this result, we next studied the impact of different Ni co-catalysts, such as NiBr2·dme, Ni(acac)2, and Ni(OAc)2·4H2O. In the absence of these Ni co-catalysts, there was no product formation, indicating the involvement of Ni in the catalytic cycle (Table 1, entry 10). Reactions using NiBr2·dme and NiCl2 gave the desired product, but the yield of the product was slightly lower (Table 1, entry 12 and 11). Other Ni-based co-catalysts showed product formation with yields in the range of 52–60%. (Table 1, 13 and 14). After finding the suitable combination of photocatalyst and metal source, we next set out to optimize the number of equivalents of DPAIPN and NiCl2·dme required for this C–N cross coupling. It was observed that a combination of 0.5 mol% of DPAIPN and 5 mol% NiCl2·dme was best for the catalytic activity. A decrease in the catalytic loading affected the product formation; even after longer reaction time, the consumption of the starting material was not complete (Table S1, ESI†). We then studied the photocatalysis reaction at different wavelengths and found that optimal yield was observed at 450 nm, followed by 427 nm. Using shorter wavelengths (370 nm), there was no product formation,17 suggesting that the coupling is driven by electronic factors as well. (Table S2, ESI†). Furthermore, solvent screening studies indicated DMA to be the best solvent for this catalysis, and the next-best solvent was ACN (Table S3, ESI†). The reaction did not proceed in other common solvents such as EtOAc, DMSO, DCM, and MeOH. Among the different bases screened, DABCO gave the corresponding product in encouraging yields, while with pyridine and DBU there was no reaction (Table S4, ESI†).
|
|
||
|---|---|---|
| SN | Deviation from standard conditions | Yieldb (%) |
| a Standard reaction conditions: 4DPAIPN (0.5 mmol%), NiCl2·dme (5 mol%), DABCO (2 equiv.), DMA (10 vol%), 450 nm blue LED, ambient temperature, 12 h. b Isolated yield. NR: no reaction | ||
| 1 | 4CzPIN | 49 |
| 2 | None | 82 |
| 3 | 3DPA2FBN | 44 |
| 4 | 9,10-Anthracenedicarbonitrile | 16 |
| 5 | Methylene blue | NR |
| 6 | Eosin-Y | NR |
| 7 | Rose Bengal | NR |
| 8 | 12-Phenyl-10H-phenothiazine | NR |
| 9 | No photocatalyst | NR |
| 10 | No NiCl2·dme | NR |
| 11 | NiCl2 | 72 |
| 12 | NiBr2·dme | 75 |
| 13 | Ni(acac)2 | 52 |
| 14 | Ni(OAc)2·4H2O | 60 |

To examine the scope of the reaction, we utilized various aryl bromo partners with different functional groups. Substrates bearing electron-donating (5) or -withdrawing (7–8) groups on the bromoarene gave the corresponding products in good-to-excellent yields. Substrates bearing an electron-withdrawing group on the aryl amine (15–18) coupled efficiently with bromoarene. Chiral aliphatic amines yielded the corresponding product without loss of the chirality (22–23). Heteroaryl bromides, such as those of pyridine, thiophene, quinoline, isoquinoline, and benzothiophene (9–15), gave the corresponding anilines in 49–56% yield under the developed reaction conditions. To our delight, the present protocol is also effective for the coupling of aryl bromides with unreactive primary and secondary aliphatic amines, such as cyclohexylamine, cyclopentylamine, benzylamine, phenylethylamine, phenylpropylamine, octylamine, azetidine, pyrrolidine, piperidine, morphine, and piperazine (24–38), which gave the corresponding arylalkylamines in modest-to-good yields (Fig. 2).
