
Fe(II) and 2-oxoglutarate-dependent dioxygenases for natural product synthesis: molecular insights into reaction diversity
Songyin
Zhao
 ,
Lunjie
Wu
,
Yan
Xu
and
Yao
Nie
*
,
Lunjie
Wu
,
Yan
Xu
and
Yao
Nie
*
Laboratory of Brewing Microbiology and Applied Enzymology, School of Biotechnology and Key Laboratory of Industrial Biotechnology, Ministry of Education, Jiangnan University, Wuxi, China. E-mail: ynie@jiangnan.edu.cn
First published on 15th October 2024
Abstract
Covering: up to 2024
Fe(II) and 2-oxoglutarate-dependent dioxygenases (Fe/2OG DOs) are a superfamily of enzymes that play important roles in a variety of catalytic reactions, including hydroxylation, ring formation, ring reconstruction, desaturation, and demethylation. Each member of this family has similarities in their overall structure, but they have varying specific differences, making Fe/2OG DOs attractive for catalytic diversity. With the advancement of current research, more Fe/2OG DOs have been discovered, and their catalytic scope has been further broadened; however, apart from hydroxylation, many reaction mechanisms have not been accurately demonstrated, and there is a lack of a systematic understanding of their molecular basis. Recently, an increasing number of X-ray structures of Fe/2OG DOs have provided new insights into the structural basis of their function and substrate-binding properties. This structural information is essential for understanding catalytic mechanisms and mining potential catalytic reactions. In this review, we summarize most of the Fe/2OG DOs whose structures have been resolved in recent years, focus on their structural features, and explore the relationships between various structural elements and unique catalytic mechanisms and their associated reaction type classification.
 Songyin Zhao | Songyin Zhao graduated from Jiangnan University with a BS in biotechnology, and obtained his master's degree in 2021 from Hubei University of Technology. He is currently studying for a PhD degree from Jiangnan University, supervised by Professor Yao Nie. His research interests include structural biology, natural product biosynthesis, biocatalysis, and enzyme engineering. |
 Lunjie Wu | Lunjie Wu studies food science and engineering at Xihua University, where he received his master's degree. He is pursuing a PhD degree under the supervision of Prof. Yao Nie and Yan Xu in Jiangnan University. His is currently a PhD student whose research interest focuses on computer-aided protein engineering and structural bioinformatics. |
 Yan Xu | Yan Xu received his PhD degree from the Jiangnan University. He is a professor in the School of Biotechnology at Jiangnan University. He was selected as a fellow of the Department of agricultural and food chemistry of the American Chemical Society. Currently, he has published about 270 international papers. His research interests are mainly focused on applied microbiology and industrial enzymology. As the first contributor, he won the National Award for Technological Invention in 2013. |
 Yao Nie | Yao Nie received his PhD degree from Jiangnan University. He studied at Rutgers University as a postdoctoral scholar. Currently, he is a professor in the School of Biotechnology at Jiangnan University. He has published about 100 international papers. His research interests are mainly focused on the theory and application of microbial molecular enzymology and catalytic engineering. |
1 Introduction
Fe(II) and 2-oxoglutarate-dependent dioxygenases (Fe/2OG DOs) were discovered as hydroxylases in the 1960s.1 Since then, research on the superfamily of Fe/2OG DOs has gradually deepened, and an increasing number of family members have been discovered. Fe/2OG DOs can not only catalyze hydroxylation but have been proven to catalyze a variety of important biochemical reactions (Table 1).25–27 Fe/2OG DOs use abundant O2 and 2OG to directly functionalize C–H at multiple sites and perform fine chemical, regional, and spatial control based on the unique binding modes between family members and substrates, which can be described as “one step to success” in chemical synthesis.27–30 The catalytic diversity of Fe/2OG DOs enables them to play different roles in various biological processes. Fe/2OG DOs are closely associated with transcriptional regulation, chromatin modification, metabolite metabolism, growth hormone synthesis, and development in animals and plants. Their functions, such as hydroxylation and demethylation, enable them to affect multiple processes in the central dogma of molecular biology.31 In humans, the inactivation of various Fe/2OG DOs is associated with cancer, such as EGLN1,32 JARID2,33 and KDM2B,34 the amplification, silencing, deletion, or mutation of their encoding genes appear in cancer cells. In addition, some other Fe/2OG DOs indirectly deregulate cells through the action of “cancer metabolites”.35 Several Fe/2OG DOs have also been found to promote or inhibit tumour growth in preclinical cancer models, suggesting the potential of Fe/2OG DOs as cancer therapeutic targets. In plants, bacteria, and fungi, Fe/2OG DOs play an important role in the biosynthesis of secondary metabolites (natural products) (Table 1),27,28,30,36 and some members also contribute to the decompose of pesticides.37 Many Fe/2OG DOs are involved in the synthesis of natural products and their derivatives used as drugs, such as penicillin and vinblastine.38,39 Fe/2OG DOs shine in the synthesis of natural products because of their unique catalytic characteristics, greatly promoting the efficient production of natural products.28,30,40 The catalysis of Fe/2OG DOs is diverse and highly efficient, and they can achieve siteselective regulation and reaction type regulation, which gives them great potential in catalytic mechanism research and biosynthesis applications.| Reaction type | Enzyme | Reaction | Biological activity | Ref. |
|---|---|---|---|---|
| Hydroxylation | VioC |
|
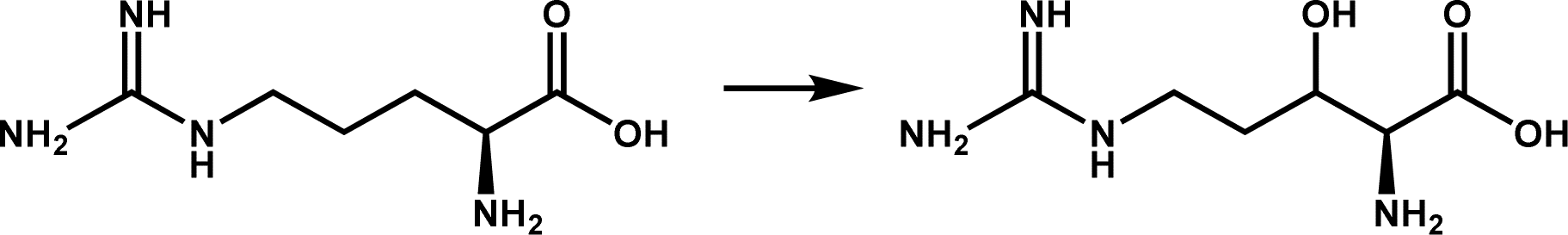
L-Arginine hydroxylation; product derivatives are precursors for the synthesis of non-ribosomal peptides, such as the tuberculin antibiotic vitamycin | 2 |
| JMJD5 |
|
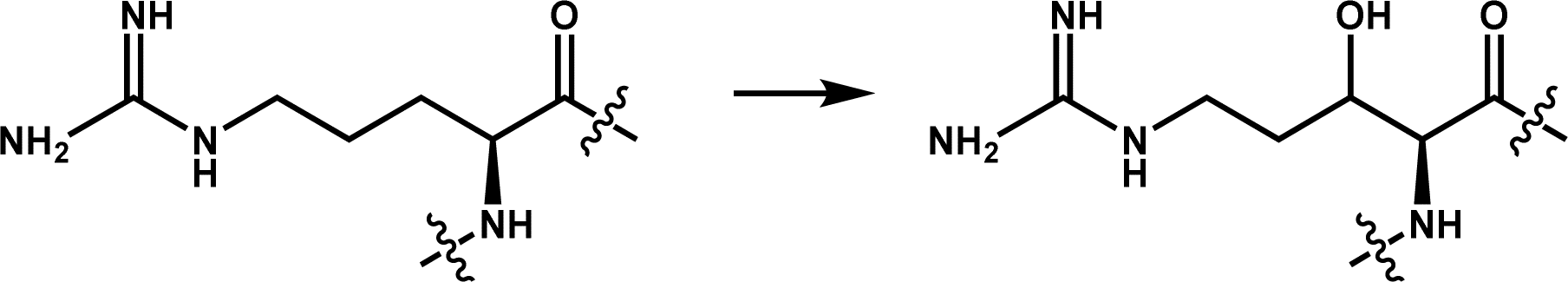
A arginyl hydroxylase that may catalyze the hydroxylation of proteins involved in translation | 3 | |
| GriE |
|
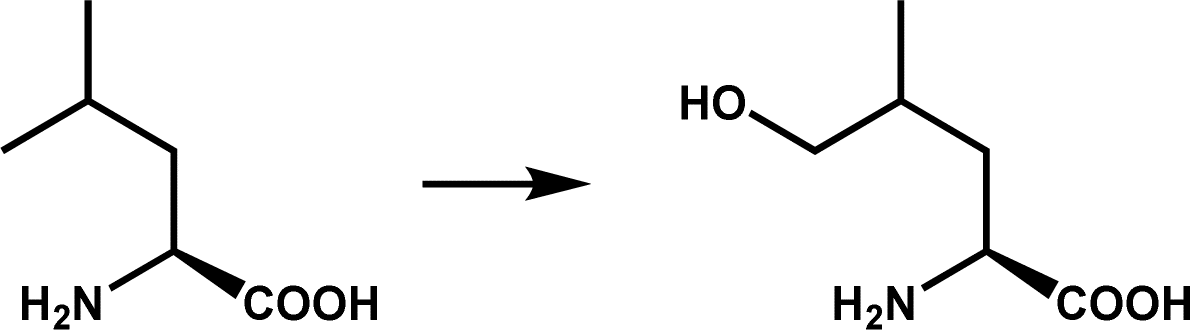
L-Leucine hydroxylation; 5-hydroxyleucine is a precursor of (2S,4R)-4-methylproline biosynthesis involved in griselimycin synthesis | 4 | |
| CsiD |
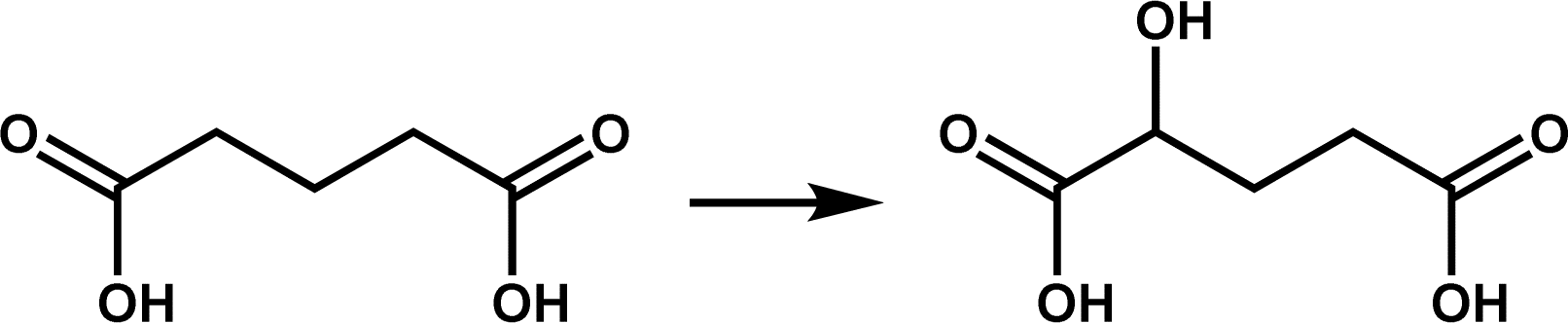
|
Participates in lysine degradation, and the product has the potential to be used in co-substrate recycling | 5 | |
| KDO1 |
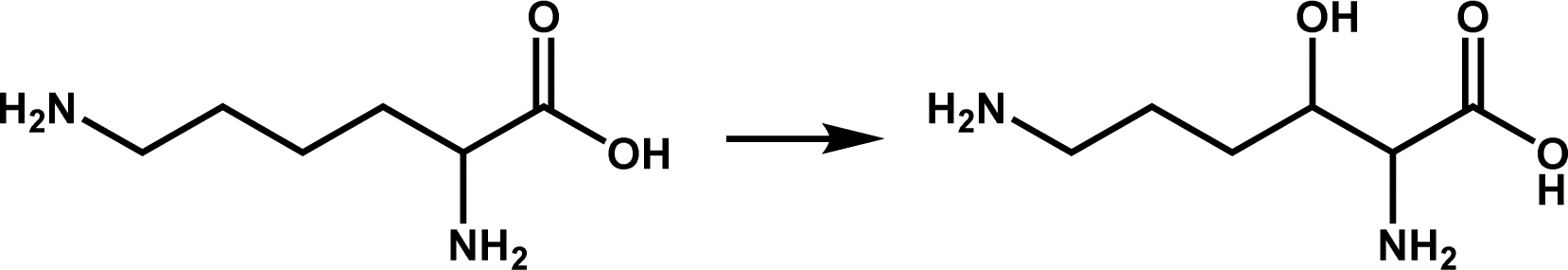
|
Lysine hydroxylation | 6 | |
| KDO5 |
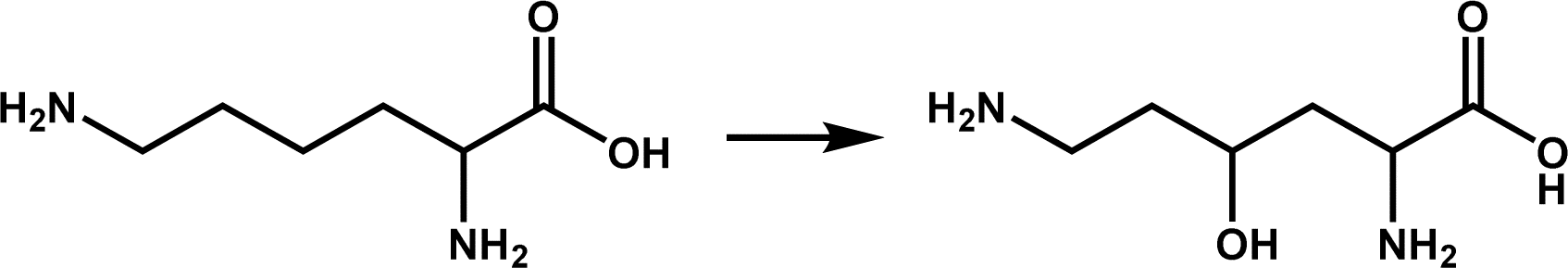
|
Lysine hydroxylation | 6 | |
| GA2ox3 |
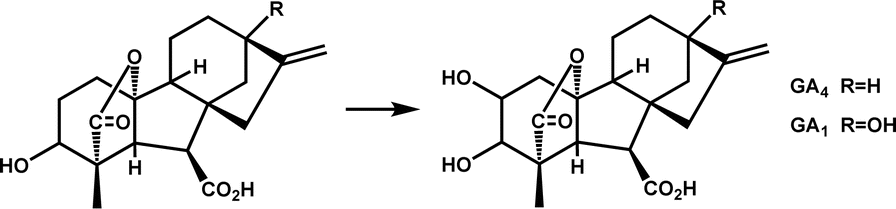
|
Participates in the synthesis and metabolism of gibberellins and regulates plant growth hormones | 7 | |
| Ring formation | DPS |
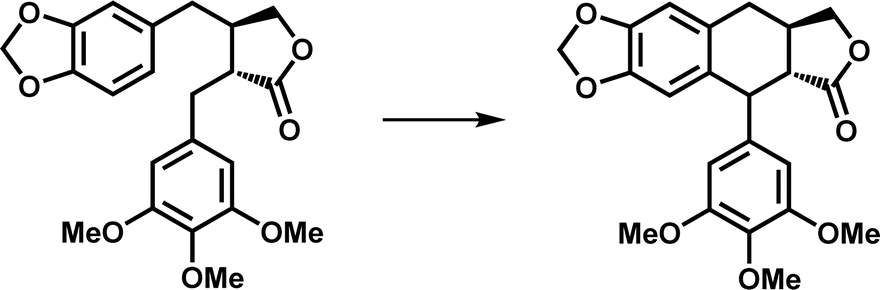
|
The product is the core skeleton of anti-tumor natural product podophyllotoxin | 8 and 9 |
| CTB9 |

|
Participates in the biosynthesis of the natural product cercosporin | 10 | |
| Ring rearrangement | DAOCS |
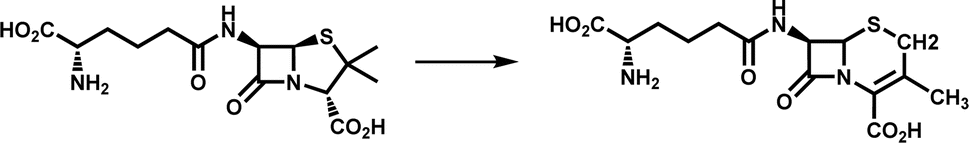
|
Participates in the biosynthesis of cephalosporin antibiotics | 11 |
| TropC |
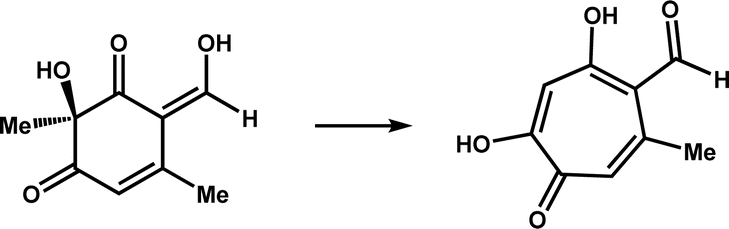
|
Participates in the biosynthesis of tropolone natural product stipitatic acid | 12 | |
| EasH |
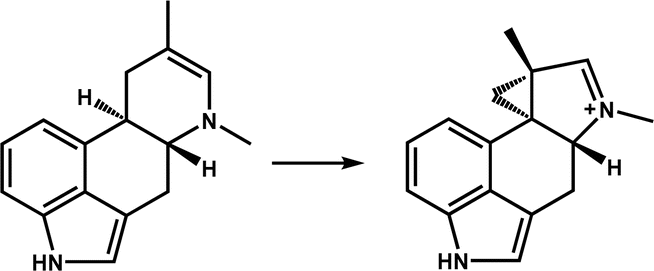
|
Participates in the biosynthesis of the ergot alkaloid cycloclavine | 13 | |
| Desaturation | NapI |

|
Arginine desaturation; participates in naphthyridinomycin biosynthetic pathway | 14 |
| VioC |

|
L-Homoarginine desaturation | 14 | |
| AsqJ |
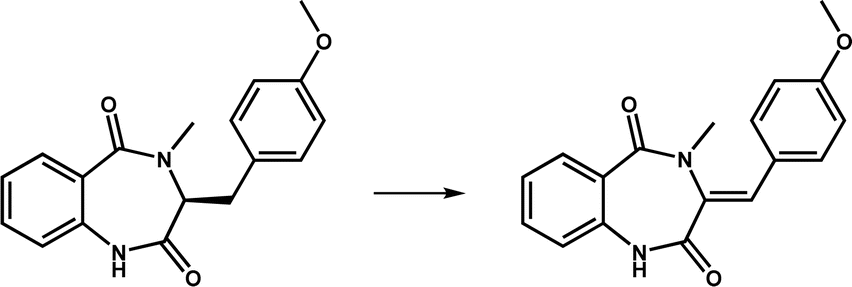
|
The catalytic product is present in a variety of quinolone alkaloids and is an important precursor of antibacterial and antitumor compounds | 15 | |
| Demethylation | ALKBH1 |

|
An important DNA demethylase that regulates genome N6-mA turnover of unpairing regions associated with dynamic chromosome regulation | 16–18 |
| Oxidative deamination | KanJ |
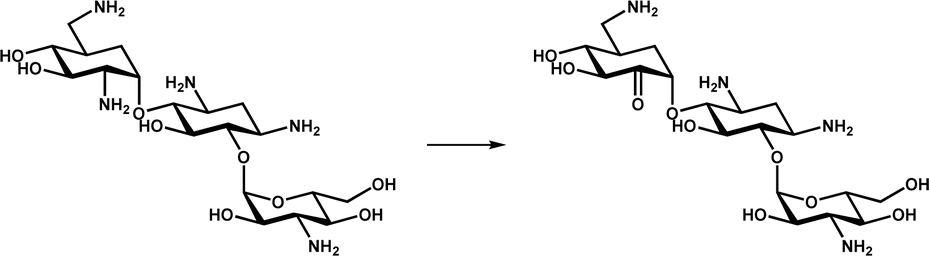
|
Participates in the biosynthesis of the aminoglycoside antibiotic kanamycin A | 19 |
| Endoperoxidation | FtmOx1 |
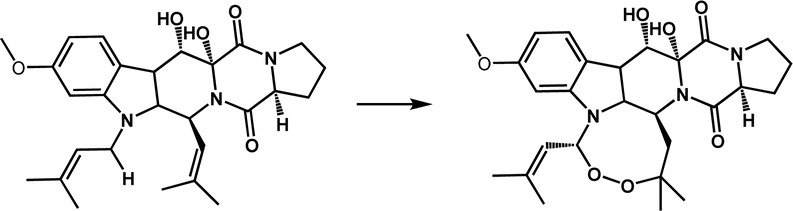
|
Participates in the biosynthesis of fumitremorgin A | 20–22 |
| NvfI |

|
Catalyzes the formation of fumigatonoid A in the biosynthesis of novofumigatonin | 23 | |
| Muti-functionalization | SptF |

|
Participates in the biosynthesis of various terpenoids and plays an important role in the synthesis of the fungal meroterpenoid emervaridone | 24 |
|
The structure of a protein determines its function; therefore, exploring the mechanism of enzymatic reactions through structural biology is an important method in biosynthesis, and structure-based enzyme engineering is an important technology for industrial applications.41,42 The catalytic diversity of the Fe/2OG DO family reflects their structural diversity. To date, the structures of approximately 100 family members have been determined. Observing the Fe/2OG DOs from the perspective of their structure, two common features can be noted. First, all Fe/2OG DOs contain a conserved double-stranded β-helix (DSBH or cupin) fold; and second, they all have crucial His1-X-Asp/Glu-Xn-His2 Fe coordination components (H-X-D/E-Xn-H motif), except for the family members that are halogenases.43–48 Halogenases use Gly/Ala residues instead of Asp/Glu in the Fe coordination components. Fe/2OG DOs use Fe(II) as a metal coenzyme and 2OG as a co-substrate and require oxygen for the reaction. The H-X-D/E-Xn-H motif forms a coordination with Fe(II), which stabilize the Fe atom and lay the foundation for subsequent catalytic reactions.46
Based on structural biology information, some reactions catalyzed by Fe/2OG DOs are understood in detail, especially hydroxylation.25,49,50 However, members of the Fe/2OG DOs family have a wide catalytic range; research on many reactions remains limited, and their structure and mechanisms lack comprehensive and systematic understanding. In this review, we focus on the structure–function relationships of Fe/2OG DOs and classify and discuss them according to their reaction types. We provide an overview of the current understanding of the structures of Fe/2OG DOs that have been addressed in recent years, discuss the structural characteristics of these enzymes, and explore their catalytic mechanisms based on these structural characteristics. We demonstrate that there is still a wide range of catalytic reactions of Fe/2OG DOs can be explored. In-depth analysis of their mechanisms will help deepen our understanding of the principles of various catalytic reactions, and provide the foundation for their future wide application, including the synthesis of natural products and derivatives.
2 General structure features
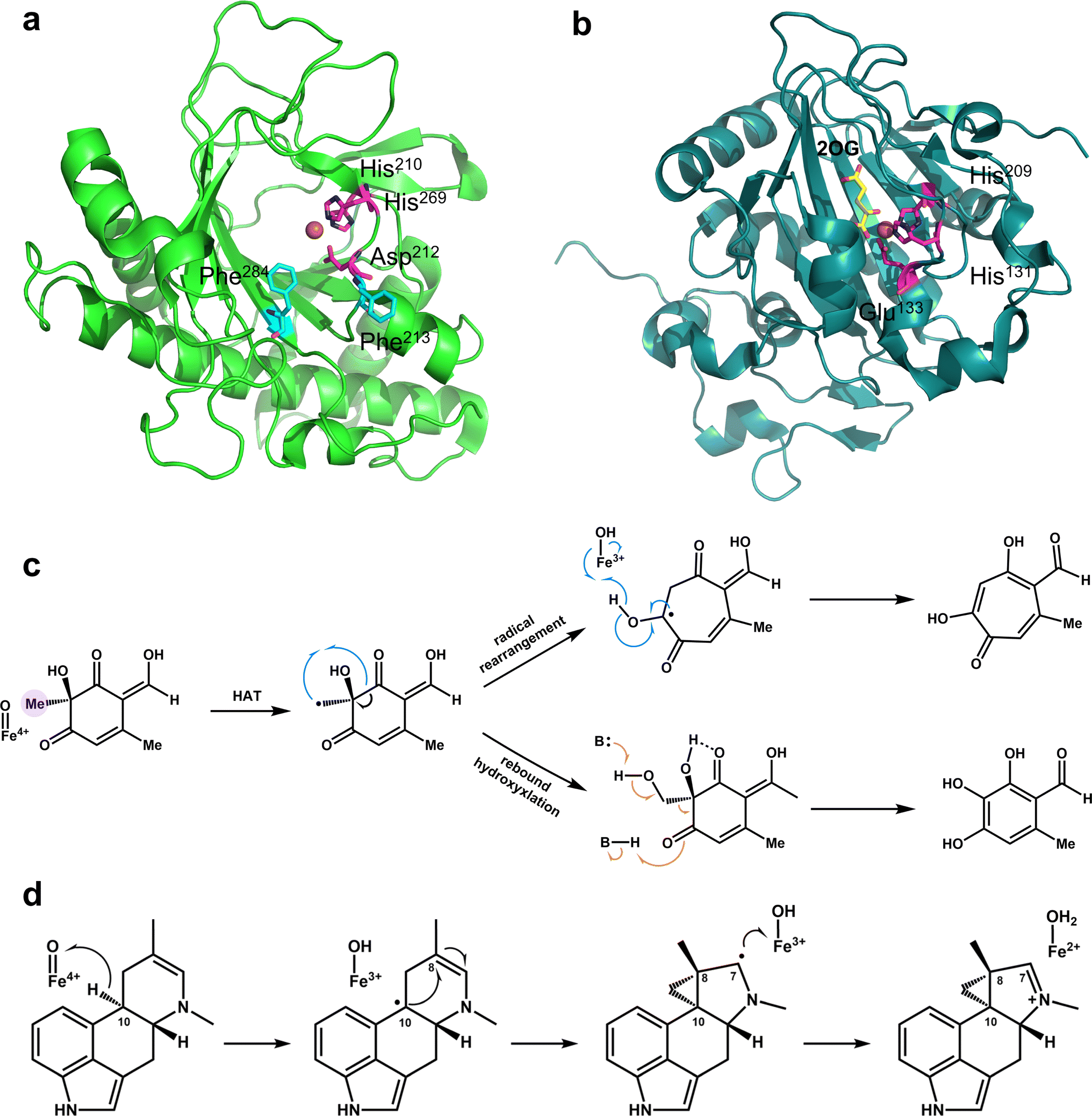
All Fe/2OG DOs have a “jelly-roll” structure, which is essentially a protein topological skeleton built with a DSBH as the core (Fig. 1). Fe/2OG DO family members are numerous, and their overall sizes differ; some members are composed of less than 300 amino acids (e.g., AlkB and RdpA),51,52 while others are composed of nearly a thousand amino acids or even more (e.g., PHF and JMJD).53,54 Despite their large differences in size, the “jelly roll fold” is characteristic of them all and is also a ubiquitous fold in mononuclear non-heme iron enzymes.46,47,55–57 The DSBH core of Fe/2OG DOs is composed of eight antiparallel β strands, forming a β sandwich structure consisting of two four-stranded antiparallel β-sheets (Fig. 1b).46,47 The two β-sheets are of varying lengths, and they support active sites and provide selectivity for binding to primary substrates. Beyond the “jelly-roll” topology, there is no significant consistency in the secondary structural modules of each Fe/2OG DOs member, with each forming its own unique function according to its specific structure. Taking members of JmjC hydroxylase as an example, JMJD5, which can function as an arginine demethylase, consists of eight β-sheets at the core, and several α-helices and small helices play stabilizing roles in the DSBH. At the N-terminus of the DSBH, there are multiple β-sheets, α-helices, and a few structural elements, such as hairpins, between β-sheets, and a flexible loop is also present.3 Compared to homologous JmjC-hydroxylase members, the DSBH-containing core framework structure is not significantly different; however, there are differences in other structural elements. TYW5, HIF-1α, and YcfD have a dimerization structural element composed of two helices,58–60 and YcfD has an additional “Winged helix” domain. PHF8 has a plant homeodomain,61 and KDM4A has multiple α-helix elements.62 These results indicate that Fe/2OG DOs have the same core structural framework, but different types have unique structural elements. | ||
| Fig. 1 Fe/2OG DOs have a DSBH core framework in their overall structure, and the conserved H-X-D/E-Xn-H motif is located inside the DSBH. (a) Crystal structure of GriE (PDB code: 5NCI). The DBSH is shown in red and orange, the remaining β-sheets are shown in blue-grey, the α helices are shown in sky blue, and the N- and C-termini are marked in green. The enlarged panel shows the binding pocket, with iron shown as a brown sphere, the D/E-Xn-H motif as purple sticks, 2OG as yellow sticks, and the substrate as cyan sticks. (b) Composite topology diagram of GriE. The main secondary structural elements follow the same colour scheme of (a). | ||
When catalysis is carried out after binding the substrate and cofactor, the “jelly-roll” core of Fe/2OG DOs does not deform,63 but the overall structure tends to condense. Taking FtmOx1 as an example, in the absence of any ligand, the active pocket size of apo-FtmOx1 is 503 Å3; after binding with 2OG, the active pocket size is 418 Å3, and when the substrates fumitremogin B and 2OG are combined, the active pocket is further compressed to 288 Å3.20
Many Fe/2OG DOs exhibited polymerization characteristics in their structure. Oligomerization is not uncommon in protein crystallization, and many of the classical crystal structures of Fe/2OG DOs also have oligomerization, such as γ-butyrobetaine hydroxylase BBOX and the asparaginyl hydroxylase factor inhibiting HIF (FIH).59,64,65 Polymerization affects the catalytic processes of some family members. Crystal structure of Oryza sativa gibberellin 2-oxidase 3 (OsGA2ox3) is composed of homotetramers. There are two main reasons for the formation of homotetramers—the disulfide bond formed by the Cys194 of the two monomers, and hydrogen bonds mediated by the substrate molecule GA4 at the interface in a face-to-face manner. Lys308 in subunits A and D forms a salt bridge with the C-6 carboxyl group of the interfacial GA4 via water molecules. K313 in subunit A interacts directly with Pro87 and Phe91 in subunit D through hydrogen bonding and vice versa. Based on enzymatic kinetic analysis of tetrameric OsGA2ox3, the Km value of the tetramer was found to be almost ten times lower than that of the monomer, and the Vmax was consistently more than three times higher than that of the monomer.7 These results indicate that polymerization significantly enhances the enzymatic activity of OsGA2ox3.
3 Binding pocket and surrounding flexible structures
The binding pocket of Fe/2OG DOs is located between two β-sheets, and the most typical feature of the binding pocket is the H-X-D/E-Xn-H motif. The H-X-D/E-Xn-H motif is located at the end of strand βII, the loop connecting βII and βIII, and the beginning of strand βVII, near the minor sheet of the “jelly-roll”.46 The entire pocket can be divided into two parts based on its spatial position: the substrate-binding pocket and the 2OG binding pocket. The conserved arginine in the 2OG binding pocket is responsible for the binding and localization of 2OG. The two pockets surround the Fe-binding motif, and the cavities overlap at a certain position but are not completely located on either side of the motif. Before catalysis, the substrate and 2OG enter the binding pocket through their respective channels. After catalysis is completed, succinic acid is located in the previous 2OG binding pocket, the corresponding product is located in the substrate-binding pocket, and the positions of the two pockets remain largely unchanged.Near the binding pocket, in addition to some conservative functional motifs, there are also some flexible elements, which play an especially important “gating” role in the regulation of catalytic reactions (Fig. 2). JMJD5, with a C-3 arginyl hydroxylation function, has two flexible loops surrounding the active site, the β3–β4 loop of the N-terminal of DSBH and the βIV–βV loop of DSBH (Fig. 2a–c). From the solved crystal structures, a change in the flexible loop can be inferred as the catalytic cycle progresses. In the structure of apoprotein JMJD5, part of the βIV–βV loop presents a hairpin structure, which is close to the β3–β4 loop, forming a “closed” conformation (Fig. 2a). As ferrous ion and 2OG enter the active pocket, the hairpin structure of the βIV–βV loop is released and becomes a partial helical conformation, which moves to the outside of the active site together with the β3–β4 loop, forming an “open” entrance (Fig. 2b). When the substrate enters the active pocket, the conformation of the β3–β4 loop changes slightly, getting closer to the βIV–βV loop, forming a “half-open” conformation (Fig. 2c).3 This conformation is also present in the complex structure of JMJD5 complex with the product. The β3–β4 loop then becomes disordered as the product leaves the binding pocket. Through the analysis of these structures, it has been deduced that these two loops play a “gating” role in the catalytic process, promoting substrate binding and product release.
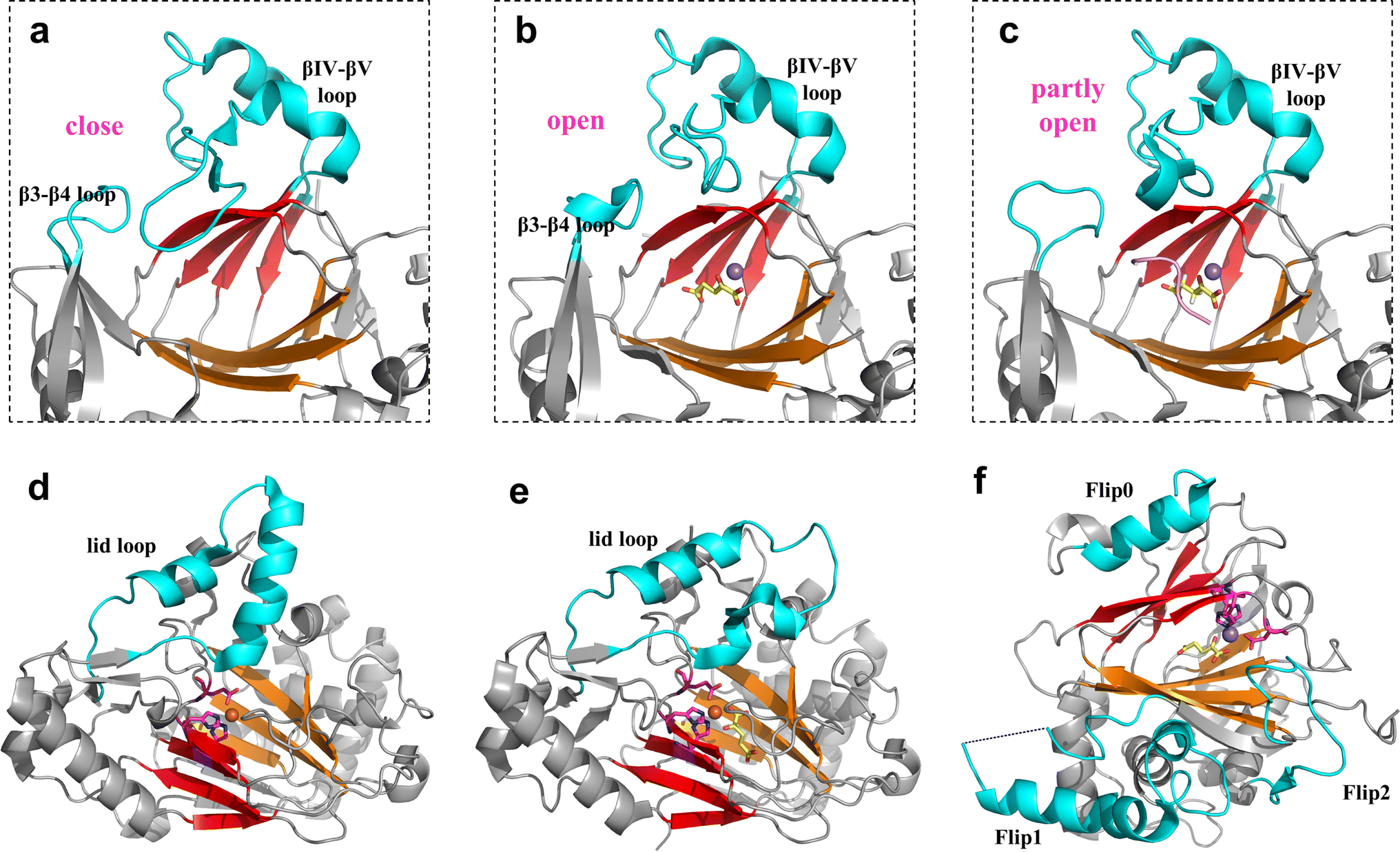 | ||
| Fig. 2 Flexible elements that play a “gating” role in Fe/2OG DOs. Flexible elements are shown in cyan. DSBH is shown in red and orange. (a–c) Crystal structure of JMJD5 (PDB code: 6F4M, 6F4N, 6F4S). Each panel shows the loop conformation of JMJD5 in different states. (d) Crystal structure of NapI (PDB code: 6DAW). (e) Crystal structure of VioC (PDB code: 6ALM). (f) Crystal structure of hALKBH1 (PDB code: 6IE2). | ||
In general, all Fe/2OG DOs contain at least one flexible loop that acts as a lid. L-Arg 3-hydroxylase VioC and L-Arg 4,5-desaturase NapI both have a lid loop in the substrate-binding pocket, which is involved in substrate binding.14 However, the lid loop does not exhibit structural consistency. The sequence identities of VioC and NapI are approximately 50%, and their overall structures are very similar. When the structures of VioC and NapI are superimposed, the DSBH framework almost overlaps, and the topology shows almost no change; however, there is a clear difference between the two at the lid loop—the flexible region of VioC is very long, while the flexible region of NapI is short and has an α helix (Fig. 2e and f). There is also no consistency in the number of flexible elements among the different Fe/2OG DO family members.
The human DNA N6-methyladenine (6mA) demethylase hALKBH1 has three flexible elements that regulate binding to the substrate DNA—Flip1, Flip2, and an N-terminal Flip0 unique to hALKBH1 (Fig. 2g). Flip1 and Flip2, which belong to the nucleotide recognition lid (NRL), are typical structures shared by members of the alkB family. However, the Flip1 region of hALKBH1 is special and long, leaving a larger binding space on the active site pocket, and Flip2 contains a pair of antiparallel β-sheets and a long loop with a high B factor, and these unique compositions and conformations may confer substrate selectivity to hALKBH1.16 Flip0 is only present in ALKBH1 in various mammalian species and is highly conserved; it is essential for hALKBH1 activity and for distinguishing single- and paired double-stranded substrates. In addition to the enzymes mentioned above, many family members of Fe/2OG DOs have unique flexible elements, such as hALKBH5, which has a unique Flip3.66 Based on these structural analyses, the “gating” role of flexible elements is pivotal for substrate recognition and control reactions.
4 Catalytic mechanisms in various reactions
4.1 Hydroxylation
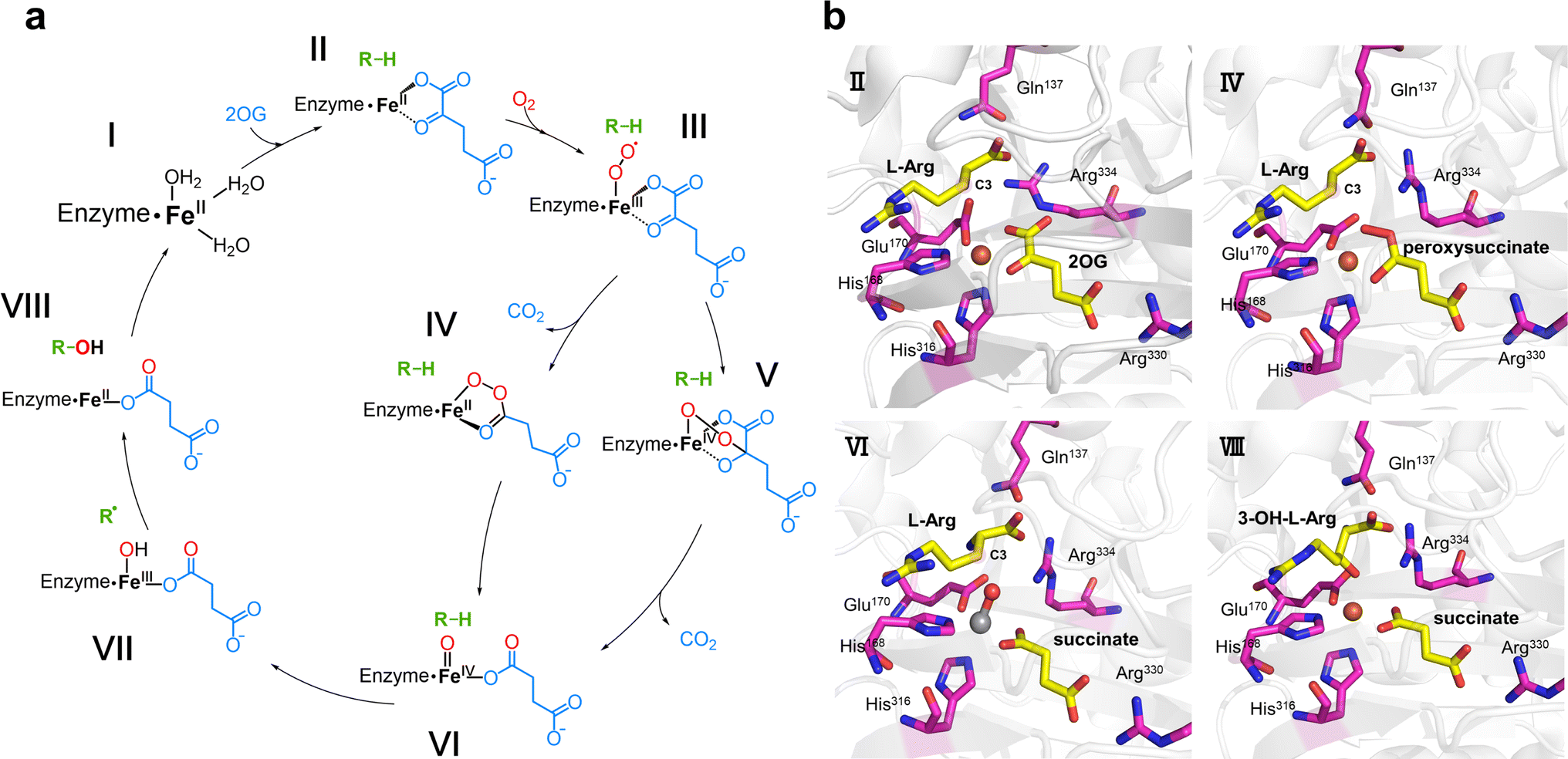
Hydroxylation is the earliest discovered reaction and is also the most widely studied function of Fe/2OG DOs.67 Since the discovery of the earliest family members and the identification of hundreds of other members, research on hydroxylation has been deepened by the expansion of the Fe/2OG DOs family. Hydroxylation is indispensable for the functional exploration of Fe/2OG DOs, and its mechanism is the basis for a variety of catalysis. Based on experimental and computational data, a consensus mechanism for the hydroxylation of Fe/2OG DOs has been established (Fig. 3a).56,68–73 At the beginning of the reaction stage, the H-X-D/E-Xn-H domain coordinates with the Fe(II) and stabilizes it at the catalytic site, and the Fe(II) also coordinates with three water molecules. After 2OG enters the reaction system, the keto group and carboxylic acid of 2OG replace two water molecules to interact with the ferrous ion and form an octahedral shape. Then the substrate and O2 enter the reaction system, O2 replaces the last coordinated water molecule to bind to Fe(II), generating an Fe(III)-superoxide intermediate. This intermediate reacts with 2OG to form a peroxohemiketal bicyclic intermediate, which then initiates subsequent oxidative decarboxylation generates succinate, CO2, and a key tetravalent iron ion intermediate, the Fe(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species (known as the ferryl intermediate). The Fe(IV)O species then abstracts a hydrogen atom from the substrate to generate a substrate radical; simultaneously, the Fe(IV)O intermediate is reduced to the Fe(III)–OH species, the hydroxyl radical rebounds to complete the hydroxylation of the substrate. Finally, the hydroxylation products dissociate and the Fe/2OG DOs return to their initial Fe(II) state, ready for the next reaction cycle.43,74–76
O species (known as the ferryl intermediate). The Fe(IV)O species then abstracts a hydrogen atom from the substrate to generate a substrate radical; simultaneously, the Fe(IV)O intermediate is reduced to the Fe(III)–OH species, the hydroxyl radical rebounds to complete the hydroxylation of the substrate. Finally, the hydroxylation products dissociate and the Fe/2OG DOs return to their initial Fe(II) state, ready for the next reaction cycle.43,74–76
 | ||
| Fig. 3 Fe/2OG DOs-mediated hydroxylation and the crystal structure of the hydroxylase VioC. (a) Proposed mechanism of VioC-catalyzed hydroxylation. (b) Crystal structure of VioC in various states (PDB code: 6ALM, 6ALO, 6ALR, 6ALP). The state of identification is consistent with (a). Fe(II) is shown as a brown sphere, while the V–O vector is shown as a grey and red ball-and-stick model. | ||
Recently, an increasing number of Fe/2OG DOs have been discovered as hydroxylases, and as such, the hydroxylation functions of Fe/2OG DOs have greatly broadened. Combined with the study of X-ray crystal structures, researchers have developed a more detailed and profound understanding of hydroxylation mechanisms beyond the classical cycle.
In the study of Fe/2OG DOs, the presence of Fe(IV)O species is very important for the catalytic process, especially for proton transfer. Mitchell et al.2 further investigated the mechanism of VioC, which can hydroxylate the C3 of L-arginine. They obtained VioC complex crystals at multiple stages in the catalytic process (Fig. 3b), particularly the use of a vanadium atom to replace the Fe atom, and realized the key Fe(IV)O state simulation, providing a detailed analysis of the mechanism of the catalytic intermediate state. In the VioC structure of the Fe(II)·2OG·L-Arg reactant complex in the absence of O2, the C1 carboxylate of 2OG shifts by approximately 35° from the equatorial plane, defined by the His168, Glu170, and 2OG ligands. The Fe(II) centre in VioC exhibits a distorted five-coordinate geometry, and the C3 of L-Arg is located 4.8 Å from the cofactor, which is a distance suitable for H atom transfer (HAT). After O2 enters the reaction system, an intermediate containing Fe(II) and peroxysuccinate is obtained. The structure shows that after decarboxylation of 2OG, C2 connects to one oxygen atom in O2, and the Fe atom of the catalytic centre coordinates with the other oxygen atom, forming a ferryl precursor. Subsequently, the structure of the ferryl state is efficiently and stably simulated by the complex crystal structure of VioC with the vanadyl ion, L-Arg, and succinate (Fig. 3b). A shift of approximately 25° in the position of the V–O vector is observed from its location in the peroxysuccinate complex, away from His316 by about 180°, so that its O is close to the C3 of the substrate. This appropriate positioning of the V–O vector is conducive to accepting the C3 pro-S hydrogen of L-Arg during the HAT step, paving the way for the subsequent hydroxylation of C3.
In addition to providing a new perspective on the intermediates, these structures also demonstrate the function of key amino acids in the catalytic pocket—most notably Arg and Gln. In the structure of VioC complexed with Fe(II)·2OG·L-Arg, the electron densities of Arg334 and Gln137 are not very clear; however, in the crystals corresponding to the subsequent catalytic process, both Arg334 and Gln137 have very complete and specific electron densities. The multiple structures indicated that the coordination mode of 2OG and the positions of Arg334 and Gln137 are significantly different in the reactant and product states. In the product complex structure, the side chain of the 3-OH-L-Arg product is significantly twisted to achieve coordination of the hydroxyl group, and the guanidine moiety is rotated and accommodated by the concomitant displacement of the protein side chain within the substrate-binding pocket. The α-NH3+ group of L-Arg moves away from the Glu170 and interacts with Gln137. The Glu170 carboxylate rotates towards the side chain of Arg334, and as it moves into the space vacated by C1 of 2OG, it becomes ordered to anchor an H-bonding network involving L-Arg carboxylate and the O1 of succinate. The structures of the vanadyl and peroxysuccinate intermediates suggest that Arg334 plays a key role in stabilizing Fe–O adducts at certain coordination sites, and the direct interaction between the O–O unit of the Fe(II)–peroxysuccinate complex and the side chain of Arg334 in its newly ordered position suggests a mechanistic role for the second-sphere residue, presumably to compensate for the different charge distributions of the nascent peroxide adducts promoting decarboxylation and C2–O bond formation. In VioC, Arg334 promotes an in-line oxo configuration that the Fe–O vector is oriented perpendicularly to the target substrate C–H bond through electrostatic interactions, steric retardation of ligand dynamics, and the maintenance of an extended active-site H-bond network. In many of the Fe/2OG DO structures resolved to date, the Arg near the active site involved in binding to 2OG and succinate is generally conserved, which is consistent with its usual role in directing the position of oxygen ligands. In other studies, the function of the enzyme may have been affected when the Arg occupying the site was transformed. For example, in the typical chlorinase WelO5, a Phe occupies the site occupied by Arg334 in VioC, and geometrically different ferryls located in the equatorial plane are used to achieve selective halogenation in this system.77 These structures indicate that Arg and Gln located in the binding pocket play important roles in the hydroxylation function of Fe/2OG DOs and provide a promising method for the subsequent reprogramming of enzyme function.
In recent years, members of the hydroxylase family have provided new insights into the recognition of similar substrates by Fe/2OG DOs. The glutarate hydroxylase CsiD can catalyze the hydroxylation of glutarate (GA), a structurally similar substrate of 2OG, to obtain the reaction product L-2-hydroxyglutarate (L2HG), which is a metabolite that inhibits Fe/2OG DOs but weakly inhibits CsiD (Fig. 4a). In the obtained CsiD crystal structure complexed with the substrate GA and the 2OG-analogue NOG (N-oxalylglycine, where atom C3 is substituted by nitrogen compared to 2OG, commonly used in the crystallization of Fe/2OG DOs), GA is located in the substrate-binding pocket through the oxygen atom of one of its terminal carboxyl groups directly coordinated to Fe(II) ions, and the carboxyl group at the other terminal forms a salt bridge with the backbone nitrogen of the conserved residues Arg311 and Gly163. NOG binds Fe(II) ion in the 2OG binding pocket through the terminal oxygen atom of its oxalyl moiety and forms a salt bridge with Arg309 (Fig. 4b). In the complex crystal structure of the product and CsiD, succinate generated by the decarboxylation of 2OG occupies the 2OG binding pocket, binds to Fe(II) in a bidentate manner through the terminal carboxyl group, and further interacts with Arg309 through a low-occupancy solvent molecule. In both crystal structures, the conformation of CsiD remains unchanged.5 The cognate ligand always occupies the corresponding site, while the other sites are not occupied. These results indicate that CsiD has a high degree of specificity in the binding of structurally similar substrates, co-substrates, and products and explain the weak inhibition efficiency of L2HG for CsiD.
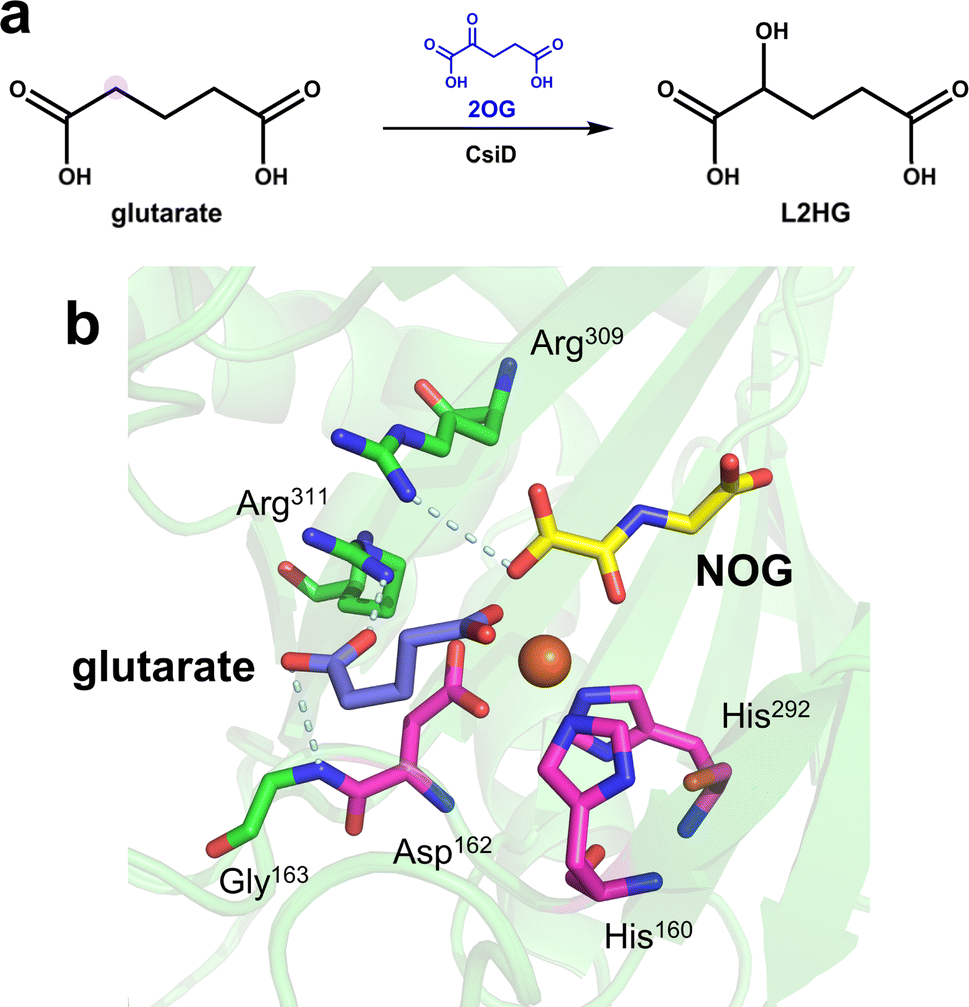 | ||
| Fig. 4 Catalytic reaction and structure of CsiD. (a) CsiD catalyzes the hydroxylation of glutarate to produce L-2-hydroxyglutarate. (b) Crystal structure of CsiD (PDB code: 6GPN, 6HL8). The model was reconstructed by aligning the individual crystal structures of CsiD bound to each ligand; the PDB code for the structure of CsiD complexed with NOG and Fe(II) is 6GPN, and the PDB code for the structure of CsiD complexed with glutarate and Fe(II) is 6HL8. | ||
Amino acid hydroxylation is also a field of extensive research and application of Fe/2OG DOs and is an important part of natural product synthesis. Family members that catalyze the hydroxylation of free amino acids include L-leucine 5-hydroxylase (GriE), L-proline hydroxylase (P4H), L-isoleucine hydroxylase (IDO), N-succinyl L-amino acid β-hydroxylase (SadA), L-enduracididine 3-hydroxylase (MPPO),4,78–80 and the L-arginine hydroxylases mentioned earlier. In addition to these classic hydroxylases, members of the Fe/2OG DO family have been found to catalyze lysine hydroxylation. The structures of the lysine-3-hydroxylase KDO1 from Catenulispora acidiphila and lysine-4-hydroxylase KDO5 from Flavobacterium have been elucidated in recent years (Table 1).6 In the KDO1 structure complex with L-lysine, the carboxylate of L-lysine forms hydrogen bonds with Ser168 and Ser236, forming a salt bridge with Arg332; the 2-amino group forms a salt bridge with Glu180, and the 6-amino group forms a salt bridge with Asp234 and Asp268. These interactions make lysine very close to the Fe coordination centre, and C3 is only 4.0 Å away from the Fe(II) ion. In the structure of KDO5, the 6-amino group of L-lysine is relatively loose, and the carboxylate forms a salt bridge with Arg338 and Arg145 to stabilize it and is positioned through a hydrogen bond with Ser167. Compared to KDO1, L-lysine is farther away from the Fe coordination centre, C2 and C3 have steric hindrance to Fe ions because of the presence of Arg338, and C4 is closer to the Fe(II) ion. Although the specific catalytic mechanisms of KDO1 and KDO5 have not yet been fully elucidated, current understanding indicates that the differences in the substrate-recognition positions of KDO1 and KDO5 lead to differences in the selective catalysis of KDO1 and KDO5, respectively. In addition, many prolyl hydroxylase structures have been reported in recent years that promote the treatment of anemia.81–84 Thus, research to date on the hydroxylation of Fe/2OG DOs has made significant progress, and further significant breakthroughs are expected.
4.2 Ring formation
Oxidative ring formation is of great value in synthetic biology, particularly for the biosynthesis of natural products; it plays a key role in the biosynthetic pathways of many drugs that have been used clinically or approved by the Food and Drug Administration, such as penicillin and etoposide. Since the end of the 20th century, considerable progress has been made in research on the structure and mechanism of Fe/2OG DOs related to oxidation ring formation.Isopenicillin N synthase (IPNS) catalyzes the transformation of L-δ-(α-aminoadipoyl)-L-cysteinyl-D-valine (ACV) (Fig. 5), the product isopenicillin N is the precursor of a variety of penicillin compounds.85–88 This unique ring-forming reaction is of great value in the biosynthesis of penicillin and cephalosporin.89 IPNS is a member of the Fe/2OG DO structural superfamily,38 Although the catalytic reaction of IPNS does not require 2OG, the characteristic structure of IPNS is consistent with Fe/2OG DO. Therefore, exploring the structural changes and mechanisms of IPNS plays an important guiding role in the ring formation catalyzed by the non-heme oxygenase family. In previous studies, some basic structural understandings of IPNS have been obtained.63,85,86,90–96 Recently, the application of time-resolved serial femtosecond crystallography (tr-SFX) technology on IPNS has deepened the study of its structure and mechanism.95,96 In the IPNS structure without a substrate or O2, Fe is coordinated to the H-X-D/E-Xn-H motif, two water molecules, and one Gln330, which is different from the classical hydroxylation mechanism starting point of Fe/2OG DOs. From the structure of IPNS compounded with ACV and Fe(II), it can be seen that when the substrate ACV enters the enzyme active centre, Gln330, and coordinated water away from the Fe(II) core region, Fe(II) coordinates with sulfur on the cysteinyl group of ACV, Arg87 produces electrostatic interaction with the L-α-aminoadipoyl carboxylate group, and multiple amino acid side chains in the active centres produce hydrophobic interactions with the valine isopropyl group—these are important for IPNS to recognize and locate ACV (Fig. 5a). As the reaction proceeds, IPNS forms Fe(III)-superoxo with the substrate, abstracts the cysteinyl C3 hydrogen atom, and then generates the intermediate Fe(II)–OOH through electron transfer. Fe(II)–OOH abstracts the amide hydrogen and mediates the formation of the monocyclic β-lactam intermediate and the Fe(IV)O species. The Fe(IV)O species mediates the abstraction of the C-3 hydrogen of ACV valine, after which the radical reacts with sulfur to generate isopenicillin N (Fig. 5d). The structure and tr-SFX studies of IPNS mutants indicate that the orderly and continuous binding of Fe(II), ACV, and NO/O2 to IPNS can affect the protein conformation in regions relatively far from the active site, including α3 and α10 helix. This evidence highlights the importance of conformational changes extending from the active site of IPNS to the protein surface and implies that dynamic changes in the overall structure of the protein may occur during catalysis, which is also critical for the reaction.
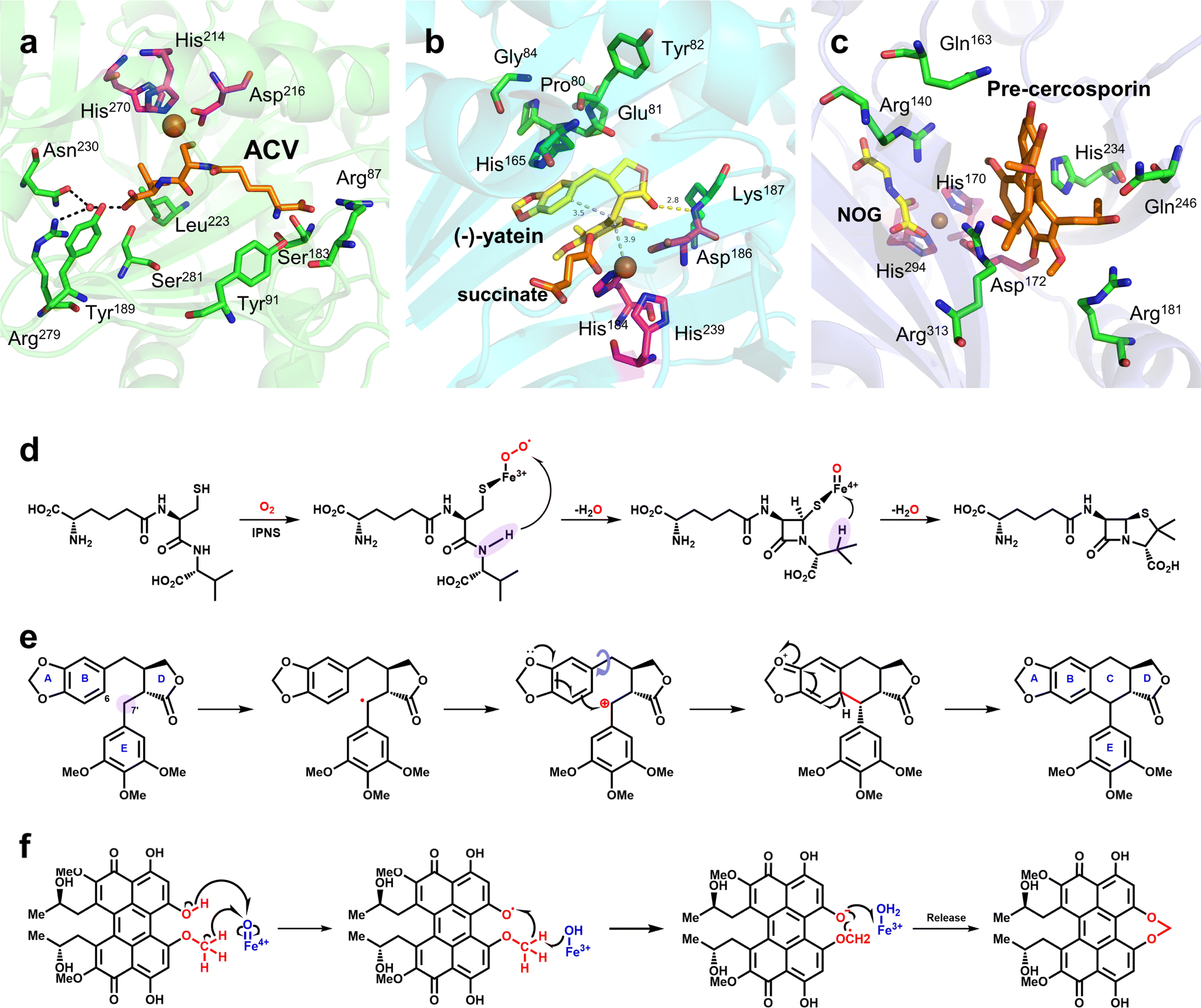 | ||
| Fig. 5 Mechanism of ring formation reaction mediated by Fe/2OG DOs and the structures of related enzymes. (a) Crystal structure of IPNS complex with ACV and Fe(II) (PDB code: 6ZAE). (b) Crystal structure of DPS complex with (−)-yatein, succinate and Fe(II) (PDB code: 7E38). (c) Crystal structure of CTB9 complex with pre-cercosporin, NOG, and Cu(II) (PDB code: 7EUU); the H-X-D/E-Xn-H motif is shown as magenta sticks. (d) Proposed mechanism of IPNS. (e) Proposed mechanism of DPS-catalyzed conversion of (−)-yatein. The site where the Fe(IV)O species abstract hydrogen is highlighted in lavender. (f) Proposed mechanism of CTB9. | ||
At present, some structures and mechanisms of Fe/2OG DOs in ring-forming reactions have been elucidated, particularly for some family members related to natural products, including deoxypodophyllotoxin synthase (DPS) and CTB9. DPS can catalyze conversion of (−)-yatein to deoxypodophyllotoxin (Fig. 5e).8,9 Its structure has been solved in recent years,97 and analysis has shown that DPS uses a mixed radical-polar pathway unseen in other members to achieve C–C linkage to form a ring, which has great significance for this type of reaction and the synthesis of related drugs. In the substrate-complexed DPS crystal structure, (−)-yatein occupies the substrate-binding pocket in a U-shaped conformation. The benzodioxole moiety (A-ring and B-ring) forms a face-to-face π–π interaction with His165, and it is parallel to a polar surface formed by the four main-chain carbonyl oxygen atoms (residues Pro80, Glu81, Tyr82, and Gly84) and a nearby water molecule. There is a buried hydrogen bond between the lactone ring (D-ring) carbonyl oxygen and the backbone amide nitrogen of Lys187. The phenyl ring (E-ring) is fixed in a space-restricted gap through hydrophobic interactions, and a hydrogen bond exists between the p-methoxy group of the E-ring and water molecule. In addition to these interactions, each ring of the substrate also has van der Waals interactions with surrounding residues.97 These complex interactions indicate that DPS has a high precision in substrate recognition and positioning, which is presumably very important for catalysis. There is π–π stacking between the E-ring and the imidazole of the proximal histidine (His184), suggesting that, in addition to its canonical role in Fe coordination, the facial triplet of 2-His-1-carboxylate may contribute to substrate recognition. In the active site, the position of the benzyl carbon (C7′) of the E-ring of the substrate is closer to the Fe centre than that of C6, and the distance between C6 and C7′ of the substrate is 3.5 Å, which predicts the position and steric preference of the resulting C6–C7′ bond. In the DPS structure of composite 2OG and Fe, 2OG is in an offline binding state, and the rearrangement of Fe(IV)O species tends to extract the hydrogen atom at C7′.44 In addition, after analyzing and comparing the structures of the substrates and products, it has been deduced that the formation of the C–C bonds requires substrate rotation. Because D-ring and E-ring are tightly fixed to the active site, this rotation may be generated by A-ring and B-ring (Fig. 5b). Based on the above structural information and analysis, it can be inferred that the DPS pathway catalyzes ring formation by (−)-yatein. DPS mainly extracts a hydrogen atom at the C7′ position, forms a C–C bond to form a ring through the free radical electrophilic aromatic substitution (rEAS) or electrophilic aromatic substitution (EAS) pathway, and finally deprotonates to form the final product (Fig. 5e). This inference is supported by the verification of substrate analogues,97 which is more convincing than the previous inference that quinone-methylated intermediates mediate C–C bond formation.98 The deeper interpretation behind this inference also shows that the cyclization reaction is regulated by many factors, and that this catalysis has strict requirements for the interaction between the substrate and the enzyme as well as the structural flexibility of the substrate.
CTB9 is a member of the Fe/2OG DOs family that catalyzes the formation of a seven-membered methylene dioxygen bridge (MDB) in cercosporin bioprocesses (Fig. 5f).10 MDB exists in a variety of natural products and plays an important antitumor and antiviral role. The seven-membered MDB is an important structural feature of natural products with various biological activities;99,100 however, its formation mechanism has not been elucidated. Recently, Liu et al.101 determined the crystal structure of CTB9 in multiple states and inferred the formation mechanism of the seven-membered methylene dioxygen bridge. By observing the crystal structure of CTB9 complexed with substrate, it was found that the loop region does not change significantly after binding 2OG and substrate; however, the conformation of multiple amino acid residues in the substrate-binding pocket changes, which is different from many Fe/2OG DO family members. To have enough space for the substrate to enter the binding pocket, Arg313 undergoes a significant conformational change to move closer to the Fe ion, and Gln246, His234, Gln163, and Arg181 undergo a slight displacement to form hydrogen bonds with the substrate. Interestingly, Asp172 in the triplet coordinated with Fe also interacts with the phenolic hydroxyl group of the substrate. These amino acid residues are important for CTB9 substrate recognition and stabilization (Fig. 5c). Throughout the catalytic process, before the substrate has entered the binding site, Fe(II) forms an octahedral hexagonal complex with the H-X-D/E-Xn-H motif, 2OG, and a water molecule. The substrate then enters the binding pocket to form a complex, and after binding to oxygen molecules, 2OG is decarboxylated to produce a highly active ferryl species. The highly active ferryl species abstracts a hydrogen atom from the phenolic hydroxyl group to form Fe(III)–OH, whereas the substrate generates an O radical. Subsequently, with the occurrence of proton-coupled electron transfer events, the hydrogen bond interaction between the Arg140 residue and the phenolic hydroxyl group of the substrate and the conjugation interaction of the pentacyclic core stabilize the negative charge on the oxygen atom of the phenolic hydroxyl group; the electrons of the methoxy group are transferred to the pentacyclic core, and the aryloxy anion is formed. Subsequently, with the approach of the methyleneoxy group, a β electron is transferred from the substrate to the Fe(III)–OH2 group, forming the MDB-containing product (Fig. 5f).
4.3 Ring rearrangement
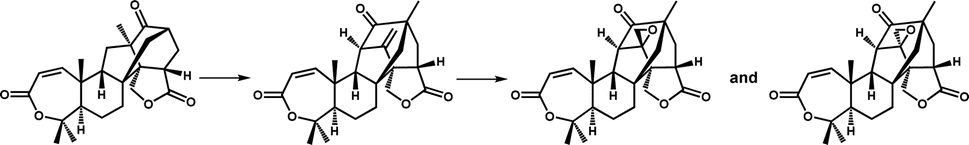
Catalytic ring rearrangement is a classic function of Fe/2OG DOs. At the end of the last century, deacetoxycephalosporin C synthase (DAOCS) was discovered to catalyze the conversion of penicillin N to deacetoxycephalosporin C (Table 1).11 The first structure of DAOCS was analyzed in 1998,11 and the structures of the mutants and other complexes were analyzed later. In addition to the classic DSBH core framework and the H-X-D/E-Xn-H motif, the change in the C-terminal arm after binding ferrous ions and 2OG is an important structural feature of the enzyme that may play an important role in isolating reactive intermediates during the catalytic process. The structure and catalytic mechanism of DAOCS have been proven and deduced in many studies.102,103 Except for DAOCS, other Fe/2OG DOs have also been found to catalyze ring rearrangement reactions in recent years, and the structures of some enzymes have been resolved, providing a deeper understanding of their catalytic processes and mechanisms.TropC is a Fe/2OG DO that contributes to ring rearrangement in the biosynthetic pathway of the natural tropolone product stipitatic acid. It catalyzes the oxidative ring expansion of its dienone-like substrates to dendrialdehyde; however, a small amount of the hydroxylated product, trihydroxybenzaldehyde, is also produced during the catalytic process.12 To explore the catalytic mechanism of TropC, its crystal structure was solved.104 The DSBH core is composed of ten antiparallel β-strands surrounded by multiple α-helices to form a compact globular structure. In comparison with structurally similar members of the family,90,105 the aromatic residues Phe284 and Phe213 are conserved in similar proteins, and it is speculated that these two residues play a crucial role in the binding and arrangement of substrates. When Phe284 is mutated to alanine, the generation of hydroxylated products significantly increases, almost equal to the yield of the ring-remodelling product. When Phe284 is mutated to tyrosine, the ring expansion activity of the enzyme is restored, indicating that the aromatic amino acid Phe284 is critical for determining the products generated by TropC (Fig. 6a). A catalytic mechanism of TropC has been proposed by combining a series of mutation experiments and computational simulations.104 TropC first mediates the extraction of the C–H atom on the six-membered ring methyl group of the dienone substrate to generate a radical intermediate, which directly mediates ring expansion of the substrate. The formation of the final product is determined by the radical termination process. The vast majority of free radicals are terminated by one-electron oxidation reactions involving Fe(III)-hydroxyl species to form stipitaldehyde ring rearrangement products, whereas a small number of free radicals are affected by rebound hydroxylation to form hydroxylated products. However, this inference does not fully explain the formation of products in the rebound hydroxylation pathway, and further verification is needed in future studies. Structural characterization, modelling, and mutagenesis analysis of TropC indicate that substrate positioning in TropC determines which radical termination step dominates the reaction, thus determining product generation (Fig. 6c). These results provide new insights into the complex reactions involved in the ring rearrangement catalyzed by Fe/2OG DOs.
 | ||
| Fig. 6 Mechanism of ring rearrangement reaction mediated by Fe/2OG DOs and the structures of related enzymes. (a) Crystal structure of TropC (PDB code: 6XJJ). (b) Crystal structure of Aj_EasH (PDB code: 5 M0T). (c) Proposed mechanism of TropC. (d) Proposed mechanism of Aj_EasH. | ||
EasH from Aspergillus japonicus (Aj_EasH) is an enzyme involved in the biosynthesis of natural products in fungi, which is responsible for catalyzing the six-membered ring substrate to generate a rare cyclopropyl product. Jakubczyk et al.13 analyzed the structure of apo-Aj_EasH for the first time. Aj_EasH stabilizes the substrate mainly by hydrophobic interaction, and it has a short helical “lid” on the surface of the active site, which acts as a “gate” to control the substrate into the binding pocket and the product release (Fig. 6b). One potential mechanism is that Aj_EasH initiates hydroxylation or halogenation at substrate C10 and then functions as a leaving group in the ring rearrangement process. However, the hydrophobicity of the active site is extremely strong, and after mutating several polar or ionizable residues close to the active centre, no key polar residues were found to protonate the leaving group, indicating that this mechanism of leaving group formation is less feasible.13 A plausible inference is that the Fe(IV)O species directly extracts hydrogen at the C10 position, followed by a free-radical-mediated ring rearrangement (Fig. 6d). In principle, this reaction mechanism does not require acid–base catalysis of the active-site residues. This inference is supported by a QM/MM calculations based on the structure.106 Aj_EasH extracts hydrogen atoms from C10, and then C10 radical C8 forms a fused ring system, this process is more reasonable. The simulation calculation results also show that the substrate-binding mode of Aj_EasH is very important for the formation of the product and the occurrence of the reaction. Only a few specific substrate binding modes can enable Fe(IV)O species to extract hydrogen atoms to promote subsequent reactions, and electron transfer may occur between the Fe centre and the substrate to promote ring rearrangement. For this unique (ring rearrangement) reaction, important further information needs to be obtained; it is necessary to further analyze the structure of the composite substrate and reveal the key substrate-binding mode of Aj_EasH, which will provide key evidence for its mechanism of deduction.
4.4 Desaturation
Desaturation reactions catalyzed by Fe/2OG DOs occur in the biosynthetic pathways of diverse natural products, including multiple flavonoids, gibberellins, and meroterpenoids.45,107 In early research, limited structures of this type of enzyme were obtained, and only CarC was thoroughly studied.108–111 This makes the mechanism of desaturation mediated by Fe/2OG DOs much less clear than hydroxylation.112 There are currently three putative desaturation pathways: (a) two hydrogen abstractions followed by diradical recombination, (b) hydroxylation and subsequent dehydration via a base, and (c) carbocation formation after hydrogen abstraction followed by the removal of H+ by a base, ultimately forming olefins (Fig. 7).14,112,113 These mechanisms are supported by a recent structural analysis of desaturases of Fe(II)/2OG DOs.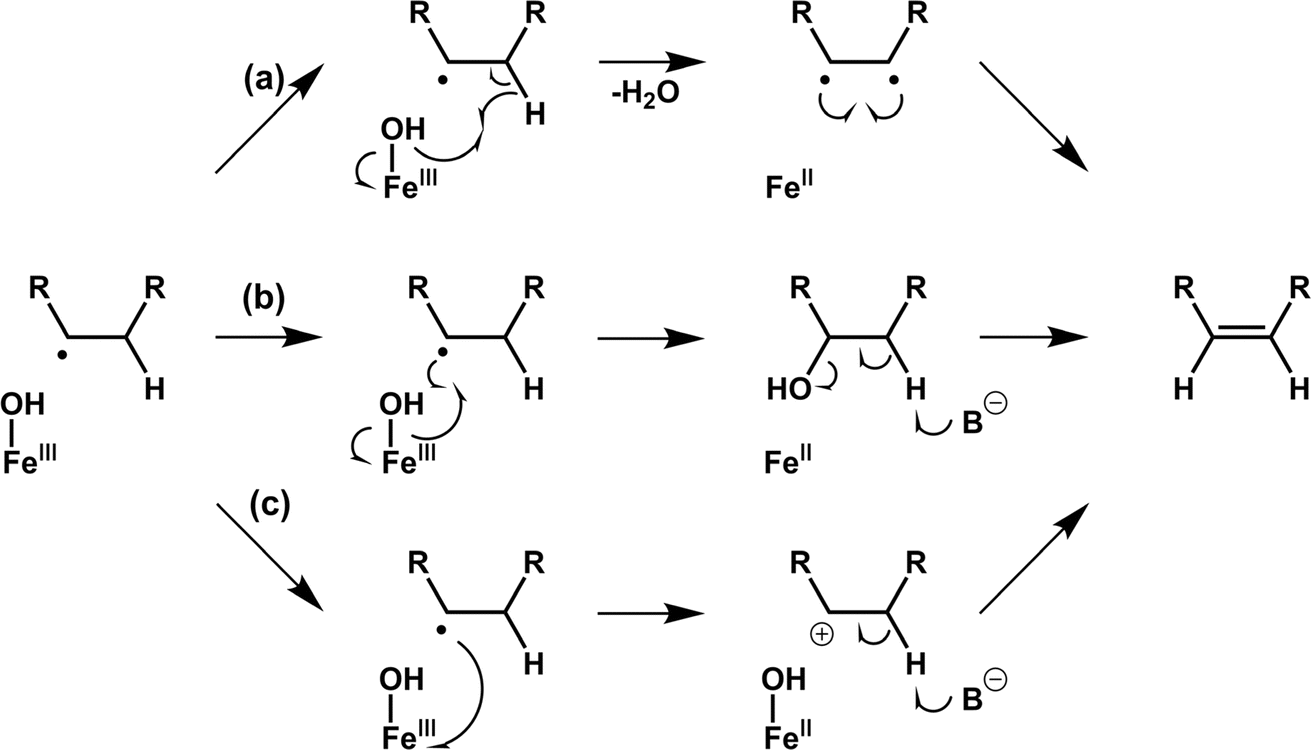 | ||
| Fig. 7 Proposed pathways for Fe/2OG DOs to achieve desaturation catalysis. | ||
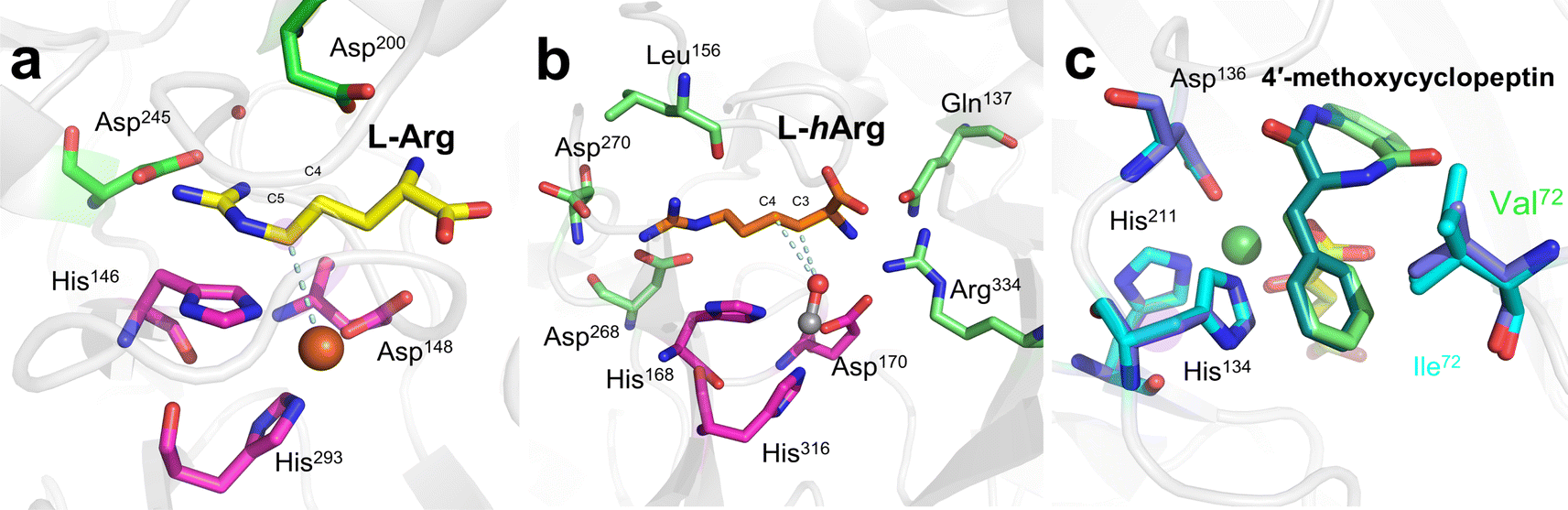
NapI is a desaturase from the naphthyridinomycin biosynthetic pathway that achieves the 4,5-desaturation of L-Arg. In the crystal structure of NapI complexed with the substrate,14 the position of C5 of L-Arg is different from that of the H donor in other Fe(II)/2OG DOs, where it is located directly above His146—provided by the H-X-D/E-Xn-H motif. This binding mode allows the α-heteroatom N6 adjacent to C5 to form a close interaction with the imidazole ring of His146, enabling the fully positively charged cation-π stabilization formed on N6. One terminal side chain nitrogen of L-Arg forms a salt bridge with Asp245 and the active site water (Fig. 8a), which presumably involves the transfer of coupled protons.14 These structural features of NapI are conducive to the cleavage of polar C4–H after HAT at the C5 position, thereby promoting 4,5-desaturation and formation of a double bond, and the catalytic mechanism is more biased towards pathways (b) or (c). In the sequential HAT mechanism of two hydrogen abstractions in pathway (a), the rebound of the hydroxyl group to C5 might compete to some extent with the second HAT step. However, deuteration of C4 of L-Arg failed to enhance C5 hydroxylation, indicating that pathway (a) is not applicable to NapI. More importantly, NapI is unable to achieve desaturation of substrate analogues that eliminate the α-heteroatom N6. In both the proposed pathways (b) and (c), an imine intermediate was generated, which required the existence of N6. This shows that the α-heteroatom nitrogen forms an iminium-like intermediate during the catalytic process,14 which plays an important role in the desaturation of NapI.
 | ||
| Fig. 8 Crystal structure of Fe/2OG DOs that mediate desaturation. (a) Binding pocket of NapI (PDB code: 6DAW). (b) Binding pocket of VioC (PDB code: 6DB2). (c) Binding pocket of AsqJ (PDB code: 5DAX, 5OA8); the wild type is shown as a slate model, and the mutant V72I is shown as a cyan model. | ||
The previously mentioned VioC also has an unexpected 3,4-desaturation effect on L-homoarginine (L-hArg), superimposing the structures of VioC and NapI, and the topologies of the two enzymes are almost identical except for the flexible lid element. Comparing the active sites of the two enzymes, it has been found that although their overall folds are almost identical, the interactions with individual groups of the substrate, including amine, carboxylate, and guanidine groups, are not identical, which leads to differences in the localization of the substrates by the two enzymes.14 Based on an analysis of structural characteristics, the desaturation of L-Arg by NapI and L-hArg by VioC may involve two different mechanisms; the former is selective and catalyze natural substrate, whereas the latter is relatively non-selective and catalyze non-natural substrate. In the catalyzed reaction by NapI, the α-heteroatom interacts more closely with the catalytic centre, and its presence promotes the polar cleavage of the second C–H bond to form a double bond. In the X-ray crystal structure of VioC complexed with L-hArg, both carbons of L-hArg are very close to the oxygen ligand (≤3.5 Å),14 which indicates that VioC is likely to use the sequential HAT mechanism of pathway (a) to complete the desaturation. However, the weak oxidative ability of Fe(III)–OH is not conducive to this mechanism.43,68 And these structural features and other research findings suggest that although the desaturation function of Fe/2OG DOs is feasible using a sequential HAT mechanism, competitive hydroxylation may be inevitable because of the structural positioning and spatial geometry of the active centre. This also explains from another perspective why α-heteroatoms are present in almost all the natural substrates of desaturase family members. To prove the correctness of this inference, more intuitive and convincing research data will be needed in the future.
Based on research on some desaturase members, it can be observed that a novel desaturation catalysis of Fe/2OG DOs. In addition to the α-heteroatom itself in the substrate, the functional groups attached to the α-heteroatom are also an important factor affecting the desaturation reaction. AsqJ from Aspergillus nidulans possesses desaturation and subsequent oxidative cyclization functions and can stereoselectively catalyze the multistep synthesis of quinolone alkaloids, which are natural products of great value for biomedical applications. In previous studies, it was observed that methylation at the N4 position of the natural substrate (4′-methoxycyclopeptin) plays a decisive role in the efficient catalytic desaturation reaction of AsqJ; when the N4-methyl group was removed, the desaturation reaction was hindered, and subsequent epoxidation products were not formed.15 However, when Val72 of AsqJ was mutated to Ile, the mutant desaturated the N4-demethylated substrate. Subsequent studies have obtained the complex structures of mutant V72K with the N4-methylated substrate and the complex structure of mutant V72I with the N4-demethylated substrate using crystallography (Fig. 8b). In the structure of mutant V72I complexed with a substrate that removes the N4-methyl group, the distance between mutant V72I and the C4′ of the demethylated analogue is reduced by 0.2 Å compared to wild-type AsqJ, and the B factor of the substrate is increased, indicating an increase in the mobility of phenyl groups. The extra electron density lobes around the benzene ring of the substrate in the mutant V72I structure also suggest that the aromatic part of the ligand is highly flexible.114 These results suggest that the π-stacking interaction between the substrate and His134 maintains the ligand at the active site for a longer time, promoting the desaturation reaction. Simultaneously, the structure of the mutant V72I complex with the N4-demethylated substrate has been compared to that of AsqJ bound to the natural substrate. It was found that the essence was to insert a methyl group into the protein to compensate for the lack of an N-methyl group in the substrate, so that the binding force between the two was enhanced and returned to a more natural binding mode, thereby restoring the desaturation function of the substrate. Importantly, such understanding can guide the subsequent design of Fe/2OG DOs.
4.5 Demethylation
Demethylation is a type of reaction catalyzed by Fe/2OG DOs, discovered approximately 20 years ago.115,116 Its main application is in genetic material repair and regulation while also contributing to the transformation of natural products.117 In terms of genetic material repair, the general mechanism of the demethylation of DNA substrates by Fe/2OG DOs has been studied;118 however, the substrate recognition methods and specific catalytic properties of some family members are not clear, and the molecular functions of some enzymes remain controversial. Now that some new structures of Fe/2OG-dependent demethylases have been resolved, the answers to these questions are gradually being revealed.Human ALKBH1 (hALKBH1), a highly conserved Fe/2OG DO in mammals, is one of the nine human homologues of the AlkB family, whose members can repair damaged DNA/RNA or other damage.17 Tian et al.16 reported the structure of hALKBH1 complexed with 2OG. After combining with 2OG, the overall structure of hALKBH1 contracted, and many conformational changes not observed in other AlkB members occurred. Furthermore, Arg344 and Tyr222 in the 2OG binding pocket directed to the active centre moved 2.5 Å and 2.3 Å, respectively, interacting with 2OG. Tyr184 and Glu236 located on Flip2 moved closer to Arg344, forming a stable triangle through hydrogen bonding, and mutation experiments further proved that this triangle interaction is crucial for substrate demethylation activity.
These DNA repair-related enzymes perform physiological functions that are largely dependent on substrate recognition. For ALKBH1, whether human or murine, there is a longer substrate channel for DNA binding. The Flip1, which is rich in a large number of lysine and arginine residues, and an additional string of basic residues around Flip2 form two extended electrostatic positive wings around the catalytic cleft, allowing the DNA-binding region to form a broad and substantially concave surface suitable for double-stranded DNA to engage with the inverted methyl base inserted into the active centre. This binding region differs from the relatively narrow binding grooves of other members of the AlkB family, making ALKBH1 more susceptible to double-stranded and bubbled DNA. These basic residues are important for the recognition of nucleic acid substrates, and mutation experiments have demonstrated that they are essential for substrate binding and enzyme activity.18 In addition to the substrate-binding channels, the size of the residues in the catalytic pocket is critical for enzyme function. S235 is a small side chain residue located within the catalytic pocket of ALKBH1 that allows for a larger N6-mA base. When this is mutated to a large sterically hindered residue, the demethylation activity of ALKBH1 towards N6-mA disappears, indicating that it plays a key role in the selection of modified bases.
4.6 Oxidative deamination
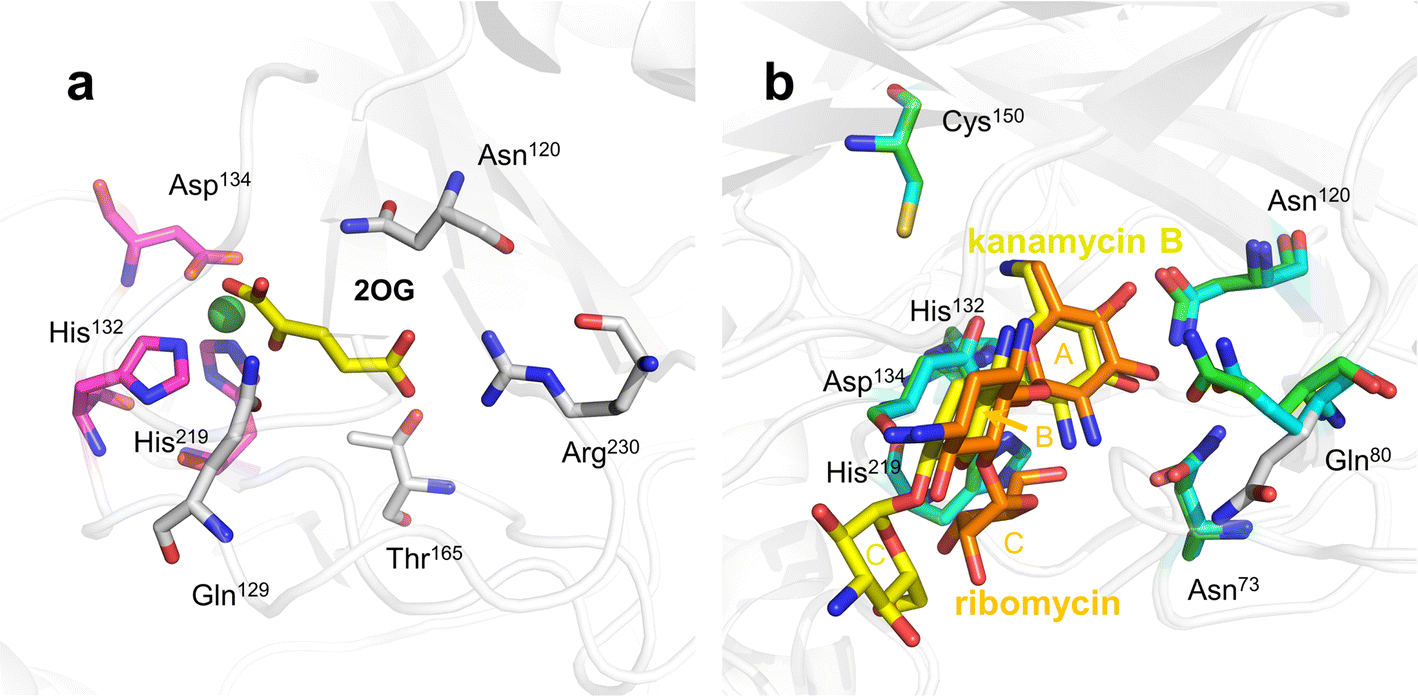
Among the 2Fe/OG DOs involved in oxidative deamination in recent years, only the crystal structure of KanJ has been determined. KanJ plays an important role in the kanamycin biosynthesis pathway and has a unique deamination activity that enables the conversion of kanamycin B to kanamycin A,19 a novel discovery for Fe/2OG DOs.Based on structural analysis, the binding pocket of KanJ is open and very spacious, allowing it to accommodate bulky substrates, such as aminoglycoside antibiotics. The characteristics of the binding pocket are similar to those of the PhyH family. The binding of 2OG is mainly caused by several hydrogen and electrostatic interactions. In the structure of kanJ complexed with 2OG (Fig. 9a), the Arg230 of the last β-sheet of the DSBH framework and Gln129 and Thr165 from two linking loops form hydrogen bonds with the C5 carboxyl group of 2OG.119 Glutamine and threonine residues are often present in Fe/2OG DOs as functional elements that recognize the C-5 group of 2OG.120 In addition, van der Waals forces between Asn120 and 2OG promote the binding of 2OG.
 | ||
| Fig. 9 Binding pocket of KanJ. (a) Structure of KanJ complexed with 2OG (PDB code: 6S0U). (b) Structure of KanJ complexed with substrates (PDB code: 6S0S, 7CL5). Structure of KanJ complexed with kanamycin B is shown as a green model; structure of KanJ complexed with ribomycin is shown as a cyan model. | ||
Substrate binding is relatively complex. The broad entry surface of the binding pocket consists of hydrophilic groups provided by four short flexible loops. The interaction between Phe282 in the C-terminal fragment and the antibiotic substrate appears to be important for substrate binding. In the closed conformation, Phe282 induces the rotation of Gln80, which is directly involved in antibiotic binding, and when KanJ binds to kanamycin, the dihedral angle around the C–C bond on the Gln80 side chain changes by approximately 30°, making the conformation more favourable for ligand binding, thereby facilitating interaction with the ligand (Fig. 9b). There are many common features among the multiple KanJ structures complexed with aminoglycoside substrates. The A- and B-rings of all antibiotics occupy the same position in the active site, and most protein–antibiotic interactions occur in ring A, which is closest to the metal ion. The hydroxyl groups bound by C3′ and C4′ form H bonds with the side chains of Gln80 and Asn120, and the positively charged NH3+ groups on C6′ form hydrogen bonds with Asp134.121 However, there are also significant differences in the spatial structure and interaction of each substrate in the binding pocket, which are mainly affected by the B- and C-rings. In the binding pocket, kanamycin B is in an extended form, whereas ribomycin has a crescent shape, bringing the C-ring of ribomycin closer to the metal cofactor, which may have implications for substrate binding and turnover.
This structural understanding and some subsequent biochemical experiments show that the necessary condition for the substrate recognition of KanJ is the presence of A-ring (6-amino-6-deoxy-α-D-glucopyranosyl), which is mainly recognized by Asn73, Gln80, Asn120, Asp134, and Cys150 at the active site. Upon binding to an antibiotic substrate, the C-terminal fragment of KanJ undergoes a conformational change that swings towards the substrate and stabilizes the binding of the substrate at the active site. However, for different antibiotic substrates, the existence of B- and C-rings creates an arrangement and steric hindrance of the substrates in the binding pocket, resulting in different substrate depths in the binding pocket and different strengths of interactions with key residues. Therefore, there are significant differences in the reactivity of kanJ with different substrates.
4.7 Endoperoxidation
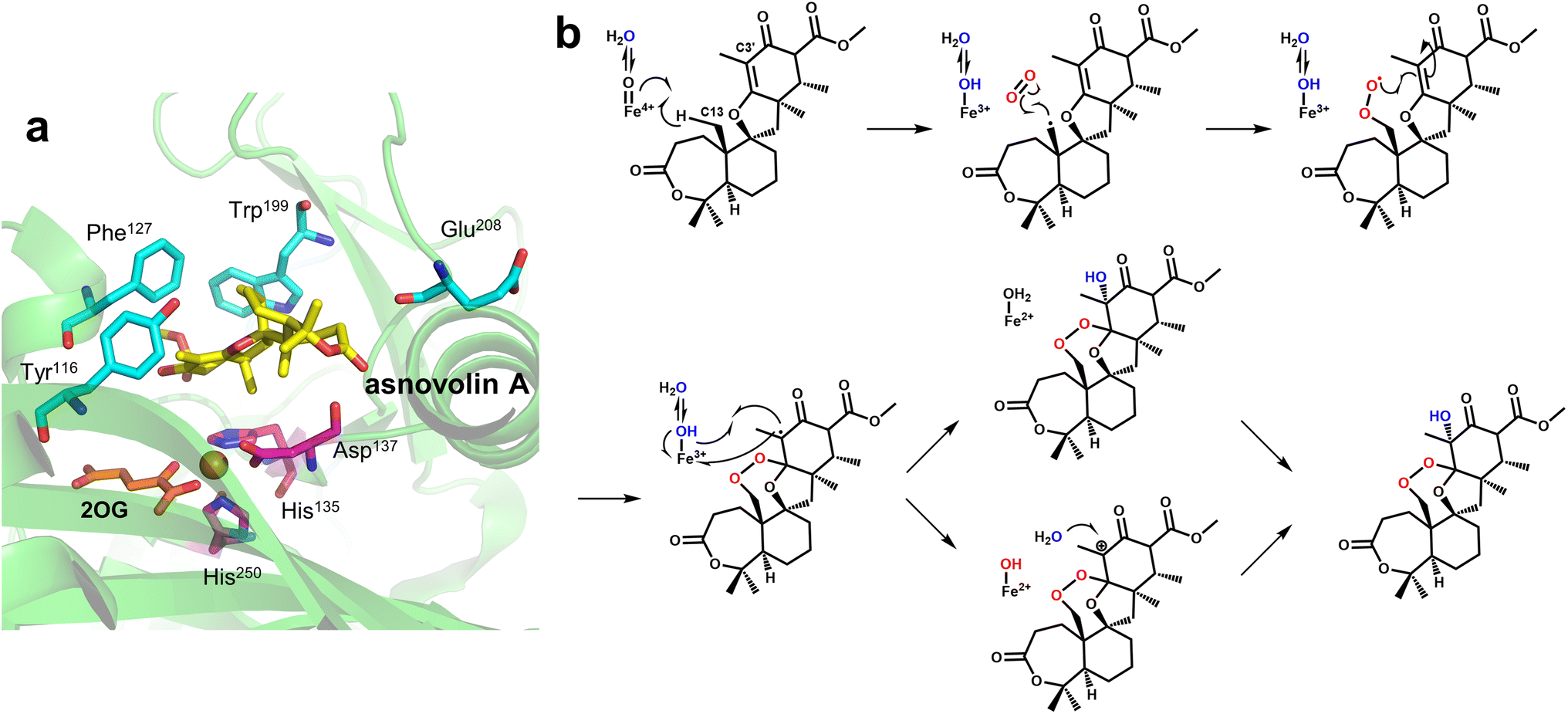
Endoperoxidation is a relatively newly discovered function of Fe/2OG DOs. FtmOx1 is a member of the Fe/2OG DOs identified as an endoperoxide synthase that catalyzes transformation of fumitremorgin B by adding an endoperoxide moiety between C21 and C27 of fumitremorgin B.21 Another family member with endoperoxidation function is NvfI, which creates a two-oxygen bridge between C13 and C2′ in asnovolin A and installs a hydroxyl group downstream of C3′ to generate fumigatonoid A.23The structure of FtmOx1 was first solved in 2015,122 but the expected complex crystals examined by electron density maps showed no substrate in the binding pocket, which rendered previous studies unclear about the catalytic mechanism. At present, researchers have two main inferences for its catalytic endoperoxidation—the Y68-mediated CarC-like mechanism and the Y224-mediated COX-like mechanism (Fig. 10a and b).123,124 Recently, Wu et al.20 solved the crystal structure of FtmOx1 containing 2OG and the natural substrate fumitremorgin B. Subsequently, Zhu et al.22 obtained complex crystals of FtmOx1 with the natural substrate analogue 13-oxo-fumitremorgin B. FtmOx1 is composed of multiple hydrophobic amino acids at the substrate binding site to form a hydrophobic pocket, which surrounds the aliphatic side chain of the substrate. When it binds to a substrate, the overall structure becomes compact (Fig. 10c). The aromatic residues His129, Tyr68, and Phe72 form π–π interactions with the indole moiety of the substrate from both sides. The carbonyl group of Pro127 and the side chain of Arg117 form hydrogen bonds with the 12-hydroxyl and 18-methoxyl groups of Fumitremorgin B, respectively. These interactions lead to the stable binding of fumiremorgin B.20 The active pocket volume of FtmOx1 is significantly compressed due to the binding of fumitremorgin B, and the Fe atom is closer to the substrate. The hydroxyl rotation of Thr134 moves backwards and pushes the methyl group of Thr134 closer to the substrate. Tyr224, Phe72, and Arg117 are deflected or displaced to make room for substrate binding. In the structure of FtmOx1 complexed with fumitremorgin B (Fig. 10d and e), all tyrosine residues are located far away from the Fe atom. Tyr224 is twisted by 115° to the outside and is pushed 8.4 Å away from the Fe atom, and Tyr74 and Tyr68 are also farther from Fe, with distances of 11.0 Å and 10.4 Å, respectively,20 and similar structural features appear in the structure complexed with 13-oxo-fumitremorgin B. These are not conducive to Tyr224, Tyr68, and other tyrosine direct HAT to form Fe(IV)O species. From another perspective, Tyr68 has a close distance to the C26 of the substrate, approximately 4 Å, which suggests that Tyr68 is likely an H-atom donor for quenching the C26 central substrate radical, favoring a Y68-mediated CarC-like mechanism. However, none of these structures show the 2OG rotation involved in the CarC-like mechanism.123 In contrast, the rotation of Tyr224 indicates the possibility of structural rearrangement. Although Tyr224 rotates and moves away from the Fe(II) centre, it is still located on the same side of the Fe(II) centre, and its rotatability around the Fe coordination center is also favourable to the extraction of hydrogen atoms and subsequent internal peroxidation of substrate C21.22 However, it is worth noting that Tyr224 forms stable hydrogen bonds with Thr134 and Q226, which prevents Tyr224 from forming oxygen radicals, which is inconsistent with the radical relay role of Y224 in previous theoretical studies.20,125 Mutation results show that Tyr68 and Tyr224 lose endoperoxidation activity after mutation to phenylalanine, which indicates that Tyr68 and Tyr224 certainly play a crucial role in the internal peroxidative function of FtmOx1. The reaction of FtmOx1 is complicated, and the reaction conditions will lead to different products. The Y68-mediated CarC-like mechanism and the Y224-mediated COX-like mechanism can reasonably explain some catalytic processes, but neither is perfect. It cannot be ruled out that there may be more complex mechanisms, and more intuitive and convincing data are needed, such as complex structures in multiple states, more kinetic experiments or molecular simulations, to reveal the mechanism of endoperoxidation catalyzed by FtmOx1.
 | ||
| Fig. 10 Mechanism of endoperoxidation reaction mediated by FtmOx1 and the structures of FtmOx1. (a) Y68-mediated CarC-like mechanism. (b) Y224-mediated COX-like mechanism. (c) Comparison of the overall structure of FtmOx1 in various states (PDB code: 7ETK, 7WSB, 4Y5S). (d) Partial details of the binding pocket of FtmOx1. (e) Further details of binding pocket FtmOx1. Fumitremogin B is shown as a dark blue stick, and oxo-Fumitremogin B is shown as a dark purple stick. | ||
Unlike FtmOx1, NvfI forms a peroxide bridge between the C13 and C2′ of asnovolin A and also forms a hydroxyl group at C3′; however, NvfI preferentially undergoes endoperoxidation rather than hydroxylation, and this mechanism is unique. Mori et al.126 obtained and analyzed multiple structural crystals to explore the mechanism of NvfI. The crystal structure of NvfI complexed with 2OG and the substrate asnovolin A showed that Arg118 and His138 form hydrogen bonds with the keto group and ester carbonyl group of the substrate, respectively, and Thr133 and Arg132 interact with the carbonyl group of the substrate via water molecules. The residues Ser122-Gly128 in loop1 and Trp199-Pro209 in loop2 exhibited obvious conformational flips when the substrate was bound, and hydrogen bond grids were formed between some residues, mainly providing steric support for substrate binding (Fig. 11a). When key residues such as Trp199 and Phe127 were mutated, enzymatic activity was almost completely lost, and the mutant structure indicated that W199F caused the substrate to not be properly accommodated in the deeper part of the binding pocket. These results suggest that the substrate-recognition mode of NvfI may be regulated by key amino acid residues in the loop. As previously mentioned, tyrosine residues are critical core residues for the catalytic activity of FtmOx1; however, this conclusion does not apply to NvfI. After the mutation of Tyr116, the only tyrosine residue located in the active site of NvfI, the catalytic specificity and activity of NvfI did not change significantly, suggesting that the catalytic mechanism of NvfI is not the same as that of FtmOx1. Based on this understanding, a unique endoperoxidation mechanism of NvfI was speculated. After NvfI binds to the substrate, the conformation of the active site changes; the C13 of the substrate is located close to the Fe centre, and the ferryl species directly abstracts hydrogen from the C13 position. Then, loop2 undergoes a conformational change again, specifically the flip of Glu208 and Phe127, and the movement of Trp199 creates a tunnel for the substrate and hides the radical intermediate inside the tunnel, preventing the Fe-coordinated hydroxyl group from rebounding to the C13 radical (Fig. 11a). Correspondingly, the C13 radical reacts with O2, which can enter the tunnel near the A-ring portion of the substrate. The generated peroxy radicals attack C2′ to form C3′-radical intermediates containing endoperoxides. As Fe(III)–OH species and C3′ are brought closer together by the repositioning of the intermediate, the hydroxy species coordinating the iron undergoes a hydroxy bounce back to the free radical, ultimately forming the product. It is also possible that an electron is directly transferred to the ferric iron to form a C3′ carbocation intermediate, which is then hydrolyzed to form the product (Fig. 11b). The exact pathway of reaction has not been clearly identified. In this presumed mechanism, O2 directly connects with C13 radicals to form peroxides; however, both subsequent C3′ hydroxylation pathways are restricted to the substrate binding mode. Therefore, the addition of water molecule and the repositioning of the free radical intermediate require further research, so that the mechanism of this conformational change that promotes the catalytic reaction can be thoroughly analyzed.
 | ||
| Fig. 11 Structure and proposed mechanism of NvfI. (a) Details of the binding pocket of NvfI (PDB code: 7DE2). (b) Proposed mechanism of NvfI. | ||
4.8 Multifunctionalization
Research on Fe/2OG DOs has shown that many family members do not have a single function; they can catalyze various reactions on the substrate. Among these, SptF is a representative Fe/2OG DO with multi-catalytic ability that can catalyze several sequential oxidation reactions, including hydroxylation, desaturation, epoxidation, and bone rearrangement (Fig. 12a–c).24,127 In addition, SptF has a broad substrate spectrum for terpenoids and a wide range of steroid hydroxylation activities; it is an enzyme with promising functional and substrate recognition features.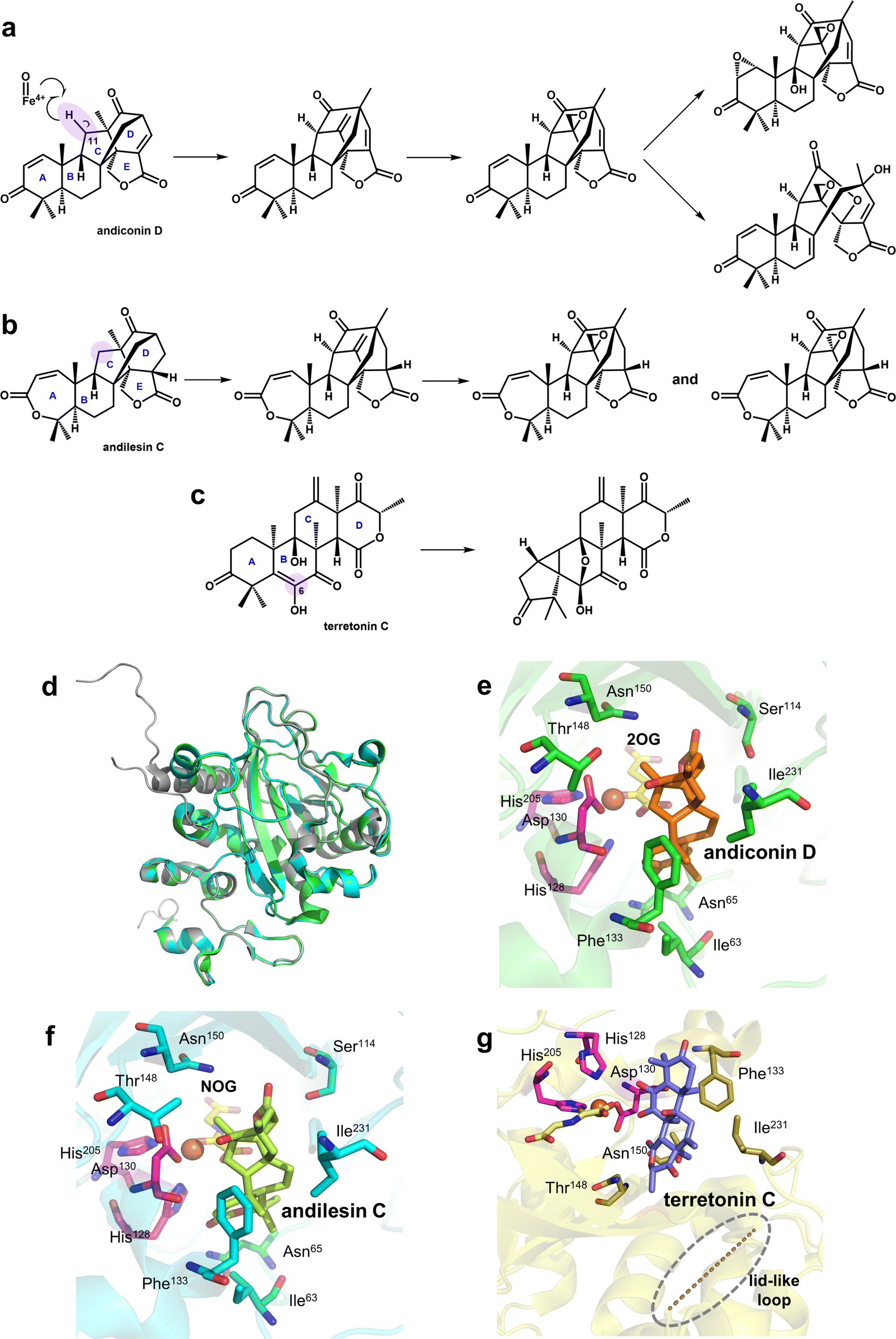 | ||
| Fig. 12 Part of the reaction catalyzed by SptF and the structure of SptF. (a) A series of reactions using andiconin D as a substrate. (b) A series of reactions using andilesin C as a substrate. (c) SptF-mediated catalytic reaction of the non-natural substrate terretonin C. (d) Overall structure of SptF; apo is shown in grey, the SptF complex with andiconin D is shown in green, and the SptF complex with andilesin C is shown in cyan. (e) Crystal structure of the SptF complex with andiconin D, 2OG, and Fe(II) (PDB code: 7EYS). (f) Crystal structure of the SptF complex with andilesin C, NOG, and Fe(II) (PDB code: 7EYT). (g) Crystal structure of the SptF complex with terretonin C, NOG, and Fe(II) (PDB code: 7EYW). | ||
Three crystal structures of SptF complexed with different terpenoid substrates have been solved,24 providing a molecular basis for exploring the reaction mechanism of SptF. A comparison of the structures of different complex substrates showed that SptF has complex interactions with substrates; it has different recognition and anchoring modes for each ring of the substrate, and natural and non-natural substrates also have different binding modes. In the structures containing the natural substrates andiconin D and andilesin C (Fig. 12e and f), the A-ring of the substrate forms only one hydrogen bond with Asn65 of SptF, and the size of the A-ring does not affect the binding mode. The B/C ring is within the van der Waals interactions range of the hydrophobic residues near the active centre, including Ile63, Phe133, and Ile231, suggesting that the hydrophobic surface within the pocket is favorable for substrate binding. Whereas the D/E ring is tightly anchored to several hydrophilic residues at the bottom of the active site cavity: the D ring interacts with Leu199 and Thr148 via water molecules, and the carbonyl oxygen of the E ring interacts with Ser114. This suggests that substrate binding does not depend on a single type of force and that both the hydrophobic and hydrophilic parts of the binding pocket play important roles in substrate binding. However, in the structure containing non-natural substrate terretonin C (Fig. 12g), a lid-like loop region is not observed, indicating that the position of SptF's interaction with the substrate partially changes, making the C6 enol group closer to the Fe(II) ion rather than C11 in the natural substrate. This also lays the groundwork for explaining the catalytic versatility of SptF.
After mutagenesis according to the structure of SptF,24 changes in the hydrophobic residues in the active centre alter the binding mode of the substrate and produce a variety of products in different proportions. The change in hydrophilic residues alters the hydrogen bonding interaction to different degrees, thereby changing the position of the hydrogen atom extraction and affecting the specificity of the product. The results of mutation and truncation of the lid-like loop region suggest that this is essential for steroid substrate recognition but not for terpenoid substrates. This behaviour is very different from that of many other Fe/2OG DOs. The flexible loop region of most Fe/2OG DOs plays a decisive role in substrate recognition and binding, whereas only Asn65 in the lid-like loop region of SptF is connected to the natural substrate through a hydrogen bond and has no decisive effect on enzyme function after the mutation, demonstrating the uniqueness of SptF substrate recognition. These suggest that the unique substrate promiscuity may be the determining factor that makes SptF multifunctional. In-depth research on the structure and catalytic reactions of related multifunctional enzymes in the future is expected to reveal the relationship between substrate complexity and catalytic function.
5 Conclusions and perspectives
Recently, an increasing number of Fe/2OG DOs have been discovered; as a result, the catalytic range that these family members can achieve has widened. In addition to the reactions mentioned in this review, others such as halogenation, dephosphorylation, and decarboxylation also exist. They can also catalyze the conversion of various types of substrates, including various amino acids, natural product precursors, and metabolic intermediates. However, there are still many family members, especially newly discovered members, whose structural features and potential mechanisms are lacking in-depth understanding, including the desaturases BcmF and BcmB in the bicyclomycin biosynthesis pathway, KabC in the kainic acid biosynthesis pathway, and chlorophyll C synthase (CHLC).128–130 Furthermore, many Fe/2OG DOs can perform multiple types of catalysis. The geometric structure and hydrophobicity of the pocket, substrate recognition and localization patterns, the position of the Fe ion coordination center and the substrate and steric hindrance greatly affect the catalysis of Fe/2OG DOs. Therefore, analysis of their structure and function is very important for deepening our understanding and exploring the potential of Fe/2OG DOs in synthetic biology and other fields.Currently, most Fe/2OG DO structures are acquired using crystallographic methods. Cryo-electron microscopy has developed vigorously in recent years and has been extensively used for the structural analysis of enzymes with large molecular weights and membrane-bound enzymes.131,132 Cryo-electron microscopy is less restricted by conditions and has the advantage of obtaining the structures of unstable enzymes and reaction intermediates. With such new technological breakthroughs in sample preparation and structural resolution, there is great potential for the structural analysis of enzymes with different molecular weights and small-molecule-enzyme complexes.133–135 Based on the development of existing structures and computer science, computing power and reliability of homology modelling and structural simulation technology have significantly improved with the help of artificial intelligence. The advent of Alphafold2 has caused a scientific revolution. A large number of protein structures have been predicted with considerable accuracy, which has led to the explosive development of protein structural bioinformatics.136–138 AlphaMissense and AlphaFold-latest aim to determine the relationship between missense mutations in human genes, the structure of the proteins they express, and the binding issues between proteins and ligands.139 The recent introduction of the RoseTTAFold all-atom method allows protein covalent modification and small molecule assembly to be reasonably predicted.140 This can not only model known structures but also predict complex interactions between biomolecules and the three-dimensional structure of the overall assembly. AlphaFold3 was recently released,141 which can predict the structures and interactions of biomolecules with unprecedented accuracy. The model is capable of joint structure prediction for complexes including proteins, nucleic acids, small molecules, ions, and modified residues. Updating these technologies can greatly advance the understanding of the structure–function relationships of the Fe/2OG DO enzyme family, thereby deepening our understanding of the related catalytic mechanisms. Molecular dynamics (MD) simulations use structural data to infer the potential conformations of molecular systems and different pathways between individual structures of molecular systems. This provides the possibility of simultaneously describing the structure and dynamics of macromolecules at atomic resolution and plays an important role in advancing the study of the dynamic processes of enzyme recognition of substrates and ligands.142,143 Multi-scale quantum mechanics/molecular mechanics (QM/MM) also provides insights into how enzymes control high-fidelity or hybrid reactions at the atomic and electronic levels. This is based on a high-precision model that considers the quantum properties of atomic motion, including free energy and reaction path methods, to provide more accurate answers for the study of enzymatic reactions.144–148 Research methods that combine multiple computing technologies can achieve multi-scale modelling, complete more accurate simulations, explain many complex processes, and enable a more in-depth analysis of various mechanisms.
Fe/2OG DOs have diverse catalytic functions, and with a full understanding of their mechanisms, their application prospects are very bright. Fe/2OG DOs have a relatively broad foundation in the production of some traditional antibiotics and hydroxylation of amino acids and have been partially well explored in the synthesis of natural products and derivatives—and there is still vast room for development. Some human diseases, including renal cancer, paraganglioma, and chronic hypoxemia, are also closely related to the dysregulation of Fe/2OG DOs, indicating that they are potentially important targets for disease treatment. In future research, it will be necessary to combine structural biology, kinetics, and computational simulation studies to determine the key factors for Fe/2OG DOs to catalyze various reactions and explore a more complete catalytic mechanism. Use more targeted transformation strategies based on enzyme engineering technology to guide functional transformation and enhancement. This is expected to achieve functional controllability and enable the construction of Fe/2OG DOs as a precise tool for the specific catalysis of target substrates, demonstrating their excellent application capabilities in chemistry, pharmacy, medicine, and other industries.
6 Data availability
All protein structural data related to this review have been deposited in the Protein Data Bank (PBD) and are available at https://www.rcsb.org/.7 Author contributions
The initiation of this project was spearheaded by Songyin Zhao and Yao Nie. The collection of references and the writing of this review were mainly completed by Songyin Zhao. Yao Nie and Yan Xu provided a lot of important guidance and careful revisions to this review the review. Lunjie Wu supplemented remainder of this review and assisted with the writing. Each author has made significant contributions to this scholarly review.8 Conflicts of interest
There are no conflicts to declare.9 Acknowledgements
Financial supports from the National Key R&D Program of China (2021YFC2102000), the National Natural Science Foundation of China (NSFC) (22378168, 22178147), the 111 Project (111-2-06), the High-end Foreign Experts Recruitment Program (G2021144005L), the National Program for Support of Top-notch Young Professionals, the Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions, Top-notch Academic Programs Project of Jiangsu Higher Education Institutions, the Jiangsu province “Collaborative Innovation Center for Advanced Industrial Fermentation” industry development program and the Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX24_2582) are greatly appreciated.10 References
- J. J. Hutton Jr, A. Kaplan and S. Udenfriend, Arch. Biochem. Biophys., 1967, 121, 384–391 CrossRef CAS.
- A. J. Mitchell, N. P. Dunham, R. J. Martinie, J. A. Bergman, C. J. Pollock, K. Hu, B. D. Allen, W. C. Chang, A. Silakov, J. M. Bollinger Jr, C. Krebs and A. K. Boal, J. Am. Chem. Soc., 2017, 139, 13830–13836 Search PubMed.
- S. E. Wilkins, M. S. Islam, J. M. Gannon, S. Markolovic, R. J. Hopkinson, W. Ge, C. J. Schofield and R. Chowdhury, Nat. Commun., 2018, 9, 1180 CrossRef.
- P. Lukat, Y. Katsuyama, S. Wenzel, T. Binz, C. Konig, W. Blankenfeldt, M. Bronstrup and R. Muller, Chem. Sci., 2017, 8, 7521–7527 RSC.
- S. Knorr, M. Sinn, D. Galetskiy, R. M. Williams, C. Wang, N. Muller, O. Mayans, D. Schleheck and J. S. Hartig, Nat. Commun., 2018, 9, 5071 CrossRef.
- K. Bastard, T. Isabet, E. A. Stura, P. Legrand and A. Zaparucha, Sci. Rep., 2018, 8, 16587 CrossRef PubMed.
- S. Takehara, S. Sakuraba, B. Mikami, H. Yoshida, H. Yoshimura, A. Itoh, M. Endo, N. Watanabe, T. Nagae, M. Matsuoka and M. Ueguchi-Tanaka, Nat. Commun., 2020, 11, 2143 CrossRef CAS PubMed.
- J. Li, X. Zhang and H. Renata, Angew Chem. Int. Ed. Engl., 2019, 58, 11657–11660 Search PubMed.
- M. Lazzarotto, L. Hammerer, M. Hetmann, A. Borg, L. Schmermund, L. Steiner, P. Hartmann, F. Belaj, W. Kroutil, K. Gruber and M. Fuchs, Angew Chem. Int. Ed. Engl., 2019, 58, 8226–8230 CrossRef CAS PubMed.
- R. de Jonge, M. K. Ebert, C. R. Huitt-Roehl, P. Pal, J. C. Suttle, R. E. Spanner, J. D. Neubauer, W. M. Jurick 2nd, K. A. Stott, G. A. Secor, B. Thomma, Y. Van de Peer, C. A. Townsend and M. D. Bolton, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E5459–E5466 CrossRef PubMed.
- K. Valegard, A. C. van Scheltinga, M. D. Lloyd, T. Hara, S. Ramaswamy, A. Perrakis, A. Thompson, H. J. Lee, J. E. Baldwin, C. J. Schofield, J. Hajdu and I. Andersson, Nature, 1998, 394, 805–809 CrossRef CAS PubMed.
- J. Davison, A. al Fahad, M. Cai, Z. Song, S. Y. Yehia, C. M. Lazarus, A. M. Bailey, T. J. Simpson and R. J. Cox, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7642–7647 CrossRef CAS PubMed.
- D. Jakubczyk, L. Caputi, C. E. Stevenson, D. M. Lawson and S. E. O'Connor, Chem. Commun., 2016, 52, 14306–14309 RSC.
- N. P. Dunham, W. C. Chang, A. J. Mitchell, R. J. Martinie, B. Zhang, J. A. Bergman, L. J. Rajakovich, B. Wang, A. Silakov, C. Krebs, A. K. Boal and J. M. Bollinger Jr, J. Am. Chem. Soc., 2018, 140, 7116–7126 CrossRef CAS PubMed.
- A. Brauer, P. Beck, L. Hintermann and M. Groll, Angew Chem. Int. Ed. Engl., 2016, 55, 422–426 Search PubMed.
- L. F. Tian, Y. P. Liu, L. Chen, Q. Tang, W. Wu, W. Sun, Z. Chen and X. X. Yan, Cell Res., 2020, 30, 272–275 Search PubMed.
- Q. Xie, T. P. Wu, R. C. Gimple, Z. Li, B. C. Prager, Q. Wu, Y. Yu, P. Wang, Y. Wang, D. U. Gorkin, C. Zhang, A. V. Dowiak, K. Lin, C. Zeng, Y. Sui, L. J. Y. Kim, T. E. Miller, L. Jiang, C. H. Lee, Z. Huang, X. Fang, K. Zhai, S. C. Mack, M. Sander, S. Bao, A. E. Kerstetter-Fogle, A. E. Sloan, A. Z. Xiao and J. N. Rich, Cell, 2018, 175, 1228–1243 CrossRef CAS PubMed.
- M. Zhang, S. Yang, R. Nelakanti, W. Zhao, G. Liu, Z. Li, X. Liu, T. Wu, A. Xiao and H. Li, Cell Res., 2020, 30, 197–210 CAS.
- H. Sucipto, F. Kudo and T. Eguchi, Angew Chem. Int. Ed. Engl., 2012, 51, 3428–3431 Search PubMed.
- L. Wu, Z. Wang, Y. Cen, B. Wang and J. Zhou, Angew Chem. Int. Ed. Engl., 2022, 61, e202112063 CrossRef CAS PubMed.
- N. Steffan, A. Grundmann, S. Afiyatullov, H. Ruan and S. M. Li, Org. Biomol. Chem., 2009, 7, 4082–4087 RSC.
- G. Zhu, W. Yan, X. Wang, R. Cheng, N. Naowarojna, K. Wang, J. Wang, H. Song, Y. Wang, H. Liu, X. Xia, C. E. Costello, X. Liu, L. Zhang and P. Liu, JACS Au, 2022, 2, 1686–1698 CrossRef CAS.
- Y. Matsuda, T. Bai, C. B. W. Phippen, C. S. Nodvig, I. Kjaerbolling, T. C. Vesth, M. R. Andersen, U. H. Mortensen, C. H. Gotfredsen, I. Abe and T. O. Larsen, Nat. Commun., 2018, 9, 2587 CrossRef.
- H. Tao, T. Mori, H. Chen, S. Lyu, A. Nonoyama, S. Lee and I. Abe, Nat. Commun., 2022, 13, 95 CrossRef CAS.
- M. S. Islam, T. M. Leissing, R. Chowdhury, R. J. Hopkinson and C. J. Schofield, Annu. Rev. Biochem., 2018, 87, 585–620 CrossRef CAS.
- C. Q. Herr and R. P. Hausinger, Trends Biochem. Sci., 2018, 43, 517–532 CrossRef CAS PubMed.
- L. F. Wu, S. Meng and G. L. Tang, Biochim. Biophys. Acta, 2016, 1864, 453–470 Search PubMed.
- S. S. Gao, N. Naowarojna, R. Cheng, X. Liu and P. Liu, Nat. Prod. Rep., 2018, 35, 792–837 RSC.
- E. King-Smith, C. R. Zwick 3rd and H. Renata, Biochemistry, 2018, 57, 403–412 CrossRef CAS.
- C. R. Zwick and H. Renata, Nat. Prod. Rep., 2020, 37, 1065–1079 RSC.
- C. Loenarz and C. J. Schofield, Trends Biochem. Sci., 2011, 36, 7–18 CrossRef CAS.
- P. L. Dahia, Cancer Cell, 2017, 31, 159–161 CrossRef CAS PubMed.
- H. Celik, W. K. Koh, A. C. Kramer, E. L. Ostrander, C. Mallaney, D. A. C. Fisher, J. Xiang, W. C. Wilson, A. Martens, A. Kothari, G. Fishberger, E. Tycksen, D. Karpova, E. J. Duncavage, Y. Lee, S. T. Oh and G. A. Challen, Cancer Cell, 2018, 34, 741–756 CrossRef CAS PubMed.
- D. Frescas, D. Guardavaccaro, F. Bassermann, R. Koyama-Nasu and M. Pagano, Nature, 2007, 450, 309–313 CrossRef CAS.
- J. A. Losman, P. Koivunen and W. G. Kaelin Jr, Nat. Rev. Cancer, 2020, 20, 710–726 CrossRef CAS PubMed.
- A. J. Mitchell and J. K. Weng, Plant Physiol., 2019, 179, 813–829 CrossRef CAS.
- M. Cheng, D. Chen, R. E. Parales and J. Jiang, Annu. Rev. Microbiol., 2022, 76, 325–348 CrossRef CAS.
- P. Rabe, J. Kamps, C. J. Schofield and C. T. Lohans, Nat. Prod. Rep., 2018, 35, 735–756 RSC.
- J. E. Sears and D. L. Boger, Acc. Chem. Res., 2015, 48, 653–662 CrossRef CAS.
- I. Abe, Chem. Pharm. Bull., 2020, 68, 823–831 CrossRef CAS PubMed.
- B. Samantaray, R. R. Behera, R. R. Mishra and H. Thatoi, Syst. Microbiol. Biomanuf., 2024, 4, 1174–1192 CrossRef CAS.
- L. Li, R. Zhang, W. Zhang and Y. Xu, Syst. Microbiol. Biomanuf., 2023, 3, 440–448 CrossRef CAS.
- C. Schofield and R. Hausinger, 2-Oxoglutarate-Dependent Oxygenases, The Royal Society of Chemistry, 2015 Search PubMed.
- R. P. Hausinger, Crit. Rev. Biochem. Mol. Biol., 2004, 39, 21–68 CrossRef CAS.
- R. P. Hausinger, in 2-Oxoglutarate-Dependent Oxygenases, ed. C. Schofield and R. Hausinger, The Royal Society of Chemistry, 2015, 10.1039/9781782621959-00001.
- W. Aik, M. A. McDonough, A. Thalhammer, R. Chowdhury and C. J. Schofield, Curr. Opin. Struct. Biol., 2012, 22, 691–700 CrossRef CAS.
- I. J. Clifton, M. A. McDonough, D. Ehrismann, N. J. Kershaw, N. Granatino and C. J. Schofield, J. Inorg. Biochem., 2006, 100, 644–669 CrossRef CAS PubMed.
- X. Jing, H. Liu, Y. Nie and Y. Xu, Syst. Microbiol. Biomanuf., 2021, 1, 275–290 CrossRef CAS.
- S. Markolovic, S. E. Wilkins and C. J. Schofield, J. Biol. Chem., 2015, 290, 20712–20722 CrossRef CAS PubMed.
- G. Zurlo, J. Guo, M. Takada, W. Wei and Q. Zhang, Biochim. Biophys. Acta, 2016, 1866, 208–220 CAS.
- P. Rabe, J. H. Beale, A. Butryn, P. Aller, A. Dirr, P. A. Lang, D. N. Axford, S. B. Carr, T. M. Leissing, M. A. McDonough, B. Davy, A. Ebrahim, J. Orlans, S. L. S. Storm, A. M. Orville, C. J. Schofield and R. L. Owen, IUCrJ, 2020, 7, 901–912 CrossRef CAS PubMed.
- J. R. Chekan, C. Ongpipattanakul, T. R. Wright, B. Zhang, J. M. Bollinger Jr, L. J. Rajakovich, C. Krebs, R. M. Cicchillo and S. K. Nair, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 13299–13304 CrossRef CAS PubMed.
- I. Alonso-de Vega, M. C. Paz-Cabrera, M. B. Rother, W. W. Wiegant, C. Checa-Rodriguez, J. R. Hernandez-Fernaud, P. Huertas, R. Freire, H. van Attikum and V. A. J. Smits, Nucleic Acids Res., 2020, 48, 4915–4927 CrossRef CAS PubMed.
- W. Xu, H. Yang, Y. Liu, Y. Yang, P. Wang, S. H. Kim, S. Ito, C. Yang, P. Wang, M. T. Xiao, L. X. Liu, W. Q. Jiang, J. Liu, J. Y. Zhang, B. Wang, S. Frye, Y. Zhang, Y. H. Xu, Q. Y. Lei, K. L. Guan, S. M. Zhao and Y. Xiong, Cancer Cell, 2011, 19, 17–30 CrossRef CAS.
- P. Hahn, J. Bose, S. Edler and A. Lengeling, BMC Genomics, 2008, 9, 293 CrossRef PubMed.
- M. A. McDonough, C. Loenarz, R. Chowdhury, I. J. Clifton and C. J. Schofield, Curr. Opin. Struct. Biol., 2010, 20, 659–672 CrossRef CAS.
- P. Rani, G. Gautam, T. Anwar, S. Gourinath and A. Datta, Int. J. Biol. Macromol., 2020, 150, 1272–1280 CrossRef CAS.
- M. Kato, Y. Araiso, A. Noma, A. Nagao, T. Suzuki, R. Ishitani and O. Nureki, Nucleic Acids Res., 2011, 39, 1576–1585 CrossRef CAS.
- J. M. Elkins, K. S. Hewitson, L. A. McNeill, J. F. Seibel, I. Schlemminger, C. W. Pugh, P. J. Ratcliffe and C. J. Schofield, J. Biol. Chem., 2003, 278, 1802–1806 CrossRef CAS PubMed.
- R. Chowdhury, R. Sekirnik, N. C. Brissett, T. Krojer, C. H. Ho, S. S. Ng, I. J. Clifton, W. Ge, N. J. Kershaw, G. C. Fox, J. R. C. Muniz, M. Vollmar, C. Phillips, E. S. Pilka, K. L. Kavanagh, F. von Delft, U. Oppermann, M. A. McDonough, A. J. Doherty and C. J. Schofield, Nature, 2014, 510, 422–426 CrossRef CAS PubMed.
- J. R. Horton, A. K. Upadhyay, H. H. Qi, X. Zhang, Y. Shi and X. Cheng, Nat. Struct. Mol. Biol., 2010, 17, 38–43 CrossRef CAS PubMed.
- S. S. Ng, K. L. Kavanagh, M. A. McDonough, D. Butler, E. S. Pilka, B. M. Lienard, J. E. Bray, P. Savitsky, O. Gileadi, F. von Delft, N. R. Rose, J. Offer, J. C. Scheinost, T. Borowski, M. Sundstrom, C. J. Schofield and U. Oppermann, Nature, 2007, 448, 87–91 CrossRef CAS PubMed.
- P. L. Roach, I. J. Clifton, C. M. Hensgens, N. Shibata, C. J. Schofield, J. Hajdu and J. E. Baldwin, Nature, 1997, 387, 827–830 CrossRef CAS.
- I. K. Leung, T. J. Krojer, G. T. Kochan, L. Henry, F. von Delft, T. D. Claridge, U. Oppermann, M. A. McDonough and C. J. Schofield, Chem. Biol., 2010, 17, 1316–1324 CAS.
- C. E. Dann 3rd, R. K. Bruick and J. Deisenhofer, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15351–15356 Search PubMed.
- C. Feng, Y. Liu, G. Wang, Z. Deng, Q. Zhang, W. Wu, Y. Tong, C. Cheng and Z. Chen, J. Biol. Chem., 2014, 289, 11571–11583 CrossRef CAS PubMed.
- B. Jia, X. Jia, K. H. Kim and C. O. Jeon, Biochim. Biophys. Acta, Gen. Subj., 2017, 1861, 323–334 CrossRef CAS PubMed.
- S. Martinez and R. P. Hausinger, J. Biol. Chem., 2015, 290, 20702–20711 CrossRef CAS.
- X. Huang and J. T. Groves, JBIC, J. Biol. Inorg. Chem., 2017, 22, 185–207 CrossRef CAS PubMed.
- S. Kal and L. Que, JBIC, J. Biol. Inorg. Chem., 2017, 22, 339–365 CrossRef CAS.
- J. C. Price, E. W. Barr, L. M. Hoffart, C. Krebs and J. M. Bollinger Jr, Biochemistry, 2005, 44, 8138–8147 CrossRef CAS PubMed.
- J. A. Hangasky, C. Y. Taabazuing, M. A. Valliere and M. J. Knapp, Metallomics, 2013, 5, 287–301 CrossRef CAS.
- T. Borowski, A. Bassan and P. E. Siegbahn, Chemistry, 2004, 10, 1031–1041 CrossRef CAS PubMed.
- D. G. Fujimori, E. W. Barr, M. L. Matthews, G. M. Koch, J. R. Yonce, C. T. Walsh, J. M. Bollinger Jr, C. Krebs and P. J. Riggs-Gelasco, J. Am. Chem. Soc., 2007, 129, 13408–13409 CrossRef CAS PubMed.
- E. I. Solomon, T. C. Brunold, M. I. Davis, J. N. Kemsley, S. K. Lee, N. Lehnert, F. Neese, A. J. Skulan, Y. S. Yang and J. Zhou, Chem. Rev., 2000, 100, 235–350 CrossRef CAS PubMed.
- P. K. Grzyska, E. H. Appelman, R. P. Hausinger and D. A. Proshlyakov, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3982–3987 CrossRef CAS.
- A. J. Mitchell, Q. Zhu, A. O. Maggiolo, N. R. Ananth, M. L. Hillwig, X. Liu and A. K. Boal, Nat. Chem. Biol., 2016, 12, 636–640 CrossRef CAS PubMed.
- T. Kodera, S. V. Smirnov, N. N. Samsonova, Y. I. Kozlov, R. Koyama, M. Hibi, J. Ogawa, K. Yokozeki and S. Shimizu, Biochem. Biophys. Res. Commun., 2009, 390, 506–510 CrossRef CAS PubMed.
- M. Hibi, T. Kawashima, T. Kasahara, P. M. Sokolov, S. V. Smirnov, T. Kodera, M. Sugiyama, S. Shimizu, K. Yokozeki and J. Ogawa, Lett. Appl. Microbiol., 2012, 55, 414–419 CrossRef CAS.
- B. Haltli, Y. Tan, N. A. Magarvey, M. Wagenaar, X. Yin, M. Greenstein, J. A. Hucul and T. M. Zabriskie, Chem. Biol., 2005, 12, 1163–1168 CrossRef CAS.
- M. Myllykoski, A. Sutinen, M. K. Koski, J. P. Kallio, A. Raasakka, J. Myllyharju, R. K. Wierenga and P. Koivunen, J. Biol. Chem., 2021, 296, 100197 CrossRef CAS.
- J. P. Holt-Martyn, A. Tumber, M. Z. Rahman, K. Lippl, W. Figg Jr, M. A. McDonough, R. Chowdhury and C. J. Schofield, Medchemcomm, 2019, 10, 500–504 RSC.
- S. D. Bembenek, H. Venkatesan, H. M. Peltier, M. D. Rosen, T. D. Barrett, K. C. Kanelakis, H. L. Palomino, T. I. Brondstetter, T. Mirzadegan and M. H. Rabinowitz, ACS Omega, 2019, 4, 6703–6708 CrossRef CAS.
- J. P. Holt-Martyn, R. Chowdhury, A. Tumber, T. L. Yeh, M. I. Abboud, K. Lippl, C. T. Lohans, G. W. Langley, W. Figg Jr, M. A. McDonough, C. W. Pugh, P. J. Ratcliffe and C. J. Schofield, ChemMedChem, 2020, 15, 270–273 CrossRef CAS.
- P. L. Roach, I. J. Clifton, V. Fulop, K. Harlos, G. J. Barton, J. Hajdu, I. Andersson, C. J. Schofield and J. E. Baldwin, Nature, 1995, 375, 700–704 CrossRef CAS PubMed.
- N. I. Burzlaff, P. J. Rutledge, I. J. Clifton, C. M. Hensgens, M. Pickford, R. M. Adlington, P. L. Roach and J. E. Baldwin, Nature, 1999, 401, 721–724 CrossRef CAS PubMed.
- W. A. Schenk, Angew Chem. Int. Ed. Engl., 2000, 39, 3409–3411 CAS.
- M. Costas, M. P. Mehn, M. P. Jensen and L. Que Jr, Chem. Rev., 2004, 104, 939–986 CAS.
- J. E. Baldwin and E. Abraham, Nat. Prod. Rep., 1988, 5, 129–145 CAS.
- H. Zhang, S. Che, R. Wang, R. Liu, Q. Zhang and M. Bartlam, Biochem. Biophys. Res. Commun., 2019, 514, 1031–1036 CAS.
- C. D. Brown, M. L. Neidig, M. B. Neibergall, J. D. Lipscomb and E. I. Solomon, J. Am. Chem. Soc., 2007, 129, 7427–7438 CrossRef CAS.
- I. J. Clifton, W. Ge, R. M. Adlington, J. E. Baldwin and P. J. Rutledge, Arch. Biochem. Biophys., 2011, 516, 103–107 CrossRef CAS.
- A. Daruzzaman, I. J. Clifton, R. M. Adlington, J. E. Baldwin and P. J. Rutledge, Arch. Biochem. Biophys., 2013, 530, 48–53 CrossRef CAS PubMed.
- M. Lundberg, P. E. Siegbahn and K. Morokuma, Biochemistry, 2008, 47, 1031–1042 CrossRef CAS PubMed.
- P. Rabe, J. Kamps, K. D. Sutherlin, J. D. S. Linyard, P. Aller, C. C. Pham, H. Makita, I. Clifton, M. A. McDonough, T. M. Leissing, D. Shutin, P. A. Lang, A. Butryn, J. Brem, S. Gul, F. D. Fuller, I. S. Kim, M. H. Cheah, T. Fransson, A. Bhowmick, I. D. Young, L. O’Riordan, A. S. Brewster, I. Pettinati, M. Doyle, Y. Joti, S. Owada, K. Tono, A. Batyuk, M. S. Hunter, R. Alonso-Mori, U. Bergmann, R. L. Owen, N. K. Sauter, T. D. W. Claridge, C. V. Robinson, V. K. Yachandra, J. Yano, J. F. Kern, A. M. Orville and C. J. Schofield, Sci. Adv., 2021, 7, eabh0250 CrossRef CAS.
- P. Rabe, C. C. Walla, N. K. Goodyear, J. Welsh, R. Southwart, I. Clifton, J. D. S. Linyard, A. Tumber, T. D. W. Claridge, W. K. Myers and C. J. Schofield, J. Biol. Chem., 2022, 298, 102249 CrossRef CAS PubMed.
- H. Tang, M. H. Wu, H. Y. Lin, M. R. Han, Y. H. Tu, Z. J. Yang, T. C. Chien, N. L. Chan and W. C. Chang, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2113770119 CrossRef CAS.
- W. Lau and E. S. Sattely, Science, 2015, 349, 1224–1228 CrossRef CAS.
- P. Wangchuk, T. Sastraruji, M. Taweechotipatr, P. A. Keller and S. G. Pyne, Nat. Prod. Commun., 2016, 11, 1801–1804 CrossRef.
- L. Calcul, W. D. Inman, A. A. Morris, K. Tenney, J. Ratnam, J. H. McKerrow, F. A. Valeriote and P. Crews, J. Nat. Prod., 2010, 73, 365–372 CrossRef CAS.
- X. Liu, Z. Yuan, H. Su, X. Hou, Z. Deng, H. Xu, B. Guo, D. Yin, X. Sheng and Y. Rao, ACS Catal., 2022, 12, 3689–3699 CrossRef CAS.
- K. Valegard, A. C. Terwisscha van Scheltinga, A. Dubus, G. Ranghino, L. M. Oster, J. Hajdu and I. Andersson, Nat. Struct. Mol. Biol., 2004, 11, 95–101 CrossRef CAS.
- H. J. Lee, M. D. Lloyd, K. Harlos, I. J. Clifton, J. E. Baldwin and C. J. Schofield, J. Mol. Biol., 2001, 308, 937–948 CrossRef CAS PubMed.
- T. J. Doyon, K. Skinner, D. Yang, L. Mallik, T. Wymore, M. Koutmos, P. M. Zimmerman and A. Narayan, ChemRxiv, 2020, preprint, DOI:10.26434/chemrxiv.12780044.v1.
- W. Li, T. Zhang and J. Ding, Nucleic Acids Res., 2015, 43, 10026–10038 CAS.
- L. Yan and Y. Liu, Inorg. Chem., 2019, 58, 13771–13781 CrossRef CAS PubMed.
- Y. Matsuda, T. Iwabuchi, T. Fujimoto, T. Awakawa, Y. Nakashima, T. Mori, H. Zhang, F. Hayashi and I. Abe, J. Am. Chem. Soc., 2016, 138, 12671–12677 CrossRef CAS PubMed.
- I. J. Clifton, L. X. Doan, M. C. Sleeman, M. Topf, H. Suzuki, R. C. Wilmouth and C. J. Schofield, J. Biol. Chem., 2003, 278, 20843–20850 CrossRef CAS PubMed.
- M. Topf, G. M. Sandala, D. M. Smith, C. J. Schofield, C. J. Easton and L. Radom, J. Am. Chem. Soc., 2004, 126, 9932–9933 CrossRef CAS.
- T. Borowski, E. Broclawik, C. J. Schofield and P. E. Siegbahn, J. Comput. Chem., 2006, 27, 740–748 CrossRef CAS PubMed.
- W. C. Chang, Y. Guo, C. Wang, S. E. Butch, A. C. Rosenzweig, A. K. Boal, C. Krebs and J. M. Bollinger Jr, Science, 2014, 343, 1140–1144 CrossRef CAS.
- A. K. Boal, J. M. Bollinger Jr and W. C. Chang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11989–11990 CrossRef CAS PubMed.
- A. Papadopoulou, F. Meyer and R. M. Buller, Biochemistry, 2023, 62, 229–240 CrossRef CAS PubMed.
- S. L. Mader, A. Brauer, M. Groll and V. R. I. Kaila, Nat. Commun., 2018, 9, 1168 CrossRef.
- P. O. Falnes, R. F. Johansen and E. Seeberg, Nature, 2002, 419, 178–182 CrossRef CAS PubMed.
- S. C. Trewick, T. F. Henshaw, R. P. Hausinger, T. Lindahl and B. Sedgwick, Nature, 2002, 419, 174–178 CrossRef CAS.
- J. M. Hagel and P. J. Facchini, Nat. Chem. Biol., 2010, 6, 273–275 CrossRef CAS PubMed.
- N. A. Kuznetsov, L. Y. Kanazhevskaya and O. S. Fedorova, Int. J. Mol. Sci., 2021, 22, 10540 CrossRef CAS PubMed.
- B. Mrugala, A. Milaczewska, P. J. Porebski, E. Niedzialkowska, M. Guzik, W. Minor and T. Borowski, FEBS J., 2021, 288, 1366–1386 CrossRef CAS PubMed.
- Z. Zhang, J. Ren, D. K. Stammers, J. E. Baldwin, K. Harlos and C. J. Schofield, Nat. Struct. Biol., 2000, 7, 127–133 CrossRef CAS.
- F. Kudo, Y. Kitayama, A. Miyanaga, M. Numakura and T. Eguchi, ChemBioChem, 2021, 22, 1668–1675 CrossRef CAS PubMed.
- W. Yan, H. Song, F. Song, Y. Guo, C. H. Wu, A. S. Her, Y. Pu, S. Wang, N. Naowarojna, A. Weitz, M. P. Hendrich, C. E. Costello, L. Zhang, P. Liu and Y. J. Zhang, Nature, 2015, 527, 539–543 CrossRef CAS.
- N. P. Dunham, J. M. Del Rio Pantoja, B. Zhang, L. J. Rajakovich, B. D. Allen, C. Krebs, A. K. Boal and J. M. Bollinger Jr, J. Am. Chem. Soc., 2019, 141, 9964–9979 CrossRef CAS.
- A. Milaczewska and T. Borowski, Dalton Trans., 2019, 48, 16211–16221 RSC.
- F. Wang, Y. Gao, C. Wang, W. Lan, J. Gan and C. Cao, Catal. Sci. Technol., 2023, 13, 6839–6849 RSC.
- T. Mori, R. Zhai, R. Ushimaru, Y. Matsuda and I. Abe, Nat. Commun., 2021, 12, 4417 CrossRef CAS.
- T. Bai, Y. Matsuda, H. Tao, T. Mori, Y. Zhang and I. Abe, Org. Lett., 2020, 22, 4311–4315 CrossRef CAS.
- S. Meng, W. Han, J. Zhao, X. H. Jian, H. X. Pan and G. L. Tang, Angew Chem. Int. Ed. Engl., 2018, 57, 719–723 CrossRef CAS.
- T.-Y. Chen, S. Xue, W.-C. Tsai, T.-C. Chien, Y. Guo and W.-c. Chang, ACS Catal., 2020, 11, 278–282 CrossRef.
- Y. Jiang, T. Cao, Y. Yang, H. Zhang, J. Zhang and X. Li, Science, 2023, 382, 92–98 CrossRef CAS PubMed.
- M. D. Tsai, W. J. Wu and M. C. Ho, Annu. Rev. Biophys., 2022, 51, 19–38 CrossRef CAS.
- L. A. Yates, R. M. Williams, S. Hailemariam, R. Ayala, P. Burgers and X. Zhang, Structure, 2020, 28, 96–104 CrossRef CAS.
- G. Weissenberger, R. J. M. Henderikx and P. J. Peters, Nat. Methods, 2021, 18, 463–471 CrossRef CAS PubMed.
- T. O. Yeates, M. P. Agdanowski and Y. Liu, Curr. Opin. Struct. Biol., 2020, 60, 142–149 CrossRef CAS.
- K. M. Yip, N. Fischer, E. Paknia, A. Chari and H. Stark, Nature, 2020, 587, 157–161 CrossRef CAS PubMed.
- M. Akdel, D. E. V. Pires, E. P. Pardo, J. Janes, A. O. Zalevsky, B. Meszaros, P. Bryant, L. L. Good, R. A. Laskowski, G. Pozzati, A. Shenoy, W. Zhu, P. Kundrotas, V. R. Serra, C. H. M. Rodrigues, A. S. Dunham, D. Burke, N. Borkakoti, S. Velankar, A. Frost, J. Basquin, K. Lindorff-Larsen, A. Bateman, A. V. Kajava, A. Valencia, S. Ovchinnikov, J. Durairaj, D. B. Ascher, J. M. Thornton, N. E. Davey, A. Stein, A. Elofsson, T. I. Croll and P. Beltrao, Nat. Struct. Mol. Biol., 2022, 29, 1056–1067 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Zidek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS.
- X. Wang, P. Yang, B. Zhao and S. Liu, Syst. Microbiol. Biomanuf., 2023, 3, 75–87 CrossRef CAS.
- J. Cheng, G. Novati, J. Pan, C. Bycroft, A. Zemgulyte, T. Applebaum, A. Pritzel, L. H. Wong, M. Zielinski, T. Sargeant, R. G. Schneider, A. W. Senior, J. Jumper, D. Hassabis, P. Kohli and Z. Avsec, Science, 2023, 381, eadg7492 CrossRef CAS PubMed.
- R. Krishna, J. Wang, W. Ahern, P. Sturmfels, P. Venkatesh, I. Kalvet, G. R. Lee, F. S. Morey-Burrows, I. Anishchenko, I. R. Humphreys, R. McHugh, D. Vafeados, X. Li, G. A. Sutherland, A. Hitchcock, C. N. Hunter, A. Kang, E. Brackenbrough, A. K. Bera, M. Baek, F. DiMaio and D. Baker, Science, 2024, eadl2528, DOI:10.1126/science.adl2528.
- J. Abramson, J. Adler, J. Dunger, R. Evans, T. Green, A. Pritzel, O. Ronneberger, L. Willmore, A. J. Ballard, J. Bambrick, S. W. Bodenstein, D. A. Evans, C.-C. Hung, M. O’Neill, D. Reiman, K. Tunyasuvunakool, Z. Wu, A. Žemgulytė, E. Arvaniti, C. Beattie, O. Bertolli, A. Bridgland, A. Cherepanov, M. Congreve, A. I. Cowen-Rivers, A. Cowie, M. Figurnov, F. B. Fuchs, H. Gladman, R. Jain, Y. A. Khan, C. M. R. Low, K. Perlin, A. Potapenko, P. Savy, S. Singh, A. Stecula, A. Thillaisundaram, C. Tong, S. Yakneen, E. D. Zhong, M. Zielinski, A. Žídek, V. Bapst, P. Kohli, M. Jaderberg, D. Hassabis and J. M. Jumper, Nature, 2024, 630, 493–500 CrossRef CAS PubMed.
- F. Barbault and F. Maurel, Expert Opin. Drug Discovery, 2015, 10, 1047–1057 CrossRef CAS.
- M. Bermudez, J. Mortier, C. Rakers, D. Sydow and G. Wolber, Drug Discovery Today, 2016, 21, 1799–1805 CrossRef CAS.
- R. Mehmood and H. J. Kulik, Methods Mol. Biol., 2022, 2397, 227–248 CrossRef CAS.
- J. Mortier, C. Rakers, M. Bermudez, M. S. Murgueitio, S. Riniker and G. Wolber, Drug Discovery Today, 2015, 20, 686–702 CrossRef CAS PubMed.
- A. Warshel and M. Levitt, J. Mol. Biol., 1976, 103, 227–249 CrossRef CAS PubMed.
- F. Zhang, T. Zeng and R. Wu, J. Chem. Inf. Model., 2023, 63, 5018–5034 CrossRef CAS PubMed.
- L. Wu, L. Qin, Y. Nie, Y. Xu and Y. L. Zhao, Biotechnol. Adv., 2022, 54, 107793 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |