
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnlocking hidden treasures: the evolution of high-throughput mass spectrometry in screening for cryptic natural products
Brett C.
Covington
 a and
Mohammad R.
Seyedsayamdost
*ab
a and
Mohammad R.
Seyedsayamdost
*ab
aDepartment of Chemistry, Princeton University, Princeton, New Jersey 08544, USA. E-mail: mrseyed@princeton.edu
bDepartment of Molecular Biology, Princeton University, Princeton, New Jersey 08544, USA
First published on 20th January 2025
Abstract
Covering: 1994 to 2024
Historically, microbial natural product discovery has been predominantly guided by biological activity from crude microbial extracts with metabolite characterization proceeding one molecule at a time. Despite decades of bioactivity-guided isolations, genomic evidence now suggests that we have only accessed a small fraction of the total natural product potential from microorganisms and that the products of the vast majority of biosynthetic pathways remain to be identified. Here we describe recent advancements that have enabled high-throughput mass spectrometry and comparative metabolomics, which in turn facilitate high-throughput natural product discovery. These advancement promise to fully unlock the reservoir of microbial natural products.
1. Introduction
Microorganisms have developed a diverse small molecule arsenal to sequester resources, communicate in complex communities, adapt to changing environments, and, in many cases, poison their neighbours. While the elaborate structures and potent biological activities of these compounds have fascinated chemists for more than a century, the discovery approaches for natural products (NPs) have undergone a profound transformation in recent years. Traditionally, NPs were identified either by visible pigmentation or by observable biological activities, which could guide compound purification. These isolations were typically carried out one molecule at a time. Advances in genomic analyses coupled with the implementation of state-of-the-art analytical instrumentation have ushered in new paradigms for NP discovery, where modern discovery approaches are largely guided by genomics and metabolomics and metabolite identification can be carried out at higher throughputs.Natural products are synthesized by dedicated genes typically organized contiguously in biosynthetic gene clusters (BGCs). The genomics revolution has provided a better understanding of the biosynthetic potential of microorganisms, with some bacteria harbouring upwards of 60 BGC.1 Hundreds of thousands of microbial genome sequences are available online, and several bioprospecting tools, such as antiSMASH2 and PRISM,3 are available to identify and analyse BGCs within each genome. With these tools, the molecular structures, physical properties, and the ecological functions of some NPs can be predicted before the products are even isolated.
Another realization gleaned from this wealth of genomic data is that there is a sizeable disparity between the number of predicted BGCs and the number of known NPs.4 The hidden biosynthetic capacity of Saccharopolyspora erythraea provides a representative case. This strain was cultured over several decades on massive scales all over the globe for the commercial production of the antibiotic erythromycin. When the genome of this producer was sequenced, bioinformatic analyses surprisingly revealed at least 25 ‘orphan’ BGCs coding for diverse natural products. Only four known classes of NPs, including erythromycin, had been observed at the time.5
While several factors contribute to this disparity, recent studies show that many BGCs are transcriptionally silent or sparingly expressed under standard laboratory growth protocols in which a species is grown in a monoculture in rich media.6 A number of strategies have been developed to activate these silent BGCs in the native producer;7 however, a given BGC may only respond to a small number of environmental factors. As there are countless exogenous stimuli, mutations, or combinations of conditions that can elicit natural product biosynthesis, and there is no way to predict which approach is best for a given cluster, technically demanding high-throughput approaches are often required to successfully identify the encoded ‘cryptic’ metabolite.
Recent advancements in analytical instrumentation provide a solution to this challenge. Mass spectrometry (MS) has been an essential tool for discovering novel NPs as well as identifying known ones, a process referred to as dereplication, from microbial extracts. While high-throughput MS data acquisition has been possible in a direct infusion or MALDI-MS format (see below), emerging UPLC-MS methods now offer high-throughput MS analysis including chromatographic separation. These advancements in rapid acquisition approaches have enabled high-throughput metabolomics-based natural product discovery in which hundreds of MS-derived metabolomes can be acquired and analysed in a single day. In this highlight we describe the tools that have facilitated this new discovery paradigm and provide select examples in which high-throughput MS has been particularly successful in unearthing novel NPs that would be inaccessible with traditional approaches. We will first summarize key approaches for endogenously inducing silent BGCs and subsequently assess the high-throughput MS methods that have been developed to uncover cryptic natural products.
2. Methods to activate silent biosynthetic gene clusters
Referring to microorganisms as ‘metabolic artists’, Zeeck and colleagues discovered throughout the 1990s that several NPs could be identified from a single strain through systematically altering cultivation parameters.8,9 By applying their one strain–many compounds (OSMAC) approach to 6 strains, they isolated over 100 compounds from ∼25 structural classes.9 Since this work, additional methods have been developed to activate silent BGCs. We have previously grouped the endogenous activation approaches, those that activate silent BGCs within the native producer, into three categories: (i) culture modalities, (ii) classical genetics, and (iii) chemical genetics (Fig. 1).7 The OSMAC approach is one of many culture modality methods which essentially modulate culture conditions to elicit NP biosynthesis. Other methods in this category include applications of rare earth metals,10 environmental stimuli,11 and coculture with multiple bacteria.12 The latter strategy, culturing combinations of bacteria together, is a particularly interesting though more complicated, as there are numerous mechanisms by which a competing organism can stimulate NP biogenesis. For instance, Onaka et al. demonstrated that a coculture of Streptomyces lividans TK23 and mycolic acid containing bacteria efficiently elicited undecylprodigiosin and actinorhodin (Fig. 1).13 This effect was abolished when the two bacteria were separated by a dialysis membrane, indicating the elicitation was contact-dependent. In another example, Pishchany et al. discovered a new anti-staphylococcal antibiotic, amycomicin, that was produced from a coculture between Amycolatopsis sp. AA4 and Streptomyces coelicolor M145.14 In this case, it was found that S. coelicolor converts glucose in the growth medium to galactose. Changing the carbohydrate content in the culture, a fundamental OSMAC style activation mechanism, ultimately activated amycomicin production. Overall, simplicity is the main advantages of the culture modalities category of approaches. These techniques can be readily applied to any microorganism; however, the drawback is that these strategies are untargeted and the alterations often subtle, with no guarantee that a silent BGC of interest will be activated by the selected condition.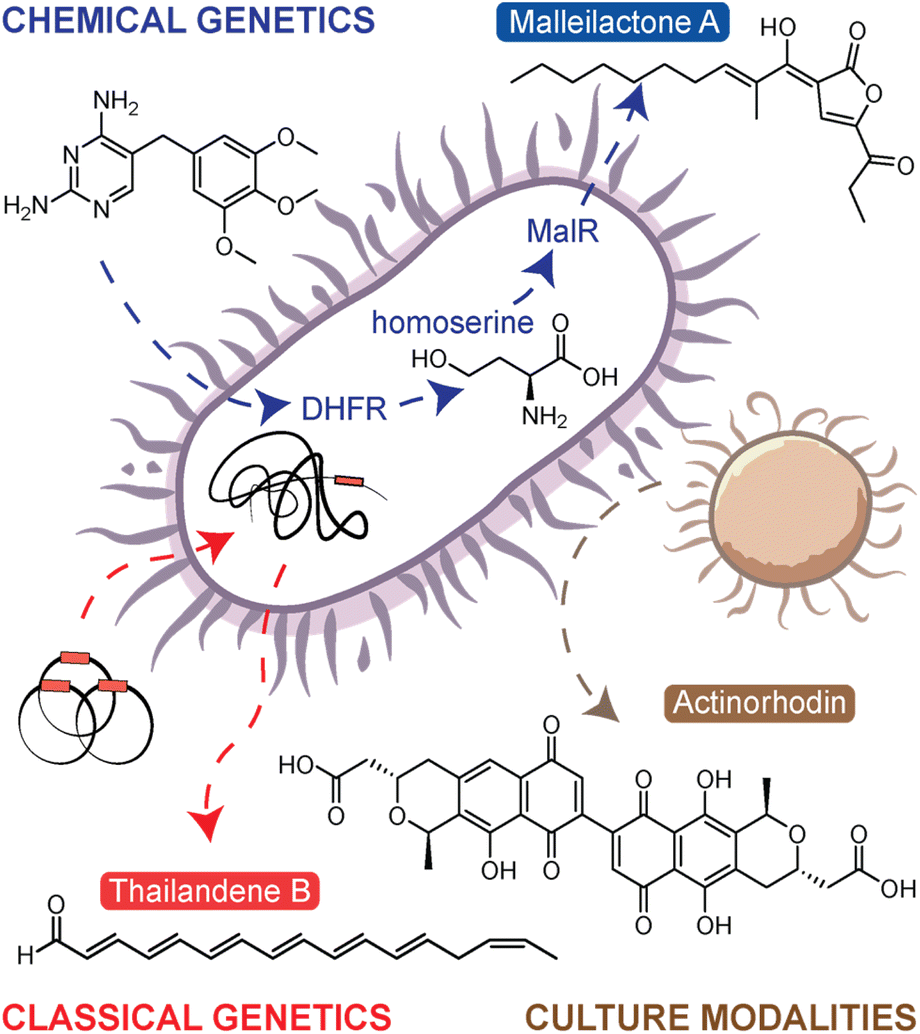 | ||
| Fig. 1 Overview of endogenous natural product activation methods. In the chemical genetics approach, trimethoprim is shown to inhibit dihydrofolate reductase (DHFR) leading to the accumulation of homoserine, which in turn activates the transcriptional regulator MalR to upregulate the malleilactone/malleicyprol BGC. The classical genetics approach shows the use of transposons to mutate the bacterial chromosome leading to increased NP production, in this case, to the activation of the thailandenes, cryptic NPs from B. thailandensis. Lastly, the culture modalities approach shows bacterial interactions leading to the overproduction of actinorhodin. | ||
Targeted and untargeted classical genetics provide an alternative approach. The targeted, or reverse genetics, approaches essentially reprogram the transcriptional regulation to artificially overexpress the selected silent BGC. This is done by modifying, overexpressing, deleting, or replacing the native regulatory genes that control BGC expression. While this is a powerful approach, it is more labour-intensive than other methods and is not applicable to organisms for which genetic methods are difficult or yet undeveloped. The untargeted, or forward genetics, approach involves generating a large mutant library either through UV-induced mutagenesis or transposon (Tn) mutagenesis and then screening the library to identify overproducing mutants.15 The screening/selection can be either reporter-guided,16 phenotype-guided,17 or metabolomics-guided,18 the latter of which will be described in detail below (see the section titled Comparative Metabolomics). The first example of using transposon mutagenesis to identify new products from a silent BGC was presented by Park et al.17 In this work, transposon mutagenesis was used to generate a library of mutants in Burkholderia thailandensis DW503. In a phenotype-guided selection strategy, pigmented mutants were selected for metabolomic investigation, leading to the discovery of new polyene NPs named the thailandenes (Fig. 1). One advantage of the transposon mutagenesis approach is it allows users to easily identify the site of mutation, and in this case, mutants with disruptions in a σ54-dependent transcriptional regulator resulted in activation of the thailandene BGC.
As classical genetics approaches can be difficult in certain organisms, the last approach, chemical genetics, offers the simple alternative of using small molecule elicitors to induce silent BGCs. Like classical genetics, chemical genetics can be either targeted or untargeted. In the targeted approach, inhibitors of the ribosome or RNA polymerase have been used to specifically alter transcriptional and translational activities, thus leading to enhanced expression of silent BGCs.19 The approach is referred to as ribosome or RNAP engineering. A notable example from application of this approach includes piperidamycin,20 a cryptic streptomycete NP identified using a combination of ribosome and RNAP engineering. In fungi, similar approaches have been used targeting epigenetic modifications.21 The untargeted, forward chemical genetics approach is known as high-throughput elicitor screening (HiTES). In HiTES, a single organism is treated with hundreds to thousands of potential elicitors and production of cryptic metabolites is then monitored with a variety of readouts. HiTES has been applied to over a dozen microbial strains and resulted in more than 150 novel, cryptic metabolites.22,23 Initially, these experiments used targeted assays such as pigmentation or genetic reporters. For instance, one of the first HiTES experiments targeted the silent malleicyprol (mal) BGC in Burkholderia thailandensis. In this work, a mal–lacZ reporter strain was elicited with a library of small molecule drugs, which revealed that subinhibitory concentrations of antibiotics, specifically trimethoprim and piperacillin, were potent transcriptional activators of the BGC.24,25 Follow-up work revealed that the induction of the mal BGC was a direct consequence of inhibition of trimethoprim's clinical target, dihydrofolate reductase.26 This disruption in the folate pathway resulted in elevated levels of homoserine and other methionine biosynthetic precursors, which in turn activated the transcriptional regulator MalR to upregulate mal expression (Fig. 1). For more information on each of these approaches, we point to our previous in-depth reviews on the subject.7,27 Despite all these powerful strategies to boost NP biosynthesis, it has remained challenging to discover new NPs from silent BGCs, and this problem is largely technical. With numerous methods to activate a given BGC, the process has been mostly limited by the throughput with which elicited NPs can be detected. Recently, solutions to this challenge have surfaced and in the next sections, we describe how MS techniques have expanded the boundaries of NP discovery by facilitating high-throughput molecular detection.
3. Ionization methods in mass spectrometry
Advanced technologies have recently emerged for acquiring high quality MS data. At a basic level, the process for MS acquisition consists of three parts: (i) ionization of the molecules in the sample, (ii) selection or separation of ions by mass-to-charge ratio (m/z), and (iii) detection of the resolved ions. For the purposes of this highlight, we will primarily discuss the electrospray family of ionization methods used for high-throughput MS analyses (Fig. 2). Electrospray ionization (ESI) is perhaps the most ubiquitous ionization method currently used for MS. In ESI, an electric field is applied across a mist of sprayed liquid sample to generate gas phase ions, which can then be directed into the MS. This approach couples well with liquid chromatography (LC) or ultrahigh-performance LC (UPLC) enabling molecules to be analysed as they elute from a column, thus generating a mass chromatogram as a function of elution time. This chromatographic coupling is especially helpful for detecting lower abundance metabolites from complex samples.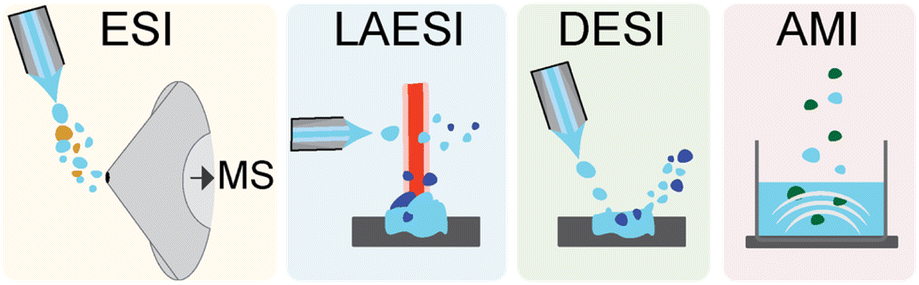 | ||
| Fig. 2 Mass spectrometer ionization techniques in the electrospray family. The coloured panels depict from left to right electrospray ionization (ESI), laser ablation electrospray ionization (LAESI), desorption electrospray ionization (DESI), and acoustic mist ionization (AMI). | ||
Typical sampling rates for UPLC-ESI-MS are between 1 and 10 minutes per sample. Another sampling strategy is to skip the chromatography and use automated, high-speed robotics to inject samples into the ESI-MS instrument directly. In these cases, an ultrafast solid phase extraction step can be used to desalt the sample and enhance ionization in the mass spectrometer. These instruments are compatible with high-throughput well plate formats such as 1536-well plates and can achieve sampling rates of 2–10 seconds per sample.28 ESI is by now ubiquitous, and the instruments leveraging this approach are readily available. The primary advantage of ESI compared to other methods is its ease of use, limited sample preparation, and LC compatibility, thus enabling the detection of thousands of metabolites across a wide range of abundances. Coupling to LC also has the benefit of limiting or eliminating ion suppression. A key disadvantage of ESI is that samples must be solubilized for analysis.
Laser ablation ESI (LAESI) is an alternative approach in which the sample is pulsed with a mid-IR laser to generate micro-explosions, ejecting predominantly neutral molecules into the gas phase.29 These molecules become ionized as they encounter a charged spray directed above the sample, and the charged ions are then shuttled into the MS. This approach was recently used to screen bacterial fatty acid production with sampling rates of <2 seconds per sample.30 The primary advantage of LAESI is that, unlike ESI, metabolites do not need to be extracted and solubilized; they can be detected directly from biological cultures without any sample preparation. However, this method is not compatible with LC separation, which limits the dynamic range of the approach for complex samples. These instruments are also less common than ESI instruments due to the cost and complexity of the laser component.
In desorption ESI (DESI) the direct impact of the ESI spray is used in place of the laser to generate aerosolized sample droplets. The charged solvent spray is directed at the sample, creating a rebounding stream of charged microdroplets that ionize and transport sample ions into the MS. Without the laser component, DESI instruments are simpler and more common than LAESI. While DESI can be coupled to LC separations, the primary advantage of DESI is that it can be performed without any sample preparation and can achieve high rates of ∼360 milliseconds per sample.31 The sensitivity of DESI varies for molecular classes (peptides, carbohydrates, lipids, etc.) depending on the adjustable spray and collection angles. This can be an advantage for screens in which a specific class of NPs is targeted, as parameters can be optimized for the targeted product. At the same time, the method is less suitable for untargeted screens, where structurally diverse NPs are monitored.
The last electrospray class approach we mention here is acoustic mist ionization (AMI), which uses an ultrasonic acoustic pulse to eject a spray or mist of droplets from the sample.32 These droplets pass through an electric field, which results in ionization of the sample as droplets desolvate on the path to the MS. AMI-MS was recently used to screen a library of 2 million compounds to identify 6745 inhibitors of a human histone deacetylase with a rate of ∼860 milliseconds per sample.33 AMI is suitable for a broad range of metabolite classes but, like LAESI, is not compatible with LC separations. AMI is a relatively new technology, and the ability of this approach to analyse NP from complex samples has not been fully evaluated. Other non-electrospray type approaches are amenable to high-throughput ionization as well. One common approach is matrix assisted laser desorption ionization (MALDI), in which the sample is spotted in a crystalline matrix on a target plate. Laser pulses are then absorbed into the matrix to generate plumes of ionized molecules, which are then funnelled into the MS. MALDI is more susceptible to interference from salts and requires additional sample preparation steps, but high-throughput MALDI-MS approaches have achieved high rates of ∼1.2 seconds per sample.34 It has been especially useful in analysing metabolomes on agar, microbial interactions on a solid surface, and isotope-incorporation experiments, which have been conducted in low to high-throughput formats.35–38 For more information on these and the many other MS ionization approaches we recommend other in-depth reviews on the subject.39,40 We next discuss methods for comparative metabolomics analysis after data acquisition.
4. Comparative metabolomics
Early implementations of comparative MS for NP discovery utilized a ‘stare-and-compare’ approach, in which users would manually inspect HPLC-ESI-MS data to identify metabolites of interest (Fig. 3A). For example, the discovery of alchivemycin A, which relied on bacterial cocultures of Streptomyces endus S-522 and Tsukamurella pulmonis, used this approach.13 While it can be effective for small datasets, it requires technical proficiency and is impractical for comparing metabolomes acquired from hundreds of differentially treated samples. Additionally, metabolites of low abundance, even if strongly elicited by a stimulus condition, are very difficult to discern by manual inspection. Instead, users now rely on feature extraction tools to identify and quantify ‘metabolomic features’ within MS data to generate comparative metabolomic profiles. The term ‘feature’ can be considered as a product of MS data pre-processing tools. These represent metabolites in the sample, though depending on the approach used, the same metabolite may be represented by multiple features. Features consist of several user-defined parameters, usually including m/z, abundance, and/or retention time. For example, nearly 2 decades ago Siuzdak and colleagues released a free MS processing tool, XCMS, that is still widely used.41 The corresponding R package, specifically, has been continually updated and improved and is compatible with high-throughput MS. The original feature extraction algorithm parsed LCMS data into extracted ion chromatogram bins, and chromatographic peaks were identified from these bins by filters distinguishing signal peaks from background noise. In this case, the features extracted from the data represent ions with a discrete m/z and retention time, and several features can stem from a single metabolite in the form of multiple charge states, in-source adducts, and isotopologues. Other MS pre-processing tools, such as the feature extraction tool in Agilent's Profinder package, condense adducts, charge states, and isotopologues into a single feature, which represents a composite of observed ions putatively stemming from a single metabolite in the sample. In either case, once features are identified, they are matched, or aligned, and quantified in each sample to generate an abundance profile. This is the most widely used method for visualizing differential metabolomes. The corresponding profiles comprise a large data matrix of feature abundances across every experimental sample, which can then be analysed to find cryptic metabolites.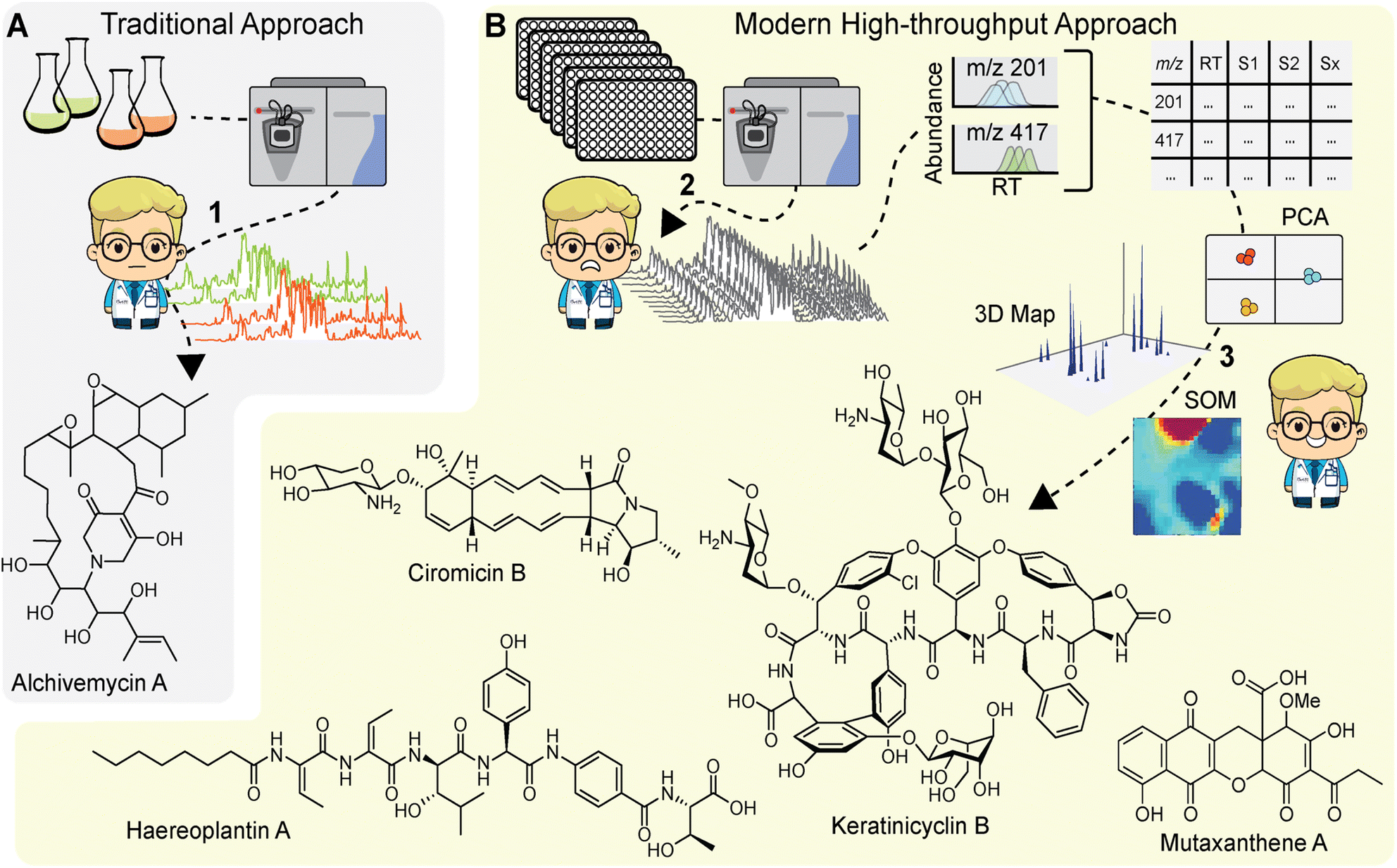 | ||
| Fig. 3 Comparison of traditional and modern comparative metabolomics approaches for NP discovery. (A) Alchivemycin A was discovered through manual inspection of LC-MS chromatograms generated from a strain grown in monoculture or in coculture with mycolic acid containing bacteria (1). While effective, manual inspection limits the throughput of this comparative MS approach. (B) Advances in MS now enable high-throughput MS data acquisition, the analysis of which can be daunting (2). Comparative metabolomics using, among other approaches, PCA and SOM analysis, now allows the multi-dimensional MS data to be interrogated. These approaches have facilitated discovery of several cryptic metabolites (3). Mutaxanthene A was uncovered by applying PCA to a collection of 10 antibiotic resistant mutants. Ciromicin B was found through a self-organizing maps (SOM) approach by comparing four coculture conditions. Haereoplantin A was similarly identified through SOM analysis by comparing metabolomes of 72 transposon mutants with that of the wild-type control keratinicyclin B was discovered via 3D map analysis upon subjecting the producer to HiTES with 502 elicitors. | ||
4.1. Principal component analysis
After generating the metabolomic data matrix, the next step is to prioritize features corresponding to new NPs. Multivariate analyses, such as principal component analysis (PCA) are routinely used to analyse metabolomics data (Fig. 3B).42 PCA transforms a rich metabolomics matrix into a simplified and dimensionally reduced data representation that retains as much of the original data variance as possible. The outputs from PCA are scores plots, which show sample groupings based on principal components and loadings plots that in turn display metabolomic features that strongly contribute to the principal component vectors. Müller and colleagues were among the first to use PCA to prioritize bacterial NPs.43 They explored the NP diversity between 98 Myxococcus xanthus isolates collected from 78 different locations around the world. PCA revealed a high degree of intraspecies diversity which was largely directed by NP variations, such as DKxanthene production. Ultimately, 7 of the 8 known M. xanthus NPs at the time were identified along with 37 putative novel NPs. In a later study, Derewacz et al. used PCA to prioritize NPs from a collection of 11 Nocardiopsis sp. FU40 mutants, leading to the discovery of a new family of NPs, the mutaxanthenes (Fig. 3B).44 More recent adaptations of this approach include application to high-resolution MS data from plant bulk tissue analysis to match metabolites to specific tissues with the ultimate goal of finding tissue-specific natural products.45 PCA is a powerful metabolomics analysis tool, and other multivariate analyses have been applied to NP prioritization,46,47 but the utility of these method decreases when the sample size is large or the samples are diverse in nature.4.2. Self-organizing maps
Another data reduction approach to prioritize metabolomic features is clustering via self-organizing maps (SOMs). In this method, features are grouped into a network of nodes based on similar abundance profiles across all experimental samples. The output is a metabolomic feature map where the nodes are coloured for each sample based on the composite intensity of features within each respective node. Bachmann and colleagues first applied the SOM approach to prioritize NPs from Streptomyces coelicolor A3(2) exposed to a series of stimuli and found that the analysis was able to prioritize all 16 observed NPs, stemming from 8 out of 22 predicted BGCs.48 The SOM metabolomics approach was then used to prioritize NPs from Nocardiopsis sp. FU40 cocultured with mycolic acid bacteria. In this study, the SOMs readily identified a new family of NPs, the ciromicins (Fig. 3B) that were only present in the coculture, demonstrating their utility for NP discovery based on comparative metabolomics.49 In a recent study we applied SOMs to prioritize NPs in a metabolomics-guided transposon mutagenesis screen. HPLC-MS metabolomic data from 72 Burkholderia plantarii ATCC43733 transposon mutant cultures and 4 wild-type control cultures were analysed using the SOM approach.18 Difference maps for mutant samples were then generated by subtracting 3× the averaged wild-type feature abundances from each mutant map. Visual inspection of the resulting difference maps led to the discovery of two new families of non-ribosomal peptide NPs, haereoplantins (Fig. 3B) and burrioplantins.4.3. 3D metabolome maps
While SOMs are very useful for clustering MS features and identifying induced NPs, they do not fully provide the throughput needed for analysing data from larger screens (>100 samples). Instead, we have recently developed a new approach that visualizes the complete elicited metabolome from any number of samples in a single plot. These 3D difference plots essentially array extracted metabolomic features across the entire dataset with peaks reflecting the features, defined by abundances and m/z, which are observed in samples after subtracting aligned features in control samples. This analytical method is best suited for data with a large number of samples, such as those generated with the HiTES approach. In the first MS-guided HiTES experiment, we used LAESI-MS to rapidly acquire MS data for more than 1000 elicited samples including ∼500 each from Streptomyces canus NRRL B3980 and Amycolatopsis keratiniphilia NRRL B24117.50 For each strain, 3D maps were constructed displaying induced metabolites, characterized by m/z and abundance, as a function of the elicitor library. For S. canus, the 3D difference plot revealed known NPs from the amphomycin family, which were strongly induced by elicitors in the screen, as well as a new family of lassopeptides, the canucins. For A. keratiniphilia, the 3D difference plot revealed several high molecular weight features that were elicited through HiTES. These induced NPs were identified as new glycopeptide antibiotics which were named the keratinimicins and keratinicyclins (Fig. 3B). These discoveries validated the utility of MS-guided HiTES to activate and prioritize new NPs. The analysis in these studies was conducted one at a time in MatLAB. We subsequently developed the Metabolomics Explorer (MetEx), an easy-to-use application for rapid and multifunctional analysis of high-dimensional MS-HiTES datasets.514.4. The metabolomics explorer (MetEx) application
MetEx provides a user-friendly interface to upload matrix-type MS-feature data from hundreds of HPLC-MS profiles. The ‘Parameters’, ‘Difference Plot Analysis’, ‘Principal Component Analysis’, and ‘Predictor’ tabs (Fig. 4) hold some of the key processes in MetEx. In the parameters tab, users can upload their datafiles, designate experimental controls, and filter data based on feature descriptors such as m/z, retention time, and abundance. Elicitor library information, when applicable, can also be uploaded to facilitate other features such as elicitor class grouping and automated sample class colouring. Elicited metabolites are then prioritized through the difference plot analysis, principal component analysis, and predictor tabs. In the difference plot analysis tab, 3D difference plots are automatically generated by subtracting from all samples the metabolomic feature abundances observed in the designated control group(s). These plots are completely interactive and can be rotated or zoomed in any direction. Any 2D component from the 3D plot can be extracted and viewed separately. For example, clicking on peaks visible in the difference plot will generate a feature abundance plot for the selected peak from which elicitor information can be observed. These 3D difference plots hold a lot of information as they essentially display the entire induced metabolome from all samples in a single plot. In the PCA tab, principal component scores and loadings plots are presented for the uploaded dataset. Users can toggle through principal components to identify trends in metabolomic responses to elicitor treatments. Like the 3D difference plot, these PCA plots are highly interactive, and clicking on a point in the loadings plot will generate an abundance plot for the selected feature. Finally, in the predictor tab, a scoring algorithm automatically identifies interesting features in the dataset. These prioritized features are presented in a series of feature abundance plots so that users can quickly evaluate the results. In our first evaluation of MetEx, we conducted a HiTES experiment with 750 elicitors in Burkholderia gladioli ATCC 10248 and collected metabolomic data using UPLC-ESI-MS. The acquisition took ∼3.5 min per sample (∼2 days for the entire analysis), and data pre-processing was conducted using XCMS to extract and align MS features. After uploading the data into MetEx, several known NPs were identified in the induced 3D difference map (shown in Fig. 4) including members of the gladiobactin, gladiolin, icosalide, and burriogladin families. Additionally, several unidentified features were identified, which provide good starting points for further cryptic metabolite discovery.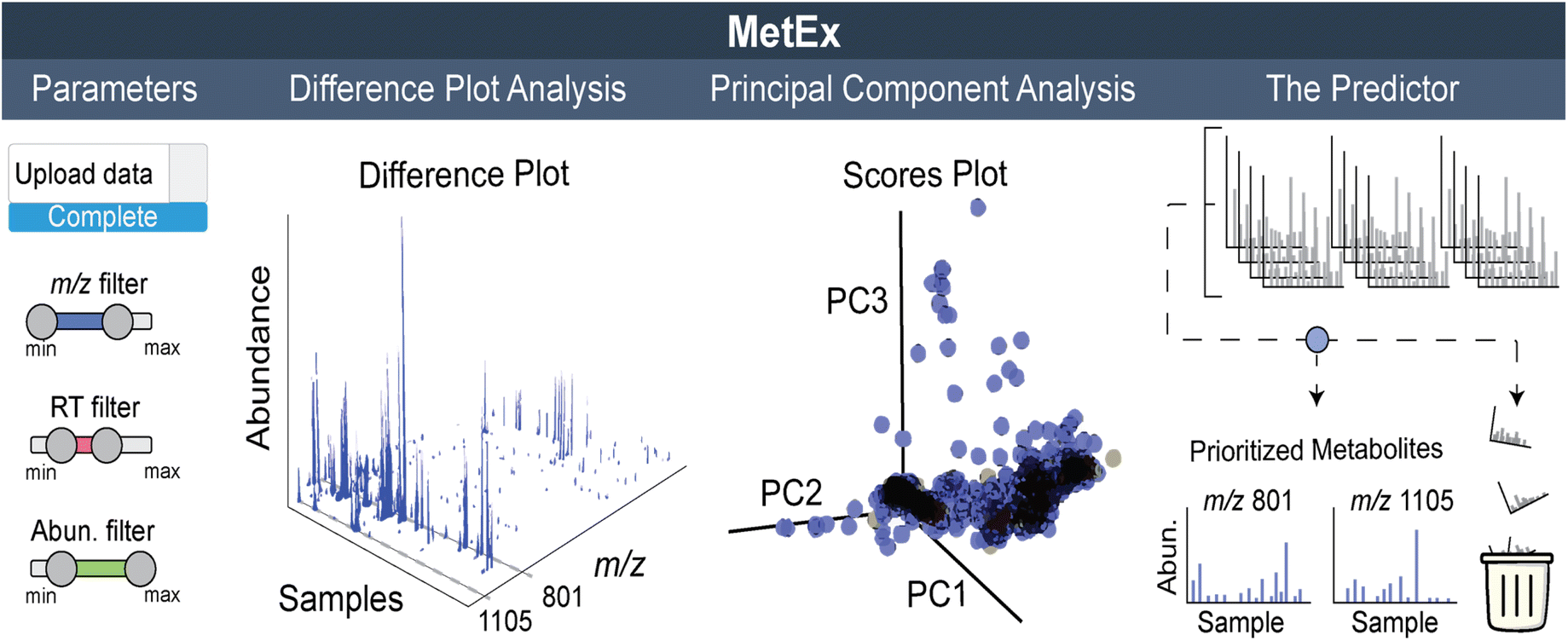 | ||
| Fig. 4 Overview of MetEx application for metabolomics-guided NP discovery. The parameters tab enables data upload and filtering. The difference plot analysis tab controls 3D plot output. The 3D plot shown here represents induced metabolomes from 750 elicited cultures with abundances at least 2-fold greater than unelicited cultures. PCA scores and loadings plots are output in the principal component analysis tab. The scores plot here represents how each of the 750 elicited samples relate to each other in relation to the first, second, and third principal components. Automatic feature prioritization is performed in the predictor tab. Here the predictor algorithm scores each metabolic feature based on their elicitation patterns. Highlighted m/z 1105 and 801 on the 3D plot correspond to gladiobactin and gladiolin respectively. | ||
MetEx is a publicly available metabolomics analysis application designed to facilitate NP discovery, and the functionality discussed here as well as additional features are described in detail in the original publication.51 Since its deployment, MetEx has been used to discover new NPs in both bacteria52 and fungi.53 In a recent example, we used UPLC-ESI-MS-based HiTES with a 442-member human endogenous metabolite library to search for cryptic NPs synthesized by Streptococcus suis ATCC 43765, a zoonotic pig pathogen that can cause severe disease in humans.54 A 3D difference plot was generated in MetEx using the ‘greatest value’ subtraction mode and a 5-fold control subtraction factor. The resulting 3D plot identified a new family of NPs, the threoglucins, which were strongly induced by pyridine containing elicitors such as nicotinic acid (Fig. 5). Ultimately, 22 threoglucins were identified from cultures elicited with nicotinic acid.
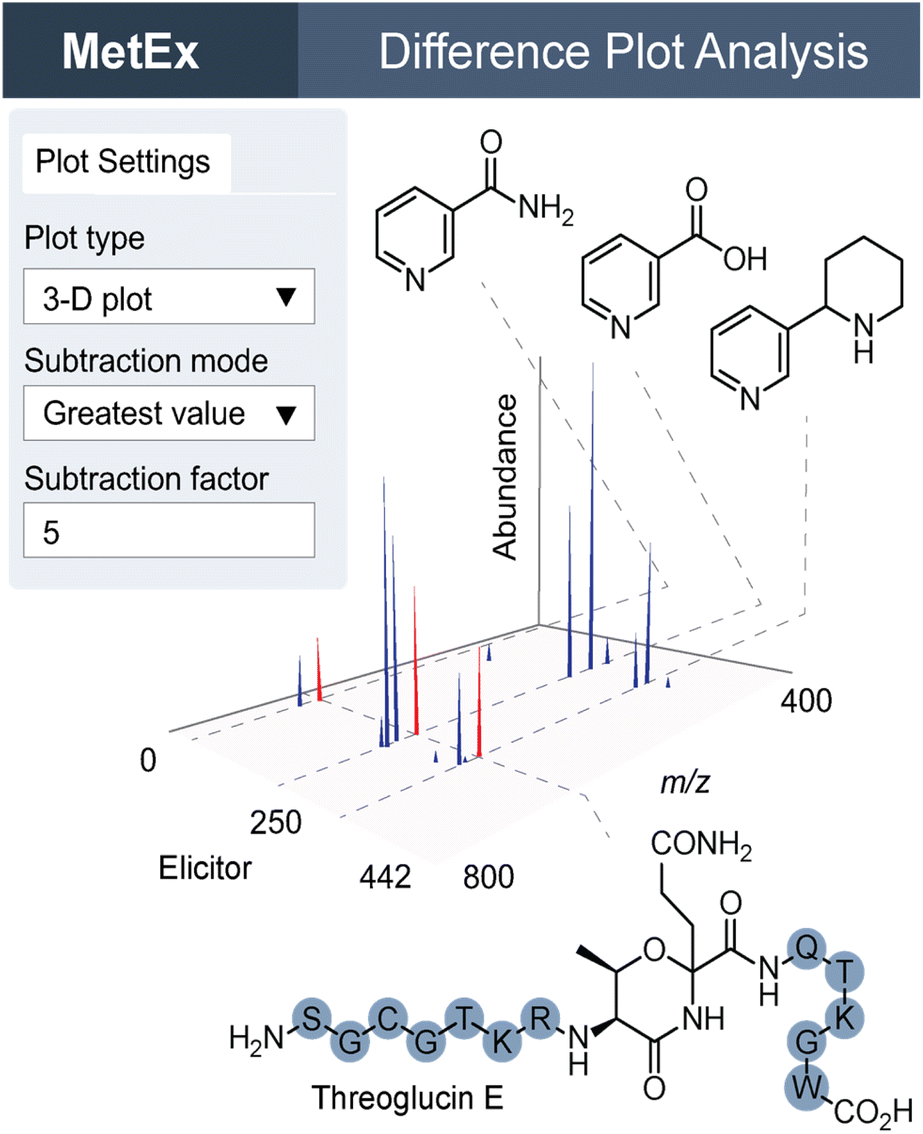 | ||
| Fig. 5 MetEx 3D difference plot of UPLC-ESI-MS HiTES with Streptococcus suis treated with 442 elicitors. The plot settings tab shows 3D plot selection parameters, which can be updated in the application to filter features by fold-change relative to control groups. Peaks in the 3D plot here represent features with greater than 5-fold abundance in the elicited condition relative to control conditions. Elicitor structures are shown for nicotinamide, nicotinic acid, and anabasine. Peaks highlighted in red on the 3D plot correspond to threoglucin E shown below the plot. | ||
Overall, the comparative metabolomics strategies described in this section offer powerful means of interrogating metabolomics data for NP discovery from silent or sparingly expressed BCSs. By facilitating analysis of hundreds of HPLC/UPLC-MS profiles, they enable NP discovery at higher throughputs. These data can be used in conjunction with innovative and now routine MS analysis and batch processing tools, such as such as the MS networking approach,55 Global Natural Product Social Molecular Networking (GNPS),56 MZmine,57 and SIRIUS 4 applications,58 which enable rapid identification of (known) compounds using MS fragmentation spectra, minimize rediscovery rates, and also aid in prioritizing novel compounds. Undoubtedly, these and other strategies will continue to be refined as further instrumental advancements become available, for example, with the recent development of ion mobility mass spectrometers.
5. Conclusions
MS has become an essential tool for the discovery and characterization of NPs. New technologies continue to push the boundaries of molecular detectability especially improvements in instrumental sensitivity, dynamic range, accuracy, and ionization efficiency. With several approaches developed to elicit biosynthesis of cryptic NPs, the present limitations do not stem from lack of methodologies but instead from the bottlenecks of acquiring and comparing elicited microbial metabolomes across the large number of approaches needed to activate cryptic NPs. Moreover, several approaches developed necessitate rapid and accurate analysis of multi-dimensional metabolomics datasets. Here we have discussed how advancements in MS-based metabolomics have aided natural product discovery and transformed a science once based on manual inspection of a handful of mass chromatograms to automated computational prioritization from several hundred samples. These advances now facilitate NP discovery with much higher throughputs. The confluence of these developments, that is methods to turn on silent BGCs and technologies to detect and analyse high-content MS data, promise to unearth the elusive reservoir of NPs from the wealth of bioinformatically predicted silent BGCs.6. Conflicts of interest
Brett C. Covington and Mohammad R. Seyedsayamdost are co-founders and shareholders of Cryptyx Bioscience, Inc.7. Acknowledgements
We thank the National Institutes of Health (R35 GM152049 and R01 GM140034), The Leona M. and Harry B. Helmsley Charitable Trust, and the Princeton Catalysis Institute for supporting our work on natural product discovery.8. Notes and references
- J. R. Doroghazi and W. W. Metcalf, BMC Genomics, 2013, 14, 611 CrossRef CAS PubMed.
- M. H. Medema, K. Blin, P. Cimermancic, V. de Jager, P. Zakrzewski, M. A. Fischbach, T. Weber, E. Takano and R. Breitling, Nucleic Acids Res., 2011, 39, W339–W346 CrossRef CAS.
- M. A. Skinnider, C. A. Dejong, P. N. Rees, C. W. Johnston, H. Li, A. L. H. Webster, M. A. Wyatt and N. A. Magarvey, Nucleic Acids Res., 2015, 43, 9645–9662 CAS.
- M. Nett, H. Ikeda and B. S. Moore, Nat. Prod. Rep., 2009, 26, 1362–1384 RSC.
- M. Oliynyk, M. Samborskyy, J. B. Lester, T. Mironenko, N. Scott, S. Dickens, S. F. Haydock and P. F. Leadlay, Nat. Biotechnol., 2007, 25, 447–453 CrossRef CAS PubMed.
- G. C. A. Amos, T. Awakawa, R. N. Tuttle, A.-C. Letzel, M. C. Kim, Y. Kudo, W. Fenical, B. S. Moore and P. R. Jensen, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E11121–E11130 CrossRef CAS PubMed.
- B. C. Covington, F. Xu and M. R. Seyedsayamdost, Annu. Rev. Biochem., 2021, 90, 763–788 CrossRef CAS PubMed.
- D. Kaiser, U. Onken, I. Sattler and A. Zeeck, Appl. Microbiol. Biotechnol., 1994, 41, 309–312 CrossRef CAS PubMed.
- H. B. Bode, B. Bethe, R. Höfs and A. Zeeck, ChemBioChem, 2002, 3, 619–627 CrossRef CAS.
- K. Kawai, G. Wang, S. Okamoto and K. Ochi, FEMS Microbiol. Lett., 2007, 274, 311–315 CrossRef CAS PubMed.
- T. Lincke, S. Behnken, K. Ishida, M. Roth and C. Hertweck, Angew Chem. Int. Ed. Engl., 2010, 49, 2011–2013 CrossRef CAS.
- K. Ueda, S. Kawai, H. Ogawa, A. Kiyama, T. Kubota, H. Kawanobe and T. Beppu, J. Antibiot., 2000, 53, 979–982 CrossRef CAS.
- H. Onaka, Y. Mori, Y. Igarashi and T. Furumai, Appl. Environ. Microbiol., 2011, 77, 400–406 CrossRef CAS PubMed.
- G. Pishchany, E. Mevers, S. Ndousse-Fetter, D. J. Horvath, C. R. Paludo, E. A. Silva-Junior, S. Koren, E. P. Skaar, J. Clardy and R. Kolter, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 10124–10129 CrossRef CAS PubMed.
- B. C. Covington and M. R. Seyedsayamdost, Methods Enzymol., 2022, 665, 305–323 CAS.
- F. Guo, S. Xiang, L. Li, B. Wang, J. Rajasärkkä, K. Gröndahl-Yli-Hannuksela, G. Ai, M. Metsä-Ketelä and K. Yang, Metab. Eng., 2015, 28, 134–142 CrossRef CAS PubMed.
- J.-D. Park, K. Moon, C. Miller, J. Rose, F. Xu, C. C. Ebmeier, J. R. Jacobsen, D. Mao, W. M. Old, D. DeShazer and M. R. Seyedsayamdost, ACS Chem. Biol., 2020, 15, 1195–1203 CrossRef CAS.
- A. Yoshimura, B. C. Covington, É. Gallant, C. Zhang, A. Li and M. R. Seyedsayamdost, ACS Chem. Biol., 2020, 15, 2766–2774 CrossRef CAS.
- K. Ochi and T. Hosaka, Appl. Microbiol. Biotechnol., 2013, 97, 87–98 CrossRef CAS PubMed.
- T. Hosaka, M. Ohnishi-Kameyama, H. Muramatsu, K. Murakami, Y. Tsurumi, S. Kodani, M. Yoshida, A. Fujie and K. Ochi, Nat. Biotechnol., 2009, 27, 462–464 CrossRef CAS PubMed.
- K. M. Fisch, A. F. Gillaspy, M. Gipson, J. C. Henrikson, A. R. Hoover, L. Jackson, F. Z. Najar, H. Wägele and R. H. Cichewicz, J. Ind. Microbiol. Biotechnol., 2009, 36, 1199–1213 CrossRef CAS PubMed.
- Y. Li, S. R. Lee, E. J. Han and M. R. Seyedsayamdost, Angew Chem. Int. Ed. Engl., 2022, 61, e202208573 CrossRef CAS PubMed.
- F. Xu, B. Nazari, K. Moon, L. B. Bushin and M. R. Seyedsayamdost, J. Am. Chem. Soc., 2017, 139, 9203–9212 CrossRef CAS.
- M. R. Seyedsayamdost, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7266–7271 CrossRef CAS.
- A. Li, B. K. Okada, P. C. Rosen and M. R. Seyedsayamdost, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2021483118 CrossRef CAS PubMed.
- A. Li, D. Mao, A. Yoshimura, P. C. Rosen, W. L. Martin, É. Gallant, M. Wühr and M. R. Seyedsayamdost, mBio, 2020, 11, e03210 CAS.
- B. K. Okada and M. R. Seyedsayamdost, FEMS Microbiol. Rev., 2017, 41, 19–33 CrossRef CAS.
- T. Bretschneider, C. Ozbal, M. Holstein, M. Winter, F. H. Buettner, S. Thamm, D. Bischoff and A. H. Luippold, SLAS Technol., 2019, 24, 386–393 CrossRef PubMed.
- P. Nemes and A. Vertes, Anal. Chem., 2007, 79, 8098–8106 CrossRef CAS PubMed.
- H. Liu, W. Gao, T. Cui, S. Wang, X. Song, Z. Wang, H. Zhang, S. Li, Y.-L. Yu and Q. Cui, Talanta, 2024, 268, 125234 CrossRef CAS PubMed.
- M. Wleklinski, B. P. Loren, C. R. Ferreira, Z. Jaman, L. Avramova, T. J. P. Sobreira, D. H. Thompson and R. G. Cooks, Chem. Sci., 2018, 9, 1647–1653 RSC.
- I. Sinclair, R. Stearns, S. Pringle, J. Wingfield, S. Datwani, E. Hall, L. Ghislain, L. Majlof and M. Bachman, SLAS Technol., 2016, 21, 19–26 CrossRef.
- I. Sinclair, M. Bachman, D. Addison, M. Rohman, D. C. Murray, G. Davies, E. Mouchet, M. E. Tonge, R. G. Stearns, L. Ghislain, S. S. Datwani, L. Majlof, E. Hall, G. R. Jones, E. Hoyes, J. Olechno, R. N. Ellson, P. E. Barran, S. D. Pringle, M. R. Morris and J. Wingfield, Anal. Chem., 2019, 91, 3790–3794 CrossRef CAS.
- C. Haslam, J. Hellicar, A. Dunn, A. Fuetterer, N. Hardy, P. Marshall, R. Paape, M. Pemberton, A. Resemannand and M. Leveridge, J. Biomol. Screening, 2016, 21, 176–186 CrossRef CAS PubMed.
- Y. L. Tang, Y. Xu, P. Straight and P. C. Dorrestein, Nat. Chem. Biol., 2009, 5, 885–887 CrossRef PubMed.
- M. F. Traxler, J. D. Watrous, T. Alexandrov, P. C. Dorrestein and R. Kolter, mBio, 2013, 4, 00459 CrossRef.
- E. Esquenazi, Y. L. Yang, J. Watrous, W. H. Gerwick and P. C. Dorrestein, Nat. Prod. Rep., 2009, 26, 1521–1534 RSC.
- R. B. Kinnel, E. Esquenazi, T. Leao, N. Moss, E. Mevers, A. R. Pereira, E. A. Monroe, An Korobeynikov, T. F. Murray, D. Sherman, L. Gerwick, P. C. Dorrestein and W. H. Gerwick, J. Nat. Prod., 2017, 80, 1541–1521 CrossRef PubMed.
- M. E. Dueñas, R. E. Peltier-Heap, M. Leveridge, R. S. Annan, F. H. Büttner and M. Trost, EMBO Mol. Med., 2022, 15, e14850 CrossRef PubMed.
- B. Challen and R. Cramer, Proteomics, 2022, 22, 2100394 CrossRef CAS.
- C. A. Smith, E. J. Want, G. O'Maille, R. Abagyan and G. Siuzdak, Anal. Chem., 2006, 78, 779–787 CrossRef CAS.
- B. Worley and R. Powers, Curr. Metabolomics, 2013, 1, 92–107 CAS.
- D. Krug, G. Zurek, B. Schneider, R. Garcia and R. Müller, Anal. Chim. Acta, 2008, 624, 97–106 CrossRef CAS PubMed.
- D. K. Derewacz, C. R. Goodwin, C. R. McNees, J. A. McLean and B. O. Bachmann, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2336–2341 CrossRef CAS PubMed.
- A. H. Vu, M. Kang, J. Wurlitzer, S. Heinicke, C. Li, J. C. Wood, V. Grabe, C. R. Buell, L. Caputi and S. E. O'Connor, J. Am. Chem. Soc., 2024, 146, 23891–23900 CrossRef CAS.
- B. C. Covington, J. A. McLean and B. O. Bachmann, Nat. Prod. Rep., 2017, 34, 6–24 RSC.
- B. C. Covington, J. M. Spraggins, A. E. Ynigez-Gutierrez, Z. B. Hylton and B. O. Bachmann, Appl. Environ. Microbiol., 2018, 84, e01125 CrossRef CAS PubMed.
- C. R. Goodwin, B. C. Covington, D. K. Derewacz, C. R. McNees, J. P. Wikswo, J. A. McLean and B. O. Bachmann, Chem. Biol., 2015, 22, 661–670 CrossRef CAS.
- D. K. Derewacz, B. C. Covington, J. A. McLean and B. O. Bachmann, ACS Chem. Biol., 2015, 10, 1998–2006 CrossRef CAS.
- F. Xu, Y. Wu, C. Zhang, K. M. Davis, K. Moon, L. B. Bushin and M. R. Seyedsayamdost, Nat. Chem. Biol., 2019, 15, 161–168 CrossRef CAS.
- B. C. Covington and M. R. Seyedsayamdost, ACS Chem. Biol., 2021, 16, 2825–2833 CrossRef CAS PubMed.
- E. J. Han, S. R. Lee, S. Hoshino and M. R. Seyedsayamdost, ACS Chem. Biol., 2022, 17, 3121–3130 CrossRef CAS.
- S. R. Lee and M. R. Seyedsayamdost, Angew Chem. Int. Ed. Engl., 2022, 61, e202204519 CrossRef CAS PubMed.
- B. C. Covington and M. R. Seyedsayamdost, J. Am. Chem. Soc., 2022, 144, 14997–15001 CrossRef CAS PubMed.
- J. Y. Yang, L. M. Sanchez, C. M. Rath, X. Liu, P. D. Boudreau, N. Bruns, E. Glukhov, A. Wodtke, R. de Felicio, A. Fenner, W. R. Wong, R. G. Linington, L. Zhang, H. M. Debonsi, W. H. Gerwick and P. C. Dorrestein, J. Nat. Prod., 2013, 76, 1686–1699 CrossRef CAS PubMed.
- N. Wang, et al. , Nat. Biotechnol., 2016, 34, 828–837 CrossRef.
- R. Schmid, et al. , Nat. Biotechnol., 2023, 41, 447–449 CrossRef CAS.
- K. Dührkop, M. Fleischauer, M. Ludwig, A. A. Aksenov, A. V. Melnik, M. Meusel, P. C. Dorrestein, J. Rousu and S. Böcker, Nat. Methods, 2019, 16, 299–302 CrossRef.
| This journal is © The Royal Society of Chemistry 2025 |