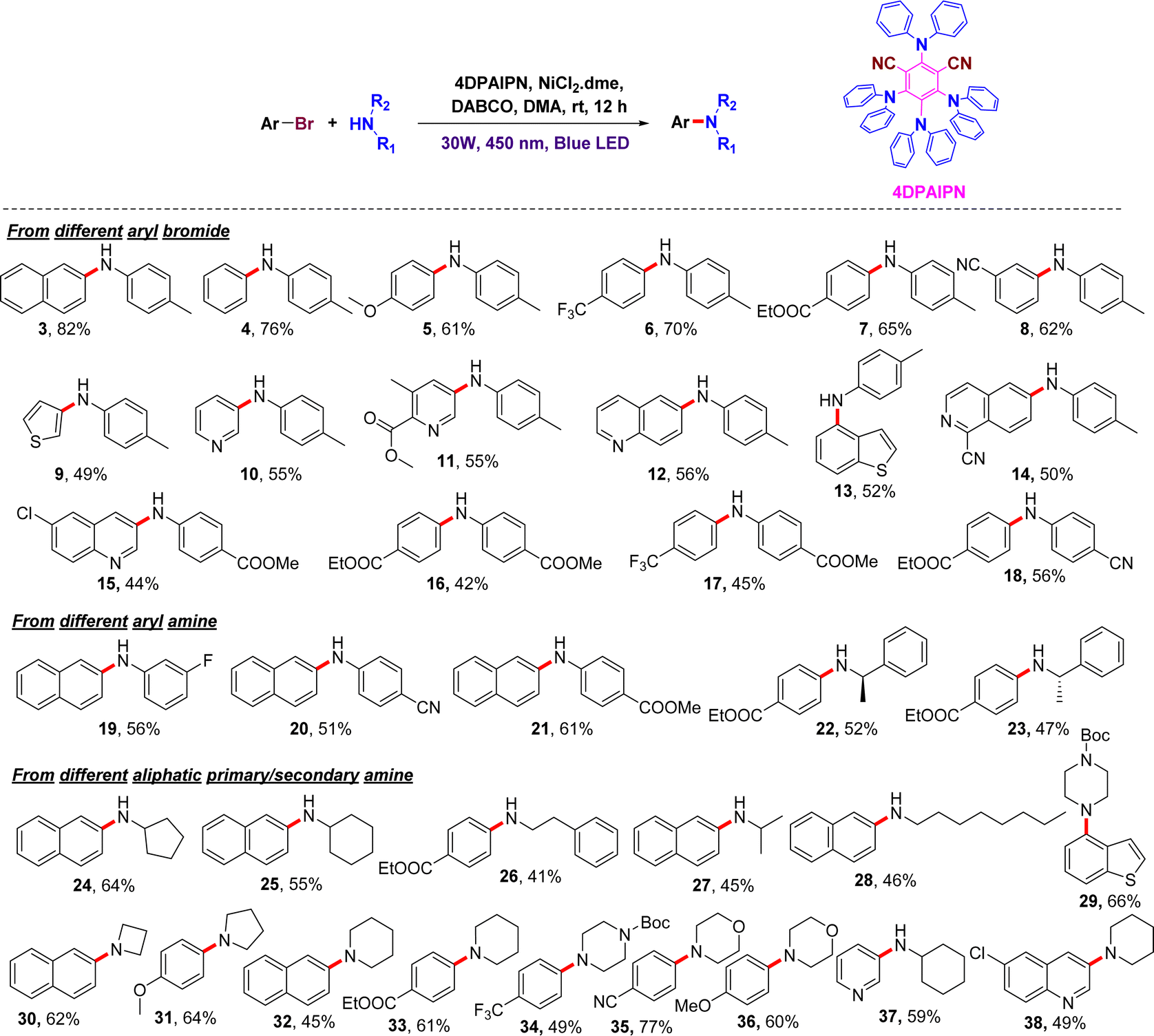 | ||
| Fig. 2 Scope of the different substrates under the photoredox C–N cross-coupling. | ||
The simplicity of the reaction conditions enticed us to check the scalability of these reaction conditions; a gram-scale reaction using 2-bromo-1-trifluromethyl benzene to obtain niflumic acid, a nonsteroidal anti-inflammatory drug (NSAID), was attempted. The reaction proceeded smoothly with just 0.002 mol% of 4-DPAIPN, affording niflumic acid in 80% isolated yield at room temperature (Scheme 2).
 | ||
| Scheme 2 Gram-scale synthesis of niflumic acid. | ||
After successfully establishing the methodology for the formation of various C–N coupled products, we extended this protocol for the synthesis of active pharmaceutical ingredients by treating the respective aryl halides and aryl/aliphatic amines; products such as mefenamic acid, meclofenac, niflumic acid, brexipiprazole, melatonin receptor ligand, β-hydroxysteroid dehydrogenase inhibitor, and BRAF inhibitors were isolated in 43–80% yields (Fig. 3).
