
Human microbiota peptides: important roles in human health
Abdul Bari
Shah
 and
Sang Hee
Shim
*
and
Sang Hee
Shim
*
Natural Products Research Institute, College of Pharmacy, Seoul National University, Seoul 08826, Republic of Korea. E-mail: sanghee_shim@snu.ac.kr
First published on 15th November 2024
Abstract
Covering: 1974 to 2024
Human microbiota consist of a diverse array of microorganisms, such as bacteria, Eukarya, archaea, and viruses, which populate various parts of the human body and live in a cooperatively beneficial relationship with the host. They play a crucial role in supporting the functional balance of the microbiome. The coevolutionary progression has led to the development of specialized metabolites that have the potential to substitute traditional antibiotics in combating global health challenges. Although there has been a lot of research on the human microbiota, there is a considerable lack of understanding regarding the wide range of peptides that these microbial populations produce. Particularly noteworthy are the antibiotics that are uniquely produced by the human microbiome, especially by bacteria, to protect against invasive infections. This review seeks to fill this knowledge gap by providing a thorough understanding of various peptides, along with their in-depth biological importance in terms of human disorders. Advancements in genomics and the understanding of molecular mechanisms that control the interactions between microbiota and hosts have made it easier to find peptides that come from the human microbiome. We hope that this review will serve as a basis for developing new therapeutic approaches and personalized healthcare strategies. Additionally, it emphasizes the significance of these microbiota in the field of natural product discovery and development.
 Abdul Bari Shah | Abdul Bari Shah is a postdoctoral research associate at the Natural Products Research Institute, College of Pharmacy, Seoul National University (SNU), South Korea. In 2023, he earned his PhD in the field of organic and natural product chemistry from Gyeongsang National University, South Korea, under the supervision of Prof. Ki Hun Park. The focus of his work mostly involves the separation and identification of bioactive compounds produced by plants, fungi, and the human microbiota, as well as studying their biological functions. He was awarded the young pioneer research award twice for his exceptional performance during his research career. |
 Sang Hee Shim | Sang Hee Shim is a Professor at the Natural Products Research Institute, College of Pharmacy, at Seoul National University (SNU), Korea. She began her career as a post-doctoral researcher at the University of Iowa and the University of Illinois at Chicago after receiving her PhD from SNU in 2004. Then she started independent work at Yeungnam University in 2007 until she transferred to Duksung Women's University in 2015. In 2021, she joined SNU as a professor. Her research group mainly focuses on the discovery of bioactive compounds from endosymbiotic microorganisms. She has published over 100 SCI(E) research papers as of 2024. |
1 Introduction
Researchers worldwide have examined the complex relationship between the intestinal microbiome and human health due to the significant progresses in sequencing technologies over the past ten years.1,2 The term “microbiota” has its roots around the past twenty years, when scientists noticed that various parts of the human body were home to several kinds of microbes, such as bacteria, yeasts, and viruses that coexisted symbiotically with the host and had a major impact on immunological response, physiological functions, and disease susceptibility.3,4 At the same time, the functional potential and metabolic diversity of these microbial communities are reflected in the human microbiome, which includes the collective genetic material of the microbiota. At an early stage of development, humans establish symbiotic associations with these microorganisms. This assemblage is influenced by various factors including the environment, closeness to other humans and animals, food, genetics, and behavioral change,5 and in turn, these complex relationships between host genetics, environmental conditions, and the dynamics of microbial colonization have led to the amazing diversity and adaptability of the human microbiota and microbiome.6Most of the complex microbial communities found in the human body are composed of bacteria, with members of well-known families, including Firmicutes, Bacteroidetes, Actinobacteria, Fusobacteria, Proteobacteria, and Verrucomicrobia, inhabiting specific physiological niches. These microbes are present in the skin, oral cavity, respiratory system, gastrointestinal tract, and urogenital regions, among other locations. Fungi are less prevalent; however, they are crucial for microbial variety and functionality. Each possesses distinct features and functions within the human body. It is becoming more and more evident that how they participate in both healthy and diseased states emphasizes how crucial it is to comprehend their roles within the human microbiota.4,7
The ability to recognize biosynthetic genes within bacterial genome sequences has been transformed by recent developments in genomic sequencing technology, which has made it easier to predict the chemical structures of a wide range of metabolites. This method, known as genome mining, has been shown to be an effective means of finding a wide spectrum of bioactive substances. Among the most important players in this process are the biosynthetic gene clusters (BGCs), which encode various regulatory elements and enzymes essential for coordinating the biosynthesis and ecological roles of secondary metabolites.8,9 The production of secondary metabolites is fundamentally guided by genetic blueprints found in these clusters. Using 16S rRNA gene, sequencing has improved our ability to comprehend different bacterial species in microbiota samples.10 By linking this method with hypotheses derived from genome sequences and the existence of secondary metabolite clusters, we can speculate about the possible metabolic strength of these bacterial species. These integrated approaches offer significant insights into the varied metabolic characteristics and physiological functions of microbial communities occupying different ecological places. Besides these developments, new bioinformatic techniques that have recently drastically changed the field of natural product discovery play a crucial role in identifying novel metabolites from the vital reservoir of the human microbiota.
With the help of this advanced and broad knowledge, a wide range of naturally occurring substances linked to human health have been isolated from microorganisms. These compounds have a wide range of chemical structures that are comparable to those of bacteria found in both aquatic and terrestrial environments.11,12 Among them are compounds that are essential mediators for interactions between different microbiological species and their hosts.13,14 The human microbiota-derived peptides play a significant role in the controlling of many human disorders, from antimicrobial to anti-inflammatory, anticancer and from obesity to diabetes and mental disorders. In addition, these peptides significantly affect metabolism, many inflammation pathways, and gut–brain axis interactions. As an example, bacteriocin helps to keep the microbiota community together by preventing harmful bacteria from multiplying. In the event of dysbiosis, which has been associated with inflammatory bowel disorders (IBD), these peptides also keep the microbiota community balanced.4,15
The primary objective of this review is to undertake a thorough investigation of peptides that come from the human microbiome. We aim to disclose the health potential and health challenges of the peptides that are derived from human microbiota, specifically bacteria by a thorough examination and synthesis through available literature. Our review starts with a discussion of the significance of the human microbiota. It then provides an extensive overview of the literature that is currently accessible on most of the peptides that are produced by these microbiota.
2 Methodology
All essential information regarding peptides originating from the human microbiome was gathered from the published literature. Research articles on human microbiota peptides were obtained from various scientific sources, including PubMed, the Web of Science, the American Chemical Society (ACS), the American Society of Microbiology (ASM), Google Scholar, and SciFinder. The process of selecting and evaluating publications for their relevance was carried out by considering their titles, abstracts, and keywords. The chemical structures were drawn using ChemDraw 22.2.0 (structure with less number of amino acids) and https://app.biorender.com/.3 Human microbiota and microbiome are hosts of important peptides
The gastrointestinal tract (GIT), an ecosystem filled with over 100 trillion microorganisms, contains an incredible assortment of approximately 3 million genes.16,17 Firmicutes, Bacteroidetes, Actinobacteria, Proteobacteria, and Verrucomicrobia are the phyla with the most dominant bacterial populations. These occupants of the GIT participate in a variety of activities that benefit the host. These activities include the breakdown of vitamins and metabolites including bile acids, amino acids, and lipids. They also regulate pH, produce peptides, and influence several cell signaling pathways.18–20 Metabolites are essential components of the human microbiome, serving different roles in sustaining health and affecting disease progression.21,22 Metabolites, particularly those peptides in the gut, play a crucial role in host–microbe interactions. They alter the local environment and influence overall health. BGCs linked with metabolite biosynthesis provide significant information on the mechanisms behind their synthesis.10,22–24The oral cavity ranks second in hosting microbiota, comprising over 700 bacterial species. Oral microbiota demonstrates remarkable diversity in predicted protein activities compared to other body locations. The oral cavity comprises two types of sites for bacterial colonization: the hard and soft tissues of the teeth and the oral mucosa. These microbiota always experience significant alterations in composition and activity, mostly influenced by nutrition, pH variations, bacterial interactions, and genetic mutations that may lead to the emergence of new strains.25,26 Moreover, the skin hosts a varied assortment of symbiotic bacteria that inhabit certain niches, which is essential for maintaining our health and provides an unexpected variety of additional benefits. The primary bacteria are Staphylococcus and Propionibacterium spp., which constitute a substantial component of the skin microbiota.27
The human microbiota produces several types of secondary metabolites such as ribosomally synthesized and post-translationally modified peptides, including lantibiotics and bacteriocins, microcins and thiazole-/oxazole-modified microcins, and heat-stable enterotoxin. Moreover, they include the byproducts of amino acid metabolism, oligosaccharides, glycolipids, terpenoids, polyketides, and nonribosomal peptides. These many groups make up the metabolic profile of the human microbiota.28,29 Although not all species within the human microbiota contribute to these substances, it is essential to thoroughly examine the other species that may yield novel bioactive compounds. The genomic loci consist of genes that encode enzymes responsible for the synthesis of various metabolites, providing a detailed plan for comprehending their production pathways. Scientists can examine BGCs to discover potential therapeutic targets for controlling metabolite production or to create probiotic strains with enhanced beneficial metabolite synthesis.30,31
It is known that several kinds of bacteria in the human microbiome are very capable of synthesizing peptides. Firmicutes and Bacteroidetes are predominant bacterial phyla in the gut. They are known for their wide range of metabolic activities. Short-chain fatty acids (SCFAs) and peptides are produced mostly by Firmicutes, which include genera like Clostridium and Lactobacillus, whereas Bacteroidetes, which include Bacteroides, are involved in the breakdown of polysaccharides and the synthesis of SCFAs. Furthermore, it is well recognized that Actinobacteria, which are represented by genera such as Bifidobacterium, produce healthy metabolites including vitamins and SCFAs.32–34 Most of the important peptides related to these groups of bacteria will be discussed in detail along with their in-depth biological role.
4 In-depth investigation of human microbiota-generated peptides and their significance in human disorders
A comprehensive analysis of the gut microbiota in a wide range of subjects, including humans and animals that have been treated with antibiotics, are germ-free, or are gnotobiotic, has revealed a strong connection between the gut microbiota and the development or advancement of several diseases. This link goes beyond a simple correlation, indicating a possible causal role of gut microbiota in the development of diseases. Various conditions such as non-alcoholic fatty liver disease, inflammatory bowel disease, allergies, obesity, Alzheimer's disease, Parkinson's disease, and depression have been linked or associated with gut microbiota. The complex relationship between gut microbiota and human health highlights the significant impact it has on disease development, emphasizing the need to comprehend its nuanced interaction with disease pathogenesis.35,36 Undoubtedly, metabolites produced by essential components within the microbiota have the ability to treat these various disorders. The six phyla of bacteria, previously mentioned, play a major role in the production of peptides, which have an enormous effect on the pathophysiology of human diseases. However, there is still a lack of thorough knowledge on the particular taxa of bacteria that produced different peptides and the frequency of similar compounds in different bacterial species. The objective of this investigation is to thoroughly classify the variety of bacterial species that live in the human microbiota and outline their individual roles in the treating of many human disorders. With broad implications for both ecological science and medical research, this endeavor attempts to provide insightful understandings of the complex interactions that shape microbial ecology by clarifying complex relationships between microbial communities and their metabolic outputs.4.1 Critical role of peptides generated from Firmicutes (Bacillota) in human disorders
Firmicutes and Bacteroidetes are two major bacterial phyla that make up the majority of the human gut microbiota. They account for more than 90% of the overall microbial population.37 Even with such a strong makeup, which is dominated by these phyla, the gut microbiota is remarkably resilient to short-term disturbances and can quickly return to its initial state.16 Different peptides produced by Firmicutes can significantly contribute to the physiology and health of a host. Firmicutes are a broad category of bacteria that include genera Bacillus, Lactobacillus, Clostridium, Ruminococcus, and Enterococcus, among others.1,32 The human gut microbiota produces a wide range of bioactive peptides, and these bacteria are well known for their adaptability in metabolism.18,384.1.1.1 Bioactive peptides from Bacillus and Lactobacillus and their biological roles. Bacillus and Lactobacillus are both found in the human microbiota, contributing significantly to the secretion of metabolites and playing a profound biological role. The number of peptides that have been isolated and characterised so far from Lactobacillus is significantly greater than that from Bacillus. For example, two peptides, Trn-α and Trn-β called thuricin CD, were extracted from the human fecal isolate Bacillus thuringiensis DPC 6431 The amino acid sequence for Trn-β consists of 49 amino acids (molecular mass = 2763), while Trn-α comprises 47 amino acids (molecular mass = 2861), demonstrating 45.3% sequence similarity and 39.6% identity, with both sequences containing three thioether linkages. The structure comprises six cycles of amino acids situated between Cys9 and Thr25, Cys5 and Thr25, and Cys13 and Ser21 in Trn-α, along with Cys9 and Ala25, Cys5 and Tyr28, and Cys13 and Thr21 in Trn-β. Subsequent NMR calculations validated the distinct D and L stereochemistry, leading to the formation of LLD isomers. These peptides have the ability to eliminate a broad spectrum of Clostridioides difficile, including ribotypes that are typically linked to Clostridium difficile-associated disease (CDAD).42,43 CDAD is a severe infection of the colon caused by the bacteria Clostridioides difficile, which typically affects people whose normal intestinal bacteria have been disturbed, generally due to recent antibiotic usage. The spectrum of this condition encompasses mild cases of diarrhoea to severe cases of sepsis and potentially fatal outcomes, with mortality rates reaching as high as 25% in vulnerable elderly individuals.44 An investigation employing flow cytometry and culture-dependent tests revealed that both Trnα and Trnβ induce membrane collapse, resulting in reduced cell size and granularity. The lytic activity occurs when thuricin CD peptides are inserted into target cells, causing the membrane to depolarize and resulting in cell death.45 One study demonstrated that thuricin CD exhibits activity at low doses when both peptides are present. A nanoformulation based on anionic lipids shows potential as a novel method for delivering thuricin CD locally through the oral route.46 Thuricin CD and several antibiotics (tigecycline, vancomycin, teicoplanin, rifampicin, and nitazoxanide) were tested on Clostridium difficile strains in biofilms and planktonic cells. The results demonstrated that Clostridium difficile strains differed in biofilm formation and antibiotic sensitivity. These findings offer new Clostridium difficile treatment techniques since strong biofilms in some strains can increase antibiotic resistance and infection recurrence.47 A comprising effect of fidaxomicin, a narrow spectrum antibiotic, on the human gut microbiome with thuricin CD and vancomycin and nisin was studied, demonstrating that thuricin CD was specific to Clostridium difficile and some Bacillus spp., with lower MICs for few other strains tested.48Fig. 1 represents a simple approach of thuricin CD isolation and its activity.
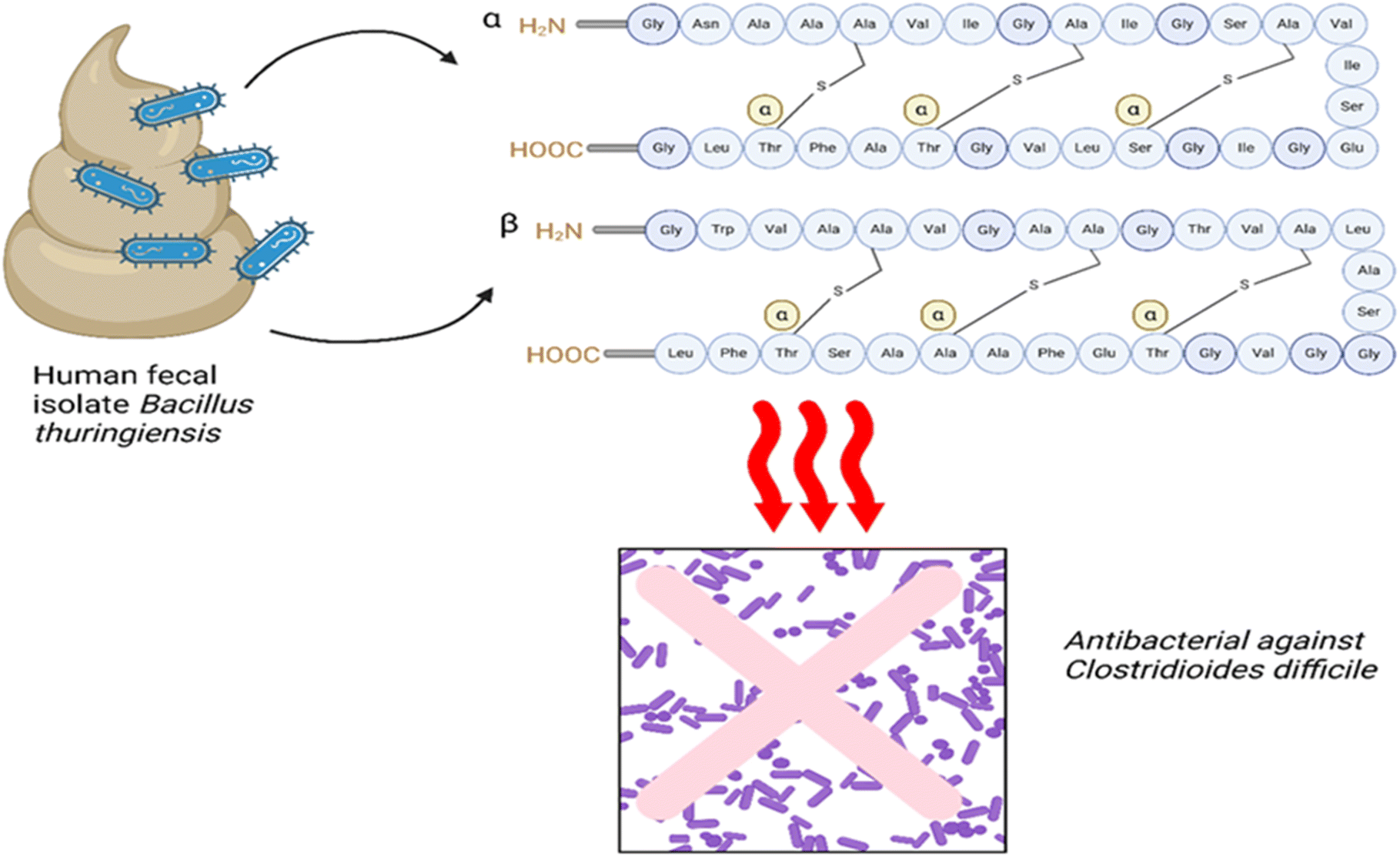 | ||
| Fig. 1 Trn-α and Trn-β isolated from Bacillus thuringiensis DPC 6431 as antibacterial agents against Clostridioides difficile (similar approach can be applied to most other bacterial metabolites). | ||
Conversely, lactobacilli, a major group of bacteria in the human body, have established themselves in several regions, particularly the digestive system, oral cavity, and female vaginal tract. They make a substantial contribution to compositions of metabolites in these places. Numerous metabolites generated by Lactobacillus, including organic acids, carboxylic acids, phenolic acids, cyclic dipeptides, hydrogen peroxide, reuterin, various proteinaceous compounds, and diverse antifungal substances, have been investigated for their potential to inhibit a wide range of fungal species.49,50 Itol et al. have studied a set of 73 strains of Lactobacillus acidophilus, in addition to one strain of Lactobacillus reuteri. The strains were obtained from human fecal samples and subsequently assessed for their ability to generate antimicrobial compounds against 16 strains that encompassed six distinct type of food-borne enteric pathogenic bacteria. Several strains of Lactobacillus gasseri exhibited notable inhibitory effects against the microorganisms that were tested. Gassericin A (1), produced by Lactobacillus gasseri LA39 isolated from human infant/adult feces on modified Lactobacillus selective agar, was found to be a powerful bacteriocin that could effectively kill bacteria without causing cell death,51 and gassericin T was also produced by Lactobacillus gasseri SBT 2055 isolated from human feces that displayed antibacterial activities.52 These bacteriocins have excellent thermal stability, favourable pH resistance, and potent bactericidal activity against numerous Gram-positive bacteria, particularly lactic acid bacteria. It is anticipated that they would serve as efficient food preservatives. Gassericins exhibit a wider range of antibacterial activities than those of nisin A. They are capable of inhibiting the growth of Gram-negative isolates when glycine is added. They can be effectively generated through the cultivation process in a food-grade medium utilising cheese whey, proteose–peptone, and surfactant yolk lecithin. The efficacy of gassericin A and glycine as preservatives was confirmed by demonstrating their ability to suppress the growth of microorganisms in custard creams.53 Furthermore, Lactobacillus gasseri SF1109 yielded a peptide of small size known as EFV12. This peptide exhibits the ability to bind to lipopolysaccharides (LPS) and efficiently mitigate their inflammatory effects through the inhibition of LPS interaction with Toll-like receptor 4. Consequently, it interferes with multiple signalling pathways, including the extracellular signal-regulated kinase, p38, and Jun N-terminal kinase mitogen-activated protein kinase pathways.54,55
Reutericin 6 with lytic activity was purified from Lactobacillus reuteri LA6b56 (reutericin 6 was the original name given to gasserinin A when it was initially reported in 1991 (ref. 57)). Reutericin 6 isolated from Lactobacillus reuteri LA 6 displayed antimicrobial activity against a range of bacteria that contain Lactobacillus acidophilus JCM 2125 and Lactobacillus delbrueckii (with many other strains).58 Lactocillin (2), a member of the thiopeptides class, has been successfully isolated and structurally characterized from Lactobacillus gasseri JV-V03, which exhibits antibiotic action towards vaginal infections within the low-to-mid nanomolar range. Nevertheless, it does not demonstrate such efficacy against vaginal commensals. It is worth noting that LFF571, another thiopeptide similar to Lactocillin, is currently undergoing phase II clinical studies.59
The investigation of the peptidome produced by Lactobacillus fermentum was undertaken by Pavlova et al. with the aim of identifying peptides that may have antibacterial effects. The Lactobacillus fermentum HF-D1, a new strain, exhibits the ability to generate antimicrobial peptides that demonstrate significant efficacy against Serratia marcescens and Pseudomonas aeruginosa. The active fraction was subjected to mass spectrometry analysis, followed by in silico prediction of antimicrobial peptides. As a result, a linear peptide sequence has been discovered as (VGAVAFGPVGAVVGGLASGFTGKQT). In addition, they discover a collection of cyclic peptides that have a high prediction score. One of them is an Epidermidin-like peptide, which is recognized for its antibacterial properties.60Lactobacillus acidophilus M46 is responsible for the production of acidocin B (3), a new bacteriocin that exhibits activities against many bacterial strains including Listeria monocytogenes, Clostridium sporogenes, Brochothrix thermosphacta, Lactobacillus fermentum, andLactobacillus delbrueckii subsp. Bulgaricus.61 The isolation and optimization of production of acidocin B were done by Brink et al., in which they find out that this peptide is heat stable and sensitive to trypsin and their in-depth antimicrobial study and mechanism reveal that it shows more inhibitory power to clostridia as compared to other lactobacilli species.62 Acidocin A, which is also a bacteriocin extracted from Lactobacillus acidophilus TK9201, display antimicrobial activity against lactic acid bacteria and also food-borne bacteria.63 Moreover, Acidocin A was administered to human erythrocytes (hRBC) obtained from two participants for testing purposes. At a dosage of 128 μM or below, it caused the destruction of only 3% of human red blood cells. Their toxic effect along with avicin A was assessed in primary human cells PBMCs and THP-1. Both acidocin A and melittin (control) but not avicin A, exhibited toxicity towards both cell lines, however PBMCs have shown more sensitivity to the peptide effects. Furthermore, the intensity of the cytotoxic impact decreased as the duration of cell incubation with resazurin extended from 2 to 24 hours. The observed harmful effects were likely attributed to a reduction in metabolic activity and cellular proliferation. It was also found out that acidocin A along with others enhanced the synthesis of inflammatory chemokines.64 The structures of peptides (1–3) are presented in Fig. 2. The identification of acidocin J1132 (alpha and beta 4) was achieved in the Lactobacillus acidophilus strain JCM1132. This demonstrates bactericidal characteristics and interferes with the membrane potential and pH gradient in vulnerable cells, thereby affecting proton motive force-dependent processes including amino acid transfer. The acidocin J1132 compound also elicits the expulsion of previously accumulated amino acids, which were first absorbed via a unidirectional transport route driven by ATP. This peptide is associated with alpha and beta components, with the beta component having an additional glycine residue and both components have inhibitory activities, with the increase in activity due to their complementary action.65,66 A bacteriocin-like peptide called rhamnosin A (5) was isolated from Lactobacillus rhamnosus 68. Rhamnosin A demonstrated inhibitory effects on Micrococcus lysodeikticus ATCC 4698; however, it did not exhibit any inhibitory effects on Lactobacillus plantarum 8014 or Lactobacillus plantarum 39![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 268. The inhibitory activity against Micrococcus lysodeikticus was observed at concentrations that demonstrated bacteriostatic effects rather than bacteriolytic or bactericidal effects. Furthermore, it maintained its biological functionality even after being subjected to heat treatment at 95 degrees Celsius for 30 minutes. However, it was susceptible to degradation by the proteolytic enzymes pepsin and trypsin.67 The investigation focused on the anticancer capabilities of recombinant bacteriocins, rhamnosin and lysostaphin (isolated from Staphylococcus simulans), which were generated in Escherichia coli, demonstrating a dose-dependent inhibition of cancer cell lines (CCA) while exhibiting lower toxicity towards normal cholangiocyte cells. Rhamnosin and lysostaphin displayed comparable or superior growth-inhibitory effects on gemcitabine-resistant cell lines compared to their parental equivalents. The simultaneous use of bacteriocins resulted in the suppression of cell growth and the promotion of cell apoptosis in both normal and gemcitabine-resistant cells, partially due to the upregulation of proapoptotic genes.68 A bactericidal and strongly hydrophobic bacteriocin amylovorin L471 also known as lactobin A (6) was isolated from Lactobacillus amylovorus DCE 471, which shared significant homologies with amino acid lactacin X, one of the two bactericidal peptides produced by Lactobacillus johnsonii VPI11088.69 The amyloyrin L471, which was later named amyloyrin L,70 exhibited no activity against the Gram-negative opportunistic pathogen Pseudomonas aeruginosa when used alone. However, it demonstrated synergistic inhibitory activity when combined with the peptide antibiotic colistin, as well as with bacteriocins pyocins S1 and S2 of Pseudomonas aeruginosa.71 The structures of peptides (4–6) are presented in Fig. 3.
268. The inhibitory activity against Micrococcus lysodeikticus was observed at concentrations that demonstrated bacteriostatic effects rather than bacteriolytic or bactericidal effects. Furthermore, it maintained its biological functionality even after being subjected to heat treatment at 95 degrees Celsius for 30 minutes. However, it was susceptible to degradation by the proteolytic enzymes pepsin and trypsin.67 The investigation focused on the anticancer capabilities of recombinant bacteriocins, rhamnosin and lysostaphin (isolated from Staphylococcus simulans), which were generated in Escherichia coli, demonstrating a dose-dependent inhibition of cancer cell lines (CCA) while exhibiting lower toxicity towards normal cholangiocyte cells. Rhamnosin and lysostaphin displayed comparable or superior growth-inhibitory effects on gemcitabine-resistant cell lines compared to their parental equivalents. The simultaneous use of bacteriocins resulted in the suppression of cell growth and the promotion of cell apoptosis in both normal and gemcitabine-resistant cells, partially due to the upregulation of proapoptotic genes.68 A bactericidal and strongly hydrophobic bacteriocin amylovorin L471 also known as lactobin A (6) was isolated from Lactobacillus amylovorus DCE 471, which shared significant homologies with amino acid lactacin X, one of the two bactericidal peptides produced by Lactobacillus johnsonii VPI11088.69 The amyloyrin L471, which was later named amyloyrin L,70 exhibited no activity against the Gram-negative opportunistic pathogen Pseudomonas aeruginosa when used alone. However, it demonstrated synergistic inhibitory activity when combined with the peptide antibiotic colistin, as well as with bacteriocins pyocins S1 and S2 of Pseudomonas aeruginosa.71 The structures of peptides (4–6) are presented in Fig. 3.
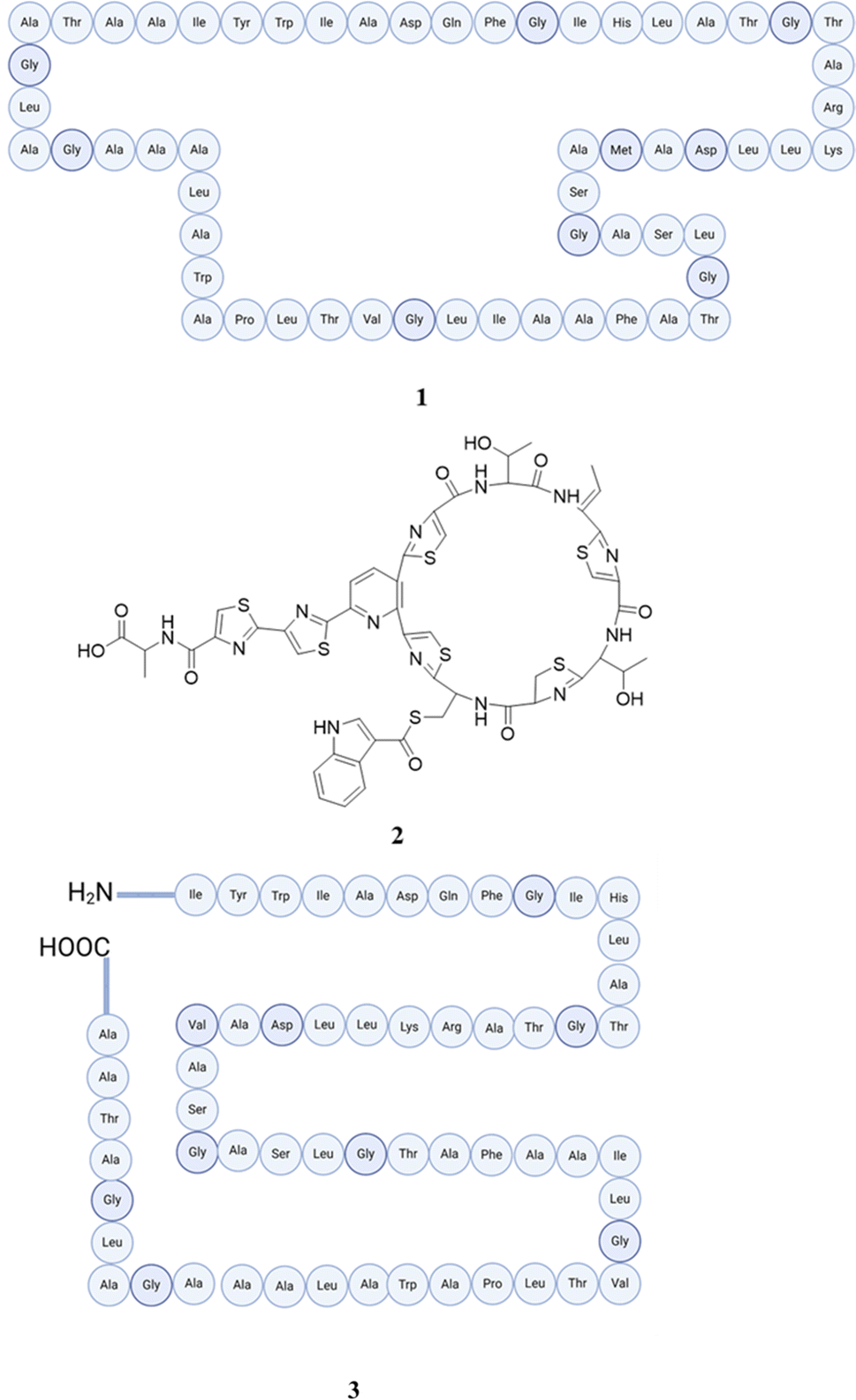 | ||
| Fig. 2 Structures of gassericin A (1), lactocillin (2), and acidocin B (3). | ||
 | ||
| Fig. 3 Structures of acidocin J1132 (alpha and beta 4), rhamnosin A (5), and lactobin A (6). | ||
A bacteriocin known as plantaricin C (7), having a 27-amino-acid sequence had one dehydroalanine, one lanthionine, and three beta-methyl-lanthionine residues. The core area of plantaricin C has a charge distribution that is suitable for an amphipathic alpha-helix. This allows the plantaricin C to be incorporated into the membrane matrix of susceptible organisms. Due to such property, it targets the cytoplasmic membrane permeability barrier, which interferes with processes that depend on the transmembrane electrochemical gradient (Δp) by disrupting it. Moreover, it causes the release of glutamate, CF, and ATP, suggesting that plantaricin C is a pore-forming bacteriocin.72,73 In order to see the mechanism, one study found out that this lantibiotic exhibits structural characteristics of both type A lantibiotic nisin and type B lantibiotic mersacidin. Lipid II was discovered to have a crucial function in the antibacterial activity of plantaricin C, specifically in pore formation. Plantaricin C was discovered to be a highly effective inhibitor of lipid II synthesis and the FemX reaction. These processes were hindered by the creation of a complex between plantaricin C and either lipid I or lipid II, leading to the inhibition of cell wall synthesis. This antimicrobial peptide is distinct because it contains lanthionine and β-methyllanthionine residues. These residues are added after the peptide is synthesised by specialised enzymes expressed by the lantibiotic operons.74 Plantaricin A (8) also enhances the antibiotic effectiveness through increasing the bacterial permeability. Another analogue, plantaricin A1, was used to improve the capacity of such peptide permeability in the membrane. OP4 (analogue) exhibited superior penetration, lower cytotoxicity, and a greater therapeutic index. Furthermore, it inhibited the development of antibiotic resistance in E. coli cells that were subjected continuously to sublethal concentrations doses of erythromycin and ciprofloxacin. OP4 demonstrated enhanced efficacy against E. coli and alleviated inflammation in in vivo experiments. The study determined that plantaricin A1 analogues, specifically OP4, decrease inherent antibiotic resistance in Gram-negative bacteria and enhance hydrophobic antibiotic susceptibility.75 The plantaricins EF (9) and JK (10) has been isolated from Lactobacillus plantarum C11, exhibiting strain-specific antagonistic activity at nanomolar concentrations when plantaricin E and plantaricin F are combined, as well as when plantaricin J and plantaricin K are combined.76 These peptides are thought to have an amphiphilic structure, which is believed to play a role in creating pores. Both plantaricin EF and plantaricin JK, which are complimentary peptides, interact with one another due to the fact that the simultaneous addition of both peptides synergistically stimulated the production of their α-helical structure in the presence of dioleoylphosphatidyl-glycerol liposomes. These peptides produce pores in the target membranes that have varying degrees of ion selectivity. The use of these bacteriocins in conjunction with one another and in a complimentary manner ensures that target cells are eliminated effectively.77,78 These bacteriocins were also recovered from a human faecal isolation of Lactobacillus plantarum LbM2a, which exhibited a broad antibacterial spectrum in addition to a high degree of stability to various conditions.79 One of the study identifies a potential membrane protein receptor for the bacteriocin plantaricin JK, by comparing Illumina sequence reads from resistant mutants to a wild-type Weissella viridescens genome, demonstrating that APC superfamily transporter is likely to serve as a target for plantaricin JK on sensitive cells.80 There have been numerous more plantaricins found to originate from this particular bacterium. In general, the primary focus of these plantaricins is on treating conditions that are caused by pathogenic bacteria, irritable bowel syndrome (IBS) and urinary tract infections (UTI).81 The structures of peptides (7–10) are presented in Fig. 4.
 | ||
| Fig. 4 Structures of plantaricin C (7), plantaricin A (8), plantaricin EF (9), and plantaricin JK (10). | ||
The cytotoxic activity of synthesised plantaricin A was investigated. The experimental results demonstrated that the application of 25 μM plantaricin A at 20 °C resulted in a significant 75% drop in cell viability. In contrast, when the treatment was conducted at 37 °C, the cell viability was reduced by 55%. Both apoptosis and necrosis were detected. Treatment with plantaricin A also led to an increase in the intracellular levels of caspase-3 in the Jurkat cell line. The study investigated the impact of the lipid composition of liposomes on the interaction between plantaricin A and liposomal membranes.82 Further, two things were demonstrated, first, that plantaricin A produces amyloid-like fibrils upon binding with negatively charged phospholipids such as phosphatidylserine; and second, that it easily leaked the fluorescent dye carboxyfluorescein from liposomes that contained negatively charged phospholipids. This suggests that the target cell membrane's negatively charged lipids may enhance plantaricin A attachment and localise it to the membrane.83 The effects of plantaricin A on anterior pituitary cells in rats were also investigated. Within 5 seconds of exposure to 10–100 μM plantaricin A, cancerous cells exhibited substantial permeabilization. The resistance dropped to a fraction of its original value as the membrane depolarized to almost 0 mV. A nonchiral mechanism was indicated by the fact that the efficacy of the peptide was similar in its D and L forms. Plantaricin A shows that it distinguishes between membrane leaflets and plasma membranes because it was insensitive to inside-out patches and main cultures of normal rat anterior pituitary cells.84 Further it was shown that how well the cationic peptide pheromone plantaricin A permeates clonal rat anterior pituitary cells (GH4 cells). While the results demonstrate that GH4 cells exhibit a high level of sensitivity to plantaricin A, this sensitivity is mitigated in solutions that partially counteract the negative surface charge of the cell. According to the research, plantaricin A must first bind to glycosylated membrane proteins electrostatically before it can bind to membrane phospholipids.85 Eukaryotic cells are protected from plasma membrane damage by the binding of positively charged bacteriocins (nisin A, plantaricin C, and pediocin PA-1/AcH) to the negatively charged glycosaminoglycan sulphate residues. Heparin, which likewise reduces the susceptibility of Lactococcus lactis to nisin A, can partially reverse this effect.86 Along with this, Lactobacillus plantarum strains are recognised for their anti-inflammatory effects, presence in the human gastrointestinal system, immunomodulatory capabilities, and potential medicinal applications. Upon isolation of peptides, the strain Lactobacillus plantarum WCFS1 has been found to produce four pyro dipeptides (pyro-phenylalanine, pyro-leucine, pyro-isoleucine, and pyro-tryptophan). The immunomodulatory characteristics of these compounds were examined by directly administering it to C57BL/6 mice in vivo, resulting in a reduction in the production of the pro-inflammatory cytokine interferon (IFN)-gamma, which is known to have a crucial function in maintaining immunological balance in the gastrointestinal tract.87
Another bacteriocin lactacin F (11) produced by Lactobacillus johnsonii also demonstrates bactericidal properties against Lactobacillus delbrueckii, Lactobacillus helveticus, and Enterococcus faecalis.65,88 Lactacin F was first derived from Lactobacillus acidophilus 11088 (NCK88). It is characterized as a heat-resistant protein that has inhibitory effects on other lactobacilli and Enterococcus faecalis.89 Lactacin F activity, which is defined by its bactericidal action on Lactobacillus delbrueckii, is dependent on the expression of two genes, namely lafA and lafX. Actually, two peptides, lactacin A (57 amino acids) and LafX (48 amino acids), make up lactacin F.90,91Lactobacillus helveticus 481 synthesized an antimicrobial compound known as helveticin J that effectively inhibited the growth of five closely related species, namely Lactobacillus helveticus 1846 and 1244, Lactobacillus bulgaricus 1373 and 1489, and Lactobacillus lactis 970. This peptide exhibited activity at a neutral pH, was susceptible to proteolytic enzymes and heat, and displayed a bactericidal mechanism of action.92 ABP-118 (12), produced by Lactobacillus salivarius subsp. salivarius UCC118, a strain isolated from the ileal-caecal area of the human gastrointestinal system, is a tiny heat-stable bacteriocin consisting of abp118alpha and abp118beta. It is noteworthy that Abp118alpha had antibacterial action, while Abp118beta exerted an improved antimicrobial effect. The gene abpIM, which provides strain UCC118 with immunity to ABP-118, has been found to be located downstream of the abp118beta gene.93 Along with genome analysis, the bacteriocin activity and the causative genes, which are homologs of salivaricin ABP-118 in Lactobacillus salivarius UCC118, have been verified.94 The structures of peptides (11–12) are presented in Fig. 5.
 | ||
| Fig. 5 Structures of lactacin F (11) and ABP-118 (12). | ||
4.1.1.2 Bioactive peptides from Clostridium and Enterococcus and their biological role. Clostridium is well acknowledged as a prominent gastrointestinal colonizer, frequently observed in individuals of all age groups, including infants and adults. Clostridium has a significant degree of genetic diversity, positioning it as the most extensive genus within the realm of anaerobic spore formation. Several significant infectious microbes including the agents causing botulism and tetanus in humans are found within this genus. In the past, Clostridium included a crucial pathogen that causes diarrhea, known as Clostridioides difficile. However, in 2016, it was reclassified into the Clostridioides genus.95–97 In the course of peptides, boticin B (13) is a bacteriocin that exhibits thermal stability and is synthesized by strain 213B of Clostridium botulinum. It demonstrates inhibitory properties against a range of strains of Clostridium botulinum and other closely related clostridia.98 The lytic activity of this bacteriocin has been reported in 1972, but the original article is in Japanese language and we are unable to review further details.99Clostridium beijerinckii ATCC 25752 produces a prepeptide comprising 72 amino acids, which undergoes processing to become a circular peptide comprised of 69 amino acid residues known as circularin A (14). Clostridium tyrobutyricum ADRIAT 932, in contrast, synthesizes a bacteriocin known as closticin 574. This bacteriocin is an 82-amino-acid peptide. In order to test this, twelve clostridial strains were analyzed to determine their ability to inhibit the growth of Clostridium tyrobutyricum B570, a widely recognized bacterium responsible for cheese spoilage, which exhibited the highest level of activity.100 Liu et al. were able to successfully carry out cloning, and heterologous expression system for clostridial circularin A and were able to produce this circular peptide in Lactococcus lactis NZ9000 successfully. Furthermore, by overlay activity experiment, the responsible strain was able to exert varied activities against various bacteria.101 The structures of peptides (13–14) are presented in Fig. 6.
 | ||
| Fig. 6 Structures of boticin B (13) and circularin A (14). | ||
Enterococci belong to the class of lactic acid bacteria and normally present in the gastrointestinal tract of humans and animals.102,103Enterococcus villorum SB2 isolated from female vagina displayed the presence of many metabolites including flavonoids, terpenoids, O-methylganoderic acid, and indole derivatives produced by tryptophan metabolism, antimicrobial compounds (fortimicin A) and fatty acids. In addition, several other metabolites were documented, including glutamate amino acid analogues (L-theanine), galactose, lactate, ketohexose deoxy sugar, acetylated glycerols, dipeptides (threoninyl-proline, valyl-glycine), and gutyrolactones; however, a peptide with several amino acids was not detached.104 A bacteriocin named bacteriocin 43 (15) was isolated from Enterococcus faecium. In such study, 636 vancomycin-resistant Enterococcus faecium isolates obtained between 1994 and 1999 from the Medical School Hospital of the University of Michigan underwent testing for bacteriocin production. Of which 44% exhibited bacteriocin production, 21 strains also showed activity against Enterococcus faecalis, Enterococcus faecium, Enterococcus hirae, Enterococcus durans, and Listeria monocytogenes.105
In a similar way, bacteriocin 32 was isolated from Enterococcus faecalis and demonstrated activity against Enterococcus faecium, Enterococcus hirae, and Enterococcus durans. However, it did not exhibit any activity against Listeria monocytogenes.106 Another bacteriocin in the same series, bacteriocin 31 (16), was found in Enterococcus faecalis, which exhibits activity against Enterococcus hirae 9790, Enterococcus faecium, and Listeria monocytogenes,107 and bacteriocin RC714 (17) was also found in Enterococcus faecalis.108 These bacteriocins typically exhibit activities against their closely related genus, which is often responsible for causing urinary tract infections, meningitis, intra-abdominal, and wound infections.109Streptococcus faecalis S-48 (Streptococcus faecalis also called Enterococcus faecalis) resulted in the production of a broad spectrum antibiotic, called AS-48 (18), which shows activities against many Gram-negative and Gram-positive bacteria.110 Gram-negative bacteria demonstrate higher resistance and exhibit a bacteriolytic action, potentially associated with initial lesions. In addition, the resistance is connected to the cell wall due to the fact that E. coli protoplasts and yeast resistance lead to an amplified sensitivity of Saccharomyces cerevisiae 3.2.111 Significant sensitivity to AS-48 was demonstrated by the bacterial strains Corynebacterium, Mycobacterium, and Nocardia, all of which contain mycolic acid in their cell walls. Nevertheless, after treatment with bacteriocin, none of them exhibited any signs of bacteriolysis. When bacteriocin was added, none of the Gram-positive bacteria, including micrococcus and Staphylococcus species, were able to be lysed. These bacteria were less susceptible than the Gram-positive bacteria. Moreover, Escherichia coli K-12 is susceptible to the bactericidal effects of this peptide; however, it does not exhibit bacteriolytic properties. As a whole, this peptide has been studied well to show both bactericidal action and bacteriolytic action.112 Cytolysin, a peptide toxin derived from Enterococcus faecalis, has attracted attention due to its ability to augment enterococcal virulence in infection models. It is secreted by both pathogenic and non-pathogenic Gram-positive bacteria. Furthermore, epidemiological investigations have linked cytolysin to patient mortality. Cytolysin is an exceptional example of a molecule that demonstrates dual activities, functioning as both an antibiotic and a catalyst for its own synthesis. The process of invasion is facilitated through the induction of macrophage and neutrophil lysis, resulting in a reduction of host immunity. The efficacy of this highly durable toxin extends to a wide range of creatures, encompassing prokaryotes, humans, and even invertebrates.113,114 Cytolysins are a type of pore-forming toxins (PFT) that are among the most numerous types of toxins. They are mostly produced as water-soluble monomers that attach to the lipid membrane of eukaryotic cells. This binding causes the passage of molecules from inside the cell to the outside, leading to the lysis of the host cell.115 The structures of peptides (15–18) are presented in Fig. 7.
 | ||
| Fig. 7 Structures of bacteriocin 43 (15), bacteriocin 31 (16), bacteriocin RC714 (17), and AS-48 (18). | ||
4.1.1.3 Bioactive peptides from Ruminococcus and Staphylococcus and their biological roles. The genus Ruminococcus comprises strictly anaerobic, Gram-positive, non-motile cocci that do not form endospores and depend on fermentable carbohydrates for their growth. Ruminococcus species are prevalent constituents of the core gut microbiome in a significant portion of the human population.116–118 In the context of peptides, Ruminococcus gnavus, an important part of the human microbiota, has been identified as the source of ruminococcin C, a sactipeptide distinguished by the presence of four Cα-thioether bridges in the L configuration. The antimicrobial activity of ruminococcin C is mainly because of the presence of thioether bridges and elimination of a leader peptide. Ruminococcin C is classified as a ribosomally synthesized and post-translationally modified peptide (RiPP) and demonstrates significant efficacy against the human pathogen Clostridium perfringens.119 The human commensal Ruminococcus gnavus E1 produced this peptide in vivo for the first time.120 A specific variant ruminococcin C1 (19) (out of C1–C5) of ruminococcin C was obtained in vivo by Chiumento et al. Ruminococcin C1 exhibits post-translational changes facilitated by a particular sactisynthase enzyme, resulting in the formation of a thioether network that gives rise to a double-hairpin folding structure. This structural characteristic is essential for its biological functionality. Ruminococcin C1 exhibits antibacterial properties against pathogenic clostridia and multidrug-resistant strains, while maintaining non-toxicity towards eukaryotic cells, which renders it a highly interesting option for therapeutic development.121 As Ruminococcus gnavus E1 produces five sactipeptides, which are designated as Ruminococcins C1 to C5, these sactipeptides are co-expressed with two radical S-adenosyl-L-methionine (SAM) maturations. With the exception of Mc2, all isoforms exhibit antibacterial activity within the same range, while C2 is solely active against eukaryotic cells. There was no observed synergistic impact between different isoforms. However, the combination of C1 with traditional antibiotics could potentially serve as a therapeutic approach against methicillin-resistant Staphylococcus aureus and vancomycin-resistant Enterococcus faecalis.122
A total of 14 bacterial strains, derived from a variety of human fecal samples, were identified as exhibiters of a trypsin-dependent antimicrobial compound that demonstrates efficacy against Clostridium perfringens, which were classified as members of species such as Ruminococcus gnavus,Clostridium nexile, and Ruminococcus hansenii, or some others. The isolation of ruminococcin A (20) has been conducted from Ruminococcus gnavus, and it has been shown that seven other strains displayed comparable properties.123 An N-acylated dipeptide aldehyde called ruminopeptin (21) has been predicated by a computational approach with in vitro biochemical characterization of biosynthetic enzymes from Ruminococcus bromii, the compound then synthesized, which inhibited Staphylococcus aureus endoproteinase GluC (SspA/V8 protease).124 The structures of peptides (19–21) are shown in Fig. 8. The genus Staphylococcus has a wide array of species that are frequently observed inhabiting Staphylococci. They are widely acknowledged as important components of the human microbiome due to their extensive application on these both niches. One of the most prominent and thoroughly researched species in this category is Staphylococcus aureus. This particular species is characterized by its coagulase-positive nature and is recognized as an opportunistic human pathogen. It can shift from a commensal state to a pathogenic state, resulting in various illnesses.125–128 Epilancin 15X (22), a lantibiotic, has been detected in Staphylococcus epidermidis from wound injury using High-Resolution Nuclear Magnetic Resonance Spectroscopy and Tandem Mass Spectrometry. Epilancin 15X exhibits three lanthionine ring structures, eleven post-translationally changed amino acids, and a hydroxy-propionyl N-terminal moiety. The structure of epilancin 15X is somewhat similar to epilancin K7 (23), having a similar mode of action.129 Epilancin K7 has been reported in Staphylococcus epidermidis K7, the structure of which was determined by NMR spectroscopy and is a lantibiotic bacteriocin.130,131 An effective self-resistance mechanism is shown by epilancin 15X, which demonstrates a powerful action against staphylococci. The structure of this lantibiotic is rather straightforward in comparison to that of other lantibiotics; nevertheless, it does contain an uncommon N-terminal D-lactate group. This group has the potential to be engineered into novel lantibiotics that exhibit additional antibacterial activities.132 It eradicates Staphylococcus carnosus and Bacillus subtilis by causing disruption to their membranes. The conservation of residues and dehydroamino acids in epilancin 15X was shown to be essential for its bioactivity, whereas the N-terminal lactyl group exhibits tolerance towards modifications. It has a detrimental impact on the process of fatty acid synthesis, RNA translation, and DNA replication, while not influencing the formation of the cell wall. Therefore, it was determined that this compound has the ability to elicit leakage from model membrane vesicles that contain negatively charged lipids in a manner regardless of lipid II.133 In the most recent study, the mechanism of action of epilancin 15X was examined by dissipating the membrane potential of intact Staphylococcus simulans cells. Antibiotic treatment decreased epilancin 15X membrane depolarization effects, and disrupting the lipid II cycle in intact bacteria led to a decrease in its activity.134Staphylococcus epidermidis Tu 3298 produces an important peptide antibiotic epidermin (24) that is synthesized in the ribosome through a precursor protein.135 Gallidermin (25), derived from Staphylococcus gallinarum, is classified as a lantibiotic and exhibits structural similarities to epidermin. However, it possesses unique sources and displays particular antibacterial activities. They displayed antimicrobial against many Gram-positive pathogenic bacteria. The mode of action of both lantibiotics is similar to that of nisin. They interact with I, II, III (undecaprenol-pyrophosphate-N-acetyl-glucosamine), and IV (undecaprenol-pyrophosphate-N-acetyl-glucosamine-N-acetyl-mannosamine), thereby inhibiting not only murein but also wall teichoic acid manufacturing. Type A lantibiotics primarily kill bacteria by permeabilizing the membrane. However, only nisin and epidermin can form a complex with [14C]-lipid II, which is incorporated into carboxyfluorescein-loaded liposomes made of phosphatidylcholine and cholesterol.136–138 The structures of peptides (22–25) are shown in Fig. 9.
 | ||
| Fig. 8 Structures of ruminococcin C1 (19), ruminococcin A (20), and ruminopeptin (21). | ||
 | ||
| Fig. 9 Structures of epilancin 15X (22), epilancin K7 (23), epidermin (24), and gallidermin (25). | ||
Epidermin along with pep5 which is also found in Staphylococcus epidermidis exhibited activity against clinical strains of Staphylococcus epidermidis and Staphylococcus aureus that were isolated from catheter-related infections. This activity was observed at a concentration of 640 activity units per ml.139 The most recent study demonstrates that the pep5-producing strain exhibits efficacy against Staphylococcus aureus, and its entire sequence was identified for the first time. The study also discovered that the antibacterial action of nisin, nukacin, and pep5 (26) (Fig. 10) is dependent on respiratory constancy, which is controlled by two-component regulatory mechanisms.140
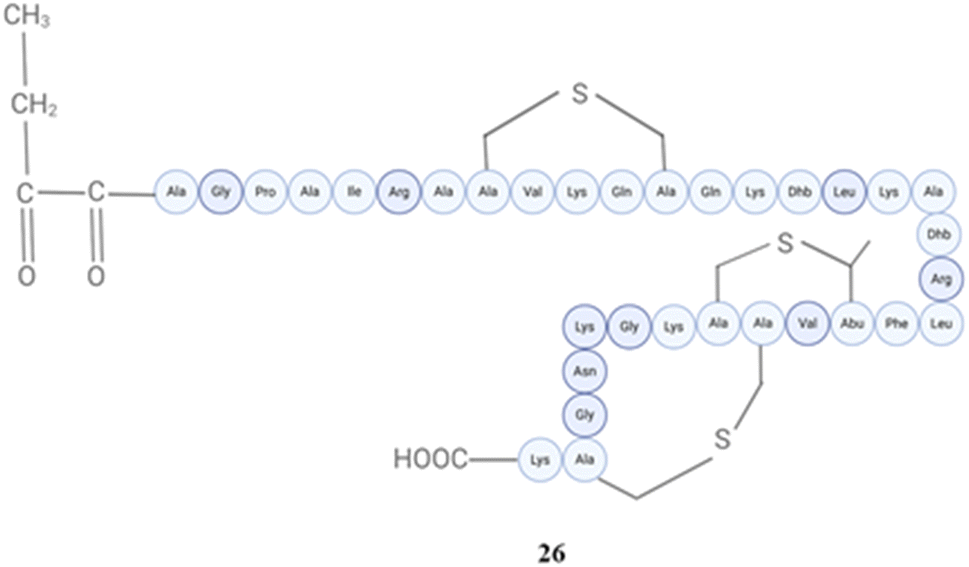 | ||
| Fig. 10 Structure of pep5 (26). | ||
Epidermin also show synergistic effects with staphylolysin LasA A against Staphylococcus aureus, Escherichia coli and Pseudomonas aeruginosa.141Staphylococcus epidermidis BN 280 leads to the extraction of a comparable peptide known as epicidin 280 (27). Epicidin 280 was shown to have a 75% similarity to pep5 based on amino acid sequence analysis.142 Epidermicin NI01 (28), an antimicrobial peptide is also produced by Staphylococcus epidermidis 224, is highly cationic, hydrophobic, and plasmid encoded. It exhibits potent antimicrobial activity against various Gram-positive bacteria, including methicillin-resistant Staphylococcus aureus (MRSA), enterococci, and biofilm-forming Staphylococcus epidermidis strains.143 One study found that single application of topical epidermicin NI01 was equally effective as twice daily administration of mupirocin for 3 days in eliminating MRSA from the nasal passages of cotton rats.144 The structures of peptides (27–28) are displayed in Fig. 11.