 | ||
| Fig. 3 Synthetic application of 4DPAIPN in the synthesis of drug molecule/active pharmaceutical ingredients. | ||
A control experiment incorporating a free-radical scavenger (TEMPO) into the standard reaction conditions was performed to determine the mechanism of this catalysis (Scheme 3). We isolated the TEMPO adduct in 6% yield (see ESI†). The formation of the TEMPO adduct without the formation of the expected coupled product suggests the involvement of a free radical in the reaction process (Scheme 3). A plausible reaction mechanism for the photoredox cross-coupling is presented in Fig. 4.16
 | ||
| Scheme 3 Control experiment for free radical trapping. | ||
 | ||
| Fig. 4 General proposed mechanism for C–N cross coupling. | ||
Upon excitation with visible light, 4-DPAIPN is promoted from the ground state to the excited singlet state 4-DPAIPN*. The reduction of Ni(II) to Ni(0) may occur through single-electron transfer from 4DPAIPN. The general mechanism follows with the oxidative addition of the aryl halide on the Ni(0) complex to give a Ni(II) aryl halide complex, which couples with the amine to result in the corresponding Ni(III) complex C. The Ni(III) complex C undergoes hydrogen atom transfer with the DABCO, resulting in Ni(II) complex D, which transfers the single electron to 4DPAIPN* to result in Ni(III) complex E. The Ni(III) complex E undergoes reductive elimination, resulting in the C–N cross-coupled product.
In conclusion, a sustainable, milder, and step-economic photocatalytic C–N cross-coupling procedure was developed using a cost-effective organic dye. The 4DPAIPN-based catalytic system showed broad applicability to a wide range of aryl bromides bearing different functional groups, such as electron-donating, electron-withdrawing, and heterocyclic groups, which coupled efficiently with aryl amines, as well as primary and secondary amines, at room temperature without additional ligands. Hopefully, this methodology will be helpful as an alternative to palladium-catalysed C–N cross-coupling in drug discovery efforts.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
M. H. thanks the analytical Department, Chemveda Life Sciences for continuous support. The authors thank Dr Bheemarao Paraselli, CEO, Chemveda Life Sciences for the encouragement. This manuscript is dedicated to Prof. Willem van Otterlo on the occasion of his 50th birthday.Notes and references
- (a) E. Vitaku, D. T. Smith and J. T. Njardarson, Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed; (b) A. E. Ricci, Amino Group Chemistry: From Synthesis to the Life Sciences, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed; (c) P. Bhutani, G. Joshi, N. Raja, N. Bachhav, P. K. Rajanna, H. Bhutani, A. T. Paul and R. Kumar, U.S. FDA Approved Drugs from 2015–June 2020: A Perspective, J. Med. Chem., 2021, 64, 2339–2381 CrossRef CAS PubMed.
- (a) J. Bariwal and E. V. D. Eycken, C–N bond forming cross-coupling reactions: an overview, Chem. Soc. Rev., 2013, 42, 9283–9303 RSC; (b) P. R. Castillo and S. L. Buchwald, Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions, Chem. Rev., 2016, 116, 12564–12649 CrossRef PubMed; (c) R. Emadi, A. B. Nekoo, F. Molaverdi, Z. Khorsandi, R. Sheibani and H. R. Aliabadi, Applications of palladium-catalyzed C–N cross-coupling reactions in pharmaceutical compounds, RSC Adv., 2023, 13, 18715–18733 RSC.
- P. Onnuch, K. Ramagonolla and R. Y. Liu, Aminative Suzuki-Miyaura coupling, Science, 2024, 383, 1019–1024 CrossRef CAS PubMed.
- R. Fan, M. Kua, D. Lin and W. Fang, A General C–N Cross-Coupling to Synthesize Heteroaryl Amines Using a Palladacyclic N-Heterocyclic Carbene Precatalyst, Org. Lett., 2022, 24(47), 8688–8693 CrossRef CAS PubMed.
- (a) W. P. Gallagher and A. Vo, Dithiocarbamates: Reagents for the Removal of Transition Metals from Organic Reaction Media, Org. Process Res. Dev., 2015, 19, 1369–1373 CrossRef CAS; (b) M. Chatzopoulou, K. S. Madden, L. J. Bromfead, C. Greaves, T. J. Cogswell, S. D. S. Pinto, S. R. G. Galan, I. Georgiou, M. S. Kennedy, A. Kennet, G. Apps, A. J. Russel and G. S. Wynne, Pilot Study to Quantify Palladium Impurities in Lead-like Com-pounds Following Commonly Used Purification Techniques, ACS Med. Chem. Lett., 2022, 13, 262–270 CrossRef CAS PubMed; (c) M. Economidou, N. Mistry, K. M. P. Wheelhouse and D. M. Lindsay, Palladium Extraction Following Metal-Catalyzed Reactions: Recent Advances and Applications in the Pharmaceutical Industry, Org. Process Res. Dev., 2023, 27, 1585–1615 CrossRef CAS.
- (a) E. B. Corcoran, M. T. Pirnot, S. Lin, S. D. Dreher, D. A. DiRocco, I. W. Davies, S. L. Buchwald and D. W. MacMillan, Aryl amination using ligand-free Ni(II) salts and photoredox catalysis, Science, 2016, 353, 279–283 CrossRef CAS PubMed; (b) P. Li, J. A. Terrett and J. R. Zbieg, Visible-Light Photocatalysis as an Enabling Technology for Drug Discovery: A Paradigm Shift for Chemical Reactivity, ACS Med. Chem. Lett., 2020, 11, 2120–2130 CrossRef CAS PubMed; (c) S. Singh, V. J. Roy, N. Dagar, P. P. Sen and S. R. Roy, Photocatalysis in Dual Catalysis Systems for Carbon-Nitrogen Bond Formation, Adv. Synth. Catal., 2020, 363, 937–979 CrossRef; (d) M. Akita, P. Ceroni, C. R. J. Stephenson and G. Masson, Progress in Photocatalysis for Organic Chemistry, J. Org. Chem., 2023, 88, 6281–6283 CrossRef CAS PubMed; (e) V. Srivastava, P. K. Singh and P. P. Singh, Recent advances of visible-light photocatalysis in the functionalization of organic compounds, J. Photochem. Photobiol., 2022, 50, 100488 CrossRef CAS.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, NY, USA, 1998; p. 30 Search PubMed.
- L. Peijun, A. J. A. Terrett and J. R. Zbieg, Visible-Light Photocatalysis as an Enabling Technology for Drug Discovery: A Paradigm Shift for Chemical Reactivity, ACS Med. Chem. Lett., 2020, 11, 2120–2130 CrossRef PubMed.
- (a) M. H. Shaw, J. Twilton and D. W. C. MacMillan, Photoredox Catalysis in Organic Chemistry, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed; (b) J. M. R. Narayanam and C. R. J. Stephenson, Visible light photoredox catalysis: applications in organic synthesis, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (c) J. Xie, H. Jin and A. S. K. Hashmi, The recent achievements of redox-neutral radical C–C cross-coupling enabled by visible-light, Chem. Soc. Rev., 2017, 6, 5193–5203 RSC; (d) C.-S. Wang, P. H. Dixneuf and J. F. Soulé, Chem. Rev., 2018, 118, 7532–7585 CrossRef CAS PubMed; (e) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed; (f) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis, Chem. Rev., 2013, 113, 5322–5563 CrossRef CAS PubMed; (g) J. Xuan and W. J. Xiao, Visible-light photoredox catalysis, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS PubMed.
- A. Denisenko, P. Garbuz, S. V. Shishkina, N. M. Voloshchuk and P. K. Mykhailiuk, Saturated Bioisosteres of ortho-Substituted Benzenes, Angew. Chem., Int. Ed., 2020, 59, 20515–20521 CrossRef CAS PubMed.
- (a) E. Meggers, Asymmetrical catalysis activated by visible light, Chem. Comm., 2015, 51, 3290–3301 RSC; (b) S. Das, C. Zhu, D. Demirbas, E. Bill, C. K. De and B. List, Asymmetric counter anion-directed photoredox catalysis, Science, 2023, 379, 494–499 CrossRef CAS PubMed.
- F. Politano and G. Oksdath-Mansilla, Light on the Horizon: Current Research and Future Perspectives in Flow Photochemistry, Org. Process Res. Dev., 2018, 22, 1045–1062 CrossRef CAS.
- (a) I. K. Sideri, E. Voutyritsa and C. G. Kokotos, Small Organic Molecules and Light in the Service of Organic Syn-thesis: The Awakening of a Sleeping Giant, Org. Biomol. Chem., 2018, 16, 4596–4614 RSC; (b) Y. Zhang, W. Schilling and S. Das, Metal-Free Photocatalysts for C-H Bond Oxygenation Reactions with Oxygen as the Oxidant, ChemSusChem, 2019, 12, 2898–2910 CrossRef CAS PubMed; (c) S. G. E. Amos, M. Garreau, L. Buzzetti and J. Waser, Photocatalysis with Organic Dyes: Facile Access to Reactive Inter-mediates for Synthesis, Beilstein J. Org. Chem., 2020, 16, 1163–1187 CrossRef CAS PubMed; (d) D. A. Nicewicz and T. M. Nguyen, Recent Applications of Organic Dyes as Photoredox Catalysts in Organic Synthesis, ACS Catal., 2014, 4, 355–360 CrossRef CAS; (e) D. Ravelli and M. Fagnoni, Dyes as Visible Light Photoredox Organocatalysts, ChemCatChem, 2012, 4, 169–171 CrossRef CAS.
- (a) M. S. Oderinde, N. H. Jones, A. Juneau, M. Frenette, B. Aquila, S. Tentarelli, D. W. Robbins and J. W. Johannes, Highly Chemoselective Iridium Photoredox and Nickel Catalysis for the Cross-Coupling of Primary Aryl Amines with Aryl Halides, Angew. Chem., Int. Ed., 2016, 55, 13219–13223 CrossRef CAS PubMed; (b) E. B. Corcoran, M. T. Pirnot, S. Lin, S. D. Dreher, D. A. Dirocco, I. W. Davies, S. L. Buchwald, D. W. C. Macmillan, S. L. Buchwald and D. W. C. Macmillan, Aryl Amination Using Ligand-Free Ni(II) Salts and Photoredox, Catal. Sci., 2016, 353, 279–283 CAS; (c) M. S. Oderinde, N. H. Jones, A. Juneau, M. Frenette, B. Aquila, S. Tentarelli, D. Robbins and J. W. Johannes, Iridium Photoredox and Nickel Catalysis for the Cross-Coupling of Primary Aryl Amines with Aryl Halides, Angew. Chem., Int. Ed., 2016, 55, 13219–13223 CrossRef CAS PubMed; (d) R. J. Key and A. K. Vannucci, Nickel Dual Photoredox Catalysis for the Synthesis of Aryl Amines, Organometallics, 2018, 37, 1468–1472 CrossRef CAS; (e) B. Konig, Photocatalysis in Organic Synthesis – Past, Present, and Future, Eur. J. Org. Chem., 2017, 1979–1981 CrossRef; (f) J. D. Bell and J. A. Murphy, Recent advances in visible light-activated radical coupling reactions triggered by (i) ruthenium, (ii) iridium and (iii) organic photoredox agents, Chem. Soc. Rev., 2021, 50, 9540–9685 RSC.
- (a) S. Senapati, S. K. Parida, S. S. Karandikar and S. Murarka, Organophotoredox-Catalyzed Arylation and Aryl Sulfonylation of Morita−Baylis−Hillman Acetates with Diaryliodonium Reagents, Org. Lett., 2023, 25, 7900–7905 CrossRef CAS PubMed; (b) Z. Yu, J. Zhao, F. Yang, X. Tang, Y. Wu, C. Ma, B. Song, L. Yun and Q. Meng, Rose bengal as photocatalyst: visible light-mediated Friedel–Crafts alkylation of indoles with nitroalkenes in water, RSC Adv., 2020, 10, 4825–4831 RSC; (c) A. Paul, A. Sengupta and S. Yadav, Organophotoredox-Catalyzed Cross-Dehydrogenative Sulfon-amidation of Indoles and Other Heterocycles, J. Org. Chem., 2023, 88, 9599–9614 CrossRef CAS PubMed.
- (a) Y. Du, R. M. Pearson, C. Lim, S. M. Sartor, M. Ryan, H. Yang, N. H. Damraur and G. M. Miyake, Strongly Reducing, Visible-Light Organic Photoredox Catalysts as Sustainable Alternatives to Precious Metals, Chem. – Eur. J., 2017, 23, 10962–10968 CrossRef CAS PubMed; (b) D. Wang, H. Huang and X. Zhu, Development of anthrazoline photocatalysts for promoting amination and amidation reactions, Chem. Commun., 2022, 58, 3529–3532 RSC; (c) D. A. Dulov and T. V. Magdesieva, N,N′-Diaryldihydrophenazines as Visible-Light Photocatalysts for Anilines’ Arylation Using a Dual Photoredox/Ni(II) Cross-Coupling Strategy, J. Org. Chem., 2023, 88, 12765–12775 CrossRef CAS PubMed; (d) F. Mohamadpour, 3DPAFIPN as a halogenated dicyanobenzene-based photosensitizer catalyzed gram-scale photosynthesis of pyrano[2,3-d]pyrimidine scaffolds, Sci. Rep., 2023, 13, 13142–13153 CrossRef CAS PubMed.
- C. Lim, M. Kudish, B. Liu and G. M. Miyake, C–N Cross-Coupling via Photoexcitation of Nickel–Amine Complexes, J. Am. Chem. Soc., 2018, 140, 7667–7673 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nj04300f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2025 |