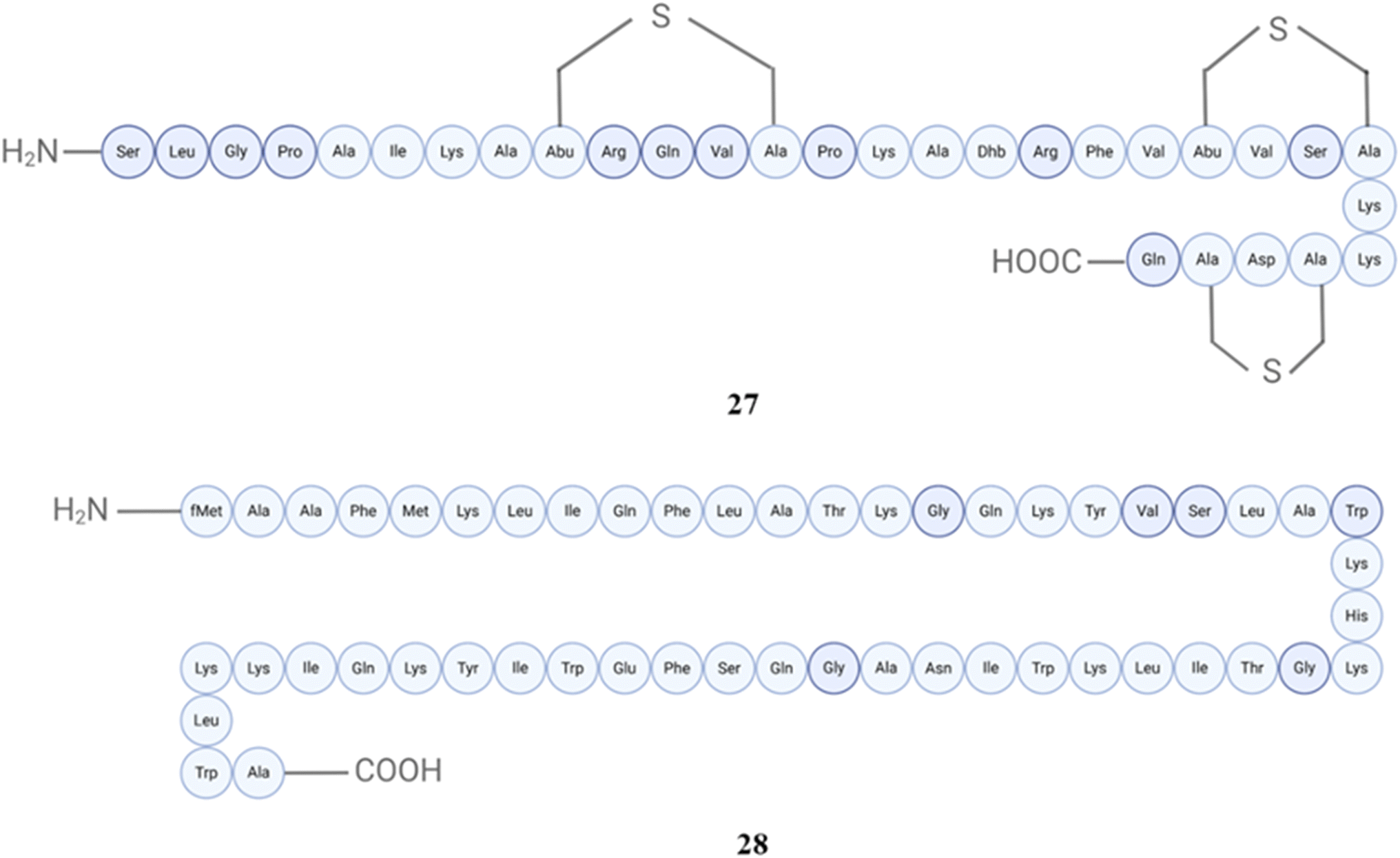 | ||
| Fig. 11 Structures of epicidin 280 (27) and epidermicin NI01 (28). | ||
Staphylococcal genomes often encode epilancin A37 (29), an antibiotic that enters the cytoplasm of Corynebacteria via a largely transmembrane-potential-driven uptake that does not disrupt cell membrane function. The antibacterial activity of epilancin is dependent on the production of intracellular membrane vesicles, which this peptide promotes upon intracellular aggregation.145 Two bacteriocin-producing genotypes of Staphylococcus epidermidis, KSE56 and KSE650, were identified in 150 isolates from the oral cavities of 287 volunteers. The epidermin-harboring plasmid pEpi56 and the nukacin IVK45-like-harboring plasmid pNuk650 were confirmed by the complete genome sequences. The amino acid sequence of epidermin from KSE56 was identical to that of previous reports; however, the genes related to epidermin synthesis were partially different. The prepeptide amino acid sequences of nukacin KSE650 (30) and IVK45 exhibited a single mismatch; however, the mature peptides were identical. In comparison to pIVK45, pNuk650 was larger and contained seven ORFs. The study observed distinct antibacterial activity profiles against different cutaneous and oral microorganisms.146 Most specifically, it exerts antibacterial activity against nasal members of the Actinobacteria, Proteobacteria, and Firmicutes.147
Staphylococcus lugdunensis strains have been recognized as synthesizers of lugdunin (31) (first reported nonribosomally synthesized antibiotic from human microbiomes), an exclusive cyclic peptide antibiotic that encompasses thiazolidine. It demonstrates bactericidal activity against important infections, demonstrates effectiveness in animal models, and displays a decreased propensity to create resistance in Staphylococcus aureus.148 Subsequently, the same group conducted more research on this peptide, discovering that prior administration of lugdunin in conjunction with components generated from the microbiota resulted in a substantial decrease in Staphylococcus aureus colonisation in both primary human keratinocytes and mouse skin. Furthermore, it enhanced the production and secretion of LL-37 and CXCL8/MIP-2 in these cells, thereby attracting monocytes and neutrophils in living organisms. In addition, it successfully eliminated Staphylococcus aureus by combining the antibacterial effects of LL-37 with dermcidin-derived peptides in a synergistic manner.149 The structures of peptides (29–31) are shown in Fig. 12.
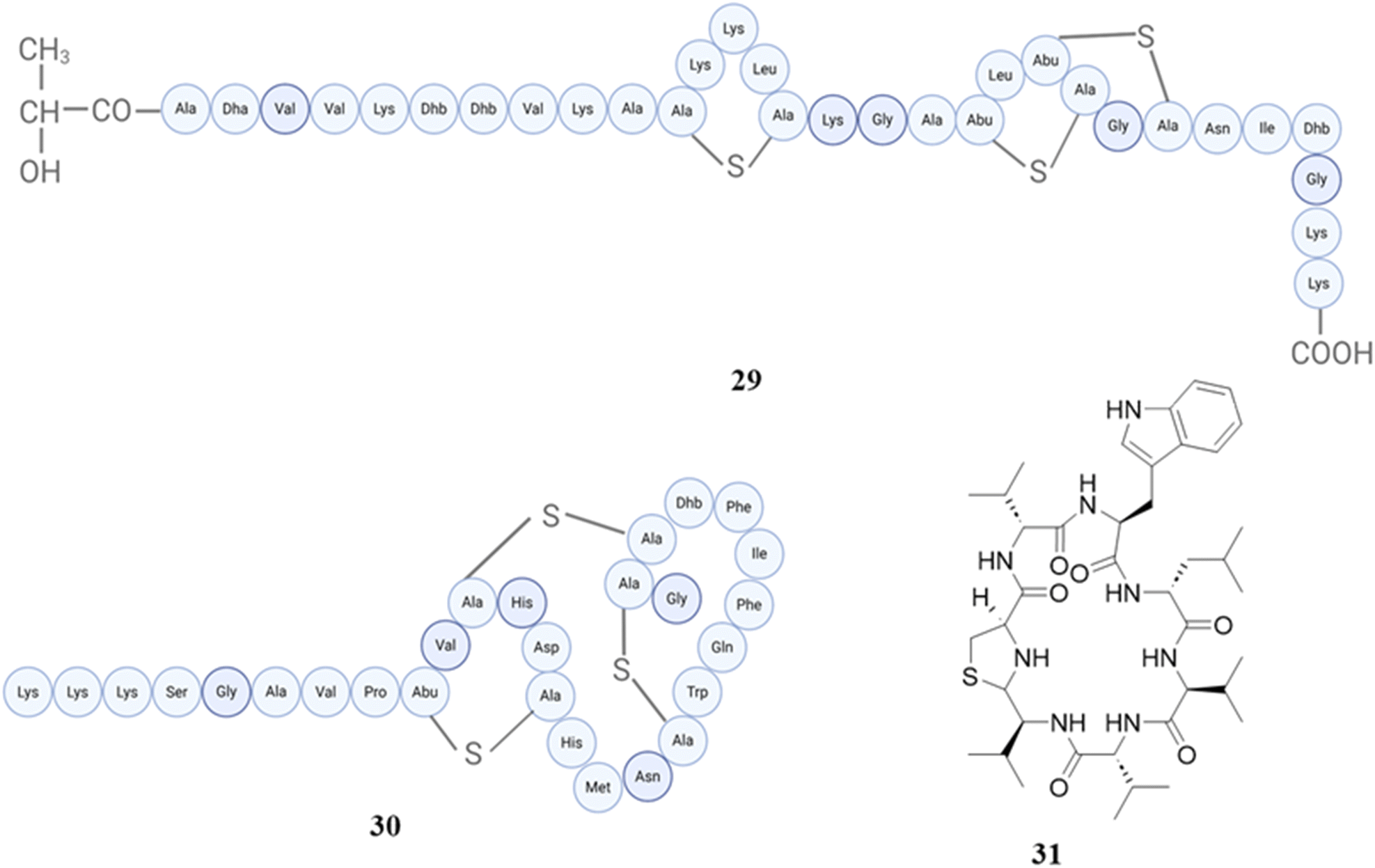 | ||
| Fig. 12 Structures of epilancin A37 (29), nukacin KSE650 (30), and lugdunin (31). | ||
The synthesised analogue and modified thiazolidine ring were used to study the antibacterial efficacy and mode of action of lugdunin. The thiazolidine ring and modifying the D- and L-amino acid backbone were the most important factors. This process involves the dissipation of membrane potentials in specific bacterial cells. The mechanism of action revealed that this peptide's antibacterial activity maintains the integrity of artificial membrane vesicles by equalising pH gradients, hence proving proton transfer. The variety of these thiazolidine cyclopeptides is further increased by adding additional tryptophan or propargyl groups.150 Its genotypic study among different strains shows that most lugdunin producing oxacillin-resistant Staphylococcus lugdunensis strains were of ST3-SCCmec V-agr II genotypes, while most lugdunin producing OSSL strains were of ST3-agr II, followed by ST1-agr I. Lugdunin exhibited weak inhibitory activity against the VISA ST239 isolate, and ST239 VSSA was more resistant to lugdunin than ST5, ST59, and ST45 VSSA.151 The same group conducted further research recently on the concept of lugdunin synthesis and determined that it is associated with molecular types, further strengthening their hypothesis.152 Most recently, it has been shown that lugdunin quickly destabilises the potential of bacterial membranes in vitro. The membranes of Gram-positive bacteria have lipid compositions that the peptide partitions into, however those containing cholesterol have a lower partitioning efficiency. The translocation of protons and monovalent cations is facilitated by the formation of hydrogen-bonded antiparallel β-sheets by lugdunin via peptide nanotubes upon insertion. According to the findings, lugdunin causes the bacterial cell's membrane potential to dissipate by functioning as a peptidic channel that forms spontaneously via a stacking mechanism.153
In the case of nisins, there are many different nisin variants isolated starting back from 1928, the first one is nisin A (32), and then nisin F (33) (Fig. 13), Q (34), Z (35) (Fig. 14), from Lactococcus lactis, nisin O1 to O4 from Blautia obeum, nisin U (36) and U2 (37) (Fig. 15) from Streptococcus uberis, nisin P (38) from Streptococcus gallolyticus subsp. pasteurianus, nisin J (39) from Staphylococcus capitis, and nisin H (40) (Fig. 16) from Streptococcus hyointestinalis.154 Nisin J is produced by Staphylococcus capitis APC 2923, a strain obtained from the microbiota of the human skin. The verification of nisin J production by Staphylococcus capitis APC 2923 has been achieved through the utilization of sophisticated analytical techniques including whole-genome sequencing and mass spectrometry. In the term of biological activity, it displayed notable antimicrobial effectiveness against a range of Gram-positive bacteria, such as methicillin-resistant Staphylococcus aureus and Cutibacterium acnes.155 Nisin H was not isolated from human microbiota (isolated from pig);156 however, nisin P was isolated from human fecal isolate, Streptococcus agalactiae DPC7040, which exhibits antimicrobial activities against many gut and food isolates because of such peptides. The antimicrobial activity of nisin P, nisin A and nisin H was studied, and it was found that nisin P was less active than the other two against Lactobacillus delbrueckii subsp. bulgaricus LMG 6901 at different time points and pH values; overall, nisin A displayed higher activity than H, and then P.157 Additionally, this peptide exhibited antibacterial activity against various clinical drug-resistant bacteria such as methicillin-resistant Staphylococcus aureus, vancomycin-resistant Enterococcus, and penicillin-resistant Streptococcus pneumoniae.158Blautia obeum A2-162, which was obtained from the human gastrointestinal tract, exhibited antibacterial properties against Clostridium perfringens, Clostridium difficile and Lactococcus lactis. This antimicrobial activity was attributed to the presence of nisin O.159 Nisin and ranalexin were tested against 40 nosocomial MRSA isolates in vitro, both alone and in combination with amoxycillin, amoxycillin-clavulanate, imipenem, clarithromycin, ciprofloxacin, rifampin, and vancomycin. At doses ranging from 1 to 32 μg ml−1, both peptides effectively blocked all isolates. Except for β-lactams, other agents showed synergy when combined.160
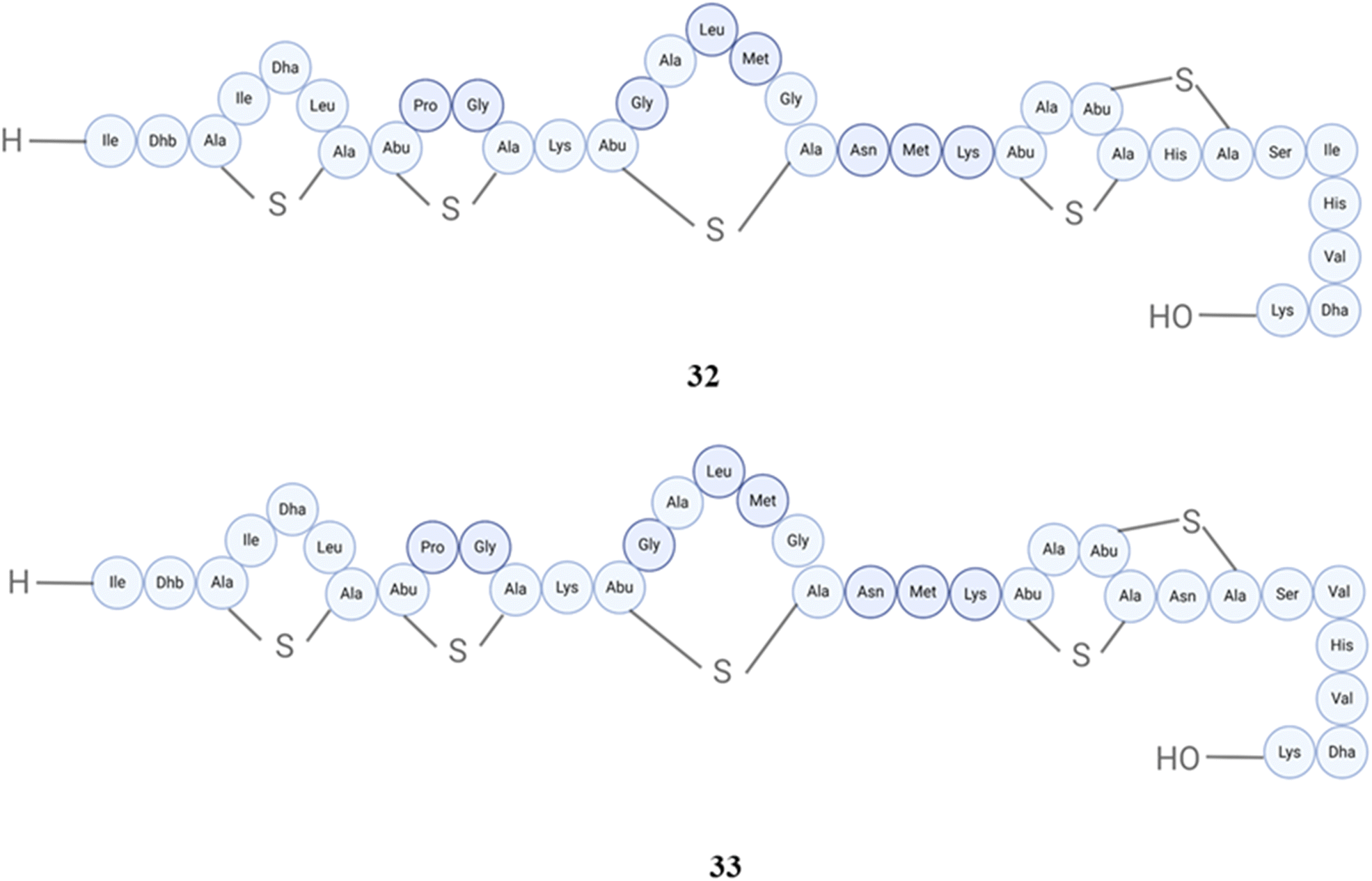 | ||
| Fig. 13 Structures of nisin A (32) and nisin F (33). | ||
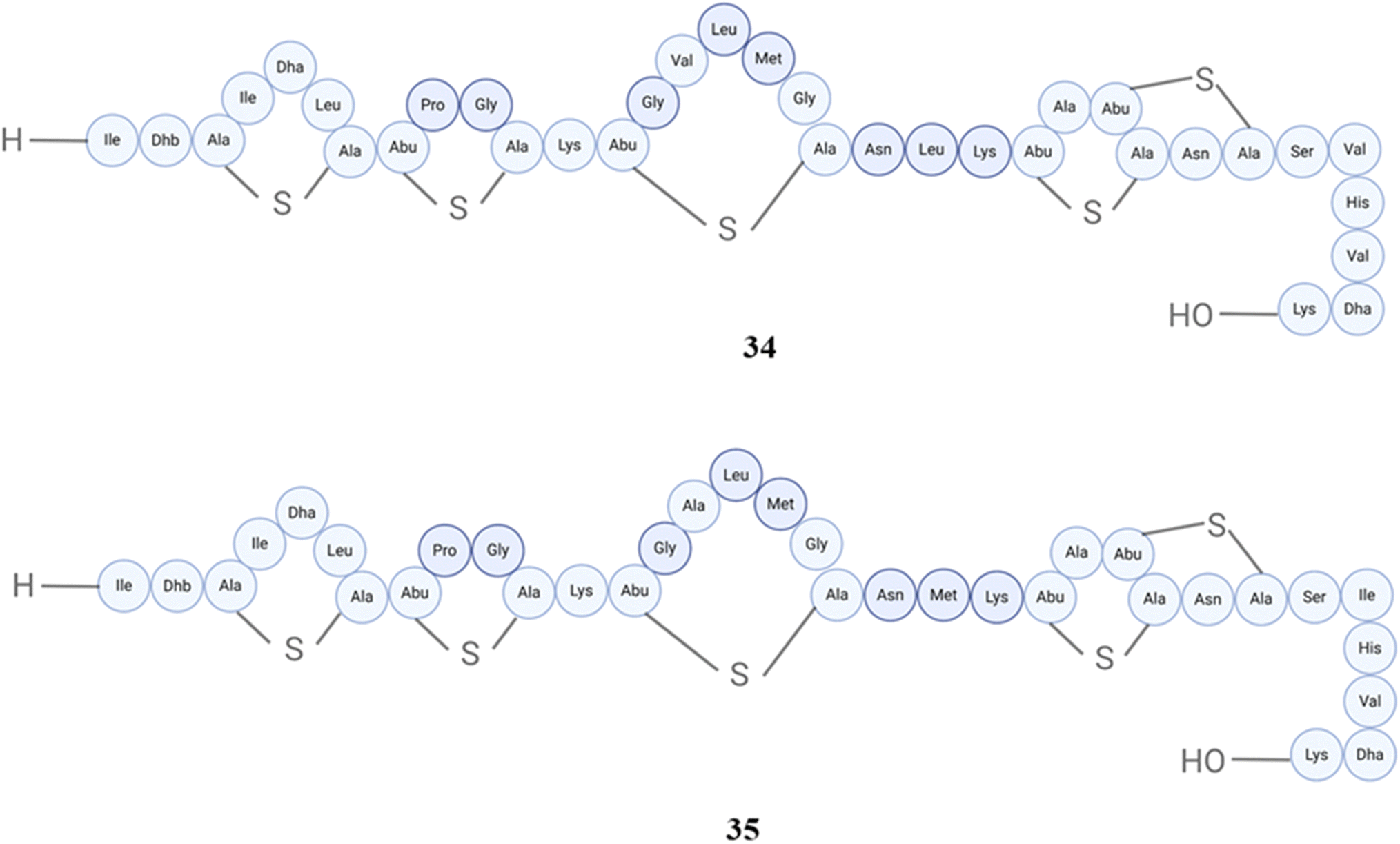 | ||
| Fig. 14 Structures of nisin Q (34) and nisin Z (35). | ||
 | ||
| Fig. 15 Structures of nisin U (36) and nisin U2 (37). | ||
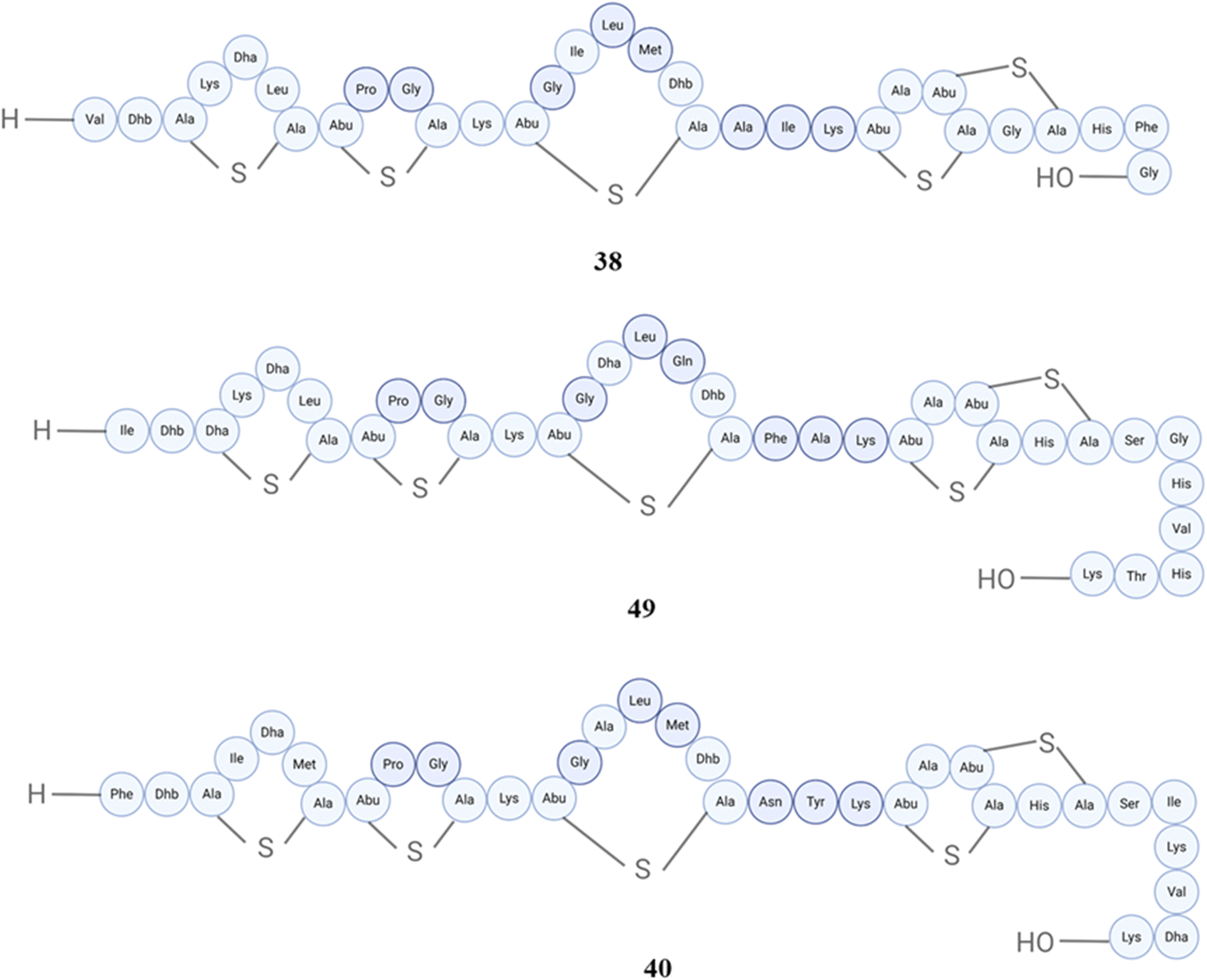 | ||
| Fig. 16 Structures of nisin P (38), nisin J (39), and nisin H (40). | ||
Streptococcus gallolyticus subsp. is the primary pathogen responsible for septicemia and infective endocarditis (IE) in older individuals and those with weakened immune systems. It is one among the few bacteria that are opportunistic and have a substantial association with colorectal cancer (CRC). One study examined the effects of cholesterol on bacteriocin activity from this species on model membranes, as well as the cytotoxic and hemolytic capabilities of bovicin HC5 (found in this species) in vitro. The reference peptide utilised was nisin. Light microscopy and the MTT assay were used to examine the vitality of three different eukaryotic cell lines that had been treated with either nisin or bovicin HC5. The haemoglobin liberation assay was used to assess the hemolytic potential. Nisin had an IC50 value of 13.48 μM and bovicin HC5 had an IC50 value of 65.42 μM against Vero cells. Neither bacteriocin was inhibited by cholesterol in the DOPC model. Cholesterol had no influence on the interaction of bovicin HC5 with model membranes, and the compound only exhibited cytotoxic effects at concentrations higher than the biologically active threshold. Previously, the same researchers showed that whereas bovicin HC5 and nisin have the same target on the membrane, their capacity to create pores in model membranes is significantly different.161,162
It has been found that loading nisin Z onto cross-linked cyclodextrin nanosponges (CDNS) successfully treated melanoma cancer both in the in vitro and in vivo studies. Nisin encased in CDNSs enhanced melanoma cancer cell line cytotoxicity and apoptosis, according to the data. Nisin placed on PMDA-NSs reduced tumour volume and weight in a mouse model of melanoma cancer, inhibiting tumour growth. Complexation with PMDA-NSs amplified nisin anti-cancer properties, which include apoptosis and antioxidant activity. Furthermore, CD31 expression in tumour tissues was reduced by nano-formulated nisin. The results indicate that the anticancer impact of nisin in melanoma cancer animal models may be enhanced by integrating it into nanosponges as a safe carrier.163 Melanoma is the most lethal form of skin cancer, hence reprogramming cellular metabolism is crucial in cancer treatment. This peptide has the ability to fight melanoma in vitro. It causes apoptosis, increases reactive oxygen species production, and negatively affects energy metabolism in melanoma cells through selective toxicity. In addition to its possible utility against melanoma-related metastasis, it reduces melanoma cell invasion and proliferation. It is possible to treat melanoma using combination therapy using recognised anti-melanoma drugs, since nisin Z puts a heavy strain on the cell energy metabolism.164 Head and neck squamous cell cancer (HNSCC) may be amenable to nisin, a bacteriocin and food preservative. When compared to primary keratinocytes, it causes HNSCC cells to undergo apoptosis, cell cycle arrest, and lessen cell growth. Moreover, in vivo, nisin decreases HNSCC carcinogenesis. A proapoptotic cation transport regulator known as CHAC1 and a calcium influx mediates this action. The work details the novel function of CHAC1 in nisin-induced cancer cell apoptosis. The results lend credence to nisin potential as a new treatment for HNSCC, and the fact that it is both safe for humans to eat and used in food preservation could speed up its introduction to clinical trials.165 Another study reported that nisin exerts potent cytotoxic effects on the breast cancer cell line MCF-7, with a higher selectivity for malignant cells relative to normal cells. Nisin and doxorubicin have synergistic antitumor effects when used together. It is possible to increase the therapeutic index and decrease adverse effects in breast cancer patients by combining doxorubicin with nisin at sub-inhibitory doses.166 The anticancer potential and mechanisms of nisin ZP in non-small cell lung cancer cells (NSCLC) were examined. Apoptosis, cell cycle arrest, and inhibition of cancer cell proliferation via non-membranolytic pathways were observed as effects of nisin ZP selective toxicity on cancer cells. Additionally, it showed promise in the fight against extremely metastatic NSCLC by reducing clonal proliferation and migration of cancer cells. Both the 3D spheroid development and the cell survival of A549 cells were significantly inhibited in the study. In sum, the data point to promising anticancer activity in vitro and call for its continued exploration as a potential new treatment for non-small cell lung cancer.167 The effects of nisin on HuH-7 and SNU182 cell lines, which are used to study liver cancer, have been found to be substantial growth inhibition and apoptosis. The epithelial-to-mesenchymal transition contributes to the development of drug resistance, a significant problem in liver cancer. Nisin treatment resulted in a down-regulation of TWIST1 expression relative to untreated cell lines, according to an analysis of the EMT transcription factors ZEB1, SNAI1, and TWIST1. Furthermore, nisin A molecular docking assessment in the Frizzled (FZD) protein binding region confirmed that nisin A could establish hydrogen bonds with critical residues. In addition to establishing nisin function in many cell models of liver cancer, this work also generated the first identification of a connection between FZD7 and nisin.168 Synergistic single platforms against many types of cancer may be possible. For such case, researchers have created a novel nanoconstruct that target DMBA/TPA-induced skin cancer in mice. This nanoconstruct is composed of oligomeric chitosan coated silver nanoparticles that are co-loaded with nisin and 5-fluorouracil. The nanostructure was created by wet reduction method, then covering it with chitosan, conjugating it with nisin, then physically loading 5-FU onto it. According to biophysical analysis, the nanostructure exhibited zeta potential, UV-visible absorption maxima, and a particle size of 72.39 nm. Restored skin histoarchitecture, improved oxidant/antioxidant balance, and a marked decrease in tumour volume and load were all observed in in vivo experiments.169 One potential substitute is the use of antimicrobial peptides in conjunction with chemotherapy medications. When tested on mice with skin cancer, 5-FU and nisin showed strong anti-cancer activity. Groups treated with nisin with 5-FU together demonstrated a reduction in tumour volume and burden in in vitro investigations. The combined group had greater apoptosis rates and histoarchitecture revealed a return to normal skin tissue. Both in vivo and in vitro experiments revealed that the combination of nisin and 5-FU was synergistic.170 Using SW480, HCT116, and PBMC from colorectal cancer patients, the researchers looked at how nisin affected the expression of HERV-K env, Syncytin-1, microRNA-9-5p, 192, and 205. A notable rise in microRNA-9-5p was observed in HCT116, SW480, and PBMC, while microRNA-192-5p was shown to be upregulated in SW480 and HCT116. Nevertheless, PBMC did not show a statistically significant upregulation of microRNA-205-5p. Consequently, it was determined that nisin has the ability to alter gene expressions and impede the advancement of colorectal cancer. The progression of colorectal cancer can be slowed by including nisin in the diet.171 Human umbilical vein endothelial cells and human keratinocytes were used to show migration and proliferation effects in vitro as part of the investigation into nisin A wound healing capabilities. The study also looked into its re-epithelization capability using an ex vivo wound healing model for porcine. Nisin A reduced proinflammatory cytokines, which may explain why it impacted cell migration but not proliferation, according to the results. The rate of re-epithelialization in porcine skin was also enhanced. Nisin A did not aid the larvae in any manner other than the direct antibacterial strategy, and it increased Galleria mellonella survival from Staphylococcus epidermidis but had no effect on E. coli. With its ability to enhance skin cell mobility, reduce bacterial growth, and mitigate the effects of lipopolysaccharide and proinflammatory cytokines, nisin A could be a promising therapeutic for wound healing.172 Using several approaches, the effects of nisin on the viability of colon cancer cells and gene expression were assessed. The results demonstrated that the apoptotic index (measured in both mRNA and protein levels) was elevated, and cell viability was markedly decreased at different nisin concentrations. The dose-dependent apoptotic effects imply that nisin has the potential to trigger apoptosis through intrinsic mechanisms, resulting in the demise of malignant cells.173 Based on these evidences, nisin can be proved as a potential anticancer agent.
In order to provide insights for the advancement of anti-infective therapies, Liu et al. undertook a comprehensive investigation involving 3000 human skin isolates, with the objective of evaluating bacterial interactions within the human skin microbiota. The findings of their study demonstrated that the Staphylococcus hominis strain showed strong and diverse efficacy against Gram-positive infections, predominantly through the action of the bacteriocin micrococcin P1 (41).174 The total synthetic form of micrococcin P1 exhibited antibacterial activity against Staphylococcus aureus, Enterococcus faecalis, Bacillus subtilis, and Streptococcus pyogenes.175 Most of the applications of this peptide have been highlighted in a previous review in more detail.176,177
There are some other genera in human microbiota that also contribute to the profile of metabolites. For instance, Streptococcus mutans UA159, an oral pathogen, has been seen to generate mutanobactins and mutanamide (42), which demonstrate inhibitory properties against the formation of hyphae in Candida albicans. This particular interaction can be considered a subsequent consequence of the mutanobactin manufacturing process. It is worth mentioning that mutanamide, which is a derivative of mutanobactins, plays a significant role in observed inhibition. Furthermore, the fully evolved mutanobactins exhibit additional immunomodulatory characteristics.178,179 Mutanobactin is also capable of inhibiting the growth of Gram-positive bacteria, including Enterococcus faecalis. The cell membrane of Enterococcus faecalis is disrupted by this antibiotic medication, which is also effective against other species of Enterococcus, including Staphylococcus aureus. The enzyme gelatinase, which is a metalloprotease that is secreted, is responsible for facilitating the resistance of Enterococcus faecalis organisms.180 The different mutanobactins include mutanobactins A, B, C and D, which are derived from Streptococcus mutans.179,181 Moreover, mutanobactins possess various other functions, including resistance to oxidative damage. It has been discovered that both wild-type and deletion mutants exhibit a significant reduction in growth rate when exposed to H2O2. Furthermore, they affirm a correlation between mutanobactin-mediated ROS resistance and biofilm development. Nevertheless, the biological significance of mutanobactin-mediated ROS tolerance in situ warrants additional investigation.182 Mutanobactins also have immunomodulatory activities, and in RAW264.7 macrophages activated with LPS, it upregulates the pro-inflammatory cytokines IL-6 and IL-12 and a downregulation of MCP-1, G-CSF, and TNF-α.183
4.1.1.4 Bioactive peptides from Streptococcus and Fusobacterium and their biological roles. Streptococcus mutans plays a crucial role in the formation of tooth decay due to its ability to form biofilms and thrive under acidic conditions. It has the ability to produce tetramic acids, mutanocyclin and reutericyclins A, B, and C through synthesis. Reutericylin A was discovered to have antibacterial properties in these molecules, while mutanocyclin exhibits anti-inflammatory benefits. Additionally, the study found that reutericyclin A effectively inhibited the development of biofilms and the production of acid even at very low concentrations. Similarly, mutanocyclin reduced the occurrence of cariogenic Limosilactobacillus fermentum.184 However, here it is to be noted that these are not actual peptides, which have smaller structures. The structures of peptides (41–42) are displayed in Fig. 17.
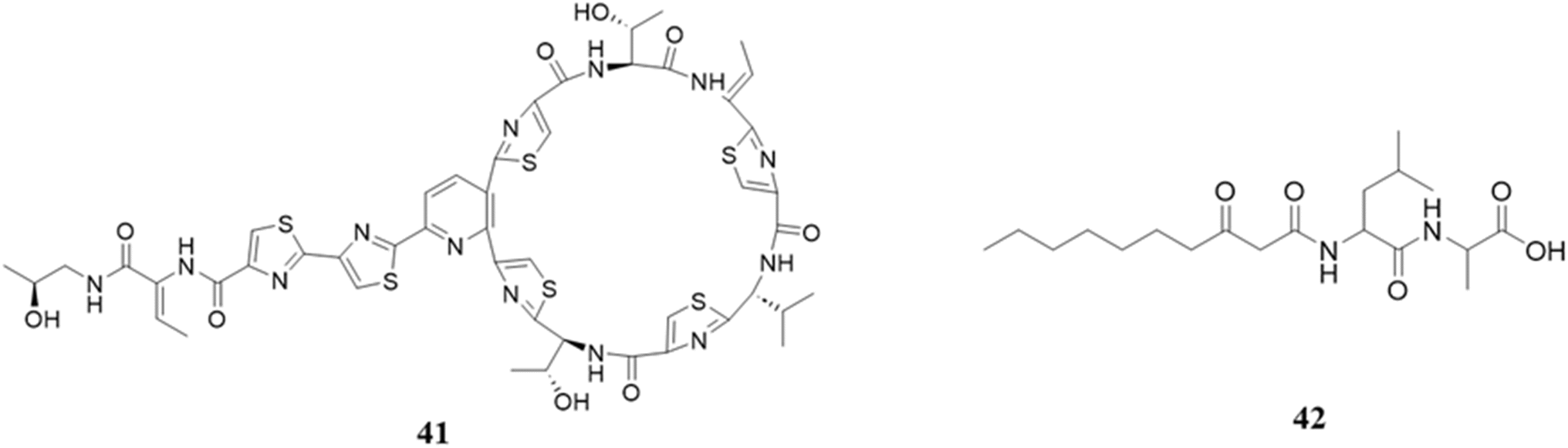 | ||
| Fig. 17 Structures of micrococcin P1 (41) and mutanamide (42). | ||
Mutacin 1140 (43) and mutacin-BNY266 (44) (Fig. 18), lantibiotics produced from Streptococcus mutans, displayed a broad spectrum of antibacterial activities.185,186 Mutacin 1140 has a thioether cage structure. Their mode of action is somewhat related to nisin. It is a potent antibacterial peptide, which specifically targets lipid II, hence impeding the formation of bacterial cell walls and ultimately resulting in cell lysis. The development of mutacin 1140 lipid II complexes indicates that these complexes can create membrane pores that allow water to pass through. A single chain of mutacin 1140 is bound to lipid II and facilitates the movement of water molecules across the membrane using a single-file water transport mechanism, thus it represents a unique method of action that enhances its antibacterial capabilities.187 A saturation mutagenesis library of 418 single-amino-acid variants of mutacin 1140, produced by Streptococcus mutans, was used to select compounds for the treatment of CDAD. OG253 (mutacin 1140, Phe1Ile) was identified as the lead compound in the safety screen 44TM in vitro pharmacological profiling assay, which indicated that these compounds had relatively low overall toxicity. In a hamster infection model, OG253 was discovered to possess superior in vivo efficacy and an evident absence of relapse.188 The extract of Streptococcus mutans JH1140 was evaluated against six bacterial species. The results indicated that the mutacin 1140 extract exhibited significantly greater inhibitory and bactericidal activities against non-mutans streptococci than mutans streptococci. Streptococcus sobrinus was discovered to be more susceptible to the extract antimicrobial action. The eradication time for microbes that were more sensitive and less sensitive was 30 minutes and 60 minutes, respectively.189 For mutacin 1140 belonging to the epidermin family, it was studied that the pharmacokinetics of mutacin 1140 was affected by charged and dehydrated residues. Dehydrated residues enhance stability, but alanine substitutions enhance the characteristics. Analogues K2A and R13A have reduced clearances and enhanced bioactivities against pathogenic microorganisms. In a model of MRSA infection, these compounds provide 100% protection at 10 mg kg−1 to all infected mice, resulting in a reduction of bacterial load.190 In the further study, a combination of Mu1140 K2A and R13A analogues is more efficient in treating MRSA bacteria compared to native Mu1140 or vancomycin. Additionally, these analogues demonstrated significant efficacy in the treatment of MRSA skin infections. The study indicates that these analogues have the potential to serve as future alternatives for the treatment of severe Gram-positive bacterial infections. The analogues exhibit prolonged drug elimination rates, resulting in elevated plasma concentrations over a period of time. The results endorse the ongoing development of therapeutic products utilising these analogues, indicating that they have the potential to enhance the outcome of severe bacterial infections.191 A study utilising the suicide vector pVA891 sought to alter the core peptide of mutacin 1140. The results revealed that out of the 14 mutants, five exhibited enhanced antibacterial activity against Micrococcus luteus ATCC 10240. The study additionally discovered that three Mutacin 1140 variants exhibited the most substantial enhancements in bioactivity against Streptococcus mutans, Streptococcus pneumoniae, Staphylococcus aureus, Clostridium difficile UK1, and Micrococcus luteus ATCC 10240, and this provides evidence that the antibacterial action might be greatly enhanced.192 Furthermore, an expression library of 418 variations was developed by conducting a study that utilised saturation mutagenesis to replace each amino acid of a lantibiotic mutacin 1140. The results indicated that the optimal activity was not dependent on all residues participating in the production of lanthionine bridges. Abu8 and Ala11 exhibited permissive substitutions, but the unsaturated bond originating from Dha5 was determined to be crucial for achieving optimal activity.193 Mutacin B-NY266 is also a highly efficient lantibiotic that has the ability to destroy a diverse range of oral streptococci.194
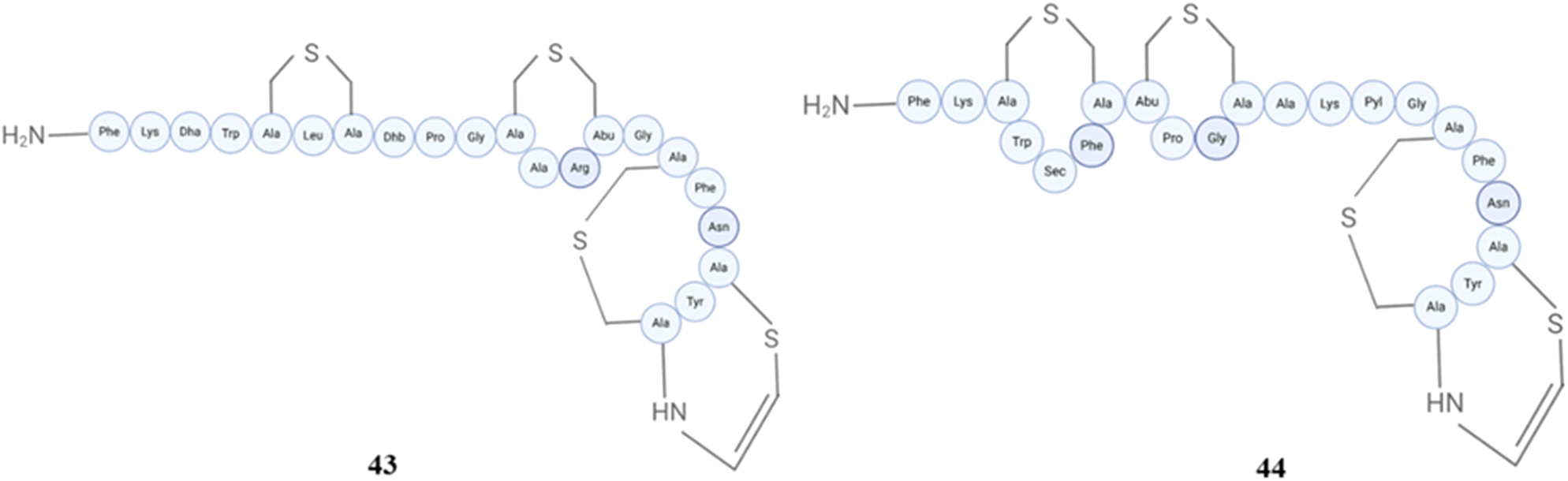 | ||
| Fig. 18 Structures of mutacin 1140 (43) and mutacin-BNY266 (44). | ||
Streptococcus salivarius is primarily located in the oral cavity, although it has also been discovered in the gastrointestinal tract. Furthermore, a particular strain of Streptococcus salivarius obtained from the faeces of a healthy child generates salivaricin D (45), indicating its presence in the gut microbiota. The producer strain also has a significant presence in the faecal microbiota, implying its potential for competitiveness and dominance in the gut flora. Salivaricin D is a lantibiotic that is naturally resistant to trypsin and shares similarities with nisin-like lantibiotics. This bacteriocin has a wide range of activities at nanomolar levels against several types of Gram-positive bacteria including Bacillus subtilis, Clostridium bifermentans, Clostridium butyricum, Streptococcus pneumoniae, Streptococcus suis, and many others.195 The structures of peptides (45, 46 and 48) are shown in Fig. 19.
 | ||
| Fig. 19 Structures of salivaricin D (45), salivaricin A (46), and salivaricin 9 (48). | ||
Another lantibiotic was isolated from Streptococcus salivarius 20P3 known as salivaricin A (46), and strains Streptococcus salivarius K12 and Streptococcus salivarius 5M6c also result in salivaricin B (47) (Fig. 20) and salivaricin D respectively. Streptococcus salivarius NU10 produced salivaricin 9 (48) which exhibited both bactericidal and bacteriolytic effects on sensitive bacterial cells while causing an increase in cytoplasmic membrane permeability. The sensitive strains exhibited pore forming activity when treated with salivaricin 9.196–198 Barbour et al. conducted a study on the bactericidal mode of action of salivaricin B against sensitive Gram-positive bacteria. Salivaricin B was discovered to require micro-molar quantities of lantibiotics, in contrast to the very powerful nano-molar lantibiotic nisin A. It disrupted the cell wall synthesis process, leading to the buildup of the ultimate soluble cell wall precursor known as UDP-MurNAc-pentapeptide. Transmission electron microscopy revealed a decrease in the thickness of the cell wall and indications of abnormal septum formation in cells exposed to salivaricin B, without any observable alterations to the integrity of the cytoplasmic membrane.199 The Streptococcus strain FF22 also produced nisin-like antibiotic streptococcin A-FF22 (49)200 (Fig. 21). Moreover, salivaricin G32 has been isolated from Streptococcus pyogenes, which is a nisin-like lantibiotic, and is different from antibiotic streptococcin A-FF22 in the absence of lysine in position 2, and it shows inhibitory effects against the Streptococcus pyogenes infection.201 A human vaginal isolate, Lactobacillus salivarius CRL, produces bacteriocin from the similar class known as Salivaricin CRL 1328, which is a heat-stable peptide. This peptide has the potential to prevent urogenital infections.202
 | ||
| Fig. 20 Structure of salivaricin B (47). | ||
 | ||
| Fig. 21 Structures of streptococcin A-FF22 (49), GllA1 (50) and GllA2 (51). | ||
Fusobacterium nucleatum, a recently identified bacterial pathogen in humans, is linked to gastrointestinal disorders such as colorectal cancer. A screening of faecal samples produces bacteriocins (salivaricin A5 and salivaricin B) with antibacterial effects against Fusobacterium nucleatum. In vitro, Streptococcus salivarius DPC6993 exhibited a limited range of antibacterial activity specifically targeting Fusobacterium nucleatum. Within a colon fermentation model, samples that were inoculated with Streptococcus salivarius DPC6993 along with Fusobacterium nucleatum DSM15643 exhibited a noteworthy decrease in the quantities of Fusobacterium nucleatum. This suggests that treatment agents aimed at these pathogens could potentially lower the likelihood of CRC development and have a favourable effect on outcomes.203 Along with this, another bacterocin, salivaricin LHM suppressing Pseudomonas aeruginosa induced inflammation by modulating immunological response in mice. In depth, salivaricin LHM exhibits anti-Pseudomonas activity and immunomodulatory effects by enhancing the production of pro-inflammatory cytokines. It also demonstrates antibiofilm properties against target bacterium urinary tract infection in both in vivo and in vitro models.204
Another Streptococcus bovis HC5 strain (Streptococcus gallolyticus subsp. gallolyticus Sgg formerly known as Streptococcus bovis type I), possesses powerful antibacterial properties. It inhibits a wide range of Gram-positive bacteria and has a spectrum comparable to monensin. Although heat, proteinase K, and α-chymotrypsin were able to deactivate the crude extracts, pronase E and trypsin were unable to do so. The pore-forming peptide (Bovicin HC5), which possessed a molecular mass of about 2440 Da, is the mediator of the antibacterial action. Similar to dehydroalanines in certain lantibiotics, the peptide ended with an amino acid sequence of VGXRYASXPGXSWKYVXF.161,205 Gallocin A is a two-peptide bacteriocin also found in Streptococcus gallolyticus subsp. gallolyticus promotes colonisation in mice. The research shows that the two peptides that make up gallocin A, GllA1 (50) and GllA2 (51) (Fig. 21), are inactive when taken individually but can kill target bacteria when combined. To provide immunity to gallocin A, the third gene in the operon, gip, is both required and sufficient. The mature peptides of GllA1 and GllA2, according to structural modelling, generate alpha-helical hairpins that are held in place by disulfide bridges that are located within the molecules themselves. The experiments validated the disulfide bond's presence in GllA1 and GllA2. There may be a new subclass of class IIb bacteriocins since the research found additional ones with comparable structures.206 Gallocin D, a two-component bacteriocin with strong action against vancomycin-resistant enterococci, is produced by Streptococcus gallolyticus LL009. The structural genes exhibit great variability and have seen gene shuffling with other streptococcal species, despite their strong gene synteny. In lab tests, gallocin D was examined and demonstrated action against the vancomycin-resistant Enterococcus strain EC300, with a MIC of 1.56 μM. These bacteriocins may aid in the colonisation process of Streptococcus gallolyticus, which has been linked to colorectal cancer.207Table 1 presents the important peptides isolated from Firmicutes along with their source and antimicrobial properties.
| No. | Peptide | Source | Active against | Ref. |
|---|---|---|---|---|
| 1 | Trn-α and Trn-β | Bacillus thuringiensis DPC 6431 | Clostridioides difficile | 42 |
| 2 | Amicoumacin A | Bacillus subtilis 3 | Helicobacter pylori | 208 and 209 |
| 3 | Gassericin A | Lactobacillus gasseri LA39 | Many species, Listeria monocytogenes, Bacillus cereus, Staphylococcus aureus | 57 |
| 4 | Gassericin T | Lactobacillus gasseri SBT 2055 | Many Bacillus sp. Staphylococcus sp. lactic acid bacteria | 53 |
| 5 | Acidocin B | Lactobacillus acidophilus M46 | Listeria monocytogenes, Clostridium sporogenes, Brochothrix thermosphacta, Lactobacillus fermentum, and Lactobacillus delbrueckii subsp. Bulgaricus | 61 |
| 6 | Acidocin J1132 | Lactobacillus acidophilus JCM1132 | Lactobacillus sp. | 66 |
| 7 | Amylovorin L471 (Lactobin-A) | Lactobacillus amylovorus DCE 471 | Pseudomonas aeruginosa | 71 |
| 8 | Plantaricin C | Lactobacillus plantarum LL441 | Lactobacillus sake, Enterococcus faecalis, Bacillus subtilis | 210 |
| 9 | Plantaricin EF and JK | Lactobacillus plantarum C11 | Escherichia coli K-12 | 211 |
| 10 | ABP-118 | Lactobacillus salivarius subsp. salivarius UCC118 | Listeria sp, Staphylococcus sp., Enterococcus sp, Bacillus sp. | 212 |
| 11 | Lactacin F | Lactobacillus johnsonii | Lactobacillus delbrueckii, Lactobacillus helveticus, and Enterococcus faecalis | 65 and 88 |
| 12 | Rhamnosin A | Lactobacillus rhamnosus 68 | Micrococcus lysodeikticus | 67 |
| 13 | Boticin B | Clostridium botulinum 213B | Clostridium botulinum | 98 |
| 14 | Closticin 574 | Clostridium tyrobutyricum ADRIAT 932 | Clostridium tyrobutyricum B570 | 100 |
| 15 | Bacteriocin 43 | Enterococcus faecium | Enterococcus faecalis, Enterococcus faecium, Enterococcus hirae, Enterococcus durans, Listeria monocytogenes | 105 |
| 16 | Bacteriocin 31 | Enterococcus faecalis | Enterococcus hirae 9790, Enterococcus faecium, Listeria monocytogenes | 107 |
| 17 | Bacteriocin 32 | Enterococcus faecalis | Enterococcus faecium, Enterococcus hirae, Enterococcus durans | 106 |
| 18 | Bacteriocin RC714 | Enterococcus faecalis | Enterococcus faecalis, Enterococcus faecium, Listeria monocytogenes | 106 |
| 19 | AS-48 | Streptococcus faecalis S-48 | Escherichia coli, Salmonella spp., Listeria monocytogenes, Staphylococcus aureus, Bacillus cereus, Paenibacillus spp. | 213 |
| 20 | Ruminococcin C | Ruminococcus gnavus | Clostridium perfringens | 119 |
| 21 | Ruminococcin A | Ruminococcus gnavus | Clostridium perfringens | 123 |
| 22 | Epilancin 15X | Staphylococcus epidermidis | Staphylococcus carnosus, Bacillus subtilis, Staphylococcus simulans | 133 and 134 |
| 23 | Nisin A | Lactococcus lactis | Lactobacillus delbrueckii subsp. bulgaricus LMG 690, 1 methicillin-resistant Staphylococcus aureus, vancomycin-resistant Enterococcus, and penicillin-resistant Streptococcus pneumoniae | 157 and 158 |
| 24 | Nisin J | Staphylococcus capitis APC 2923 | Staphylococcus aureus, Cutibacterium acnes | 155 |
| 25 | Nisin P | Streptococcus gallolyticus subsp. Pasteurianus, Streptococcus agalactiae DPC7040 | Lactobacillus delbrueckii subsp.bulgaricus LMG 6901 | 154 and 157 |
| 26 | Nukacin IVK45 | Staphylococcus epidermidis IVK45 | Many nasal members of the Actinobacteria, Proteobacteria, and Firmicutes | 147 |
| 27 | Lugdunin | Staphylococcus lugdunensis | Staphylococcus aureus | 148 and 149 |
| 28 | Mutacin 1140 | Streptococcus mutans 1140 | Clostridium difficile, Micrococcus luteus ATCC 10240, Streptococcus pneumoniae, Staphylococcus aureus, Clostridium difficile UK1 | 188 and 192 |
| 29 | Mutacin-BNY266 | Streptococcus mutans NY266 | Many oral streptococci | 194 |
| 30 | Salivaricin A | Streptococcus salivarius 20P3 | All strains of Streptococcus pyogenes | 214 |
| 31 | Salivaricin B | Streptococcus salivarius K12 | Fusobacterium nucleatum, Corynebacterium spp. GH17, Lactococcus lactis subsp. cremoris HP, Lactococcus lactis ATCC11454, Micrococcus luteus ATCC10240, Staphylococcus aureus RF122, many Streptococcus | 199 and 203 |
| 32 | Salivaricin D | Streptococcus salivarius 5M6c | Bacillus subtilis, Clostridium bifermentans, Clostridium butyricum, Streptococcus pneumoniae, Streptococcus suis | 195 |
| 33 | Salivaricin 9 | Streptococcus salivarius NU10 | Streptococcus pyogenes | 215 |
| 34 | Gallocin D | Streptococcus gallolyticus LL009 | Vancomycin-resistant Enterococcus EC300 | 207 |
4.2 In-depth analysis of Proteobacteria-derived peptides
Proteobacteria, also called pseudomonadota, is one of the major phyla of bacteria. This phylum contains one of the most studied and well-known genus of bacteria, Escherichia. Proteobacteria exhibit a vast distribution across multiple anatomical regions of the human body, encompassing the skin, oral cavity, tongue, vaginal canal, and gastrointestinal systems, in addition to their frequent presence in stool samples.216,217 Proteobacteria are found in the gut microbiota; however, they are generally less abundant than other phyla like Firmicutes and Bacteroidetes. Numerous health disorders have been linked to changes in the relative abundance of Proteobacteria. Proteobacteria comprises several genera, including Escherichia, Helicobacter, and Campylobacter, which consist of both commensal and pathogenic species. E. coli is a prevalent resident of the human gut microbiota and can play advantageous roles such as synthesizing vital vitamins. Nevertheless, specific strains of E. coli present pathogenicity and have the potential to induce gastrointestinal illnesses. Helicobacter pylori, a bacterium belonging to the Helicobacter genus, is known to inhabit the gastric region of a significant section of the human populace. Helicobacter pylori infection has been linked to the occurrence of stomach ulcers and gastric cancer in certain individuals. However, it has also been observed to potentially have protective effects against specific esophageal illnesses and asthma.217–219 | ||
| Fig. 22 Structures of microcin L (52), microcin M (53), and microcin H47 (54). | ||
Different colicins are produced by and also toxic for some strains of E. coli, including microcin V (previously colicin V).232 Different types of comparison were used to examine strains that produce microcin V, colicin E3, and colicin E7. Although microcin V was determined to be the least potent toxin in paired interactions, it consistently outperformed colicin E3 in competitions involving these strains.233 A lasso peptide, which has a lasso knot structure, has also been isolated with the name microcin J25 (55) (Fig. 23) from E. coli AY25 isolated from human feces and it is active against E. coli, Salmonella spp. and Shigella flexneri.234 To pass through the cell membrane, the majority of microcins and colicins that rely on TonB use siderophore receptors. Only microcin 24 (also known as microcin N) relies on FhuA, the receptor for the hydroxamate siderophore ferrichrome. In E. coli, microcin J25 is also uptaken by FhuA. However, microcin 24 is sensitive to Salmonella enterica serovar Typhimurium, whereas microcin J25 is resistant to it. Proving that microcin 24 recognises a wider range of hosts than microcin J25.224 Thirty years of research on microcin J25 has been reviewed recently by Baquero et al.235 A IId peptide with unmodified, linear, non-pediocin-like, single-peptide bacteriocins was isolated from E. coli G3/10 and is known as microcin S, which also exhibited antibacterial activities.236
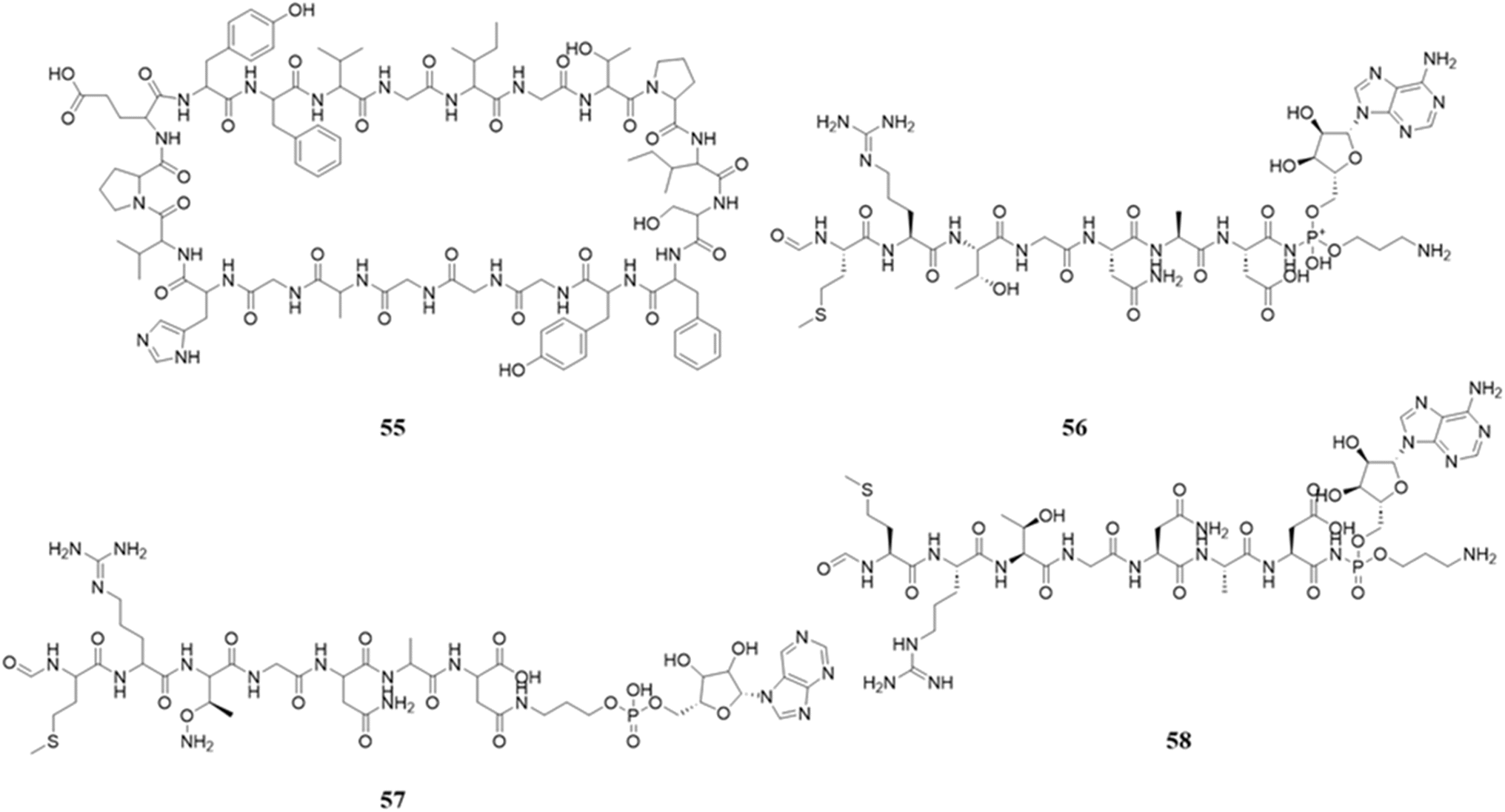 | ||
| Fig. 23 Structures of microcin J25 (55), microcin C7 (56), microcin C51 (57), and microcin C (58). | ||
Microcins C7/C51 (56, 57), B, and C (58) (Fig. 23) are also reported to be isolated from E. coli with antibacterial properties.28,237 An amide bond connects the carbonyl of the C-terminal aspartic acid residue to a modified nucleotide unit, which forms the main chain of microcin C7. In vitro, protein translation is inhibited by microcin C7 and the peptide, while a synthetic n-aminopropanol substituent has no effect. Microcin C7 antibiotic action is due to the peptide, and transport requires the C-terminal substituent.238 The mechanism involves the mature microcin C7 passing through the pore protein OmpF on the outer membrane of the bacterial cell and subsequently through the inner membrane of the bacterial cell, once the translation changes are finished. The YejABEF ABC transporter specifically identifies f-Met at the N-terminus of the microcin C7 peptide and facilitates the transport of microcin C7 into cells. Once this peptide enters a target bacterial cell, the process of microcin C7 processing commences. The complete mechanism and structural description have been reviewed recently by Yang et al.239
The genus Enterobacter, part of the Enterobacteriaceae family, is predominantly associated with infections in healthcare settings. Enterobacter species are primarily associated with nosocomial infections, although they can also infrequently cause community-acquired diseases including urinary tract infections, pulmonary infections, soft tissue infections, osteomyelitis, and endocarditis, among other ailments. Nevertheless, it is crucial to acknowledge that not all are implicated in human suffering.269 This genus possess a specific gene for the synthesis of endotoxin and uraemic toxins such as 4-hydroxy phenyl lactate, p-cresol sulfate, indoxyl sulfate, indole-3 acetic acid, trimethylamine-N-oxide, and imidazole propionate.270 As mentioned earlier, Colibactin, a bacterial toxin, was also found in the clusters of Enterobacter aerogenes. However, as a whole this is very less studied in case of metabolites.114 There are many other bacteria from phylum Proteobacteria that are a part of human microbiota. However, their contribution to the metabolite profile is quite low. Some of them have made little contribution other than discussed before. The ability to create virulence factors and withstand high temperatures is an evolutionary hallmark of human infections. Recently, it has been found that blood of a sick child yielded the pathoadapted bacterium Luteibacter anthropi. Genome comparisons showed that compared to other bacteria, Luteibacter anthropi possesses a distinct NRPS gene locus that codes for metallophore production, and it also has a greater metabolic capacity. A new family of nonribosomal peptides called anthrochelins A–D (61–64) were discovered, and they are derived from salicylates. A homocysteine tag, likely added during peptide termination, is located at the C-terminus of these peptides.271
Other genera in human microbiota from Proteobacteria include Helicobacter, Campylobacter, and Vibrio. They are mostly acting as pathogens to humans. For instance, the relationship between Helicobacter pylori infection and changes in the gut microbiota has been the subject of multiple studies. A considerable body of research has been dedicated to investigating the impact of eradication therapy on the gut flora. The prevalence of Helicobacter pylori infection exceeds 50% among the worldwide population. Vacuolating cytotoxin A is a prominent virulence factor synthesized by Helicobacter pylori, and such toxin is composed of the p33 and p55 subunits, attaches to host cells and causes significant vacuolation after being released through a type V auto transport secretion system.272–274Campylobacter is a widespread cause of diarrheal disease worldwide. Butyric acid, a metabolite produced by microorganisms, plays a crucial role in inhibiting the colonization of Campylobacter jejuni by the gut microbiota. Furthermore, both butyric acid and deoxycholic acid play a role in mitigating colitis produced by Campylobacter jejuni.275–277 The harmful bacterium known as Vibrio cholerae is the reason for the severe diarrheal illness known as cholera, which affects millions of people every year and is responsible for more than 100000 deaths each year. At the beginning of the infection, Vibrio clings to epithelial cells and begins to multiply.
This is where the infection begins. Vibrio, once it has entered the human digestive system, launches a wide variety of complex pathogenic factors. An important factor that contributes to the severity of diarrheal sickness is the production of cholera toxin by Vibrio cholerae, which is one of these. The cholera toxin, which is classified as a heat-labile enterotoxin and is a member of the AB toxin family, is an essential component in the evolution of the disease called cholera.278–280 The structures of (59–64) are shown in Fig. 24.
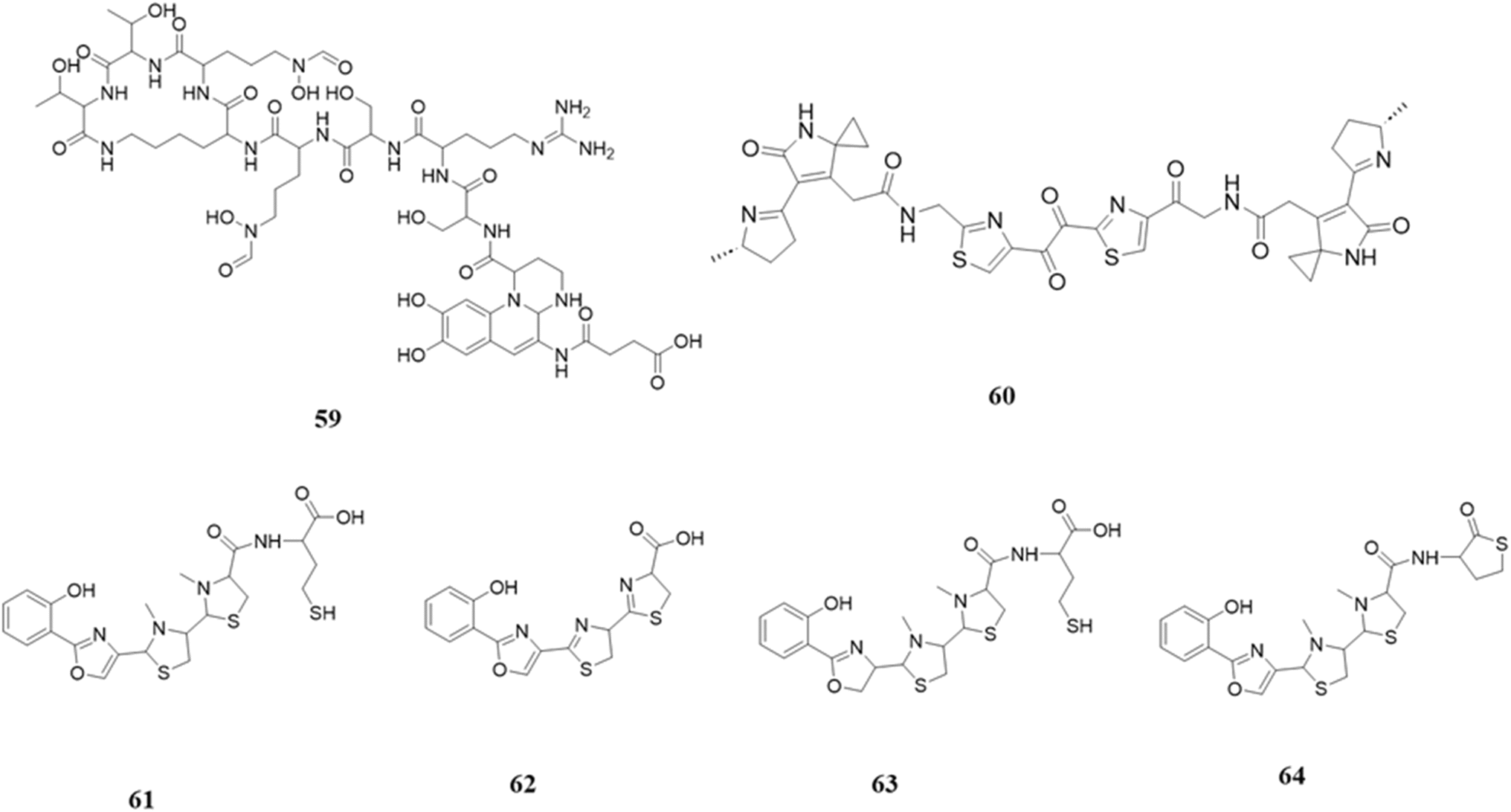 | ||
| Fig. 24 Structures of pyoverdin (59), colibactin (60), and anthrochelins A–D (61–64). | ||
4.3 Biological roles of Actinobacteria (Actinomycetota)-derived peptides
Actinobacteria have been detected in various sites of the human body, including mucosal surfaces and the skin. They are essential components of a healthy microbiota. Their presence and abundance in these specific sites correlate with the individual's health status. This phylum also contains those bacteria that cause tuberculosis and diphtheria. Although they make up only a small percentage of the gut microbiota, they play a key role in maintaining gut homeostasis.281–283 They comprise numerous taxa that are highly significant, such as Bifidobacterium, Corynebacterium, Rhodococcus, and Mycobacterium, in terms of extracting peptides.Moreover, several strains have demonstrated the capacity to mitigate cutaneous inflammations, such as eczema and atopic dermatitis, in newborns. Within the human gut microbiota, Bifidobacterium is among the most prevalent genera. They make up 3–7% of the adult digestive tract population and as much as 91% of the entire population in newborns.284–288 Yu et al. have recently studied, where they re-evaluated numerous Bifidobacterium species that were previously believed to have limited capacities to produce bacteriocins. The identification of bacteriocin genes in these species indicates that they have a distinct advantage in establishing themselves in the intestinal tract of infants. The presence of many biosynthetic gene clusters was observed in Bifidobacterium longum subsp. infantis, encompassing various groups responsible for the synthesis of lanthipeptides, lasso peptides, thiopeptides, and class IId bacteriocins. The potential for bacteriocin production was demonstrated by confirming the antibacterial potency of the crude bacteriocins by bioassays and transcriptional analysis.289 A lantibiotic, BLD 1648, has been reported from bifidobacterium longum DJO10A.65Bifidobacterium longum BL-10 also possess 15 antioxidant genes, which displayed strong reducing power activity and scavenging activity of DPPH, hydroxyl radicals, and superoxide anions,290 which is a clue that it contains specific metabolites that have the antioxidant potential, in such case, an amino acid metabolite, phenylacetic acid, has been reported from Bifidobacterium.28 The bacteriocin production of five different strains of Bifidobacterium bifidum was examined in MRS broth supplemented with 0.05% cysteine. The only bacteriocin that NCFB 1454 released was bifidocin B (65) (Fig. 25). Bifidocin B inhibited the growth of certain Gram-positive and Gram-negative bacteria, including those responsible for food poisoning and spoilage, such as Listeria, Enterococcus, Bacillus, Lactobacillus, Leuconostoc, and Pediococcus species.291 Furthermore, it was shown that bifidocin B exhibited adsorption specifically to Gram-positive bacteria. The adsorption exhibited pH dependency but not time dependency. Bifidocin B exhibited fast adsorption and lethal activity against susceptible cells. Cellular binding of bifidocin B was not reduced by pre-treatment with detergents, organic solvents, or enzymes. The ability of cell wall preparations to absorb bifidocin B was diminished when treated with methanol:chloroform and hot 20% (w/v) TCA. Bifidocin B adsorption was inhibited by the introduction of pure heterologous lipoteichoic acid.292 Bifidin, bacteriocin generated by Bifidobacterium bifidum NCDC 1452, is the first bacteriocin thought to be associated with Bifidobacterium. When it is cultured in skim milk, its antibacterial activity is at its peak, also its activity become high even after being partially cleansed and stored in the cold.293 Bifidin I, a novel bacteriocin derived from Bifidobacterium infantis BCRC 14602, was purified using a three-step purification procedure. Bifidin I exhibits potent antilisterial activity and effectively inhibits the development of both Gram-positive and Gram-negative bacteria, which are responsible for food deterioration and foodborne illnesses. Consequently, it has considerable potential for application in ensuring food safety.294 Bifilong from bifidobacteria also possess bacteriocin characteristics. This compound possesses heat stability, is composed of proteins, and exhibits activity within a pH range of 2–10. It can efficiently fight against various Gram-positive species, such as bifidobacteria, lactobacilli, clostridia strains, and certain streptococci strains.295
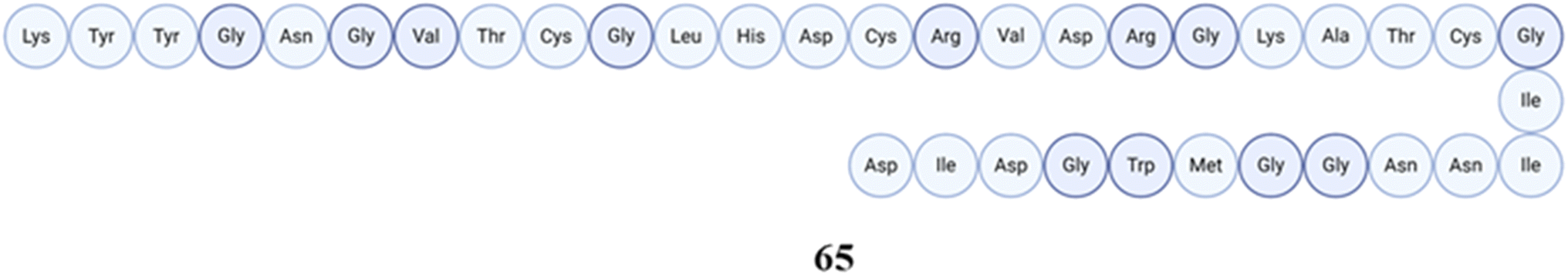 | ||
| Fig. 25 Structure of bifidocin B (65). | ||
The analysis of biosynthetic gene clusters in Rhodococcus erythropolis and Rhodococcus equi revealed the existence of the requisite genetic material for the manufacture of humimycin, resulting in the generation of two distinct humimycin A (66) and B (67). In mouse models, these humimycins demonstrate the capacity to impede lipid II flippase and augment the efficacy of β-lactam antibiotics against methicillin-resistant Staphylococcus aureus. This finding exhibits potential for the advancement of innovative therapeutic approaches.296 Furthermore, Rhodococcus erythropolis is capable of synthesizing siderophores, such as heterobactins A (68) and B (69), consisting of tripeptide sequence ((N-OH)-L-Orn-Gly-D-Orn- (delta-N-dihydroyxbenzoate)). Experiments on growth promotion with different transport mutants showed that the catecholate receptor Cir is the sole E. coli heterobactin A receptor, whereas a hydroxamate transport system is responsible for the uptake of heterobactin B in both E. coli and Microbacterium flavescens JG9.297 The structures of (66–67) and (68–69) are shown in Fig. 26 and 27, respectively.
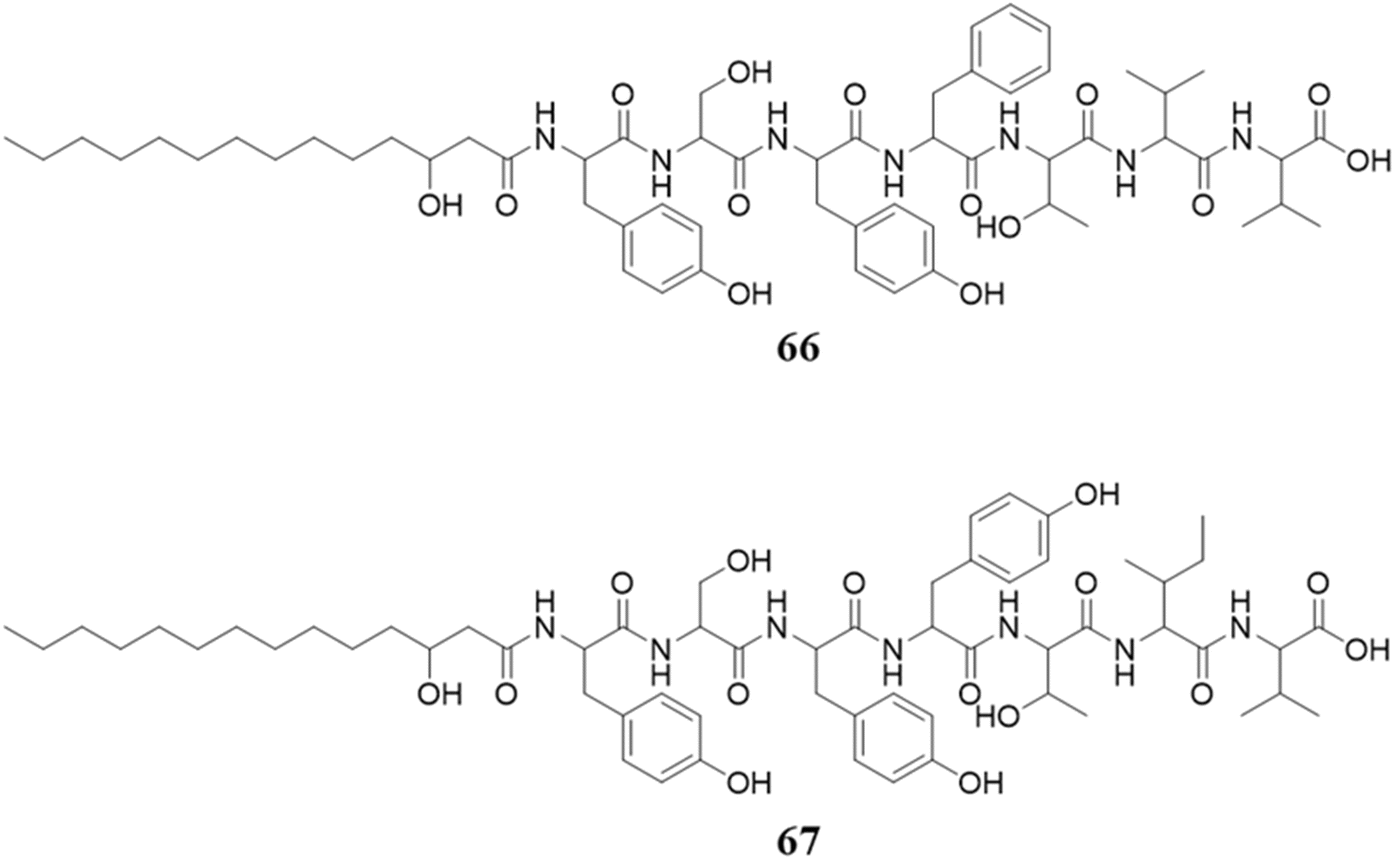 | ||
| Fig. 26 Structures of humimycin A (66) and humimycin B (67). | ||
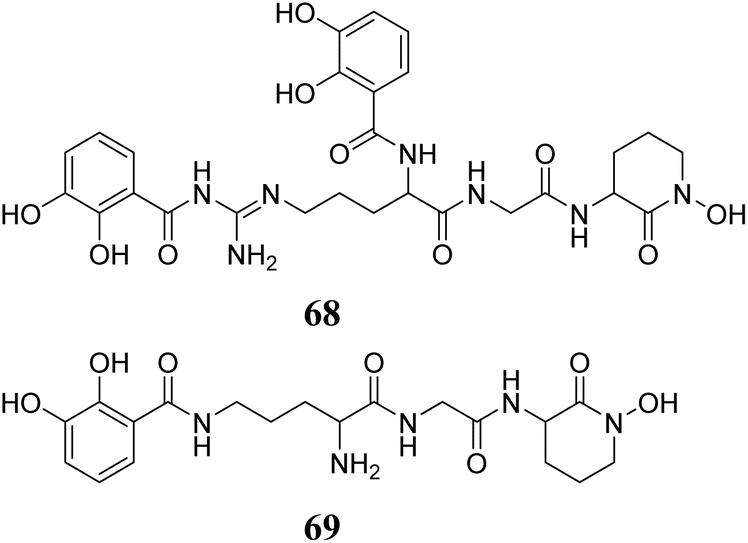 | ||
| Fig. 27 Structures of heterobactin A (68) and heterobactin B (69). | ||
Mycobacterium species encompasses a wide range of acid-fast, aerobic bacteria that are distinguished by their sluggish growth rate. There are more than 70 different species in this genus, and around 30 of them have been associated with diseases in humans. Mycobacterium tuberculosis is of particular importance as the major etiological agent responsible for tuberculosis.298 Phthiocerol dimycocerosates are a notable category of lipids that have been identified as significant factors in the pathogenicity of Mycobacterium tuberculosis. The exterior surface of virulent strains within the Mycobacterium tuberculosis complex has a notable abundance of glycolipids, which serve crucial functions in the pathogenicity of the bacterium.299,300 Lipoarabinomannan is a glycolipid that plays a crucial role in the structural composition of the cell wall of Mycobacterium tuberculosis, the pathogen responsible for tuberculosis. Such glycolipid operates as a virulence factor through its ability to hinder the activities of monocytes and eliminate oxidative radicals. Lipoarabinomannan has been observed in the urine of specific individuals diagnosed with tuberculosis.301 Mycolic acids have also been reported from Mycobacterium with immunomodulatory activities.28
There are many other genera of Actinobacteria which reside in humans and represent metabolites profile and many other biological activities. Nocardia belongs to Actinobacteria is pathogenic bacteria to humans and it causes human infection called nocardiosis. It is also known to produce a variety of compounds with antitumor, and antimicrobial activities. Brasilinolide A (70), brasilinolide B (71) (Fig. 28), and transvalencin Z (72) have been reported from Nocardia.302–304 Brasilinode A exhibits significant antifungal activity against Aspergillus niger, with a minimum inhibitory concentration of 3.13 μg ml−1. Furthermore, it demonstrated immunosuppressive effect in the mixed lymphocyte reaction test technique.305 Transvalencin Z exhibits significant antibacterial activity against Gram-positive bacteria, while without any cytotoxic effects. It exhibits specific antimycobacterial effects against Mycobacterium smegmatis. Furthermore, it exhibited antimicrobial properties against fungi such as Trichophyton mentagrophytes and Cryptococcus neoformans. The antibiotic exhibits activity against Gram-positive bacteria, including Micrococcus luteus.303,306,307 Nocardithiocin (73), a thiopeptide compound synthesised by Nocardia pseudobrasiliensis strain IFM 0757, demonstrates potent activity against Mycobacterium and Gordonia species. It effectively inhibits both rifampicin-resistant and sensitive strains of Mycobacterium tuberculosis at concentrations ranging from 0.025 to 1.56 μg ml−1.308
 | ||
| Fig. 28 Structures of brasilinolide A (70), and brasilinolide B (71). | ||
Many siderophores have been isolated from Nocardia spp. For instance, JBIR-16, brasilibactin A, asterobactin, nocobactin NA and nocardamine (74–78).309 JBIR-100 exhibits significant antibacterial properties. When Bacillus subtilis is treated with JBIR-100, it causes the permeabilization and depolarization of the cell membrane.310 Treatment with ferric chloride diminished the antibacterial properties of brasilibactin A against Staphylococcus aureus and Micrococcus luteus, with MIC values of 0.73 and 4.5 mg ml−1, respectively. There was a concentration-dependent increase (0.3–3 μM) in caspase-3 activity in HL60 cells, while it also showed strong cytotoxicity against murine leukaemia L1210 and human epidermoid carcinoma KB cells (IC50, 0.02 and 0.04 μg ml−1, respectively).311 At concentrations ranging from 0.2 to 3.2 μg ml−1, asterobactin demonstrated anticancer efficacy against a number of cultured cell lines, including HL-60 and HeLa cells.312 The structures of (72–77) and (78) are shown in Fig. 29 and 30, respectively.
 | ||
| Fig. 29 Structures of transvalencin Z (72), nocardithiocin (73), JBIR-16 (74), brasilibactin A (75), asterobactin (76), and nocobactin NA (77). | ||
 | ||
| Fig. 30 Structure of nocardamine (78). | ||
Collinsella is a genus of Gram-positive anaerobic bacteria that are classified within the Actinomycetota phylum and are commonly found in the human gut microbiome. Additionally, it contributes to the development of health conditions such as rheumatoid arthritis and irritable bowel syndrome. Collinsella has the potential to impact metabolism through its ability to modify the absorption of cholesterol in the intestines, reduce glycogenesis in the liver and enhance the production of triglycerides.313 In the human skin microbiota, Propionibacterium acnes is a Gram-positive bacterium that is considered a normal constituent. Additionally, it can be observed throughout the mouth cavity, large intestine, conjunctiva, and external ear canal. Propionibacterium acnes is the predominant bacterium found on the surface of the human skin, especially in the sebaceous regions. It is widely postulated that it exerts an influence on the pathogenesis of acne vulgaris, a persistent inflammatory dermatological disorder that impacts around 10% of the global populace.314 Many more species within this phylum are likely to produce many metabolites, given their close relationship to other species in the same phylum.
4.4 In-depth analysis of Bacteroidetes-derived peptides
The Bacteroidaceae family is widely distributed in the gut microbiota and exhibits a greater ability to engage with their host and break down complex polysaccharides. Their metabolic processes frequently yield acetate, propionate, and succinate.315,316 The metabolite profile of human microbiota is significantly influenced by most genera within the Bacteroidetes group. As previously stated, both Firmicutes and Bacteroidetes constitute most of the human gut microbiota, comprising over 90% of the total microbial population. However, the addition of peptides to metabolic profile from this phylum is not as substantial compared to Firmicutes.The obligately anaerobic bacteria of the genus Bacteroides, which are a significant component of the Bacteroidetes phylum, have drawn considerable attention in the field of human gut microbiology since their original identification. The strong adaptation of these microbes to their specific biological niche is highlighted by their exclusive localization and proliferation within the gastrointestinal tracts of mammals. These microbes possess the ability to form long-lasting and mutually advantageous relationships with their human hosts, functioning as both commensals and mutualists. As a result, they provide a wide range of health-enhancing effects.317–319
In the case of metabolites, Bacteroides fragilis or its cell-free supernatant was used to inhibit in vitro Salmonella Heidelberg translocation, and such friction was then analyzed by Liquid Chromatography High-Resolution Tandem Mass Spectrometry along with the Molecular networking to display the possible compounds in such activities, and cholic acid and deoxycholic acid were found to be the responsible metabolites.320 Different species, i.e., Bacteroides thetaiotaomicron, Bacteroides eggerthii, Bacteroides ovatus, Bacteroides fragilis, Parabacteroides distasonis, were tested for the metabolite production, but these are not the peptides we are searching for.321
Commendamide, or N-(3-hydroxypalmitoyl)-glycine (79) (Fig. 31), is a biologically active compound that is synthesized by the gut microbiota, Bacteroides vulgatus. It exhibits a similar structure to long-chain N-acyl-amino acids, which are components of the complex lipid signalling system known as the endocannabinoidome. It plays essential roles in mammals by stimulating different receptors such as G-protein-coupled receptors.322,323 Coyne et al. performed a comprehensive analysis of 140 distinct strains of Bacteroides and Parabacteroides found in the human stomach. Out of them, a total of 15 unique species were identified, and four of them were shown to produce a metabolite that specifically targets many species of Bacteroides and Parabacteroides. There are four strains of Bacteroides ovatus (BoCL02T12C04), Bacteroides thetaiotaomicron (BtCL15T12C11), and two strains of Bacteroides vulgatus (BvCL01T12C17 and BvCL14T03C19) that can generate toxins. Furthermore, the confirmation of the existence of bacteroidetocin A (80) and B (81) (Fig. 32) were obtained in many strains.324 As there are many species of Bacteroides present in humans, very few have been studied for metabolites so far.
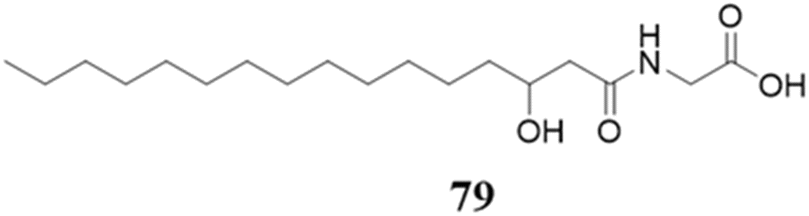 | ||
| Fig. 31 Structure of commendamide (79). | ||
 | ||
| Fig. 32 Structures of bacteroidetocin A (80) and bacteroidetocin B (81). | ||
4.5 Biological functions of Fusobacteria-derived peptides with their role in human disorders
The phylum Fusobacteria comprises bacteria that exhibit obligate anaerobic characteristics, possess a Gram-negative nature, and do not form spores. They are rod-shaped, cylindrical bacilli with sharp tips. This species is quite prevalent in the oral cavity, found in both individuals with diseases and healthy. Periodontal illnesses, ranging from moderate reversible gingivitis to severe irreversible periodontitis, are associated with its involvement. Fusobacteria are a constituent of the indigenous microbiota within the human body and commonly present in the gastrointestinal system and female vaginal tract. Nevertheless, these microbes have the potential to induce various infections and abscesses, such as Lemierre's syndrome, acute and chronic mastoiditis, chronic otitis and sinusitis, tonsillitis, peritonsillar and retropharyngeal abscesses, and postanginal cervical lymphadenitis.325–327Fusobacteria are commonly present in the oral, gastrointestinal, and vaginal microbiota, but they can cause septic thrombophlebitis in the adjacent neck arteries when the infection is linked to an oropharyngeal abscess. Several reliable studies conducted on Fusobacterium have reported an annual incidence of bacteremia at a rate of 0.55 cases per 100000 individuals, along with a diverse range of clinical manifestations. Fusobacterium nucleatum and Fusobacterium necrophorum are the predominant species that have been isolated within this particular genus.328,329 The immunostimulatory molecules known as muramyl dipeptide (82) and muramyl tripeptide (83) (Fig. 33) are produced from the cell walls of bacteria. These entities play a crucial role in the composition of the bacterial peptidoglycan layer, which serves as a defensive shield enveloping bacterial cells. N-Acetylmuramic acid and L-alanine are the constituent amino acids of muramyl dipeptide, whereas muramyl tripeptide include an extra amino acid, D-isoglutamine. Muramyl dipeptide and tripeptide have been reported from the Fusobacterium nucleatum, which displayed immunomodulatory activity.28,330 Short-chain fatty acids are also released from Fusobacterium nucleatum as end-products of its metabolism. These Short-chain fatty acids act as neutrophil chemoattractants through free fatty acid receptor 2.331 Another genus within the same phylum that is found in the human microbiota is Leptotrichia, which are non-motile facultative anaerobic/anaerobic microorganisms mostly distributed in the mouth cavity and certain regions of the human body.332 There exist several additional genera within this phylum that have not been extensively investigated in relation to the isolation and purification of metabolites. A similar situation is also observed in the context of the Verrucomicrobia phylum.
 | ||
| Fig. 33 Structures of as muramyl dipeptide (82) and muramyl tripeptide (83). | ||
5 Future challenges and conclusive remarks
The human gut microbiota consist of trillions of symbiotic microbial cells in every individual. Genomics has facilitated the identification of a wide range of natural chemicals using a technique to which we can say “genomic guided isolation”. The metabolic function of gut microbial species is influenced by the presence of these essential substances derived from the diet, which in turn affect the functioning of the host organism. These metabolites include various significant categories of secondary metabolites, such as lantibiotics and bacteriocins, microcins and thiazole, enterotoxin, glycolipids, terpenoids, and, polyketides and nonribosomal peptides.28,33,333,334After performing an extensive analysis of existing research, it is evident that Firmicutes, followed by Actinobacteria, and Proteobacteria are the three phyla of the human microbiota that have gained considerable interest, particularly in relation to their peptides. Nevertheless, there is a dearth of research on the investigation of metabolites in the other phyla. Although Firmicutes and Bacteroidetes make up the largest portion of the human gut microbiota, accounting for over 90% of the total microbial population, Bacteroidetes has received less attention in terms of its metabolites. In addition, there has been limited focus on studying the metabolites associated with Fusobacteria and Verrucomicrobia, resulting in a significant knowledge gap in the connection between human microbiota and metabolites. To fill this gap, it is necessary to make progress in current chromatographic, spectroscopic (including mass spectrometric), genomic, and computational techniques. Due to the persistent challenges faced by these primary techniques, such as restricted metabolite detection, current methodologies require enhancement to successfully identify numerous bioactive chemicals that may remain undetected due to their low abundance or complex chemical structures. Along with this, the dynamic nature of microbiota, which is impacted by several aspects such as health status, environment, and nutrition, necessitates further investigation, as existing methodologies are not progressively addressing this topic over time. The integration of multi-omics data presents a substantial difficulty in elucidating the microbiota characteristics and intricacies. Torres et al. have recently examined 444054 small protein families derived from 1773 human metagenomes to assess their antibacterial capabilities. Their findings revealed the existence of many compounds in the human microbiome that may be confirmed as possible antibacterial agents using contemporary methods.335 Another key necessity to address is the improvement of preclinical and clinical trials that can effectively bridge this gap.
These enhancements are essential for further exploration into the complex realm of metabolites linked to the human microbiota, ultimately enhancing our comprehension of its functional significance in human health and disease. The pursuit of new antimicrobial drugs has become a crucial effort in addressing the significant challenges presented by antibiotic resistance. Studying the environments where harmful bacteria thrive and investigating how their ecological rivals control them have produced encouraging findings. The application of this theoretical framework to develop practical treatments for severe infections and widespread disorders can be enhanced by identifying and studying various antibacterial substances obtained from the human microbiome.
Since the advent of antibiotics in the twentieth century, several medications have become potent instruments in the treatment of a wide range of diseases and ailments. Furthermore, antibiotic resistance has emerged as a serious problem due to improper and incorrect use; if not addressed, this pandemic might wipe out millions of people. It is one of the leading causes of death worldwide in 2019, with 1.27 million fatalities attributable to AMR and 4.95 million deaths, according to the World Health Organization (WHO). Significant monetary expenditures accompany AMR, which also causes death and disability. By 2050, AMR would drive up healthcare expenditures by $1 trillion and reduce GDP by $1 trillion to $3.4 trillion annually by 2030, according to the World Bank.
There is a requirement for a new and natural antibiotic that can effectively combat these harmful bacteria. The human microbiota have demonstrated distinct and promising structures that can play a vital role in addressing this problem. In this particular scenario, it was necessary to present the existing literature on peptides derived from these bacteria, which paves the path for future research. Here, we effectively addressed this information gap by meticulously collecting and analysing a significant number of crucial peptides from the human microbiota, as well as exploring their potential biological functions in relation to human health. We also presented the mode of action of the majority of the peptides, along with a structural description where necessary. We have demonstrated that peptides exhibit not just antibacterial properties, but also powerful activity in the context of inflammation and cancer. This paves the door for further study to comprehensively explore the potential of these peptides not only as antimicrobial agents, but also in addressing various other human disorders. Acquiring this valuable information is essential for uncovering new molecular formations originating from the human microbiota, which may lead to a diverse array of metabolites with different biological effects. These findings not only improve our understanding of microbial ecology but also open up new possibilities for therapeutic approaches and the formulation of medications.
6 Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.7 Conflicts of interest
There are no conflicts to declare.8 Acknowledgements
This research was supported by the Bio and Medical Technology Development Program of the National Research Foundation (NRF) funded by the Korean government (MSIT) (No. RS-2024-00352229) and the National Research Foundation (NRF) of Korea (NRF-2021R1A2C1004958 and NRF-2022R1A4A3022401).9 References
- N. Aggarwal, S. Kitano, G. R. Y. Puah, S. Kittelmann, I. Y. Hwang and M. W. Chang, Chem. Rev., 2022, 123, 31–72 CrossRef PubMed.
- S. El-Sahhar and P. Varga-Weisz, in Advances in Ecological Research, ed. D. A. Bohan and A. Dumbrell, Academic Press, 2022, vol. 67, pp. 289–330 Search PubMed.
- S. Altveş, H. K. Yildiz and H. C. Vural, Biosci. Microbiota, Food Health, 2020, 39, 23–32 CrossRef.
- K. Hou, Z.-X. Wu, X.-Y. Chen, J.-Q. Wang, D. Zhang, C. Xiao, D. Zhu, J. B. Koya, L. Wei, J. Li and Z.-S. Chen, Signal Transduction Targeted Ther., 2022, 7, 1–28 CrossRef PubMed.
- L. K. Ursell, H. J. Haiser, W. Van Treuren, N. Garg, L. Reddivari, J. Vanamala, P. C. Dorrestein, P. J. Turnbaugh and R. Knight, Gastroenterology, 2014, 146, 1470–1476 CrossRef CAS.
- C. A. Lozupone, J. I. Stombaugh, J. I. Gordon, J. K. Jansson and R. Knight, Nature, 2012, 489, 220–230 CrossRef CAS.
- M. S. Kennedy and E. B. Chang, Prog. Mol. Biol. Transl. Sci., 2020, 176, 1–42 Search PubMed.
- A. H. Russell and A. W. Truman, Comput. Struct. Biotechnol. J., 2020, 18, 1838–1851 CrossRef CAS PubMed.
- R. Chen, H. L. Wong and B. P. Burns, Medicines, 2019, 6, 32 CrossRef CAS.
- G. M. Weinstock, Nature, 2012, 489, 250–256 CrossRef CAS.
- A. G. Atanasov, S. B. Zotchev, V. M. Dirsch and C. T. Supuran, Nat. Rev. Drug Discovery, 2021, 20, 200–216 CrossRef CAS PubMed.
- J. V. Pham, M. A. Yilma, A. Feliz, M. T. Majid, N. Maffetone, J. R. Walker, E. Kim, H. J. Cho, J. M. Reynolds, M. C. Song, S. R. Park and Y. J. Yoon, Front. Microbiol., 2019, 10, 1404, DOI:10.3389/fmicb.2019.01404.
- L. Niehaus, I. Boland, M. Liu, K. Chen, D. Fu, C. Henckel, K. Chaung, S. E. Miranda, S. Dyckman, M. Crum, S. Dedrick, W. Shou and B. Momeni, Nat. Commun., 2019, 10, 2052 CrossRef.
- Y. Zhang, R. Chen, D. Zhang, S. Qi and Y. Liu, Biomed. Pharmacother., 2023, 160, 114295 CrossRef CAS.
- J. Ji, W. Jin, S. Liu, Z. Jiao and X. Li, MedComm, 2023, 4, e420 CrossRef PubMed.
- E. Rinninella, P. Raoul, M. Cintoni, F. Franceschi, G. A. D. Miggiano, A. Gasbarrini and M. C. Mele, Microorganisms, 2019, 7, 14, DOI:10.3390/microorganisms7010014.
- E. Thursby and N. Juge, Biochem. J., 2017, 474, 1823–1836 CrossRef CAS.
- I. Rowland, G. Gibson, A. Heinken, K. Scott, J. Swann, I. Thiele and K. Tuohy, Eur. J. Nutr., 2018, 57, 1–24 CrossRef CAS.
- N. M. Swer, B. S. Venkidesh, T. S. Murali and K. D. Mumbrekar, Mol. Biol. Rep., 2023, 50, 1663–1675 CrossRef CAS PubMed.
- J. E. Belizário and M. Napolitano, Front. Microbiol., 2015, 6, 1050 Search PubMed.
- J. Wu, K. Wang, X. Wang, Y. Pang and C. Jiang, Protein Cell, 2021, 12, 360–373 CrossRef.
- J. Liu, Y. Tan, H. Cheng, D. Zhang, W. Feng and C. Peng, Aging Dis., 2022, 13, 1106–1126 CrossRef.
- N. Koppel and E. P. Balskus, Cell Chem. Biol., 2016, 23, 18–30 CrossRef CAS.
- Y. Sugimoto, F. R. Camacho, S. Wang, P. Chankhamjon, A. Odabas, A. Biswas, P. D. Jeffrey and M. S. Donia, Science, 2019, 366, eaax9176 CrossRef CAS PubMed.
- J. S. McLean, Front. Cell. Infect. Microbiol., 2014, 4, 98 Search PubMed.
- P. N. Deo and R. Deshmukh, J. Oral Maxillofac. Pathol., 2019, 23, 122–128 CrossRef.
- E. A. Grice and J. A. Segre, Nat. Rev. Microbiol., 2011, 9, 244–253 CrossRef CAS.
- M. S. Donia and M. A. Fischbach, Science, 2015, 349, 1254766 CrossRef.
- S. A. Fobofou and T. Savidge, Am. J. Physiol.: Gastrointest. Liver Physiol., 2022, 322, G535–G552 CrossRef CAS PubMed.
- G. N. Tenea and P. Ascanta, Front. Microbiol., 2022, 13, 868025, DOI:10.3389/fmicb.2022.868025.
- A. Milshteyn, D. A. Colosimo and S. F. Brady, Cell Host Microbe, 2018, 23, 725–736 CrossRef CAS.
- W. Fusco, M. B. Lorenzo, M. Cintoni, S. Porcari, E. Rinninella, F. Kaitsas, E. Lener, M. C. Mele, A. Gasbarrini, M. C. Collado, G. Cammarota and G. Ianiro, Nutrients, 2023, 15, 2211 Search PubMed.
- Z. Y. Kho and S. K. Lal, Front. Microbiol., 2018, 9, 1835, DOI:10.3389/fmicb.2018.01835.
- M. Afzaal, F. Saeed, Y. A. Shah, M. Hussain, R. Rabail, C. T. Socol, A. Hassoun, M. Pateiro, J. M. Lorenzo, A. V. Rusu and R. M. Aadil, Front. Microbiol., 2022, 13, 999001, DOI:10.3389/fmicb.2022.999001.
- I. Sekirov, S. L. Russell, L. C. M. Antunes and B. B. Finlay, Physiol. Rev., 2010, 90, 859–904 CrossRef CAS.
- L. H. Morais, H. L. Schreiber and S. K. Mazmanian, Nat. Rev. Microbiol., 2021, 19, 241–255 CrossRef CAS PubMed.
- F. Magne, M. Gotteland, L. Gauthier, A. Zazueta, S. Pesoa, P. Navarrete and R. Balamurugan, Nutrients, 2020, 12, 1474 CrossRef CAS PubMed.
- A. Koh and F. Bäckhed, Mol. Cell, 2020, 78, 584–596 CrossRef CAS.
- S. Gebhard, Mol. Microbiol., 2012, 86, 1295–1317 CrossRef CAS PubMed.
- M. Zasloff, Nature, 2002, 415, 389–395 CrossRef CAS PubMed.
- J. Talapko, T. Meštrović, M. Juzbašić, M. Tomas, S. Erić, L. Horvat Aleksijević, S. Bekić, D. Schwarz, S. Matić, M. Neuberg and I. Škrlec, Antibiotics, 2022, 11, 1417 CrossRef CAS.
- M. C. Rea, C. S. Sit, E. Clayton, P. M. O'Connor, R. M. Whittal, J. Zheng, J. C. Vederas, R. P. Ross and C. Hill, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9352–9357 CrossRef CAS.
- Y. Chen, J. Wang, G. Li, Y. Yang and W. Ding, Front. Chem., 2021, 9, 595991 CrossRef CAS.
- A. Hassoun, BMJ, 2018, 363, k4369 CrossRef PubMed.
- H. Mathur, V. Fallico, P. M. O'Connor, M. C. Rea, P. D. Cotter, C. Hill and R. P. Ross, Front. Microbiol., 2017, 8, 696, DOI:10.3389/fmicb.2017.00696.
- C. Viera Herrera, P. M. O'Connor, P. Ratrey, R. Paul Ross, C. Hill and S. P. Hudson, Int. J. Pharm., 2024, 654, 123918 CrossRef CAS PubMed.
- H. Mathur, M. C. Rea, P. D. Cotter, C. Hill and R. P. Ross, Gut Pathog., 2016, 8, 20 CrossRef PubMed.
- L. Walsh, A. Lavelle, P. O'Connor, C. Hill and R. P. Ross, Gut Microbes, 2024, 16, 2342583 CrossRef CAS PubMed.
- S. Crowley, J. Mahony and D. van Sinderen, Trends Food Sci. Technol., 2013, 33, 93–109 CrossRef CAS.
- A. Abdel-Nasser, A. S. Hathout, A. N. Badr, O. S. Barakat and H. M. Fathy, Biotechnol. Rep., 2023, 38, e00799 CrossRef CAS.
- T. Itoh, Y. Fujimoto, Y. Kawai, T. Toba and T. Saito, Lett. Appl. Microbiol., 1995, 21, 137–141 CrossRef CAS.
- Y. Kawai, B. Saitoh, O. Takahashi, H. Kitazawa, T. Saito, H. Nakajima and T. Itoh, Biosci., Biotechnol., Biochem., 2000, 64, 2201–2208 CrossRef CAS.
- K. Arakawa, Y. Kawai, H. Iioka, M. Tanioka, J. Nishimura, H. Kitazawa, K. Tsurumi and T. Saito, J. Dairy Sci., 2009, 92, 2365–2372 CrossRef CAS PubMed.
- A. Zanfardino, G. Criscuolo, B. Di Luccia, E. Pizzo, M. L. Ciavatta, E. Notomista, A. Carpentieri, A. Pezzella and M. Varcamonti, Benefic. Microbes, 2017, 8, 133–141 CAS.
- M. D. Napoli, B. D. Luccia, G. Vitiello, G. D'Errico, A. Carpentieri, A. Pezzella, E. Pizzo, E. Notomista, M. Varcamonti and A. Zanfardino, Benefic. Microbes, 2020, 11, 815–824 Search PubMed.
- T. Kabuki, T. Saito, Y. Kawai, J. Uemura and T. Itoh, Int. J. Food Microbiol., 1997, 34, 145–156 CrossRef CAS PubMed.
- N. Pandey, R. K. Malik, J. K. Kaushik and G. Singroha, World J. Microbiol. Biotechnol., 2013, 29, 1977–1987 CAS.
- T. Toba, S. K. Samant, E. Yoshioka and T. Itoh, Lett. Appl. Microbiol., 1991, 13, 281–286 CAS.
- M. S. Donia, P. Cimermancic, C. J. Schulze, L. C. Wieland Brown, J. Martin, M. Mitreva, J. Clardy, R. G. Linington and M. A. Fischbach, Cell, 2014, 158, 1402–1414 CrossRef CAS PubMed.
- A. S. Pavlova, G. D. Ozhegov, G. P. Arapidi, I. O. Butenko, E. S. Fomin, N. A. Alemasov, D. A. Afonnikov, D. R. Yarullina, V. T. Ivanov, V. M. Govorun and A. R. Kayumov, Protein J., 2020, 39, 73–84 Search PubMed.
- R. J. Leer, J. M. B. M. van der Vossen, M. van Giezen, M. van Noort Johannes and P. H. Pouwels, Microbiology, 1995, 141, 1629–1635 CAS.
- B. ten Brink, M. Minekus, J. M. B. M. van der Vossen, R. J. Leer and J. H. J. H. in't Veld, J. Appl. Bacteriol., 1994, 77, 140–148 CAS.
- K. Kanatani, M. Oshimura and K. Sano, Appl. Environ. Microbiol., 1995, 61, 1061–1067 CrossRef CAS.
- D. V. Antoshina, S. V. Balandin, I. V. Bogdanov, M. A. Vershinina, E. V. Sheremeteva, I. Y. Toropygin, E. I. Finkina and T. V. Ovchinnikova, Membranes, 2022, 12, 1253 CrossRef CAS.
- E. Garcia-Gutierrez, M. J. Mayer, P. D. Cotter and A. Narbad, Gut Microbes, 2019, 10, 1–21 CrossRef CAS.
- T. Tahara, M. Oshimura, C. Umezawa and K. Kanatani, Appl. Environ. Microbiol., 1996, 62, 892–897 CrossRef CAS.
- R. Dimitrijević, M. Stojanović, I. Zivković, A. Petersen, R. M. Jankov, L. Dimitrijević and M. Gavrović-Jankulović, J. Appl. Microbiol., 2009, 107, 2108–2115 CrossRef.
- K. Kerdkumthong, W. Chanket, P. Runsaeng, S. Nanarong, K. Songsurin, P. Tantimetta, C. Angsuthanasombat, A. Aroonkesorn and S. Obchoei, Probiotics Antimicrob. Proteins, 2024, 16, 713–725 CrossRef CAS.
- R. Callewaert, H. Holo, B. Devreese, J. Van Beeumen, I. Nes and L. De Vuyst, Microbiology, 1999, 145(Pt 9), 2559–2568 CrossRef CAS PubMed.
- L. De Vuyst, L. Avonts, P. Neysens, B. Hoste, M. Vancanneyt, J. Swings and R. Callewaert, Int. J. Food Microbiol., 2004, 90, 93–106 CrossRef CAS PubMed.
- M. R. F. Moreno, B. Baert, S. Denayer, P. Cornelis and L. De Vuyst, FEMS Microbiol. Lett., 2008, 286, 199–206 CrossRef CAS.
- D. L. Turner, L. Brennan, H. E. Meyer, C. Lohaus, C. Siethoff, H. S. Costa, B. Gonzalez, H. Santos and J. E. Suárez, Eur. J. Biochem., 1999, 264, 833–839 CrossRef CAS PubMed.
- B. Gonzalez, E. Glaasker, E. Kunji, A. Driessen, J. E. Suarez and W. N. Konings, Appl. Environ. Microbiol., 1996, 62, 2701–2709 CrossRef CAS.
- I. Wiedemann, T. Böttiger, R. R. Bonelli, T. Schneider, H.-G. Sahl and B. Martínez, Appl. Environ. Microbiol., 2006, 72, 2809–2814 CrossRef CAS.
- F. Meng, Y. Liu, T. Nie, C. Tang, F. Lyu, X. Bie, Y. Lu, M. Zhao and Z. Lu, Appl. Environ. Microbiol., 2022, 88, e003711 Search PubMed.
- E. L. Anderssen, D. B. Diep, I. F. Nes, V. G. H. Eijsink and J. Nissen-Meyer, Appl. Environ. Microbiol., 1998, 64, 2269–2272 CrossRef CAS.
- G. N. Moll, E. van den Akker, H. H. Hauge, J. Nissen-Meyer, I. F. Nes, W. N. Konings and A. J. M. Driessen, J. Bacteriol., 1999, 181, 4848–4852 CrossRef CAS PubMed.
- H. H. Hauge, D. Mantzilas, V. G. H. Eijsink and J. Nissen-Meyer, J. Bacteriol., 1999, 181, 740–747 CrossRef CAS.
- A. Chaalel, A. Riazi, R. Dubois-Dauphin and P. Thonart, Asian Pac. J. Trop. Dis., 2015, 5, 474–482 CrossRef CAS.
- C. Oppegård, M. Kjos, J.-W. Veening, J. Nissen-Meyer and T. Kristensen, MicrobiologyOpen, 2016, 5, 700–708 CrossRef.
- R. Abdulhussain Kareem and S. H. Razavi, J. Food Saf., 2020, 40, e12735 CrossRef.
- H. Zhao, R. Sood, A. Jutila, S. Bose, G. Fimland, J. Nissen-Meyer and P. K. J. Kinnunen, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 1461–1474 CrossRef CAS.
- S. Kaur and S. Kaur, Front. Pharmacol, 2015, 6, 272 Search PubMed.
- S. L. Sand, T. M. Haug, J. Nissen-Meyer and O. Sand, J. Membr. Biol., 2007, 216, 61–71 CrossRef CAS PubMed.
- S. L. Sand, J. Nissen-Meyer, O. Sand and T. M. Haug, Biochim. Biophys. Acta, Biomembr., 2013, 1828, 249–259 CrossRef CAS PubMed.
- R. Martín, S. Escobedo, C. Martín, A. Crespo, L. M. Quiros and J. E. Suarez, Antimicrob. Agents Chemother., 2014, 59, 677–681 CrossRef.
- R. Zvanych, N. Lukenda, J. J. Kim, X. Li, E. O. Petrof, W. I. Khan and N. A. Magarvey, J. Antibiot., 2014, 67, 85–88 CrossRef CAS.
- T. Abee, T. R. Klaenhammer and L. Letellier, Appl. Environ. Microbiol., 1994, 60, 1006–1013 CrossRef CAS PubMed.
- P. M. Muriana and T. R. Klaenhammer, Appl. Environ. Microbiol., 1991, 57, 114–121 CrossRef CAS.
- C. Fremaux, C. Ahn and T. R. Klaenhammer, Appl. Environ. Microbiol., 1993, 59, 3906–3915 CrossRef CAS.
- Y. Héchard and H.-G. Sahl, Biochimie, 2002, 84, 545–557 CrossRef.
- M. C. Joerger and T. R. Klaenhammer, J. Bacteriol., 1986, 167, 439–446 CrossRef CAS PubMed.
- S. Flynn, D. van Sinderen, G. M. Thornton, H. Holo, I. F. Nes and J. K. Collins, Microbiology, 2002, 148, 973–984 CrossRef CAS.
- Y.-J. Cho, J. K. Choi, J.-H. Kim, Y.-S. Lim, J.-S. Ham, D.-K. Kang, J. Chun, H.-D. Paik and G.-B. Kim, J. Bacteriol., 2011, 193, 5021–5022 CrossRef CAS PubMed.
- J. Bien, V. Palagani and P. Bozko, Ther. Adv. Gastroenterol., 2013, 6, 53–68 CrossRef PubMed.
- M. G. Dieterle, K. Rao and V. B. Young, Ann. N. Y. Acad. Sci., 2019, 1435, 110–138 CrossRef.
- P. Guo, K. Zhang, X. Ma and P. He, J. Anim. Sci. Biotechnol., 2020, 11, 24 CrossRef PubMed.
- S. S. Dineen, M. Bradshaw and E. A. Johnson, Appl. Environ. Microbiol., 2000, 66, 5480–5483 CrossRef CAS PubMed.
- F. Kato, S. Ogata and M. Hongo, Agric. Biol. Chem., 1976, 40, 1101–1105 CAS.
- R. Kemperman, A. Kuipers, H. Karsens, A. Nauta, O. Kuipers and J. Kok, Appl. Environ. Microbiol., 2003, 69, 1589–1597 CrossRef CAS.
- F. Liu, A. J. van Heel, J. Chen and O. P. Kuipers, Front. Microbiol., 2022, 13, 1026290, DOI:10.3389/fmicb.2022.1026290.
- O. Ben Braïek and S. Smaoui, BioMed Res. Int., 2019, 2019, 5938210 Search PubMed.
- H. Hanchi, W. Mottawea, K. Sebei and R. Hammami, Front. Microbiol., 2018, 19, 1791, DOI:10.3389/fmicb.2018.01791.
- S. S. Gaur and U. S. Annapure, World J. Microbiol. Biotechnol., 2022, 38, 219 CrossRef CAS PubMed.
- D. Todokoro, H. Tomita, T. Inoue and Y. Ike, Appl. Environ. Microbiol., 2006, 72, 6955–6964 CrossRef CAS.
- T. Inoue, H. Tomita and Y. Ike, Antimicrob. Agents Chemother., 2006, 50, 1202–1212 CrossRef CAS PubMed.
- H. Tomita, S. Fujimoto, K. Tanimoto and Y. Ike, J. Bacteriol., 1996, 178, 3585–3593 CrossRef CAS.
- R. del Campo, C. Tenorio, R. Jiménez-Díaz, C. Rubio, R. Gómez-Lus, F. Baquero and C. Torres, Antimicrob. Agents Chemother., 2001, 45, 905–912 CrossRef CAS PubMed.
- M. S. Said, E. Tirthani and E. Lesho, in StatPearls, StatPearls Publishing, Treasure Island (FL), 2024 Search PubMed.
- A. Gálvez, M. Maqueda, E. Valdivia, A. Quesada and E. Montoya, Can. J. Microbiol., 1986, 32, 765–771 CrossRef.
- A. Gálvez, M. Maqueda, M. Martínez-Bueno and E. Valdivia, Res. Microbiol., 1989, 140, 57–68 CrossRef.
- M. J. Grande Burgos, R. Pérez Pulido, M. d. C. López Aguayo, A. Gálvez and R. Lucas, Int. J. Mol. Sci., 2014, 15, 22706–22727 CrossRef.
- P. S. Coburn and M. S. Gilmore, Cell. Microbiol., 2003, 5, 661–669 CrossRef CAS PubMed.
- W. K. Mousa, B. Athar, N. J. Merwin and N. A. Magarvey, Nat. Prod. Rep., 2017, 34, 1302–1331 RSC.
- P. Sathyanarayana, S. Maurya, A. Behera, M. Ravichandran, S. S. Visweswariah, K. G. Ayappa and R. Roy, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E7323–E7330 CrossRef CAS PubMed.
- A. J. La Reau and G. Suen, J. Microbiol., 2018, 56, 199–208 CrossRef CAS PubMed.
- A. J. La Reau, J. P. Meier-Kolthoff and G. Suen, Microb. Genomics, 2016, 2, e000099 Search PubMed.
- E. H. Crost, E. Coletto, A. Bell and N. Juge, FEMS Microbiol. Rev., 2023, 47, fuad014 CrossRef CAS PubMed.
- C. Balty, A. Guillot, L. Fradale, C. Brewee, M. Boulay, X. Kubiak, A. Benjdia and O. Berteau, J. Biol. Chem., 2019, 294, 14512–14525 CrossRef CAS.
- E. H. Crost, E. H. Ajandouz, C. Villard, P. A. Geraert, A. Puigserver and M. Fons, Biochimie, 2011, 93, 1487–1494 CrossRef CAS.
- S. Chiumento, C. Roblin, S. Kieffer-Jaquinod, S. Tachon, C. Leprètre, C. Basset, D. Aditiyarini, H. Olleik, C. Nicoletti, O. Bornet, O. Iranzo, M. Maresca, R. Hardré, M. Fons, T. Giardina, E. Devillard, F. Guerlesquin, Y. Couté, M. Atta, J. Perrier, M. Lafond and V. Duarte, Sci. Adv., 2019, 5, eaaw9969 CrossRef CAS PubMed.
- L. Shamseddine, C. Roblin, I. Veyrier, C. Basset, L. De Macedo, A. Boyeldieu, M. Maresca, C. Nicoletti, G. Brasseur, S. Kieffer-Jaquinod, É. Courvoisier-Dezord, A. Amouric, P. Carpentier, N. Campo, M. Bergé, P. Polard, J. Perrier, V. Duarte and M. Lafond, iScience, 2023, 26, 107563 CrossRef CAS PubMed.
- F. Marcille, A. Gomez, P. Joubert, M. Ladiré, G. Veau, A. Clara, F. Gavini, A. Willems and M. Fons, Appl. Environ. Microbiol., 2002, 68, 3424–3431 CrossRef CAS PubMed.
- B. A. Schneider and E. P. Balskus, Tetrahedron, 2018, 74, 3215–3230 CrossRef CAS.
- F. Götz, T. Bannerman and K.-H. Schleifer, Prokaryotes, 2006, 5–75 Search PubMed.
- B. O. Torres Salazar, S. Heilbronner, A. Peschel and B. Krismer, Microb. Physiol., 2021, 31, 198–216 CrossRef CAS PubMed.
- R. Coates, J. Moran and M. J. Horsburgh, Future Microbiol., 2014, 9, 75–91 CrossRef CAS.
- C. Laux, A. Peschel and B. Krismer, Microbiol. Spectrum, 2019, 7, 10–1128, DOI:10.1128/microbiolspec.gpp3-0029-2018.
- M. B. Ekkelenkamp, M. Hanssen, S.-T. Danny Hsu, A. de Jong, D. Milatovic, J. Verhoef and N. A. J. van Nuland, FEBS Lett., 2005, 579, 1917–1922 CrossRef CAS PubMed.
- M. Van De Kamp, H. W. Van Den Hooven, R. N. H. Konings, G. Bierbaum, H.-G. Sahl, O. P. Kuipers, R. J. Siezen, W. M. De Vos, C. W. Hilbers and F. J. M. Van De Ven, Eur. J. Biochem., 1995, 230, 587–600 CrossRef CAS PubMed.
- M. Van De Kamp, L. M. Horstink, H. W. Van Den Hooven, R. N. H. Konings, C. W. Hilbers, A. Frey, H.-G. Sahl, J. W. Metzger and F. J. M. Van De Ven, Eur. J. Biochem., 1995, 227, 757–771 CrossRef CAS PubMed.
- J. E. Velásquez, X. Zhang and W. van der Donk, Chem. Biol., 2011, 18, 857–867 CrossRef.
- C. Wu, B. A. Lower, R. Moreira, D. Dorantes, T. Le, C. Giurgiu, Y. Shi and W. A. van der Donk, Front. Microbiol., 2023, 14, 1247222, DOI:10.3389/fmicb.2023.1247222.
- X. Wang, Y. Xu, N. I. Martin and E. Breukink, Biochim. Biophys. Acta, Biomembr., 2024, 1866, 184282 CrossRef CAS PubMed.
- H. Allgaier, G. Jung, R. G. Werner, U. Schneider and H. Zähner, Eur. J. Biochem., 1986, 160, 9–22 CrossRef CAS.
- F. Götz, S. Perconti, P. Popella, R. Werner and M. Schlag, Int. J. Med. Microbiol., 2014, 304, 63–71 CrossRef PubMed.
- H. Brötz, M. Josten, I. Wiedemann, U. Schneider, F. Götz, G. Bierbaum and H.-G. Sahl, Mol. Microbiol., 1998, 30, 317–327 CrossRef.
- R. R. Bonelli, T. Schneider, H.-G. Sahl and I. Wiedemann, Antimicrob. Agents Chemother., 2006, 50, 1449–1457 CrossRef CAS PubMed.
- M. B. C. Fontana, M. d. C. F. de Bastos and A. Brandelli, Curr. Microbiol., 2006, 52, 350–353 CrossRef CAS.
- Y. Suzuki, M. Kawada-Matsuo, M. N.-T. Le, S. Eng, J. Hisatsune, M. Sugai, T. Sakaguchi and H. Komatsuzawa, Appl. Environ. Microbiol., 2024, e00300–e00324 Search PubMed.
- M. K. Al-Malkey, A. H. Ibrahim, M. C. Ismeel, F. O. Sameer, I. A. Beram and I. A. Jasim, Biomed. Pharmacol. J., 2020, 13, 199–206 CrossRef CAS.
- C. Heidrich, U. Pag, M. Josten, J. Metzger, R. W. Jack, G. Bierbaum, G. Jung and H.-G. Sahl, Appl. Environ. Microbiol., 1998, 64, 3140–3146 CrossRef CAS PubMed.
- S. Sandiford and M. Upton, Antimicrob. Agents Chemother., 2012, 56, 1539–1547 CrossRef CAS PubMed.
- S. Halliwell, P. Warn, A. Sattar, J. P. Derrick and M. Upton, J. Antimicrob. Chemother., 2017, 72, 778–781 CAS.
- J.-S. Puls, B. Winnerling, J. J. Power, A. M. Krüger, D. Brajtenbach, M. Johnson, K. Bilici, L. Camus, T. Fließwasser, T. Schneider, H.-G. Sahl, D. Ghosal, U. Kubitscheck, S. Heilbronner and F. Grein, ISME J., 2024, 18, wrae044 CrossRef PubMed.
- K. Nakazono, M. N.-T. Le, M. Kawada-Matsuo, N. Kimheang, J. Hisatsune, Y. Oogai, M. Nakata, N. Nakamura, M. Sugai and H. Komatsuzawa, PLoS One, 2022, 17, e0258283 CrossRef CAS PubMed.
- D. Janek, A. Zipperer, A. Kulik, B. Krismer and A. Peschel, PLoS Pathog., 2016, 12, e1005812 CrossRef.
- A. Zipperer, M. C. Konnerth, C. Laux, A. Berscheid, D. Janek, C. Weidenmaier, M. Burian, N. A. Schilling, C. Slavetinsky, M. Marschal, M. Willmann, H. Kalbacher, B. Schittek, H. Brötz-Oesterhelt, S. Grond, A. Peschel and B. Krismer, Nature, 2016, 535, 511–516 CrossRef CAS.
- K. Bitschar, B. Sauer, J. Focken, H. Dehmer, S. Moos, M. Konnerth, N. A. Schilling, S. Grond, H. Kalbacher, F. C. Kurschus, F. Götz, B. Krismer, A. Peschel and B. Schittek, Nat. Commun., 2019, 10, 2730 CrossRef PubMed.
- N. A. Schilling, A. Berscheid, J. Schumacher, J. S. Saur, M. C. Konnerth, S. N. Wirtz, J. M. Beltrán-Beleña, A. Zipperer, B. Krismer, A. Peschel, H. Kalbacher, H. Brötz-Oesterhelt, C. Steinem and S. Grond, Angew. Chem., Int. Ed., 2019, 58, 9234–9238 CrossRef PubMed.
- S.-C. Chang, C.-Y. Kao, L.-C. Lin, J. H. Hidrosollo and J.-J. Lu, Microbiol. Spectrum, 2023, 11, e01298 Search PubMed.
- L.-C. Lin, C.-Y. Kao, S.-C. Chang, J. H. Hidrosollo and J.-J. Lu, J. Microbiol., Immunol. Infect., 2024, 57, 278–287 CrossRef CAS.
- D. Ruppelt, M. F. W. Trollmann, T. Dema, S. N. Wirtz, H. Flegel, S. Mönnikes, S. Grond, R. A. Böckmann and C. Steinem, Nat. Commun., 2024, 15, 3521 CrossRef CAS.
- J. Reiners, M. Lagedroste, J. Gottstein, E. T. Adeniyi, R. Kalscheuer, G. Poschmann, K. Stühler, S. H. J. Smits and L. Schmitt, Front. Microbiol., 2020, 11, 573614 CrossRef.
- J. N. O’Sullivan, P. M. O’Connor, M. C. Rea, O. O’Sullivan, C. J. Walsh, B. Healy, H. Mathur, D. Field, C. Hill and R. P. Ross, J. Bacteriol., 2020, 202, e00639 CrossRef PubMed.
- P. M. O'Connor, E. F. O'Shea, C. M. Guinane, O. O'Sullivan, P. D. Cotter, R. P. Ross and C. Hill, Appl. Environ. Microbiol., 2015, 81, 3953–3960 CrossRef.
- E. Garcia-Gutierrez, P. M. O'Connor, G. Saalbach, C. J. Walsh, J. W. Hegarty, C. M. Guinane, M. J. Mayer, A. Narbad and P. D. Cotter, Sci. Rep., 2020, 10, 3738 CrossRef CAS.
- A. Aldarhami, A. Felek, V. Sharma and M. Upton, J. Med. Microbiol., 2020, 69, 605–616 CrossRef CAS PubMed.
- D. Hatziioanou, C. Gherghisan-Filip, G. Saalbach, N. Horn, U. Wegmann, S. H. Duncan, H. J. Flint, M. J. Mayer and A. Narbad, Microbiology, 2017, 163, 1292–1305 CrossRef CAS PubMed.
- A. Giacometti, O. Cirioni, F. Barchiesi and G. Scalise, Diagn. Microbiol. Infect. Dis., 2000, 38, 115–118 CrossRef CAS.
- E. Pasquereau-Kotula, M. Martins, L. Aymeric and S. Dramsi, Front. Microbiol., 2018, 9, 614, DOI:10.3389/fmicb.2018.00614.
- A. D. Paiva, M. D. de Oliveira, S. O. de Paula, M. C. Baracat-Pereira, E. Breukink and H. C. Mantovani, Microbiology, 2012, 158, 2851–2858 CrossRef CAS PubMed.
- Y. Khazaei Monfared, M. Mahmoudian, F. Caldera, A. R. Pedrazzo, P. Zakeri-Milani, A. Matencio and F. Trotta, J. Drug Delivery Sci. Technol., 2023, 79, 104065 CrossRef CAS.
- A. Lewies, J. F. Wentzel, H. C. Miller and L. H. Du Plessis, Biochimie, 2018, 144, 28–40 CrossRef CAS PubMed.
- N. E. Joo, K. Ritchie, P. Kamarajan, D. Miao and Y. L. Kapila, Cancer Med., 2012, 1, 295–305 CrossRef CAS PubMed.
- A. Avand, V. Akbari and S. Shafizadegan, Iran. J. Biotechnol., 2018, 16, e1867 Search PubMed.
- S. M. Patil and N. K. Kunda, Pharm. Res., 2022, 39, 2859–2870 CrossRef CAS.
- P. Balcik-Ercin and B. Sever, Chem.-Biol. Interact., 2022, 366, 110152 CrossRef CAS.
- K. Rana, S. Kumar Pandey, S. Chauhan and S. Preet, Int. J. Pharm., 2022, 620, 121744 CrossRef CAS PubMed.
- K. Rana, R. Sharma and S. Preet, Biochem. Biophys. Res. Commun., 2019, 520, 551–559 CrossRef CAS PubMed.
- S. Tavakoli, M. Gholami, K. Ghorban, F. Nojoumi, E. Faghihloo, M. Dadmanesh and N. H. Rouzbahani, Gene Rep., 2021, 23, 101025 CrossRef CAS.
- M. V. Mouritzen, A. Andrea, K. Qvist, S. S. Poulsen and H. Jenssen, Wound Repair Regen., 2019, 27, 650–660 CrossRef PubMed.
- S. Ahmadi, M. Ghollasi and H. M. Hosseini, Microb. Pathog., 2017, 111, 193–197 CrossRef CAS PubMed.
- Y. Liu, Y. Liu, Z. Du, L. Zhang, J. Chen, Z. Shen, Q. Liu, J. Qin, H. Lv, H. Wang, L. He, J. Liu, Q. Huang, Y. Sun, M. Otto and M. Li, Microbiome, 2020, 8, 85 CrossRef CAS.
- S. Akasapu, A. B. Hinds, W. C. Powell and M. A. Walczak, Chem. Sci., 2019, 10, 1971–1975 RSC.
- M. A. Ciufolini and D. Lefranc, Nat. Prod. Rep., 2010, 27, 330–342 RSC.
- W. Hameister, R. Bergmann and H. Wahlig, Arzneimittelforschung, 1975, 25, 1365–1369 CAS.
- R. Zvanych, N. Lukenda, X. Li, J. J. Kim, S. Tharmarajah and N. A. Magarvey, Mol. BioSyst., 2014, 11, 97–104 RSC.
- P. M. Joyner, J. Liu, Z. Zhang, J. Merritt, F. Qi and R. H. Cichewicz, Org. Biomol. Chem., 2010, 8, 5486–5489 RSC.
- E. B. Robertson and J. L. E. Willett, bioRxiv, 2023, preprint, DOI:10.1101/2023.09.12.557362.
- S. Hamada and H. D. Slade, Microbiol. Rev., 1980, 44, 331–384 CrossRef CAS PubMed.
- C. C. Barber and W. Zhang, J. Ind. Microbiol. Biotechnol., 2021, 48, kuab010 CrossRef CAS PubMed.
- R. Zvanych, N. Lukenda, X. Li, J. J. Kim, S. Tharmarajah and N. A. Magarvey, Mol. BioSyst., 2014, 11, 97–104 RSC.
- C. Uranga, K. E. Nelson, A. Edlund and J. L. Baker, Front. Oral Health, 2022, 2, 796140, DOI:10.3389/froh.2021.796140..
- J. D. Hillman, J. Novák, E. Sagura, J. A. Gutierrez, T. A. Brooks, P. J. Crowley, M. Hess, A. Azizi, K.-P. Leung, D. Cvitkovitch and A. S. Bleiweis, Infect. Immun., 1998, 66, 2743–2749 CrossRef CAS PubMed.
- M. Mota-Meira, C. Lacroix, G. LaPointe and M. C. Lavoie, FEBS Lett., 1997, 410, 275–279 CrossRef CAS PubMed.
- R. Pokhrel, N. Bhattarai, P. Baral, B. S. Gerstman, J. H. Park, M. Handfield and P. P. Chapagain, Phys. Chem. Chem. Phys., 2019, 21, 12530–12539 RSC.
- J. A. Kers, R. E. Sharp, A. W. Defusco, J. H. Park, J. Xu, M. E. Pulse, W. J. Weiss and M. Handfield, Front. Microbiol., 2018, 9, 415, DOI:10.3389/fmicb.2018.00415.
- C. P. na Ayudhya, S. Luengpailin and V. Lulitanond, Khon Kaen Dent. J., 2017, 20, 1–10 Search PubMed.
- M. Geng, A. Ravichandran, J. Escano and L. Smith, Antimicrob. Agents Chemother., 2018, 62(12), e01626, DOI:10.1128/aac.01626-18.
- M. Ju, T. Joseph, N. Hansanant, M. Geng, M. Williams, A. Cothrell, A. R. Buhrow, F. Austin and L. Smith, Front. Microbiol., 2022, 13, 1067410, DOI:10.3389/fmicb.2022.1067410.
- S. Chen, S. Wilson-Stanford, W. Cromwell, J. D. Hillman, A. Guerrero, C. A. Allen, J. A. Sorg and L. Smith, Appl. Environ. Microbiol., 2013, 79, 4015–4023 CrossRef CAS.
- J. A. Kers, R. E. Sharp, S. Muley, M. Mayo, J. Colbeck, Y. Zhu, A. W. DeFusco, J. H. Park and M. Handfield, Chem. Biol. Drug Des., 2018, 92, 1940–1953 CrossRef CAS.
- D. Dufour, A. Barbour, Y. Chan, M. Cheng, T. Rahman, M. Thorburn, C. Stewart, Y. Finer, S.-G. Gong and C. M. Lévesque, J. Bacteriol., 2020, 202, e00762 CrossRef CAS PubMed.
- D. J. Birri, D. A. Brede and I. F. Nes, Appl. Environ. Microbiol., 2012, 78, 402–410 CrossRef CAS PubMed.
- K. F. Ross, C. W. Ronson and J. R. Tagg, Appl. Environ. Microbiol., 1993, 59, 2014–2021 CrossRef CAS PubMed.
- A. Barbour, K. Philip and S. Muniandy, PLoS One, 2013, 8, e77751 CrossRef CAS PubMed.
- O. Hyink, P. A. Wescombe, M. Upton, N. Ragland, J. P. Burton and J. R. Tagg, Appl. Environ. Microbiol., 2007, 73, 1107–1113 CrossRef CAS PubMed.
- A. Barbour, J. Tagg, O. K. Abou-Zied and K. Philip, Sci. Rep., 2016, 6, 31749 CrossRef CAS PubMed.
- J. R. Tagg and L. W. Wannamaker, Antimicrob. Agents Chemother., 1978, 14, 31–39 CrossRef CAS.
- P. A. Wescombe, K. H. Dyet, K. P. Dierksen, D. A. Power, R. W. Jack, J. P. Burton, M. A. Inglis, A. L. Wescombe and J. R. Tagg, Int. J. Microbiol., 2012, 2012, 738503 Search PubMed.
- E. Vera Pingitore, E. M. Hébert, M. E. Nader-Macías and F. Sesma, Res. Microbiol., 2009, 160, 401–408 CrossRef PubMed.
- G. W. Lawrence, N. McCarthy, C. J. Walsh, T. M. Kunyoshi, E. M. Lawton, P. M. O'Connor, M. Begley, P. D. Cotter and C. M. Guinane, Gut Microbes, 2022, 14, 2100203 CrossRef PubMed.
- L. H. Mahdi, H. S. Jabbar and I. G. Auda, Int. J. Biol. Macromol., 2019, 134, 1132–1144 CrossRef CAS PubMed.
- H. C. Mantovani, H. Hu, R. W. Worobo and J. B. Russell, Microbiology, 2002, 148, 3347–3352 CrossRef CAS.
- A. Proutière, L. du Merle, M. Garcia-Lopez, C. Léger, A. Voegele, A. Chenal, A. Harrington, Y. Tal-Gan, T. Cokelaer, P. Trieu-Cuot and S. Dramsi, Microbiol. Spectrum, 2023, 11, e05085 CrossRef.
- D. Hill, P. M. O'Connor, E. Altermann, L. Day, C. Hill, C. Stanton and R. P. Ross, Sci. Rep., 2020, 10, 13431 CrossRef CAS.
- P. Malfertheiner, M. C. Camargo, E. El-Omar, J.-M. Liou, R. Peek, C. Schulz, S. I. Smith and S. Suerbaum, Nat. Rev. Dis. Prim., 2023, 9, 1–24 CrossRef.
- I. V. Pinchuk, P. Bressollier, B. Verneuil, B. Fenet, I. B. Sorokulova, F. Mégraud and M. C. Urdaci, Antimicrob. Agents Chemother., 2001, 45, 3156–3161 CrossRef CAS PubMed.
- D.-F. Song, M.-Y. Zhu and Q. Gu, PLoS One, 2014, 9, e105549 CrossRef PubMed.
- G. Pal and S. Srivastava, World J. Microbiol. Biotechnol., 2014, 30, 2829–2837 CrossRef CAS PubMed.
- P. Therdtatha, C. Tandumrongpong, K. Pilasombut, H. Matsusaki, S. Keawsompong and S. Nitisinprasert, SpringerPlus, 2016, 5, 1060 CrossRef.
- M. Sánchez-Hidalgo, M. Montalbán-López, R. Cebrián, E. Valdivia, M. Martínez-Bueno and M. Maqueda, Cell. Mol. Life Sci., 2011, 68, 2845–2857 CrossRef.
- M. Upton, J. R. Tagg, P. Wescombe and H. F. Jenkinson, J. Bacteriol., 2001, 183, 3931–3938 CrossRef CAS PubMed.
- P. A. Wescombe, M. Upton, P. Renault, R. E. Wirawan, D. Power, J. P. Burton, C. N. Chilcott and J. R. Tagg, Microbiology, 2011, 157, 1290–1299 CrossRef CAS.
- S. Saksena, K. Forbes, N. Rajan and D. Giles, Biochem. Biophys. Rep., 2023, 35, 101504 CAS.
- G. Rizzatti, L. R. Lopetuso, G. Gibiino, C. Binda and A. Gasbarrini, BioMed Res. Int., 2017, 2017, e9351507 CrossRef PubMed.
- J. N. V. Martinson and S. T. Walk, EcoSal Plus, 2020, 9, ESP-0003-2020, DOI:10.1128/ecosalplus.ESP-0003-2020.
- C. Zhou, T. M. Bisseling, R. S. van der Post and A. Boleij, Comput. Struct. Biotechnol. J., 2023, 23, 186–198 CrossRef PubMed.
- J. B. Kaper, J. P. Nataro and H. L. Mobley, Nat. Rev. Microbiol., 2004, 2, 123–140 CrossRef CAS PubMed.
- M. Mueller and C. R. Tainter, in StatPearls, StatPearls Publishing, Treasure Island (FL), 2024 Search PubMed.
- S. Gaillard-Gendron, D. Vignon, G. Cottenceau, M. Graber, N. Zorn, A. van Dorsselaer and A.-M. Pons, FEMS Microbiol. Lett., 2000, 193, 95–98 CrossRef CAS.
- N. Morin, I. Lanneluc, N. Connil, M. Cottenceau, A. M. Pons and S. Sablé, Antimicrob. Agents Chemother., 2011, 55, 997–1007 CrossRef CAS.
- S. I. Patzer, M. R. Baquero, D. Bravo, F. Moreno and K. Hantke, Microbiology, 2003, 149, 2557–2570 CrossRef CAS PubMed.
- M. Laviña, C. Gaggero and F. Moreno, J. Bacteriol., 1990, 172, 6585–6588 CrossRef.
- Y. Ma, W. Fu, B. Hong, X. Wang, S. Jiang and J. Wang, Int. J. Mol. Sci., 2023, 24, 11688 CAS.
- J. D. Palmer, E. Piattelli, B. A. McCormick, M. W. Silby, C. J. Brigham and V. Bucci, ACS Infect. Dis., 2018, 4, 39–45 CAS.
- M. Trujillo, E. Rodríguez and M. Laviña, Antimicrob. Agents Chemother., 2001, 45, 3128–3131 CAS.
- E. Rodríguez and M. Laviña, Antimicrob. Agents Chemother., 2003, 47, 181–187 CrossRef.
- J. D. Palmer, B. M. Mortzfeld, E. Piattelli, M. W. Silby, B. A. McCormick and V. Bucci, ACS Infect. Dis., 2020, 6, 672–679 CrossRef CAS PubMed.
- Y. Cherrak, M. A. Salazar, K. Yilmaz, M. Kreuzer and W.-D. Hardt, PLoS Biol., 2024, 22, e3002616 CrossRef CAS PubMed.
- E. Cascales, S. K. Buchanan, D. Duché, C. Kleanthous, R. Lloubès, K. Postle, M. Riley, S. Slatin and D. Cavard, Microbiol. Mol. Biol. Rev., 2007, 71, 158–229 CrossRef CAS PubMed.
- J. K. Parker and B. W. Davies, Microbiology, 2022, 168, 001175 CrossRef CAS.
- R. A. Salomón and R. N. Farías, J. Bacteriol., 1992, 174, 7428–7435 CrossRef PubMed.
- F. Baquero, K. Beis, D. J. Craik, Y. Li, A. J. Link, S. Rebuffat, R. Salomón, K. Severinov, S. Zirah and J. D. Hegemann, Nat. Prod. Rep., 2024, 41, 469–511 RSC.
- C. Beimfohr, Int. J. Bacteriol., 2016, 2016, 3535621 Search PubMed.
- I. A. Khmel, V. M. Bondarenko, I. M. Manokhina, E. I. Basyuk, A. Z. Metlitskaya, V. A. Lipasova and Y. M. Romanova, FEMS Microbiol. Lett., 1993, 111, 269–274 CrossRef CAS PubMed.
- J. I. Guijarro, J. E. González-Pastor, F. Baleux, J. L. S. Millán, M. A. Castilla, M. Rico, F. Moreno and M. Delepierre, J. Biol. Chem., 1995, 270, 23520–23532 CrossRef CAS PubMed.
- F. Yang, F. Yang, J. Huang, H. Yu and S. Qiao, Int. J. Mol. Sci., 2024, 25, 7213 CrossRef CAS PubMed.
- L. Ruiz-Roldán, B. Rojo-Bezares, M. de Toro, M. López, P. Toledano, C. Lozano, G. Chichón, L. Alvarez-Erviti, C. Torres and Y. Sáenz, Sci. Rep., 2020, 10, 11667 CrossRef PubMed.
- M. P. Silverio, G. B. Kraychete, A. S. Rosado and R. R. Bonelli, Antibiotics, 2022, 11, 985 CrossRef PubMed.
- T. de Sousa, M. Hébraud, M. L. N. E. Dapkevicius, L. Maltez, J. E. Pereira, R. Capita, C. Alonso-Calleja, G. Igrejas and P. Poeta, Int. J. Mol. Sci., 2021, 22, 12892 CrossRef CAS PubMed.
- P. V. Liu, J. Infect. Dis., 1974, 130, S94–S99 CrossRef PubMed.
- D. Kang, D. R. Kirienko, P. Webster, A. L. Fisher and N. V. Kirienko, Virulence, 2018, 9, 804–817 CrossRef CAS.
- N. Tabassum, F. Khan, G.-J. Jeong, D.-M. Jo and Y.-M. Kim, Biofilm, 2024, 7, 100192 CrossRef.
- Y. A. Jassim and E. H. S. Aniz, J. Appl. Nat. Sci., 2024, 16, 777–785 Search PubMed.
- Z. Alsarraf, M. Ahmed, Z. H. A, M. F. Mohammad, H. B. Saadon and M. J. Al-Jassani, Rev. Electron. Vet., 2023, 24, 69–76 Search PubMed.
- S. Ito, M. Kageyama and F. Egami, J. Gen. Appl. Microbiol., 1970, 16, 205–214 CrossRef CAS.
- T. Watanabe and H. Saito, Biochim. Biophys. Acta, Gen. Subj., 1980, 633, 77–86 CrossRef CAS PubMed.
- H. Charkhian, E. Soleimannezhadbari, A. Bodaqlouei, L. Lotfollahi, H. Lotfi, N. Yousefi, E. Shojadel and Z. Gholinejad, Microb. Cell Factories, 2024, 23, 175 CrossRef CAS.
- L. Blasco, M. G. de Aledo, C. Ortiz-Cartagena, I. Blériot, O. Pacios, M. López, L. Fernández-García, A. Barrio-Pujante, M. Hernández-Garcia, R. Cantón and M. Tomás, Sci. Rep., 2023, 13, 117 CrossRef CAS.
- B. A. Iwalokun, K. A. Akinsinde, O. Lanlenhin and C. C. Onubogu, Afr. J. Biotechnol., 2006, 5, 1072–1077 CAS.
- R. Onori, S. Gaiarsa, F. Comandatore, S. Pongolini, S. Brisse, A. Colombo, G. Cassani, P. Marone, P. Grossi, G. Minoja, C. Bandi, D. Sassera and A. Toniolo, J. Clin. Microbiol., 2015, 53, 2861–2868 CrossRef CAS.
- T. Le, L. Wang, C. Zeng, L. Fu, Z. Liu and J. Hu, Antimicrob. Resist. Infect. Control, 2021, 10, 41 CrossRef.
- R. Podschun and U. Ullmann, Clin. Microbiol. Rev., 1998, 11, 589–603 CrossRef CAS.
- T. Wu, F. Xu, C. Su, H. Li, N. Lv, Y. Liu, Y. Gao, Y. Lan and J. Li, Front. Immunol., 2020, 11, 1331, DOI:10.3389/fimmu.2020.01331.
- E. M. Nolan, M. A. Fischbach, A. Koglin and C. T. Walsh, J. Am. Chem. Soc., 2007, 129, 14336–14347 CrossRef CAS.
- V. I. Holden, P. Breen, S. Houle, C. M. Dozois and M. A. Bachman, mBio, 2016, 7, e01397 CrossRef CAS.
- N. Strakova, K. Korena and R. Karpiskova, Toxicon, 2021, 197, 126–135 CrossRef CAS PubMed.
- M. Xue, C. S. Kim, A. R. Healy, K. M. Wernke, Z. Wang, M. C. Frischling, E. E. Shine, W. Wang, S. B. Herzon and J. M. Crawford, Science, 2019, 365, eaax2685 CrossRef CAS.
- W. K. Mousa, Front. Pharmacol, 2022, 13, 958012 CrossRef CAS.
- J.-W. Tang, X. Liu, W. Ye, Z.-R. Li and P.-Y. Qian, Nat. Prod. Rep., 2022, 39, 991–1014 RSC.
- K. M. Wernke, M. Xue, A. Tirla, C. S. Kim, J. M. Crawford and S. B. Herzon, Bioorg. Med. Chem. Lett., 2020, 30, 127280 CrossRef CAS PubMed.
- P. C. Williams, K. M. Wernke, A. Tirla and S. B. Herzon, Nat. Prod. Rep., 2020, 37, 1532–1548 RSC.
- E. P. Balskus, Nat. Prod. Rep., 2015, 32, 1534–1540 RSC.
- H. B. Bode, Angew. Chem., Int. Ed., 2015, 54, 10408–10411 CrossRef CAS.
- T. Faïs, J. Delmas, N. Barnich, R. Bonnet and G. Dalmasso, Toxins, 2018, 10, 151 CrossRef.
- E. Addington, S. Sandalli and A. J. Roe, Microbiology, 2024, 170, 001427 CrossRef CAS PubMed.
- D. Ramirez and M. Giron, in StatPearls, StatPearls Publishing, Treasure Island (FL), 2024 Search PubMed.
- S. Boopathi, R. M. S. Kumar, P. S. Priya, B. Haridevamuthu, S. P. R. R. Nayak, L. Chulenbayeva, K. Almagul and J. Arockiaraj, Exp. Gerontol., 2023, 173, 112088 CrossRef.
- H. Büttner, J. Hörl, J. Krabbe and C. Hertweck, ChemBioChem, 2023, 24, e202300322 CrossRef.
- N. R. Dash, G. Khoder, A. M. Nada and M. T. Al Bataineh, PLoS One, 2019, 14, e0218274 CrossRef CAS PubMed.
- S. L. Palframan, T. Kwok and K. Gabriel, Front. Cell. Infect. Microbiol., 2012, 2, 92 CrossRef PubMed.
- C. Iino and T. Shimoyama, World J. Gastroenterol., 2021, 27, 6224 CrossRef CAS PubMed.
- G. H. Fischer, M. F. Hashmi and E. Paterek, in StatPearls, StatPearls Publishing, Treasure Island (FL), 2024 Search PubMed.
- K. Du, M. S. Foote, S. Mousavi, A. Buczkowski, S. Schmidt, E. Peh, S. Kittler, S. Bereswill and M. M. Heimesaat, Front. Microbiol., 2023, 14, 1128500 CrossRef.
- N. O. Kaakoush, N. Castaño-Rodríguez, H. M. Mitchell and S. M. Man, Clin. Microbiol. Rev., 2015, 28, 687–720 CrossRef CAS.
- J. A. Ojeda Rodriguez and C. I. Kahwaji, in StatPearls, StatPearls Publishing, Treasure Island (FL), 2024 Search PubMed.
- D. Vanden Broeck, C. Horvath and M. J. S. De Wolf, Int. J. Biochem. Cell Biol., 2007, 39, 1771–1775 CrossRef CAS PubMed.
- J. Y. Cho, R. Liu, J. C. Macbeth and A. Hsiao, Gut Microbes, 13, 1937015 CrossRef.
- C. Wu, in Encyclopedia of Metagenomics, ed. K. E. Nelson, Springer, New York, NY, 2013, pp. 1–7 Search PubMed.
- C. Binda, L. R. Lopetuso, G. Rizzatti, G. Gibiino, V. Cennamo and A. Gasbarrini, Dig. Liver Dis., 2018, 50, 421–428 CrossRef PubMed.
- E. Dekaboruah, M. V. Suryavanshi, D. Chettri and A. K. Verma, Arch. Microbiol., 2020, 202, 2147–2167 CrossRef CAS PubMed.
- R. Tojo, A. Suárez, M. G. Clemente, C. G. de los Reyes-Gavilán, A. Margolles, M. Gueimonde and P. Ruas-Madiedo, World J. Gastroenterol., 2014, 20, 15163–15176 CrossRef.
- A. O'Callaghan and D. van Sinderen, Front. Microbiol., 2016, 7, 925 Search PubMed.
- M. Derrien, F. Turroni, M. Ventura and D. van Sinderen, Trends Microbiol., 2022, 30, 940–947 CrossRef CAS.
- C. Hidalgo-Cantabrana, S. Delgado, L. Ruiz, P. Ruas-Madiedo, B. Sánchez and A. Margolles, Microbiol. Spectrum, 2017, 5(3), BAD-0010-2016, DOI:10.1128/microbiolspec.bad-0010-2016.
- A. Cheikhyoussef, N. Pogori, H. Chen, F. Tian, W. Chen, J. Tang and H. Zhang, Food Control, 2009, 20, 553–559 CrossRef CAS.
- D. Yu, Z. Pei, Y. Chen, H. Wang, Y. Xiao, H. Zhang, W. Chen and W. Lu, Appl. Environ. Microbiol., 2023, 89(9), e00979 CrossRef PubMed.
- J. Dong, L. Ping, Y. Meng, K. Zhang, H. Tang, D. Liu, B. Li and G. Huo, J. Agric. Food Chem., 2022, 70, 8680–8692 CrossRef CAS PubMed.
- Z. Yildirim and M. G. Johnson, J. Food Prot., 1998, 61, 47–51 CrossRef CAS.
- Z. Yildirim, D. K. Winters and M. G. Johnson, J. Appl. Microbiol., 1999, 86, 45–54 CrossRef CAS PubMed.
- F. A. C. Martinez, E. M. Balciunas, A. Converti, P. D. Cotter and R. P. de Souza Oliveira, Biotechnol. Adv., 2013, 31, 482–488 CrossRef CAS PubMed.
- A. Cheikhyoussef, N. Cheikhyoussef, H. Chen, J. Zhao, J. Tang, H. Zhang and W. Chen, Food Control, 2010, 21, 746–753 CrossRef CAS.
- J. Meghrous, P. Euloge, A. M. Junelles, J. Ballongue and H. Petitdemange, Biotechnol. Lett., 1990, 12, 575–580 CrossRef CAS.
- J. Chu, X. Vila-Farres, D. Inoyama, M. Ternei, L. J. Cohen, E. A. Gordon, B. V. B. Reddy, Z. Charlop-Powers, H. A. Zebroski, R. Gallardo-Macias, M. Jaskowski, S. Satish, S. Park, D. S. Perlin, J. S. Freundlich and S. F. Brady, Nat. Chem. Biol., 2016, 12, 1004–1006 CrossRef CAS PubMed.
- C. J. Carrano, M. Jordan, H. Drechsel, D. G. Schmid and G. Winkelmann, BioMetals, 2001, 14, 119–125 CrossRef CAS.
- J. L. Cloud, H. Neal, R. Rosenberry, C. Y. Turenne, M. Jama, D. R. Hillyard and K. C. Carroll, J. Clin. Microbiol., 2002, 40, 400–406 CrossRef CAS.
- A. M. Block, S. B. Namugenyi, N. P. Palani, A. M. Brokaw, L. Zhang, K. B. Beckman and A. D. Tischler, mSystems, 2023, 8, e0069922 CrossRef PubMed.
- P. Draper, S. N. Payne, G. Dobson and D. E. Minnikin, Microbiology, 1983, 129, 859–863 CAS.
- J. Flores, J. C. Cancino and L. Chavez-Galan, Front. Microbiol., 2021, 12, 638047, DOI:10.3389/fmicb.2021.638047.
- Y. Mikami, H. Komaki, T. Imai, K. Yazawa, A. Nemoto, Y. Tanaka and U. Gräefe, J. Antibiot., 2000, 53, 70–74 CrossRef CAS.
- A. Mukai, T. Fukai, Y. Matsumoto, J. Ishikawa, Y. Hoshino, K. Yazawa, K. Harada and Y. Mikami, J. Antibiot., 2006, 59, 366–369 CrossRef CAS PubMed.
- Y. Tanaka, U. Gräfe, K. Yazawa, Y. Mikami and M. Ritzau, J. Antibiot., 1997, 50, 822–827 CrossRef CAS PubMed.
- Y. Tanaka, H. Komaki, K. Yazawa, Y. Mikami, A. Nemoto, T. Tojyo, K. Kadowaki, H. Shigemori and J. Kobayashi, J. Antibiot., 1997, 50, 1036–1041 CrossRef CAS PubMed.
- Y. Hoshino, A. Mukai, K. Yazawa, J. Uno, J. Ishikawa, A. Ando, T. Fukai and Y. Mikami, J. Antibiot., 2004, 57, 797–802 CrossRef CAS PubMed.
- K. M. Nelson, C. E. Salomon and C. C. Aldrich, J. Nat. Prod., 2012, 75, 1037–1043 CrossRef CAS.
- A. Mukai, T. Fukai, Y. Hoshino, K. Yazawa, K. Harada and Y. Mikami, J. Antibiot., 2009, 62, 613–619 CrossRef CAS PubMed.
- A. Mukai, H. Komaki, M. Takagi and K. Shin-ya, J. Antibiot., 2009, 62, 601–603 CrossRef CAS PubMed.
- E. M. Molloy, J. I. Tietz, P. M. Blair and D. A. Mitchell, Bioorg. Med. Chem., 2016, 24, 6276–6290 CrossRef CAS PubMed.
- M. Tsuda, M. Yamakawa, S. Oka, Y. Tanaka, Y. Hoshino, Y. Mikami, A. Sato, H. Fujiwara, Y. Ohizumi and J. Kobayashi, J. Nat. Prod., 2005, 68, 462–464 CrossRef CAS.
- A. Nemoto, Y. Hoshino, K. Yazawa, A. Ando, Y. Mikami, H. Komaki, Y. Tanaka and U. Gräfe, J. Antibiot., 2002, 55, 593–597 CrossRef CAS.
- M. Bilen, M. Beye, M. D. Mbogning Fonkou, S. Khelaifia, F. Cadoret, N. Armstrong, T. T. Nguyen, J. Delerce, Z. Daoud, D. Raoult and P. Fournier, Microbiologyopen, 2018, 7, e00580 CrossRef PubMed.
- J. McLaughlin, S. Watterson, A. M. Layton, A. J. Bjourson, E. Barnard and A. McDowell, Microorganisms, 2019, 7, 128 CrossRef CAS.
- S. Amini Khiabani, S. Haghighat, H. Tayebi Khosroshahi, M. Asgharzadeh and H. Samadi Kafil, Health Promot. Perspect., 2023, 13, 237–242 CrossRef.
- M. Kollarcikova, M. Faldynova, J. Matiasovicova, E. Jahodarova, T. Kubasova, Z. Seidlerova, V. Babak, P. Videnska, A. Cizek and I. Rychlik, Microorganisms, 2020, 8, 1483 CrossRef CAS.
- A. G. Wexler and A. L. Goodman, Nat. Microbiol., 2017, 2, 1–11 Search PubMed.
- J. C. Andrade, D. Almeida, M. Domingos, C. L. Seabra, D. Machado, A. C. Freitas and A. M. Gomes, Front. Bioeng. Biotechnol., 2020, 8, 550, DOI:10.3389/fbioe.2020.00550.
- S. Stojanov, A. Berlec and B. Štrukelj, Microorganisms, 2020, 8, 1715 CrossRef CAS.
- T. Gautier, N. Oliviero, S. Ferron, P. Le Pogam, S. David-Le Gall, A. Sauvager, P. Leroyer, I. Cannie, S. Dion, A. Sweidan, O. Loréal, S. Tomasi and L. Bousarghin, Front. Microbiol., 2022, 13, 1023315 Search PubMed.
- W. R. Russell, S. H. Duncan, L. Scobbie, G. Duncan, L. Cantlay, A. G. Calder, S. E. Anderson and H. J. Flint, Mol. Nutr. Food Res., 2013, 57, 523–535 CrossRef CAS.
- L. J. Cohen, H.-S. Kang, J. Chu, Y.-H. Huang, E. A. Gordon, B. V. B. Reddy, M. A. Ternei, J. W. Craig and S. F. Brady, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E4825–E4834 CAS.
- R. Villano, F. Tinto and V. Di Marzo, Front. Chem., 2022, 10, 858854 CrossRef CAS.
- M. J. Coyne, N. Béchon, L. M. Matano, V. L. McEneany, M. Chatzidaki-Livanis and L. E. Comstock, Nat. Commun., 2019, 10, 3460 CrossRef PubMed.
- Y. W. Han, Curr. Opin. Microbiol., 2015, 23, 141–147 CrossRef CAS PubMed.
- W. E. Moore and L. V. Moore, Periodontol, 2000, 1994(5), 66–77 Search PubMed.
- C. A. Field, M. D. Gidley, P. M. Preshaw and N. Jakubovics, J. Periodontal. Res., 2012, 47, 470–478 Search PubMed.
- R. Garcia-Carretero, M. Lopez-Lomba, B. Carrasco-Fernandez and M. T. Duran-Valle, J. Crit. Care Med., 2017, 3, 141–147 Search PubMed.
- K. Afra, K. Laupland, J. Leal, T. Lloyd and D. Gregson, BMC Infect. Dis., 2013, 13, 264 CrossRef PubMed.
- C. Ogawa, Y.-J. Liu and K. S. Kobayashi, Curr. Bioact. Compd., 2011, 7, 180–197 CrossRef CAS PubMed.
- A. Dahlstrand Rudin, A. Khamzeh, V. Venkatakrishnan, A. Basic, K. Christenson and J. Bylund, Cell. Microbiol., 2021, 23, e13348 CrossRef CAS PubMed.
- E. R. K. Eribe and I. Olsen, J. Oral Microbiol., 2017, 9, 1368848 CrossRef PubMed.
- B. Wang, M. Yao, L. Lv, Z. Ling and L. Li, Engineering, 2017, 3, 71–82 CrossRef.
- L. K. Ursell, J. L. Metcalf, L. W. Parfrey and R. Knight, Nutr. Rev., 2012, 70, S38–S44 CrossRef PubMed.
- M. D. T. Torres, E. F. Brooks, A. Cesaro, H. Sberro, M. O. Gill, C. Nicolaou, A. S. Bhatt and C. de la Fuente-Nunez, Cell, 2024, 187, 5453–5467 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |